
Cu-Catalyzed O-alkylation of phenol derivatives with alkylsilyl peroxides†
Shunya
Sakurai
 a,
Taichi
Kano
a and
Keiji
Maruoka
*abc
a,
Taichi
Kano
a and
Keiji
Maruoka
*abc
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Sakyo, Kyoto 606-8502, Japan. E-mail: maruoka.keiji.4w@kyoto-u.ac.jp
bGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo, Kyoto 606-8501, Japan
cSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, China
First published on 25th November 2020
Abstract
A Cu-catalyzed O-alkylation of phenol derivatives using alkylsilyl peroxides as alkyl radical precursors is described. The reaction proceeds smoothly under mild reaction conditions and the use of two different ligands with a Cu catalyst provides a wide range of products. A mechanistic study suggested that the reaction proceeds via a radical mechanism.
Alkyl aryl ethers represent a very important chemical motif because they are found in a variety of natural products, pharmaceuticals, and bioactive molecules (Fig. 1a).1,2 Traditional approaches for the synthesis of alkyl aryl ethers such as the Williamson ether synthesis3 involve the nucleophilic substitution of alkyl halides with phenoxides. This often requires a base and thus β-elimination can readily proceed to afford an alkene as a major side product (Fig. 1b). A complementary approach that involves a nucleophilic aromatic substitution (SNAr reaction) using alcohols and aryl halides is also well-known.4 However, this method generally requires the presence of electron-withdrawing groups on the aromatic ring. To address these issues, a number of researchers have established strategies for the formation of C–O bonds by cross-coupling reactions under transition-metal or photoredox catalysis.5,6 Although tremendous efforts have been dedicated into the development of these strategies, there are fewer methods for transition-metal-catalyzed phenolic O-alkylations than there are for C(sp2)–O bond coupling reactions using a transition-metal catalyst, aryl halides, and alcohols.
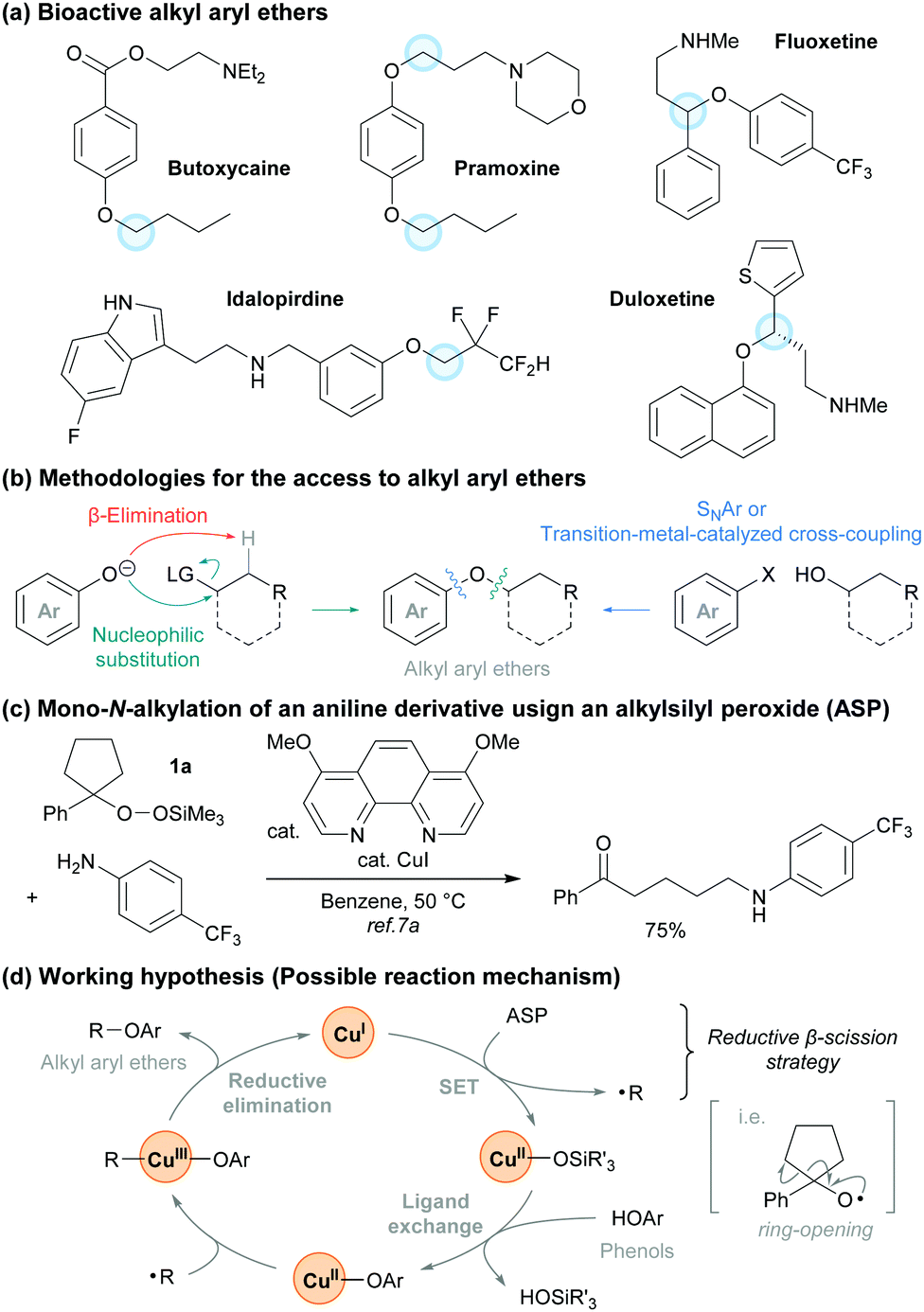 | ||
| Fig. 1 (a) Bioactive alkyl aryl ethers. (b) Methods to access alkyl aryl ethers. (c) Working hypothesis for this study. | ||
We have recently developed and applied a Cu-catalyzed method for the generation of alkyl radicals from alkylsilyl peroxides (ASPs) that proceeds under mildly reductive conditions (reductive β-scission strategy).7 ASPs are easily synthesized and handled,8 and become a variety of alkyl radical precursors under Cu, Fe, and Ni catalysis.9 During our studies on the transformations with ASPs, we reported the Cu-catalyzed mono-N-alkylation of aniline derivatives using a cyclic ASP 1a (Fig. 1c).7a On the basis of these results, we hypothesized that our reductive β-scission strategy could be applied to the radical-mediated O-alkylation of phenol derivatives in the presence of a Cu catalyst (Fig. 1d).
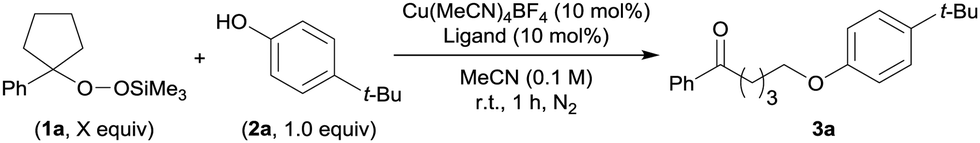
To verify our hypothesis, we optimized the conditions of the Cu-catalyzed O-alkylation using ASP 1a and p-tert-butylphenol (2a) (Table 1). Using 1.5 equiv. of 1a and 1.0 equiv. of 2a with a Cu(MeCN)4BF4/2,2′-bipyridyl (bpy) catalyst system in MeCN as a solvent, the C(sp3)–O cross-coupling proceeded at room temperature and the desired product 3a was observed in 37% NMR yield. Neither the use of THF as a solvent instead of MeCN nor the addition of N,N-diisopropylethylamine (i-Pr2NEt) as a base were effective (entries 2 and 3). Next, we attempted to improve the product yield and examined a variety of ligands for the Cu catalyst (entries 4–9). While the use of L110 or L2 improved the product yield (41% and 61% NMR yield, respectively), the use of L3–L511 produced 3a in lower yields (<26% NMR yields) than when bpy was used. When we tested ligands L6–L14, a dramatic improvement in the yield of 3a was not observed (see also ESI† for details). On the basis of these results, we determined L2 to be the optimal ligand and 3a was obtained in 85% isolated yield by using L2 and 2.5 equiv. of 1a (entry 10).
|
|
|||
|---|---|---|---|
| Entry | X | Ligand | Yieldb (%) |
a The reactions were performed on a 0.2 mmol scale in MeCN (2.0 mL) under a N2 atmosphere.
b
1H NMR yield using 1,3,5-trimethoxybenzene as an internal standard.
c THF (2.0 mL) was used as the solvent instead of MeCN.
d i-Pr2NEt (1.5 equiv.) was used as an additive reagent.
e See ESI for details.
f Isolated yield.
|
|||
| 1 | 1.5 | bpy | 37 |
| 2c | 1.5 | bpy | 9 |
| 3d | 1.5 | bpy | <1 |
| 4 | 1.5 | L1 | 41 |
| 5 | 1.5 | L2 | 61 |
| 6 | 1.5 | L3 | <1 |
| 7 | 1.5 | L4 | 23 |
| 8 | 1.5 | L5 | 26 |
| 9e | 1.5 | Other ligands (L6–L14) | <52 |
| 10 | 2.5 | L2 | 91 (85f) |
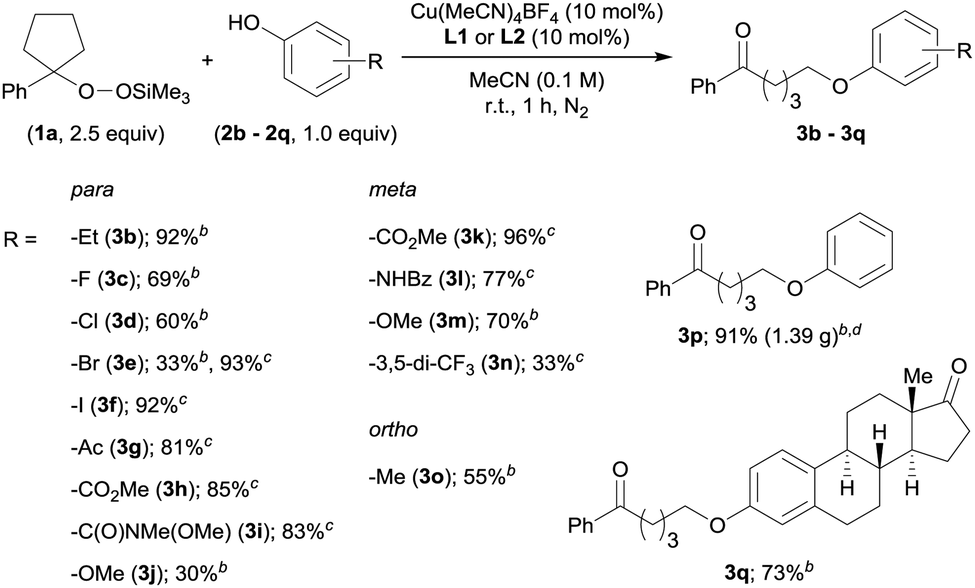
With the optimized reaction conditions in hand, we investigated the substrate scope of the present O-alkylation with ASP 1a and a series of phenol derivatives (2b–2q) (Table 2). Initially, we examined the effect of varying the para-substituents on the phenols. The use of p-ethylphenol (2b) afforded 3b in 92% yield, indicating that the presence of a benzylic proton is tolerated in our reaction. While halides such as F and Cl at the para-position were also well tolerated (3c and 3d), the reaction of p-bromophenol (2e) furnished 3e in low yield (33%). Therefore, we rescreened the ligands and found L1 to be more effective (93%). From this point on, we continued to investigate the substrate scope with either L1 or L2, depending on the structure of the phenol derivatives. The use of L1 for the reaction of p-iodophenol (2f) furnished 3f in 92% yield. The reactions with the phenols bearing a carbonyl group such as a ketone, an ester, or a Weinreb amide, at the para-position proceeded smoothly in the presence of L1, and desired products 3g–3i were obtained in high yields (81–85%). However, the combination of p-methoxyphenol (2j) and L2 produced 3j only in low yield. In this case, the use of L1 did not improve the yield of 3j. Substituents at the meta- or ortho-position were also tolerated (3k–3o), albeit that the product yield was low in the case of 3,5-bis(trifluoromethyl)phenol (2n). We were also able to conduct the O-alkylation on a gram-scale (6.0 mmol scale), which furnished the coupling product 3p from 1a and phenol (2p) in 91% yield (1.39 g). We also tested estrone (2q), which has a phenolic hydroxy group, and the desired O-alkylated product 3q was obtained in 73% yield.
We then examined the scope of the ASPs tolerated by the reaction. A variety of ASPs were reacted with 2e or 2h using the Cu(MeCN)4BF4/L1 catalyst system. We discovered that both cyclic and acyclic ASPs were applicable to the reaction (Table 3). Cyclic ASPs (1b–1d) provided the corresponding products 4a–4c in acceptable yields (64–9%), while a higher reaction temperature (50 °C) was required in the case of 1b. The methyl-substituted five-membered cyclic ASP 1e furnished the desired product 4d regioselectively in 95% yield. The selective transfer of secondary alkyl groups such as isopropyl, cyclohexyl and tetrahydropyranyl from 1f–1h was also possible, and the desired products 4e–4g were obtained in excellent yields (94–96%). It should be noted here that these products are difficult to access via the traditional Williamson ether synthesis due to the ease with which β-elimination occurs. Additionally, the reaction with ASP 1i, which was prepared by the Co-catalyzed air-oxidation of (S)-(−)-limonene in the presence of triethylsilane,8 proceeded at 50 °C to afford 4h in 31% yield. The use of bicyclic ASP 1j, which is derived from the corresponding norbornene derivative, gave 4i regioselectively in 92% yield as a separable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture. This result suggests that our Cu-catalyzed O-alkylation proceeds via a radical process.
:1 diastereomeric mixture. This result suggests that our Cu-catalyzed O-alkylation proceeds via a radical process.
| a The reactions were performed on a 0.2 mmol scale in MeCN (2.0 mL) under a N2 atmosphere. |
|---|

|
To better understand the underlying reaction mechanism and to confirm the radical process of this reaction, a control experiment was carried out (Scheme 1). We added an excess amount of tetrachloromethane (CCl4) as a chlorine-atom donor12 to the reaction using 1a and 2h as substrates. The product 3h was observed in 61% NMR yield, together with 5-chloro-1-phenylpentan-1-one (5) in 75% yield. It is feasible to assume that 5 would be generated via the chlorine-atom abstraction by the ring-opened alkyl radical 6.
 | ||
| Scheme 1 Control experiment. | ||
Finally, we were pleased to find that in preliminary studies our O-alkylation can be applied to an asymmetric Cu-catalyzed reaction (Scheme 2). The use of a chiral bis(oxazoline) (BOX) ligand L1513 in the reaction between 1e and 2i afforded 4d in 25% yield with 33% ee. Further studies to improve the yield and enantioselectivity are currently in progress in our laboratory.
 | ||
| Scheme 2 Enantioselective synthesis of 4d. | ||
In summary, we have developed a Cu-catalyzed O-alkylation of phenol derivatives using alkylsilyl peroxides (ASPs) as alkyl radical sources. The reaction proceeded smoothly under mild reaction conditions and the use of two different ligands with the Cu catalyst afforded a wide range of products. A control experiment using CCl4 suggested that this reaction proceeds via a radical mechanism. Further improvements to realize the highly enantioselective formation of C(sp3)–O bonds are currently in progress and a more extensive report of these investigations will be disclosed in due course.
We gratefully acknowledge financial support via JSPS KAKENHI Grants JP26220803, JP17H06450 (Hybrid Catalysis), and JP18J22096. S. S. is thankful for the Grant-in-Aid for JSPS Fellowships for Young Scientists.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) F. Műller, Agrochemicals: Composition, Production, Toxicology, Applications, Wiley-VCH, Weinheim, 1999 Search PubMed; (b) J. J. Li, D. S. Johnson, D. R. Sliskovic and B. D. Roth, Contemporary Drug Synthesis, Wiley, Hoboken, 2004 CrossRef.
- For selected reports, see: (a) F. P. Bymaster, E. E. Beedle, J. Findlay, P. T. Gallagher, J. H. Krushinski, S. Mitchell, D. W. Robertson, D. C. Thompson, L. Wallace and D. T. Wong, Bioorg. Med. Chem. Lett., 2003, 13, 4477 CrossRef CAS; (b) T. Nakata, Chem. Rev., 2005, 105, 4314 CrossRef CAS; (c) P. Das, M. D. Delost, M. H. Qureshi, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2019, 62, 4265 CrossRef CAS.
- (a) A. Williamson, Justus Liebigs Ann. Chem., 1851, 77, 37 CrossRef CAS; (b) E. Fuhrmann and J. Talbiersky, Org. Process Res. Dev., 2005, 9, 206 CrossRef CAS.
- (a) S. Caron and A. Ghosh, Practical Synthetic Organic Chemistry, Wiley, Hoboken, 2011, 237 CrossRef; (b) A. S. Henderson, S. Medina, J. F. Bower and M. C. Galan, Org. Lett., 2015, 17, 4846 CrossRef CAS.
- For selected reports, see: (a) R. A. Widenhoefer, H. A. Zhong and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 6787 CrossRef CAS; (b) G. Mann and J. F. Hartwig, J. Org. Chem., 1997, 62, 5413 CrossRef CAS; (c) C.-G. Yang and C. He, J. Am. Chem. Soc., 2005, 127, 6966 CrossRef CAS; (d) S. Ueno and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 1928 CrossRef CAS; (e) X. Wu, B. P. Fors and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9943 CrossRef CAS; (f) J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS; (g) S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136 CrossRef CAS; (h) R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 13624 CrossRef CAS; (i) J. Derosa, M. L. O’Duill, M. Holcomb, M. N. Boulous, R. L. Patman, F. Wang, M. Tran-Dubé, I. McAlpine and K. M. Engle, J. Org. Chem., 2018, 83, 3417 CrossRef CAS; (j) L. Yang, H.-H. Lu, C.-H. Lai, G. Li, W. Zhang, R. Cao, F. Liu, C. Wang, J. Xiao and D. Xue, Angew. Chem., Int. Ed., 2020, 59, 12714 CrossRef CAS; (k) X.-Y. Yu, J. Chen, H.-W. Chen, W.-J. Xiao and J.-R. Chen, Org. Lett., 2020, 22, 2333 CrossRef CAS; (l) H. Zhang, P. Ruiz-Castillo, A. W. Schuppe and S. L. Buchwald, Org. Lett., 2020, 22, 5369 CrossRef CAS.
- A recent report on the electrochemical C(sp2)–O bond formation, see: S.-K. Zhang, J. Struwe, L. Hu and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 3178 CrossRef CAS.
- For our previous studies on alkylsilyl peroxides under Cu catalysis, see: (a) R. Sakamoto, S. Sakurai and K. Maruoka, Chem. − Eur. J., 2017, 23, 9030 CrossRef CAS; (b) R. Sakamoto, T. Kato, S. Sakurai and K. Maruoka, Org. Lett., 2018, 20, 1400 CrossRef CAS; (c) S. Sakurai, T. Kato, R. Sakamoto and K. Maruoka, Tetrahedron, 2019, 75, 172 CrossRef CAS; (d) T. Seihara, S. Sakurai, T. Kato, R. Sakamoto and K. Maruoka, Org. Lett., 2019, 21, 2477 CrossRef CAS; (e) S. Sakurai, S. Tsuzuki, R. Sakamoto and K. Maruoka, J. Org. Chem., 2020, 85, 3973 CrossRef CAS; (f) S. Sakurai, A. Matsumoto, T. Kano and K. Maruoka, J. Am. Chem. Soc., 2020, 142, 19017 CrossRef CAS.
- (a) S. Isayama, Bull. Chem. Soc. Jpn., 1990, 63, 1305 CrossRef CAS; (b) T. Tokuyasu, S. Kunikawa, K. J. McCullough, A. Masuyama and M. Nojima, J. Org. Chem., 2005, 70, 251 CrossRef CAS; (c) T. G. Driver, J. R. Harris and K. A. Woerpel, J. Am. Chem. Soc., 2007, 129, 3836 CrossRef CAS; (d) S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912 CrossRef CAS.
- For applications of alkylsilyl peroxides under Fe or Ni catalysis, see: (a) P. Gao, H. Wu, J.-C. Yang and L.-N. Guo, Org. Lett., 2019, 21, 7104 CrossRef CAS; (b) L. Chen, J.-C. Yang, P. Xu, J.-J. Zhang, X.-H. Duan and L.-N. Guo, J. Org. Chem., 2020, 85, 7515 CrossRef CAS; (c) Y. Shiozaki, S. Sakurai, R. Sakamoto, A. Matsumoto and K. Maruoka, Chem. − Asian J., 2020, 15, 573 CrossRef CAS.
- For an example for the use of L1 under transition-metal catalysis, see: L. Li, M. Zeng and S. B. Herzon, Angew. Chem., Int. Ed., 2014, 53, 7892 CrossRef CAS.
- For an example for the use of L3 under transition-metal catalysis, see: A. J. Hickman and M. S. Sanford, ACS Catal., 2011, 1, 170 CrossRef CAS.
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794 CrossRef CAS.
- For selected reports on the Cu/chiral bis(oxazoline) (BOX) catalysis, see: (a) D. Rechavi and M. Lemaire, Chem. Rev., 2002, 102, 3467 CrossRef CAS; (b) J. Thorhauge, M. Roberson, R. G. Hazell and K. A. Jørgensen, Chem. – Eur. J., 1888, 2002, 8 Search PubMed; (c) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561 CrossRef CAS; (d) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2011, 111, PR284 CrossRef CAS; (e) F.-D. Lu, D. Liu, L. Zhu, L.-Q. Lu, Q. Yang, Q.-Q. Zhou, Y. Wei, Y. Lan and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 6167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc07305a |
| This journal is © The Royal Society of Chemistry 2021 |