
Electroanalytical study of five carbosilane dendrimers at the interface between two immiscible electrolyte solutions†
Karolina
Kowalewska
 a,
Tamara
Rodriguez-Prieto
bcd,
Slawomira
Skrzypek
a,
Jesús
Cano
bcd,
Rafael Gómez
Ramírez
*bcd and
Lukasz
Poltorak
*a
a,
Tamara
Rodriguez-Prieto
bcd,
Slawomira
Skrzypek
a,
Jesús
Cano
bcd,
Rafael Gómez
Ramírez
*bcd and
Lukasz
Poltorak
*a
aDepartment of Inorganic and Analytical Chemistry, Electroanalysis and Electrochemistry Group, Faculty of Chemistry, University of Lodz, Tamka 12, 91-403 Lodz, Poland. E-mail: lukasz.poltorak@chemia.uni.lodz.pl
bDepartment of Organic and Inorganic Chemistry, Chemical Research Institute “Andrés M. Del Río” (IQAR), University of Alcalá, 28805 Madrid, Spain. E-mail: rafael.gomez@uah.es
cRamón y Cajal Health Research Institute (IRYCIS), 28034 Madrid, Spain
dNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), 28029 Madrid, Spain
First published on 10th December 2020
Abstract
This work is focused on the electroanalytical study of a family of five imidazolium-terminated carbosilane dendrimers (from generation G1 to G3) at the polarized liquid–liquid interface formed between water and 1,2-dichloroethane solutions. All dendrimers with permanently and positively charged imidazolium groups located at the periphery within the branched carbosilane core were found to be electrochemically active. Based on the concentration and scan rate dependencies we have concluded that these molecules undergo interfacial ion transfer processes accompanied by interfacial adsorption/desorption rather than the electrochemically induced interfacial formation of the macromolecule–anion (tetrakis(4-chlorophenyl)borate) from the organic phase complex. Also, we report several physicochemical and electroanalytical parameters (e.g. diffusion coefficients, LODs, and detection sensitivities) for the studied family of dendrimers. Our work aims to contribute to the understating of the interaction between branched macromolecules and biomimetic interfaces.
1. Introduction
Since Flory′s report in 1952, hyperbranched polymers have been studied and significantly developed with the aim to be applied in several fields of science.1–3 In the beginning, just two types of traditional architectures were recognized, which are linear and crosslinked polymers. Branched and dendritic systems were discovered at a later stage.4 Dendritic systems are one of the most important systems among four classes of polymers holding a number of potential and emerging applications, especially in biomedicine.3 This is mainly due to their unique properties: (i) unlike other polymers, these chemical species are ideally monodisperse; (ii) dendrimers are nano-sized and versatile macromolecules with a noteworthy structural precision; (iii) the organic chemistry toolbox offers a number of existing protocols that allow for the precise control of resulting macromolecule chemical properties, well-defined structures, sizes, and shapes.5,6 In the dendritic architecture, it is possible to differentiate three parts: the core or focal point, where all the branches emerge; the branches, which determine the size or generation of the dendritic system; and the periphery, where the functional groups are localized. Among the dendrimer typologies available,7 this work is focused on carbosilane dendrimers,8 whose structure is based on C–Si bonds that provide chemical and thermal stability and biocompatibility (especially desired when it comes to biomedical applications). Furthermore, the periphery of this family of dendrimers, here reported, is made from imidazolium salts being positively charged within a full conventional pH scale. Normally, the application of the dendrimer is determined by the peripheral groups; however, the core or focal point or the cavities in the internal structure of the dendrimer can also play an important role. Focusing on biomedical applications, anionic dendrimers are deeply studied as antivirals, as these can interact with the virus, avoiding its subsequent infection. Cationic ones are used as potent antibacterial species, damaging the bacterial membrane and provoking the death of the microorganism.9,10 Dendrimers can also be used as carriers of metals, drugs, or biological molecules, and hence, are extensively studied in e.g. anticancer treatment, diagnosis, and gene transfection.11–14 Imidazolium salts are used in a great variety of biological applications, for instance, anticancer agents, antioxidants, antimicrobials, drug delivery, or biosensors.15 Therefore, the inclusion of these groups in the periphery of the dendrimer can result in properties and behaviours, such as multivalence, and needs to be studied. Reaching the full potential of all these applications require a good fundamental understanding on the interaction between dendrimers and bio-interfaces. Model bio-mimicking systems operating under electrochemical control are especially useful in this respect.Electrified liquid–liquid interfaces (LLIs) have many similarities with the real bio-interfaces (e.g. discontinuous properties originating from the existence of contacted hydrophobic and hydrophilic domains, potential drops defined by the ions, and/or intrinsic sieving properties being the result of the size, charge, and chemical nature of studied chemical species). It was proposed already in the 80s that this system can be considered as a model of a half of a lipid bilayer (analogous to a lipid monolayer).16 As a matter of fact, this belief is still valid to date.17 Electrified LLIs together with the electrochemical studies devoted to supported18–20 and black lipid bilayers21,22 offer an impressive toolbox focused on studying various bio-important interactions.
Polarization of the LLI is possible when contacting phases contain highly hydrophilic (the aqueous phase) and hydrophobic (the organic phase) salts with ideally zero mutual partitioning.23,24 In such configurations, this system is known as the interface between two immiscible electrolyte solutions (ITIES) which can be electrochemically studied in dedicated glass (and recently 3D printed25) cells. This system is especially exciting since in addition to electron transfer reactions between redox couples placed in both phases it allows for the direct electroanalytical probing of the simple ion transfer crossing the ITIES.26 Correspondingly, chemical species (e.g. proteins,27,28 drugs,29 biologically important molecules,30 or polyelectrolytes31) with ionizable or permanently charged chemical functional groups are potentially active at the electrified LLI. Due to the still emerging biotechnological applications of dendrimers, these chemical species have attracted significant attention when it comes to the ITIES. Various generations of poly(propylenimine) (DABAM-n), poly(amidoamine) (PAMAM),32 and poly-L-lysine33,34 dendrimers were found to be electrochemically active (when charged, pH < pKa) at the polarized water–1,2-dichloroethane interface. It was found that these species undergo interfacial adsorption and facilitate the transfer of hydrophobic anions (usually tetrakis(4-chlorophenyl)borate) initially dissolved in the organic phase rather than undergoing simple interfacial ion transfer reactions.35 PAMAM dendrimers were also used to study photoinitiated interfacial electron transfer between PAMAM − Zinc(II) porphyrin associates from the aqueous phase and ferrocene dissolved in 1,2-dichloroethane.36,37 Finally, smaller generations of PAMAM dendrimers (G0 and G1) that may undergo simple ion transfer reactions were used to assess the charge and size sieving properties of silica membranes in situ synthesized at the ITIES.38
In this work, we show that each member of the studied family (from G1 to G3) of self-synthesized imidazolium terminated carbosilane dendrimers initially dissolved in the aqueous phase gives rise to ionic currents recorded at the ITIES. Based on our results we have concluded that these molecules, rather than facilitating the transfer of the organic phase background electrolyte anion, undergo simple ion transfer accompanied by interfacial adsorption. Our work provides a number of electroanalytical parameters such as detection sensitivities and LOD values. The easiness in size tunability of the studied molecules may give them a new application in the evaluation of the size and charge sieving properties of modified ITIESs.
2. Methods and materials
2.1. Chemicals
BDTR-1, BDTR-2, BDTR-3, BDTR-4, and BDTR-5 were synthesized according to the protocol described below and reported elsewhere.39 Sodium chloride (NaCl, for analysis, ChemPur) and 1,2-dichloroethane (1,2-DCE, for analysis, POCH) were used as received. The organic phase electrolyte bis(triphenylphosphoranylidene)ammonium tetrakis(4-chlorophenyl)borate (BTPPA+TPBCl−) was prepared via a metathesis reaction between bis(triphenylphosphoranylidene)ammonium chloride (BTPPA+Cl−, 97%, Sigma-Aldrich) and K+TPBCl− potassium tetrakis(4-chlorophenyl)borate (K+TPBCl−, >98%, Sigma-Aldrich) salts. The aqueous phase was prepared using water distilled in triplicate.2.2. Carbosilane dendrimers
In this research, we have studied a self-synthesized new family of spherical carbosilane dendrimers with two types of moieties and generations. BDTR-1, BDTR-3, and BDTR-5 bear a methyl group as the N-substituent in the imidazolium fragments, whereas compounds BDTR-2 and BDTR-4 hold a mesityl unit. All compounds used to perform these experiments were prepared by our research group. Briefly, the procedure consists of the N-alkylation reaction of the imidazolium salts using iodine-terminated precursor dendrimers under mild conditions.39 Their characterization included 1H and 13C NMR spectroscopy performed using a Varian Unity VXR-300 NMR spectrometer at ambient temperature, along with elemental analyses (PerkinElmer 240C) and mass spectrometry (Agilent 6210 spectrometer (ESI) in the positive mode. Data are in agreement with those reported in the literature.2.3. Electrochemical experiments
Electrochemical characterization of all dendrimers was performed in a macroscopic voltammetric glass cell (the interface radius equals 0.7 cm) equipped with a set of appropriate electrodes (see Fig. 1). Two Ag/AgCl and two Pt wires were used as the reference and counter electrodes, respectively. One pair (Ag/AgCl + Pt) was placed in each phase. All voltammograms were recorded with EmStat3+ (PalmSens) equipped with a differential pulse amplifier or Autolab 302n (Metrohm).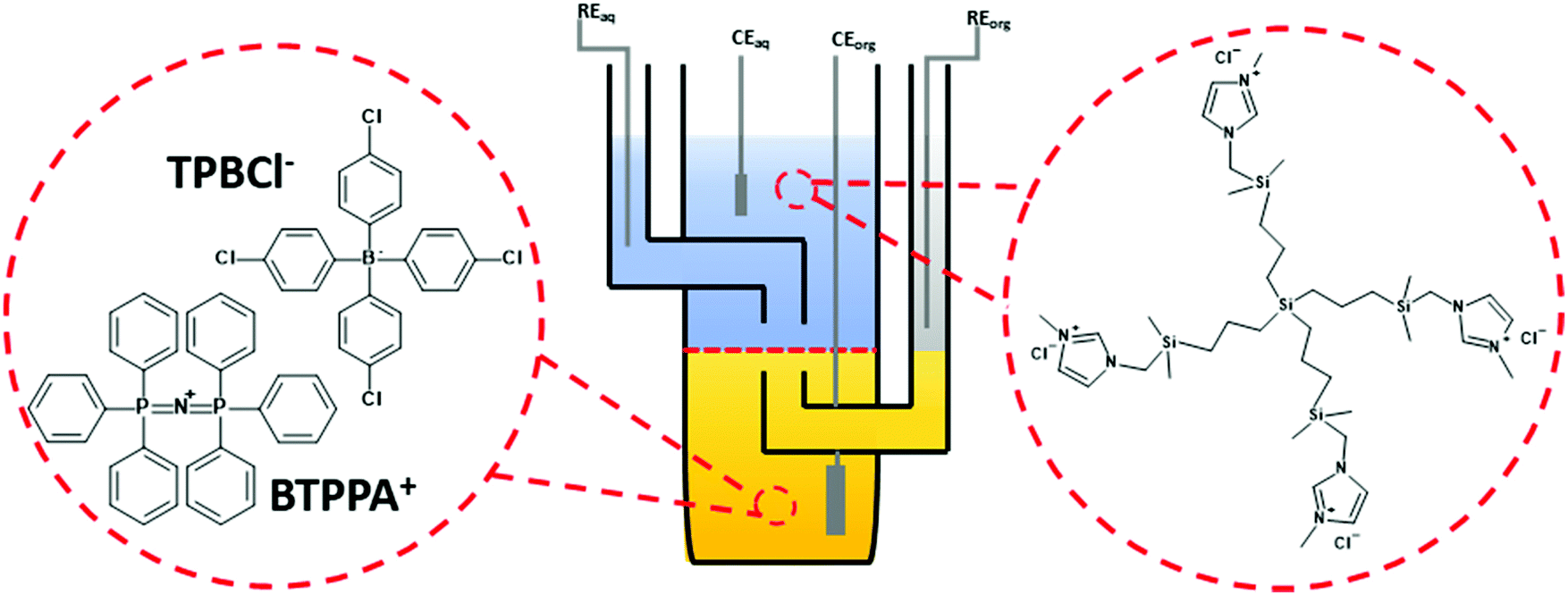 | ||
| Fig. 1 The electrochemical cell which was used to study the interfacial behavior of imidazolium terminated carbosilane dendrimers. Left: structure of the BTPPA+TPBCl− being the organic phase (bottom phase) background electrolyte. Right: structure of the G1 dendrimer, which similarly to other members of the family, was always initially dissolved in the aqueous phase. RE – reference electrode, CE – counter electrode, aq – the aqueous phase, and org – the organic phase. The red dashed line in between two Luggin capillaries indicates the position of the ITIES. | ||
The following electrochemical cells were applied during experiments:
Cell I:
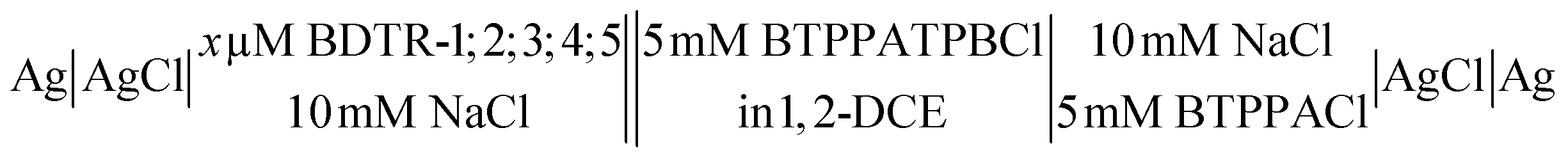
Cell II

Unless otherwise mentioned 20 mV s−1 was used to record all voltammograms. The pH of the aqueous phase in all experiments, except for ITVs shown in ESI Fig. 1,† was equal to around 6 (pH of the non-buffered 10 mM NaCl solution affected by the atmospheric CO2 uptake).
3. Results and discussion
Synthesized dendrimers had the appearance of yellowish oil that was dissolved in 10 mM NaCl aqueous phase, serving as the stock solution. Fig. 2A–E show ion transfer voltammograms recorded before (red dash–dot curves) and after (solid black curves) the addition of the corresponding dendrimers to electrochemical cell I. We have found that the addition of each studied compound to the aqueous phase gave rise to ionic currents. The interfacial activity is expected and governed by the existence of quaternary ammonium cations located within imidazolium groups at the periphery of the carbosilane core, assuring electrochemical activity on the entire conventional pH scale (see ESI Fig. 1†). According to the convention, positive currents are attributed to the positively charged dendrimers transferring from the water to the organic phase, whereas negative currents are recorded on their back transfer from the organic phase to the aqueous phase. Studied molecules differ in size (the hydrodynamic radius grows together with the generation), the number of charges located at the dendrimer periphery (BDTR-1, 4(+); BDTR-2, 4(+); BDTR-3, 8(+); BDTR-4, 8(+); BDTR-5, 16(+)), and the existence of the mesitylene substituent attached to a nitrogen atom located within the imidazolium ring (BDTR-2 and BDTR-4) or methyl substituent for the rest. For the chemical structures refer to Fig. 2 – right section. Molecular hydrophilicity–hydrophobicity can be directly inferred from the voltammetry data. All studied species transfer across the interface, within the less positive side of the potential window indicating their increased hydrophobicity (hydrophilic species require higher applied potential difference values to be transferred from the aqueous to the organic phase).40 Occasionally, especially lower dendrimer generations gave irregular current patterns similar to those shown in Fig. 2A (second positive peak with a strange geometry). We believe that these signals originate from the phenomenon known as the electrochemical instability41–43 caused by the surface-active species triggering local changes (gradients) in the interfacial tension upon adsorption to the LLI. Formal Gibbs energy of the ion transfer reaction is directly related to the formal Galvani potential of ion transfer (see eqn (1)).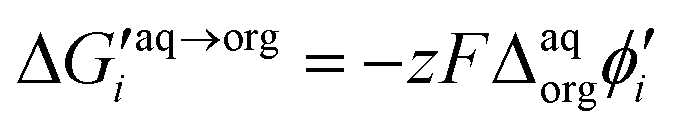 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1) and
485 C mol−1) and 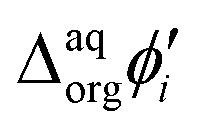 is the formal Galvani potential difference of the ion transfer reaction. Based on the shape of the recorded irregular ionic currents we concluded that upon interface polarization dendrimers undergo an interfacial ion transfer process accompanied by an interfacial adsorption (this is also discussed later in the manuscript).
is the formal Galvani potential difference of the ion transfer reaction. Based on the shape of the recorded irregular ionic currents we concluded that upon interface polarization dendrimers undergo an interfacial ion transfer process accompanied by an interfacial adsorption (this is also discussed later in the manuscript).
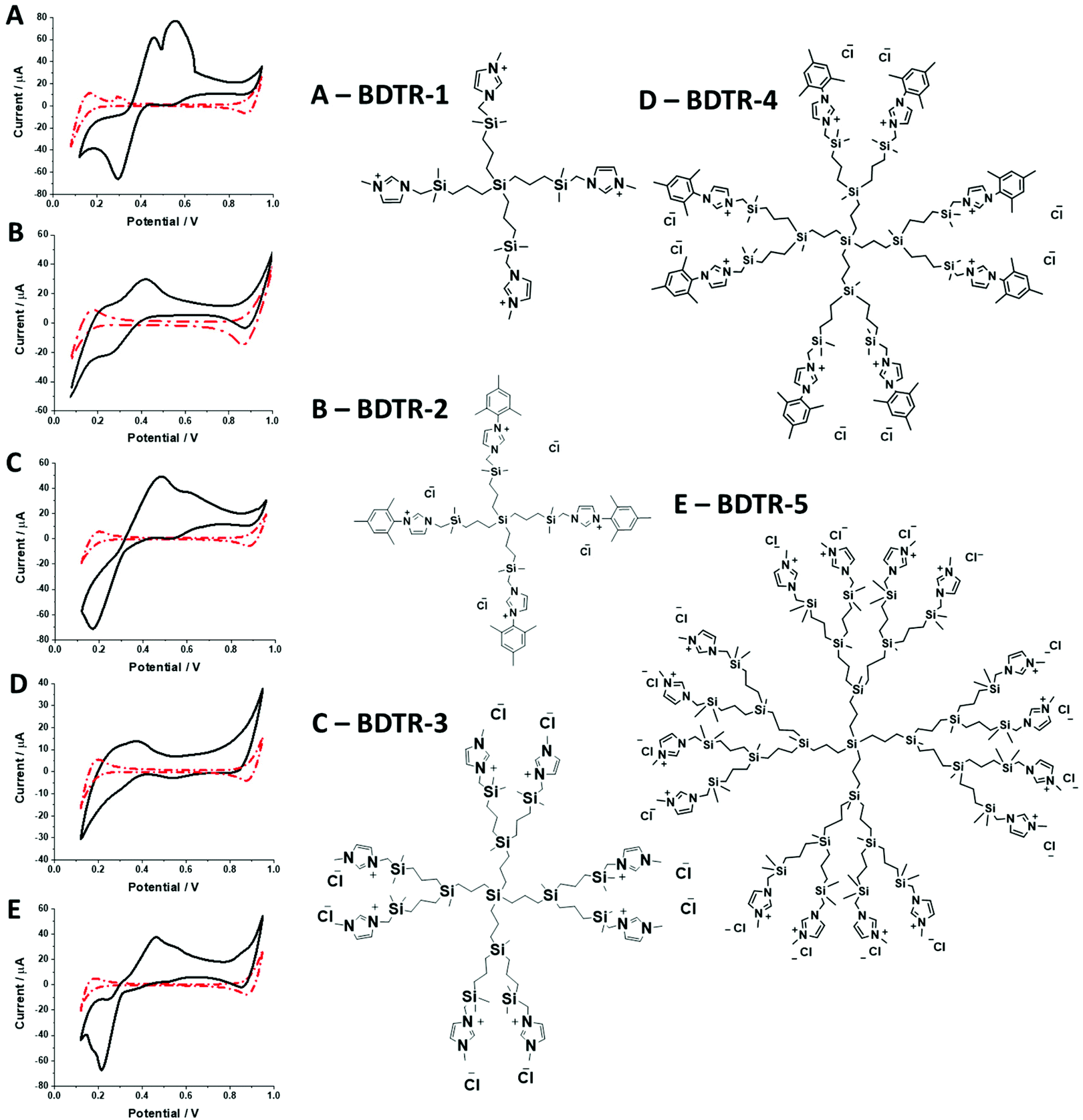 | ||
| Fig. 2 Ion transfer voltammograms recorded in the left panel correspond to the following dendrimer generations: A – G1 (BDTR-1); B – G2 (BDTR-2); C – G3 (BDTR-3); D – G4 (BDTR-4); E – G5 (BDTR-5). The black solid lines were recorded in cell I for x = 150 μM. The red dash–dot lines correspond to the blank voltammograms recorded in the absence of a dendrimer (10 mM NaCl). The right panel depicts dendrimer structures. | ||
It is therefore difficult to unambiguously determine the 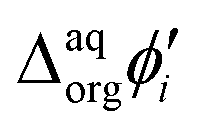 values. However, for evenly charged molecules (for BDTR-1 and BDTR-2 the charge equals 4; for BDTR-3 and BDTR-4 the charge equals 8) we can deduce the change in the free Gibbs energy (ΔΔG) according to
values. However, for evenly charged molecules (for BDTR-1 and BDTR-2 the charge equals 4; for BDTR-3 and BDTR-4 the charge equals 8) we can deduce the change in the free Gibbs energy (ΔΔG) according to
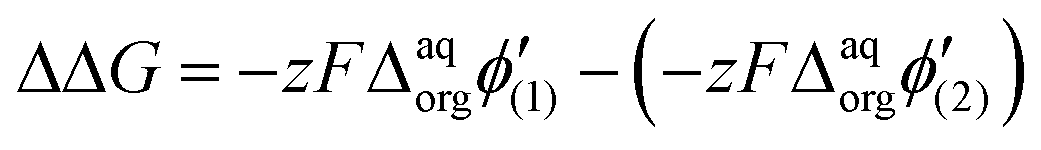 | (2) |
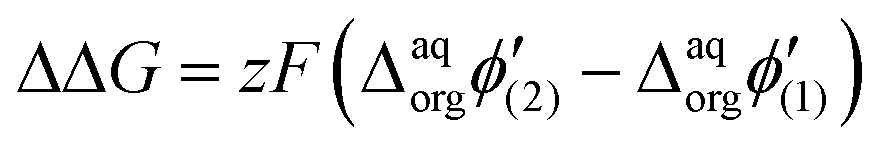 | (3) |
Since the difference in the formal Galvani potential difference of the ion transfer reaction between evenly charged dendrimers (1 – more hydrophilic and 2 – more hydrophobic) should be equal to the difference in the corresponding forward (or reverse) peak potential values we can simplify eqn (3) to
| ΔΔG = zF(Eforward or reveresedp(2) − Eforward or reveresedp(1) | (4) |
Like so, we can calculate the additional fraction of energy needed to trigger interfacial ion transfer for evenly charged dendrimers decorated with mesitylene and methyl substituents. In both cases, the presence of mesitylene groups within the structures of dendrimers BDTR-2 and BDTR-4 caused a shift in the recorded peak position towards less positive potential difference values with respect to their structural analogs deprived of this substituent (BDTR-1 and BDTR-3, respectively). According to our calculations (eqn (4)), the ΔΔG between BDTR-1 and BDTR-2 equals around 18.5 kJ mol−1, whereas for BDTR-3 and BDTR-4 it is 53.3 kJ mol−1. If we further divide it by the dendrimer charge, we get similar values (holding the same order of magnitudes) of 4.6 kJ mol−1 and 6.7 kJ mol−1 per mesitylene branch for pairs BDTR-1/BDTR-2 and BDTR-3/BDTR-4, respectively.
As shown in Fig. 3 and ESI Fig. 2 (see ESI†) increasing concentration of each dendrimer in the aqueous phase resulted in an increasing recorded peak current. Ionic currents started appearing at dendrimer concentrations equal to around 5–20 μM which agrees well with the calculated limit of detection defined as 3 times the standard deviation of the intercept divided by the slope of the calibration curve (see Table 1, column 11, for details). The calibration curves from Fig. 3B and ESI Fig. 2B, D† recorded for the BDTR-2, BDTR-1 and BDTR-3, respectively, exhibit two linear dynamic ranges. Sensitivity for the first part of the data set (from the first studied concentration to around 50 μM) for the first three studied dendrimer generations was always higher than the sensitivity of the second dynamic range. Surprising is the second part of the calibration curve which levels off stating from [dendrimer] > 50 μM. This most probably indicates an interfacial adsorption process which to some extent affects the simple ion transfer reaction. The pattern of the calibration curve for BDTR-5 is different. This chemical compound exhibits a behaviour that was reported for some of the positively charged macromolecules that were found to be adsorbed to the LLI, further facilitating the transfer of the organic phase anion.44,45 At this point we can speculate that the interfacial adsorption of BDTR-5, leading to the positive charge accumulation within the interface at higher dendrimer concentration values, may induce the interfacial transfer of TPBCl− (being the anionic part of the organic phase background electrolyte) from the organic phase to the water phase. This, in turn, may explain the higher than expected recorded ionic currents.
 | ||
| Fig. 3 A and C show the ion transfer voltammograms recorded in the presence of increasing (indicated next to the black arrow) concentrations of BDTR-2 and BDTR-5, respectively. Calibration curves are given as part B (for BDTR-2) and part D (BDTR-5). Red dash–dot lines are the blank readings recorded in the absence of dendrimers. The scan rate was equal to 20 mV s−1. Linear fit equations are displayed next to calibration curves. 1st – corresponds to the first linear dynamic range and 2nd – corresponds to the second linear dynamic range. | ||
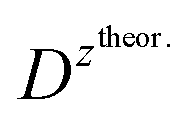 and
and 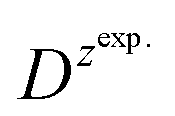 – diffusion coefficients calculated using the Randles–Sevcik equation with either ztheor. or zexp.; r – the hydrodynamic radius calculated using the Stokes–Einstein equation; LOD – the limit of detection calculated as 3 times the standard deviation of the intercept divided by the slope of the calibration curve; Saq or Sorg – the slope of the calibration curve of the forward and reverse signal, respectively. SlogI vs. log[v] – the slope of the logarithmic current versus logarithmic scan rate dependency. Aq and org stand for the aqueous phase and the organic phase, respectively. (+) and (−) pertain to the positive (forward) and negative (reverse) peak currents, respectively
– diffusion coefficients calculated using the Randles–Sevcik equation with either ztheor. or zexp.; r – the hydrodynamic radius calculated using the Stokes–Einstein equation; LOD – the limit of detection calculated as 3 times the standard deviation of the intercept divided by the slope of the calibration curve; Saq or Sorg – the slope of the calibration curve of the forward and reverse signal, respectively. SlogI vs. log[v] – the slope of the logarithmic current versus logarithmic scan rate dependency. Aq and org stand for the aqueous phase and the organic phase, respectively. (+) and (−) pertain to the positive (forward) and negative (reverse) peak currents, respectively
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | z theor. | z exp. | ΔEp/mV |
|
M/g mol−1 |
|
|
|
|
LOD/μM | S aq/A M−1 | S org/A M−1 |
S
logI vs. log[v]
|
| BDTR-1 | 4 | 2.1 | 28.1 | 0.95 | 955.36 | (+) 4.8 × 10−7 | (+) 3.8 × 10−6 | (+) 4.55 | (+) 0.6 | (+) 2.24 | 1st 0.576 | 1st 0.665 | (+) 0.56 |
| (−) 3.1 × 10−7 | (−) 2.5 × 10−6 | (−) 7.0 | (−) 0.9 | (−) 6.96 | 2nd 0.317 | 2nd 0.395 | (−) 0.45 | ||||||
| BDTR-2 | 4 | 1.2 | 51.0 | 0.97 | 1371.97 | (+) 7.6 × 10−8 | (+) 1.4 × 10−6 | (+) 28.7 | (+) 1.7 | (+) 5.36 | 1st 0.322 | 1st 0.247 | (+) 0.49 |
| (−) 7.2 × 10−8 | (−) 1.3 × 10−7 | (−) 30.3 | (−) 1.8 | (−) 4.09 | 2nd 0.139 | 2nd 0.144 | (−) 0.51 | ||||||
| BDTR-3 | 8 | 2.9* | 20.5 | 0.92 | 2223.44 | (+) 1.2 × 10−8 | (+) 2.5 × 10−7 | (+) 182.0 | (+) 8.7 | (+) 17.27 | 1st 0.508 | 1st 0.877 | (+) 0.54 |
| (−) 3.9 × 10−8 | (−) 8.3 × 10−7 | (−) 560.0 | (−) 26.3 | (−) 21.89 | 2nd 0.265 | 2nd 0.538 | (−) 0.54 | ||||||
| BDTR-4 | 8 | — | — | — | 3056.66 | (+) 7.5 × 10−8 | — | (+) 29.1 | — | (+) 5.8 | 1st 0.082 | 1st - | (+) 0.58 |
| (−) - | (−) - | (−) – | 2nd - | 2nd - | (−) - | ||||||||
| BDTR-5 | 16 | — | — | 0.55 | 4756.60 | (+) 2.2 × 10−10 | — | (+) 9930 | — | (+) 16.98 | 1st 0.159 | 1st 0.216 | (+) 0.56 |
| (−) 2.4 × 10−10 | (−) 9100 | (−) 11.00 | 2nd 0.458 | 2nd 0.559 | (−) 0.26 |
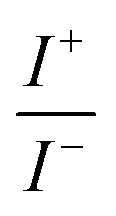
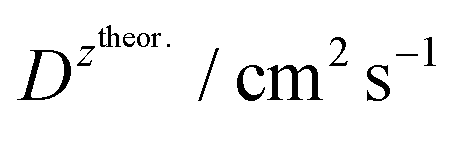
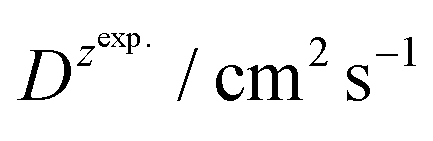

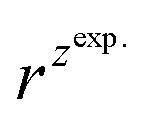
For BDTR-1, BDTR-2, and BDTR-3 the forward and reverse peak current ratio was close to unity, indicating the reversibility of the ion transfer reaction (see Table 1, column 5). Since the transfer of BDTR-4 was severely overlapped with the interfacial transfer of the potential window limiting ion (in that case Cl−aq↔org) we were unable to extract most of its physicochemical and analytical parameters summarized in Table 1. For BDTR-5 the forward and reverse peak current ratio was equal to 0.55. However, the very broad forward (positive current) peak indicates that more than one charge transfer process occurs within the given potential range (from e.g. 0.3 to 0.7 V). The integrated area below forward and reverse peak currents equals 0.13 μC and 0.17 μC, respectively, and the corresponding ratio is 0.76. We think that dendrimer BDTR-5 is first adsorbed to the LLI, followed by its gradual transfer to the organic phase, manifested as the complex, and a multistep charge transfer reaction coupled to the interfacial transfer of the TPBCl− ion. On the back transfer, the current is first slightly increased to finally give a sharp and well-defined peak with a characteristic diffusion-limited tail.
For all studied dendrimers we have found that the forward and reverse peak currents, for a fixed dendrimer concentration, increase linearly with the square root of the scan rate (see Fig. 4 and ESI Fig.2†). The rearranged Randles–Sevcik equation
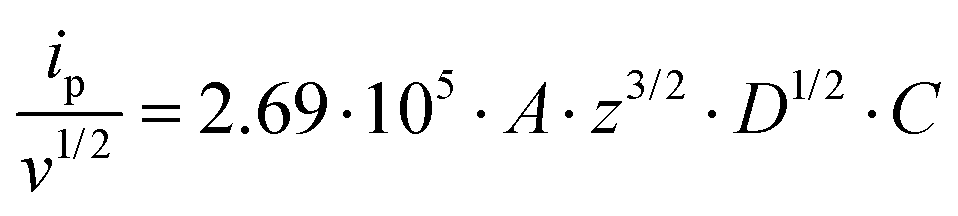 | (5) |
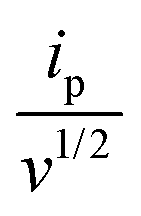 is the slope of the linear fit equation (see Fig. 4B, D and ESI Fig. 3B, D and F†), 2.69 × 105 is a result of an operation on physicochemical constants (for 25 °C), A is the surface area of the LLI (1.33 cm2), z is the charge crossing the LLI, and C is the dendrimer concentration (mol cm−3), was used to calculate the diffusion coefficients (D, cm2 s−1) for studied molecules. The resulting values are summarized in Table 1 (columns 7 and 8). For the calculations, we have used the theoretical number of charges deduced from the chemical structure of each dendrimer and equal to the number of imidazolium groups within the molecular periphery and the charge that was estimated based on the forward and reverse peak to peak separation (Table 1, columns 2 and 3, respectively). When theoretical molecular charge values were substituted in eqn (5) we have obtained diffusion coefficient values 1–2 orders of magnitude lower than expected 10−6 cm2 s−1 for lower and 10−7 cm2 s−1 for higher dendrimer generations.35,38,46 This means that the interfacial charge transfer reactions recorded in the presence of dendritic molecules are not a one-step process. Reasonable values were obtained, which were 3.8 × 10−6 cm2 s−1 for BDTR-1, 1.4 × 10−6 cm2 s−1 for BDTR-2, and 2.5 × 10−7 cm2 s−1 for BDTR-3, when we performed the charge calculated based on the forward and reverse peak to peak separation taken from ITVs recorded at [dendrimer] < 100 μM at 2 mV s−1 (due to uncompensated resistance the peak to peak separation values were increasing during the measurements together with the increasing [dendrimer] and the experimental scan rate). Assuming that the interfacial adsorption processes affect the dendrimer ion transfer to some extent the utility of the Randles–Sevcik equation is most probably limited, and hence, obtained values should be treated as approximations. The calculated zexp. values were significantly lower than the ztheor.. This discrepancy may be due to a few reasons, including sluggish kinetics, the presence of the uncompensated resistance, and/or the occurrence of the interfacial adsorption processes. Diffusion coefficient values for higher generations (which are BDTR-4 and BDTR-5) are not available since we were unable to accurately define the peak to peak separation, and we assumed that these species (especially BDTR-5) do not undergo reversible ion transfer reactions. Also, based on the calculated diffusion coefficients and the Einstein-Stokes equation we have estimated the values of hydrodynamic radii for BDTR-1, BDTR-2, and BDTR-3 which were equal to 0.6, 1.7, and 8.7 nm, respectively. We have found that the first two values are in good agreement (BDTR-1: 0.9 nm and BDTR-2: 0.7 nm) with the hydrodynamic radii calculated based on diffusion coefficients measured with diffusion oriented spectroscopy-NMR. The same technique provided the hydrodynamic radius for BDTR-3 equal to 1.7 nm, still within the same order of magnitude (see Table S1 from ESI† for more details) as the value calculated using the electrochemically deduced diffusion coefficient. This underlines that the complex interfacial behaviour of higher dendrimer generations hampers proper evaluation of the charge values crossing the interface during single ion transfer. The values of the slope of the logarithmic dependence of the peak current versus experimental scan rate were in the range from 0.45 to 0.58, indicating that the charge transfer reaction is diffusion-limited. Also, we have noticed that BDTR-5, see Fig. 4A and 3C, gave very broad positive and negative peaks (especially for [dendrimer] < 50 μM), which suggests the occurrence of a multistep charge transfer process. At this point, we believe that BDTR-5, to some extent, can facilitate the transfer of TPBCl− from the organic to the aqueous phase. All these indicate that the voltammetric behavior of all studied dendrimers is complex. The ion transfer reaction for higher generations is surely coupled to the interfacial adsorption/desorption step. This is especially prominent for BDTR-3–BDTR-5 and is in line with other reports.47
is the slope of the linear fit equation (see Fig. 4B, D and ESI Fig. 3B, D and F†), 2.69 × 105 is a result of an operation on physicochemical constants (for 25 °C), A is the surface area of the LLI (1.33 cm2), z is the charge crossing the LLI, and C is the dendrimer concentration (mol cm−3), was used to calculate the diffusion coefficients (D, cm2 s−1) for studied molecules. The resulting values are summarized in Table 1 (columns 7 and 8). For the calculations, we have used the theoretical number of charges deduced from the chemical structure of each dendrimer and equal to the number of imidazolium groups within the molecular periphery and the charge that was estimated based on the forward and reverse peak to peak separation (Table 1, columns 2 and 3, respectively). When theoretical molecular charge values were substituted in eqn (5) we have obtained diffusion coefficient values 1–2 orders of magnitude lower than expected 10−6 cm2 s−1 for lower and 10−7 cm2 s−1 for higher dendrimer generations.35,38,46 This means that the interfacial charge transfer reactions recorded in the presence of dendritic molecules are not a one-step process. Reasonable values were obtained, which were 3.8 × 10−6 cm2 s−1 for BDTR-1, 1.4 × 10−6 cm2 s−1 for BDTR-2, and 2.5 × 10−7 cm2 s−1 for BDTR-3, when we performed the charge calculated based on the forward and reverse peak to peak separation taken from ITVs recorded at [dendrimer] < 100 μM at 2 mV s−1 (due to uncompensated resistance the peak to peak separation values were increasing during the measurements together with the increasing [dendrimer] and the experimental scan rate). Assuming that the interfacial adsorption processes affect the dendrimer ion transfer to some extent the utility of the Randles–Sevcik equation is most probably limited, and hence, obtained values should be treated as approximations. The calculated zexp. values were significantly lower than the ztheor.. This discrepancy may be due to a few reasons, including sluggish kinetics, the presence of the uncompensated resistance, and/or the occurrence of the interfacial adsorption processes. Diffusion coefficient values for higher generations (which are BDTR-4 and BDTR-5) are not available since we were unable to accurately define the peak to peak separation, and we assumed that these species (especially BDTR-5) do not undergo reversible ion transfer reactions. Also, based on the calculated diffusion coefficients and the Einstein-Stokes equation we have estimated the values of hydrodynamic radii for BDTR-1, BDTR-2, and BDTR-3 which were equal to 0.6, 1.7, and 8.7 nm, respectively. We have found that the first two values are in good agreement (BDTR-1: 0.9 nm and BDTR-2: 0.7 nm) with the hydrodynamic radii calculated based on diffusion coefficients measured with diffusion oriented spectroscopy-NMR. The same technique provided the hydrodynamic radius for BDTR-3 equal to 1.7 nm, still within the same order of magnitude (see Table S1 from ESI† for more details) as the value calculated using the electrochemically deduced diffusion coefficient. This underlines that the complex interfacial behaviour of higher dendrimer generations hampers proper evaluation of the charge values crossing the interface during single ion transfer. The values of the slope of the logarithmic dependence of the peak current versus experimental scan rate were in the range from 0.45 to 0.58, indicating that the charge transfer reaction is diffusion-limited. Also, we have noticed that BDTR-5, see Fig. 4A and 3C, gave very broad positive and negative peaks (especially for [dendrimer] < 50 μM), which suggests the occurrence of a multistep charge transfer process. At this point, we believe that BDTR-5, to some extent, can facilitate the transfer of TPBCl− from the organic to the aqueous phase. All these indicate that the voltammetric behavior of all studied dendrimers is complex. The ion transfer reaction for higher generations is surely coupled to the interfacial adsorption/desorption step. This is especially prominent for BDTR-3–BDTR-5 and is in line with other reports.47
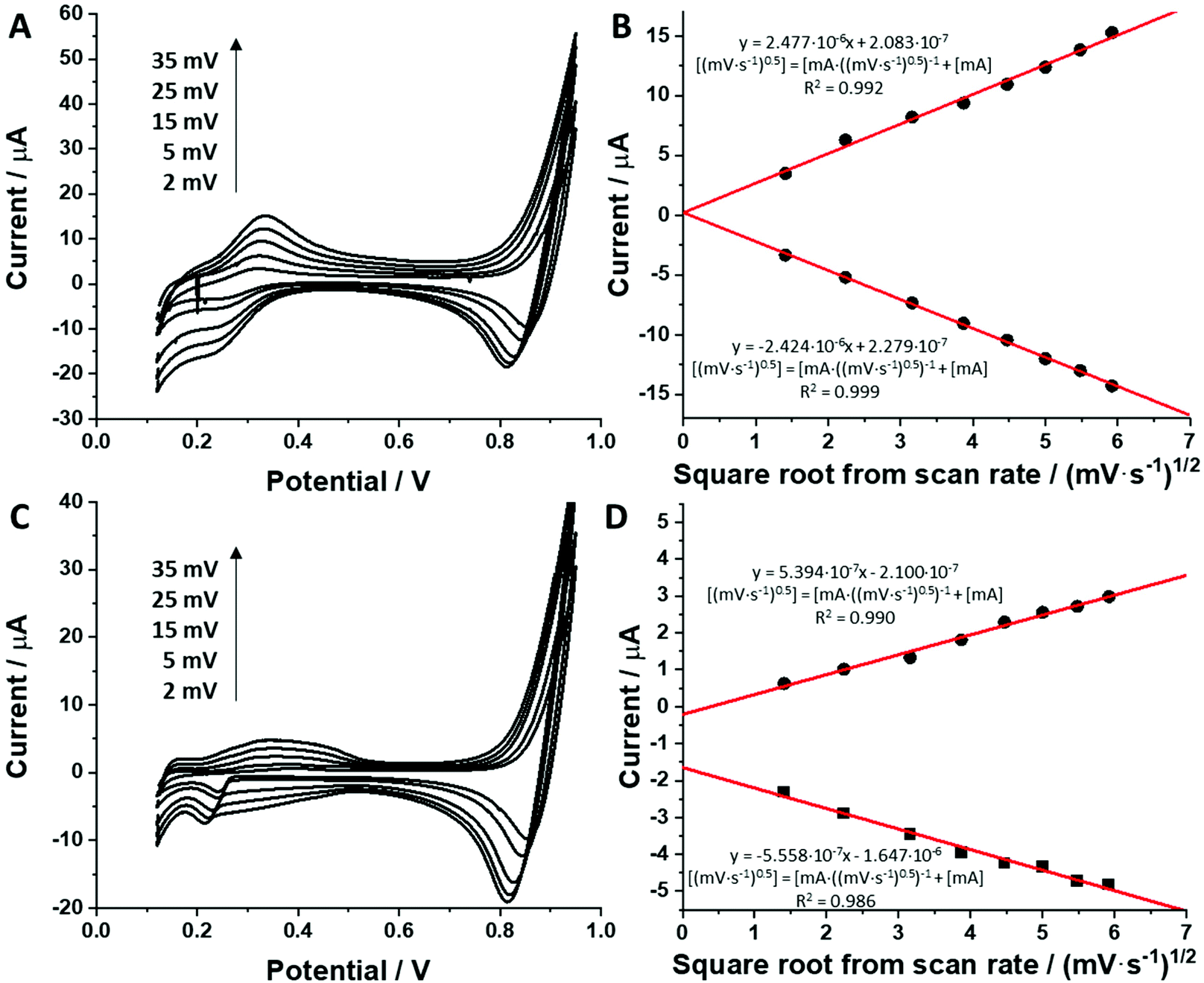 | ||
| Fig. 4 A and C are ion transfer voltammograms recorded at 2; 5; 15; 25 and 35 mV s−1, whereas B and D show current versus square root of the scan rate dependency. Graphs A and B correspond to the BDTR-2 (100 μM) dendrimer. Graphs C and D correspond to the BDTR-5 dendrimer (50 μm). Linear fit equations are displayed next to the corresponding experimental data points. | ||
The behaviour of charged macromolecules (proteins, polyelectrolytes, and dendrimers) at the electrified LLI shares many mechanistic aspects.32,45,48–50 Upon interfacial polarization, these molecules were found to form a complex with the organic phase anion at the LLI, giving characteristic voltammetric behaviour. (i) Instead of a diffusion-limited current tail, a sharp drop is recorded for the reverse peak current after reaching its maximum (attributed to the partial back transfer of the organic phase anion from the interface to the organic phase). Here, even for the highest dendrimer generation, this behaviour was absent. The complex shape of the (especially) forward peak patterns indicates that the interfacial charge transfer is coupled to the dendrimer adsorption/desorption to/from the LLI. (ii) Macromolecules, upon consecutive voltammetric cycling, tend to form multilayer deposits at the LLI, shown by the increasing forward and reverse peak currents. For all studied dendrimers, the opposite dependency was observed, with the currents dropping over the first few cycles (see the insets of Fig. 5A and B). At this point, we can speculate that the dendrimers studied here (especially smaller molecules) are initially (before LLI polarization) adsorbed to, or in other words pre-concentrated at, the LLI. This could explain the high ionic currents recorded during the first scan, which may be due to the analyte stripping from the LLI. (iii) The position and intensity of the faradaic signals for interfacially active macromolecules are highly dependent on the nature and concentration of the organic phase background electrolyte anion.28,51 For the interfacial complex formation (charged macromolecule–hydrophobic anion) it is expected to observe dropping faradaic current signals as a function of the dropping concentration of the background electrolyte. Fig. 5C and E show the ion transfer voltammograms recorded for BDTR-2 and BDTR-5, respectively, in cell II for the organic phase containing either 1 mM or 10 mM BTPPATPBCl in the 1,2-dichloroethane. Fig. 5D and F show the positive and negative peak current intensities for other studied concentrations of the BTPPATPBCl dissolved in the organic phase (ITVs are not shown). We have observed that the decreasing concentration of the organic phase background anion has a minimal effect on the positive and negative peak current intensities for BDTR-2. The increasing forward and reverse peak to peak separation (see the red dash–dotted curve recorded for 1 mM BTPPATPBCl) is due to the increasing uncompensated resistance of the system rather than due to the interaction between BDTR-2 and the anionic part of the organic phase background electrolyte. Based on this and the above observations, we concluded that the interfacial charge transfer reaction for smaller dendrimers is mainly governed by the simple and reversible ion transfer reaction. The positive and negative peak current intensities for BDTR-5, although slightly more scattered, (see Fig. 5F), gave lower peak current values for decreasing concentration of BTPPATPBCl dissolved in the organic phase. As such, we have concluded that branched generations with higher dimensionality undergo interfacial ion transfer coupled to interfacial adsorption (when the direction of the ion transfer is from the aqueous to the organic phase). This is evidenced by (i) broad forward positive peaks, (ii) overlaid signals indicating multistep interfacial charge transfer reactions and (iii) irregular variations in the peak current intensity and forward peak position recorded at different pH values (ESI Fig. 1†). Our study reveals that all five carbosilane dendrimers can cross the LLI under external polarization. In addition to the insight into the interaction between studied macromolecules and the mimetic interface, we think that these molecules can be used to study the permeability of the electrified LLI modified with porous materials that we wish to pursue in the future.
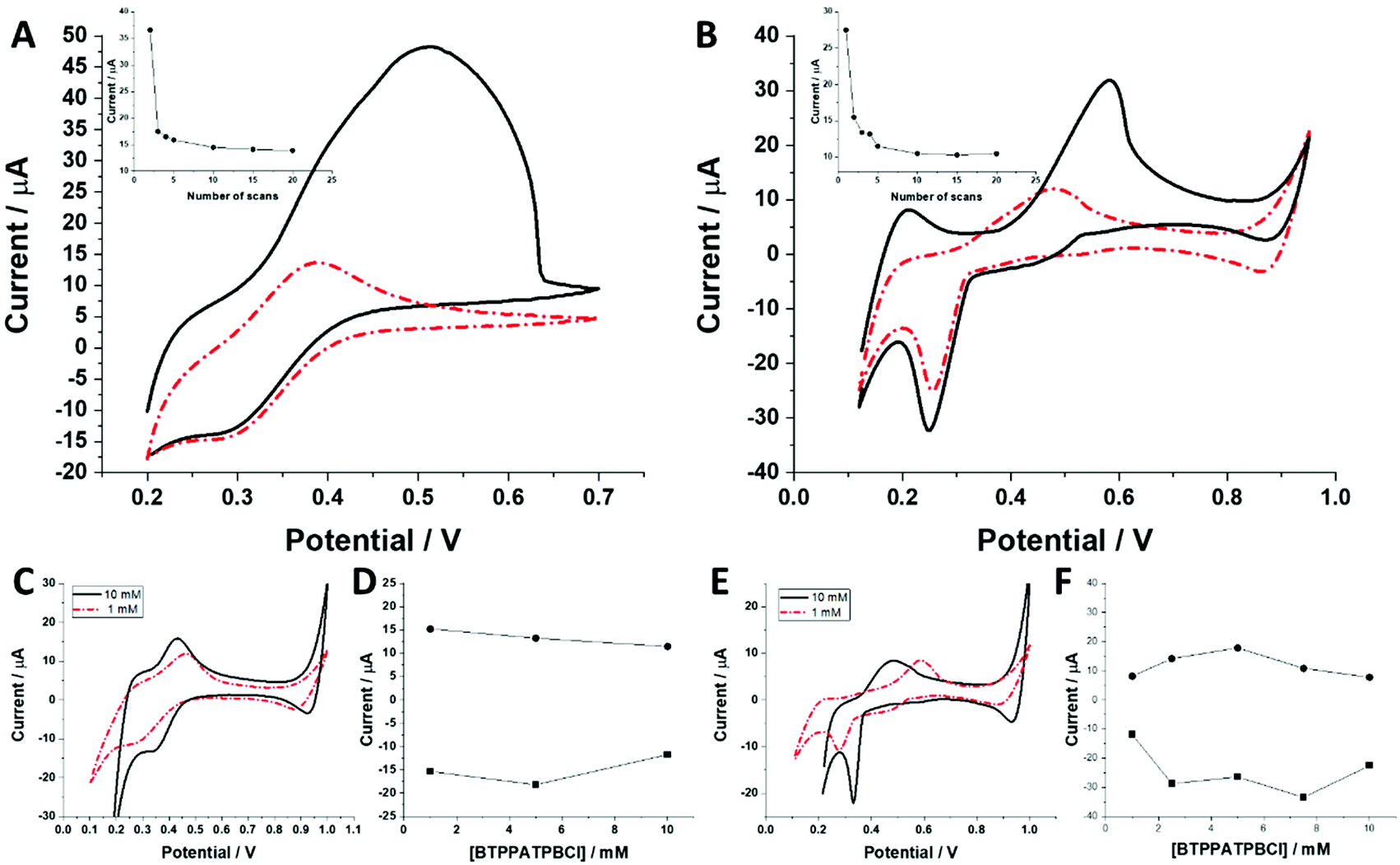 | ||
| Fig. 5 A and B show the effect of consecutive voltammetric cycling for BDTR-2 and BDTR-5, respectively. Black solid lines represent scan 2 for BDTR-2 and scan 1 for BDTR-5. The red dot–dashed line is scan 20. Insets are the forward peak currents versus scan number 1 (or 2 for BDTR-2), 2, 3, 4, 5, 10, 15, and 20. The dendrimer concentration was set to 100 μM, and the scan rate was 20 mV s−1. C (BDTR-2) and E (BDTR-5) are ion transfer voltammograms recorded in cell II with 1 mM (red dot–dash line) and 10 mM (black solid line) BTPPATPBCl in the organic phase. D and F are the corresponding forward and reverse peak current intensities for the varying concentration of the organic phase background electrolyte. The concentration of BDTR-2 was equal to 100 μM and for BDTR-5 it was 50 μM. The scan rate was set to 20 mV s−1. Positive peak currents are given as a result of the signal subtracted from a blank. | ||
4. Conclusions
In this work, we have studied the behaviour of different generations of carbosilane dendrimers terminated with methyl- (G1–G3) or mesityl-imidazolium (G1–G2) groups. We have found that all studied species are electrochemically active at the polarized water–1,2-dichloroethane interface. The studied compounds could be detected starting from concentrations equal to around 5–20 μM. Our data indicate that small changes in the molecular structure affecting molecular lipophilicity can be directly visualized using ion transfer voltammetry. The interfacial behaviour of the studied species is complex and recorded ionic currents originate from a diffusion-limited ion transfer reaction coupled to the interfacial adsorption process. The latter was significantly more prominent for higher dendrimer generations. We have excluded the possibility of facilitated transfer of the organic phase anion (usually responsible for interfacial charge transfer phenomena recorded in the presence of positively charged macromolecules) triggered by the BDTR dendrimers (from BDTR-1 to BDTR-3) adsorbed to the liquid–liquid interface. We are convinced that these species can be used in the future for the characterization of liquid–liquid interfaces modified with porous membranes or lipid monolayers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. P. is grateful for financial support from the National Science Center (NCN) in Krakow, Poland (Grant No. UMO-2018/31/D/ST4/03259). This work was also supported by CTQ2017-86224-P (MINECO) and by a EUROPARTNER: strengthening and spreading international partnership activities of the Faculty of Biology and Environmental Protection for interdisciplinary research and innovation of the University of Lodz Programme: NAWA International Academic Partnership Programme (PPI/APM/2018/1/00007/U/001). CIBER-BBN is an initiative funded by the VI National R&D&i Plan 2008–2011, Iniciativa Ingenio 2010, Consolider Program, CIBER Actions, and financed by the Instituto de Salud Carlos III with assistance from the European Regional Development Fund.References
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257–1281 CrossRef CAS.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS.
- M. Seiler, Fluid Phase Equilib., 2006, 241, 155–174 CrossRef CAS.
- J. M. Frechet, Science, 1994, 263, 1710–1715 CrossRef CAS.
- D. A. Tomalia, A. M. Naylor and W. A. Goddard, Angew. Chem., Int. Ed. Engl., 1990, 29, 138–175 CrossRef.
- R. S. Ambekar, M. Choudhary and B. Kandasubramanian, Eur. Polym. J., 2020, 126, 109546 CrossRef CAS.
- S. W. Krska and D. Seyferth, J. Am. Chem. Soc., 1998, 120, 3604–3612 CrossRef CAS.
- D. Maciel, C. Guerrero-Beltrán, R. Ceña-Diez, H. Tomás, M. Á. Muñoz-Fernández and J. Rodrigues, Nanoscale, 2019, 11, 9679–9690 RSC.
- I. Heredero-Bermejo, N. Gómez-Casanova, S. Quintana, J. Soliveri, F. J. de la Mata, J. Pérez-Serrano, J. Sánchez-Nieves and J. L. Copa-Patiño, Pharmaceutics, 2020, 12, 918 CrossRef CAS.
- N. Weber, P. Ortega, M. I. Clemente, D. Shcharbin, M. Bryszewska, F. J. de la Mata, R. Gómez and M. A. Muñoz-Fernández, J. Controlled Release, 2008, 132, 55–64 CrossRef CAS.
- E. Pedziwiatr-Werbicka, K. Milowska, V. Dzmitruk, M. Ionov, D. Shcharbin and M. Bryszewska, Eur. Polym. J., 2019, 119, 61–73 CrossRef CAS.
- B. Noriega-Luna, L. A. Godínez, F. J. Rodríguez, A. Rodríguez, G. Zaldívar-Lelo de Larrea, C. F. Sosa-Ferreyra, R. F. Mercado-Curiel, J. Manríquez and E. Bustos, J. Nanomater., 2014, 2014, 507273 Search PubMed.
- P. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268–307 CrossRef CAS.
- L. Zhao, C. Zhang, L. Zhuo, Y. Zhang and J. Y. Ying, J. Am. Chem. Soc., 2008, 130, 12586–12587 CrossRef CAS.
- J. Koryta, M. Březina, A. Hofmanová, D. Homolka, L. Q. Hung, W. Khalil, V. Mareček, Z. Samec, S. K. Sen, P. Vanýsek, J. Weber and J. Heyrovský, Bioelectrochem. Bioenerg., 1980, 7, 61–68 CrossRef CAS.
- H. A. Santos, V. García-Morales and C. M. Pereira, ChemPhysChem, 2010, 11, 28–41 CrossRef CAS.
- L. Poltorak, M. L. Verheijden, D. Bosma, P. Jonkheijm, L. C. P. M. de Smet and E. J. R. Sudhölter, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 2669–2680 CrossRef CAS.
- O. Purrucker, H. Hillebrandt, K. Adlkofer and M. Tanaka, Electrochim. Acta, 2001, 47, 791–798 CrossRef CAS.
- C. Steinem, A. Janshoff, H. J. Galla and M. Sieber, Bioelectrochem. Bioenerg., 1997, 42, 213–220 CrossRef.
- M. Naumowicz and Z. A. Figaszewski, J. Membr. Biol., 2005, 205, 29–36 CrossRef CAS.
- M. Naumowicz and Z. A. Figaszewski, J. Membr. Biol., 2011, 240, 47–53 CrossRef CAS.
- P. Vanysek and L. B. Ramirez, J. Chil. Chem. Soc., 2008, 2, 1455–1464 Search PubMed.
- J. Koryta, Electrochim. Acta, 1979, 24, 293–300 CrossRef CAS.
- L. Poltorak, K. Rudnicki, V. Kolivoška, T. Sebechlebská, P. Krzyczmonik and S. Skrzypek, J. Hazard. Mater., 2021, 402, 123411 CrossRef CAS.
- Z. Samec, Pure Appl. Chem., 2004, 76, 2147–2180 CAS.
- F. Kivlehan, Y. H. Lanyon and D. W. M. Arrigan, Langmuir, 2008, 24, 9876–9882 CrossRef CAS.
- H. Sakae, Y. Toda and T. Yokoyama, Electrochim. Acta, 2018, 90, 83–86 CAS.
- J. A. Ribeiro, F. Silva and C. M. Pereira, Anal. Chem., 2013, 85, 1582–1590 CrossRef CAS.
- M. D. Scanlon, J. Strutwolf and D. W. M. Arrigan, Phys. Chem. Chem. Phys., 2010, 12, 10040–10047 RSC.
- J. S. Riva, C. I. Cámara, A. V. Juarez and L. M. Yudi, J. Appl. Electrochem., 2014, 44, 1381–1392 CrossRef CAS.
- A. Berduque, M. D. Scanlon, C. J. Collins and D. W. M. Arrigan, Langmuir, 2007, 23, 7356–7364 CrossRef CAS.
- G. Herzog, S. Flynn and D. W. M. Arrigan, J. Phys.: Conf. Ser., 2011, 307, 1–6 CrossRef.
- H. Nagatani, M. Fujisawa and H. Imura, Langmuir, 2018, 34, 3237–3243 CrossRef CAS.
- H. Nagatani, T. Ueno and T. Sagara, Electrochim. Acta, 2008, 53, 6428–6433 CrossRef CAS.
- H. Sakae, H. Nagatani, K. Morita and H. Imura, Langmuir, 2014, 30, 937–945 CrossRef CAS.
- H. Nagatani, H. Sakae, T. Torikai, T. Sagara and H. Imura, Langmuir, 2015, 31, 6237–6244 CrossRef CAS.
- L. Poltorak, K. Morakchi, G. Herzog and A. Walcarius, Electrochim. Acta, 2015, 179, 9–15 CrossRef CAS.
- T. Rodríguez-Prieto, A. Fattori, C. Camejo, F. Javier de la Mata, J. Cano, M. Francesca Ottaviani and R. Gómez, Eur. Polym. J., 2020, 133, 109748 CrossRef.
- L. Poltorak, I. Eggink, M. Hoitink, M. De Puit, E. J. R. Sudholter and M. De Puit, Anal. Chem., 2018, 90, 8–13 CrossRef.
- T. Kakiuchi, M. Chiba, N. Sezaki and M. Nakagawa, Electrochem. Commun., 2002, 4, 701–704 CrossRef CAS.
- T. Kakiuchi, J. Electroanal. Chem., 2004, 569, 287–291 CrossRef CAS.
- Y. Kitazumi and T. Kakiuchi, Langmuir, 2009, 25, 8062–8068 CrossRef CAS.
- L. Poltorak, N. Van Der Meijden, S. Oonk, E. J. R. Sudhölter and M. De Puit, Electrochem. Commun., 2018, 94, 27–30 CrossRef CAS.
- G. Herzog, V. Kam and D. W. M. Arrigan, Electrochim. Acta, 2008, 53, 7204–7209 CrossRef CAS.
- M. A. van Dongen, B. G. Orr and M. M. B. Holl, J. Phys. Chem. C, 2014, 118, 7195–7202 CrossRef CAS.
- D. L. Cheung and P. Carbone, Soft Matter, 2013, 9, 6841–6850 RSC.
- G. Herzog, S. Flynn, C. Johnson and D. W. M. Arrigan, Anal. Chem., 2012, 84, 5693–5699 CrossRef CAS.
- E. Alvarez de Eulate and D. W. M. Arrigan, Anal. Chem., 2012, 84, 2505–2511 CrossRef CAS.
- S. Ulmeanu, H. J. Lee and H. H. Girault, Electrochem. Commun., 2001, 3, 539–543 CrossRef CAS.
- G. Herzog, W. Moujahid, J. Strutwolf and D. W. M. Arrigan, Analyst, 2009, 134, 1608–1613 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an02101f |
| This journal is © The Royal Society of Chemistry 2021 |