
Meyer–Schuster-type rearrangement for the synthesis of α-selanyl-α,β-unsaturated thioesters†
Lucas L.
Baldassari
a,
Anderson C.
Mantovani
a,
Micaela
Jardim
a,
Boris
Maryasin
 *bc and
Diogo S.
Lüdtke
*a
*bc and
Diogo S.
Lüdtke
*a
aInstituto de Química, Universidade Federal do Rio Grande do Sul, UFRGS, Av. Bento Gonçalves 9500, 91501-970, Porto Alegre, RS, Brazil. E-mail: dsludtke@iq.ufrgs.br
bInstitute of Organic Chemistry, University of Vienna,
Währinger Straße 38
, 1090 Vienna, Austria. E-mail: boris.maryasin@univie.ac.at
cInstitute of Theoretical Chemistry, University of Vienna, Währinger Straße 17, 1090 Vienna, Austria
First published on 24th November 2020
Abstract
A new approach to prepare α-selanyl-α,β-unsaturated thioesters from propargylthioalkynes and an electrophilic selenium species is reported. Pivotal is the intermediacy of a sulfur-stabilized vinyl cation, which is captured intramolecularly and ultimately enables 37 examples of the target compounds to be prepared in good yields. Computational studies shed light on the nature of intermediates in this transformation.
α,β-Unsaturated thioesters are a class of significant molecules and their importance is highlighted by their use in several areas, such as polymer chemistry,1 enoyl-CoA carboxylase/reductases substrate in enzymatic catalysis,2 and as an intermediate for the synthesis of commercially important fragrances phenoxanol and hydroxycitronellal.3 Indeed, this is a useful synthetic building block, being a common substrate for 1,4-additions,4,5 α-trifluoromethylations,6 cycloadditions,7 hydrogenations,8 and rearrangements.9,10 The vast majority of the methods available for their synthesis involves the use of acryloyl chlorides and thiols, a route that commonly presents issues related to the handling of unstable acryloyl chlorides and competing thiol 1,4-addition side reactions (Scheme 1A).11 Other methodologies are based on the use of carbonyl compounds12,13 epoxides14 and thioacetylenes. Rearrangement of propargyl sulfoxides,15 thiol 1,4-addition,16 olefinations17,18, aldol reaction,19 decomposition of α-sulfinyl-thioesters,20 thiocarbonylation of acetylenes21 and thioesterification of aldehydes with disulfides catalyzed by N-heterocyclic carbenes22 have also been reported. Despite of the available methods, α,β-unsaturated thioesters are on several occasions not easy to synthesize, presenting regioselectivity problems and formation of difficult separation mixtures, leaving its general synthesis still a challenge to synthetic chemists.
 | ||
| Scheme 1 Key precedents and this work. | ||
One elegant method for the synthesis of α,β-unsaturated compounds is the Meyer–Schuster rearrangement of propargyl acetylenes to the corresponding enones (Scheme 1B).23,24
Recently, our research group has been studying the potential use of heteroatom-substituted alkynes atoms in the stabilization of vinyl cations and their reactivity towards a number of different reactions.25 In particular, we have reported the use of selenoalkynes for the synthesis of α-arylated selenol esters in a redox-neutral oxyarylation (Scheme 1C).26 In continuation of our studies, we hypothesized that the concept of vinyl cation stabilization could be extended to propargyl thioalkynes, which would be suitable substrates for electrophilic activation.27 Therefore, an ensuing sulfur-stabilized vinyl cation would result from the reaction of the alkyne with an appropriate soft selenium electrophile (e.g. PhSeX). This intermediate might then be intercepted by an intramolecular nucleophilic attack of oxygen, leading to the formation of an oxetene ring, which would then decompose, delivering the final α-selanyl-α,β-unsaturated thioesters in a Meyer–Schuster-type rearrangement (Scheme 1D). Besides the above-mentioned utility of α,β-unsaturated thioesters organic selenium compounds, including vinyl selenides, have found widespread application in several areas of science, including organic synthesis, material sciences, and pharmacology.28 Therefore, mild methods that are able to deliver functionalized alkenes bearing an organoselenium moiety are desirable.
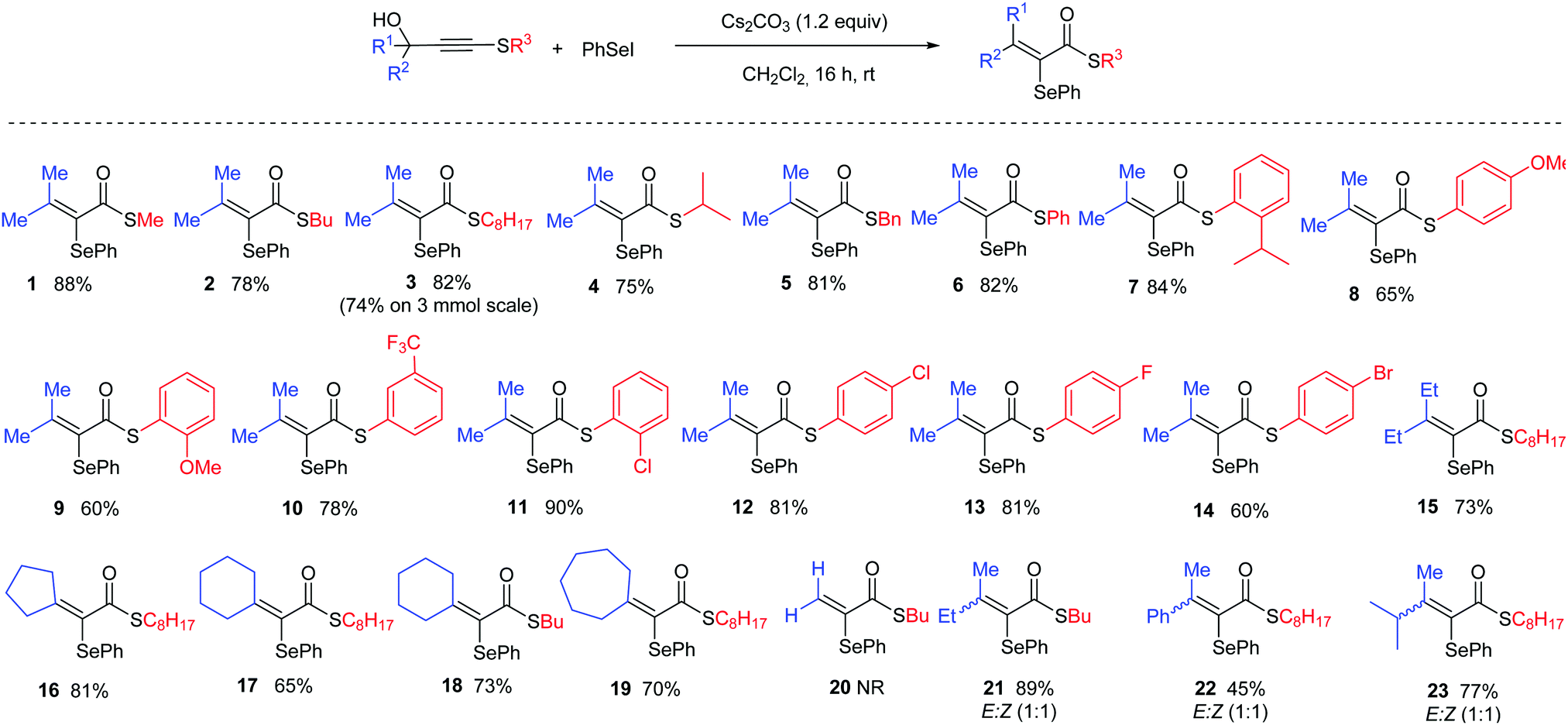
Reducing our plan to practice, we started our studies by reacting the propargyl thioalkyne with an in situ generated selenium electrophile. The results are summarized in Table 1. The first attempt was performed in the absence of base, using 2 equiv. of PhSeI (generated in situ by the reaction of 1 equiv. Ph2Se2 with 1 equiv. I2) as the electrophilic selenium source, and led to the formation of the desired α-selanyl-α,β-unsaturated thioester 1 in 39% yield (entry 1). Subsequently, the influence of the use of base in the reaction was verified. Carrying out the experiment with 2 equiv. of Cs2CO3 resulted in product 1 in 64% yield. Additional studies have revealed that NaH is also a suitable base for promoting the reaction (entry 3). A decrease in the amount of Cs2CO3 to 1.2 equivalents resulted in a slight increase in the yield (entry 4, 70%). A solvent screening was performed and the best yield was obtained using dichloromethane (entries 4–8). Increasing the temperature to 40 °C led to a drop in the yield to 44% (entry 9). Longer reaction times proved beneficial and an increase in the yield was observed by keeping the reaction for 16 h (entry 10). Finally, we have found that changing the stoichiometry from 2 to 4 equiv. of PhSeI led to an increase in the yield of 1 (entries 10–12, 75 to 88% yield). It is worth pointing out that the excess of the selenium compound is recovered at the end of the reaction as Ph2Se2 and reused. Importantly, the PhSeI electrophile is conveniently generated in situ by the reaction of Ph2Se2 with I2, and after 30 minutes, potassium persulfate is added to reduce any remaining molecular iodine and suppress the formation of iodinated side-products. After establishing the optimal conditions for the α,β-unsaturated thioester synthesis 1, we proceeded to examine the reaction scope in respect of the propargylic thioalkynes (Scheme 2). A number of thioacetylenes with different substitution patterns have been studied. Variations at the R3 attached to the sulfur atom have shown that linear alkyl chains of different sizes (1 = methyl; 2 = n-butyl and 3 = octyl), branched alkyl chain (4), benzyl (5) and aromatic groups bearing electron-donating (6–9) and electron-withdrawing groups (10–14) groups have been well tolerated under the reaction conditions. It's worth pointing out that compound 7, bearing a bulky iso-propyl group at the ortho position was obtained in 84% isolated yield.
|
|
|||||
|---|---|---|---|---|---|
| (PhSe)2 (equiv.) | I2 (equiv.) | Base (equiv.) | Solvent, t (h) | Yieldb (%) | |
| a Reaction was performed using 0.25 mmol of thioalkyne, at 25 °C. PhSeI was generated in situ by the reaction of (PhSe)2 with I2, and after 30 min, Na2S2O3 was added to reduce the unreacted I2. b Yields determined by 1H-NMR using mesitylene as internal standard. c Reaction performed at 40 °C. d 18% of unreacted starting material. e Starting material was fully consumed after 16 h. | |||||
| 1 | 1.0 | 1.0 | — | CH2Cl2, 3 | 39 |
| 2 | 1.0 | 1.0 | Cs2CO3 (2.0) | CH2Cl2, 3 | 64 |
| 3 | 1.0 | 1.0 | NaH (2.0) | CH2Cl2, 3 | 61 |
| 4 | 1.0 | 1.0 | Cs2CO3 (1.2) | CH2Cl2, 3 | 70 |
| 5 | 1.0 | 1.0 | Cs2CO3 (1.2) | THF, 3 | NR |
| 6 | 1.0 | 1.0 | Cs2CO3 (1.2) | DCE, 3 | 67 |
| 7 | 1.0 | 1.0 | Cs2CO3 (1.2) | PhCH3, 24 | 40 |
| 8 | 1.0 | 1.0 | Cs2CO3 (1.2) | CH2Cl2/THF, 3 | 29 |
| 9 | 1.0 | 1.0 | Cs2CO3 (1.2) | CH2Cl2, 3 | 44c |
| 10 | 1.0 | 1.0 | Cs2CO3 (1.2) | CH2Cl2, 16 | 75d |
| 11 | 1.5 | 1.5 | Cs2CO3 (1.2) | CH2Cl2, 16 | 80d |
| 12 | 2.0 | 2.0 | Cs 2 CO 3 (1.2) | CH 2 Cl 2 , 16 | 88 |
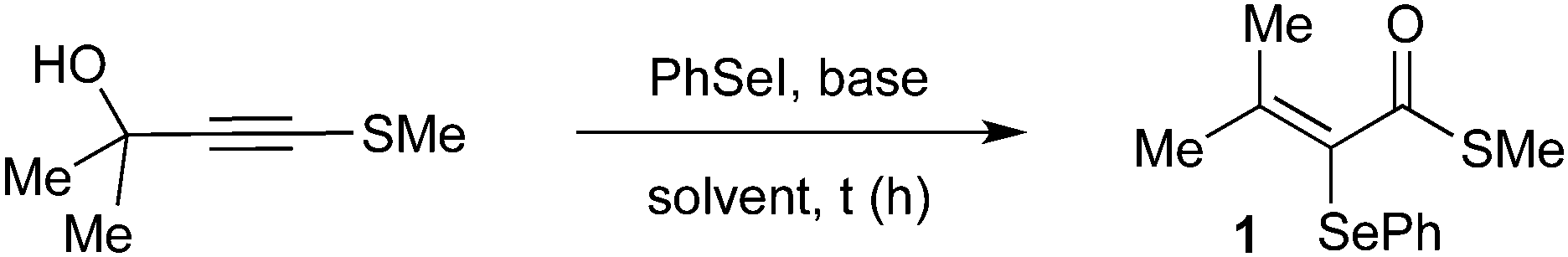
 | ||
| Scheme 2 Scope of thioacetylenes. | ||
In addition, variations at the R1 and R2 groups were also examined. When R1 and R2 = ethyl, the corresponding thioester 15 was obtained in 73% yield. Cyclic moieties have also been well tolerated and the corresponding products bearing 5-, 6- and 7-membered rings (16–19) have been obtained in good yields. Unfortunately, when unsubstituted propargyl thioacetylene was used, product 20 was not observed, despite full consumption of the starting thioalkyne. In order to study the selectivity of the reaction, compounds bearing different R1 and R2 substituents have been evaluated. In these cases, albeit products 21, 22 and 23 have been isolated in good yields, unfortunately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z mixtures of have been obtained. Importantly, the reaction is amenable to scale-up and when the reaction was performed on a 3 mmol scale only a small decrease in the isolated yield for product 3 was observed (82% on 0.25 mmol to 74% on a 3 mmol scale).
:1 E/Z mixtures of have been obtained. Importantly, the reaction is amenable to scale-up and when the reaction was performed on a 3 mmol scale only a small decrease in the isolated yield for product 3 was observed (82% on 0.25 mmol to 74% on a 3 mmol scale).
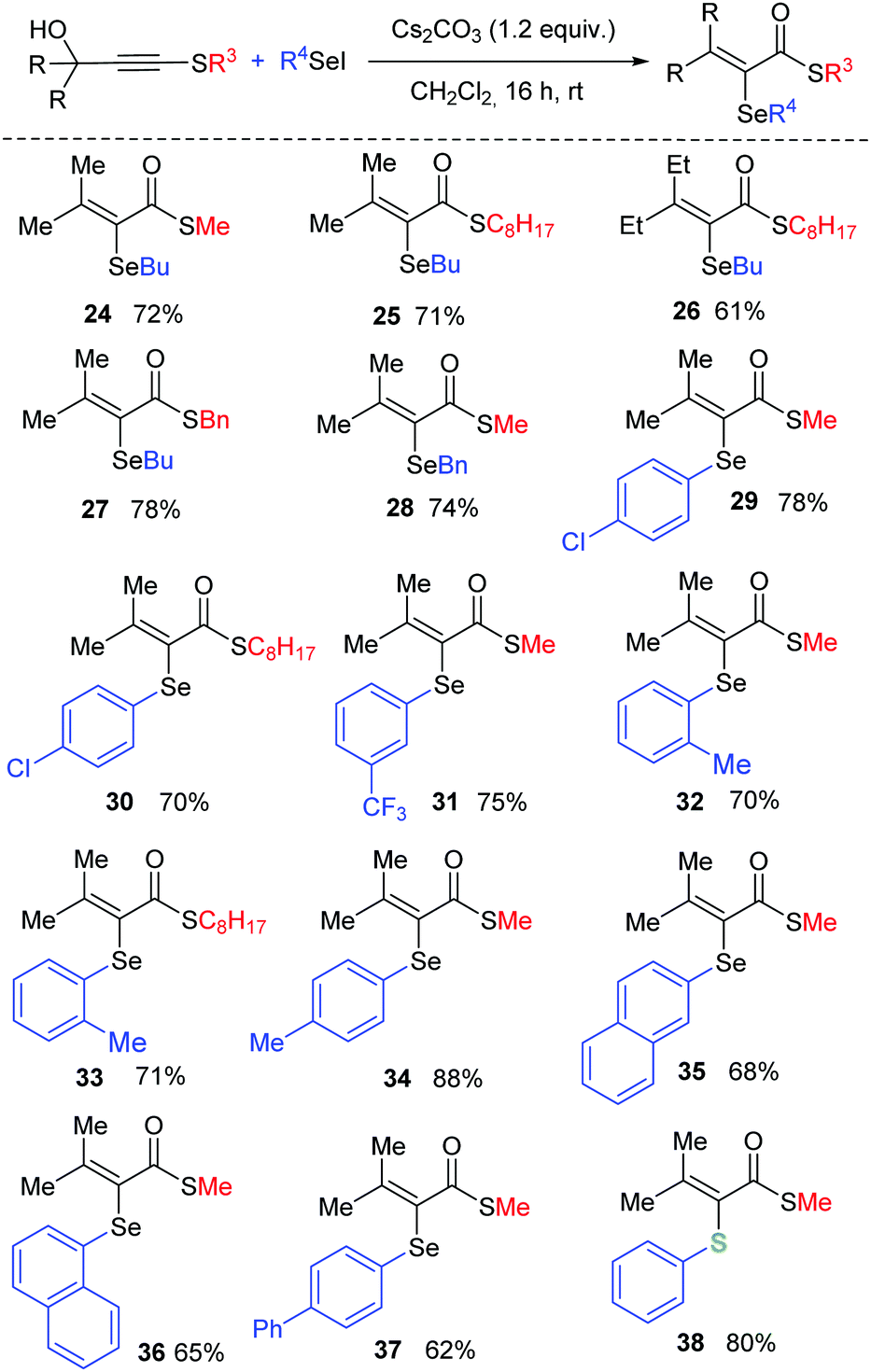
Aiming to study the versatility in the synthesis of α,β-unsaturated thioesters, we then turned our attention to the scope of the reaction with respect to the substitution pattern on selenium (Scheme 3). When R4 group is n-butyl, the corresponding products 24–27 were isolated in good yields, for different substitution at the remaining positions of the molecule. An electrophilic Se species bearing a benzyl group was also tested, delivering product 28 in 74% yield. Diverse arylseleno derivatives are competent activators and both electron-donating and electron-withdrawing groups are suitable substituents (29–38). For example, products bearing p-Cl (29 and 30), m-CF3 (31), o-Me (32–33), p-Me (34), 2-naphthyl (35), 1-naphthyl (36) and p-Ph (37) were successfully obtained. Finally, the reaction is also amenable to the use of a sulfur electrophile and product 38 was isolated in 80% yield, highlighting the versatility of the method for the synthesis of α,β-unsaturated thioesters with different chalcogen atoms at the alpha position.
 | ||
| Scheme 3 Scope of diselenides. | ||
In order to get additional information on the reaction pathway, quantum chemical calculations have been performed (Fig. 1). The first step A → B is the SN2 type attack of phenylselenyl iodide on the alkyne with the formation of a ketenethionium intermediate B. The next stage is the cyclisation of B to generate the oxetene intermediate C. The oxetene ring-opens via cleavage of the C–O bond forming the intermediate D. This step, C → D, is highly exergonic (ΔG(C → D) = −27.4 kcal mol−1) and thus it compensates the endergonic formation of intermediate B (ΔG(A → B) = +15.2 kcal mol−1) at the first stage of the reaction pathway. Finally, D is deprotonated by the base, yielding the final product. The whole process is both thermodynamically (ΔG(A → D) = −17.2 kcal mol−1) and kinetically favorable (the highest transition state TSB–C is only 17.8 kcal mol−1 less stable energetically compared to the sum of the reactants A). Therefore, the computed mechanism suggests that the reaction can readily proceed at room temperature, in accordance with the experimental evidence.
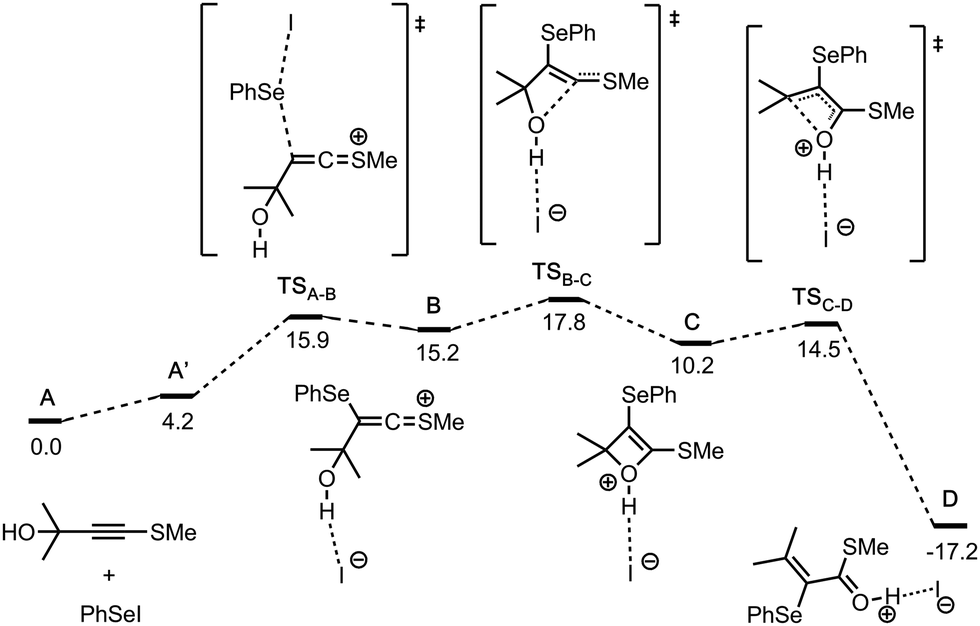 | ||
| Fig. 1 Computed relative free energy profile (DLPNO-CCSD(T)/def2-TZVP//B3LYP-D3/def2-SVP, ΔG298,DCM, kcal mol−1) for the conversion of the reactants A (taken as a reference 0.0 kcal mol−1) to the final intermediate D. | ||
Finally, as an application, we have performed a derivatization of the thioester products by a Fukuyama coupling reaction29 with Et2Zn, smoothly delivering the corresponding ketone in moderate yield (Scheme 4).
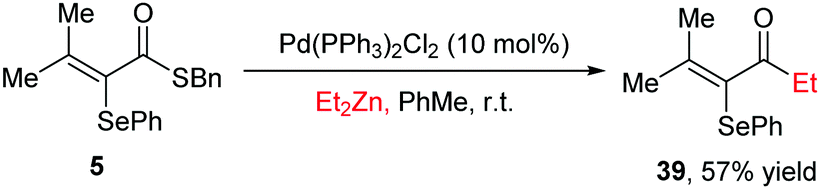 | ||
| Scheme 4 Derivatization of the product. | ||
In summary, we have presented a new methodology for the synthesis of a broad range of α-selanyl-α,β-unsaturated thioesters by a Meyer–Schuster-type rearrangement of propargylic thioalkynes and selenium electrophilic species. The reaction proceeds through the initial activation of the thioalkyne with a soft selenium electrophile, leading to a sulfur-stabilized vinyl cation, which undergoes an intramolecular attack, forming an oxetene intermediate, which in turn, decomposes to the final product. Support for the mechanism was obtained from quantum chemical calculations underlining the kinetic and thermodynamic favored nature of the process.
We thank CNPq, CAPES, and INCT-Catálise for financial support. Calculations were partially performed at the Vienna Scientific Cluster (VSC). Profs. Nuno Maulide and L. González (Univ. Vienna) are gratefully acknowledged for support and helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Aksakal and C. R. Becer, Polym. Chem., 2016, 7, 7011 RSC
.
-
(a) D. M. Peter, L. S. Borzyskowski, P. Kiefer, P. Christen, J. A. Vorholt and T. J. Erb, Angew. Chem., Int. Ed., 2015, 54, 13457 CrossRef CAS
- Z. Gao and S. P. Fletcher, Chem. Commun., 2017, 53, 10216 RSC
-
(a) L. Marzorati, M. C. de Mattos, B. Wladislaw and C. Di Vitta, Synth. Commun., 2002, 32, 1427 CrossRef CAS
-
(a) F. López, A. J. Minnaard and B. L. Feringa, Acc. Chem. Res., 2007, 40, 179 CrossRef
- Z. Fang, Y. Ning, P. Mi, P. Liao and X. Bi, Org. Lett., 2014, 16, 1522 CrossRef CAS
-
(a) F. F. Camilo and J. Gruber, Synth. Commun., 2012, 42, 394 CrossRef CAS
-
(a) N. Li, J. Ou, M. Miesch and P. Chiu, Org. Biomol. Chem., 2011, 9, 6143 RSC
-
(a) H. Lu, J.-Y. Zhang, J.-Y. Liu, H.-Y. Li and P.-F. Xu, ACS Catal., 2017, 7, 7797 CrossRef CAS
-
(a) T. Niwa, H. Ochiai, M. Isoda and T. Hosoya, Chem. Lett., 2017, 46, 1315 CrossRef CAS
-
(a) B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522 CrossRef
- L. B. Bos and J. F. Arens, Rec. Trav. Chim., 1963, 82, 157 CrossRef CAS
-
(a) M. P. C. Mulder, P. Fodran, J. Kemmink, E. J. Breukink, J. A. W. Kruijtzer, A. J. Minnaard and R. M. J. Liskamp, Org. Biomol. Chem., 2012, 10, 7491 RSC
- H. Zhu, W. Jin, J. He, Y. Zhang and G. Zhu, Adv. Synth. Catal., 2016, 358, 3730 CrossRef CAS
- R. Zheng, Y. Wang and L. Zhang, Tetrahedron Lett., 2015, 56, 3144 CrossRef CAS
- A. R. Mohite, T. B. Mete and R. G. Bhat, Tetrahedron Lett., 2017, 58, 770 CrossRef CAS
- E. Schaumann, B. Mergardt and S. Fittkau, Synthesis, 1990, 47 CrossRef CAS
- G. E. Keck, E. P. Boden and S. A. Mabury, J. Org. Chem., 1985, 50, 709 CrossRef CAS
- M. Sugiura, Y. Ashikari and M. Nakajima, J. Org. Chem., 2015, 80, 8830 CrossRef CAS
- B. Wladislaw, L. Marzorati and J. Gruber, Synth. Commun., 1990, 20, 2937 CrossRef CAS
- A. Ogawa, J.-I. Kawakami, M. Mihara, T. Ikeda, N. Sonoda and T. Hirao, J. Am. Chem. Soc., 1997, 119, 12380 CrossRef CAS
- S. Singh and L. D. S. Yadav, Tetrahedron Lett., 2012, 53, 5136 CrossRef CAS
-
(a) S. Puri, N. Thirupathi and M. S. Reddy, Org. Lett., 2014, 16, 5246 CrossRef CAS
- A few, low yielding examples of PPSE-catalyzed synthesis of α,β-unsaturated thioesters have been reported. M. Yoshimatsu, M. Naito, M. Kawahigashi, H. Shimizu and T. Kataoka, J. Org. Chem., 1995, 60, 4798 CrossRef CAS
- L. L. Baldassari, A. de la Torre, J. Li, D. S. Lüdtke and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 15723 CrossRef CAS
- L. L. Baldassari, A. C. Mantovani, S. Senoner, B. Maryasin, N. Maulide and D. S. Lüdtke, Org. Lett., 2018, 20, 5881 CrossRef CAS
-
(a) L.-G. Xie, S. Niyomchon, A. J. Mota, L. González and N. Maulide, Nat. Commun., 2016, 7, 10914 CrossRef
-
(a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS
- H. Tokuyama, S. Yokoshima, T. Yamashita and T. Fukuyama, Tetrahedron Lett., 1998, 39, 3189 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, copies of NMR spectra, and computational details. See DOI: 10.1039/d0cc07019j |
| This journal is © The Royal Society of Chemistry 2021 |