
Palladium catalyzed annulation of 2-iodobiphenyl with a non-terminal alkene enabled by neighboring group assistance†
Bo-Cheng
Tang
a,
Cai
He
a,
Xiang-Long
Chen
a,
Jin-Tian
Ma
a,
Miao
Wang
b,
Yan-Dong
Wu
*a and
An-Xin
Wu
 *a
*a
aKey Laboratory of Pesticide & Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China. E-mail: chwuyd@mail.ccnu.edu.cn; chwuax@mail.ccnu.edu.cn
bHenan Key Laboratory of Function-Oriented Porous Materials, College of Chemistry and Chemical Engineering, Luoyang Normal University, Luoyang 471934, P. R. China
First published on 24th November 2020
Abstract
In this work, an efficient palladium catalyzed annulation of 2-iodobiphenyl with a non-terminal alkene was developed. The key factor in this transformation was the formation of a highly reactive oxo-palladacycle intermediate, which was enabled by a neighboring hydroxyl group, and remarkably restrained the β-H elimination process. Mechanistically, control experiments demonstrated that the hydroxyl group may act as an anionic ligand, which was irreplaceable in this reaction. This transformation presented good reactivity and selectivity, and no simple Heck coupling products were detected for all of the explored substrates.
2-Iodobiphenyl is a building block that has great research value in the field of transition-metal catalyzed reactions, particularly in palladium chemistry, for the synthesis of diverse and complex compounds.1 One important reason for its good reactivity is the formation of dibenzopalladacyclopentadiene, a key intermediate that is obtained via C–H activation of 2-iodobiphenyl with a palladium catalyst.2 Notably, two carbon–palladium bonds can be functionalized in this highly reactive complex. Therefore, various novel transformations can be realized by combining dibenzopalladacyclopentadiene with other appropriate substrates (Scheme 1).3 Many notable reports have been published by Zhang and co-workers in this field. For instance, an efficient disilylation of 2-iodobiphenyl was reported by Zhang and Liang in 2018,4 as well as a synthesis of carbazoles using diaziridinone as the N1 source.5 Additionally, Larock et al. reported the annulation of 2-iodobiphenyl with alkynes and arynes (Scheme 2a).6 Comparatively, the annulation of 2-iodobiphenyl with alkenes was not feasible because of the inevitable β-H elimination process. In this case, mono-functionalization products of 2-iodobiphenyl with the alkene via a Heck-type coupling are obtained (Scheme 2b and c).7 Therefore, realizing the [4+2] annulation of 2-iodobiphenyl with an alkene-containing compound is a difficult task. The key to solving this issue is avoiding the potential β-H elimination process. In previous reports, a typical solution was to introduce a blocking group at the β-C position of the alkenes.8 Additionally, special alkenes such as Norbornene (NBE) derivatives could also restrain the β-H elimination process.9 However, because of the diversity of the alkene family, the development of more efficient and specific solutions to suppress the β-H elimination of alkenes during the transition-metal catalyzed annulation process is necessary.
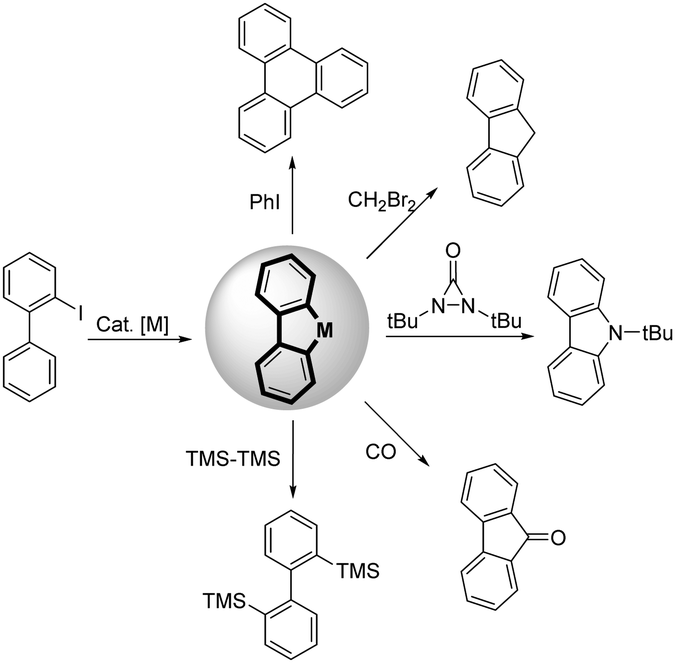 | ||
| Scheme 1 The typical reactions of 2-iodobiphenyl. | ||
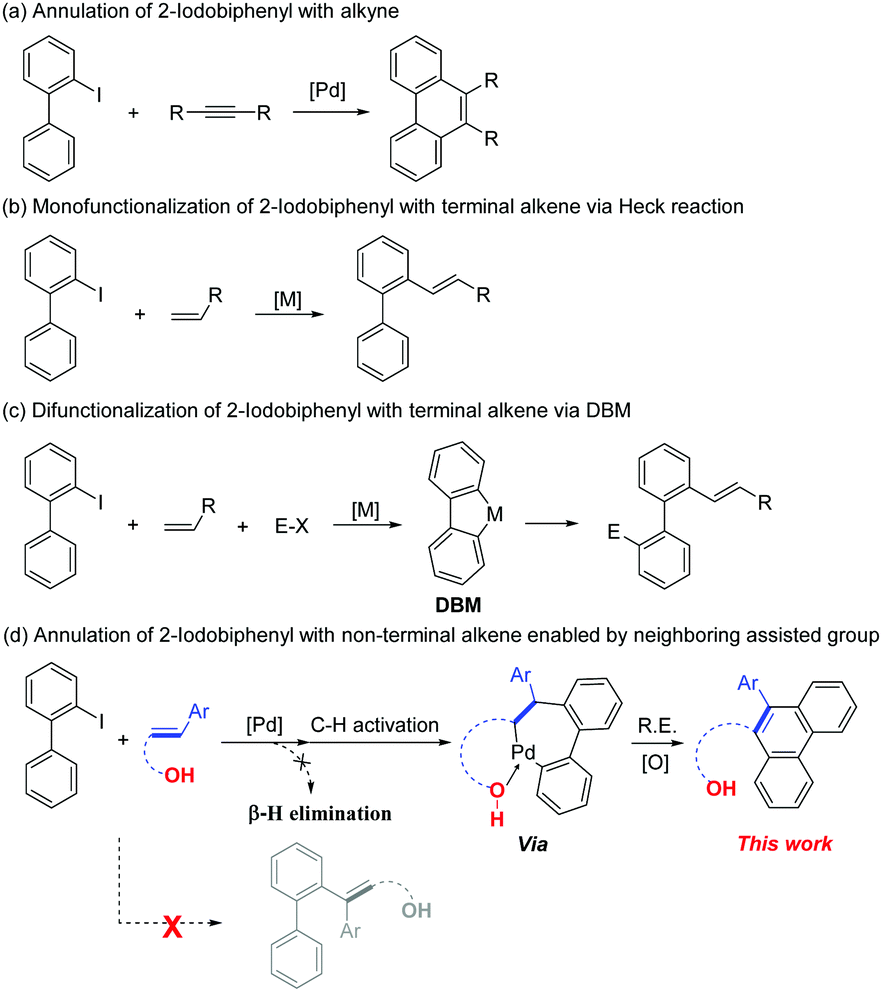 | ||
| Scheme 2 Functionalization of 2-iodobiphenyl with alkyne and alkene. | ||
The strategy of neighboring group assistance is an important method for exploiting the new reaction. These assistance groups can interact with potential reactive intermediate, to suppress the inherent reaction process and thereby to create a new reaction pathway.10 For instance, in the transition-metal-catalyzed difunctionalization of alkenes, the pyridine-containing groups, such as 8-aminoquinoline, are usually installed on the substrate to suppress the potential β-H elimination process.11 Comparatively, we hope to avoid the further installation of the assisted group by using a simpler substrate, which vary the reaction pathway by the inherent functional group of the substrate. In this regard, we envisaged that the hydroxyl group of the phenolic compounds would present a significant difference. Inspired by these works12 and our previous efforts13 on phenolic compounds, as well as their important biological and chemical properties.14 Here we report a palladium catalyzed annulation of 2-iodobiphenyl with non-terminal alkene enabled by neighboring group assistance (Scheme 2d).
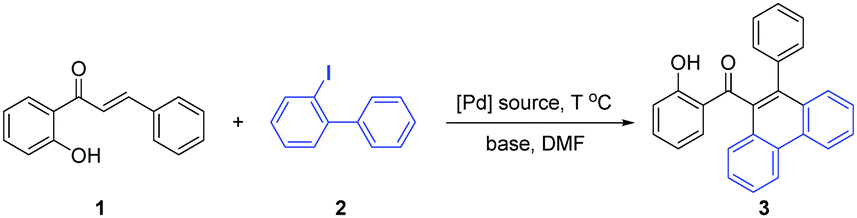
Initially, we focused on the model reaction of 2′-OH chalcone (1) and 2-iodobiphenyl (2) with Na2CO3 and Pd(PPh3)4 in DMF at 100 °C. Pleasingly, this reaction afforded compound 3 in 37% yield (Table 1, entry 1). Encouraged by this promising result, we then systematically screened the reaction conditions to improve the yield of 3. First, different palladium catalysts were examined, such as Pd2(dba)3, Pd(OAc)2, Pd(TFA)2, PdCl2 and K2PdCl6, and the results suggested that Pd(OAc)2 was the best palladium catalyst for this reaction (entries 2–6). Next, a series of bases were also investigated to further improve the yield. However, no positive results were obtained neither inorganic bases (entries 7–11) nor organic base were used (entries 12–13). Subsequently, various temperatures were screened, showing that 100 °C was the optimal choice (entries 14–16). It should be noted that while the yield of 3 did not decline significantly at 90 °C, it was suboptimal in additional tests of the substrate scope. Additionally, the necessity of water was evaluated (entries 17–19), with the results showing that a trace-amount of water is necessary in the solvent for this reaction. Finally, the 1 mmol scale reaction was performed in the standard condition, and the yield was slightly decreased. (entry 20).
|
|
||||
|---|---|---|---|---|
| Entry | [Pd] source | Base | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: 1 (0.1 mmol), 2 (1.5 equiv.), base (2.0 equiv.) and Pd source (5 mmol%) at T °C in DMF (1.0 mL) under Ar for 12 h. b Isolated yields based on 1. c dry DMF was used. d Extra H2O (2.0 equiv.). e Extra H2O (5.0 equiv.). f 1 mmol scale. | ||||
| 1 | Pd(PPh3)4 | Na2CO3 | 100 | 37 |
| 2 | Pd2(dba)3 | Na2CO3 | 100 | 25 |
| 3 | Pd(OAc) 2 | Na 2 CO 3 | 100 | 76 |
| 4 | Pd(TFA)2 | Na2CO3 | 100 | 66 |
| 5 | PdCl2 | Na2CO3 | 100 | 52 |
| 6 | K2PdCl6 | Na2CO3 | 100 | 40 |
| 7 | Pd(OAc)2 | K2CO3 | 100 | 71 |
| 8 | Pd(OAc)2 | Cs2CO3 | 100 | 30 |
| 9 | Pd(OAc)2 | NaHCO3 | 100 | 55 |
| 10 | Pd(OAc)2 | K3PO4 | 100 | 69 |
| 11 | Pd(OAc)2 | tBuOK | 100 | 68 |
| 12 | Pd(OAc)2 | Et3N | 100 | 22 |
| 13 | Pd(OAc)2 | DBU | 100 | Trace |
| 14 | Pd(OAc)2 | Na2CO3 | 90 | 72 |
| 15 | Pd(OAc)2 | Na2CO3 | 70 | 32 |
| 16 | Pd(OAc)2 | Na2CO3 | 50 | n.d. |
| 17c | Pd(OAc)2 | Na2CO3 | 100 | 29 |
| 18d | Pd(OAc)2 | Na2CO3 | 100 | 75 |
| 19e | Pd(OAc)2 | Na2CO3 | 100 | 58 |
| 20f | Pd(OAc)2 | Na2CO3 | 100 | 70 |
We then investigated the substrate scope of this reaction (Scheme 3). Substitutions of the alkene moiety were examined first. The reaction was applicable for various alkene substrates. As shown in Scheme 3, alkyl- and halogen substituents at the R1 moiety were well-tolerated, delivering corresponding products in moderate to excellent yields (46–85%, 3–11). Next, the substituents at the R2 moiety were evaluated. As expected, the substrates bearing an electron-donating (–Me, –OMe, –OEt), electron-withdrawing (–F, –Cl, –Br, –CF3), and multi-substituted groups all reacted well and afforded the corresponding compounds in moderate to excellent yields (43–86%, 12–24). Additionally, heterocyclic groups such as 2-thienyl- and 2-furyl groups were well-tolerated (41–74%, 25, 26). Nevertheless, fused-ring alkene substrates were not compatible with this reaction and afforded an inseparable complex mixture. The exact structure of product 5 was verified by X-ray single crystal diffraction analysis.
 | ||
| Scheme 3 Substrate scope.a,b.aReaction conditions: alkene (0.1 mmol), aryl iodide (1.5 equiv.) Na2CO3 (2.0 equiv.) and Pd(OAc) (5 mmol%) at 100 °C in DMF (1.0 mL) under Ar for 12 h. bIsolated yields based on alkene. cPartially separable. r.r. = regioisomeric ratio. | ||
Encouraged by these results, we extended this transformation from alkene derivatives to aryl iodide substrates. Nevertheless, an inseparable complex mixture was obtained for many of the variations at the aryl iodide; this phenomenon may be partially attributed to the potential intramolecular 1,4-palladium shift of 2-iodobiaryls, which produced positional isomers with an approximately equivalent ratio.7 To our satisfaction, for the R3 groups, alkyl-, alkoxyl-, halogen-, and electron-withdrawing substituents (–CF3, –OCF3) at the para-, and meta-positions reacted smoothly with good selectivity to afford the corresponding products in moderate yield (43–72%, 27–32). However, the reaction was quite sensitive to the aryl iodide containing ortho-substituted groups, which resulted in a complex mixture or no reaction, probably because of the steric effect.
To gain insight into the reaction mechanism, several control experiments were taken into consideration (Scheme 4). First, no reaction occurred when the hydroxyl group was removed (Scheme 4a-1) or replaced by –OMe group (Scheme 4a-2). Moreover, a complex mixture was observed for 2′-aminochalcone (Scheme 4a-3), and no reaction occurred when pyridyl was used as an alternative assisted group (Scheme 4b). These results indicated that the hydroxyl group maybe essential in this transformation. To explore the potential coordination mode between the hydroxyl group and the palladium center, the cesium salt of 1 was synthesized to test the reaction. Interestingly, 65% yield of 3 was obtained under standard conditions (Scheme 4a-4). Similar to the model reaction (Table 1, entry 17), the yield of 3 also clearly decreased when using dry-DMF (Scheme 4c). These results suggested that a trace-amount of water is important and that hydroxyl groups may act as an anionic ligand in this reaction.
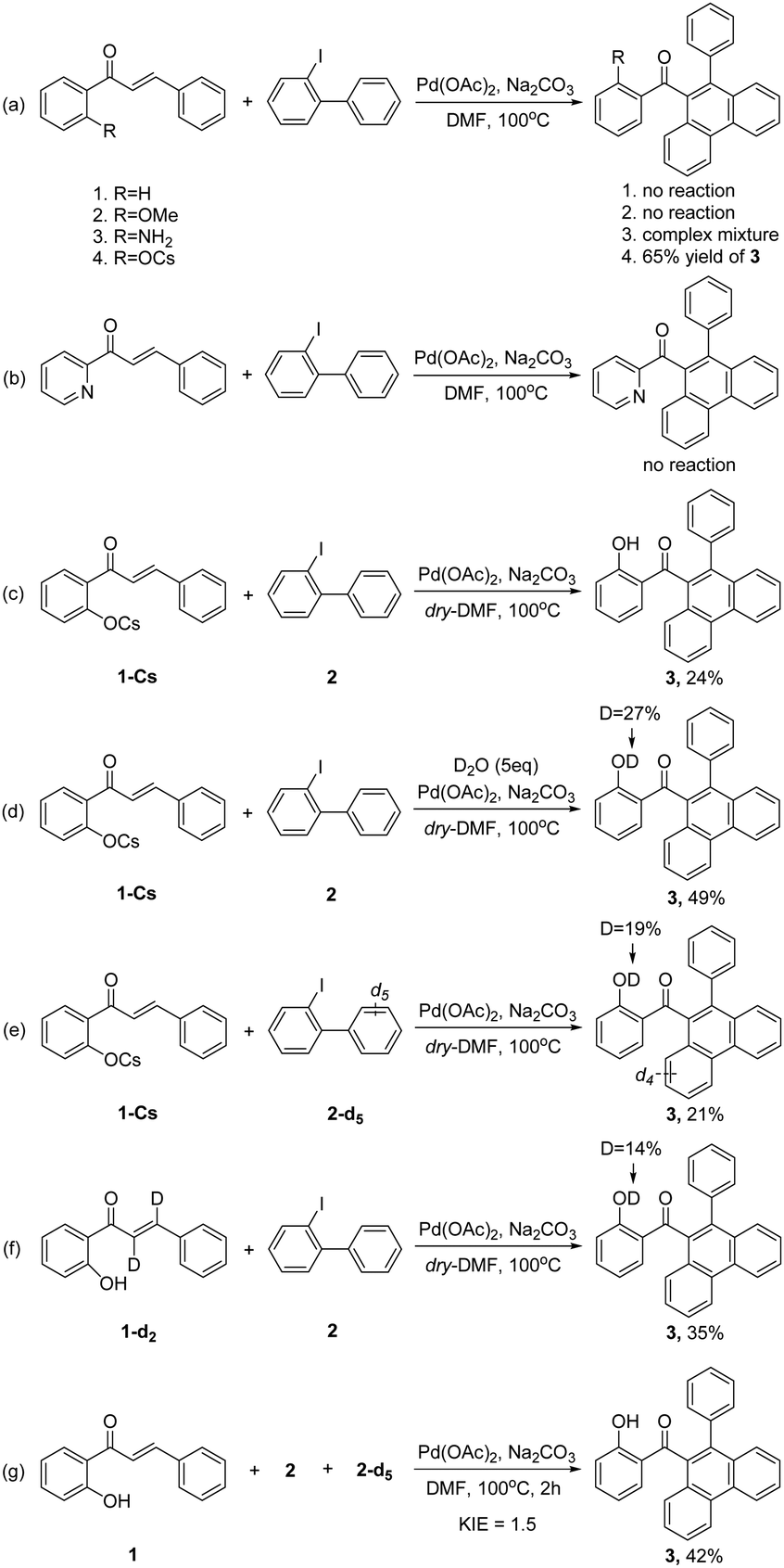 | ||
| Scheme 4 Control experiments. | ||
To further understand the origin of the proton of the final hydroxyl group, several isotopic labelling experiments were conducted (Scheme 4d–f). The combined experimental results suggested that the final proton may originate from various moieties and that a specific hydrogen atom transfer is probably not involved in this reaction. Finally, KIE study from intermolecular competition conditions using 2 and 2-d5 gave kH/kD = 1.5, indicating that C–H bond cleavage is probably not a turnover-limiting step (Scheme 4g).
A plausible mechanism for this hydroxyl dominated annulation between alkene and 2-iodobiphenyl is proposed based on the results of the control experiments and previous reports of Saegusa-Ito oxidation15 (Scheme 5). Initially, the oxidative addition from the aryl iodide to Pd(0) gives Pd(II) complex, which then coordinates with 1 to form intermediate A. A undergoes a migratory insertion to affords intermediate B. The β-H elimination process for B is not feasible because cyclizing with a hydroxyl delivers a thermodynamically more stable intermediate C, with the extrusion of the HI. Then, an anion exchange, followed by a concerted metalation-deprotonation leads to intermediate D. Reductive elimination of D affords complex E. Oxidative addition from hydroxyl to Pd(0) gives intermediate F, which undergoes a sequential transmetalation and deprotonation delivers intermediated G. Further ligand exchange gives complex H. Finally, sequential β-H elimination and protonation afford the product 3, with the release of Pd(0) to complete the catalytic cycle (the amount of biphenyl detected by GC-MS is less than the amount of product. Other possible dehydrogenation processes cannot be excluded).
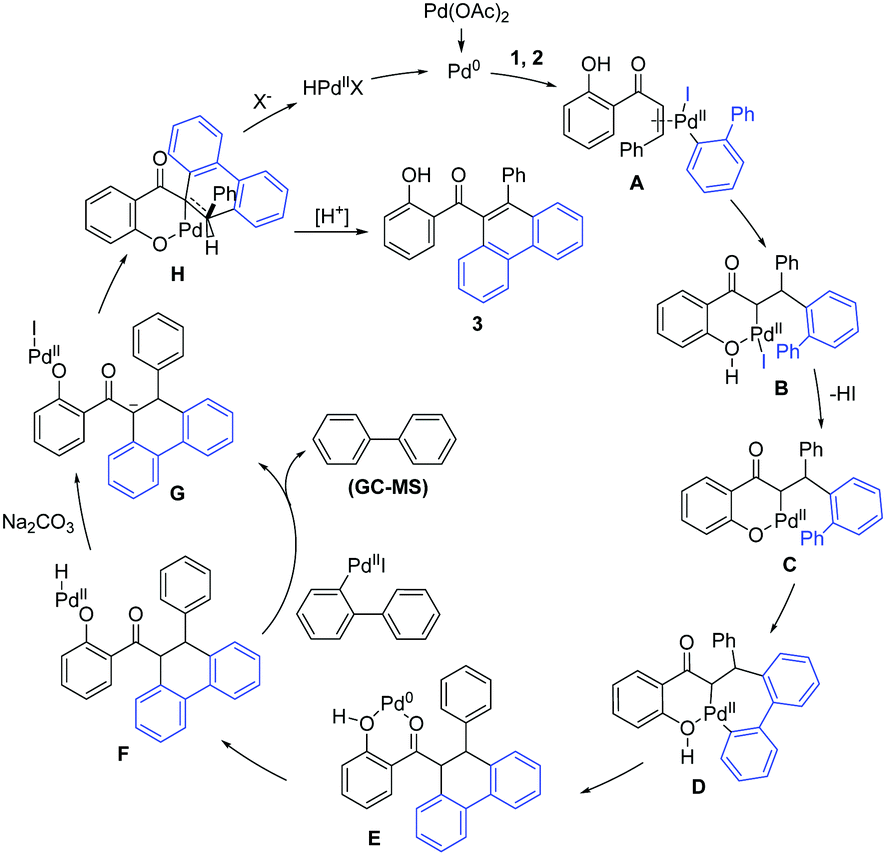 | ||
| Scheme 5 Proposed mechanism. | ||
In conclusion, we developed a palladium catalyzed annulation of 2-iodobiphenyl with non-terminal alkene using a neighboring group assistance strategy. The formation of the key six-membered oxo-palladacycle guided this transformation from a classic Heck coupling to a highly selective C–H activation. This transformation provides a potential strategy for annulation between aryl halide and alkene via an oxo-palladacycle intermediate. Further studies toward possible expansion to other alkene-derived substrates using this strategy are currently underway.
We are grateful to the National Natural Science Foundation of China (Grant No. 21971080, 21971079, 21602070 and 21772051) for financial support. This work was also supported by the 111 Project B17019.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Nishino, Y. Ogiwara and N. Sakai, Eur. J. Org. Chem., 2017, 5892 CrossRef CAS; (b) H. Konishi, S. Futamata and K. Manabe, Adv. Synth. Catal., 2018, 360, 1805 CrossRef CAS; (c) X. Wu, J. Han and L. Wang, J. Org. Chem., 2018, 83, 49 CrossRef CAS; (d) J. Bortoluzzi, V. Jha and F. Leroux, J. Org. Chem., 2018, 83, 7751 CrossRef CAS.
- (a) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950 CrossRef CAS; (b) B. L. Edelbach, R. J. Lachicotte and W. D. Jones, Organometallics, 1999, 18, 4660 CrossRef CAS; (c) N. Simhai, C. N. Iverson, B. L. Edelbach and W. D. Jones, Organometallics, 2001, 20, 2759 CrossRef CAS; (d) D. Masselot, J. P. Charmant and T. Gallagher, J. Am. Chem. Soc., 2006, 128, 694 CrossRef CAS; (e) G. Shi, D. Chen and Y. H. Zhang, Org. Lett., 2016, 18, 2958 CrossRef CAS.
- (a) T. Matsuda and H. Kirikae, Organometallics, 2011, 30, 3923 CrossRef CAS; (b) M. Campo and R. Larock, J. Org. Chem., 2002, 67, 5616 CrossRef CAS; (c) M. Tobisu, S. Imoto and N. Chatani, J. Org. Chem., 2010, 75, 4835 CrossRef CAS; (d) M. Iwasaki, Y. Araki and Y. Nishihara, J. Org. Chem., 2015, 80, 9247 CrossRef CAS; (e) S. Pan, H. Jiang and Y. H. Zhang, Org. Lett., 2016, 18, 5192 CrossRef CAS; (f) H. Jiang, Y. Zhang and Y. H. Zhang, Org. Lett., 2016, 18, 2032 CrossRef CAS; (g) Y. Ou and N. Jiao, Chem. Commun., 2013, 49, 3473 RSC.
- (a) W. Li, G. Xiao and Y. Liang, Org. Chem. Front., 2018, 5, 1488 RSC; (b) A. Lu, X. Ji and Y. H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3233 CrossRef CAS.
- C. Shao, B. Zhou and Y. H. Zhang, Adv. Synth. Catal., 2018, 360, 887 CrossRef CAS.
- (a) R. Larock, M. Doty and Q. Tian, J. Org. Chem., 1997, 62, 7536 CrossRef CAS; (b) Z. Liu, X. Zhang and R. Larock, J. Am. Chem. Soc., 2005, 127, 15716 CrossRef CAS; (c) K. Kanno, Y. Liu and T. Takahashi, Org. Lett., 2005, 7, 5453 CrossRef CAS; (d) Z. Liu and R. Larock, J. Org. Chem., 2007, 72, 223 CrossRef CAS.
- (a) M. Campo and R. Larock, J. Am. Chem. Soc., 2002, 124, 14326 CrossRef CAS; (b) Q. Huang, M. Campo and R. Larock, J. Org. Chem., 2004, 69, 8251 CrossRef CAS; (c) M. Campo, W. Jenks and R. Larock, J. Am. Chem. Soc., 2007, 129, 6298 CrossRef CAS.
- (a) D. Petrone, A. Yen and M. Lautens, Org. Lett., 2015, 17, 4838 CrossRef CAS; (b) R. Liu, T. Xu and Y. Jia, Chem. Commun., 2016, 52, 13664 RSC; (c) S. Chen, H. Jing and Y. Liang, Org. Lett., 2016, 18, 4016 CrossRef CAS; (d) X. Xu, F. Wang and B. Yin, Chem. Commun., 2017, 53, 7796 RSC.
- (a) M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119 CrossRef CAS; (b) Y. Xu, Y. Liang and Y. Yang, J. Org. Chem., 2018, 83, 13930 CrossRef CAS.
- X. Geng, C. Wang and A. X. Wu, Org. Lett., 2020, 22, 140 CrossRef CAS.
- (a) Z. Liu, T. Zeng and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 15122 CrossRef CAS; (b) C. Wang, G. Xiao and T. Loh, J. Am. Chem. Soc., 2018, 140, 9332 CrossRef CAS.
- (a) K. M. Engle, T. S. Mei and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS; (b) G. Li, D. Leow and J. Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS; (c) C. Zhang, J. Ji and P. P. Sun, J. Org. Chem., 2014, 79, 3200 CrossRef CAS.
- (a) J. C. Xiang, Y. D. Wu and A. X. Wu, Org. Lett., 2016, 18, 4360 CrossRef CAS; (b) B. C. Tang, Y. D. Wu and A. X. Wu, Adv. Synth. Catal., 2018, 360, 4023 CrossRef CAS; (c) M. Wang, Y. D. Wu and A. X. Wu, J. Org. Chem., 2018, 83, 3409 CrossRef CAS.
- (a) M. M. Koganov, O. V. Dueva and B. L. Tsorin, J. Nat. Prod., 1999, 62, 481 CrossRef CAS; (b) R. A. Davis, C. T. Supuran and S. A. Poulsen, J. Med. Chem., 2011, 54, 1682 CrossRef CAS.
- (a) Y. Ito, T. Hirao and T. Saegusa, J. Org. Chem., 1978, 43, 1011 CrossRef CAS; (b) Y. Lu, P. L. Nguyen and H. Lebel, J. Org. Chem., 2013, 78, 776 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2024940. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc07133a |
| This journal is © The Royal Society of Chemistry 2021 |