
Design, synthesis, and bioimaging applications of a new class of carborhodamines†
Xin
Lv
 *,
Taihe
Han
,
Xia
Yuan
,
Hu
Shi
and
Wei
Guo
*
*,
Taihe
Han
,
Xia
Yuan
,
Hu
Shi
and
Wei
Guo
*
School of Chemistry and Chemical Engineering, Shanxi University, Taiyuan 030006, China. E-mail: guow@sxu.edu.cn; lvxin@sxu.edu.cn
First published on 27th October 2020
Abstract
We herein developed a new class of carborhodamines (CRs), i.e. 10-methoxy-substituted carborhodamines MCRs, by a simple synthesis procedure, which have absorption and emission wavelengths longer than classical CRs while retaining their excellent photophysical properties. Based on the MCR platform, we constructed the mitochondria-targeted fluorescent probe MCR-DMA and demonstrated its potential for sensing singlet oxygen (1O2) in living cells during the photodynamic therapy process.
Fluorescent dyes have become invaluable tools for bioimaging assays, environmental analysis, and materials sciences. With regard to bioimaging applications, fluorescent dyes that have absorption and emission in the near-infrared region (NIR) are particularly prized due to the minimum photodamage to biological samples, deep tissue penetration, and reduced photobleaching and light scattering.1,2 Among various fluorescent dyes, rhodamines have been widely used as labeling reagents in biological studies due to their excellent photophysical properties including large extinction coefficients, high quantum yields, and good photostability.3,4 However, typical rhodamines have short excitation and emission wavelengths (below 600 nm), thus being disadvantageous for in vivo bioimaging applications in many cases. Thus, in recent years, considerable efforts have been made to develop NIR rhodamine derivatives to overcome the shortcoming, typically including carbon-, silicon-, and phospha-rhodamine.5–11
One of the attractive strategies to develop NIR rhodamine derivatives is replacing the 10-position oxygen atom of traditional rhodamines with a carbon atom.5,6 The as-prepared carborhodamines (CRs) commonly contain a geminal dimethyl group [C(CH3)2] in the 10-position and have an emission maximum around 640 nm, an ∼50 nm bathochromic shift when compared with classical rhodamine B, while inheriting the excellent photophysical properties of rhodamines, such as high fluorescence quantum yields, large molar extinction coefficients, and excellent photostability. However, the preparation of CRs is a challenging task, partly because the synthetic routes are not completely described in the early publications.12–14 Until recent years, a general synthetic route to CRs was exploited by Hell's group,15 and the resulting CRs have been used at the forefront of modern microscopy methods.6,16,17 However, the reported synthesis is still complex when compared with the other NIR rhodamine derivatives, which to some extent limits their wide applications. Thus, the development of a simpler synthesis route for this class of dyes is urgently demanded.
In this work, we developed a new and simple synthetic routine for CRs, and obtained a series of 10-methoxy-substituted CRs MCR1–5 (Scheme 1A). MCR1–5 not only retain the excellent photophysical properties of classical CRs including high fluorescence quantum yields, large molar extinction coefficients, and good photo- and chemostability, but also show an ∼25 nm bathochromic shift in both absorption and emission maxima in comparison with typical CRs. Moreover, due to their cationic character, MCR1–5 could rapidly penetrate cell membranes and specifically localize in the mitochondria of living cells, thus being suitable for cellular bioimaging. Based on the MCR platform, we further developed a NIR fluorescent probe MCR-DMA for singlet oxygen (1O2) and confirmed its ability to sensitively trace 1O2 in the mitochondria of living cells during the photodynamic therapy process (Scheme 1B).
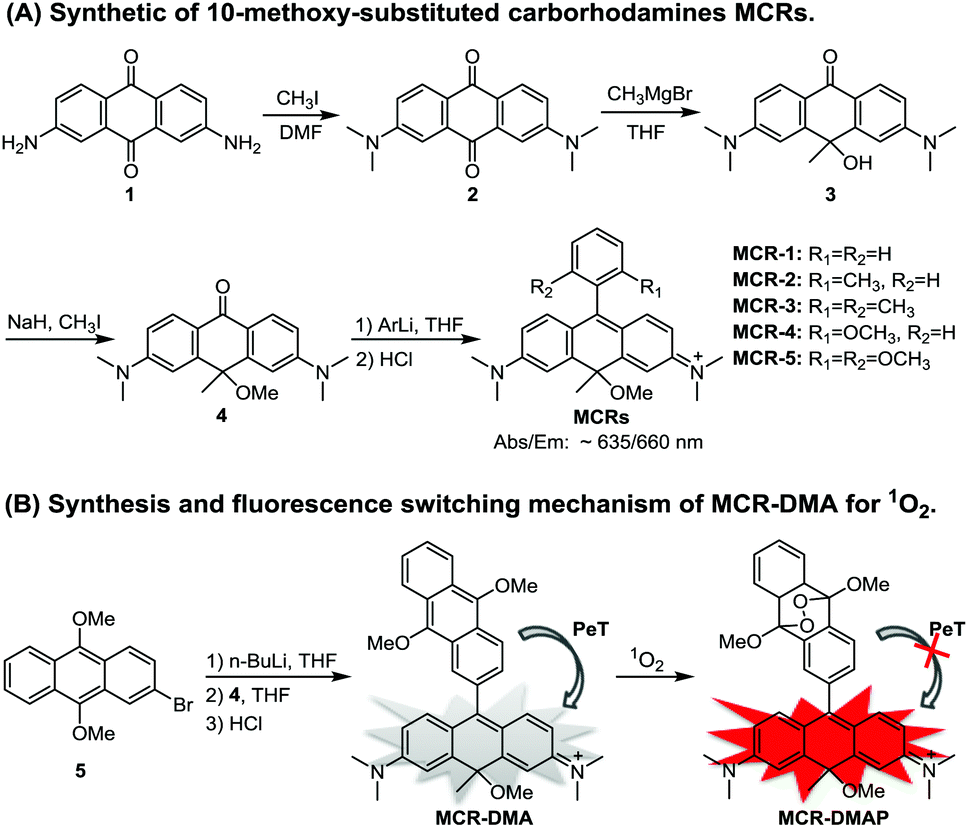 | ||
| Scheme 1 (A) Synthesis of 10-methoxy-substituted carborhodamines MCRs. (B) Synthesis and proposed sensing mechanism of MCR-DMA for 1O2. | ||
MCR1–5 were synthesized via a simple four-step procedure starting from 2,7-diamino anthraquinone 1,18 which is indeed more convenient and operable than the previously reported procedure for typical CRs.15 As shown in Scheme 1, N-alkylation of 1 with CH3I in DMF gave 2,7-dimethylamino anthraquinone 2; the reaction of 2 with a methyl-Grignard reagent (CH3MgBr) occurred regioselectively at the 10-position carbon atom to furnish 3, which was then alkylated with CH3I to provide intermediate 4; and the desired carborhodamines MCR1–5 were finally obtained by a typical organometallic addition to 4. The detailed synthetic procedures are shown in the ESI.†
With MCR1–5 in hand, we first studied their photophysical properties in CH2Cl2, EtOH and PBS (pH 7.4, 10 mM). The absorption and emission profiles are shown in Fig. 1 and Fig. S1 and 2 (ESI†), and the corresponding photophysical data are presented in Table 1. As can be seen, in CH2Cl2, MCR1–5 all displayed similar spectra profiles with the absorption and emission maxima located at average 636 nm and 660 nm (∼25 nm longer than those of classical CRs), respectively, along with large extinction coefficients [ε = (1.20–1.78) × 105 M−1 cm−1] and high fluorescence quantum yields (Φfl = 0.47–0.56). Due to structural symmetries, the maximal absorption and emission wavelengths of MCR1–5 were hardly affected by solvent polarity, as indicated by the minor spectra differences in CH2Cl2, EtOH, and PBS. As with the classical rhodamines and their derivatives, MCR1–5 also showed a gradual decrease in the quantum yield as the solvent polarity increased, most likely due to the formation of the twisted intramolecular charge transfer (TICT) state (a nonemissive species) that can greatly be stabilized by polar solvent molecules.19–21 Even so, their quantum yields in PBS (ca. 0.30) were still high enough for bioimaging applications. Taken together, these results reveal that MCR1–5 not only retain the excellent photophysical properties of classical CRs but also display ∼25 nm longer absorption and emission wavelengths, thus being more advantageous for bioimaging applications.
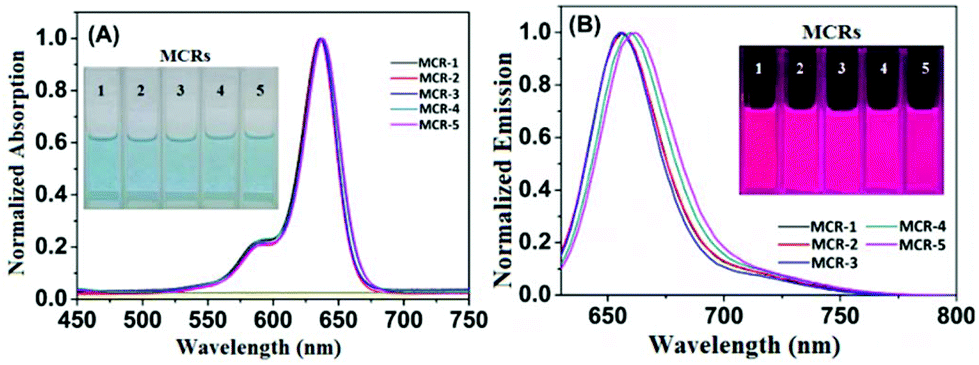 | ||
| Fig. 1 Normalized absorption (A) and emission (B) spectra of MCR1–5 in CH2Cl2 (for those in EtOH and PBS, see Fig. S1 and S2†) at 25 °C. λex = 620 nm. Inset: Colour (A) and fluorescence (B) images of MCR1–5 in CH2Cl2. | ||
| Dyes | Solvents | λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Φ |
|---|---|---|---|---|---|
| a Quantum yields were determined using Cy5 as a reference (Φf = 0.27 in PBS). | |||||
| MCR-1 | CH2Cl2 | 636 | 1.36 × 105 | 656 | 0.50 |
| EtOH | 635 | 1.20 × 105 | 661 | 0.33 | |
| PBS | 634 | 0.95 × 105 | 659 | 0.28 | |
| MCR-2 | CH2Cl2 | 636 | 1.78 × 105 | 656 | 0.55 |
| EtOH | 636 | 1.54 × 105 | 662 | 0.42 | |
| PBS | 635 | 1.09 × 105 | 659 | 0.28 | |
| MCR-3 | CH2Cl2 | 637 | 1.20 × 105 | 656 | 0.56 |
| EtOH | 637 | 1.03 × 105 | 662 | 0.43 | |
| PBS | 636 | 0.97 × 105 | 659 | 0.31 | |
| MCR-4 | CH2Cl2 | 637 | 1.35 × 105 | 660 | 0.50 |
| EtOH | 637 | 0.91 × 105 | 664 | 0.39 | |
| PBS | 638 | 0.96 × 105 | 663 | 0.31 | |
| MCR-5 | CH2Cl2 | 638 | 1.40 × 105 | 662 | 0.47 |
| EtOH | 637 | 1.22 × 105 | 665 | 0.40 | |
| PBS | 641 | 0.97 × 105 | 668 | 0.32 | |
Subsequently, we evaluated the pH stabilities of MCR1–5 given that in typical mammalian cells, the pH values commonly range from 4.5 to 8.0 (about 4.5 in the lysosome and 8.0 in the mitochondria). It was found that the fluorescence intensities of MCR1–5 remained stable in the pH range of 2–12 (Fig. S3, ESI†), confirming the pH-independent emission of these dyes at a wide pH range. Furthermore, the photostability of MCR1–5 was evaluated in PBS (10 mM, pH 7.4) and compared with the widely used NIR labeling agent Cy5 due to their similar spectral profiles. As shown in Fig. S4 (ESI†), when irradiated with a LED lamp (620 nm, 45 mW cm−2) for 30 min, more than 97% of the initial emission intensities of MCR1–5 was retained, while Cy5 only retained 30% of its initial fluorescence intensity, suggesting that MCR1–5 have sufficient photostability for bioimaging applications. In addition, we also evaluated the chemical stabilities of MCR1–5 toward reactive oxygen species (ROS) including HClO, H2O2, TBHP, ˙OH and 1O2. As shown in Fig. S5 (ESI†), when treated with the above ROS in excess for 30 min, MCR1–5 retained over 90% of the initial fluorescence intensities, whereas under the same conditions, commercial Cy5 was degraded completely by HClO. Besides, MCR1–5 all showed good water solubilities (at least 20 μM), as indicated by the concentration-dependent absorption spectra (Fig. S6, ESI†).
Encouraged by the above results, we examined the bioimaging ability of MCR1–5 in living cells using confocal fluorescence microscopy. As shown in Fig. S7 (ESI†), when HeLa cells were incubated with MCR1–5 for 15 min, intense NIR fluorescence in the cytoplasm region was observed. Moreover, the intracellular fluorescence remained stable when the MCR1–5 loaded HeLa cells were irradiated continuously for 20 min with a semiconductor laser (633 nm; 2% of laser power) (Fig. S8, ESI†). Thus, MCR1–5 are cell membrane-permeable and have enough photostability for long-term bioimaging applications. Furthermore, co-localization experiments revealed that MCR1–5 were able to preferably accumulate in the mitochondria of living cells (Fig. 2) but not in the lysosomes (Fig. S9, ESI†), consistent with their cationic character. Moreover, MCR1–5 all had low cytotoxicity in the concentration range of 2–10 μM (cell viability: >85% for 24 h of incubation) as indicated by the standard MTT assays (Fig. S10, ESI†). Thus, MCR1–5 should be good dye platforms for constructing fluorescent probes to track mitochondrial species.
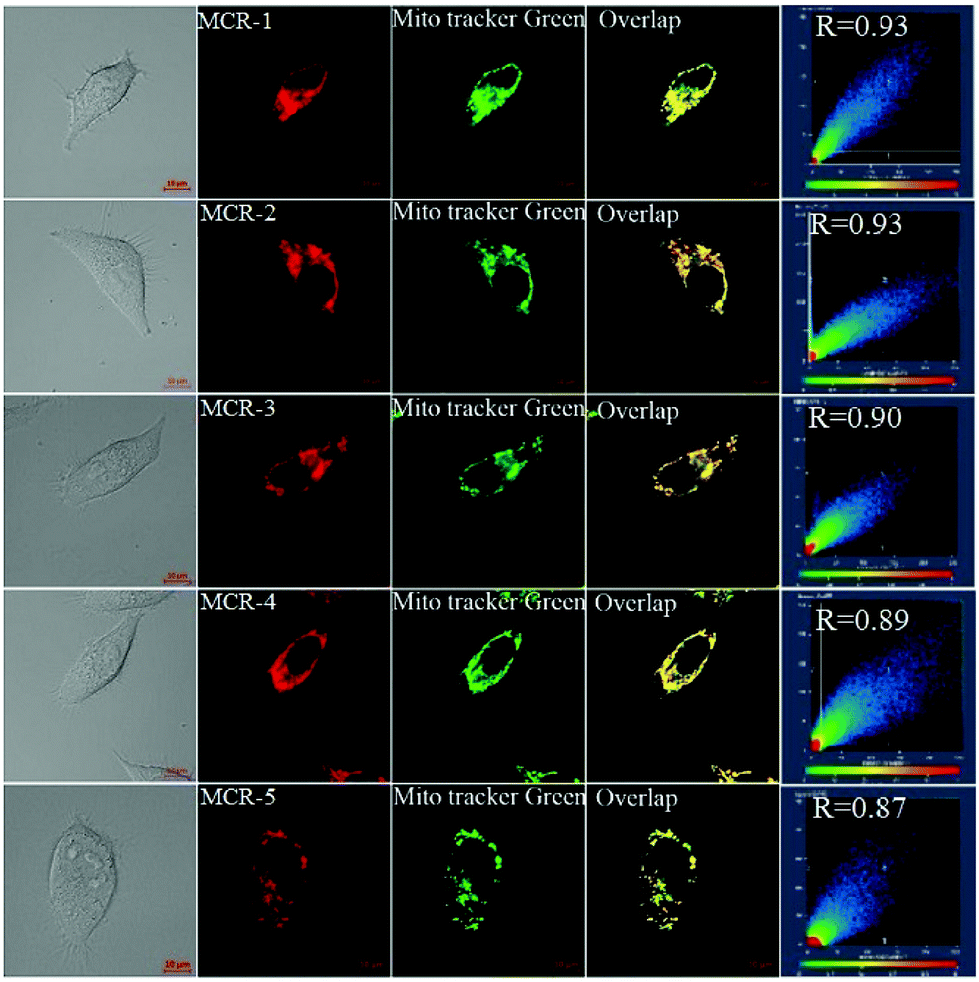 | ||
| Fig. 2 Fluorescence images of HeLa cells costained with MCRs (0.5 μM, 15 min) and MitoTracker Green (0.2 μM, 15 min) in PBS at 37 °C. For MCRs, emissions were collected at 650–750 nm (λex = 633 nm); for MitoTracker Green, at 500–600 (λex = 488 nm). Scale bar: 10 μm. | ||
Towards this end, we designed and synthesized a new NIR fluorescent probe MCR-DMA through the integration of the MCR fluorophore and the 9,10-dimethoxyanthracene (DMA) group for sensing mitochondrial singlet oxygen (1O2) (Scheme 1B), a highly reactive oxygen species that participates in various pathological and physiological processes22–24 and also is a key killer in type-II photodynamic therapy (PDT).25,26 The detailed synthetic procedure and characteristic data are shown in the ESI.† In our probe design, the DMA moiety functions not only as a 1O2 trapper but also as a fluorescence quencher via photoinduced electron transfer (PET).27–29 It is worth noting that the high π-electron density of DMA arising from the strong electron-donating 9,10-dimethoxy groups will improve not only the reactivity of DMA with 1O2 but also its HOMO energy level to facilitate a PeT process from the DMA moiety to the MCR core. Thus, compared with the previously reported fluorescent 1O2 probes that commonly bear classical 9-methylanthracene or 9,10-dimethylanthracene moieties,27–30MCR-DMA was expected to have higher detection sensitivity and lower background fluorescence. Moreover, the long excitation and emission wavelengths of MCR-DMA will also significantly minimize photodamage and autofluorescence interference during the bioimaging process. Besides, due to the mitochondria-targeted character of MCR dyes, MCR-DMA was also expected to be mitochondria-targetable and could image 1O2 generation within the mitochondria. This nature is indeed especially interesting given that the mitochondria are the main target organelles in PDT, the actions and dysfunctions of which are closely associated with programmed cell death.27
As shown in Table S1,†MCR-DMA was found to be nearly non-fluorescent in CH2Cl2, EtOH and PBS with Φfl = 0.007/0.004/0.002, respectively, indicating the highly efficient PET process from the DMA group to the MCR core. The PET process in MCR-DMA was also supported by density functional theory (DFT) calculations (Fig. S11, ESI†). The fluorescence response of MCR-DMA to 1O2 was subsequently examined using a commercial type-II photosensitizer TMPyP4 (ΦΔ = 0.74)31 in PBS solution (pH 7.4, 10 mM). As shown in Fig. 3A, MCR-DMA showed a time-dependent fluorescence enhancement at 667 nm (λex = 620 nm) upon photoirradiation of the co-incubated TMPyP4 with a LED lamp (410 nm, 30 mW cm−2), indicating that the DMA moiety of MCR-DMA converted into its endoperoxide form by reacting with 1O2,27 which inhibits the PET process and results in a large fluorescence enhancement (nearly 100-fold, Φfl = 0.18). The transformation of the DMA moiety to its endoperoxide was also supported by absorption spectra study under the same conditions, where a gradual disappearance of DMA absorbance was observed due to the disruption of DMA π-conjugation by 1O2 (Fig. 3B). In contrast, in the absence of the photosensitizer TMPyP4, MCR-DMA only showed negligible fluorescence enhancement (Fig. 3C), indicating that the observed fluorescence enhancement is indeed caused by 1O2 produced by photoirradiation of TMPyP4, as well as the negligible self-oxidation of MCR-DMA during the process of photoirradiation. Similar spectral changes were also observed when 1O2 was induced by a NaClO–H2O2 system (Fig. S12, ESI†), and under these conditions the detection limit for 1O2 was calculated to be 5.6 μM (S/N = 3) (Fig. S13, ESI†). The reaction product MCR-DMAP was confirmed by the HRMS assay of MCR-DMA treated with 1O2 (produced by photoirradiation of the photosensitizer TMPyP4), where the peak at m/z = 577.2695 corresponding to MCR-DMAP was clearly observed (Fig. S14, ESI†). In addition, the fluorescence responses of MCR-DMA toward other biological species were also investigated. As shown in Fig. 3D, the representative species, including anions, cations, biothiols and various ROS, induced only negligible fluorescence enhancement of MCR-DMA, indicative of the superior selectivity of the probe for 1O2. Also, MCR-DMA displayed a stable fluorescence response to 1O2 in the pH range of 7–9, indicative of its suitability for imaging applications at physiological pH (Fig. S15, ESI†).
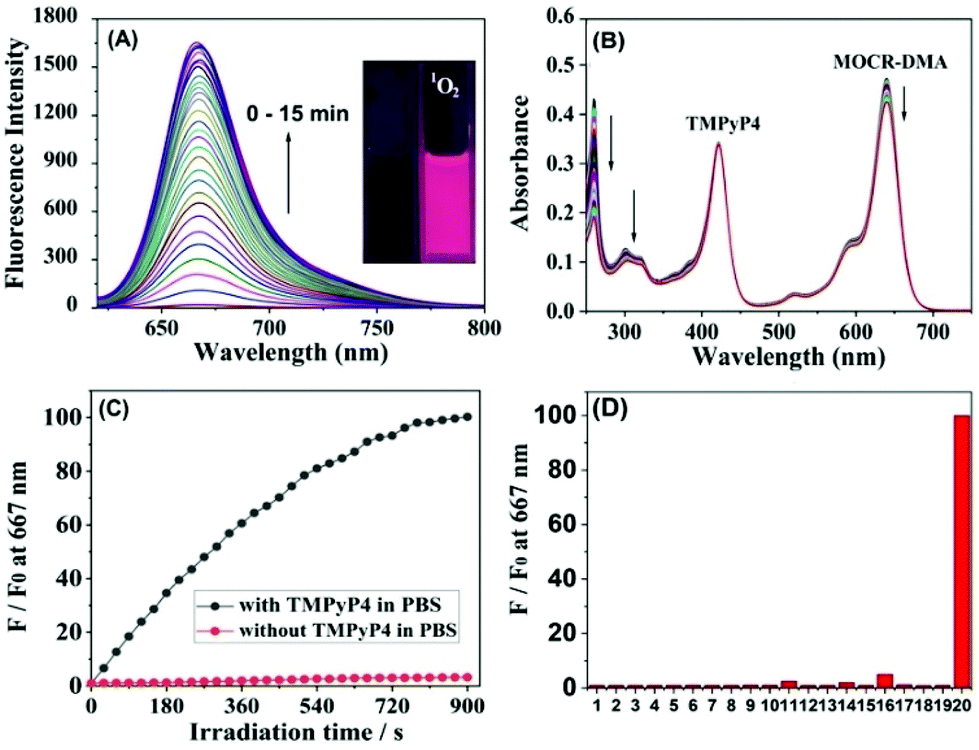 | ||
| Fig. 3 (A) Fluorescence and (B) absorption spectral changes of MCR-DMA (5 μM) in PBS (pH 7.4, 10 mM) upon photoirradiation of the co-dissolved TMPyP4 (5 μM) for 15 min. (C) Fluorescence intensity changes of MCR-DMA at 667 nm during 15 min of photoirradiation in the presence and absence of TMPyP4. (D) Fluorescence responses of MCR-DMA toward various biological species including various ions (1 mM), biothiols (1 mM), and ROS (200 μM). 1–20: blank, Na+, K+, Mg2+, Fe3+, Cu2+, Zn2+, Cl−, Br−, CO32−, ˙OH, O2−, NO, HClO, H2O2, ONOO−, Cys, GSH, Hcy and 1O2. For generating 1O2, a 410 nm LED light source (0.03 W cm−2) was used. Fluorescence spectra were recorded with a time interval of 30 s (λex = 620 nm). | ||
Finally, we evaluated the ability of MCR-DMA in imaging 1O2 production within living cells during the PDT process using confocal fluorescence microscopy. Before the experiments, we confirmed by MTT assays that the probe has low cytotoxicity to living cells in the concentration range of 2–10 μM (cell viability: >80% for 24 h of incubation) (Fig. S16, ESI†), and by co-localization assays that the probe mainly accumulates in the mitochondria of living cells (Fig. S17, ESI†). In order to produce 1O2 intracellularly, a commercial PDT drug 5-aminolevulinic acid (5-ALA), a precursor for the biosynthesis of the mitochondrial-targeted photosensitizer protoporphyrin IX (PpIX),31–33 was used as the 1O2 donor. After incubation with 5-ALA (150 μg mL−1) for 4 h, we observed obvious red fluorescence from 5-ALA-derived PpIX in HeLa cells upon excitation at 405 nm, which overlapped well with the green fluorescence of commercial MitoTracker (Fig. S18, ESI†), consistent with the mitochondria-targeted ability of 5-ALA-derived PpIX. The fluorescence imaging of intracellular 1O2 during the PDT process was then investigated. In the assay, HeLa cells were first incubated with 5-ALA for 4 h and then stained with MCR-DMA for another 30 min. As shown in Fig. 4A, photoirradiation of 5-ALA-derived PpIX with a 405 nm laser induced an obvious fluorescence enhancement of MCR-DMA within 40 s (emissions were collected at 650–750 nm with λex = 633 nm). In contrast, photoirradiation of HeLa cells incubated with MCR-DMA alone only caused a negligible fluorescence change under the same conditions (Fig. 4B). Notably, when the 5-ALA treated cells were stained with MCR-DMA and NaN3 (a cell-permeable 1O2 quencher),27 almost no fluorescence enhancement was found under photoirradiation (Fig. 4C). These results clearly demonstrate that MCR-DMA can serve as an effective imaging tool for tracing 1O2 generation in the mitochondria of living cells during the PDT process.
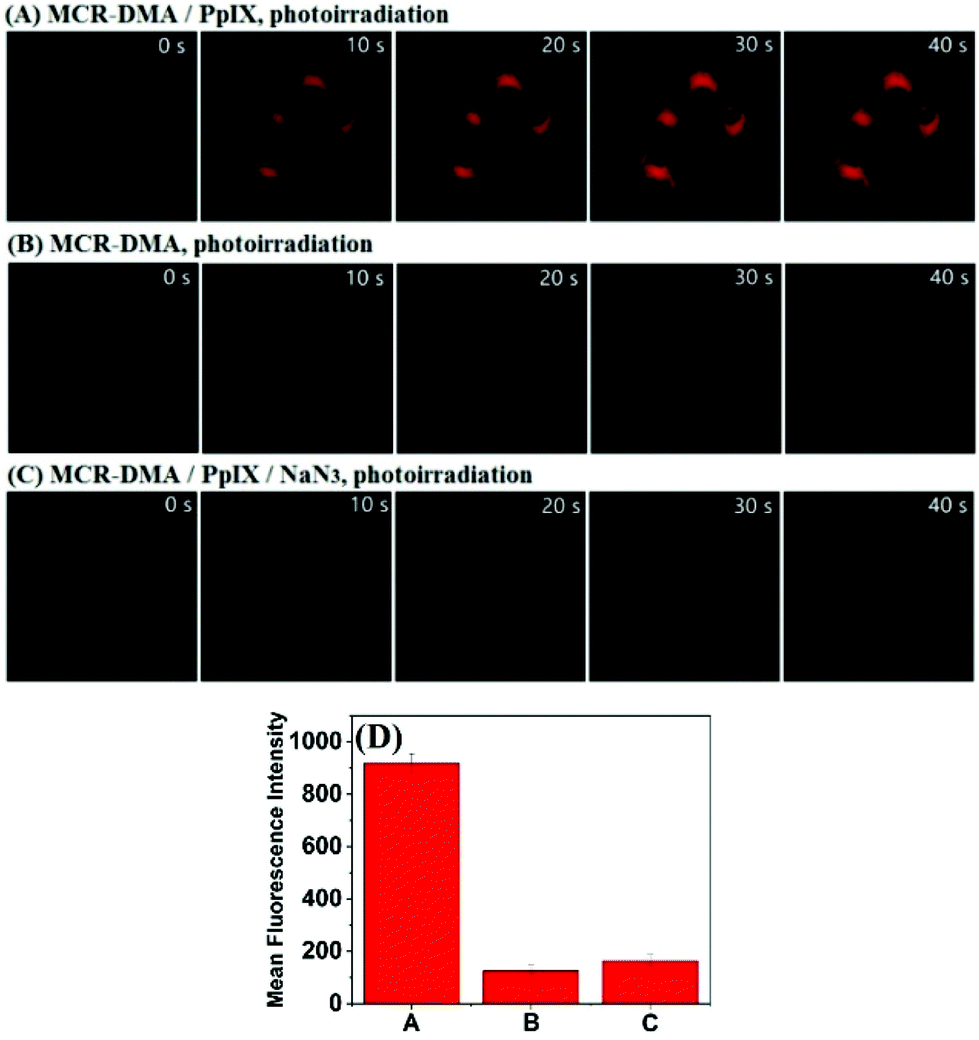 | ||
| Fig. 4 Imaging 1O2 during the simulated PDT process under confocal fluorescence microscopy. (A) HeLa cells were pre-treated with 5-ALA (150 μg mL−1, 4 h) and MCR-DMA (0.5 μM, 30 min) and then photoirradiated. (B) HeLa cells were pre-treated with MCR-DMA (0.5 μM, 30 min) and then photoirradiated. (C) HeLa cells were pre-treated with 5-ALA (150 μg mL−1, 4 h), MCR-DMA (0.5 μM, 30 min), and NaN3 (200 μM) and then photoirradiated. (D) Quantification of the fluorescence signals from (A–C) at the time point of 40 s of photoirradiation. For generating 1O2, a 405 nm laser was used. For imaging 1O2, a 633 nm laser was used and emissions were collected at 650–750 nm. Scale bar: 20 μm. | ||
In summary, we have designed and synthesized a new class of carborhodamine dyes MCR1–5 by a simple and convenient procedure, which showed longer absorption and emission wavelengths than classical carborhodamines while retaining their excellent photophysical properties. Moreover, MCR1–5 can rapidly penetrate the cell membrane and specifically localize in the mitochondria of living cells. Based on the dye platform, we further developed a mitochondria-targeted NIR fluorescent 1O2 probe, MCR-DMA, which could highly selectively detect 1O2 with an approximately 100-fold fluorescence enhancement and has successfully been utilized to image 1O2 generation in the mitochondria of living cells during the PDT process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21778036, 21877077, 21572121, and 21502108) and the Program for the Outstanding Innovative Teams of Higher Learning Institutions of Shanxi.Notes and references
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- F. Amat-Guerri, A. Costela, J. M. Figuera, F. Florido and R. Sastre, Chem. Phys. Lett., 1993, 209, 352–356 CrossRef CAS.
- X. Chen, T. Pradhan, F. Wang, J. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS.
- K. Kolmakov, C. Wurm, M. V. Sednev, M. L. Bossi, V. N. Belov and S. W. Hell, Photochem. Photobiol. Sci., 2012, 11, 522–532 RSC.
- J. B. Grimm, A. J. Sung, W. R. Legant, P. Hulamm, S. M. Matlosz, E. Betzig and L. D. Lavis, ACS Chem. Biol., 2013, 8, 1303–1310 CrossRef CAS.
- M. Fu, Y. Xiao, X. Qian, D. Zhao and Y. Xu, Chem. Commun., 2008, 15, 1780–1782 RSC.
- Y. Koide, Y. Urano, K. Hanaoka, W. Piao, M. Kusakabe, N. Saito, T. Terai, T. Okabe and T. Nagano, J. Am. Chem. Soc., 2012, 134, 5029–5031 CrossRef CAS.
- X. Zhou, R. Lai, J. R. Beck, H. Li and C. I. Stains, Chem. Commun., 2016, 52, 12290–12293 RSC.
- X. Chai, X. Cui, B. Wang, F. Yang, Y. Cai, Q. Wu and T. Wang, Chem. – Eur. J., 2015, 21, 16754–16758 CrossRef CAS.
- M. Grzybowski, M. Taki, K. Senda, Y. Sato, T. Ariyoshi, Y. Okada, R. Kawakami, T. Imamura and S. Yamaguch, Angew. Chem., Int. Ed., 2018, 57, 10137–10141 CrossRef CAS.
- J. Arden-Jacob, J. Frantzeskos, N. U. Kemnitzer and A. Zilles, Spectrochim. Acta, Part A, 2001, 57, 2271–2283 CrossRef CAS.
- C. Aaron and C. C. Barker, J. Chem. Soc. B, 1971, 319–324 RSC.
- R. Neill and P. V. Fischer, Guava Tech. Inc., WO2004/003510, 08. Jan. 2004.
- K. Kolmakov, V. N. Belov, C. A. Wurm, B. Harke, M. Leutenegger, C. Eggeling and S. W. Hell, Eur. J. Org. Chem., 2010, 3593–3610 CrossRef CAS.
- M. V. Sednev, C. A. Wurm, V. N. Belov and S. W. Hell, Bioconjugate Chem., 2013, 24, 690–700 CrossRef CAS.
- A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. H. Meineke, E. D'Este, P. T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 3290–3294 CrossRef CAS.
- H. Huang, US Pat., 0253709, 2009 Search PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS.
- X. Liu, Q. Qiao, W. Tian, W. Liu, J. Chen, M. J. Lang and Z. Xu, J. Am. Chem. Soc., 2016, 138, 6960–6963 CrossRef CAS.
- X. Lv, C. Gao, T. Han, H. Shi and W. Guo, Chem. Commun., 2020, 56, 715–718 RSC.
- J. Cadet, T. Douki, J. Pouget and J. Ravanat, Methods Enzymol., 1999, 319, 143–153 Search PubMed.
- S. Zhuang, J. Demirs and I. Kochevar, J. Biol. Chem., 2000, 275, 25939–25948 CrossRef CAS.
- S. Ryter and R. Tyrrell, Free Radical Biol. Med., 1998, 24, 1520–1534 CrossRef CAS.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–538 CrossRef CAS.
- X. Li, N. Kwon, T. Guo, Z. Liu and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 11522–11531 CrossRef CAS.
- S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707–11715 CrossRef CAS.
- N. Umezawa, K. Tanaka, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, Angew. Chem., Int. Ed., 1999, 38, 2899–2901 CrossRef CAS.
- K. Tanaka, T. Miura, N. Umezawa, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, J. Am. Chem. Soc., 2001, 123, 2530–2536 CrossRef CAS.
- B. Song, G. Wang, M. Tan and J. Yuan, J. Am. Chem. Soc., 2006, 128, 13442–13450 CrossRef CAS.
- R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391–475 CrossRef CAS.
- Z. Zhang, S. Long, J. Cao, J. Du, J. Fan and X. Peng, ACS Sens., 2020, 5, 1411–1418 CrossRef CAS.
- L. Long, X. Yuan, S. Cao, Y. Han, W. Liu, Q. Chen, A. Gong and K. Wang, Chem. Commun., 2019, 55, 8462–8465 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplemental spectra, and 1H-, 13C-NMR, and MS spectra. See DOI: 10.1039/d0an01916j |
| This journal is © The Royal Society of Chemistry 2021 |