
From tissue engineering to engineering tissues: the role and application of in vitro models†
Daniela
Peneda Pacheco
 a,
Natalia
Suárez Vargas‡
a,
Sonja
Visentin
b and
Paola
Petrini
*ac
a,
Natalia
Suárez Vargas‡
a,
Sonja
Visentin
b and
Paola
Petrini
*ac
aDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta” – Politecnico di Milano, Italy. E-mail: paola.petrini@polimi.it
bMolecular Biotechnology and Health Sciences Department, University of Torino, Torino, Italy
cInter-University Center for the Promotion of the 3Rs Principles in Teaching & Research (Centro 3R), Politecnico di Milano Unit, Italy
First published on 10th November 2020
Abstract
Engineered models have emerged as relevant in vitro tools to foresee the translational potential of new therapies from the bench to the bedside in a fast and cost-effective fashion. The principles applied to the development of tissue-engineered constructs bring the foundation concepts to engineer relevant in vitro models. Engineered models often face scepticism, because regularly these do not include the extreme complexity of nature, but rather a simplification of a phenomenon. While engineering in vitro models, a hypothesis is imposed towards which defined parameters are included to assess the degree of similarity between the in vitro model and the native phenomenon, keeping in mind their intrinsic limitations. The development of in vitro models has been highly supported and disseminated by different regulatory agencies. This review aims at defining and exploring the multifaceted potential of tangible, not theoretical, models within the biomedical field to represent physiological tissues and organ-related phenomena.
 Daniela Peneda Pacheco | Daniela Peneda Pacheco obtained a BSc in bioengineering – biomedical engineering at Catholic University of Portugal, which was followed by a MSc in pharmaceutical technology from University of Porto. Recently, Daniela obtained a PhD in materials engineering from Politecnico di Milano in which she has mostly developed material-based in vitro models of the respiratory tract that enable to determine permeability coefficients and support the in vitro growth of human microbiota. Daniela is currently the CTO and founder member of Bac3Gel Lda, a startup based on human mucus models and substrates for culturing microbiota. |
 Natalia Suarez Vargas | Natalia Suarez Vargas is a mechanical engineer from Universidad de los Andes with a minor degree in product design and a MSc in biomedical engineering from Politecnico di Milano with a specialisation in cell, tissue engineering and biotechnology. Natalia is currently a PhD student in biomedical engineering at Universidad de los Andes in Bogota, studying translational medicine strategies for regenerative medicine devices. She also works as a Professor of Biodesign in Escuela Colombiana de Ingeniería Julio Garavito and as a research assistant in Universidad de los Andes within the research group of cardiovascular dynamics and R&D in medical devices. |
 Sonja Visentin | Sonja Visentin is an assistant Professor belonging to the scientific disciplinary sector of Medicinal Chemistry at the University of Turin. Her research activities have focused on the synthesis and pharmacological characterization of NO and calcium blocker donors. Subsequently her scientific research has expanded towards other innovative fields and now her main research interests are focused on the synthesis and pharmacological characterization of photosensitizers for photodynamic therapy, on nanotechnologies applied for medical purposes and on the study of the interaction between drugs and mucus. Sonja is one of the founder members of Bac3Gel Lda. |
 Paola Petrini | Paola Petrini is an Associate professor at Politecnico di Milano. She holds a MSc in Chemistry and a PhD in Materials for Engineering and Bioengineering. Her research interest focus on the chemical and structural design of biomaterials and technologies to face the challenges of regenerative medicine, drug delivery systems, and engineered tissue models. Her research keywords include natural materials, hydrogels, polysaccharides. In her academic life, she prioritizes mentoring students and young researchers in the early stage of their career. She is active in technology transfer and innovation and founder of Bac3Gel Lda. |
“All models are wrong, but some are useful” – George Box
1. Introduction
The replication of tissues and organs is a challenge deriving from the extremely intricate interrelation of different parameters within the complexity of nature, including mechanisms that are still unknown. This challenge was faced, in the last years, by tissue engineering (TE) that aims to regenerate failed tissues and organs.On the other side, models of tissues and physiological mechanisms can be engineered for diverse purposes. A model is a schematic representation to account selected aspects of a multifactorial phenomenon, and it is never intended as a replica of the entire complexity of any system in nature. Instead, they can be illuminating and useful tools for research when applying them for the specific purpose for which they were engineered, keeping in mind their intrinsic limitations.1 Models can be applied to many different fields, covering different semantic aspects. Indeed, one key application falls on the development of new drugs and medical devices by providing models of tissues and organs.
Both TE and engineered tissue models base their designs on the native tissue components and interactions, and both benefit from the biological and biochemical growing knowledge of the human body (Fig. 1). They both aim to replicate desired relations that could provide the closest possible approximation to the physiological situation. In the case of TE, this is done to induce repair or regeneration of a failing tissue for therapeutic purposes. On the other perspective, engineered models aim to recreate an in vitro exemplary that could serve either as a test bench for several issues, including drug effectiveness, or to unravel fundamental research questions about physiological or pathological phenomena for diagnostic or high throughput screening (HTS) purposes.
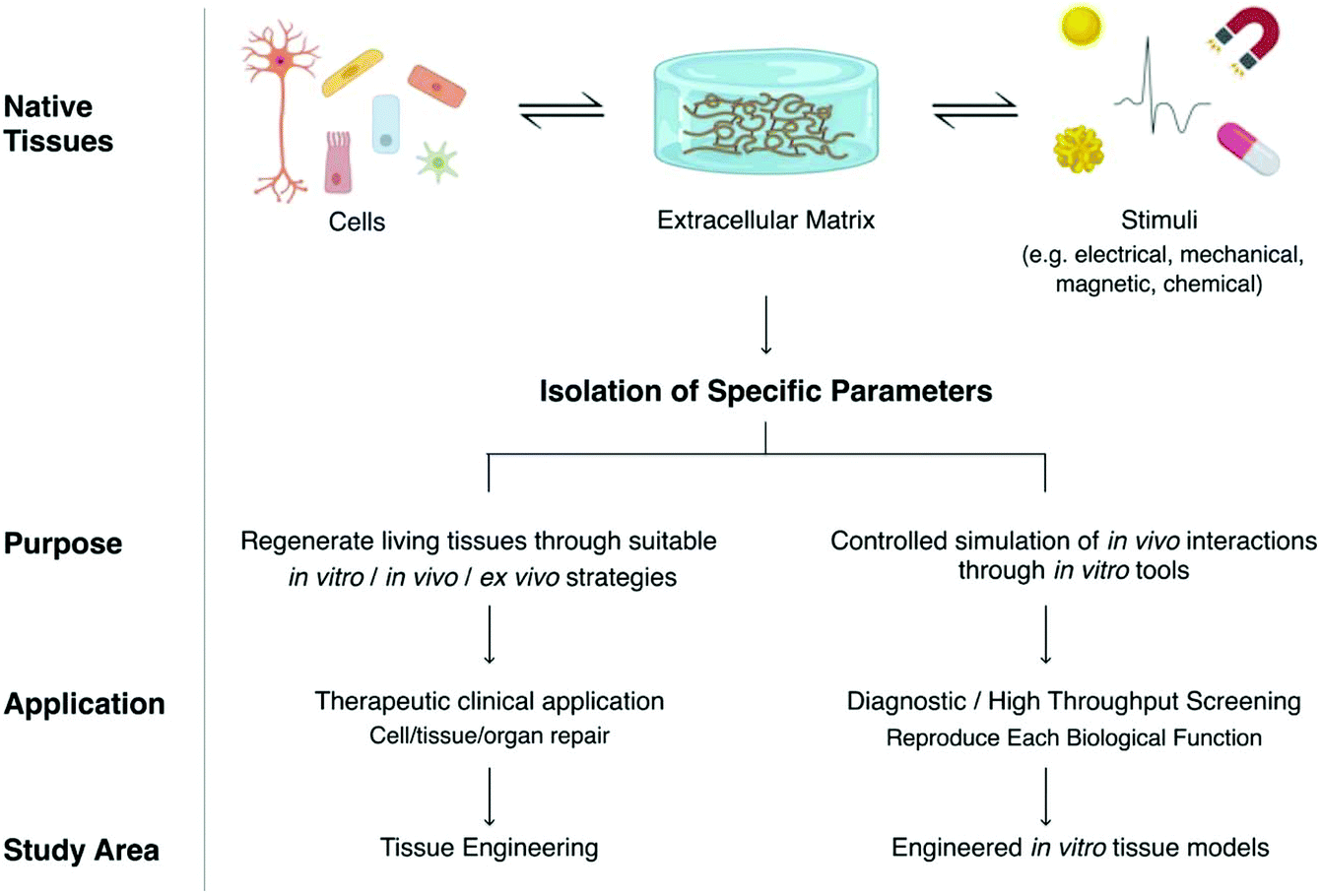 | ||
| Fig. 1 Differences between engineering tissue constructs and engineered tissue models according to TE terminology.78 | ||
Although tissue engineering and engineered tissue models share similar foundations, their development follows different strategies. Tissue-engineered constructs aim at providing proper physiological cues for cells to create an artificial tissue, which, when implanted, will integrate with the surrounding tissues. Engineered models draw inspiration from human tissues to build something that may be different in shape, materials, and stimuli, but, within their limitations, provide a similar response to that of human tissues when interacting with drugs and molecules. For this reason, tissue-engineered constructs cannot be simply upcycled as engineered models as their purpose and engineering strategies are different.
2. The power of a model within the biomedical field
Intrinsic to the concept of a model lays the fact that there is no point in using a sophisticated, complex prototype to test a single factor affecting a phenomenon. Instead, each factor should be better studied independently at earlier stages using simpler models and then projected into the full prototype. For both drug discovery and medical device development, the use of relevant models is fundamental. These models comprise in silico, in chemico, in vitro material- and cell-based, as well as in vivo, and are employed at different stages of drug discovery and device development bearing different degrees of complexity. Each of these models is a filter that selects the most suitable and promising drug candidates and device characteristics, as well as give cues over efficacy and safety prior to clinical trials. Both the selection of the most relevant models according to the initial hypothesis and data interpretation are crucial at a pre-clinical stage for the translation and application of new therapies.2Dimensional analysis theories in fluid mechanics define a model as a scaled design that accurately represents a prototype. “We do not build a million-dollar airplane and see whether it has enough lift force. We measure the lift on a small model and use a scaling law to predict the lift on the full-scale prototype airplane”.3 Under the biomedical context, a relevant model has the ability to generate predictive data of certain aspects of the human response. Under the same nature of stimuli or external factors, the model responds in an analogous fashion to the original phenomenon. In dimensional analysis, this condition is known as similarity.3 Similarity is evaluated through dimensionless coefficients that group the physical parameters that influence the phenomenon. Dimensionless coefficients serve as indicators of the ability of the model to simulate the studied phenomenon. If they are equal to the original prototype, the model accurately represents the condition. Since all parameters do not affect the phenomenon in the same manner, this means that a model is not necessarily a reduced size of the prototype and both the innate characteristics of the model or the intensity of the stimuli need to be adjusted to achieve similarity.
A fundamental point is that the design of a model greatly depends on the study that one wants to perform. It is important to define which parameters need to be studied so that only the ones affecting the response of the system are included with proper indexes (i.e. values that enable to validate the similarity of the model to the studied phenomenon) of evaluation. A paradigmatic example is the vascular model designed to study the effectiveness of intravenous permeability of a therapy to the surrounding cells.4 Taking into account, the vascular permeability during nanoparticles (NPs) extravasation, the essential parameters that were considered to be similar to the native tissue were those affecting mass transport from the intravascular compartment towards the external environment, such as the permeability of the vascular tissue.4 A polydimethylsiloxane (PDMS) microfluidic device cultured with human umbilical vein endothelial cells served to test the effectiveness of extravasation of NPs as vehicles for drug delivery, obtaining comparable permeability behaviour as that observed in in vivo studies. For permeability assays within this model, a number of separated rectangular surfaces of endothelial tissue under dynamic flow were used, leaving aside the native geometry, continuity of the vascular tissue, and viscoelastic properties of the conducting fluid, as well as hydrostatic and interstitial fluid pressures, as these were not essential to the permeability assay. This allowed to control the surface area of diffusion and to standardise flows. By submitting the cells to different treatments, it was possible to independently model the effect of pharmacological therapies under health or disease states. This model is produced with totally different design requirements that would have been needed for the development of a tissue-engineered vascular graft.
Trying to mimic all the components exhibited by a multifactorial phenomenon in one single model, especially within the biomedical context, is not only far to be achieved but also counteracts the efficiency of separating the parameters. Different parameters may compete against each other and create a dependency between the indexes that would make the phenomenon difficult to study. Sometimes, complex models, like modified animal models, end up giving results that cannot be related to a specific parameter, owing to the complexity of interactions intrinsic to the experiment. To test the effectiveness of bone marrow stem cells (BMSCs) on restoring thyroidal activity after radiation treatment with 131Iodine, an experiment over four groups of irradiated mice was conducted, two of which were treated with BMSCs, while the others were used as positive and negative controls.5 At the end of the experiment, it was not possible to determine if the BMSCs had any positive action. Despite exhibiting a regulatory effect at early stages (two days), at long term, both treated and untreated groups restored TSH and T4 levels, questioning the actual level of effectiveness of the BMSCs in the recovery process. The fact that it was not possible to isolate the effect of the BMSCs from the surrounding regulatory processes is inherent to the complexity of the animal model, since it gave the experiment too many parameters acting at the same time, and therefore the specific effect of the one out of the many to be studied could not be observed.
Orthogonal experimental design has risen as an answer to the urge to control the number of variables affecting an experiment. It aims to reduce the number of variables in every experiment by giving each variable a level of importance. By only testing the combinations with higher influence, the number of experiments can be reduced. This method implies the knowledge of the significance of the different parameters in the studied phenomenon.6 As a consequence, when designing a model, careful attention should be given to the choice of appropriate parameters that have enough meaning to provide reliable results without clouding each other. In this sense, a model comprises: a scaled setup with defined parameters; a set of indexes that evaluate similarity (the capacity of the model to represent the phenomenon); a set of defined variables to be set as the input for the experiments; and their relation to the native phenomenon (Fig. 2). Before being accepted and continuously applied, any model needs to be validated by comparing the results of the model to the original/native phenomenon. On the example of Fig. 2, the similarity index is related to NPs permeability regardless of shape and length of the veins4 (Table 1). The depicted vascular model exhibited size-selectivity towards both dextrans with different molecular weights (10 and 70 kDa) and polystyrene NPs (20, 40, 100 and 200 nm) as observed in physiologic vasculature in vivo. The diffusion permeability coefficients (Pd) obtained while using this model slightly differ from those reported in in vivo experiments, yet Pd decreased in a comparable manner with the increase of molecular weight. Indeed, the authors suggested that these differences could be corrected by inserting a suitable constant correction (Table 1). A similar approach was adopted to develop an in vitro model of osteoarthritis.7 Both physiological (10%) and hyperphysiological compression (30%) were selected as input parameters, to mechanically simulate articular chondrocytes embedded within poly(ethylene glycol) hydrogels contained in a PDMS-based device. The degree of similarity between the osteoarthritis cartilage-on-a-chip model and human osteoarthritic states was evaluated through immunofluorescent and gene expression analyses. The simple application of hyperphysiological compression induced the production of pro-inflammatory cytokines and hypertrophic differentiation by/of articular chondrocytes characteristic of osteoarthritis.
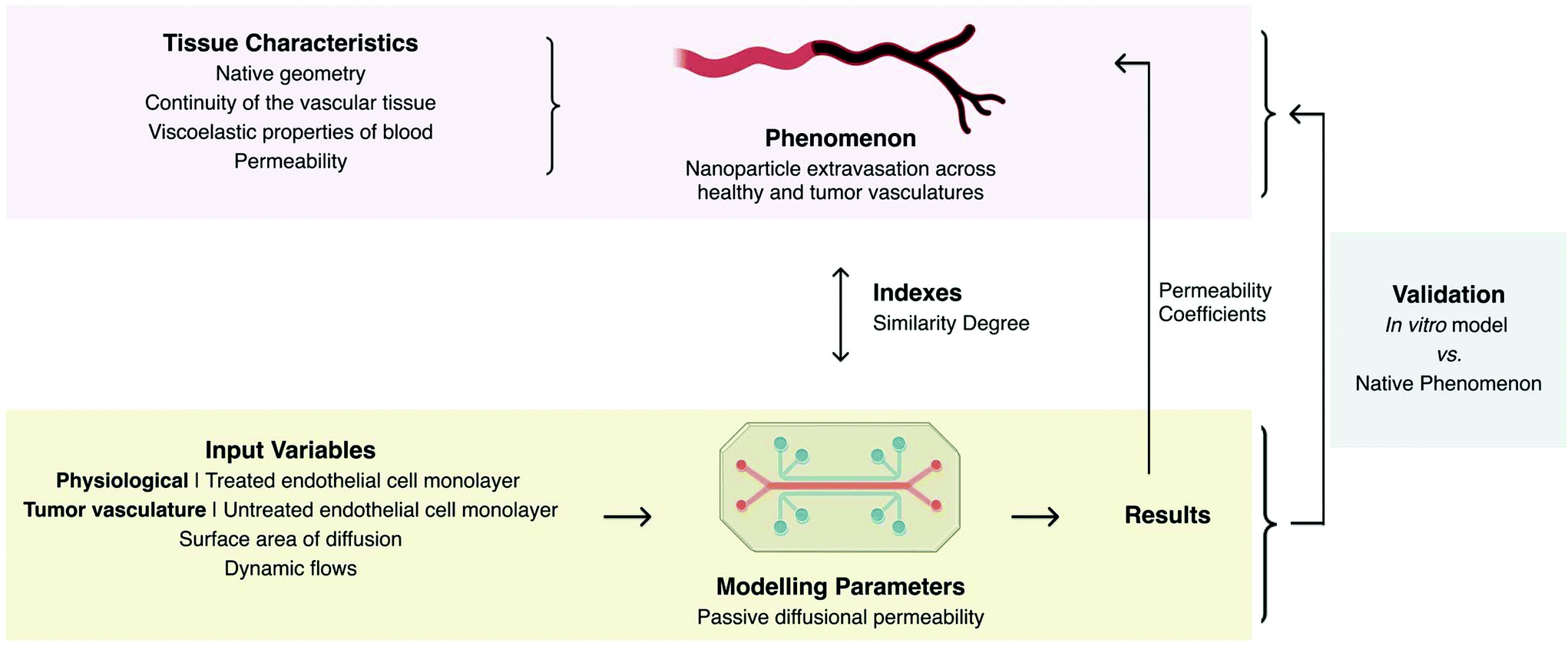 | ||
| Fig. 2 Designing of an engineered in vitro model depicting parameters, input variables, and indexes taking as example the model of the permeability of the vasculature engineered by Ho et al.4 | ||
| Type of model | Model description | Objective | Application | Parameters | Indexes | Ref. |
|---|---|---|---|---|---|---|
| Fluid model | Simulated body fluid | Study the deposition of calcium phosphate on biomaterial surfaces (bone bioactivity). | Evaluation of orthopaedic devices performance | Composition of plasma – ionic concentrations, pH | Validation through apatite deposition on surface | 18 |
| FeSSID/FaSSID (fed and fasted state intestinal fluid) | Model gastrointestinal fluids | Evaluate drug solubility through gastrointestinal fluids | pH, buffer capacity and osmolarity | Salt concentration and surface tension | 23 | |
| Simulated gastric fluid | Study nanoparticle delivery for antimicrobial activity | Evaluation of the effect of physiologic conditions on AgNPs growth | pH | pH | 27 | |
| Tissue model | Rectangular surfaces of endothelial tissue in microfluidic devices | Model de vascular wall permeability to nanoparticles under different pathological states of vascular wall cells | Drug delivery | Those affecting mass transport: permeability | Permeability coefficients Pd – particle size sensitivity | 4 |
| Material-based models of phospholipidic cell membrane | Study permeability of physiological barriers | Evaluation of gastrointestinal absorption, blood brain barrier permeability, skin permeability, among others | Affinity of the membrane and porosity | Passive permeability coefficients | 31–33 | |
| Dual-channel microfluidic intestine-on-a-chip | Study the effect of microbiota to the integrity of intestinal barrier | Disease modelling | Oxygen availability/type of bacteria | Bacteria culture sustainability | 44 | |
| Tissue model | Lung-on-a-chip | Study mass transfer through epithelial membrane from alveoli to vasculature | Study of lung physiology | Cell types in each side of the membrane and stress strain conditions of the epithelia | Cell alignment and shape distortion | 45 |
| Cell monolayer over protein matrix | Model epidermal layer | Perform irritation studies of cosmetic products | Cell type and/or stress | Morphology, lipid composition, and biochemical markers, as well as photo-toxicity, irritancy, transport, and corrosion | 47 | |
| Full thickness skin model containing keratinocytes in collagen coated transwells | Model skin physiology | Test in vitro skin irritation | Mass transport characteristics and cell physiology | Cell morphology and tissue stratification, barrier action of the tissue, stability in phosphate buffered saline solution and tissue integrity under controlled exposition to cytotoxic elements | 48 | |
| Dermo-epidermal human skin equivalent | Model epidermal layer | Test in vitro skin irritation | Cell type and extracellular matrix composition | Permeability and sensitivity to irritants | 49 | |
| Pathophysiological model | Mechanically stimulated hydrogel seeded with chondrocytes | Study the effect of mechanical stress on chondrocyte behaviour in osteoarthritis | Disease modelling | Mechanical properties and compression levels | Immunofluorescence and gene expression of chondrocytes | 7 |
| Lipid monolayers | Determine the effect of acyl chain length in skin permeability | Disease modelling of atopic dermatitis | Acyl chain length | Permeability coefficients | 46 | |
| Multicellular tissue spheroids composed of hepatic stellate cells | Study cancer cell response to anti-tumour drugs | Test anti-cancer therapies | Cluster size – cell distribution within the spheroid | Spheroid size | 52 |
The key within designing an engineered in vitro model is to include in the model only the desired interactions between the principal parameters. These parameters can be inputted alone or one-by-one, so that it is possible to evaluate the single effect of each parameter in the model. But also, the model should allow the combination of the input parameters to evaluate their combined effect over a phenomenon, as well as their effect over each other.
3. Biomimicking and bioinspiring from natural tissues
For bioengineering, natural tissues are the source of inspiration to engineer tissue-constructs and in vitro models, but also represent the objective that needs to be mimicked when designing an engineered tissue or an engineered model (Fig. 3). When bioinspiring, the characteristics of natural tissues should be extracted, evaluated and parameterised into engineering components that could be designed, manufactured and controlled. When biomimicking, combinations of non-native components should be tailored so that under different circumstances, they produce situations that resemble those of natural tissues. The balance between bioinspiring and biomimicking allows both engineered tissue-constructs and in vitro models to be complete enough to model pathophysiological scenarios under the limits of available engineering technologies (Fig. 3).8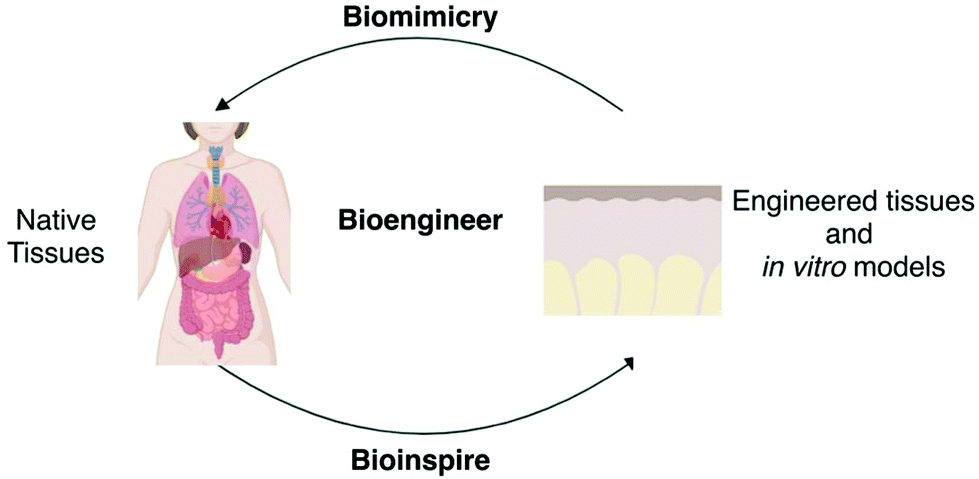 | ||
| Fig. 3 Biomimicking and bioinspiring from natural tissues towards the development of engineered tissues and in vitro models. | ||
To recreate tissues in vitro, the native components that need to be added are known as the “tissue engineering triad” and include cells and scaffolds that act as extracellular matrix (ECM) with or without signalling cues.9 Cells are the main active parameter and both scaffolds and signals provide the physical and chemical stimuli required to maintain cell viability and functionality to perform specific physiological tasks. ECM comprehends the structural support for cells resultant of their genotypic expression and plays a key role in initiating biochemical and biomechanical cues that induce specific cell behaviour.10 Signals are the communication channels between cells, and between cells and the surrounding microenvironment. Signals can be either chemical or physical cues that give cells the correct programming and ionic balances to control cellular processes. Chemical signals such as nutrients, growth factors or oxygen (O2) tension can be provided in the culture medium and atmosphere or by engineering structures bearing precise control over the spatial location of these factors. Internal and external stimuli impact cellular behaviour and morphology, and therefore differentiation patterns and physiological activity. Bioreactors are efficient means in recreating the dynamic conditions of cell microenvironments. These may provide culture media under dynamic conditions with the necessary biochemical regulatory signals while creating proper stress-states by applying external forces that simulate the physiological activity of tissues. Bioreactors are envisioned to be the key to convert cell constructs into actual large-scale medical devices that can be applied clinically.11 Bioreactors can also be exploited when engineering models, as these could recreate microenvironmental conditions that need to be included in the model, especially if the designed model includes cellular components or particular physiological or pathologic distinctive conditioning.12,13 None of these parameters can be considered independent or separated in their activity from its “partners”, as complex communication channels and interactions affect each of their physiology and make their specific characteristics depending on the others. For example, the majority of cells are anchorage dependent, and their morphology is strictly related to the rigidity of the ECM where they reside, while in pathophysiology or ageing, cells can induce ECM stiffening and/or weakening.14 Thus, engineered models should be designed for the modular building of interactive components to face the complexity of physiological phenomena.
3.1. Learning from tissue engineering
TE aims to support and stimulate regeneration by creating tissue-like substitutes (tissue-engineered constructs). This is only possible if cells are allowed to live and proliferate in a biomimetic extracellular environment.10 For this purpose, cells are cultured in the presence of an engineered scaffold with designed physical and chemical stimuli and, if needed, under dynamic perfusion with the aid of bioreactors.A common limitation of cell-derived products is the amount and type of cells required. Unfortunately, the proliferation of mature cells in in vitro culture may be limited and excessive manipulation could lead to a considerable loss in phenotypic characteristics. Additionally, poor inter- and intra-laboratory reproducibility is commonly associated with the use of cells as a model. Specific conditions such as subject health or age also affect the capacity of his own cells to proliferate, which makes it difficult to use isolated cells. With this limit, stem cells rise as the most viable options. Adult stem cells (ASCs) that natively reside in every self-renewing tissue have tremendous regenerative capacity. In the past decade, the increased understanding of the niche factors that favour stem cell maintenance, such as niche architecture and stress-states, has permitted to grow ASCs in vitro into organoids.15 Current strategies rely on standardized cell lines, which have been amply manipulated and therefore exhibit altered phenotypes that allow easy culture and proliferation, but do not necessarily behave in a physiological manner and therefore are not suitable for implantation.
In TE, scaffolds serve as ECM that provide cells with structural support to attach, grow, proliferate and migrate ex vivo and stability to the defect intended to be regenerated. These should offer proper mechanical properties to the engineered tissue, favour cell proliferation and differentiation, serve as reservoir and delivery vehicle for chemical cues and therapeutic molecules when needed, and the volume of the tissue for new vascularisation and regeneration.16 Different materials and three-dimensional (3D) structures have been proposed for TE. Hydrogels have also given a great push to the development of biomimetic/bioinspired structures because they can be produced from natural biocompatible sources and can be designed to exhibit chemical moieties that resemble the characteristics of ECM. In some cases, this strategy allows interaction in semi-physiological ways between the cells and the artificial support.17
The development of engineered models can take advantage of the mature knowledge developed for tissue regeneration behind the choice of materials, their modification and processing, and the reproduction of proper microenvironmental conditions. Engineered models take many forms from model fluids to tissue models of pathophysiological phenomena, which can be either under static or dynamic conditions (Fig. 4). Model fluids may provide insightful information over possible physicochemical interactions of drugs, particles and medical devices with human body fluids. Engineered tissue models, in their turn, can recreate 3D chemical, structural, biomechanics and/or biological features of human tissues that are relevant to determine diffusional permeability coefficients, toxicokinetics, to study the efficacy of anti-tumour, antiviral and antimicrobial agents, induce pathological events, among others. These can be further complicated to reproduce tissue dynamics by the aid of bioreactors and simple or integrated microfluidics to engineer significant tissue pathophysiological aspects, or more complex organs-on-a-chip by including fluid dynamics, hydrostatic and interstitial fluid pressures and consequent shear stresses, and so forth.
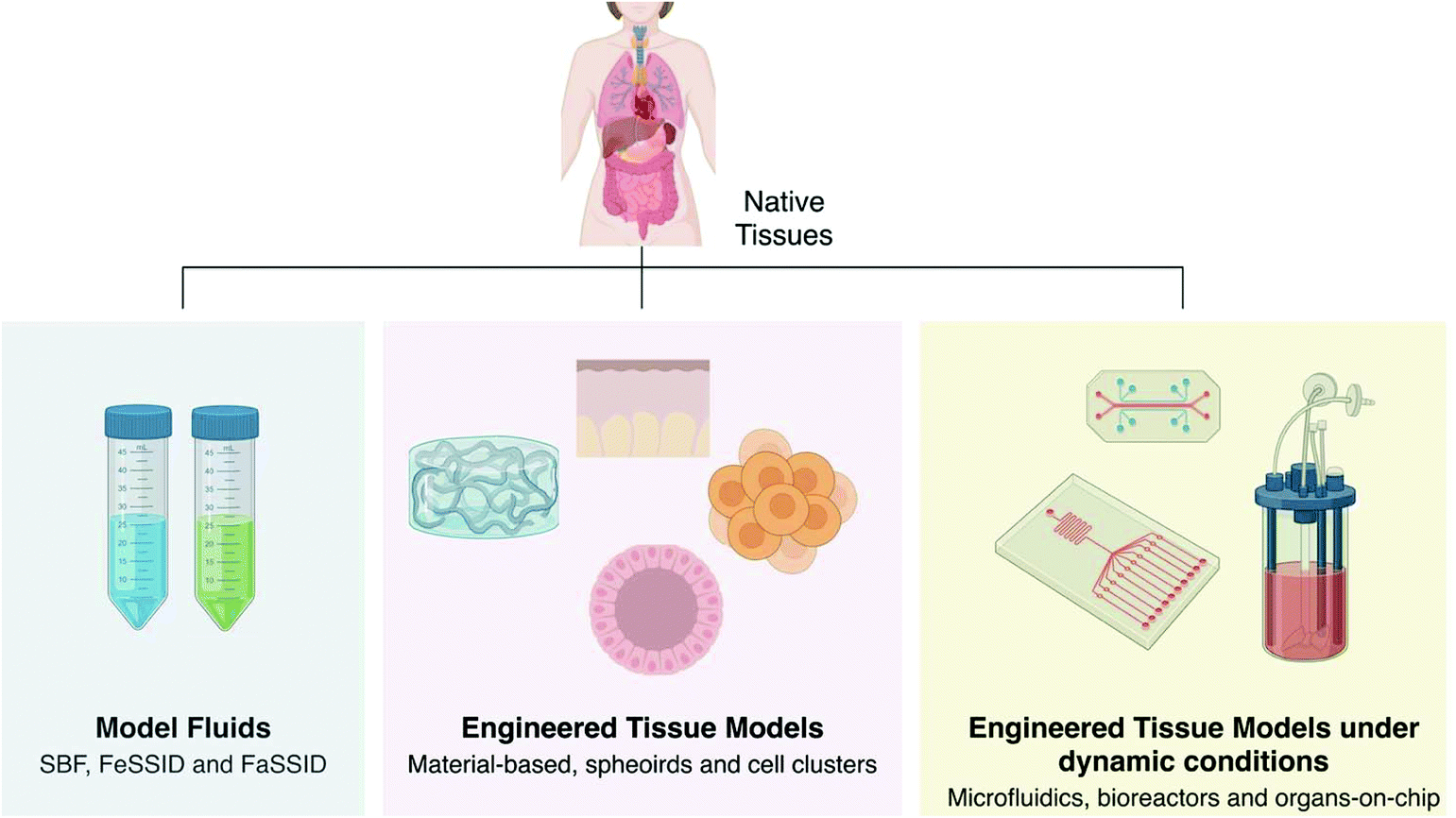 | ||
| Fig. 4 Current engineered models of physiological fluids and tissues in both healthy and disease states. | ||
3.2. Model fluids: empowering simplicity or oversimplification?
A widely established approach to gain a preliminary overview of the interaction of new technologies or materials with the organism is the use of fluids, which simulate the effect of body fluids on materials (Fig. 4). This represents an enormous simplification of the natural environment, yet they demonstrated to function as useful tools to mimic complex phenomena (Table 1). An example is the apatite deposition on biomaterial surfaces by incubation in Simulated Body Fluid (SBF). The first formulation of SBF was used to test the deposition of calcium phosphate onto Bioglass after implantation.18 From that moment on, it has been reformulated and corrected to model the composition of the plasma in terms of ionic concentrations and physiological pH. Kokubo et al. reviewed the viability to extend its use to test the formation of apatite on the surface of biomaterials applied in bone regeneration. Materials that supported apatite growth in SBF showed similar capacity when implanted, and therefore SBF is currently used as a model fluid to screen bone bioactivity.19–21 Although different compositions of SBF have been used as equivalents for bioactivity tests, they do not necessarily comply with the physiological thermodynamic status validated by Kokubo. This increases the probability to produce false positive or false negative results in terms of bioactivity.22 Also, the validated SBF only considers apatite formation as the sole possibility of bone binding, discarding its relevance for testing materials that are intended to be applied outside the field of orthopaedics.Other bio-relevant media have been designed to model the physiological solubility and dissolution of newly developed drugs, including simulated gastric, intestinal and colonic fluids (Table 1). The intestinal fluid is simulated under fed (FeSSID) and fasted states (FaSSID). The first FeSSID and FaSSID aimed to reflect the pH, buffer capacity and osmolality of human intestinal fluid. Improved solutions allowed the development of media that better mimic the bile salts concentration and surface tension. These fluids are now at use for drug development as it has been proved that blank buffers underestimate solubility, while previously used sodium dodecyl sulphate solution overestimates it.23 Bio-relevant FeSSIF and FasSSIF were used to test bioequivalence between a fixed dose combination of atorvastatin and amlodipine of an oral disintegration tablet and their independent delivery. Atorvastatin and amlodipine are a common therapy for cardiovascular disease.24 Despite obtaining no difference between the fixed dose composition tablet and the separated molecules within one state, the dissolution profile of single amlodipine was different when tested in FeSSIF and FaSSIF. This means that the modelled microenvironment (intestinal fluid) displayed different effects when state parameters were changed (fed or fasted), reflecting the need to carefully address similar conditions when designing a model.
The development of new technologies and techniques is always accompanied by a model that follows their developmental stages to evaluate if the new drugs properly fulfil the requirements. This aspect could be important not only for molecules, but also for NPs. Model fluids can play a crucial role in the activity of drugs or drug delivery systems. For example, silver nanoparticles (AgNPs) have been exploited as antimicrobial materials25 or delivery systems and most recently have been studied for many biomedical applications26 (Table 1). Assuming oral delivery, the change in diameter of the AgNPs was followed using simulated gastric fluid at different pH, covering a range of 2 to 5.27 It was observed that AgNPs growth was, in fact, dependent on its initial size and the pH to which these are submitted, with a higher and faster NPs growth for more acidic pHs that might be associated to the formation of Ag+ within acidic environments. In a follow up study, AgNPs of the same size did not show any toxic effect or tissue accumulation in mice with 70.5 to 98.6% Ag+ retrieved from the animal faeces and less than 0.5% detected in liver, spleen, intestines, as well as urine after 48 h, which was independent of particle size.28
Fluid models combine simplicity with usefulness. Fluid models are active tools to predict which chemical reactions are triggered when medical devices and microparticles are implanted/administered. They also support drug dissolution studies to enlighten the path of a drug in different body parts. Detailed description of the different available fluid models, their formulation and applications can be found in Marques et al. (2011).29 In spite of their relevance, fluid models present some limitations. Most of the administered drugs have to overcome the steric and interactive barriers. Currently, most studies conducted to predict drug permeability through mucus rely on mucin-based solutions. These solutions can disclose if a drug interacts with mucin or not, but they do not ensure that it will cross the mucus. Given the importance of this barrier to drug diffusion, our research group has recently developed mucus models that reproduce the chemical composition, viscoelastic properties and both steric and interactive barriers of mucus. After the preparation of the mucus, mucin was able to interact with drugs that had documented affinity in solution (Table 1).30 Other examples are parallel artificial membrane permeability assay (PAMPA) systems, which are material-based models of the phospholipidic cell membrane. PAMPA systems are commercially available for a wide number of tests, including gastrointestinal absorption31,32 and blood brain barrier permeability,33 Skin-PAMPA (PAMPA membranes including certramide, free fatty acid and cholesterol) to assess permeability through the stratum corneum,34 and Corneal-PAMPA (PAMPA incorporating different ratios of phospholipids) to measure corneal permeability.35 These material-based models exhibit high correlation to passive permeability across the phospholipidic cell membrane, in some cases higher than 90%, without including cells.36
3.3. Engineered tissue models
The development of in vitro tissue models is highly encouraged by both pharmacological research and regulatory fields, following the reduction of animal testing in many applicative fields. Tissue models of human tissues commonly exhibit higher fidelity than two-dimensional (2D) models and therefore can be used as novel tools in drug delivery to assess/screen toxicity and effectiveness in vitro. Some attempts of 3D models have been designed to test specific treatments, such as cancer drug testing (tumour models), anti-arthritic inflammation drug delivery, efficacy testing (cartilaginous tissue), drug toxicity (liver model) and permeability (skin model).37Tissue models are usually developed applying the same components as those defined by TE, which work as the foundation knowledge of the field. Yet, considering that engineered models are intended as in vitro platforms, special attention needs to be given to signals and cell microenvironment to compensate the interactions at physiological or pathological conditions. However, since a model should be engineered to enable the study of a specific process it must possess clear indexes and parameters for validation, and it does not require all the components and the same type of interactions that tissue-engineered constructs entail. In this sub-chapter, in vitro models proposed in the literature are taken as example to highlight how simplicity may closer recapitulate a biological phenomenon or how complexity can cloud the individual effect of a drug or compound on the overall phenomenon.
3.4. Cell-based engineered models
In the case of engineered in vitro models, the risk of tumour formation upon implantation of undifferentiated PSCs in TE strategies is absent, because models are not intended to be implanted. In this way, PSCs are a suitable source of cells that could provide accurate models and even mimic the native proliferation potential of different cell types in the same tissue. Further details on the relevance, affecting parameters, and applications of PSCs in in vitro modelling can be found in Ebert et al. (2012),38 Cochrane et al. (2019)39 and Srivastava et al. (2019).40 Altered cell lines may lack the production of specific factors that have important interaction on the tested mechanisms, and therefore result in non-trustworthy outcomes in early stage experiments. For modelling purposes, it is important to validate if any important interacting factor is missing in the selected cell line. There is a wide library of materials that can be employed in the development of in vitro models since these do not require approval by any health regulatory agency and unless the model is intended to study specific interactions with cells, considerations of biocompatibility are lighter.Several organoid-based studies have also been conducted for different tissue types, while addressing a variety of parameters that play a specific role in the corresponding human tissue. In fact, different organoid-based structures have already been translated to the market by MIMETAS, 3D Biomatrix, Inc., Microtissues, Inc., InSphero AG, and Cyfuse Biomedical, among others, that simulate liver, gut, osteochondral and cardiac tissues. Other than eukaryotic cells, new in vitro models that include other cell types, like bacteria, fungi and yeasts should be developed, as there is a higher awareness over microorganisms dysbiosis-associated diseases, like Parkinson's, Alzheimer's, depression, among others.41 In fact, the effect of the resident microbiota in the gastrointestinal tract has proven to have a higher impact over organ physiology than the cellular components and specific ECM interactions. Indeed, perturbations on host-microbe interactions may lead to severe pathologies. Therefore, models of the gastrointestinal tract should include the microorganisms present in this system42,43 (Table 1). With this in mind, a dual-channel microfluidic intestine-on-a-chip device was engineered to sustain the growth of Caco2 and endothelial cells, as well as complex human intestinal microbiota under anaerobic conditions.44 During culture, the cells were submitted to a hypoxic gradient that did not affect cell viability up to 7 days and that maintained the ability of Caco2 cells to secrete mucus and endothelial cells to form a hollow vascular lumen. After culturing the human microbiota within the microfluidic intestine-on-a-chip device, it was observed that bacteria cultured under anaerobic conditions did not affect the integrity of the intestinal barrier, contrary to those cultured under aerobic conditions. Additionally, anaerobic conditions sustained the growth of different bacteria, which variability and abundance were similar to those reported on the Human Microbiome Project.
3.5. Simplifying the complexity
The biomimetic microsystem that models alveolar capillary interface is one clear example of modelling indispensable parameters without having to recreate the precise native environment45 (Table 1). The microsystem provides pulsatile strain to epithelial cells, typical of alveolar physiology during ventilation, not by providing oscillating air pressure in the air interface, but by externally stretching the monolayer membrane where epithelial cells reside. The validation of the model showed that the vacuum applied externally to the membrane effectively deformed the cell layer causing cell shape distortion and cell alignment. The model was posteriorly used to study the passage of NPs from alveoli to lung vasculature.The stratum corneum is the outermost layer of the skin, and it is composed of cornified cells and a very organised lipidic ECM. Several chronic skin diseases show alterations in the lipidic components, specifically the shortening of ceramide acyl chains. This has led to the hypothesis that the very large ceramide acyl chains are involved in the skin barrier development, affecting the permeability of the stratum corneum. However, owing to the numerous factors affecting atopic dermatitis, this hypothesis has been difficult to confirm46 (Table 1). This complexity inspired the development of a model that directly compared the effect of lipidic layers with very long ceramide acyl chains (CerNS24) on the permeability of the stratum corneum in comparison with long ceramide acyl chains (CerNS16). The model consisted of two lipid monolayers formed by the ceramide chain cholesterol, cholesterol sulphates and either one single type of free fatty acids or a heterogeneous mixture of free fatty acids. This model showed higher permeability for both CerNS16 lipid monolayers in one single type of fatty acid and in heterogeneous mixtures proving that very long CerNS24 contributes to the barrier development independently of the fatty acid composition.
3.6. Approaching physiological complexity
Different 3D skin constructs have been developed to model disease conditions or as testing platforms for the cosmetic industry to carry on irritation studies. These models implement increasing levels of complexity, starting from the early models, now commercially available, to the more recent ones (Table 1). For tests over the epidermal layer, keratinocytes sheets (differentiated cells seeded on a protein surface) are commercially available and widely accepted.47 Netzlaff et al. tested some of these commercially available products in terms of morphology, lipid composition, and biochemical markers, as well as photo-toxicity, irritancy, transport, and corrosion. Although these represent more complex/complete models when compared with standard (2D) cultures with similar toxicological behaviour in respect to human skin, the 3D skin constructs failed to model permeability, probably associated to the lack of a dermis structure (Table 1). Aiming at establishing a reliable protocol for the development of an in-house reconstructed human epidermis as models to test in vitro skin irritation, normal human epidermal keratinocytes were seeded on top of collagen type IV-coated transwells.48 The model was then validated by testing its ability to discriminate irritants from non-irritants finding 100% sensitivity for irritant substances with 87.5% specificity, showing false positive for one of the non-irritant substances, giving a net accuracy of 92.3%. The model complied with all the quality parameters from the Organization for Economic Co-operation and Development to perform in vitro skin irritation testing, among which are cell morphology and tissue stratification, barrier action of the tissue, stability in phosphate-buffer saline solution and tissue integrity under controlled exposition to certain cytotoxic elements. Full-thickness models are expected to mimic mass transport and “artificial” sensibility that enable the sensitisation and permeability testing of new biomedical devices. With this in mind, a cell coating technology was developed by Akagi et al. aiming to produce Dermo-Epidermal Human Skin Equivalent (DEHSEs)49 (Table 1). In this way, dermal fibroblasts were incubated with either fibronectin or gelatin, and thin films were further evolved by interchangeable depositing the cell solutions layer-by-layer. Upon coating the previously established thin films with collagen type IV, keratinocytes were deposited on top intending to reproduce the epidermis. The permeability of the DEHSEs was assessed in a diffusion chamber using molecules of different sizes, while its sensibility was determined by testing known, irritable and non-irritable molecules, and further compared to excised human skin. Finally, the DEHSEs showed its ability to model both permeability and irritation, according to the European Centre for Validation of Alternative Testing.493.7. Engineered disease models
The available time to provide a regenerative solution for a patient with a critic pathology is limited. In contradiction, the time scale for the development and approval of a new therapy is up to 10–15 years, and it is longer and more expensive when dealing with innovative therapies and new drugs.50 Despite the long process, the effectiveness of reaching a solution is limited by very low efficiency during the testing phase, which is translated in a low percentage of approved drugs for clinical (11.8%).51For new pharmaceutical therapies, the low efficiency evidences a very small number of successful therapies in contrast to the initial number of options presented. This is a consequence of the discontinuity in the study conditions through the process, which makes it difficult to predict possible outcomes of more complex stages from the initial screening results. Thus, the process generates unexpected results that discard most possibilities, which may also occur at later stages of testing. An optimal screening for a new therapy should put in evidence the majority of problems at early stages, filtering the most promising outcomes to be studied with higher complexity.52
There are two dramatic jumps along the approval process for a new therapy from in vitro to animal models and real interaction with patients during clinical trials. Firstly, 2D cultures provide a cellular response that may be far from real 3D tissue interactions. Bioreactors have given significant improvement in bridging the gap between in vitro culture and native cell microenvironment by adding physical stimulation and providing feeding and chemical signals in a modulatory dynamic manner. To increase the efficiency of pre-clinical studies, culture systems that better recapitulate physiological microenvironments must be implemented. Culture systems should mimic the architecture of cellular actors, their physical microenvironment and provide proper chemical stimulation to the districts that the pharmaceutical therapy intends to address52 (Table 1). Multicellular tumour spheroids (MTS) have risen as superior tools to extend the possibilities of in vitro research for cancer. MTS are spherical structures of cell clusters that in correspondence to their size display different behaviours. In particular, MTS with diameters higher than 500 μm are able to resemble tumours because they have a proliferative surface, a quiescent interior, and a necrotic core. This cell distribution, caused by the O2 and nutrient exchange gradients, mimics the cell microenvironment of tumours.53 For this reason, the power of the model relies on the capacity to produce homogeneous large spheroids, as different sizes or shapes can induce different responses to tested pharmaceuticals. Spheroids formed by hepatocytes and hepatic stellate cells (HSCs) are an example of a disease model that serves as a test bench for pharmaceutical development. HCSs are responsible for the production of fibrotic tissue in the liver, which becomes active due to an injury or as a response to toxic molecules. Leite et al. used HSCs in a quiescent state to perform simple hepatotoxicity tests and activated HSCs for pharmacological tests in fibrotic conditions.54 HSCs inducers were used to modulate the activation of HSCs within the spheroids in two independent settings, which allowed the same drug to be tested in two different conditions. The spheroid nature of this model made this modulation possible because when co-culturing hepatocytes and HSCs in monolayers, HSCs immediately activate and exhibit fibrotic features. In this way, spheroid-based tissue models can model certain parameters that are key players on a defined pathology, and therefore offer superior therapies accordingly to the application. Deeper information on methods of development, composition, applications and limitations of spheroid-based models can be found in Chua et al. (2019),55 Nunes et al. (2019),56 Zanoni et al. (2019)57 and Zanoni et al. (2020).58
The second jump occurs when moving into pre-clinical trials. Only one-third of the animal experiments enter clinical trials and as little as 8% of the tested drugs finish Phase I of trials with success.59 Even if animal models enable a closer understanding of the interactions of the therapy with living tissues, it has been proven that the immune responses of different species are significantly different from humans.60 For example, the TGN1412, a CD28 superagonist antibody that activates and promotes the proliferation of regulatory T cells, was designed as immunomodulatory treatment of diseases like rheumatoid arthritis, multiple sclerosis and some types of cancer. Upon rigorous testing, TGN1412 demonstrated safety in mice in terms of toxicity and effectiveness using doses hundreds of times higher than those administered to humans during clinical trials. Nevertheless, in clinical trials, a systematic organ failure was observed in patients submitted to this therapy.61
Animal models fail to mimic the extremely complex process of human carcinogenesis, physiology and disease progression, as well as infections. Despite being able to replicate specific processes within the disease, these models lack the whole spectrum of pathological changes that occur in humans. As a consequence, drugs that have proven to be effective and safe in animal models may either induce dangerous responses when administered to patients or have no positive effect on treating the pathology. One evident example concerns the development of drugs to tackle Alzheimer's disease. In the last two years, many were the clinical trials being halted with otherwise molecules that have shown promising results in animal models, some examples include the Aducanumab proposed by the partnership Biogen – Eisai, Crenezumab by Roche, and Atabecestat by Jansen.62,63 Many reasons have been suggested either the underlying mechanisms of Alzheimer's disease are still to uncover, or mice are not a suitable filter during pre-clinical studies.64,65 Cancer research has also witnessed cases in which animal models do not accurately represent the processes that take place in human pathology. The most commonly used models in cancer research are experimental tumours grown in rodents (human tumour xenografts). For example, IPI-926, an antagonist to the pathway that provides key growth and survival signals for some tumours, displayed increasing survival rates in mice with advanced brain tumours, but showed no difference with respect to placebo in humans during clinical trials.66,67 Similar evidence was experienced using matrix metalloproteinases inhibitors (MMPIs). MMPIs were designed to reduce tumour invasion and metastasis in pathologies such as cancer and arthritis. From 16 types of MMPIs that have entered clinical trials, only one has been approved for clinical use, the other 15 showed poor selectivity. These outcomes were attributed to the poorly defined animal models that were not able to predict the effect of such inhibitors over the physiological pathways that include matrix metalloproteinases. As inhibitors to physiologically relevant molecules, MMPIs evidenced toxicity.68 In another example, norovirus, a virus that causes common diarrhoea in humans, could not be analysed in mice, because these were innate immune to the virus. Once it was understood that only human B cells were involved in the infection, a proper modified mice model with cultured B cells was defined for posterior studies, which are nowadays used as models for norovirus infections.69
4. Endorsing the development and translation of in vitro models
A new drug or medical device takes 10–15 years and 3–7 years, respectively, to reach the market, which costs millions of euros for both pharmaceutical and Biotech companies.70,71 The long time and economic burden are in part associated with the strict steps that one has to cross to comply with the requirements imposed by regulatory agencies, including the U.S. Food and Drug Administration and the European Medicines Agency. A successful new drug or medical device has to overcome sequential pre-clinical studies before entering clinical trials. The later stage of pre-clinical studies relies on relevant animal testing. Though, as previously described, the mismatch between animal models and the human body not only has been translated into poor screening at the pre-clinical stage, mainly due to physiological differences, but also into ethical concerns. In fact, only 1 in each 10 new tested drugs that reach clinical trials successfully reaches the market,70 because animal testing is not able to properly filter the effective compounds. This is a huge step back in terms of money and time for both Pharma and Biotech industries, which often discourages investment in new treatments and technologies. Low inter-laboratory reproducibility is another drawback of animal testing. To tackle this, the National Centre for the Replacement, Refinement and Reduction of Animals in Research, a UK government-sponsored organisation, has developed and proposed the ARRIVE (Animal Research: Reporting of in vivo Experiment) guidelines as a way to improve standardisation of animal-testing and higher reproducibility among laboratories.72 These guidelines are expected to maximise the published scientific outcomes, while minimising unnecessary animal studies. With this in mind, governmental organisations have encouraged the replacement, reduction and refinement (3Rs) of animal testing.The 3Rs paradigm is raising awareness of the intrinsic problems associated with animal testing, while opening doors for alternative and disruptive alternative technologies. This paradigm is also extended to studies that include products from animals, as those frequently used in cell culture and histological protocols (e.g. foetal bovine serum, trypsin, antibodies, among others). The Replacement term falls within the last point, its focus is to whenever possible, adopt relevant alternative methods to animal testing methods that bear comparable validity.73 In Europe, the validation and promotion of alternative methods are performed by the EU Reference Laboratory for alternatives to animal testing (EURL ECVAM).74 Different alternative methods are proposed accordingly to the area of application. Recently, EURL ECVAM have created the EURL ECVAM Database Service on Alternative Methods to Animal Experimentation,75 in which current validated alternative methods are recommended to determine toxicity, carcinogenicity and irritation, including acute oral toxicity, aquatic toxicity, phototoxicity, eye irritation/serious eye damage, skin corrosion, irritation and sensitisation, among others.76 As previously stressed, a model aims at recreating a specific phenomenon rather than the whole complexity of any system in nature. This means that one model is not capable of substituting an animal. Instead, many relevant models that address the phenomenon intended to be tackled must be adopted, and the collective information must be gathered to phase animals out of research or at least reduce the number of animals required to reach a conclusion or to answer to the initial hypothesis. In similarity to what happens when choosing an animal model, the fundamental point when choosing or designing an in vitro or in silico model is to define the hypothesis of the study, and which parameters need to be taken into consideration so that only the ones affecting the response of the system are included with proper indexes of evaluation. In other words, which is the hypothesis or objective of the study and which parameters affect or take part in the phenomenon that must be addressed by the alternative methods. The Reduction term, in its turn, concerns the use of the minimum number of animals that does not compromise the quality and the outcomes of the study, including statistical relevance.73 Sharing/transferring the knowledge obtained with a higher number of animals between laboratories may, at long term, reduce the number of animals adopted in experimentation, as this knowledge, if standardised, may be the basis of other studies. Using alternative in vitro and in silico methods is also expected to highly decrease the number of animals required, as a small number of drugs and medical devices would be tested in animals. The last foundation of 3Rs policies is the Refinement. Refinement specifies that if animals cannot be replaced, then the pain/suffering associated with experimentation should be minimised.77In vitro models can provide the means to reduce and ultimately replace animal testing, when these are designed to answer a defined hypothesis bearing in mind their intrinsic limitations. The transitional process from animal testing to in vitro models will depend on many parameters, including the potential of the model to address the initial hypothesis, developmental stage, validation state, type of disease being modelled and scepticism by the end-user. All these factors, contribute to a gradual rather than immediate transition. Some of these cumbersome points may be overcame by providing end-users the regulatory endorsement of in vitro models, and thus accelerate this transition. This is an open challenge and still far to be answered. Yet, several agencies, as EURL ECVAM, support researchers since early stage of development, from concept to validation and translation of in vitro models.
5. The role and application of in vitro models
The development of engineered models provides promising in vitro tools for the development of new drugs and medical devices, but also to unravel unanswered fundamental pathophysiological phenomena. The key components of engineered models are often shared with tissue-engineered constructs, including cells, materials and/or signalling molecules, which can be further integrated within bioreactors to introduce more realistic dynamic conditions. In fact, the gathered knowledge within the tissue engineering field offers important insights on the engineering process of in vitro models.Recalling the theory of dimensional analysis, the process of engineering in vitro models must start with a hypothesis to which defined parameters with integrated proper indexes of evaluation to assess the degree of similarity between the engineered in vitro model and the phenomenon intended to be modeled. A set of defined variables is then built-in to assess their relation in respect to the native phenomenon. Within in vitro models, phenomenon is either related to physiological responses or pathological states in any specific tissue or system. Parameters usually consider chemical, physical or biological characteristics of the native tissue intended to be modelled. For this matter, depending on the relative effect of the characteristic to the phenomena, a decision to whether to include each characteristic needs to be made. The engineered model will most often not be a representation of the whole complexity of the native phenomenon, but rather one piece of the puzzle that when combine with other engineered models give more complete information of intricate processes. Indexes are generally associated with measurable characteristics and once their effect has been validated, they become the criteria for the acceptance of a model as a suitable substrate to be used for the desired purpose. The validation of these indexes usually requires a comparison of the in vitro model with in vivo studies or known mechanisms.
The ability of engineered models to provide more relevant, faster, and, sometimes, high-throughput information than animal-testing without bearing physiological mismatch, inter-laboratory variability and ethical concerns has been highly supported and disseminated by regulatory agencies across the world under the 3Rs paradigm. The path to phase animal-testing out of drug and medical device development most certainly lies in the integration of different alternative methods, including in vitro and in silico models, with a combined effort from experts of different fields (materials science and engineering, cell and molecular biology, chemistry, biomedical engineering, pharmacy and regulation). The engineered models produced so far have been well-received by both Pharma and Biotech industries as these are improved filters at early stages of drug and medical device development, which highly reduces the time and cost of initial investment required with the potential for a faster turnover.
Conflicts of interest
There are no conflicts to declareAcknowledgements
D.P.P. would like to thank the European Commission Joint Research Centre for the Summer School on “Non-Animal Approaches in Science: Challenges and Future Directions” for providing valuable information that strengthen this review.Notes and references
- G. E. P. Box, Robustness Stat, 1979, pp. 201–236 Search PubMed.
- D. Zhang, G. Luo, X. Ding and C. Lu, Acta Pharm. Sin. B, 2012, 2, 549–561 CrossRef.
- F. M. White, in Fluid Mechanics, ed. F. M. White, McGraw-Hill, New York, 8th edn, 2016, pp. 277–324 Search PubMed.
- Y. T. Ho, G. Adriani, S. Beyer, P.-T. Nhan, R. D. Kamm and J. C. Y. Kah, Sci. Rep., 2017, 7, 707 CrossRef.
- G. E. Guajardo-Salinas, J. A. Carvajal, Á. A. Gaytan-Ramos, L. Arroyo, A. G. López-Reyes, J. F. Islas, B. G. Cano, N. Arroyo-Currás, A. Dávalos, G. Madrid and J. E. Moreno-Cuevas, J. Negat. Results Biomed., 2007, 6, 1 CrossRef.
- G. Zou, J. Xu and C. Wu, Int. J. Pavement Res. Technol., 2017, 10, 282–288 CrossRef.
- P. Occhetta, A. Mainardi, E. Votta, Q. Vallmajo-Martin, M. Ehrbar, I. Martin, A. Barbero and M. Rasponi, Nat. Biomed. Eng., 2019, 3, 545–557 CrossRef CAS.
- J. Laurent, G. Blin, F. Chatelain, V. Vanneaux, A. Fuchs, J. Larghero and M. Théry, Nat. Biomed. Eng., 2017, 1, 939–956 CrossRef CAS.
- B. P. Chan and K. W. Leong, Eur. Spine J., 2008, 17, 467–479 CrossRef.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS.
- J. Zhao, M. Griffin, J. Cai, S. Li, P. E. M. Bulter and D. M. Kalaskar, Biochem. Eng. J., 2016, 109, 268–281 CrossRef CAS.
- Z. Ma, N. Huebsch, S. Koo, M. A. Mandegar, B. Siemons, S. Boggess, B. R. Conklin, C. P. Grigoropoulos and K. E. Healy, Nat. Biomed. Eng., 2018, 2, 955–967 CrossRef CAS.
- L. A. MacQueen, S. P. Sheehy, C. O. Chantre, J. F. Zimmerman, F. S. Pasqualini, X. Liu, J. A. Goss, P. H. Campbell, G. M. Gonzalez, S.-J. Park, A. K. Capulli, J. P. Ferrier, T. F. Kosar, L. Mahadevan, W. T. Pu and K. K. Parker, Nat. Biomed. Eng., 2018, 2, 930–941 CrossRef CAS.
- M.-O. Lee, S. H. Moon, H.-C. Jeong, J.-Y. Yi, T.-H. Lee, S. H. Shim, Y.-H. Rhee, S.-H. Lee, S.-J. Oh, M.-Y. Lee, M.-J. Han, Y. S. Cho, H.-M. Chung, K.-S. Kim and H.-J. Cha, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3281–E3290 CrossRef CAS.
- S. Bartfeld, Dev. Biol., 2016, 420, 262–270 CrossRef CAS.
- P. X. Ma, Mater. Today, 2004, 7, 30–40 CrossRef CAS.
- J. M. Saul and D. F. Williams, in Handbook of Polymer Applications in Medicine and Medical Devices, eds. K. Modjarrad and S. Ebnesajjad, Elsevier, USA, 1st edn, 2011, pp. 279–302 Search PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS.
- H.-M. Kim, T. Himeno, T. Kokubo and T. Nakamura, Biomaterials, 2005, 26, 4366–4373 CrossRef CAS.
- M. Kawashita, M. Nakao, M. Minoda, H.-M. Kim, T. Beppu, T. Miyamoto, T. Kokubo and T. Nakamura, Biomaterials, 2003, 24, 2477–2484 CrossRef CAS.
- A. L. B. Maçon, T. B. Kim, E. M. Valliant, K. Goetschius, R. K. Brow, D. E. Day, A. Hoppe, A. R. Boccaccini, I. Y. Kim, C. Ohtsuki, T. Kokubo, A. Osaka, M. Vallet-Regí, D. Arcos, L. Fraile, A. J. Salinas, A. V. Teixeira, Y. Vueva, R. M. Almeida, M. Miola, C. Vitale-Brovarone, E. Verné, W. Höland and J. R. Jones, J. Mater. Sci. Mater. Med., 2015, 26, 115 CrossRef.
- M. Bohner and J. Lemaitre, Biomaterials, 2009, 30, 2175–2179 CrossRef CAS.
- A. Fuchs, M. Leigh, B. Kloefer and J. B. Dressman, Eur. J. Pharm. Biopharm., 2015, 94, 229–240 CrossRef CAS.
- T. J. Dennison, J. Smith, R. K. S. Badhan and A. R. Mohammed, Drug Des., Dev. Ther., 2017, 11, 811–826 CrossRef CAS.
- M. K. Rai, S. D. Deshmukh, A. P. Ingle and A. K. Gade, J. Appl. Microbiol., 2012, 112, 841–852 CrossRef CAS.
- A. Lateef, S. A. Ojo, J. A. Elegbede, M. A. Azeez, T. A. Yekeen and A. Akinboro, J. Cluster Sci., 2017, 28, 1379–1392 CrossRef CAS.
- J. L. Axson, D. I. Stark, A. L. Bondy, S. S. Capracotta, A. D. Maynard, M. A. Philbert, I. L. Bergin and A. P. Ault, J. Phys. Chem. C, 2015, 119, 20632–20641 CrossRef CAS.
- I. L. Bergin, L. A. Wilding, M. Morishita, K. Walacavage, A. P. Ault, J. L. Axson, D. I. Stark, S. A. Hashway, S. S. Capracotta, P. R. Leroueil, A. D. Maynard and M. A. Philbert, Nanotoxicology, 2016, 10, 352–360 CrossRef CAS.
- M. R. C. Marques, R. Loebenberg and M. Almukainzi, Dissolution Technol., 2011, 18, 15–28 CrossRef CAS.
- D. P. Pacheco, C. S. Butnarasu, F. Briatico Vangosa, L. Pastorino, L. Visai, S. Visentin and P. Petrini, J. Mater. Chem. B, 2019, 7, 4940–4952 RSC.
- A. Avdeef and O. Tsinman, Eur. J. Pharm. Sci., 2006, 28, 43–50 CrossRef CAS.
- A. Avdeef, S. Bendels, L. Di, B. Faller, M. Kansy, K. Sugano and Y. Yamauchi, J. Pharm. Sci., 2007, 96, 2893–2909 CrossRef CAS.
- O. Tsinman, K. Tsinman, N. Sun and A. Avdeef, Pharm. Res., 2011, 28, 337–363 CrossRef CAS.
- B. Sinkó, T. M. Garrigues, G. T. Balogh, Z. K. Nagy, O. Tsinman, A. Avdeef and K. Takács-Novák, Eur. J. Pharm. Sci., 2012, 45, 698–707 CrossRef.
- G. Dargó, A. Vincze, J. Müller, H. J. Kiss, Z. Z. Nagy and G. T. Balogh, Eur. J. Pharm. Sci., 2019, 128, 232–239 CrossRef.
- E. Naderkhani, J. Isaksson, A. Ryzhakov and G. E. Flaten, J. Pharm. Sci., 2014, 103, 1882–1890 CrossRef CAS.
- Y. Peck and D.-A. Wang, Expert Opin. Drug Delivery, 2013, 10, 369–383 CrossRef CAS.
- A. D. Ebert, P. Liang and J. C. Wu, J. Cardiovasc. Pharmacol., 2012, 60, 408–416 CrossRef CAS.
- A. Cochrane, H. J. Albers, R. Passier, C. L. Mummery, A. van den Berg, V. V. Orlova and A. D. van der Meer, Adv. Drug Delivery Rev., 2019, 140, 68–77 CrossRef CAS.
- P. Srivastava and K. A. Kilian, Front. Bioeng. Biotechnol., 2019, 7, 1–12 CrossRef.
- T. C. Fung, C. A. Olson and E. Y. Hsiao, Nat. Neurosci., 2017, 20, 145–155 CrossRef CAS.
- A. Lechanteur, J. das Neves and B. Sarmento, Adv. Drug Delivery Rev., 2018, 124, 50–63 CrossRef CAS.
- L. Sardelli, D. P. Pacheco, A. Ziccarelli, M. Tunesi, O. Caspani, A. Fusari, F. Briatico Vangosa, C. Giordano and P. Petrini, RSC Adv., 2019, 9, 15887–15899 RSC.
- S. Jalili-Firoozinezhad, F. S. Gazzaniga, E. L. Calamari, D. M. Camacho, C. W. Fadel, A. Bein, B. Swenor, B. Nestor, M. J. Cronce, A. Tovaglieri, O. Levy, K. E. Gregory, D. T. Breault, J. M. S. Cabral, D. L. Kasper, R. Novak and D. E. Ingber, Nat. Biomed. Eng., 2019, 3, 520–531 CrossRef CAS.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS.
- P. Pullmannová, L. Pavlíková, A. Kováčik, M. Sochorová, B. Školová, P. Slepička, J. Maixner, J. Zbytovská and K. Vávrová, Biophys. Chem., 2017, 224, 20–31 CrossRef.
- F. Netzlaff, C.-M. Lehr, P. W. Wertz and U. F. Schaefer, Eur. J. Pharm. Biopharm., 2005, 60, 167–178 CrossRef CAS.
- T. do N. Pedrosa, C. M. Catarino, P. C. Pennacchi, S. R. de Assis, F. Gimenes, M. E. L. Consolaro, S. B. de M. Barros and S. S. Maria-Engler, Toxicol. In Vitro, 2017, 42, 31–37 CrossRef CAS.
- T. Akagi, M. Nagura, A. Hiura, H. Kojima and M. Akashi, Tissue Eng., Part A, 2017, 23, 481–490 CrossRef CAS.
- J. A. DiMasi, R. W. Hansen and H. G. Grabowski, J. Health Econ., 2003, 22, 151–185 CrossRef.
- J. A. DiMasi, R. W. Hansen and H. G. Grabowski, J. Health Polit. Policy Law, 2008, 33, 319–324 CrossRef.
- G. Ugolini, D. Cruz-Moreira, R. Visone, A. Redaelli and M. Rasponi, Micromachines, 2016, 7, 233 CrossRef.
- M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua and A. Tesei, Sci. Rep., 2016, 6, 19103 CrossRef CAS.
- S. B. Leite, T. Roosens, A. El Taghdouini, I. Mannaerts, A. J. Smout, M. Najimi, E. Sokal, F. Noor, C. Chesne and L. A. van Grunsven, Biomaterials, 2016, 78, 1–10 CrossRef CAS.
- A. C. Y. Chua, A. Ananthanarayanan, J. J. Y. Ong, J. Y. Wong, A. Yip, N. H. Singh, Y. Qu, L. Dembele, M. McMillian, R. Ubalee, S. Davidson, A. Tungtaeng, R. Imerbsin, K. Gupta, C. Andolina, F. Lee, K. S.-W. Tan, F. Nosten, B. Russell, A. Lange, T. T. Diagana, L. Rénia, B. K. S. Yeung, H. Yu and P. Bifani, Biomaterials, 2019, 216, 119221 CrossRef CAS.
- A. S. Nunes, A. S. Barros, E. C. Costa, A. F. Moreira and I. J. Correia, Biotechnol. Bioeng., 2019, 116, 206–226 CrossRef CAS.
- M. Zanoni, S. Pignatta, C. Arienti, M. Bonafè and A. Tesei, Expert Opin. Drug Discov., 2019, 14, 289–301 CrossRef CAS.
- M. Zanoni, M. Cortesi, A. Zamagni, C. Arienti, S. Pignatta and A. Tesei, J. Hematol. Oncol., 2020, 13, 1–15 CrossRef.
- I. W. Mak, N. Evaniew and M. Ghert, Am. J. Transl. Res., 2014, 6, 114–118 Search PubMed.
- T. Hasenberg, S. Mühleder, A. Dotzler, S. Bauer, K. Labuda, W. Holnthoner, H. Redl, R. Lauster and U. Marx, J. Biotechnol., 2015, 216, 1–10 CrossRef CAS.
- H. Attarwala, J. Young Pharm., 2010, 2, 332–336 CrossRef CAS.
- P. S. Aisen, J. Prev. Alzheimers Dis., 2019, 6, 150 CAS.
- J. Cummings, G. Lee, A. Ritter and K. Zhong, Alzheimers Dement (NY), 2018, 4, 195–214 CrossRef.
- S. E. Cavanaugh, ALTEX, 2014, 31, 279–302 CrossRef.
- K. Mullane and M. Williams, Curr. Protoc. Pharmacol., 2019, 84, e57 CrossRef.
- M. J. Lee, B. A. Hatton, E. H. Villavicencio, P. C. Khanna, S. D. Friedman, S. Ditzler, B. Pullar, K. Robison, K. F. White, C. Tunkey, M. LeBlanc, J. Randolph-Habecker, S. E. Knoblaugh, S. Hansen, A. Richards, B. J. Wainwright, K. McGovern and J. M. Olson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7859–7864 CrossRef CAS.
- K. Sasaki, J. R. Gotlib, R. A. Mesa, K. J. Newberry, F. Ravandi, J. E. Cortes, P. Kelly, J. L. Kutok, H. M. Kantarjian and S. Verstovsek, Leuk. Lymphoma, 2015, 56, 2092–2097 CrossRef CAS.
- J. Cathcart, A. Pulkoski-Gross and J. Cao, Genes Dis., 2015, 2, 26–34 CrossRef CAS.
- M. K. Jones, M. Watanabe, S. Zhu, C. L. Graves, L. R. Keyes, K. R. Grau, M. B. Gonzalez-Hernandez, N. M. Iovine, C. E. Wobus, J. Vinje, S. A. Tibbetts, S. M. Wallet and S. M. Karst, Science, 2014, 346, 755–759 CrossRef CAS.
- G. A. Van Norman, JACC Basic Transl. Sci., 2016, 1, 170–179 CrossRef.
- G. A. Van Norman, JACC Basic Transl. Sci., 2016, 1, 277–287 CrossRef.
- C. Kilkenny, W. J. Browne, I. C. Cuthill, M. Emerson and D. G. Altman, PLoS Biol., 2010, 8, e1000412 CrossRef.
- J. MacArthur Clark, Br. J. Nutr., 2018, 120, S1–S7 CrossRef CAS.
- EU Reference Laboratory for alternatives to animal testing, https://ec.europa.eu/jrc/en/eurl/ecvam, (accessed 6 May 2019).
- EURL ECVAM, EURL ECVAM Database Service on Alternative Methods to Animal Experimentation, http://cidportal.jrc.ec.europa.eu/ftp/jrc-opendata/EURL-ECVAM/datasets/DBALM/LATEST/online/dbalm.html, (accessed 6 October 2020).
- Validated Test Methods, https://ec.europa.eu/jrc/en/eurl/ecvam/alternative-methods-toxicity-testing/validated-test-methods, (accessed 6 May 2019).
- The 3Rs, https://www.nc3rs.org.uk/the-3rs, (accessed 6 May 2019).
- ASTM International, F2312-11 Standard Terminology Relating to Tissue Engineered Medical Products, Pennsylvania, USA, 2011 Search PubMed.
Footnotes |
| † All schematic representations included in the manuscript were designed with BioRender.com. |
| ‡ The current address of Natalia Suárez Vargas is the Department of Biomedical Engineering at Universidad de los Andes. |
| This journal is © The Royal Society of Chemistry 2021 |