
Next-generation derivatization reagents optimized for enhanced product ion formation in photodissociation-mass spectrometry of fatty acids†
Venkateswara R.
Narreddula
 a,
Benjamin I.
McKinnon
b,
Samuel J. P.
Marlton
b,
David L.
Marshall
cd,
Nathan R. B.
Boase
ad,
Berwyck L. J.
Poad
cd,
Adam J.
Trevitt
b,
Todd W.
Mitchell
ef and
Stephen J.
Blanksby
*acd
a,
Benjamin I.
McKinnon
b,
Samuel J. P.
Marlton
b,
David L.
Marshall
cd,
Nathan R. B.
Boase
ad,
Berwyck L. J.
Poad
cd,
Adam J.
Trevitt
b,
Todd W.
Mitchell
ef and
Stephen J.
Blanksby
*acd
aSchool of Chemistry and Physics, Science and Engineering Faculty, Queensland University of Technology, Brisbane, QLD 4000, Australia. E-mail: stephen.blanksby@qut.edu.au
bMolecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, NSW 2522, Australia
cCentral Analytical Research Facility, Institute for Future Environments, Queensland University of Technology, Brisbane, QLD 4000, Australia
dCentre for Materials Science, Queensland University of Technology, Brisbane, QLD 4000, Australia
eSchool of Medicine, University of Wollongong, Wollongong, NSW 2522, Australia
fIllawarra Health and Medical Research Institute, Wollongong, NSW 2522, Australia
First published on 24th October 2020
Abstract
Ultraviolet-photodissociation (UVPD) mass spectrometry is an emerging analytical tool for structural elucidation of biomolecules including lipids. Gas phase UVPD of ionised fatty acids (FAs) can promote fragmentation that is diagnostic for molecular structure including the regiochemistry of carbon–carbon double bonds and methyl branching position(s). Typically, however, lipids exhibit poor conversion to photoproducts under UVPD and thus require longer integration times to achieve the signal-to-noise required for structural assignments. Consequently, the integration of UVPD into liquid-chromatography mass spectrometry (LC-MS) workflows for FAs has been limited. To enhance photofragmentation efficiency, an alternative strategy has been devised using wet-chemical derivatization of FAs to explicitly incorporate photolabile groups. FA derivatives that include an aryl-iodide motif have photodissociation conversions of up to 28% when activated by a single 266 nm photon. The radical-directed dissociation product ions resulting from UVPD of these derivatives provide key details of molecular structure and discriminate between lipid isomers. Herein, we describe the structure–activity guided development of new FA derivatives capable of photoproduct yields of up to 97%. UVPD-action spectroscopy demonstrates that photodissociation for FAs derivatized with N-(2-aminoethyl)-4-iodobenzamide (NIBA) is maximised near 266 nm and highlights the key role of the 4-iodobenzamide motif in the efficient formation of [M − I]˙+ radical cations (and diagnostic secondary product ions). The high photodissociation yield of NIBA-derivatized lipids is maintained across 37 commonly observed FAs with the resulting UVPD mass spectra shown to be effective in the discrimination of isomeric FAs that differ in the position(s) of carbon–carbon double bonds. Integration of this strategy with reversed-phase LC-MS workflows is confirmed with high-quality UVPD mass spectra acquired across each chromatographic peak.
1. Introduction
Fatty acids (FAs) are a category of lipids with a variety of physiological functions in living systems.1 Of the more than 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lipid structures currently catalogued in the LIPID MAPS database, approximately 7000 are fatty acyl lipids with structures varying in carbon-chain length; degree of unsaturation; location and stereochemistry of carbon–carbon double bond(s); chain branching; functionalisation (e.g., hydroxylation, nitration, etc.); and the incorporation of carbocyclic motifs.2–4 These structural variations define the physical and chemical properties of each lipid and inform their physiological role. Dysregulation in lipid metabolism that results in changes in FA structure or composition of the FA ensemble has been implicated in various pathologies such as cancer, cardiovascular disease and diabetes.5–7 As a consequence, the identification and quantification of FAs in complex biological samples is essential for understanding their roles in health and disease.
000 lipid structures currently catalogued in the LIPID MAPS database, approximately 7000 are fatty acyl lipids with structures varying in carbon-chain length; degree of unsaturation; location and stereochemistry of carbon–carbon double bond(s); chain branching; functionalisation (e.g., hydroxylation, nitration, etc.); and the incorporation of carbocyclic motifs.2–4 These structural variations define the physical and chemical properties of each lipid and inform their physiological role. Dysregulation in lipid metabolism that results in changes in FA structure or composition of the FA ensemble has been implicated in various pathologies such as cancer, cardiovascular disease and diabetes.5–7 As a consequence, the identification and quantification of FAs in complex biological samples is essential for understanding their roles in health and disease.
Hyphenated chromatography–mass spectrometry (MS) techniques have been the most extensively used for structural identification and quantification of FAs from biological samples. Among these, gas chromatography (GC) coupled to electron ionization (EI)-MS is most widely employed for quantitative FA analysis from biological samples, due to its high sensitivity, chromatographic resolution and excellent reproducibility.8,9 In GC-MS protocols, FAs are typically converted to their corresponding fatty acid methyl esters (FAMEs) to promote volatilisation and enhance chromatographic separations. EI mass spectra of FAMEs, however, provide little structurally-diagnostic information beyond the sum composition (i.e., total number of carbons and degree of unsaturation). As a result, FAMEs identification relies principally on retention time alignment with reference standards and thus makes structure determination of unusual, or previously unreported FAs, more challenging. To overcome this limitation, alternative GC-MS derivatives have been developed specifically to localise the charge and promote radical-directed dissociation. The best-known examples of this are picolinyl esters and dimethyloxazolines. EI mass spectra derived from FAs conjugated with these functional groups are exquisitely sensitive to variations in molecular structure and can thus be used for lipid identification, including the assignment of sites of unsaturation and chain branching, based on de novo interpretation of product ions.10,11
In parallel with developments in GC-MS, there has been growing interest in using liquid chromatography (LC) coupled with electrospray ionization (ESI)-MS for FA analysis.12,13 These strategies harness the wide dynamic range of LC-MS and, importantly, can access higher molecular weight lipids along with more functionalised and thermally labile species compared with conventional GC-MS. In typical LC-MS workflows, the ESI-MS is operated in negative ion mode to generate [FA − H]− anions by deprotonation at the carboxylic acid. While negative ion mode LC-MS can be extremely sensitive,14 the limited structural information derived from low-energy collision-induced dissociation (CID) of deprotonated FA anions makes identification of unexpected FAs challenging.15,16 This problem is compounded by the lower chromatographic resolution of LC compared with GC whereby isomeric FAs often co-elute.13 In response, derivatization strategies specific for LC-MS have been developed that conjugate a readily ionizable or fixed-charge moiety onto the carboxylic acid group of the FA and thus alter its chromatographic, ionization and CID characteristics.17–19 Among this class of derivatives, conjugation of FAs with the fixed-charge N-(4-aminomethylphenyl) pyridinium (AMPP+) moiety20,21 is effective in enhancing both detection and identification of FAs across a broad range of structures extracted from a wide variety of biological sources.22–24 Importantly, the optimized combination of the fixed pyridinium charge carrier conjugated via an amide linkage to the FA mean that, upon CID, these derivatives undergo charge-remote fragmentation of the hydrocarbon chain which facilitates the spectral identification of site(s) of unsaturation and other functionalisation.25 In general however, charge-remote fragmentation is rather inefficient under low energy CID (<100 eV) conditions, which has prompted the exploration of alternative modes of ion activation to promote carbon–carbon bond cleavage in ionised lipids.26
Ultraviolet photodissociation (UVPD) is emerging as a viable alternative to low-energy CID for structural elucidation of lipids (and other biomolecules) due to the characteristic mass spectra arising from either dissociation channels through excited states or the induction of radical-directed dissociation.27–30 In the context of FA analysis, UVPD of ionised lipids exploiting irradiation at 157 nm or 193 nm induce dissociation of carbon–carbon bonds within the hydrocarbon chain and thus enable discrimination between isomers. For example, Reilly and co-workers used 157 nm radiation to reveal structural differences in leukotriene isomers27 while, more recently, the Reid group has undertaken 193 nm irradiation of the [FA − H + 2Li]+ cations to discriminate between monounsaturated FA isomers.31 Similar UVPD strategies have also been deployed on a broad range of lipid classes including phospholipids, glycosphingolipids, gangliosides and lipopolysaccharides demonstrating broad applicability in assigning key structural features of FA chains carried by these complex lipids.28,29,32–34 Common among these UVPD mass spectra however, is a relatively low conversion of precursor ions to photoproducts –often just a few percent– and, as a consequence, multiple laser shots are deployed leading to longer spectral integration times. Accordingly, this ion activation modality is better suited to direct infusion (or shotgun) lipidomics analysis compared to hyphenated LC-MS workflows where tandem mass spectra must be acquired more rapidly.
An alternative UVPD strategy has been pioneered by Julian and co-workers and deploys 266 nm radiation for targeted dissociation of an aryl iodide motif introduced by chemical derivatization. In this approach, photolytic cleavage of the carbon–iodine bond produces a localized radical within the ion which undergoes subsequent dissociation to reveal key elements of molecular structure.35 Initially developed for protein and peptide analysis, this strategy complements the shorter wavelength approaches described above by providing excellent regio-selectivity and high photo-conversions of up to 97% in some instances.36 Application of this so-called radical-directed dissociation (RDD) in lipidomics has been effective in structure determination for classes of complex lipids including glycerophospholipids, triacylglycerols and glycosphingolipids, wherein the aryl iodide was introduced via non-covalent complexation during ESI.30,37,38 Our group has further adapted this strategy for FA analysis using ester and amide conjugation of aryl-iodide groups to the carboxylic acid moiety.26,39 The recently introduced aminomethyl-4-iodophenyl pyridin-1-ium (I-AMPP+) FA derivative carried both the photolabile aryl-iodide motif and the fixed charge pyridinium and successfully enhanced both ionization and photodissociation events. Importantly, 266 nm UV photodissociation (PD266) of FAs modified with I-AMPP+ gives rise to diagnostic product ions that enabled the assignment of position(s) of unsaturation, hydroxyl and ester substitutions, methyl branch points and even carbocyclic structures.26,40,41 While effective, the overall sensitivity of these PD266 strategies in FA analysis is intrinsically limited by the photodissociation yield. Complete conversion of precursor to product ions by a single laser pulse would increase the signal-to-noise of UVPD mass spectra, which is particularly important for assigning the molecular structures of lesser abundant lipids within complex extracts. Herein we report a structure–activity investigation aimed at understanding the critical structural elements of derivatization reagents for enhancing PD266 efficiency. Maintaining the aryl-halide core as the photo-caged radical motif, structural features including the position of the halogen, the type of halogen and the derivatization linker were systematically varied (Fig. 1). Based on these investigations and inspired by p-iodobenzamide derivatives of peptides and proteins that exhibit quantitative radical cation formation from a single laser pulse at 266 nm,36 we describe the synthesis of FA derivatives based on N-(2-aminoethyl)-4-iodobenzamide (NIBA). The PD266 of NIBA modified FAs reveals consistently high photo-conversion efficiencies across a diverse range of molecular weights and functionality. The high yield of structurally diagnostic product ions arising from a single photon event present the opportunity to incorporate PD266 into LC-MS workflows for FA analysis.
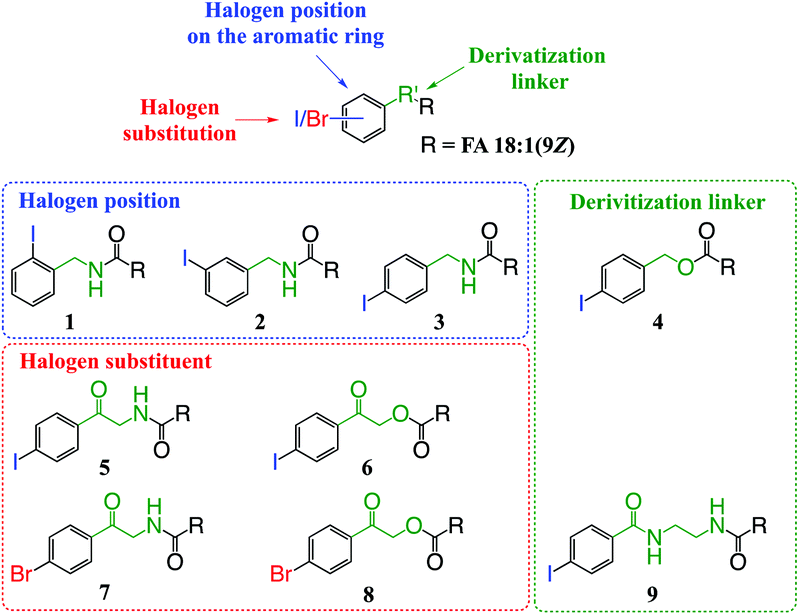 | ||
| Fig. 1 Structural features of derivatives to be optimized for enhanced photofragmentation conversion at 266 nm. Structures 1–9 of derivatives conjugated to FA 18:1(9Z) (represented as R) used for structure–activity optimization. | ||
2. Materials and methods
2.1. Nomenclature and general reagents
Fatty acids are described using recommended shorthand notation.42 Oleic acid, FA 18:1(9Z), was purchased from Tokyo Chemical Industry (Tokyo, Japan) while FA 19:0 was purchased from Chem Service, Inc. (Pennsylvania, USA). Food Industry 37 FAMEs mix was purchased from RESTEK (Bellefonte, PA, USA) with details provided as ESI (Table S4†). 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate (HATU) was purchased from Tokyo Chemical Industry (Tokyo, Japan). N,N-Diisopropylethylamine (DIPEA); N,N-dimethylformamide (DMF); tetrabutylammonium hydroxide solution (40 wt% in water); and 18-crown-6 ether (18-C-6) were purchased from Sigma-Aldrich (Sydney, Australia). Ammonium chloride (Analytical grade); formic acid (Analytical grade); and potassium bicarbonate (Analytical grade) were purchased from Ajax Finechem, Pty. Ltd (Scoresby, Australia). Acetonitrile (ACN, Optima LC-MS grade); methanol (HPLC grade); water (HPLC grade); hexane (HPLC grade) and dichloromethane (HPLC grade); were purchased from Thermo Fisher Scientific (Scoresby, Australia). Methyl tert-butyl ether (MTBE, HPLC grade) was purchased from RCI Labscan Ltd (Bangkok, Thailand). Additional chemicals, reagents and solvents used for synthesis and purification are provided in ESI.†2.2. Derivatization reagents
The derivatization reagents 2-, 3-, and 4-iodobenzylamine hydrochloride (10, 11, 12); 2-bromo-1-(4-bromophenyl)ethan-1-one (13); and 2-amino-1-(4-bromophenyl)ethan-1-one hydrochloride (14) were purchased from Sigma-Aldrich (Sydney, Australia) and were used without further purification or modification. Details of the synthesis, purification and characterisation protocols for 2-bromo-1-(4-iodophenyl)ethan-1-one (16), 2-amino-1-(4-iodophenyl)ethan-1-one hydrochloride (18), and N-(2-aminoethyl)-4-iodobenzamide (NIBA, 23) are provided in ESI.†2.3. Derivatization of FAs
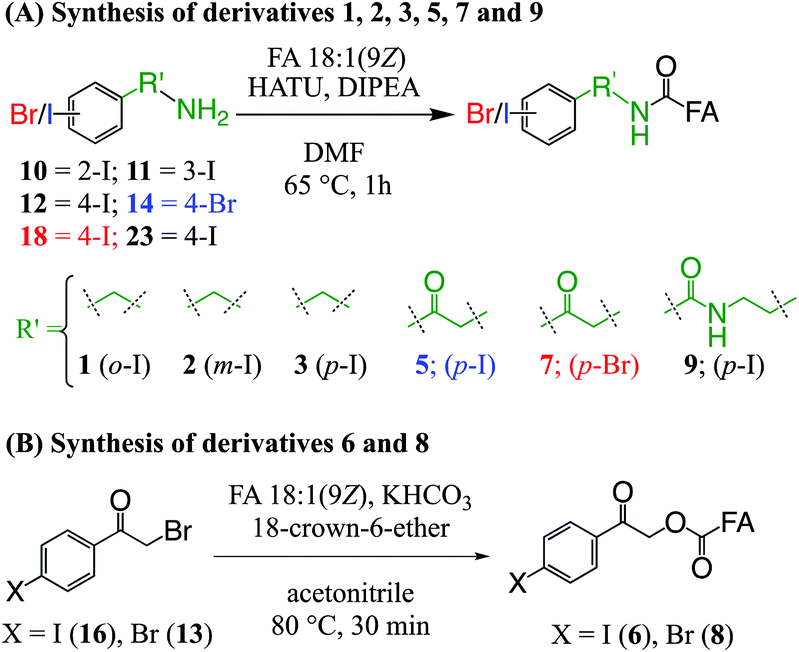 | ||
| Scheme 1 Preparation of derivatives 1, 2, 3, 5, 6, 7, 8 and 9 from the non-esterified FA 18:1(9Z). | ||
Ester derivative 4 (see Fig. 1) was prepared in accordance with a prior report,39 while ester conjugated derivatives 6 and 8 (see Fig. 1) were prepared from their respective bromides (16 and 13) by a procedure based on the work of Mehta et al. and summarised in Scheme 1B.45 Briefly, non-esterified FA 18:1(9Z) (0.0035 mmol, 1.0 equiv.) was dissolved in ACN (0.5 mL) before KHCO3 (0.017 mmol, 5 equiv.) was added at room temperature. To this mixture, derivatization reagents 16 or 13 (0.010 mmol, 3 equiv.) and 18-crown-6 ether (0.001 mmol, 0.3 equiv.) were added and the mixture was heated to 80 °C for 45 min. After cooling to room temperature, ACN was removed by a stream of nitrogen before the residue was dissolved in MTBE (1.5 mL) and filtered on a bed of silica gel, which was further washed with MTBE (1.5 mL). The MTBE filtrate was dried under a stream of nitrogen to give a pale-orange solid (6, 81% crude yield) or white solid (8, 95% crude yield) that was taken up in methanol for analysis.
:methanol (20:80, 30 mg L−1) with FA 19:0 (1 μg) as an internal standard. Hydrolysis of the 37 FAMEs mix and derivatization of FAs with NIBA (23) was carried out using protocols summarised in Scheme 2. Briefly, the 37 FAMEs mix stock solution (50 μL, 1 equiv.) and 50 μL of dichloromethane:methanol (20:80) were added to a 4 mL glass screw-capped vial. To this solution, 200 μL of methanol and 200 μL of tert-butylammonium hydroxide solution (40 wt% in water) were added. The resulting mixture was vortexed for 1 min prior to heating to 75 °C for 2 h. After this, the mixture was cooled to room temperature and 1.5 mL of cold water and 1.5 mL of hexane were added. The reaction mixture was acidified to pH = 2–3 with hydrochloric acid (5 M) and the sample was vortexed for 1 min. The organic layer was removed into another vial, and the extraction was repeated with another 1.5 mL of hexane. The combined hexane extracts were dried under nitrogen. The FA mixture obtained was derivatized with the NIBA reagent (23) following the procedure described above. The derivatized extract was dissolved in dichloromethane:methanol (1:4) and then diluted further with methanol, as required, for analysis.
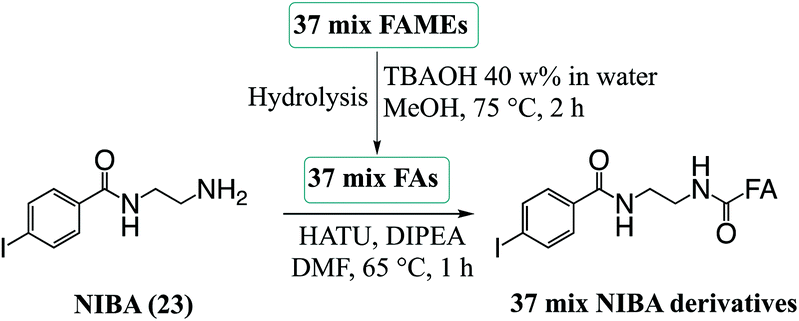 | ||
| Scheme 2 Procedure for hydrolysis of esterified FAs (Food Industry 37 FAMEs mix) followed by amide coupling of FA to the derivatization reagent NIBA (23). | ||
2.4. Instrumentation
High-performance liquid chromatography (HPLC) experiments were carried out using a Dionex Ultimate 3000 RSLC with a quaternary pump, column oven and split loop autosampler (Thermo Fisher Scientific, San Jose, CA, USA) controlled by Chromeleon™ 7.2 Chromatography Data System software. LC was carried out using an Acclaim™ C30 reverse-phase column (150 mm × 2.1 mm, 3.0 μm, Thermo Fisher Scientific) maintained at 45 °C. Solvent A was 0.1% formic acid in water and solvent B was 0.1% formic acid in acetonitrile, delivered at 0.3 mL min−1. The mobile phase gradient is provided in the ESI (see, Table S1†).
2.5. Electronic structure calculations
Initial structural optimizations of compounds 3, 5 and 9 were undertaken in the ORCA/4.2.1 program package employing the density functional theory (DFT) M06-2X method and the def2-SVP basis set treating the iodine atom with an effective core potential (ECP).48–51 Lowest energy structures were re-optimized using SCS-CC2 with the def2-SVP basis set treating the iodine atom with an ECP in the TURBOMOLE (V7.2) program package.52–54 Using the SCS-CC2/def2-SVP optimised geometries, the carbon iodine bond length was scanned keeping all other atoms frozen and these geometries were employed to calculate SCS-CC2 excitation energies with the def2-SVPD basis set. Additional details of energy calculations and structure visualizations are provided in the ESI (Fig. S1†).3. Results and discussion
3.1. Derivatization reagent design
The aim of this work was to design and develop photo-labile derivatization reagents for FAs capable of generating high abundances of hydrogen deficient radical cations upon PD at 266 nm. For simplicity, the structural elements considered in the design and optimization of candidate reagents were classified into three components: (i) the photo-labile aryl-halogen bond; (ii) the position of halogen substitution relative to the derivatization linker and; (iii) the composition of the linker conjugating the reagent to the FA (Fig. 1). Based on systematic variation of these three elements and the commercial availability or synthetic accessibility, the test set of derivatives shown in Fig. 1 was devised. Beyond the optimization of reagent structure for photodissociation, other key properties were also considered including the ease of derivatization; the influence of different cation adducts; and the modulation of photochemistry by the FA structure (i.e., the length and degree(s) of unsaturation of the hydrocarbon chain).3.2. Position of substitution on the aromatic ring
To examine the effect on PD266 efficiency of the iodine position on the aromatic ring, the FA 18:1(9Z) was conjugated to each of the 2-, 3- and 4-iodobenzylamine isomers to yield the derivatized FAs 1, 2 and 3, respectively (Fig. 1). Methanolic solutions of each derivatized FA were infused into the electrospray ionization mass spectrometer yielding abundant [M + Na]+ ions at m/z 520. Each of the sodiated precursor ions was mass-selected and irradiated with a single laser pulse at 266 nm and the resulting PD266 mass spectra are displayed in Fig. 2. For all three derivatized FAs, [M + Na − I]˙+ radical cations resulting from photolytic cleavage of the carbon–iodine bond can be observed at m/z 393 (Fig. 2A–C). Closer inspection of the PD266 mass spectra of these derivatives also reveals a series of less abundant product ions, contributing 3–4% of the total ion signal, that arise from prompt radical-directed dissociation of the [M + Na − I]˙+ radical cation. The ion at m/z 295 is prominent among these secondary products and is consistent with dissociation at the allylic position relative to the carbon–carbon double bond of FA18:1(9Z). This preference for fragmentation at the allylic position is analogous to previous reports of PD266 for FAs esterified with iodobenzyl alcohols.39 Analysis of the PD266 mass spectra shown in Fig. 2A–C reveal differences in the relative depletion of the precursor ion as summarised in Fig. 2D. In this representation, PD266 conversion is considered for both the abundance of primary [M + Na − I]˙+ radical cation and the secondary product ions each presented as a percentage of total ion abundance. Combining the normalised product ion abundance for each of these channels gives the overall photo-conversion for the respective derivatives. As illustrated in Fig. 2D, the derivatized FA 3 displayed the highest PD266 conversion at ∼28% compared to 2 at 21% and 1 at 13% indicating a preference for para over meta and ortho substitution patterns around the aromatic ring (with respect to the derivatization linker). The PD266 conversions obtained from [M + Na]+ precursor ions of 3 also compared favourably with alternative cations formed from the same compound such as [3 + Li]+ (18%) and [3 + K]+ (15%) (ESI, Fig. S2 and S3†). In contrast, the PD266 mass spectra of protonated [3 + H]+ ions showed photo-conversion of up to 47% (Fig. S2 and S3†). To investigate this further, both the primary PD product ions [3 + Na − I]˙+ and [3 + H − I]˙+ were re-isolated and subjected to CID in MS3 experiments. The resulting PD266/CID mass spectra are compared in Fig. S4 (ESI†) and indicate that dissociation of the [3 + Na − I]˙+ radical cation gives a significantly improved signal-to-noise ratio for product ions associated with the position of the carbon–carbon double bond compared with the protonated analogue. Given the over-arching goal of robust identification of FAs, the preference for product ions diagnostic of molecular structure represents a key advantage. Based on this assessment all subsequent PD266 and PD266/CID experiments were acquired with [M + Na]+ cations, while iodine substitution was maintained on the para-position of the aromatic ring relative to the FA derivatization linker.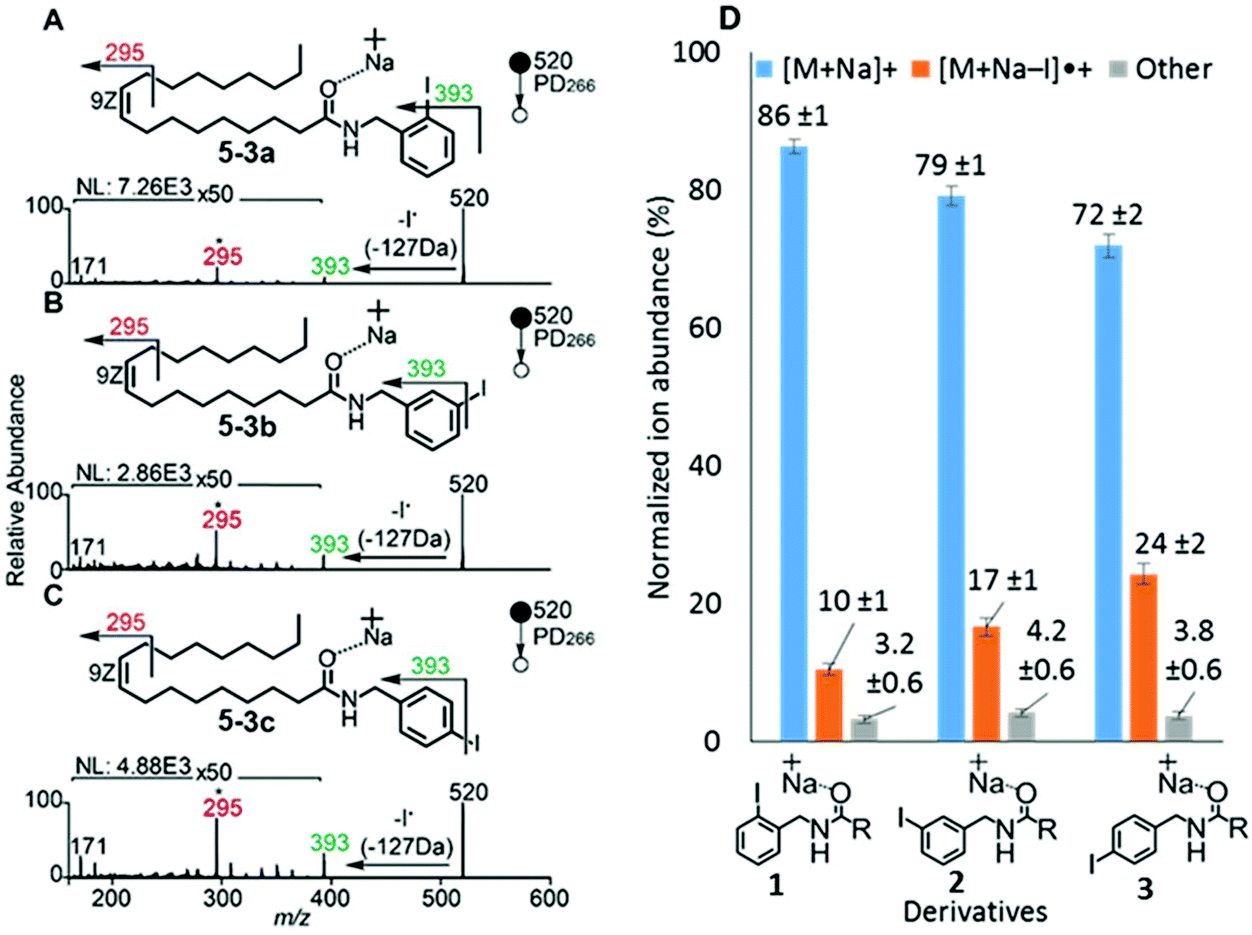 | ||
| Fig. 2 PD266 mass spectra of [M + Na]+ ions derived from FA 18:1(9Z) derivatized with (A) 2-iodobenzylamine (1), (B) 3-iodobenzylamine (2), and (C) 4-iodobenzylamine (3). ‘NL’ represents the ion count for the base peak. (D) Normalized product ion abundance from the PD mass spectra; R = FA 18:1(9Z), averaged over 80 scans. Error bars represent 1 standard deviation from the mean. | ||
3.3. Selection of the derivatization linker and aryl-halogen motif
The composition of the covalent linkage between the derivatization reagent and carboxylic acid moiety in the FA also plays an important role in the overall optimization of photodissociation behaviour. Among the isomeric iodobenzylamine derivatives tested above, the para-substituted derivative 3 exhibited the highest photoproduct yield, with 28% of the precursor ion population converted to products. To compare the amide to the corresponding ester-linkage, the 4-iodobenzyl ester derivative of FA 18:1(9Z) (4, Fig. 1) was prepared and the [M + Na]+ ion was subjected to PD266. The resulting PD266 mass spectrum is represented in Fig. 3A and a direct comparison of these data (and the corresponding PD266/CID mass spectra) with compound 3 is provided as ESI (Fig. S5 and S6†). Analysis of these results shows a photo-conversion of 26% for the ester 4, which is slightly lower than that for the corresponding amide, 3 (Fig. 4). Julian and co-workers previously achieved near quantitative conversion of derivatized proteins at 266 nm using p-iodobenzoate and p-iodobenzamide groups.36 This report guided the design of similar derivatives in our study through incorporation of a benzoyl functional group as part of the linker connecting the photo-labile reagent and the FA. As illustrated in Fig. 1, this test set included derivatives with a p-iodoacetophenone core and either an amide (5) or ester (6) linkage to FA 18:1(9Z). Relative to 3, derivatives based on p-iodoacetophenone displayed significantly increased abundance of [M + Na − I]˙+ photoproduct ions at 36% (5) and 33% (6) (shown as orange bars in Fig. 4). Moreover, the summed relative abundance of secondary product ions arising from prompt radical-directed dissociation also increase relative to 3 (grey bars Fig. 4). In each case, characteristic product ions are observed arising from radical-driven allylic cleavage that pinpoint the position of unsaturation (Fig. 3) and overall 57% (5) and 62% (6) of the total ion population is accounted for by photo-product ions; a marked enhancement relative to derivatives 3 and 4 (Fig. 4).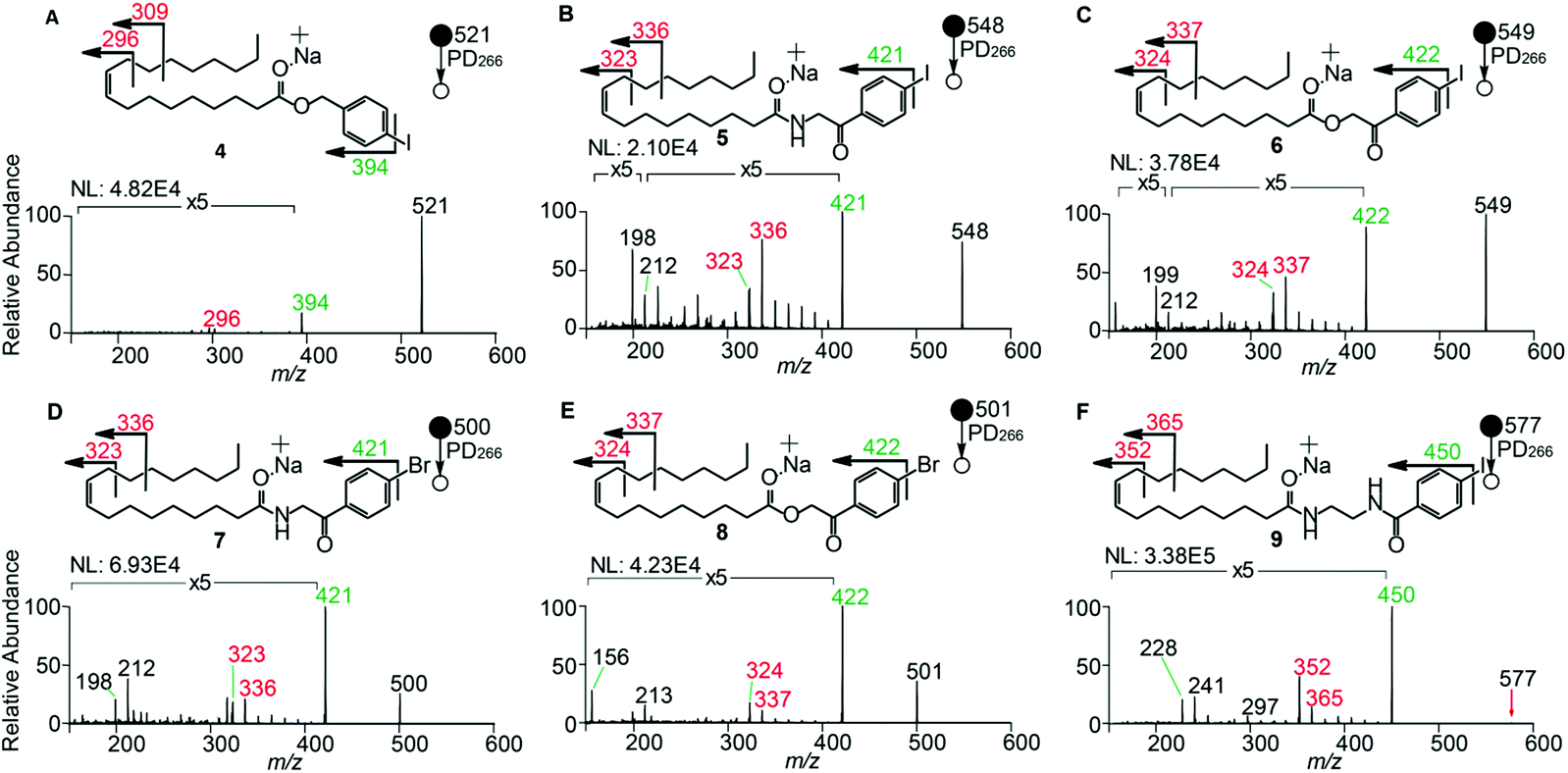 | ||
| Fig. 3 PD266 mass spectra of FA 18:1(9Z) derivatized as structures (A) 4, (B) 5, (C) 6, (D) 7, (E) 8, and (F) 9. Methanolic solutions of these compounds were subjected to direct infusion. Mass spectra represent an average of 80 scans. ‘NL’ represents the ion count for the base peak. The [M + Na − I]˙+ product ions are labelled in green and are not included in the magnified regions. | ||
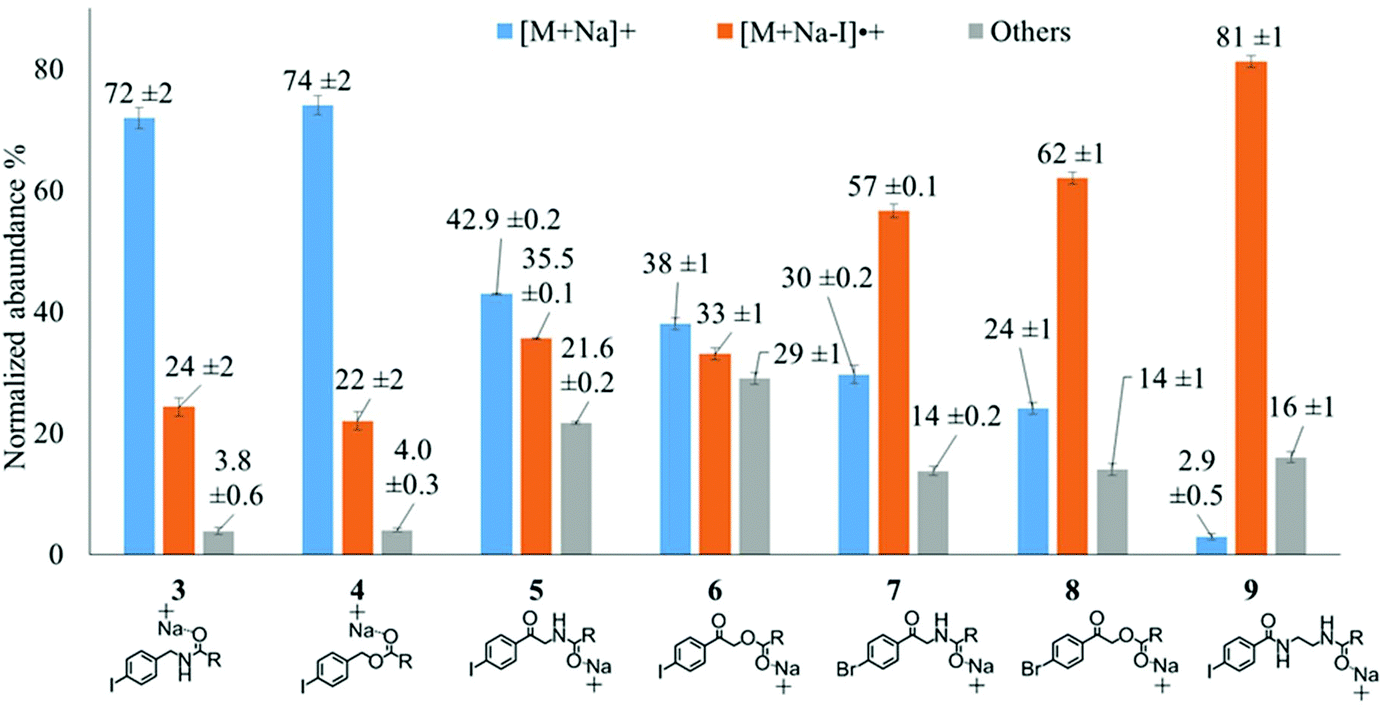 | ||
| Fig. 4 Analysis of the PD266 photo-conversion of FA 18:1(9Z) as derivatives 3–9. All PD266 mass spectra were acquired via direct infusion of a methanolic solution of the compound with the bars representing the mean normalized ion counts over 80 individual scans and the error bars representing one standard deviation in the mean. | ||
Given the availability of suitable precursors, the p-bromoacetophenone core was also investigated as a photo-labile functional group. The analogues 7 and 8 (Fig. 1) are based on commercially available derivatives employed for improving chromatographic resolution and quantification of FAs in HPLC workflows.45,55 Interestingly, the [M + Na]+ ions formed from these derivatives displayed photo-conversions of 71% (7) and 76% (8) of their precursor ion populations; greater than that of their iodinated analogues 5 and 6 (Fig. 3). This result was surprising given the bond dissociation energies of the aryl–bromide bonds (BDE ∼ 84 kcal mol−1) are typically greater than the corresponding aryl-iodides (BDE ∼ 67 kcal mol−1).56 These relative thermochemistries could account for the greater proportion of secondary product ions in the PD266 mass spectra of the iodides 5 (22%) and 6 (29%) compared with the bromides 7 (14%) and 8 (14%). In the latter case, the higher BDE of the aryl–bromide bond means less residual energy in the dissociation partners and, presumably, less internal energy in the radical ion after photolysis (cf., 266 nm ∼ 107.5 kcal mol−1). The extent of nascent product ion activation will, however, depend on the partitioning of energy into the various degrees of freedom, which is unknown for these systems. The observations of overall greater conversion of the bromides compared with the iodides also stands in contrast to the previously reported photodissociation of the archetypal 4-bromoanilinium and 4-iodoanilium cations.47 In their study, Hansen et al. found more efficient photodissociation of the iodo- over the bromo-anilinium cations for the spectral region around 266 nm. Interestingly however, the relative photoproduct yield of the two congeners inverts with just a small shift to the blue with the photofragmentation of 4-bromoanilinium favoured at wavelengths shorter than 245 nm. These findings stress the importance of the photodissociation dynamics in the selection and optimization of the photolabile functional group and prompted the investigation of the PD action spectroscopy of selected compounds from the test set (vide infra). On balance however, the more even distribution of photo-products over both the [M + Na − X]˙+ (where X = I or Br) and the secondary RDD channels (cf. orange and grey bars in Fig. 4) led us to select the aryl-iodide scaffold 5 for further optimization.
While displaying significant improvements in PD266 conversion, the iodo- and bromo-acetophenone derivatives 5–8 still showed significant abundance of residual precursor ions following irradiation (24–43%, see Fig. 4). Considering again the derivatized peptides investigated by Ly et al. we elected to further modify the covalent linker between the chromophore and the lipid to incorporate a benzamide group.36 The N-(2-aminoethyl)-4-iodobenzamide (NIBA) reagent was thus prepared and conjugated to FA 18:1(9Z) to yield compound 9 (Fig. 1). In the NIBA reagent, the carbonyl functionality appended to the aryl-halide is an amide moiety rather than the ketone examined previously (cf.9 and 5 in Fig. 1). The PD266 mass spectrum of 9 is shown in Fig. 3F and reveals near-complete conversion (97%) of the [M + Na]+ precursor ion at m/z 577 to the [M + Na − I]˙+ radical cation (m/z 450) and secondary product ions arising from RDD. Partitioning between the photo-products is quantified in Fig. 4 with the [M + Na − I]˙+ species representing the majority of the product ion population at 81% with the remaining product ions contributing 16%. The efficient photodissociation of the ionized derivative 9 at 266 nm could be due to strong absorption at this wavelength and/or favourable excited state dissociation dynamics. To further rationalise the gas phase PD266 observations, solution phase UV-absorption measurements were undertaken for 9 along with analogues 7 and 5. These results are summarised in the ESI (Fig. S7 and S8†) and reveal that in acidified aqueous acetonitrile (95:5 acetonitrile:water with 0.1% formic acid) derivative 9 has a maximum absorption of λmax = 250 nm compared with 255 nm (7) and 265 nm (5). Moreover, at 266 nm the molar absorptivity of 9 is the lowest among these three derivatives. Taken together, these observations suggest that the enhanced photo-conversion of 9 is more likely to be due to efficient dissociation on an excited state surface rather than as a result of stronger absorption at this wavelength.
To further explore the exceptional photo-conversion of the NIBA derivatized FA 18:1(9Z), 9, a tuneable laser system was deployed to measure its PD action spectrum and to contrast its photofragmentation behaviour with the structurally related analogue 5. The PD action spectra of the gas phase [M + Na]+ ions formed from 9 and 5 are plotted in Fig. 5 and reveal that for the NIBA derivative (9) aryl-iodide photolysis is maximised near 266 nm compared to the analogue 5 that is significantly red-shifted with maximum near 305 nm. These data indicate that the gas phase absorption/dissociation characteristics of derivative 9 are optimized for fixed wavelength photo-fragmentation at 266 nm while 5 could be deployed to greater effect with an alternative light source at longer wavelengths. Electronic structure calculations show the predicted electronic transitions for 9 and 5 are in good agreement with experiment when an empirical 0.3 eV red-shift is applied (Fig. 5). Calculations also predict that for compound 3, in the complete absence of the carbonyl linker motif, excitation will shift significantly to higher photon energies. Exploring the aryl carbon–iodine coordinate on ground and excited state potentials provides some insights into these trends.57 Notably for the computational model of the sodiated NIBA derivative 9 (rigid-body scan, referenced at 0 Å S0 equilibrium geometry, in Fig. 5B), the bound ππ* state (green) curve is cut by dissociative πσ* and nσ* states (orange and red). Crucially, these results suggest that efficient internal conversion from the ππ* to the πσ*/nσ* states is suitably arranged for efficient carbon–iodine dissociation, as the latter states are unbound with respect to the carbon–iodine coordinate. These computed potential energy surfaces therefore indicate that the efficient photodissociation of derivative 9 at 266 nm is facilitated by S0 → ππ* photo-excitation, followed by internal conversion to either the πσ* or nσ* states leading to facile bond homolysis. Importantly for the other analogues investigated either the S0 → ππ* transition is shifted to higher energies (i.e., the blue shift predicted for 3) and/or the crossing point from ππ* → πσ*/nσ* states is further displaced from the equilibrium geometry on the S0 surface. That is, the crossing points are displaced by ca. 0.2 Å in the case of 5 but by 0.0–0.1 Å for 9 (cf. vertical arrows in Fig. 5B and C, respectively). This might also be a cause of the extraordinarily efficient aryl–iodine photolysis yield for compound 9. Ultimately, the combination of PD action spectroscopy and excited state electronic structure calculations provides a clear framework for the structure–activity optimization undertaken here. Moreover, they predict that the size and structure of the FA will play a limited role in modulating the absorption/dissociation characteristics at 266 nm.
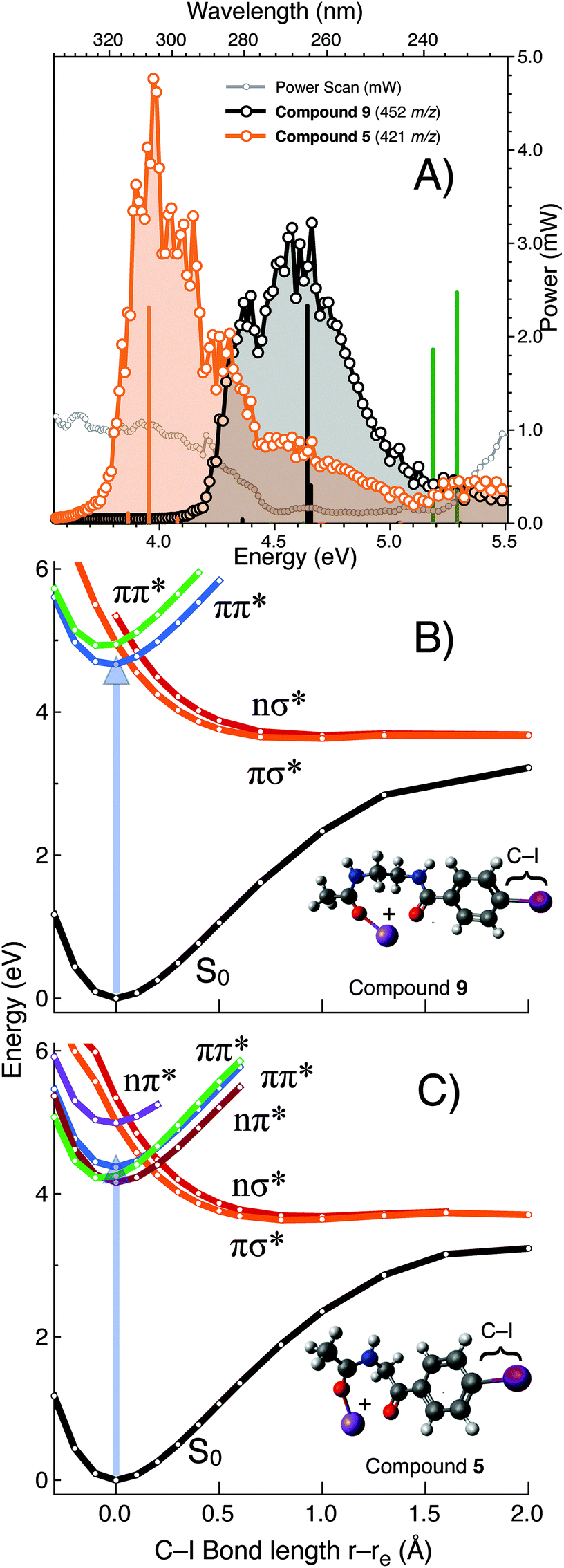 | ||
| Fig. 5 (A) Photodissociation action spectra of FA 18:1(9Z) derivatized as 5 and 9 between 230–350 nm. Normalized abundance of PD product ions [M + Na − I]˙+ are corrected for power fluctuations and plotted as a function of incident wavelength. Electronic vertical transition energies computed using the SCS-CC2/def2-SVPD method for derivatives 3 (green), 5 (orange) and 9 (black) are shown as vertical lines (with a redshift of 0.3 eV). Computed potentials for dissociation along the carbon–iodine coordinate on ground (S0) and excited state singlet surfaces for models of (B) compound 9 and (C) compound 5. In the model systems used in these calculations the FA is represented as a methyl group. For reference 266 nm ∼ 4.66 eV. | ||
3.4. Radical-directed dissociation of NIBA derivatized FAs
As described above, PD266 of the sodium adduct ion of NIBA derivatized FA 18:1(9Z) gave efficient conversion to the [M + Na − I]˙+ along with other photoproducts arising from RDD of the FA chain. Among the latter fragments, the most abundant product ions are observed at m/z 241, 228 and 352 (Fig. 3F) and can be rationalised as arising from an initial hydrogen atom abstraction from the FA chain by the newly formed phenyl radical (Scheme 3). As previously described, the thermodynamics of the hydrogen-atom transfer likely dictates the abundance of product ions observed in the PD266 and PD266/CID mass spectra.30,39 This is illustrated for the most abundant fragments in Scheme 3 where RDD is favoured in instances where the hydrogen is abstracted from an activated position (e.g., m/z 241) or where bond cleavage itself leads to a stabilised radical population (e.g., m/z 228). The prominent ion pair at m/z 241 and 228 are thus assigned as markers for the NIBA derivatization reagent and can be used as strong transitions for identifying thus tagged FAs in complex mixtures (vide infra). The preference for fragmentation at activated positions also confirms that the PD266 and PD266/CID mass spectra will be highly sensitive to differences in molecular structure including the position(s) of carbon–carbon double bonds. For example, the abundant product ion at m/z 351 (Fig. 3F) corresponds to scission of the C11–C12 bond at the allylic position and thus serves a marker for the site of unsaturation in FA 18:1(9Z). Interestingly, upon re-isolation and activation of the [M + Na − I]˙+ in a PD266/CID experiment the mechanism for cleavage at this allylic position shifts to favour formation of the analogous radical cation at m/z 352. This shift reflects the modified internal energy distribution of the radical ion population opening up new possibilities for hydrogen-atom migration and associated bond cleavage mechanisms (cf.Scheme 3). Importantly, both the prompt dissociation arising directly from PD266 and that arising from subsequent CID give fragmentation patterns that are sensitive to molecular structure and can therefore be deployed to discriminate between isomeric FAs. While there is a small cost in instrument duty cycle in undertaking the ion trap MS3 (PD266/CID) compared to the MS2 (PD266) scan, this is well compensated by the signal-to-noise enhancement in the former experiment. The combination of the high photo-conversion of the NIBA derivative with the efficiency of the radical-directed dissociation upon CID means that ca. 50% of the [M + Na]+ precursor can be converted through to structurally diagnostic product ions (see ESI, Fig. S9†).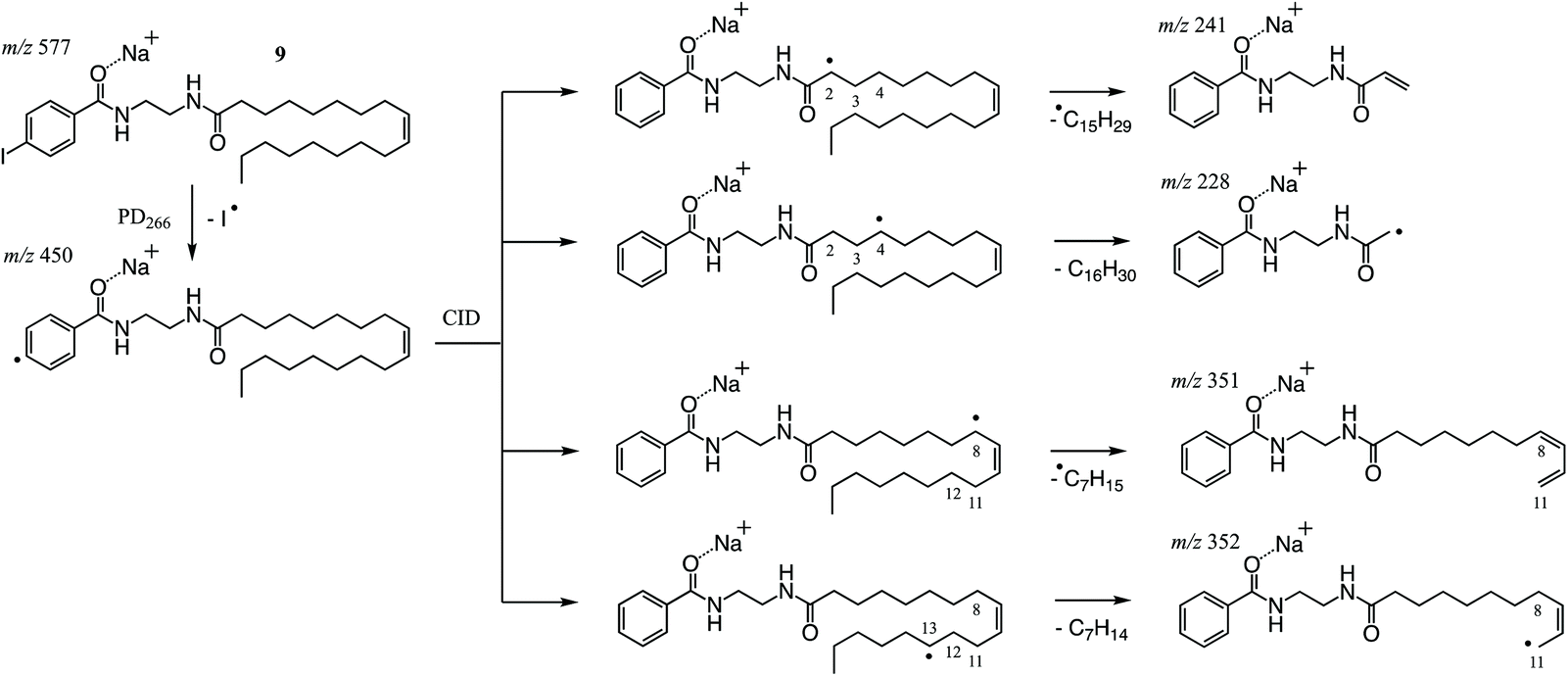 | ||
| Scheme 3 Proposed mechanism for radical-directed dissociation leading to the abundant product ions at m/z 241, 228, 351 and 352 observed in PD266 and PD266/CID mass spectra of the NIBA derivatized FA 18:1(9Z), 9. | ||
3.5. LC-PD266 performance of NIBA derivatives
In order to evaluate the compatibility of the optimized reagent NIBA with a reversed-phase LC-PD266 workflow, a commercially available mixture of 37 FAs was selected. This widely used reference material includes FAs of differing chain lengths, degrees of unsaturation, stereo- and regio-chemistries with each component present in the mixture as its methyl ester (i.e., FAMEs). For the evaluation, the 37 FAMEs mixture was hydrolysed to release the FAs, which were subsequently derivatized with the NIBA reagent and loaded onto a C30 reverse-phase column coupled with the ion trap mass spectrometer. The ion trap was configured for a data-dependent workflow where a target list of precursor masses corresponding to the sodium adduct ions of each NIBA-modified FA were programmed across the appropriate segment of the chromatographic run. Full experimental details of chromatographic methods, including the schedule of MS/MS events, are provided as ESI (Tables S1, S2, and S4†). Under these chromatographic conditions all FAs could be detected over a 36 min gradient and followed the expected trends of longer retention times for longer chain FAs (see ESI, Fig. S10†). As typically observed on reversed-phase columns unsaturated FAs eluted earlier than their saturated counterparts with increasing unsaturation leading to earlier elution. As a consequence of these behaviours, polyunsaturated FAs were often found to co-elute with shorter chain more saturated species (e.g., FA 20:5 co-eluted with FA 18:3 and FA 14:0), however, mass discrimination removed any ambiguity from the chromatographic analysis. Under these conditions 10–30 targeted PD266 spectra were obtained across the chromatographic peak for each derivatized FA. Combined with the effective photo-conversion of the derivatized FAs to their corresponding [M + Na − I]˙+ radical cations, this provided excellent selectivity in assigning chromatographic features to individual FAs as illustrated in Fig. 6 (with further examples in ESI, Fig. S10†).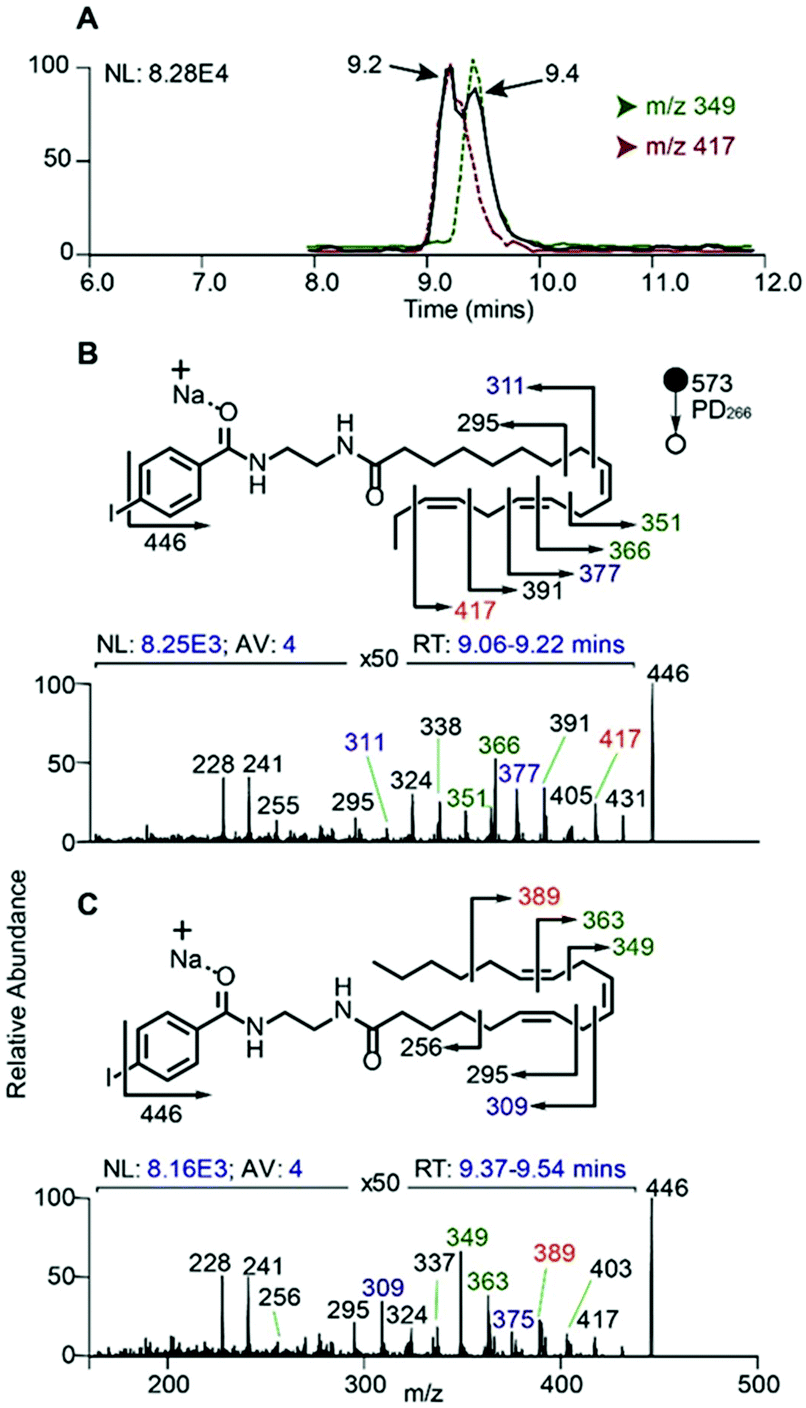 | ||
| Fig. 6 (A) Extracted ion chromatograms of the [M + Na − I]˙+ ions at m/z 446 (black trace) arising from PD266 of FA 18:3 isomers as NIBA derivatives as well as the RDD product ions m/z 349 (green) and m/z 417 (red) characteristic for FA 18:3(6Z,9Z,12Z) and FA 18:3(9Z,12Z,15Z), respectively. PD266 mass spectra of NIBA derivatives of (B) FA 18:3(9Z,12Z,15Z) eluting at 9.2 min and (C) FA 18:3(6Z,9Z,12Z) eluting at 9.4 min. | ||
In addition to providing diagnostic [M + Na − I]˙+ radical cations, the LC-PD266 analysis of NIBA derivatized FAs yielded structurally diagnostic product ions arising from prompt RDD of the FA structure. For example, the FA mixture included two isomers of linolenic acid namely, α-linolenic acid, FA 18:3(9Z,12Z,15Z) and γ-linolenic acid, FA 18:3(6Z,9Z,12Z). NIBA derivatives of these double bond positional isomers were partially resolved by the reversed-phase chromatography as illustrated by the extracted ion chromatogram of the [M + Na − I]˙+ at m/z 446 that is common to both isomers (black trace, Fig. 6A). PD266 mass spectra obtained from the partially resolved features centred at 9.2 and 9.4 min in the chromatogram (Fig. 6B and C, respectively) show distinctive fragmentation patterns that facilitate de novo assignment of the two FA isomers. For example, the PD266 mass spectrum of the peak at 9.2 min displays prominent product ions at m/z 431, 417 and 391 (Fig. 5B), which can be assigned to radical-directed cleavage either side of a Δ15 double bond and consistent with the assignment of this chromatographic feature to α-linolenic acid, FA 18:3(9Z,12Z,15Z). These same product ions are of significantly lower abundance in the PD266 mass spectrum obtained across the chromatographic peak eluting at 9.4 min (Fig. 6C). Instead, this spectrum displays a higher abundance of ions at m/z 389 and 349 associated with the Δ12 double bond along with an abundant ion at m/z 309 resulting from allylic cleavage adjacent to a Δ6 double bond, thereby confirming the assignment of this feature to γ-linolenic acid, FA 18:3(6Z,9Z,12Z). While chromatographic resolution of the two derivatized FA 18:3 isomers was incomplete, the diagnostic PD266 fragmentation of the two species enabled the generation of extracted ion chromatograms based on unique radical-directed dissociation product ions that could uniquely define their individual elution profiles (Fig. 6A).
3.6. Influence of FA chain length and unsaturation on photo-conversion
Acquiring the LC-PD266 across a test set of 37 structurally distinct FAs derivatized with the NIBA reagent allowed the evaluation of the influence of the FA structure on both photo-conversion and the distribution of the photoproducts over different dissociation channels. Across this test set, with carbon chain lengths ranging from FA 6:0 up to FA 24:0, conversion efficiency of the [M + Na]+ precursor ions to photoproducts at 266 nm ranged between 93 and 99% (ESI Fig. S11†). Interestingly, the abundance of photoproducts other than the [M + Na − I]˙+ ions vary more markedly, ranging from 7% up to 35%. In general, the contribution of products arising from prompt RDD to the total ion abundance decreases from FA 8:0, where it represents up to 35% of the signal, up to longer chains such as FA 17:0 where it declines to 17% and remains reasonably stable at this level across the longer chains in the test set. This trend likely reflects the more facile redistribution of the excess energy from photolysis of aryl–iodine bond (vide supra) with increasing chain length. Internal redistribution of the excess energy competes with intra-molecular rearrangement (e.g., hydrogen atom transfer) and thus radical-directed dissociation. An interesting exception to this trend however, is observed for NIBA derivatized FA 6:0 which has a 93% photo-conversion of the precursor but –despite its short chain– partitions just 7% of its product ion population into channels arising from RDD. One possible explanation for this contrast is a minimal steric requirement for hydrogen-atom transfer from the FA chain to the phenyl radical site in the [M + Na − I]˙+ ion population. The reactions of dioxygen with radical ions have previously been used to probe radical migration58 and so, in the present study, [M + Na − I]˙+ radical ions formed by PD226 of NIBA derivatives from FA 6:0 and FA 8:0 were mass-selected in the ion trap and stored in the presence of background dioxygen for variable time periods (1000–10000 ms, ESI, Fig. S12†). Under these conditions, the radical cation formed from the FA 6:0 derivative at m/z 284 showed evidence of undergoing a reaction with dioxygen to form abundant product ions at m/z 316 of the form [M + Na − I + O2]˙+. This reaction is consistent with addition of dioxygen to a phenyl-type radical to produce a phenylperoxyl radical. This chemistry has been studied extensively in the gas phase59 and, in this context, suggests that much of the radical population comprising m/z 284 remains localised at the phenyl position (i.e., the site of the aryl–iodine homolysis). In contrast, isolation of the [M + Na − I]˙+ radical cations formed from derivatized FA 8:0 (m/z 312) in the presence of dioxygen produced minimal [M + Na − I + O2]˙+ product ions at m/z 344 but rather favoured reaction to an even-electron [M + Na − I + O2 − HO]+ species at m/z 327. This reaction chemistry has been reported for distonic radical ions incorporating alkyl-type radicals60,61 and strongly suggests that for the FA 8:0 the nascent phenyl radical formed upon photolysis undergoes facile migration onto the hydrocarbon chain; a process facilitated by the additional two methylene units in its carbon chain.
Unsaturation within the hydrocarbon chains has a significant influence on the physical and chemical properties of FAs. Therefore, the influence of unsaturation on photo-conversion of FA 18:X derivatives (where X = 0, 1, 2 and 3) was systematically explored. PD266 mass spectra of these derivatives consistently exhibited photo-conversion of the precursor ion populations exceeding 98%, and apparently irrespective of the degree of unsaturation (Fig. 7). However, closer inspection of these data reveals subtle differences in the partitioning of photoproducts between [M + Na − I]˙+ and product ions arising from FA chain cleavage. As illustrated by the grey bars in Fig. 7, positions of unsaturation proximate to the amide linkage induce a higher relative abundance of ions arising from radical-directed dissociation of the FA chain. Taking FA 18:3(6Z,9Z,12Z) for example, 19% of the photo-products are partitioned into chain-cleavage product ions compared to just 13% for the FA 18:3(9Z,12Z,15Z) isomer. We speculate that the curvature in the FA chain induced at the Δ6 position in FA 18:3(6Z,9Z,12Z) promotes more facile hydrogen-atom transfer from the carbon chain to the aromatic ring. Consistent with this hypothesis, the relative abundance of FA cleavage product ions was moderately higher for derivatives of the polyunsaturates FA 20:4, FA 20:5 and FA 22:6, due to the presence of Δ5 or Δ4 double bonds (Fig. S11†).
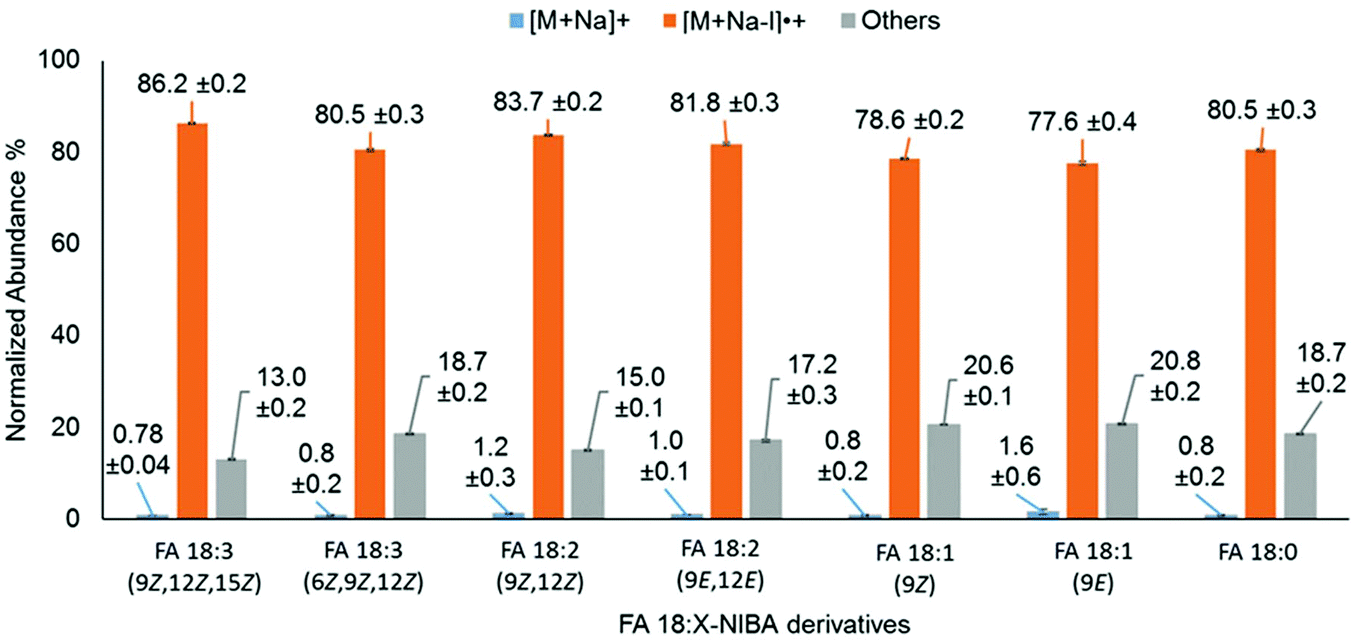 | ||
| Fig. 7 Normalized product ion abundances from PD266 of FA18:X isomers derivatized with NIBA reagent (where X = 0, 1, 2 and 3). | ||
4. Conclusions
Through a systematic structure–activity investigation, we have developed a novel derivatization reagent, NIBA (9), which, when conjugated with FAs, undergoes highly efficient photodissociation at 266 nm enabling selective detection and structural characterisation of these lipids. Important structural features of the NIBA reagent include the para-substitution on the aromatic chromophore and incorporation of the benzamide functional group that results in a significant blue shift in the absorption/dissociation characteristics and alignment with the 266 nm light source. When conjugated to FAs, the NIBA reagent was efficiently ionized by ESI to yield [M + Na]+ ions that exhibited photo-conversions to [M + Na − I]˙+ (and secondary radical-directed dissociation products) above 95%. Importantly, the efficient photolysis of derivatized FAs was found to be highly conserved across a large test set of structurally diverse lipids with PD266 (and PD266/CID) generating characteristic fragmentation patterns that enabled discrimination between isomeric FAs even when reversed-phase chromatographic resolution was incomplete. Based on these traits, the combination of NIBA derivatization with the LC-PD266 workflow has significant potential for the identification of FAs in complex biological mixtures including the possibility for the discovery of new lipids through de novo structural assignment. Finally, the framework for tuning the absorption/dissociation characteristics of derivatization reagents described herein can be deployed in the future to optimize reagents to be used in combination with other light sources such as the PD213 systems which have recently been commercialised.Abbreviations
| ACN | Acetonitrile |
| AMPP+ | N-(4-Aminomethylphenyl)pyridinium |
| CID | Collision-induced dissociation |
| DIPEA | N,N-Diisopropylethylamine |
| DMF | Dimethyl formamide |
| ESI | Electrospray ionization |
| EI | Electron ionization |
| FA | Fatty acid |
| FAME | Fatty acid methyl ester |
| FAIBA | Fatty acid iodo-benzyl amide |
| GC | Gas chromatography |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate |
| HPLC | High performance liquid chromatography |
| LC | Liquid chromatography |
| NCE | Normalized collision energy |
| NIBA | N-(2-Aminoethyl)-4-iodobenzamide |
| MS | Mass spectrometry |
| MS/MS | Tandem mass spectrometry |
| MTBE | Methyl tert-butyl ether |
| NIBA | N-(2-Aminoethyl)-4-iodobenzamide |
| PD | Photodissociation |
| RDD | Radical-directed dissociation |
| UVPD | Ultra-violet photodissociation |
Author contributions
S. J. B., N. R. B. and T. W. M. conceived and designed the project. Experiments and calculations were performed by V. R. N., B. I. M., D. L. M. and S. J. P. M. while B. L. J. P. and A. J. T. guided instrument modification and data interpretation. V. R. N., D. L. M., S. J. B. and N. R. B. B. wrote the manuscript. All authors reviewed the manuscript and have given approval to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support for this research from the Australian Research Council Discovery Program (DP150101715 and DP190101486). S. J. B., V. R. N., N. R. B., B. L. J. P. and D. L. M. acknowledge the support of the Central Analytical Research Facility operated by the Institute for Future Environments (QUT). V. R. N. is grateful for a QUT Postgraduate Research Award. This work was supported by computational resources provided by the Australian Government through the National Computation Infrastructure under the National Computational Merit Allocation Scheme.References
- M. Gurr, J. Harwood and K. Frayn, Lipid biochemistry, Blackwell Publishing Company, Oxford, 5th edn, 2002 Search PubMed.
- E. Fahy, S. Subramaniam, H. A. Brown, C. K. Glass, A. H. Merrill Jr., R. C. Murphy, C. R. Raetz, D. W. Russell, Y. Seyama and W. Shaw, A comprehensive classification system for lipids, Eur. J. Lipid Sci. Technol., 2005, 107(5), 337–364 CrossRef CAS.
- E. Fahy, S. Subramaniam, R. C. Murphy, M. Nishijima, C. R. Raetz, T. Shimizu, F. Spener, G. van Meer, M. J. Wakelam and E. A. Dennis, Update of the LIPID MAPS comprehensive classification system for lipids, J. Lipid Res., 2009, 50(Supplement), S9–S14 CrossRef.
- M. Sud, E. Fahy, D. Cotter, A. Brown, E. A. Dennis, C. K. Glass, A. H. Merrill Jr., R. C. Murphy, C. R. Raetz and D. W. Russell, LMSD: Lipid maps structure database, Nucleic Acids Res., 2007, 35(suppl_1), D527–D532 CrossRef CAS.
- A. P. Simopoulos, Essential fatty acids in health and chronic disease, Am. J. Clin. Nutr., 1999, 70(3), S560–S569 CrossRef.
- A. Z. Fernandis and M. R. Wenk, Membrane lipids as signaling molecules, Curr. Opin. Lipidol., 2007, 18(2), 121–128 CrossRef CAS.
- F. Röhrig and A. Schulze, The multifaceted roles of fatty acid synthesis in cancer, Nat. Rev. Cancer, 2016, 16(11), 732 CrossRef.
- W. W. Christie, Gas chromatography-mass spectrometry methods for structural analysis of fatty acids, Lipids, 1998, 33(4), 343–353 CrossRef CAS.
- O. Quehenberger, A. M. Armando and E. A. Dennis, High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography–mass spectrometry, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2011, 1811(11), 648–656 CrossRef CAS.
- W. W. Christie, E. Y. Brechany, S. B. Johnson and R. T. Holman, A comparison of pyrrolidide and picolinyl ester derivatives for the identification of fatty acids in natural samples by gas chromatography-mass spectrometry, Lipids, 1986, 21(10), 657–661 CrossRef CAS.
- G. Dobson and W. W. Christie, Structural analysis of fatty acids by mass spectrometry of picolinyl esters and dimethyloxazoline derivatives, TrAC, Trends Anal. Chem., 1996, 15(3), 130–137 CrossRef CAS.
- C. Pettinella, S. H. Lee, F. Cipollone and I. A. Blair, Targeted quantitative analysis of fatty acids in atherosclerotic plaques by high sensitivity liquid chromatography/tandem mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 850(1–2), 168–176 CrossRef CAS.
- H. Takahashi, H. Suzuki, K. Suda, Y. Yamazaki, A. Takino, Y.-I. Kim, T. Goto, Y. Iijima, K. Aoki and D. Shibata, Long-chain free fatty acid profiling analysis by liquid chromatography–mass spectrometry in mouse treated with peroxisome proliferator-activated receptor α agonist, Biosci., Biotechnol., Biochem., 2013, 77(11), 2288–2293 CrossRef CAS.
- E. Hewawasam, G. Liu, D. W. Jeffery, B. S. Muhlhausler and R. A. Gibson, A validated method for analyzing polyunsaturated free fatty acids from dried blood spots using LC–MS/MS, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2017, 125, 1–7 CrossRef CAS.
- J. L. Kerwin, A. M. Wiens and L. H. Ericsson, Identification of fatty acids by electrospray mass spectrometry and tandem mass spectrometry, J. Mass Spectrom., 1996, 31(2), 184–192 CrossRef CAS.
- R. C. Murphy, Tandem mass spectrometry of lipids: molecular analysis of complex lipids, Royal Society of Chemistry, 2014 Search PubMed.
- S. M. Lamos, M. R. Shortreed, B. L. Frey, P. J. Belshaw and L. M. Smith, Relative quantification of carboxylic acid metabolites by liquid chromatography–mass spectrometry using isotopic variants of cholamine, Anal. Chem., 2007, 79(14), 5143–5149 CrossRef CAS.
- W.-C. Yang, J. Adamec and F. E. Regnier, Enhancement of the LC/MS analysis of fatty acids through derivatization and stable isotope coding, Anal. Chem., 2007, 79(14), 5150–5157 CrossRef CAS.
- X. E. Zhao, S. Zhu and H. Liu, Recent progresses of derivatization approaches in the targeted lipidomics analysis by mass spectrometry, J. Sep. Sci., 2020, 43(9–10), 1838–1846 CrossRef CAS.
- J. G. Bollinger, G. Rohan, M. Sadilek and M. H. Gelb, LC/ESI-MS/MS detection of FAs by charge reversal derivatization with more than four orders of magnitude improvement in sensitivity, J. Lipid Res., 2013, 54(12), 3523–3530 CrossRef CAS.
- J. G. Bollinger, W. Thompson, Y. Lai, R. C. Oslund, T. S. Hallstrand, M. Sadilek, F. Turecek and M. H. Gelb, Improved sensitivity mass spectrometric detection of eicosanoids by charge reversal derivatization, Anal. Chem., 2010, 82(16), 6790–6796 CrossRef CAS.
- R. V. Tatituri, B. J. Wolf, M. B. Brenner, J. Turk and F.-F. Hsu, Characterization of polar lipids of Listeria monocytogenes by HCD and low-energy CAD linear ion-trap mass spectrometry with electrospray ionization, Anal. Bioanal. Chem., 2015, 407(9), 2519–2528 CrossRef CAS.
- F.-F. Hsu, Characterization of hydroxyphthioceranoic and phthioceranoic acids by charge-switch derivatization and CID tandem mass spectrometry, J. Am. Soc. Mass Spectrom., 2016, 27(4), 622–632 CrossRef CAS.
- C. Hu, M. Wang, Q. Duan and X. Han, Sensitive analysis of fatty acid esters of hydroxy fatty acids in biological lipid extracts by shotgun lipidomics after one-step derivatization, Anal. Chim. Acta, 2020, 1105, 105–111 CrossRef CAS.
- J. Adams and M. L. Gross, Tandem mass spectrometry for collisional activation of alkali metal-cationized fatty acids: a method for determining double bond location, Anal. Chem., 1987, 59(11), 1576–1582 CrossRef CAS.
- V. R. Narreddula, N. R. Boase, R. Ailuri, D. L. Marshall, B. L. Poad, M. J. Kelso, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Introduction of a fixed-charge, photolabile derivative for enhanced structural elucidation of fatty acids, Anal. Chem., 2019, 91(15), 9901–9909 CrossRef CAS.
- A. Devakumar, D. K. O'Dell, J. M. Walker and J. P. Reilly, Structural analysis of leukotriene C4 isomers using collisional activation and 157 nm photodissociation, J. Am. Soc. Mass Spectrom., 2008, 19(1), 14–26 CrossRef CAS.
- D. R. Klein and J. S. Brodbelt, Structural characterization of phosphatidylcholines using 193 nm ultraviolet photodissociation mass spectrometry, Anal. Chem., 2017, 89(3), 1516–1522 CrossRef CAS.
- E. Ryan, C. Q. N. Nguyen, C. Shiea and G. E. Reid, Detailed structural characterization of sphingolipids via 193 nm ultraviolet photodissociation and ultra high resolution tandem mass spectrometry, J. Am. Soc. Mass Spectrom., 2017, 28(7), 1406–1419 CrossRef CAS.
- H. T. Pham, T. Ly, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Differentiation of complex lipid isomers by radical-directed dissociation mass spectrometry, Anal. Chem., 2012, 84(17), 7525–7532 CrossRef CAS.
- M. Fang, Y. Rustam, M. Palmieri, O. M. Sieber and G. E. Reid, Evaluation of ultraviolet photodissociation tandem mass spectrometry for the structural assignment of unsaturated fatty acid double bond positional isomers, Anal. Bioanal. Chem., 2020, 1–13 Search PubMed.
- D. R. Klein, D. D. Holden and J. S. Brodbelt, Shotgun analysis of rough-type lipopolysaccharides using ultraviolet photodissociation mass spectrometry, Anal. Chem., 2016, 88(1), 1044–1051 CrossRef CAS.
- P. E. Williams, D. R. Klein, S. M. Greer and J. S. Brodbelt, Pinpointing double bond and sn-positions in glycerophospholipids via hybrid 193 nm ultraviolet photodissociation (UVPD) mass spectrometry, J. Am. Chem. Soc., 2017, 139(44), 15681–15690 CrossRef CAS.
- M. Blevins, V. James, C. Herrera, A. Purcell, M. S. Trent and J. S. Brodbelt, Unsaturation Elements and other Modifications of Phospholipids in Bacteria: New Insight from UVPD Mass Spectrometry, Anal. Chem., 2020, 92(13), 9146–9155 CrossRef CAS.
- T. Ly and R. R. Julian, Residue-specific radical-directed dissociation of whole proteins in the gas phase, J. Am. Chem. Soc., 2008, 130(1), 351–358 CrossRef CAS.
- T. Ly, X. Zhang, Q. Sun, B. Moore, Y. Tao and R. R. Julian, Rapid, quantitative, and site specific synthesis of biomolecular radicals from a simple photocaged precursor, Chem. Commun., 2011, 47(10), 2835–2837 RSC.
- H. T. Pham and R. R. Julian, Mass shifting and radical delivery with crown ether attachment for separation and analysis of phosphatidylethanolamine lipids, Anal. Chem., 2014, 86(6), 3020–3027 CrossRef CAS.
- H. T. Pham and R. R. Julian, Characterization of glycosphingolipid epimers by radical-directed dissociation mass spectrometry, Analyst, 2016, 141(4), 1273–1278 RSC.
- H. T. Pham, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Rapid differentiation of isomeric lipids by photodissociation mass spectrometry of fatty acid derivatives, Rapid Commun. Mass Spectrom., 2013, 27(7), 805–815 CrossRef CAS.
- S. E. Hancock, R. Ailuri, D. L. Marshall, S. H. Brown, J. T. Saville, V. R. Narreddula, N. R. Boase, B. L. Poad, A. J. Trevitt and M. D. Willcox, Mass spectrometry-directed structure elucidation and total synthesis of ultra-long chain (O-acyl)-ω-hydroxy fatty acids, J. Lipid Res., 2018, 59(8), 1510–1518 CrossRef CAS.
- V. R. Narreddula, P. Sadowski, N. R. Boase, D. L. Marshall, B. L. Poad, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Structural elucidation of hydroxy fatty acids by photodissociation mass spectrometry with photolabile derivatives, Rapid Commun. Mass Spectrom., 2020, 34(9), e8741 CrossRef CAS.
- G. Liebisch, J. A. Vizcaíno, H. Köfeler, M. Trötzmüller, W. J. Griffiths, G. Schmitz, F. Spener and M. J. Wakelam, Shorthand notation for lipid structures derived from mass spectrometry, J. Lipid Res., 2013, 54(6), 1523–1530 CrossRef CAS.
- B. Aquila, L. Dakin, J. Ezhuthachan, S. Lee, P. Lyne, T. Pontz and X. Zheng, Quinazolinone derivatives and their use as B-RAF inhibitors, US2010/0216791A1, 2009.
- H. Wu, C. J. Kelley, A. Pino-Figueroa, H. D. Vu and T. J. Maher, Macamides and their synthetic analogs: evaluation of in vitro FAAH inhibition, Bioorg. Med. Chem., 2013, 21(17), 5188–5197 CrossRef CAS.
- A. Mehta, A. M. Oeser and M. G. Carlson, Rapid quantitation of free fatty acids in human plasma by high-performance liquid chromatography, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 719(1–2), 9–23 CrossRef CAS.
- T. Ly, B. B. Kirk, P. I. Hettiarachchi, B. L. Poad, A. J. Trevitt, G. da Silva and S. J. Blanksby, Reactions of simple and peptidic alpha-carboxylate radical anions with dioxygen in the gas phase, Phys. Chem. Chem. Phys., 2011, 13(36), 16314–16323 RSC.
- C. S. Hansen, B. B. Kirk, S. J. Blanksby, R. A. O'Hair and A. J. Trevitt, UV photodissociation action spectroscopy of haloanilinium ions in a linear quadrupole ion trap mass spectrometer, J. Am. Soc. Mass Spectrom., 2013, 24(6), 932–940 CrossRef CAS.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(1), 73–78 CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120(1–3), 215–241 Search PubMed.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding and C. Hättig, TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations, J. Chem. Phys., 2020, 152(18), 184107 CrossRef CAS.
- O. Christiansen, H. Koch and P. Jørgensen, The second-order approximate coupled cluster singles and doubles model CC2, Chem. Phys. Lett., 1995, 243(5–6), 409–418 CrossRef CAS.
- S. Grimme, Improved second-order Møller–Plesset perturbation theory by separate scaling of parallel-and antiparallel-spin pair correlation energies, J. Chem. Phys., 2003, 118(20), 9095–9102 CrossRef CAS.
- H.-Y. Liu, L. Ding, Y. Yu, Y. Chu and H. Zhu, Comparison of three derivatization reagents for the simultaneous determination of highly hydrophilic pyrimidine antitumor agents in human plasma by LC–MS/MS, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 893, 49–56 CrossRef.
- S. J. Blanksby and G. B. Ellison, Bond dissociation energies of organic molecules, Acc. Chem. Res., 2003, 36(4), 255–263 CrossRef CAS.
- A. G. Sage, T. A. Oliver, D. Murdock, M. B. Crow, G. A. Ritchie, J. N. Harvey and M. N. Ashfold, nσ* and πσ* excited states in aryl halide photochemistry: a comprehensive study of the UV photodissociation dynamics of iodobenzene, Phys. Chem. Chem. Phys., 2011, 13(18), 8075–8093 RSC.
- B. N. Moore, S. J. Blanksby and R. R. Julian, Ion–molecule reactions reveal facile radical migration in peptides, Chem. Commun., 2009,(33), 5015–5017 RSC.
- B. B. Kirk, D. G. Harman, H. I. Kenttämaa, A. J. Trevitt and S. J. Blanksby, Isolation and characterization of charge-tagged phenylperoxyl radicals in the gas phase: direct evidence for products and pathways in low temperature benzene oxidation, Phys. Chem. Chem. Phys., 2012, 14(48), 16719–16730 RSC.
- B. B. Kirk, D. G. Harman and S. J. Blanksby, Direct observation of the gas phase reaction of the cyclohexyl radical with dioxygen using a distonic radical ion approach, J. Phys. Chem. A, 2010, 114(3), 1446–1456 CrossRef CAS.
- B. B. Kirk, A. J. Trevitt, B. L. Poad and S. J. Blanksby, Characterisation of the ionic products arising from electron photodetachment of simple dicarboxylate dianions, Int. J. Mass Spectrom., 2013, 351, 81–94 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: LC-MS conditions and expanded experimental details; detailed description of synthesis and characterization of derivatization reagents; additional details of electronic structure calculations including molecular orbital representations and; additional figures including PD266, PD266/CID mass spectra and chromatograms. See DOI: 10.1039/d0an01840f |
| This journal is © The Royal Society of Chemistry 2021 |