
Palladium-catalysed annulative allylic alkylation for the synthesis of benzannulated heteroarenes†
Sonu
Yadav
and
S. S. V.
Ramasastry
 *
*
Organic Synthesis and Catalysis Lab, Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Sector 81, Manauli PO, S. A. S. Nagar, Punjab 140306, India. E-mail: ramsastry@iisermohali.ac.in; ramsastrys@gmail.com
First published on 20th November 2020
Abstract
A conceptually novel intramolecular allylic alkylation strategy is developed for the synthesis of carbazoles and dibenzothiophenes. In an unusual event, palladium catalyses the formation of π-allylpalladium complexes of the respective (2-methylindol-3-yl)allyl acetates and subsequently facilitates the benzannulation process.
The seminal contributions of Tsuji and Trost have led to the establishment of metal-catalysed allylic alkylations as an incredibly broad research area.1 In particular, the intramolecular variants are quite popular in the synthesis of various types of cyclic structures.2 In this context, we have recently reported palladium-catalysed annulative transformation of allyl acetates to an array of cyclopentene-fused (hetero)arenes, incorporated with an all-carbon quaternary/spiro stereocenter, Scheme 1a.3
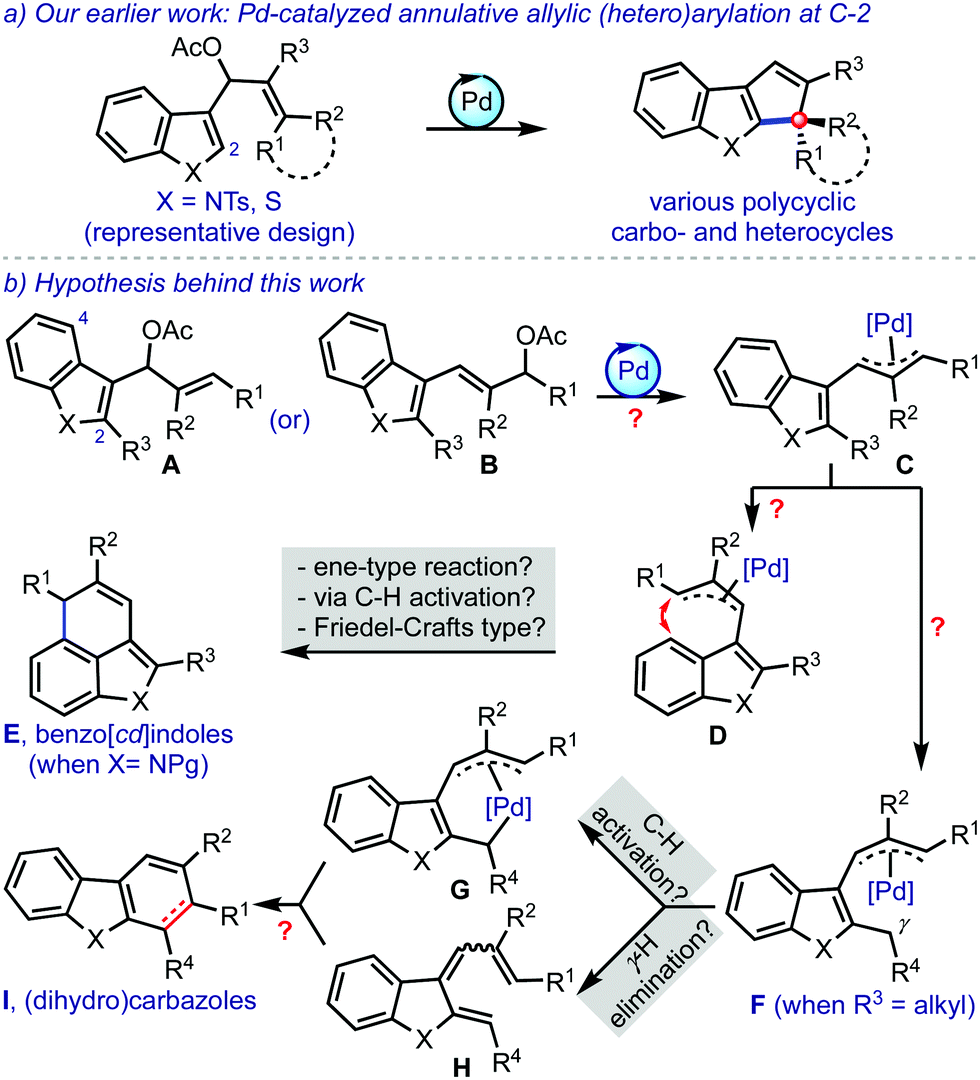 | ||
| Scheme 1 Background and hypothesis for the synthesis of benzo[cd]indoles and/or (dihydro)carbazoles. | ||
With this background, it was hypothesised that the introduction of a substituent at C-2 (R3) in A or B could prompt the π-allylpalladium species C to reorganise to D (due to steric considerations between R2 and R3), Scheme 1b.4 A subsequent C-4 cyclisation could possibly deliver benzo[cd]indoles E.5 On the other hand, a potential alternate pathway was envisioned with the presence of an alkyl group at R3. The π-allylpalladium complex F could facilitate a C(sp3)–H activation6 for the formation of a species such as G. The intermediate F could also generate the triene H by the elimination of the γ-hydrogen. Then, Gvia reductive C–C bond formation, and H by undergoing 6π-electrocyclisation provide (dihydro)carbazoles I.
Benzo[cd]indole and carbazole scaffolds are frequently encountered in several architecturally intriguing bioactive molecules.7 However, the synthesis of such complex targets is often complicated due to the lack of broadly applicable synthetic methods. Therefore, we intended to come up with a novel strategy for their synthesis based on the Pd-catalysed annulative allylic (hetero)arylation strategy.3
Based on the aforementioned hypothesis, the allyl acetates 1a and 2a were prepared and treated with a catalytic amount of palladium chloride in toluene, Scheme 2. To our surprise, the carbazole 3a was isolated in good yield in both cases. There was no indication of the formation of the respective benzo[cd]indole. It was indeed an encouraging result because carbazoles are privileged heterocycles, which are prevalent in several bioactive compounds.7 Carbazoles find applications as optoelectronic materials, conducting polymers, and dyes.8 In addition, carbazole derivatives are also employed in sensing applications.9
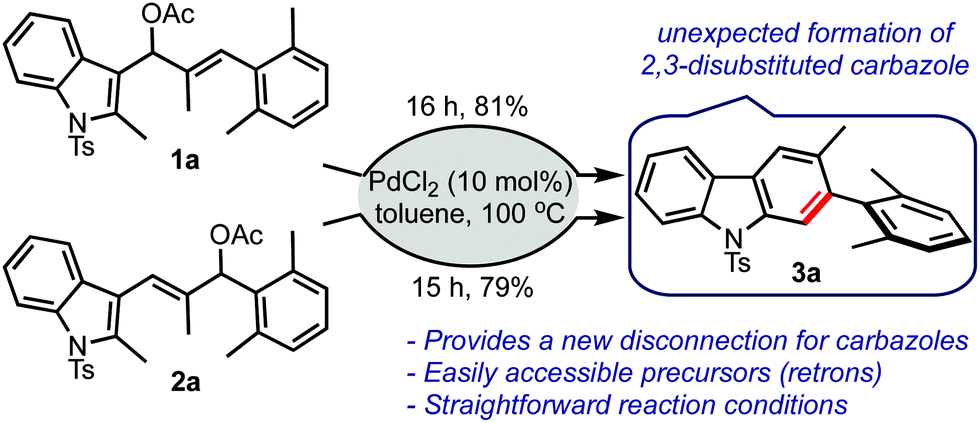 | ||
| Scheme 2 Preliminary results. | ||
Therefore, we proceeded to optimise the reaction of the allyl acetate 2a by evaluating the role of different metal catalysts, solvents and temperature. Some of the important results are summarised in Table 1. Since the initial result was obtained with a Pd(II) catalyst, a few other Pd(II) complexes were screened (entries 1–3). The reaction of 2a in the presence of Pd(PPh3)2Cl2 gave no product, while Pd(OAc)2 and [PdCl(allyl)]2 provided 3a only in poor yields. Next, Pd(0)-based complexes were also screened (entries 4 and 5). Among them, only the reaction catalysed by Pd2(dba)3 generated 3a, albeit in poor yield. Our efforts to enhance the efficiency of the reaction with Ir- or Ni-catalysts were not encouraging (entries 6 and 7). With these unfavourable results, we resumed the optimisation with PdCl2 as the catalyst. Accordingly, a brief solvent screening was undertaken, which revealed that toluene was optimal for the conversion of 2a to 3a (entries 8–13).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Solvent | Time (h) | Yieldc (%) |
| a Reaction conditions: see the ESI for details. b Unless mentioned otherwise, all the reactions were performed at 100 °C. c Isolated yields after column chromatography. d 2a remained as such. e At 90 °C. f At 70 °C. g 5 mol% PdCl2 was employed. h 20 mol% PdCl2 was employed. i Reaction of 2a′ possessing the OBoc group instead of OAc. | ||||
| 1d | Pd(PPh3)2Cl2 | Toluene | 24 | — |
| 2 | Pd(OAc)2 | Toluene | 24 | 35 |
| 3 | [PdCl(allyl)]2 | Toluene | 22 | 47 |
| 4 | Pd2(dba)3 | Toluene | 48 | 29 |
| 5d | Pd(PPh3)4 | Toluene | 48 | — |
| 6 | [Ir(cod)Cl]2 | Toluene | 24 | 10 |
| 7d | Ni(cod)2 | Toluene | 48 | — |
| 8 | PdCl2 | Toluene | 16 | 81 |
| 9e | PdCl2 | 1,2-DCE | 24 | 26 |
| 10 | PdCl2 | DMF | 16 | 75 |
| 11 | PdCl2 | CH3CN | 19 | 71 |
| 12 | PdCl2 | 1,4-Dioxane | 24 | 32 |
| 13df | PdCl2 | THF | 24 | — |
| 14g | PdCl2 | Toluene | 24 | 74 |
| 15h | PdCl2 | Toluene | 12 | 83 |
| 16i | PdCl2 | Toluene | 15 | 69 |

Next, the requirement of the catalyst loading was also studied. When 5 mol% of PdCl2 was employed, 3a was obtained in marginally less yield with increased reaction time (Table 1, entry 14). Whereas, the reaction with higher catalyst loading (20 mol%), although it required less time for completion, did not translate into higher yield of the product (entry 15). On the other hand, the reaction of 2a′ (2a with OBoc group) under the best yielding conditions afforded 3a in 69% yield (entry 16).
Thus, 10 mol% of the cheap and readily available palladium chloride was realised to be the optimal catalyst for the conversion of 2a to 3a. An interesting feature of this reaction is that it does not require any additives, re-oxidants, or bases, unlike typical Pd-catalysed transformations.
To evaluate the scope and generality of the method, the optimised conditions were applied to a range of allyl acetates 2, Table 2. A variety of 2,3-disubstituted carbazoles 3b–g were obtained in good yields regardless of the presence of electron-donating (3c–f) or electron-withdrawing (3g) aryl groups at the allylic position (R1). Even sterically encumbered arene rings were also well-tolerated (3c–f). Interestingly, the reaction of the allyl acetate 4 with the N-Boc indole backbone straightaway delivered 5, by undergoing simultaneous carbazole formation and in situ Boc-deprotection.10
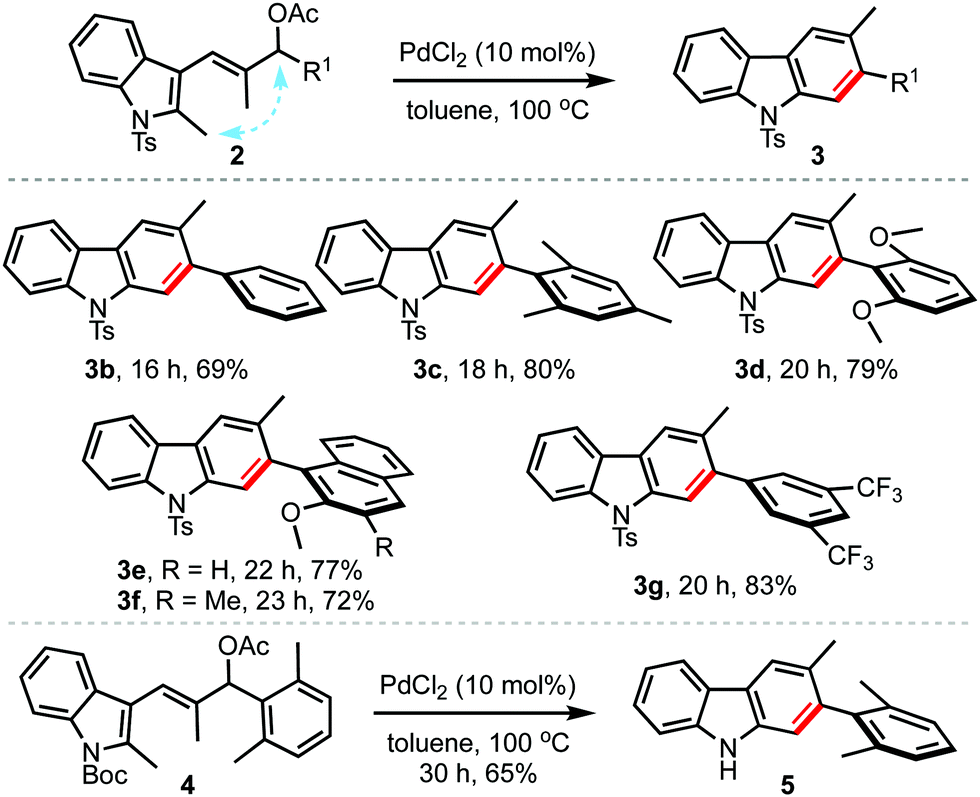
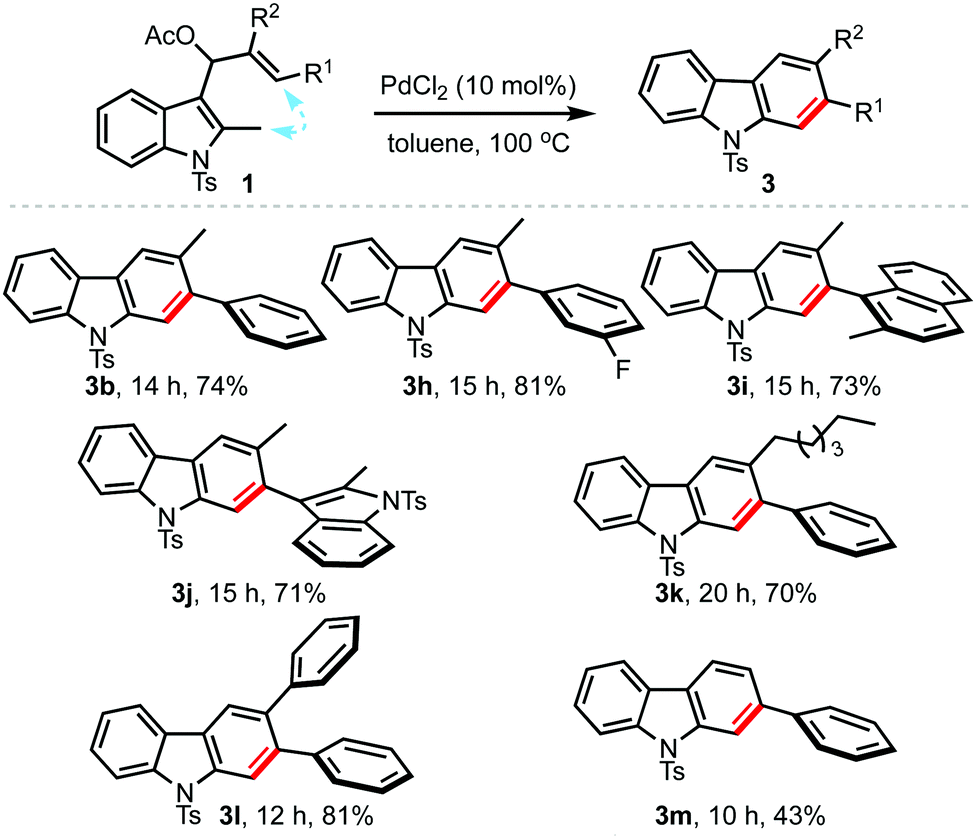
Next, the scope with regard to the allyl acetates 1 was also evaluated under the optimised conditions, Table 3. By this means, a wide variety of 2,3-disubstituted carbazoles possessing electron-donating as well as electron-withdrawing arenes, and heteroarenes at R1 could be accessed in good yields (3b, 3h–m).11 Even carbazoles substituted with alkyl and aryl groups at R2 (3h–k and 3l) could be obtained in an efficient manner. Further, mono-substituted carbazoles such as 3m were also conveniently prepared, although in moderate yield. Thus, it is evident from Tables 2 and 3 that both types of allyl acetates (1 or 2) can be employed to access carbazoles of choice.
This method can even be customised for the synthesis of polycyclic carbazoles, Scheme 3. For example, the chiral dihydroquino[4,3-b]carbazole 3n could be prepared with an appropriate choice of the precursor (1n). Even the oxa- and carbocyclic counterparts of quino[4,3-b]carbazole (3o and 3p, respectively) could also be synthesised in a similar manner. Thus, the present strategy can be utilised for the synthesis of analogues of the calothrixin and isocryptolepine family of natural products,12 and medicinally important quinocarbazoles.13
 | ||
| Scheme 3 Synthesis of polycyclic carbazoles. | ||
Having established a general method for the facile synthesis of carbazoles,14 the feasibility extending this concept to access related carbo- and heterocycles such as naphthalenes, dibenzofurans and dibenzothiophenes was also explored, Scheme 4. Accordingly, the allylic acetates 6a, 7a and 8a were prepared and subjected to the optimised conditions. Among them, 6a and 7a did not yield the desired products, but 8a generated the dibenzothiophene 9a in good yield. Owing to the significance of dibenzothiophenes in medicinal chemistry as well as in materials science,15 a few other analogues were prepared. Apart from 2,3-disubstituted dibenzothiophenes 9a and 9b, this method was improvised to afford 1,2,3-trisubstituted dibenzocarbazoles such as 9c in an efficient manner. This is achievable by the introduction of branching at the benzylic position (R1). However, a similar trend was not observed with indole-based substrates.
 | ||
| Scheme 4 Attempts to synthesise naphthalenes, dibenzofurans and dibenzothiophenes. | ||
Next, we turned our attention to gain insights about the mechanism, Scheme 5. Important evidence was obtained when 2r under the optimised conditions generated predominantly the olefin 10 in about 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioisomeric ratio (r.r.), Scheme 5a. This result can be explained by considering the intermediacy of the triene K or the palladacycle L, both originating from the π-allylpalladium species J.
:1 regioisomeric ratio (r.r.), Scheme 5a. This result can be explained by considering the intermediacy of the triene K or the palladacycle L, both originating from the π-allylpalladium species J.
 | ||
| Scheme 5 Insights about the reaction mechanism. | ||
Furthermore, the value of the kinetic isotope effect (KIE) was ascertained by conducting an intermolecular competition experiment between 2a and 2a-D3, Scheme 5b.16 The kH/kD was realised to be about 1.85, supporting the γ-hydrogen elimination during the conversion of J to K as well as the γ-C(sp3)–H activation process during the transformation of J to L.
Based on the experimental evidence and related literature reports,6b,17 a plausible mechanism is presented in Scheme 6. The reaction commences with the oxidative addition of Pd(0) to the substrate to generate the (η3-allyl)palladium complex F.3,18 A subsequent γ-C(sp3)–H activation possibly generates the seven-membered palladacycle N, which can exist in equilibrium with the five-membered palladacycle M. Next, N undergoes reductive elimination to afford the dihydrocarbazole O. On the other hand, the 6π-electrocyclisation of the triene H, formed via the γ-hydrogen elimination, can also provide O. An eventual aerobic oxidation of O provides benzannulated products.
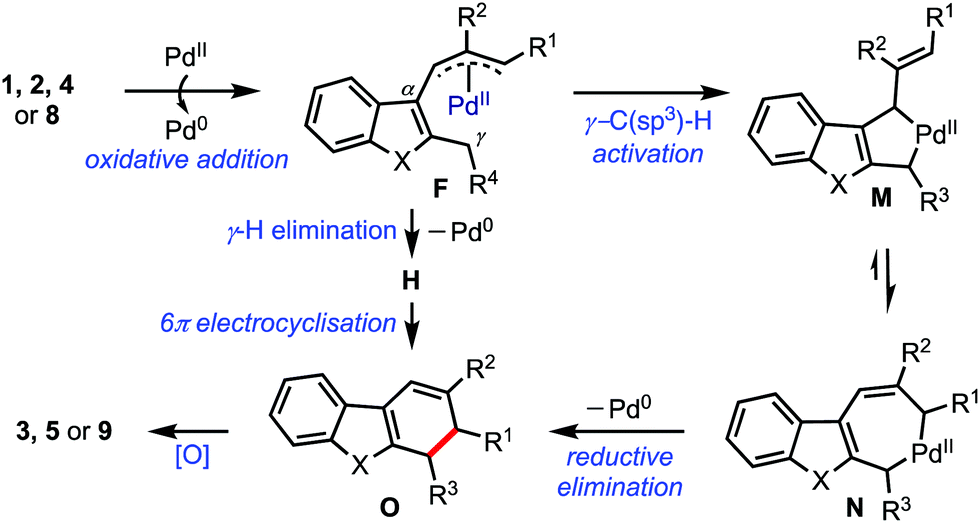 | ||
| Scheme 6 The proposed mechanism. | ||
In conclusion, we discovered a Pd(II)-catalysed annulative allylic alkylation reaction, through which the synthesis of carbazoles and dibenzothiophenes was achieved. The method was realised to be practical and scalable,16 and offers great potential for the synthesis of novel heterocycles. The available data suggests a C(sp3)–H activation pathway or the 6π-electrocyclisation pathway for this transformation. We are currently working on ascertaining the mechanism by conducting in-depth DFT studies. We are also extending this strategy for the atroposelective synthesis of carbazoles, and the results will be communicated in due course.
We thank IISER Mohali for the financial support, and for the NMR, X-ray and mass facilities. S. S. V. R. thanks DST for the Swarnajayanti fellowship (DST/SJF/CSA-01/2017-18), and SERB for the Core Research Grant (CRG/2018/000016). S. Y. thanks IISER Mohali for a research fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Few selected articles: (a) J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 6, 4387 CrossRef; (b) B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292 CrossRef CAS; (c) B. M. Trost, Tetrahedron, 2015, 71, 5708 CrossRef CAS; (d) B. M. Trost and C. A. Kalnmals, Chem. – Eur. J., 2020, 26, 1906 CrossRef CAS.
- Few selected articles: (a) H. Kinoshita, H. Shinokubo and K. Oshima, Angew. Chem., Int. Ed., 2005, 44, 2397 CrossRef CAS; (b) B. Schulte and A. Studer, Synthesis, 2006, 2129 CAS; (c) X. Wu, S.-S. Chen, L. Zhang, H. J. Wang and L.-Z. Gong, Chem. Commun., 2018, 54, 9595 RSC; (d) M. Faltracco, V. Sukowski, M. van Druenen, T. A. Hamlin, F. M. Bickelhaupt and E. Ruijter, J. Org. Chem., 2020, 85, 9566 CrossRef CAS.
- B. Singh, S. K. Bankar, K. Kumar and S. S. V. Ramasastry, Chem. Sci., 2020, 11, 4948 RSC.
- For an instance where product selectivity was switched by the introduction of a C-2 substituent, see: T. Itahara and T. Sakakibara, Synthesis, 1978, 607 CrossRef CAS.
- In analogy to: (a) G. Henrion, T. E. J. Chavas, X. L. Goff and F. Gagosz, Angew. Chem., Int. Ed., 2013, 52, 6277 CrossRef CAS; (b) D. H. Dethe, S. K. Sau and S. Mahapatra, Org. Lett., 2016, 18, 6392 CrossRef CAS.
- Selected cases where π-allylpalladium complexes promote C(sp3)–H activation: (a) T. Piou, A. Bunescu, Q. Wang, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 12385 CrossRef CAS; (b) K. H. Kim, H. R. Moon, J. Lee and J. N. Kim, Adv. Synth. Catal., 2015, 357, 701 CrossRef CAS; (c) P. V. Santhini, A. S. Smrithy, C. P. I. Jesin, S. Varughese, J. John and K. V. Radhakrishnan, Chem. Commun., 2018, 54, 2982 RSC; (d) R. A. Fernandes and J. L. Nallasivam, Org. Biomol. Chem., 2019, 17, 8647 RSC.
- For bioactive benzo[cd]indoles, see: (a) I.-K. Park, J. Park and C. G. Cho, Angew. Chem., Int. Ed., 2012, 51, 2496 CrossRef CAS; (b) S. Romanini, E. Galletti, L. Caruana, A. Mazzanti, F. Himo, S. Santoro, M. Fochi and L. Bernardi, Chem. – Eur. J., 2015, 21, 17578 CrossRef CAS For bioactive carbazoles, see: ; (c) C. J. Moody, Synlett, 1994, 681 CrossRef CAS; (d) J. Roy, A. K. Jana and D. Mal, Tetrahedron, 2012, 68, 6099 CrossRef CAS; (e) C. R. Reddy, R. R. Velleti and U. Dilipkumar, Chem. – Eur. J., 2016, 22, 2501 CrossRef; (f) S. Yadav, R. Hazra, A. Singh and S. S. V. Ramasastry, Org. Lett., 2019, 21, 2983 CrossRef CAS.
- (a) J. Li and A. C. Grimsdale, Chem. Soc. Rev., 2010, 39, 2399 RSC; (b) C. Teng, X. Yang, C. Yuan, C. Li, R. Chen, H. Tian, S. Li, A. Hagfeldt and L. Sun, Org. Lett., 2009, 11, 5542 CrossRef CAS.
- (a) P. A. Gale, Chem. Commun., 2008, 4525 RSC; (b) D. A. Kim, P. Kang, M.-G. Choi and K.-S. Jeong, Chem. Commun., 2013, 49, 9743 RSC.
- Substrates with N-methyl or N-benzyl indole backbones were not successful under the reaction conditions.
- The reaction of 1 or 2 with aliphatic groups at R1 gave predominantly β-eliminated products. See the ESI† for details.
- (a) B. M. Ramalingam, N. D. Moorthy, S. R. Chowdhury, T. Mageshwaran, E. Vellaichamy, S. Saha, K. Ganesan, B. N. Rajesh, S. Iqbal, H. K. Majumder, K. Gunasekaran, R. Siva and A. K. Mohanakrishnan, J. Med. Chem., 2018, 61, 1285 CrossRef; (b) C. Chen, Y. Wang, X. Shi, W. Sun, J. Zhao, Y.-P. Zhu, L. Liu and B. Zhu, Org. Lett., 2020, 22, 4097 CrossRef CAS.
- (a) A. K. Mohanakrishnan and P. C. Srinivasan, J. Org. Chem., 1995, 60, 1939 CrossRef CAS; (b) D. Liu, J. Huang, Z. Fu and W. Huang, Green Chem., 2019, 21, 968 RSC.
- The allyl acetates appended to the C-2 position of the indoles were also validated. See the ESI† for details.
- (a) R. Sanz, Y. Fernandez, M. P. Castroviejo, A. Perez and F. J. Fananas, J. Org. Chem., 2006, 71, 6291 CrossRef CAS; (b) M. J. Barrett, P. W. Davies and R. S. Grainger, Org. Biomol. Chem., 2015, 13, 8676 RSC; (c) M. Tobisu, Y. Masuya, K. Baba and N. Chatani, Chem. Sci., 2016, 7, 2587 RSC; (d) K. Nogi and H. Yorimitsu, Chem. Commun., 2017, 53, 4055 RSC; (e) K. Li, A. Yu and X. Meng, Org. Lett., 2018, 20, 1106 CrossRef CAS.
- See the ESI† for details.
- (a) M.-C. A. Cordonnier, S. B. J. Kan, B. Gockel, S. S. Goh and E. A. Anderson, Org. Chem. Front., 2014, 1, 661 RSC; (b) A. Mekareeya, P. R. Walker, A. C. Rios, C. D. Campbell, A. Steven, R. S. Paton and E. A. Anderson, J. Am. Chem. Soc., 2017, 139, 10104 CrossRef CAS.
- The substrate itself may reduce Pd(II) to Pd(0) during the reaction, and consequently, the in situ formed Pd(0) serves as the active metal center to promote the subsequent reaction. See: (a) F.-Q. Yuan, L.-X. Gao and F.-S. Han, Chem. Commun., 2011, 47, 5289 RSC; (b) F.-Q. Yuan, F.-Y. Sun and F.-S. Han, Tetrahedron, 2012, 68, 6837 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06695h |
| This journal is © The Royal Society of Chemistry 2021 |