
Isolation of extracellular vesicles with multivalent aptamers
Fei
Xue†
a,
Yundi
Chen†
a,
Yi
Wen
a,
Komal
Abhange
a,
Wenlong
Zhang
b,
Gong
Cheng
c,
Zachary
Quinn
a,
Wenjun
Mao
*d and
Yuan
Wan
 *a
*a
aThe Pq Laboratory of Micro/Nano BiomeDx, Department of Biomedical Engineering, Binghamton University-SUNY, Binghamton, NY 13902, USA. E-mail: ywan@binghamton.edu; Fax: +1 607-777-5780; Tel: +1 607-777-5477
bDepartment of Biomedical Engineering, Micro and Nano Integrated Biosystem (MINIBio) Laboratory, Carnegie Mellon University, Pittsburgh, PA 15213, USA
cDepartment of Earth and Planetary Sciences and Origins of Life Initiative, Harvard University, Cambridge, MA 02138, USA
dDepartment of Cardiothoracic Surgery, Wuxi People's Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu 214023, China. E-mail: maowenjun1@njmu.edu.cn; Tel: +86 510 82700775
First published on 7th October 2020
Abstract
Extracellular vesicles (EVs) are lipid-enclosed submicron-sized vesicles that are secreted by all eukaryotic cells. EVs can selectively encapsulate tissue-specific small molecules from parent cells and efficiently deliver them to recipient cells. As signal mediators of intercellular communication, the molecules packaged in EVs play critical roles in the pathophysiology of diseases. In relevant clinical translation, EV contents have been used for cancer diagnosis and treatment monitoring. To further promote EV-based cancer liquid biopsy toward large-scale clinical implementation, the efficient and specific isolation of pure tumor-derived EVs from body fluids is a prerequisite. However, the existing EV isolation methods are unable to address certain technical challenges, such as lengthy procedures, low throughput, low specificity, heavy protein contamination, etc., and thus, new approaches for EV isolation are required. Here, we report a multivalent, long single-stranded aptamer with repeated units for EV enrichment and retrieval. After short incubation of biotin-labeled multivalent aptamers (MAs) with the samples, EVs can be quickly secured by MAs, anchored onto streptavidin-coated microspheres, and further retrieved via digestion of the DNA aptamer. Approximately 45% of EVs can be isolated from the spiked samples in 40 min with a depletion of 84.7% of albumin contamination. In addition, 93.1% of the isolated EVs can be retrieved via DNase-mediated aptamer degradation in 10 min for downstream molecular analyses. Our findings suggest that MAs can efficiently and specifically isolate EVs derived from malignant lymphocytes, and this simple method could facilitate the EV-centered study of acute lymphoblastic leukemia.
Introduction
Extracellular vesicles (EVs) are submicron-sized, lipid bilayer-enclosed vesicles released by all eukaryotic cells which contain a variety of the parental cell's nucleic acids and proteins.1 Once released, EVs can travel to tissues or organs both near and far via a variety of bodily fluids such as blood, lymph, cerebrospinal fluid, and urine. There, they interact with surrounding cells through surface adhesion proteins, delivering their contents to recipient cells through membrane fusion or endocytosis.2–5 As novel mediators of cell–cell communication, EVs play important roles in various physiological and pathological processes.6 In cancer, growing evidence demonstrates that EVs are involved in immunosuppression, extracellular matrix remodeling, tumor growth, metastasis, chemoresistance, etc.7–9 Therefore, the isolation of tumor-derived EVs and comprehensive analyses of the encapsulated molecules can improve our understanding of tumor biology. Simultaneously, tumor-derived EVs hold great potential for clinical cancer molecular diagnosis, treatment monitoring, and prognosis.10–13 Nevertheless, the efficient isolation of pure tumor-derived EVs for liquid biopsy is a prerequisite. The challenges faced here are two-tiered. First, the low abundance of tumor-derived EVs compared to EVs originating from unrelated cells adversely impacts the enrichment efficiency of the targeted EVs.14 Fortunately, this scarcity can be mitigated by increasing the amount of specimen initially obtained. On the other hand, the isolation specificity of the existing approaches is, at best, modest. Currently, size, density, surface charge, membrane proteins, and lipid membranes have been explored for the isolation of EVs. However, there is a significant overlap between tumor-derived and somatic cell-derived EVs,15–19 which invariably decreases the isolation specificity. Additional challenges include low sample throughput and heavy protein contamination which further hinder the prospect of clinical translation.20,21 Among these approaches, bioaffinity-mediated EV isolation may achieve relatively high isolation specificity;22 however, it relies on the premise that EV membrane molecules must be tumor-specific. Although typical EV protein markers such CD9, CD63, CD81, and HSP70 have been commercialized for EV isolation, these commonly used kits fail to specifically isolate tumor-derived EVs.23–26 Therefore, bioaffinity-based EV isolation targeting unique EV membrane molecules requires further improvement. In order to increase the capture specificity for tumor-related EVs, surface antigens, such as EpCAM, EGFR, HER2, PSA, Tim4, and Annexin V, have been designed as targets for immunoisolation.27–29Protein tyrosine kinase-7 (PTK7) is a biomarker that is overexpressed in acute lymphoblastic leukemia, lung cancer, and colorectal cancer, but is otherwise poorly expressed in normal cells.30–32 Moreover, PTK7-based isolation of cancer cells from peripheral blood specimens has been demonstrated in previous studies.33–35 Hence, we speculate that PTK7 can be used for the isolation of cancer cell-derived EVs. In this study, we use rolling circle amplification (RCA) to prepare biotin-labeled, long single-stranded DNA aptamers containing repeated units that can specifically recognize and bind to PTK7.36 The pile of thousands of aptamer units in a long single DNA chain forms a 3D aptamer network in a suspension, and thus it is expected to increase the binding efficiency of EVs bearing PTK7 (Fig. 1). Subsequently, the captured EVs on the multivalent aptamer (MA) are introduced to streptavidin-coated microspheres (SMs), allowing quick precipitation of beads for EV collection. Furthermore, nuclease treatment can be used to release the isolated EVs via digestion of the DNA aptamers for downstream analyses of the EV contents. Our results demonstrate that MAs can specifically isolate ∼45% of EVs from the spiked samples in minutes with a depletion of 84.7% of albumin. This isolation efficiency is similar to what can be achieved by the much slower and bulkier method of ultracentrifugation.37 Our approach is also faster and simpler than the majority of EV isolation approaches. In addition, MAs can be abundantly prepared by PCR, and thus the cost is much lower than that of commercial antibody-based EV isolation kits. Altogether, MAs could be translated into clinical use for the isolation of cancer-derived EVs from complex body fluids for clinical diagnostics and cancer research.
 | ||
| Fig. 1 Schematic of the multivalent aptamers prepared by RCA for EV enrichment and downstream analysis. Biotin-modified multivalent aptamers with ∼800 repeated aptamer units are prepared by RCA. These anti-PTK7 multivalent aptamers can form a 3D aptamer network in a suspension for the capture of PTK7-overexpressing EVs derived from tumor cells followed by enrichment with streptavidin-coated microspheres. Subsequently, the isolated EVs can be retrieved by degradation of the aptamers with DNA nucleases. Alternatively, the EV contents can be extracted by direct lysis of the isolated EVs on microspheres and then analyzed by different methods, such as PCR, Sanger sequencing, and western blot. This approach could be translated into clinical use for EV-based clinical diagnostics. | ||
Methods
Cell culture
Acute lymphoblastic leukemia cell line CCRF-CEM cells and MCF-7 cells (ATCC) passed Mycoplasma testing, confirming no contamination during the present experiments. The CCRF-CEM cells were cultured in RPMI-1640 supplemented with 5% FBS and 100 IU ml−1 penicillin–streptomycin at 37 °C with 5% CO2 and 95% humidity. The MCF-7 cells were cultured in EMEM supplemented with 5% FBS and 0.01 mg ml−1 human recombinant insulin. Before EV collection, the cells were maintained in FBS-free medium for at least 24 h before EV harvest.EV harvest and characterization
In total, 200 ml of the cell culture supernatant were collected followed by centrifugation at 500g at 4 °C for 5 min to remove the intact cells, at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C for 15 min to discard cellular debris, and at 180000g at 4 °C for 2 hours to harvest the EVs. The EV pellets were resuspended in 30 μL of PBS and cryopreserved at −80 °C. The EV concentration and size distribution were determined with a Nanosight NS300 system. Before the measurement, we introduced PBS into the chamber using a 1 ml syringe and checked that it was free from particles, i.e., no light scattering. Five μL of EV samples were placed on 300 mesh grids and incubated for 3 minutes at room temperature (RT). Excess samples were blotted with filter paper and stained with 1% uranyl acetate for 5 minutes. The samples were then examined under a TEM (Hitachi). Western blotting was also routinely performed. After lysis with RIPA buffer, the EV protein quantities were determined using a BCA assay. The Mini-PROTEAN Tetra Handcast System (Bio-Rad) and Trans-Blot Turbo Transfer System (Bio-Rad) were used for electrophoresis and subsequent transferring. The protein blot was blocked for 1 hour with 5% nonfat dry milk in PBS/0.05% Tween 20 and incubated for 6 hours at 4 °C with Santa Cruz Biotechnology HRP conjugated antibodies against TSG-101 (sc-7964, 1:500), CD81 (sc-166029, 1:500), PTK7 (sc-100304, 1:500) and GAPDH (sc-47724, 1:1000). Samples were washed with PBS/0.05% Tween 20 for 10 minutes thrice. The blots were developed with chemiluminescence (Bio-Rad).
000g at 4 °C for 15 min to discard cellular debris, and at 180000g at 4 °C for 2 hours to harvest the EVs. The EV pellets were resuspended in 30 μL of PBS and cryopreserved at −80 °C. The EV concentration and size distribution were determined with a Nanosight NS300 system. Before the measurement, we introduced PBS into the chamber using a 1 ml syringe and checked that it was free from particles, i.e., no light scattering. Five μL of EV samples were placed on 300 mesh grids and incubated for 3 minutes at room temperature (RT). Excess samples were blotted with filter paper and stained with 1% uranyl acetate for 5 minutes. The samples were then examined under a TEM (Hitachi). Western blotting was also routinely performed. After lysis with RIPA buffer, the EV protein quantities were determined using a BCA assay. The Mini-PROTEAN Tetra Handcast System (Bio-Rad) and Trans-Blot Turbo Transfer System (Bio-Rad) were used for electrophoresis and subsequent transferring. The protein blot was blocked for 1 hour with 5% nonfat dry milk in PBS/0.05% Tween 20 and incubated for 6 hours at 4 °C with Santa Cruz Biotechnology HRP conjugated antibodies against TSG-101 (sc-7964, 1:500), CD81 (sc-166029, 1:500), PTK7 (sc-100304, 1:500) and GAPDH (sc-47724, 1:1000). Samples were washed with PBS/0.05% Tween 20 for 10 minutes thrice. The blots were developed with chemiluminescence (Bio-Rad).
Preparation and characterization of MAs
All DNA (Table 1) was obtained from Sangon (Shanghai, China). Following the well-established protocol, RCA products were prepared in solution as usual. In brief, 10 μL each of 2 μM circle and 2 μM ligation templates were mixed and denatured at 95 °C for 5 min followed by incubation at 37 °C for 30 min to allow for hybridization. Subsequently, 10 μl of the mixed solution was mixed with 10 μl of the 5× T4 ligation buffer, 1 μl of 5 U μl−1 T4 ligase (Thermo Fisher, EL0011), and 29 μl of DI water. The ligation mixture was incubated at 37 °C for 30 min followed by 65 °C for 10 min to deactivate the ligase. Finally, 40 μl of the prepared ligation solution, 10 μl of 10× Phi29 buffer (Thermo Fisher EP0092), 10 μl of 10 mM dNTPs (Thermo Fisher, R0242), 2 μl of 10 U μl−1 Phi29 polymerase (Thermo Fisher EP0092) and 38 μl DI water were mixed for RCA. The mixture was incubated at 37 °C for 10 min, 30 min, 1 h, and 2 h, followed by denaturation of Phi29 polymerase at 65 °C for 10 min. The RCA products were further characterized using 0.5% agarose gel. To confirm that the biotin-labeled RCA products can be conjugated onto SMs, 50 μl of biotin-labeled MAs were hybridized with 10 μl of 100 pM AF488-modified polyA probes at RT for 2 h followed by incubation with 10 μl of SMs (Spherotech SVP-10-5, 1.0% w/v) at RT for 30 min. The microspheres were rinsed with PBS and centrifuged at 3000g for 1 min thrice to thoroughly remove surplus fluorescent probes. The fluorescence of the microspheres was determined by fluorescence microscopy (Nikon Eclipse Ti2).| CCRF-CEM ligation template | 3′-CTG CGC CGC CGG GAA AAT ACT G-5′ |
| CCRF-CEM unit aptamer | 3′-Biotin-ATC TAA CTG CTG CGC CGC CGG GAA AAT ACT GTA CGG TTA GA |
| CCRF-CEM circular template | 3′-CGG CGG CGC AGC AGT TAG ATA AAA AAA AAA AAA AAA AAA TCT AAC CGT ACA GTA TTT TCC-phosphorylation-5′ |
| CCRF-CEM RCA primer | 3′-CTG CGC CGC CGG GAA AAT ACT G-biotin-5′ |
| CCRF-CEM RCA probe | 3′-AF488-AAA AAA AAA AAA AAA-5′ |
| Mock aptamer | 3′-TAC CCC TTT AAT CCC AAA CCC-biotin-5′ |
| KRAS F-primer | 5′-AAG GCC TGC TGA AAA TGA CTG-3′ |
| KRAS R-primer | 5′-TCA CAA TAC CAA GAA ACC CAT-3′ |
EV isolation and retrieval
The amount of MAs, the amount of SMs, and the incubation time of MAs with EVs were all optimized. The default operation parameters are defined as follows: 100 μL of the EV model sample was mixed with 100 μL of MAs for 30 min at RT; the mixture was further incubated with 20 μL of SMs at RT for 10 min. Assuming no interdependency among the parameters, the following optimization was performed. Accordingly, 100 μl of the CCRF-CEM cell-derived EV model samples were incubated with 10, 50, 100, or 200 μl of MAs at RT for 30 min, 1 h, 2 h, and 4 h. Next, 10, 20, and 40 μl of SMs were added and incubated at RT for 10 min followed by RNA extraction from the isolated EVs. In the negative control group, 100 μl of the CCRF-CEM cell-derived EV model samples were incubated with an equal amount of mock aptamers. In the positive control group, 100 μl of the CCRF-CEM cell-derived EV model samples were directly used without processing. The concentration of the extracted RNA derived from the isolated EVs was measured using a Nanodrop 1000 system (Thermo Fisher). The isolation efficiency of the EVs was determined by the mass fraction of the RNA extracted from the captured EVs over that of the total EVs. Then, under the optimized conditions, the EV isolation efficiency from the spiked samples was measured. 100 μl of CCRF-CEM EVs or MCF-7 EVs were incubated with 100 μl of MAs and SMs, followed by the extraction of the RNA cargo. In the control group, MCF-7 EVs were incubated with an equal amount of mock aptamers. The isolation efficiency was calculated as previously described. Meanwhile, the respective isolation efficiency of EVs with MAs and an equal amount of monovalent aptamers was measured. An equal amount of the biotin-modified anti-PTK7 monoclonal antibody (Miltenyl Biotec, 130-100-176) was used as a positive control. Similarly, 100 μL of the EV model sample were incubated with 20 pmol of antibodies (∼3 μg). After thorough rinsing, the isolation efficiency of the EVs using antibodies was measured. Furthermore, the EV retrieval efficiency and depletion of protein contaminants were determined. After EV enrichment via MAs and SMs, DNase (Sigma, DN25) was used to digest the MAs according to the manufacturer's instructions to retrieve the EVs. The SMs with or without the isolated EVs were visualized by SEM. The morphology and size distribution of the retrieved CCRF-CEM EVs were further characterized with a SEM, TEM, and Nanosight NS300 system following the aforementioned protocols. The retrieved EVs in the suspension were directly lysed and RNA was extracted using the miRNeasy Mini Kit (Qiagen) following the manufacturer's instruction. In the control group, DNase was not used. The retrieval efficiency was calculated from the RNA amounts extracted from the supernatant containing the released EVs divided by the total amount of RNA from the isolated EVs. In addition, the amount of protein contaminants in the isolated EVs was determined with a BCA assay according to the manufacturer's instructions. Approximately 3.5 × 1010 CCRF-CEM EVs were spiked into 100 μl of 100 mg ml−1 BSA forming plasma-mimicking samples. CCRF-CEM EVs were isolated with the optimized parameters followed by protein measurement. In the control group, 100 mg ml−1 BSA without the spiked CCRF-CEM EVs was used. To ensure that the measured concentration of the samples is well covered by the linear range of a BCA assay, unprocessed samples and the supernatant of processed samples were diluted 10×. Subsequently, 25 μL of the diluted sample was incubated with 200 μL of the reagent at 37 °C for 30 min followed by absorbance reading. In the control group, 25 μL of DI water (Sigma, 38796) was used. The reported data represented the measured value. An equal amount of the protein cargo derived from the isolated CCRF-CEM EVs and MCF-7 EVs was analyzed with western blot following the above-mentioned protocol. The DNA was extracted from the isolated CCRF-CEM EVs using the QIAamp DNA Micro Kit (Qiagen, Germany) according to the manufacturer's instructions. The KRAS fragment (466 bp) was amplified with primers (Table 1) followed by Sanger sequencing. The detailed protocol can be found elsewhere.37 CCRF-CEM EVs harbor inherent KRASG12D mutations, while MCF-7 EVs have wild-type KRAS.38 To evaluate the reliability of EV isolation using MAs and SMs, we mixed CCRF-CEM EVs and MCF EVs in ratios of 100%, 10%, 1% and 0.1%. A total of 5 × 1010 mixed EVs were spiked in 100 μL BSA forming model samples with a gradient mutation allele frequency of KRASG12D. Subsequently, each sample was processed with MAs and SMs following the optimized protocol. After isolation, the EV DNA was extracted, followed by the detection of KRAS mutations with a commercial mutation assay (Thermo Fisher, 4465804). In the control groups, an equal volume of DI water was used as the control. The curves were plotted using GraphPad Prism 8.Results
Characterization of MAs and EVs
RCA was performed for 10 min, 30 min, 1 h and 2 h followed by characterization with 0.5% agarose gel (Fig. 2a). The respective signal intensity in each group was 6592, 10190, 20333, and 37341. The signal intensity continuously doubled from 30 min to 2 h, reflecting that the total amount of RCA products is proportional to the reaction time. Of note, the signal intensities in each group only reached 10%, 16%, 31% and 57% of the saturation signal, respectively, further indicating that RCA did not fully enter the PCR plateau phase even after 2 h of reaction. Due to the relatively short reaction time, a smeared band showing small DNA fragments can be observed in the first three channels. By contrast, in the 2 h channel, the overwhelming majority of extremely long RCA products were unable to easily maneuver through the gel pores and thus tended to stay within the sample loading well. Given that 0.5% agarose gel can efficiently separate 2000–50000 bp DNA fragments, the length of most RCA products in the 2 h channel would be larger than 50000 bp. Given that the length of the circular template is 60 bp, a single MA contains at least 800 repeated aptamer units. Moreover, 100 μl of the RCA product contains 20 pmol of MA; therefore, approximately 1 × 1016 aptamer units are available for PTK7 binding. The biotin-labeled MAs can be fluorescently labeled with a short dye-modified polyA that is complementary to the polyT spacer. Once the complex was conjugated onto SMs via the biotin–streptavidin interaction, the non-fluorescent polystyrene microspheres displayed green fluorescence (Fig. 2b and c). The EV size measured using a Nanosight system ranged from 30 nm to ∼300 nm.
 | ||
| Fig. 2 Characterization and measurement of MAs and SMs. (a) 0.5% agarose gel electrophoresis characterization of the MAs prepared with various amplification times and relevant quantitative data. The size of the MAs is typically thousands of nucleotides, which correlates to the amplification time. (b) The hybridized fluorescent probe/MA complex grafted onto the SMs (left: fluorescence; middle: phase; right: merged). The scale bar is 2 μm. (c) The SMs do not possess self-fluorescence, and only show green fluorescence after the immobilization of the probe/MA complex (left, fluorescent image of the microspheres with the hybridized fluorescent probe/MA complex under a 20× objective lens; middle: microspheres with and without the hybridized fluorescent probe/MA complex; right: fluorescent image of the microspheres without the hybridized fluorescent probe/MA complex under a 20× objective lens). | ||
The average and mode sizes of the CCRF-CEM cell-derived EVs were 114.1 ± 2.5 and 87.5 ± 4.0 nm, respectively (Fig. 3a). The EVs from the CCRF-CEM cells were isolated by ultracentrifugation and identified by TEM (Fig. 3a inset). The EVs exhibited a typical saucer-shaped morphology. The 100 μl EV sample re-suspended in PBS as a model sample contained ∼3.5 × 1010 EVs. The housekeeping protein GAPDH, characteristic EV markers CD81 and TSG101, and PTK7 extracted from EV protein lysates and CCRF-CEM cell lysates were routinely identified by western blot (Fig. 3b). The four proteins derived from the MCF-7 cell and EV lysates were also measured. In comparison, the PTK7 expression in either MCF-7 cell lysates or EV lysates was very low (Fig. 3c).
 | ||
| Fig. 3 Characterization of EVs derived from CCRF-CEM cells and MCF-7 cells. (a) Size distribution of the EVs measured using a Nanosight NS300 system (inset: TEM image of the CCRF-CEM cell-derived EV). The scale bar is 200 nm. (b) Western blot analysis of CCRF-CEM EV surface marker proteins (CD81 and TSG101), targeted protein PKT7, and housekeeping protein GAPDH extracted from EV lysates and cellular lysates. (c) Western blot analysis of CD81, TSG101, PKT7, and GAPDH from MCF-7 EV lysates and cell lysates. | ||
EV isolation and retrieval
To determine the isolation efficiency of EVs, 100 μL of EVs in PBS containing ∼3.5 × 1010 CCRF-CEM EVs were used as model samples. Meanwhile, 100 μl of MAs (equivalent to 20 pmol of MAs) contained ∼1 × 1016 aptamer units. It was also determined that 20 μL of the SM suspension contained ∼2 × 108 microspheres (equivalent to 0.2 mg). In addition, according to the manufacturer's manual, 0.2 mg of SMs can bind ∼140 pmol of biotin. Accordingly, the amounts of MAs and SMs were theoretically sufficient for the isolation of 3.5 × 1010 EVs. We first evaluated the effect of the amount of MAs, ranging from 10, 50, 100, to 200 μl, on the EV isolation efficiency. EVs were incubated with 10–200 μl of MAs for 30 min at RT. The mixture was further incubated with 20 μL of SMs at RT for 10 min followed by brief centrifugation and RNA extraction. The isolation efficiency increased gradually with increasing amounts of MAs. The efficiency was 14.5 ± 2.3%, 31.9 ± 1.8%, 43.1 ± 4.5%, and 47.7 ± 3.6%, respectively (Fig. 4a). No significant difference was found between the last two groups (ANOVA, n = 5, p > 0.05). An additional amount of MAs did not increase the isolation efficiency. Hence, 100 μl of MAs was used for further optimization. In the control group, 100 μL of CCRF-CEM EVs were directly incubated with ∼20 pmol of mock aptamers followed by incubation with SMs and RNA extraction. In the control group, RNA concentration was undetectable with a Nanodrop system, indicating that mock aptamers cannot bind to CCRF-CEM EVs. Similarly, in the following optimization and comparison, we were unable to determine the RNA concentration or further calculate the RNA mass in the control groups as the isolated EVs were very scarce. Next, we determined the effect of incubation time, ranging from 0.5 h to 4 h on EV isolation with 100 μl of MAs at RT. Later, the mixture was further incubated with 20 μL of SMs at RT for 10 min followed by RNA extraction. The EV isolation efficiencies at 30 min, 1 h, 2 h, and 4 h time points were 44.8 ± 2.2%, 46.0 ± 5.3%, 46.4 ± 1.6%, and 48.3 ± 4.1%, respectively (Fig. 4b). Although there was a gentle increase in the average isolation efficiency with incubation time, prolonged incubation failed to yield any statistically significant benefit (ANOVA, n = 5, p > 0.05). As a result, 30 minutes was used for incubation of EVs with MAs. Furthermore, we investigated the effect of the amount of SMs, ranging from 10, 20, to 40 μl, on the EV isolation efficiency. EVs were incubated with 10–200 μl of MAs for 30 min at RT followed by incubation with various amounts of SMs at RT for 10 min. The isolation efficiency was 36.2 ± 4.2%, 44.8 ± 2.7%, and 46.2 ± 3.6%, respectively (Fig. 4c). A significant difference was found between the first group and the other two groups (ANOVA, n = 5, p < 0.05). However, no significant difference was found between groups 2 and 3 (ANOVA, n = 5, p > 0.05). Therefore, 20 μl of SMs was selected. The isolation efficiency of MCF-7 EVs was only 2.4 ± 1.8%. Compared to that of CCRF-CEM EVs, the isolation efficiency of MCF-7 EVs was much lower (t-test, p < 0.05), which was also in line with the PTK-7 expression level in the two cell lines. Of note, we did not optimize the incubation time of biotin with streptavidin, since the mutual interaction is very efficient. Our previous study demonstrated that the interaction reaches equilibrium within 10 minutes.37 Moreover, based on the EV RNA mass, we determined that the average isolation efficiency of EVs with prevalent ultracentrifugation is only ∼17%, which is in line with the reported values.39,40 Altogether, approximately 45% of EVs from the model sample can be isolated using 100 μL of MAs (equivalent to 20 pmol) and 20 μL of SMs (equivalent to 2 × 108 SMs), with the whole isolation procedure taking about 40 minutes.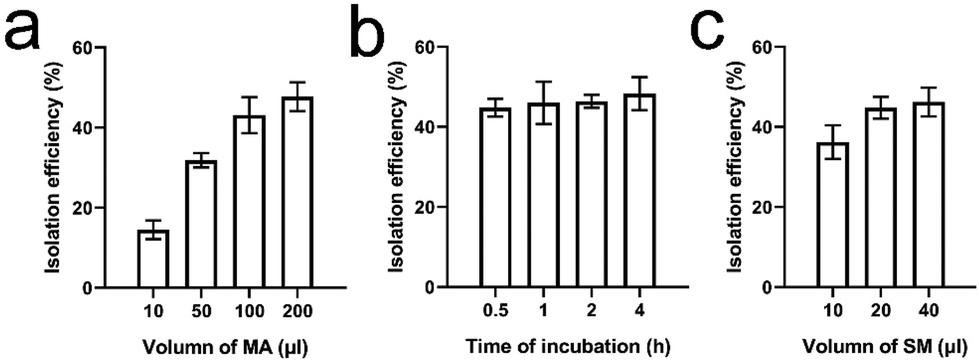 | ||
| Fig. 4 Optimization for the isolation efficiency. (a) The isolation efficiency of EVs from model samples as a function of the MA amount (n = 5). (b) The isolation efficiency of EVs from model samples as a function of the incubation time of the EVs with MAs (n = 5). (c) The isolation efficiency of EVs from model samples as a function of the SM amount (n = 5). | ||
Subsequently, we measured the isolation efficiency of EVs from plasma-mimicking samples. Given that the concentration of albumin in plasma is ∼5% (w/v), we mixed 100 μL of EVs with 100 μL of 10% BSA. Under the optimized conditions, the isolation efficiency of EVs from the spiked samples was 45.4 ± 4.7%, indicating that MAs can specifically recognize and bind to PTK7 on the EV membrane regardless of BSA and further enrich EVs onto the SMs. By contrast, 20 pmol of biotin-labeled monovalent aptamers only isolated 28.1 ± 2.1% of EVs from the spiked samples, demonstrating that MAs are superior to monovalent aptamers in EV isolation. In the positive control group, the anti-PTK7 monoclonal antibody isolated 30.9 ± 4.5% of EVs. The Kd of the anti-PTK7 aptamer is 0.8 nM.33 The binding affinity between the aptamer and PTK7 is sufficient for EV isolation, although it is still approximately 1–2 orders of magnitude lower than that of the monoclonal antibody. The isolation efficiency of EVs using antibodies slightly increased. However, no significant difference in the EV isolation efficiency was found between monovalent aptamers and monoclonal antibodies (ANOVA, n = 5, p > 0.05). The antibody-based isolation efficiency is still lower than that of MAs (ANOVA, n = 5, p < 0.005), further suggesting that multivalent ligands are ideal for EV isolation. The BCA assay determined the average protein concentration of unprocessed samples and the supernatant of processed samples (n = 5) to be 1.41 mg ml−1 and 0.216 mg ml−1, respectively. Therefore, the average mass of proteins was ∼1269 μg in 100 μL of unprocessed samples. After isolation with MAs and SMs, only ∼194.4 μg of protein contaminants were detected, indicating that ∼84.7% of BSA was depleted, providing relatively more pure EVs in comparison with that from ultracentrifugation.37 We used Dnase to digest MAs for the retrieval of the isolated EVs from the SM surface. The RNA mass in the unprocessed sample was 313.6 ng. After only 10 minutes of digestion, ∼292.1 ng of RNA was collected from the retrieved EVs, and thus the EV retrieval efficiency was determined as 93.1 ± 3.5%. Fig. 5a shows the SEM photomicrographs of SMs before incubation with EVs and after retrieval of EVs. The morphology of the retrieved CCRF-CEM EVs was visualized with a SEM and TEM (Fig. 5b and c, respectively). No significant difference in the morphology of EVs in the initial sample and retrieved EVs was observed. Although the degraded nucleic acid fragments, salt components, and deactivated nucleases as “background noises” influenced the observation, the size of the retrieved EVs and nanospherical morphology still can be determined. The retrieved CCRF-CEM EVs with a typical saucer-shaped morphology were observed in the TEM image. A minority of EVs adhered to each other due to the incomplete degradation of MAs. These aggregated or agglomerated EVs could be dissociated by extended digestion time and vortexing. However, a long incubation time at 37 °C may impair EV integrity, to a certain extent. The average size of the retrieved CCRF-CEM EVs from SMs was 153.9 nm (Fig. 5d), falling within the EV size range of the unprocessed samples. It is noteworthy that a minority of the retrieved CCRF-CEM EVs still aggregated or agglomerated, and thus relatively large particles were detected. Moreover, EV markers CD81, TSG101, PTK7, and GAPDH extracted from the retrieved CCRF-CEM EVs and MCF-7 EVs can be detected by western blot (Fig. 5e). The signal intensity of proteins in the retrieved CCRF-CEM EVs was significantly higher than those of the retrieved MCF-7 EVs, indicating that MAs can efficiently isolate PTK7-overexpressing CCRF-CEM EVs. Of note, an equal mass of the EV protein was loaded in the two groups. Furthermore, Sanger sequencing of the DNA extracted from the retrieved CCRF-CEM EVs confirmed the inherent heterozygous KRAS G12D mutation (Fig. 5f left), while the wild-type KRAS exon 12 was found in the DNA derived from MCF-7 EVs (Fig. 5f right).41 Next, the EVs isolated from the spiked sample of mixed EVs were lysed, and KRAS mutation analysis was performed by qPCR. The slope value of the standard curve is −3.423. The amplification efficiency is 95.95%, which falls within the range between 90% and 110%, and thus it is generally considered as a quality capable of generating reliable data. The plotted curve shows that the experimental data were in line with the spike ratio (the R2 value is 0.986), indicating good linearity (Fig. 5g).
 | ||
| Fig. 5 Characterization of the isolated EVs and retrieved EVs. (a) SEM images of CCRF-CEM EVs in the model sample, MA-grafted SMs before EV isolation, isolated EVs on the MA-grafted SMs, and MA-grafted SMs after EV retrieval by the digestion of MAs via nuclease. All scale bars are 1 μm. (b) SEM images of the retrieved CCRF-CEM EVs after nuclease treatment. The scale bar is 200 nm. (c) TEM images of the retrieved CCRF-CEM EVs. The scale bar is 200 nm. (d) Size distribution of the retrieved CCRF-CEM EVs. (e) Western blot of CD81, TGS101, PTK7, and GAPDH proteins derived from the retrieved CCRF-CEM EVs (1) and MCF-7 EVs (2). (f) Sanger sequencing of the inherently harbored KRASG12D mutation in the CCRF-CEM EV DNA (left) and wild-type KRAS in the MCF-7 EV DNA (right). (g) Spiked samples of EVs derived from CCRF-CEM and MCF-7 cells at various allele frequencies measured by qPCR. Error bars correspond to the standard deviation (n = 5). (h) The flocculent precipitation comprises MAs and captured EVs. | ||
Discussion
EVs as a new addition to the cancer liquid biopsy portfolio have been previously demonstrated.42,43 The isolation of tumor-derived EVs from body fluids and analyses of the EV contents could facilitate tumor detection, characterization of live tumor cells in vivo, and our understanding of cancer pathophysiology. To translate EVs into clinical diagnostics, improving the technologies to isolate EVs is critical.44,45 Current EV isolation approaches, including differential ultracentrifugation, density gradient ultracentrifugation, polymer-based precipitation, charge-mediated electrostatic adsorption, ultrafiltration, size exclusion, lipid anchors, and antibody-based immunoisolation, have been developed for the isolation of EVs from body fluids toward potential clinical implementation.46 Generally, these approaches have a combination of both good and bad qualities, including lengthy protocols, low isolation efficiency, and low purity.47,48 In particular, due to the overlap between EVs derived from tumor cells and normal cells in the size, density, surface charge, surface proteins, plasticity, and lipid envelope, it is extremely difficult to isolate pure, low-abundance tumor-derived EVs from the large amount of EVs released from non-tumor cells. Among the existing approaches, only immunoisolation is promising for the specific isolation of tumor-derived EVs from body fluids, while also possessing a higher isolation efficiency than that of the more prevalent ultracentrifugation.48 However, current immunoisolation approaches mainly utilize common membrane protein markers of EVs, which fail to enjoy such advantages and lead to just ordinary specificity. Hence, only tumor-specific antigens or overexpressed tumor-associated antigens are recommended for the isolation of pure tumor-derived EVs. Therefore, in this study, we used aptamers targeting overexpressed PTK7 as the target protein for the isolation of tumor-derived EVs.We used RCA to prepare MAs for the isolation of EVs. Compared to monovalent aptamers, an equal molar amount of MAs is more efficient in EV isolation due to three advantages. First, in comparison with the monovalent counterparts, the repeated aptamer units in MAs provide thousands of additional binding sites for EV recognition and interaction. Moreover, the extremely long length of the single-stranded MAs causes considerable overlap, forming nets in a suspension,13 thus allowing for more efficient trapping of the EVs. In fact, after 30 min of incubation of CCRF-CEM EVs with MAs, flocculent precipitation was observed (Fig. 5h), reflecting the potential crosslinking among MAs and EVs. By contrast, in the control group, CCRF-CEM EVs incubated with roughly an equal amount of monovalent aptamers (∼16 nmol) still homogeneously dispersed in 10% BSA. This finding further inspires the future design of a “mesh” equipped with numerous binding sites for the isolation of EVs. The EV/“mesh” complex may not require additional microspheres for EV collection, as the complex itself can spontaneously precipitate, and thus the “mesh” could further minimize the operational steps and processing time. Furthermore, due to the >50000 nt length of MAs, the MA/EV complex is less likely to block the interaction between biotin and streptavidin. In contrast, the monovalent aptamer/EV complex can overlay many streptavidin sites on the SMs, decreasing its binding capacity.
It is worth noting that the isolation efficiency of the EVs in both the model sample and spiked samples mimicking plasma was approximately 45%, indicating that the aptamer can specifically recognize and interact with PTK7 regardless of the presence of high-abundance BSA. Generally, the isolation efficiency of EVs using the existing approaches is approximately 30–40%.49–51 Among these approaches, depending on the isolation strategy, the design of the microfluidic device, sample processing speed, or sample types, the isolation efficiency of the EVs may range from 1.5% to 99%.19,49–51 By contrast, the performance of MAs in EV isolation is moderate and comparable. However, we admit that this isolation efficiency might decrease in processing body fluids in real-life settings due to their complex composition. The retrieval of the EVs can also be fulfilled with nuclease-mediated digestion of DNA. The extended digestion time could completely destroy the MAs, while the relatively high temperature might also cause the deterioration of the EVs. Hence, we selected 10 minutes, which is frequently used for the detachment of captured tumor cells, for the retrieval of the isolated EVs.52 Moreover, BSA contamination was depleted by ∼85%. Compared to our previous study,18,19 the depletion efficiency is relatively low. We speculate that it may be a result of the physical entrapment of BSA within the crosslinked MAs and EVs.
Overall, this simple and inexpensive approach utilizing MAs and SMs can efficiently isolate ∼45% EVs and deplete ∼85% of protein contaminants from plasma-mimicking spiked samples in 40 minutes without using cumbersome ultracentrifuges, magnets, or microfluidic devices. In addition, ∼93% of the isolated EVs can be retrieved in 10 minutes, allowing various downstream molecular analyses. The notable advantages of this method potentially enable its translation into clinical use. In future studies, patients’ blood specimens will be used to further validate the feasibility and reliability of MAs in EV isolation. In addition, towards steady clinical translation, the length of MAs, the length of the polyT spacer, the size of SMs, and the density of streptavidin on SMs would require rigorous optimization to achieve the highest isolation efficiency and repeatability.
Author contributions
F. X., Y. C., W. M. and Yuan W. participated in the conceptualization and protocols; F. X., Y. C., K. A., Yi W., W. Z., and G. C. produced the data; all authors analyzed and interpreted the data; F. X., Y. C., Z. Q., W. M., and Yuan W. prepared the manuscript. All the authors approved the final version of the manuscript.Conflicts of interest
All authors declare that they have no conflicts of interest and no competing interest.Acknowledgements
This work was partially supported by the Binghamton University Faculty Startup Fund 910252-35 and Binghamton University S3IP award ADLG195.References
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS.
- N. Arraud, R. Linares, S. Tan, C. Gounou, J. M. Pasquet, S. Mornet and A. R. Brisson, J. Thromb. Haemostasis, 2014, 12, 614–627 CrossRef CAS.
- A. Gámez-Valero, S. I. Lozano-Ramos, I. Bancu, R. Lauzurica-Valdemoros and F. E. Borràs, Front. Immunol., 2015, 6, 6 Search PubMed.
- D. Chiasserini, J. R. van Weering, S. R. Piersma, T. V. Pham, A. Malekzadeh, C. E. Teunissen, H. de Wit and C. R. Jiménez, J. Proteomics, 2014, 106, 191–204 CrossRef CAS.
- A. Milasan, N. Tessandier, S. Tan, A. Brisson, E. Boilard and C. Martel, J. Extracell. Vesicles, 2016, 5, 31427 CrossRef.
- L. Wang, K. K. Abhange, Y. Wen, Y. Chen, F. Xue, G. Wang, J. Tong, C. Zhu, X. He and Y. Wan, ACS Omega, 2019, 4, 22638–22645 CrossRef CAS.
- A. Becker, B. K. Thakur, J. M. Weiss, H. S. Kim, H. Peinado and D. Lyden, Cancer Cell, 2016, 30, 836–848 CrossRef CAS.
- M. Nawaz, N. Shah, B. R. Zanetti, M. Maugeri, R. N. Silvestre, F. Fatima, L. Neder and H. Valadi, Cells, 2018, 7, 167 CrossRef CAS.
- P. D. Robbins and A. E. Morelli, Nat. Rev. Immunol., 2014, 14, 195–208 CrossRef CAS.
- A. L. S. Revenfeld, R. Bæk, M. H. Nielsen, A. Stensballe, K. Varming and M. Jørgensen, Clin. Ther., 2014, 36, 830–846 CrossRef.
- M. Yin, X. Loyer and C. M. Boulanger, Eur. J. Pharmacol., 2015, 763, 90–103 CrossRef CAS.
- Y. Wan, B. Liu, H. Lei, B. Zhang, Y. Wang, H. Huang, S. Chen, Y. Feng, L. Zhu, Y. Gu, Q. Zhang, H. Ma and S. Y. Zheng, Ann. Oncol., 2018, 29, 2379–2383 CrossRef CAS.
- Y. Wan, L. Wang, C. Zhu, Q. Zheng, G. Wang, J. Tong, Y. Fang, Y. Xia, G. Cheng, X. He and S. Y. Zheng, Cancer Res., 2018, 78, 798–808 CrossRef CAS.
- J. Van Deun, P. Mestdagh, R. Sormunen, V. Cocquyt, K. Vermaelen, J. Vandesompele, M. Bracke, O. De Wever and A. Hendrix, J. Extracell. Vesicles, 2014, 3 DOI:10.3402/jev.v3.24858.
- Y. Yuana, J. Levels, A. Grootemaat, A. Sturk and R. Nieuwland, J. Extracell. Vesicles, 2014, 3, 23262 CrossRef.
- K. Iwai, T. Minamisawa, K. Suga, Y. Yajima and K. Shiba, J. Extracell. Vesicles, 2016, 5, 30829 CrossRef.
- D. W. Greening, R. Xu, H. Ji, B. J. Tauro and R. J. Simpson, in Proteomic Profiling, Springer, 2015, pp. 179–209 Search PubMed.
- W. Mao, Y. Wen, H. Lei, R. Lu, S. Wang, Y. Wang, R. Chen, Y. Gu, L. Zhu, K. K. Abhange, Z. J. Quinn, Y. Chen, F. Xue, M. Zheng and Y. Wan, Anal. Chem., 2019, 91, 13729–13736 CrossRef CAS.
- Y. Wan, M. Maurer, H. Z. He, Y. Q. Xia, S. J. Hao, W. L. Zhang, N. S. Yee and S. Y. Zheng, Lab Chip, 2019, 19, 2346–2355 RSC.
- K. Reiter, P. P. Aguilar, V. Wetter, P. Steppert, A. Tover and A. Jungbauer, J. Chromatogr., A, 2019, 1588, 77–84 CrossRef CAS.
- T. Yoshida, T. Ishidome and R. Hanayama, Curr. Protoc. Cell Biol., 2017, 77, 3.45.1–3.45.18 Search PubMed.
- W. Norde and C. A. Haynes, Proteins at Interfaces II, ACS Publications, 1995, ch. 2, pp. 26–40, DOI:10.1021/bk-1995-0602.ch002.
- D. Freitas, M. Balmana, J. Pocas, D. Campos, H. Osorio, A. Konstantinidi, S. Y. Vakhrushev, A. Magalhaes and C. A. Reis, J. Extracell. Vesicles, 2019, 8, 1621131 CrossRef CAS.
- P. S. Sung, T. F. Huang and S. L. Hsieh, Nat. Commun., 2019, 10, 2402 CrossRef.
- J. H. Kim, C. H. Lee and S. W. Lee, Mol. Ther.–Nucleic Acids, 2019, 14, 483–497 CrossRef CAS.
- M. Capello, J. V. Vykoukal, H. Katayama, L. E. Bantis, H. Wang, D. L. Kundnani, C. Aguilar-Bonavides, M. Aguilar, S. C. Tripathi, D. S. Dhillon, A. A. Momin, H. Peters, M. H. Katz, H. Alvarez, V. Bernard, S. Ferri-Borgogno, R. Brand, D. G. Adler, M. A. Firpo, S. J. Mulvihill, J. J. Molldrem, Z. Feng, A. Taguchi, A. Maitra and S. M. Hanash, Nat. Commun., 2019, 10, 254 CrossRef.
- L. Balaj, N. A. Atai, W. Chen, D. Mu, B. A. Tannous, X. O. Breakefield, J. Skog and C. A. Maguire, Sci. Rep., 2015, 5, 10266 CrossRef CAS.
- W. Nakai, T. Yoshida, D. Diez, Y. Miyatake, T. Nishibu, N. Imawaka, K. Naruse, Y. Sadamura and R. Hanayama, Sci. Rep., 2016, 6, 33935 CrossRef CAS.
- Y. T. Kang, E. Purcell, C. Palacios-Rolston, T. W. Lo, N. Ramnath, S. Jolly and S. Nagrath, Small, 2019, 15, 1903600 CrossRef CAS.
- H. Zhang, B. Qiao, Q. Guo, J. Jiang, C. Cai and J. Shen, Analyst, 2020, 145, 3557–3563 RSC.
- H. Zhang, A. Wang, S. Qi, S. Cheng, B. Yao and Y. Xu, Int. J. Mol. Sci., 2014, 15, 11665–11677 CrossRef CAS.
- D.-S. Choi, D.-Y. Choi, B. Hong, S. Jang, D.-K. Kim, J. Lee, Y.-K. Kim, K. Pyo Kim and Y. Gho, J. Extracell. Vesicles, 2012, 1, 18704 CrossRef CAS.
- Y. Zhao, D. Xu and W. Tan, Integr. Biol., 2017, 9, 188–205 CrossRef.
- W. Zhao, C. H. Cui, S. Bose, D. Guo, C. Shen, W. P. Wong, K. Halvorsen, O. C. Farokhzad, G. S. Teo, J. A. Phillips, D. M. Dorfman, R. Karnik and J. M. Karp, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19626–19631 CrossRef CAS.
- P. Jung, C. Sommer, F. M. Barriga, S. J. Buczacki, X. Hernando-Momblona, M. Sevillano, M. Duran-Frigola, P. Aloy, M. Selbach, D. J. Winton and E. Batlle, Stem Cell Rep., 2015, 5, 979–987 CrossRef CAS.
- M. M. Ali, F. Li, Z. Zhang, K. Zhang, D.-K. Kang, J. A. Ankrum, X. C. Le and W. Zhao, Chem. Soc. Rev., 2014, 43, 3324–3341 RSC.
- Y. Wan, G. Cheng, X. Liu, S. J. Hao, M. Nisic, C. D. Zhu, Y. Q. Xia, W. Q. Li, Z. G. Wang, W. L. Zhang, S. J. Rice, A. Sebastian, I. Albert, C. P. Belani and S. Y. Zheng, Nat. Biomed. Eng., 2017, 1, 0058 CrossRef CAS.
- E. Dirican, Asian Pac. J. Cancer Prev., 2016, 17, 2731 Search PubMed.
- J. Caradec, G. Kharmate, E. Hosseini-Beheshti, H. Adomat, M. Gleave and E. Guns, Clin. Biochem., 2014, 47, 1286–1292 CrossRef CAS.
- M. Oksvold, Arch. Med., 2015, 6, 18 Search PubMed.
- S. A. Moharram, K. Shah and J. U. Kazi, J. Cancer, 2017, 8, 4124 CrossRef.
- Y. H. Soung, S. Ford, V. Zhang and J. Chung, Cancers, 2017, 9, 8 CrossRef.
- T. An, S. Qin, Y. Xu, Y. Tang, Y. Huang, B. Situ, J. M. Inal and L. Zheng, J. Extracell. Vesicles, 2015, 4, 27522 CrossRef.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS.
- D. Wu, J. Yan, X. Shen, Y. Sun, M. Thulin, Y. Cai, L. Wik, Q. Shen, J. Oelrich, X. Qian, K. L. Dubois, K. G. Ronquist, M. Nilsson, U. Landegren and M. Kamali-Moghaddam, Nat. Commun., 2019, 10, 3854 CrossRef.
- A. Liga, A. D. Vliegenthart, W. Oosthuyzen, J. W. Dear and M. Kersaudy-Kerhoas, Lab Chip, 2015, 15, 2388–2394 RSC.
- S. Kuang, J. Zheng, H. Yang, S. Li, S. Duan, Y. Shen, C. Ji, J. Gan, X. W. Xu and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10642–10647 CrossRef CAS.
- N. Zarovni, A. Corrado, P. Guazzi, D. Zocco, E. Lari, G. Radano, J. Muhhina, C. Fondelli, J. Gavrilova and A. Chiesi, Methods, 2015, 87, 46–58 CrossRef CAS.
- G. K. Patel, M. A. Khan, H. Zubair, S. K. Srivastava, M. D. Khushman, S. Singh and A. P. Singh, Sci. Rep., 2019, 9, 5335 CrossRef.
- L. L. Yu, J. Zhu, J. X. Liu, F. Jiang, W. K. Ni, L. S. Qu, R. Z. Ni, C. H. Lu and M. B. Xiao, BioMed Res. Int., 2018, 2018, 3634563 Search PubMed.
- P. Li, M. Kaslan, S. H. Lee, J. Yao and Z. Q. Gao, Theranostics, 2017, 7, 789–804 CrossRef CAS.
- Q. Zheng, S. M. Iqbal and Y. Wan, Biotechnol. Adv., 2013, 31, 1664–1675 CrossRef.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |