
Hydrates of adenosine 3′,5′-cyclic monophosphate sodium and their transformation†
Pengpeng
Yang
 a,
Junyang
Jin
a,
Qingshi
Wen
b,
Chenguang
Lin
a,
JinQiu
Fu
c,
Wei
Zhuang
a,
Jinglan
Wu
a,
Dong
Liu
*a,
Chenjie
Zhu
*a and
Hanjie
Ying
a
a,
Junyang
Jin
a,
Qingshi
Wen
b,
Chenguang
Lin
a,
JinQiu
Fu
c,
Wei
Zhuang
a,
Jinglan
Wu
a,
Dong
Liu
*a,
Chenjie
Zhu
*a and
Hanjie
Ying
a
aNational Engineering Technique Research Center for Biotechnology, State Key Laboratory of Materials-Oriented Chemical Engineering, College of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, Nanjing 210009, China. E-mail: liudong@njtech.edu.cn; zhucj@njtech.edu.cn
bIndustrial Biotechnology Institute of Jiangsu Industrial Technology Research Institute, Nanjing, China
cSchool of Chemical Engineering, Zhengzhou University, Zhengzhou 450001, China
First published on 15th October 2020
Abstract
Almost all salt forms of nucleotides exist as crystalline hydrates. In this study, the hydrates of adenosine-3′,5′-cyclic phosphate sodium (cAMPNa·nH2O, n = 2, 4, 5, including its methanol solvate of n = 3) were systematically investigated for the crystal structures, their humidity sensibility, the transformation behaviors mediated by humidity, and the relationship between crystal water behaviors and specific structures. It was found that there are some major differences in crystal morphologies, molecular conformation, bonding profiles, and crystal water distribution in the lattices of different hydrates. The difference in the desorption dynamics of crystal water for the different hydrates was mainly attributed to their steric effects in the crystal lattice despite the relation to the interaction of water molecules with sodium ions and cAMP anions. The reversible conversions between the hydrates and their corresponding low hydrates or even anhydrates occur along with desorption/uptake of crystal water mediated by humidity, yet the reversible transformation capability would be disabled once the host structures were destroyed due to excessive loss of crystal water.
1. Introduction
According to the Food and Drug Administration (FDA), over 80% of active pharmaceutical ingredients (APIs) are developed in the anhydrous form, while 8% of solid APIs are developed as hydrates and 8% as solvates.1 In general terms, the anhydrous APIs are preferable due to their ease of exploitability and as it has been proven that they are more stable than their hydrates or solvates, yet there may be an exception for nucleotides. Nucleotide hydrates are very common and universal, and almost all their salt forms exist as crystalline hydrates.2–6 In addition, the diversity of their hydrates is another feature, including more than one form of hydrate for a specified compound.7,8 This indicates that the hydrates of these compounds tend to be more stable than their anhydrous form.In contrast to common APIs, nucleotides contain many hydroxyl groups and dissociation sites (Fig. 1a),9 which provide recognition and binding sites with water molecules. In addition, several rotation bonds, such as the glycosidic bond, which is the bond connecting the phosphate group with pentose, provide additional molecular degrees of flexibility that might offer more possibilities for the specific mode of host molecules.
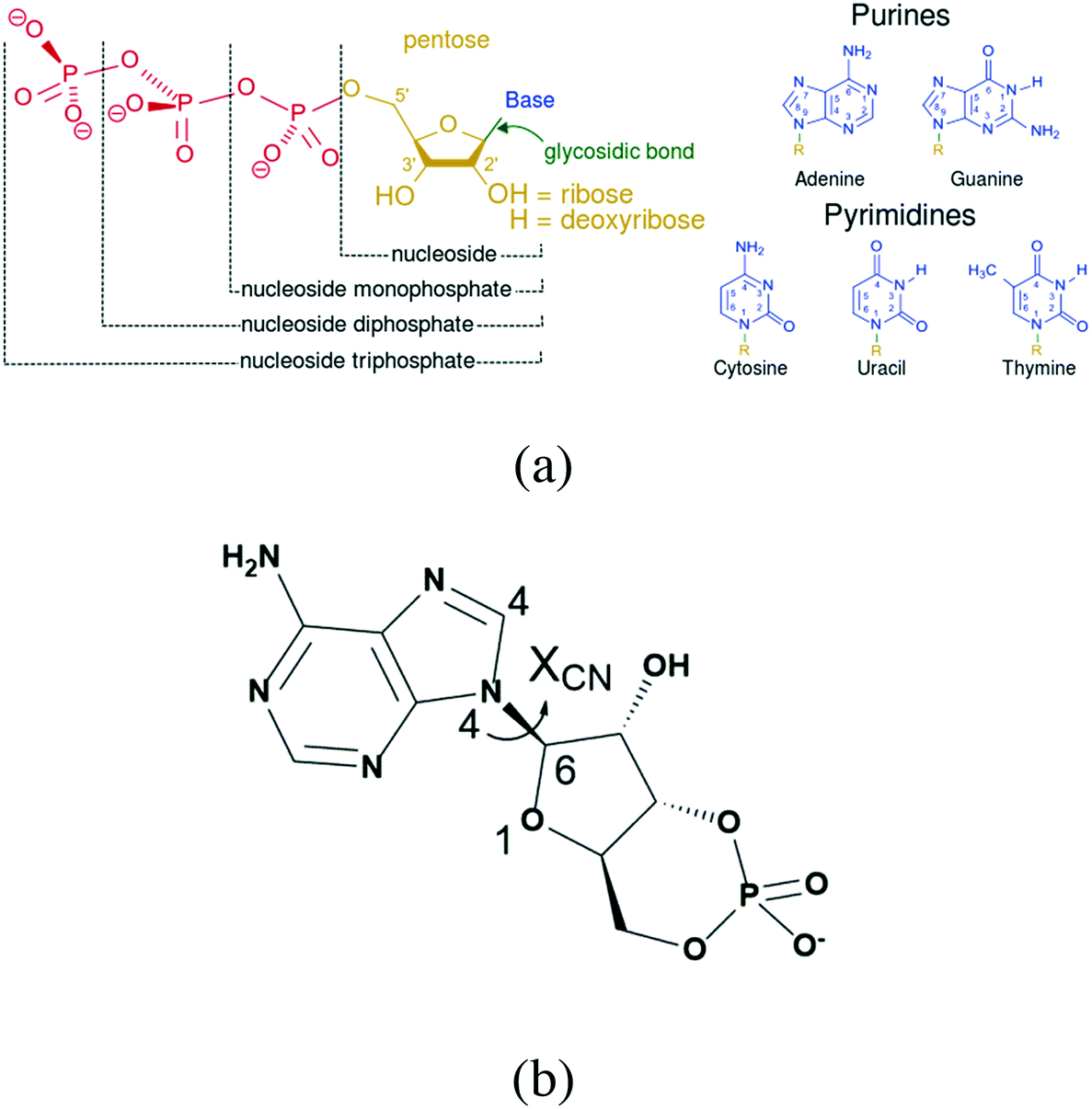 | ||
| Fig. 1 (a) Structural elements of common nucleotides; (b) structural formula of cAMP−. | ||
The incorporation of water molecules into the crystal lattices of an anhydrate or a relatively low hydrate alters the intermolecular interactions within the solid, thereby modifying the internal energy, and subsequently, the enthalpy of the solid. As a result of the hydration of the solid, changes in the shape and symmetry of the unit cell affect the entropy of the solid, resulting in changes in the free energy and chemical potential of the solid. Consequently, the thermodynamic activity of the solid was modified,10 including the crystal morphology and stability, which are of crucial importance in the downstream processing operability and quality of the end products. Then conversely, the water molecules could be affected by the host molecules and their arrangement, which reflects in the behavior of the crystal water.
The existence of water in the crystallization system is a prerequisite but not a sufficient condition for the formation of hydrates.11 Water molecules can be readily incorporated into the crystalline lattices due to their small size and ability to form multiple hydrogen bonds, acting as both a hydrogen bond donor and acceptor,12,13 widely involving intermolecular interactions with most compounds, particularly strong polar molecules or species.35,36 Lourdes Infantes et al. performed a database study of 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770 accurate organic crystal structures, and pointed out that the most important factor for determining a relatively high frequency of hydrate formation is the sum of the average donor and acceptor counts for functional groups.14 Dračínský et al. investigated the dynamics of water molecules in the solid hydrates of sodium salts of guanosine monophosphate (GMP) and uridine monophosphate (UMP) by combining X-ray diffraction (XRD) and nuclear magnetic resonance (NMR) experiments with molecular dynamics (MD) simulations. They found that the structure of the GMP system is relatively rigid with a well-ordered nucleotide hydration shell, while the water molecules in the UMP system are extremely mobile even at −80 °C.15 In addition, these crystals have been shown to exhibit humidity-dependent phase transitions. Sugawara et al. stated that the number of crystal water molecules per cytidine monophosphate (CMP) in disodium cytidine 5′-monophosphate (Na2CMP) hydrates varies approximately from one to nine in the relative humidity (RH) range from 0 to 90%.2
770 accurate organic crystal structures, and pointed out that the most important factor for determining a relatively high frequency of hydrate formation is the sum of the average donor and acceptor counts for functional groups.14 Dračínský et al. investigated the dynamics of water molecules in the solid hydrates of sodium salts of guanosine monophosphate (GMP) and uridine monophosphate (UMP) by combining X-ray diffraction (XRD) and nuclear magnetic resonance (NMR) experiments with molecular dynamics (MD) simulations. They found that the structure of the GMP system is relatively rigid with a well-ordered nucleotide hydration shell, while the water molecules in the UMP system are extremely mobile even at −80 °C.15 In addition, these crystals have been shown to exhibit humidity-dependent phase transitions. Sugawara et al. stated that the number of crystal water molecules per cytidine monophosphate (CMP) in disodium cytidine 5′-monophosphate (Na2CMP) hydrates varies approximately from one to nine in the relative humidity (RH) range from 0 to 90%.2
Adenosine-3′,5′-cyclic phosphate sodium (cAMPNa, Fig. 1b), a representative nucleotide that acts as a second messenger in organisms, has good therapeutic and preventive effects on cardiovascular and tumor diseases.16–18 It has been reported that there are various hydrates, including the tetrahydrate discovered by Varughese et al. in 1982 and the pentahydrate and methanol trihydrate reported in our previous study.19,20 This study used cAMPNa as an example and systematically investigated the structure features of its different hydrates and the relationship between crystal water behaviors and crystal structures when hydrates were subjected to heat and moisture, as well as the transformation behaviors among its different solid forms.
2. Experimental section
2.1. Materials
The raw material cAMPNa (CAS number: 37839-81-9, purity >99% as anhydrous state calculation) was purchased from Biotogether (Nanjing, China) and used after re-crystallization. Inorganic compounds and organic solvents of analytical grade were purchased from commercial sources and used without further purification. Ultrapure water (ELGA PURELABE classic 18.2 MΩ cm) was used in all the experiments.2.2. Preparation of different crystal forms of cAMPNa
Single crystal or crystalline samples of different crystal forms of cAMPNa were obtained by controlling the crystallization temperature of several common laboratory solvents through plenty of screening experiments. The solubility of cAMPNa in water + (ethanol, methanol, and acetone) is referred to in our previous report.21 Dihydrate: a saturated cAMPNa solution in the 0.2 molar fraction of ethanol–water at 65 °C was prepared, which was fully dissolved at 70 °C, and then it was transferred to a 60 °C water bath and cultured for one week. Tetrahydrate: the solid powder of 3.00 g cAMPNa was fully dissolved in 3.98 g pure water at 50 °C, and 4.36 g ethanol was added to the resulting solution, and then it was transferred to a 30 °C water bath and cultured for 20 hours. Methanol trihydrate: the solid powder of 3.67 g cAMPNa was fully dissolved in 4.94 g water at 50 °C, and 3.76 g of methanol was added to the resulting solution, and then it was transferred to a 25 °C water bath and cultured for 20 hours. pentahydrate: the solid powder of 2.82 g cAMPNa was fully dissolved in 4.77 g water at 50 °C, and 3.84 g of acetone was added to the resulting solution, and then it was transferred to a 30 °C water bath and cultured for 20 hours. The obtained crystals were collected by filtration, washed, air-dried at 35 °C, ground and used for subsequent experiments.2.3. Single-crystal XRD
The single-crystal XRD data of cAMPNa were collected on a Bruker APEX-II CCD diffractometer using Mo Kα radiation (graphite monochromator, λ = 0.71073 Å). The structures were solved by direct methods and refined by full-matrix least-squares on F2 for all data using SHELX-97.22 The N-bound and O-bound H atoms were located in a difference Fourier map and refined freely, with bond-length restraints of N–H = 0.86(1) Å and O–H = 0.82–1.05 Å. The C-bound H atoms were positioned geometrically and refined using a riding model, with C–H = 0.93–0.98 Å. Materials Studio 6.0, Diamond 3.2e, and Mercury 3.3 software23 were used for the crystal structure analysis and visualization.2.4. Powder X-ray diffraction
The specimens were first ground gently into a fine powder and then identified using a D8 Advance powder X-ray diffractometer (Bruker AXS GmbH, Germany) with Cu Kα radiation (λ = 1.54059 Å). The voltage and current applied were 40 kV and 40 mA, respectively. The samples were gently ground to a fine powder prior to analysis and were placed on a sample holder with a diameter of 1.5 cm. The sample was scanned within the scan range of 2θ = 5° to 60° continuous scan with a scan rate of 0.2 s per 0.02°. The variable-temperature powder XRD (VT-PXRD) experiment for the samples was performed in the range from room temperature to 200 °C at a heating rate of 5 °C min−1, and the data at the original temperature, 120 °C, and 200 °C were collected for the PXRD analysis.2.5. Composition analysis
The water content in the hydrates or solvate crystals was analyzed using a Mettler Toledo 870 KF Titrino plus a Karl Fischer titrator. The methanol content in the solvates was determined by gas chromatography analysis, which was carried out on an Agilent 7890A instrument. These two methods were used for the quantification of water and methanol in the experimental samples.2.6. Thermal analysis
Thermogravimetry (TG) and differential scanning calorimetry (DSC) were performed simultaneously using a Netzsch STA-409 system. The experiments were carried out at a heating rate of 10 K min−1 from 30 °C to 400 °C using a 20 mL min−1 nitrogen flow. Approximately 5 mg of each sample was added to an aluminium oxide crucible. The instrument was calibrated using indium as the reference material.3. Results and discussion
3.1. Crystal structures and structural features
A series of crystallization experiments on cAMPNa at different temperatures and with different solvents was performed. Several single crystals were obtained and determined by single-crystal XRD, and they were shown to have four crystal forms: trigonal dihydrate cAMPNa·2H2O (form I), monoclinic tetrahydrate cAMPNa·4H2O (form II), orthorhombic pentahydrate cAMPNa·5H2O (form III), and orthorhombic methanol trihydrate cAMPNa·CH3OH·3H2O (form IV). They all belong to the stoichiometric hydrates or solvates.24 Their crystal morphologies are shown in Fig. 2, and the crystallographic data are summarized in Table 1. It was found that there are different crystal habits for different hydrates, quadrangular for form I, trilateral for form II, and block-shaped for form III, which seems to be consistent with their respective crystal system. Form III and form IV display similar crystal habits, which may be related to their parallel molecular arrangement. | ||
| Fig. 2 Morphologies of the four cAMPNa crystal forms. | ||
| Crystal form | Dihydrate (form I) | Tetrahydrate (form II) | pentahydratea (form III) | Methanol trihydratea (form IV) |
|---|---|---|---|---|
| Note: the data with labelled “a” was referred to in our previous report.27 | ||||
| Sum formula | C10H15N5NaO8P | C20H38N10Na2O20P2 | C10H21N5NaO11P | C11H21N5NaO10P |
| Simplified formula | cAMPNa·2H2O | cAMPNa·4H2O | cAMPNa·5H2O | cAMPNa·CH3OH·3H2O |
| Formula weight (g mol−1) | 387.23 | 846.52 | 441.28 | 437.29 |
| Crystal system | Trigonal | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P3221 (no.154) | P21 (no. 4) | P212121 (no. 19) | P212121 (no. 19) |
| a (Å) | 10.9535(15) | 5.8453(12) | 6.5585(12) | 6.5021(10) |
| b (Å) | 10.9535(15) | 21.435(5) | 7.0470(13) | 7.0943(11) |
| c (Å) | 22.449(5) | 13.955(3) | 38.869(7) | 39.198(6) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 90 | 95.506(3) | 90 | 90 |
| γ (°) | 120 | 90 | 90 | 90 |
| V (Å3) | 2332.6(10) | 1740.4(7) | 1796.4(6) | 1808.1(5) |
| Z/Z′ | 6/1 | 2/2 | 4/1 | 4/1 |
| Density (calc) (g cm−3) | 1.654 | 1.615 | 1.632 | 1.606 |
| Crystal size (mm) | 0.10 × 0.20 × 0.30 | 0.21 × 0.24 × 0.26 | 0.22 × 0.24 × 0.26 | 0.19 × 0.23 × 0.26 |
| Tot. data | 3230 | 9699 | 12569 |
10127 |
| Unique data | 1633 | 5586 | 3174 | 3198 |
| R(int) | 0.099 | 0.018 | 0.062 | 0.022 |
| Observed data [I > 2.0 sigma(I)] | 1178 | 5446 | 3059 | 3029 |
| R_factor_gt | 0.0547 | 0.0260 | 0.0499 | 0.0480 |
| wR2 | 0.1582 | 0.0665 | 0.1397 | 0.1279 |
| Goodness-of-fit (on F2) | 1.00 | 1.07 | 1.10 | 1.16 |
| CCDC | 976996 | 1030129 | 1026499 | 1026507 |
| Coordination number of Na+ | 5 | 6 | 6 | 6 |
| Combined water in hydrogen bond | 1 | 1 | 3 | 1 |
| Coordinated water | 1 | 3 | 2 | 2 |
| Torsion (χCN, C4–N4–C6–O1, °) | 32.94° | 9.93°, 29.97° | 63.29° | 59.75° |
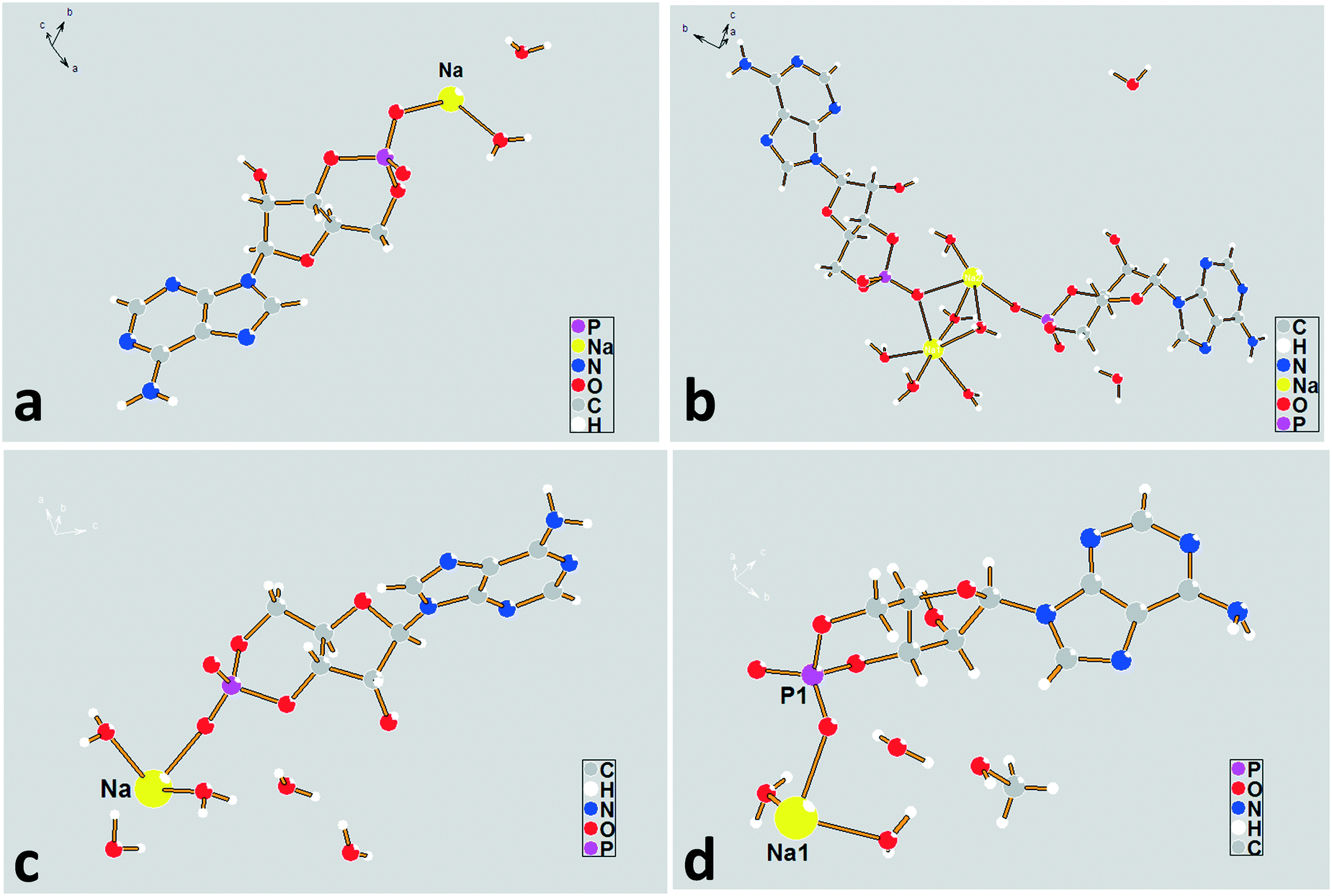 | ||
| Fig. 3 The asymmetric unit diagrams for forms I–IV. (a) Form I, (b) form II, (c) form III, and (d) form IV. | ||
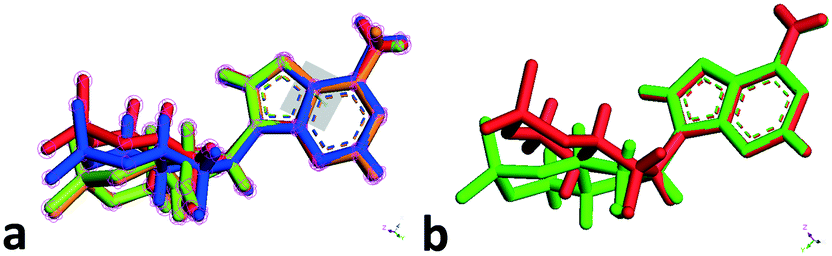 | ||
| Fig. 4 Overlaps and comparisons of conformers of the cAMP species in the lattices of forms I–IV. (a) Four conformers in forms I–IV, red (one of the conformers in form II), blue (form I), green (form IV) and yellow (form III); (b) two conformers in form II. | ||
To investigate the flexibility of torsion χCN, we run a potential energy surface scan (PES scan) with a relaxed scan approach for the cAMP− species from the dihydrate structure at the B3LYP/6-31G level with a step size of 15° with Gaussian 09,28 and the relative energy with respect to the global energy minimum is shown in Fig. 5. The results show that the energy minimum was located at approx. −50°, and the energy fluctuation in the range from 9° to 64° was less than 5 kJ mol−1, which indicates that the cAMP− species of different conformations in all of the crystal structures were all of the same conformer,29 just with some conformation adjustment, as they are all linked to the same conformation minimum.
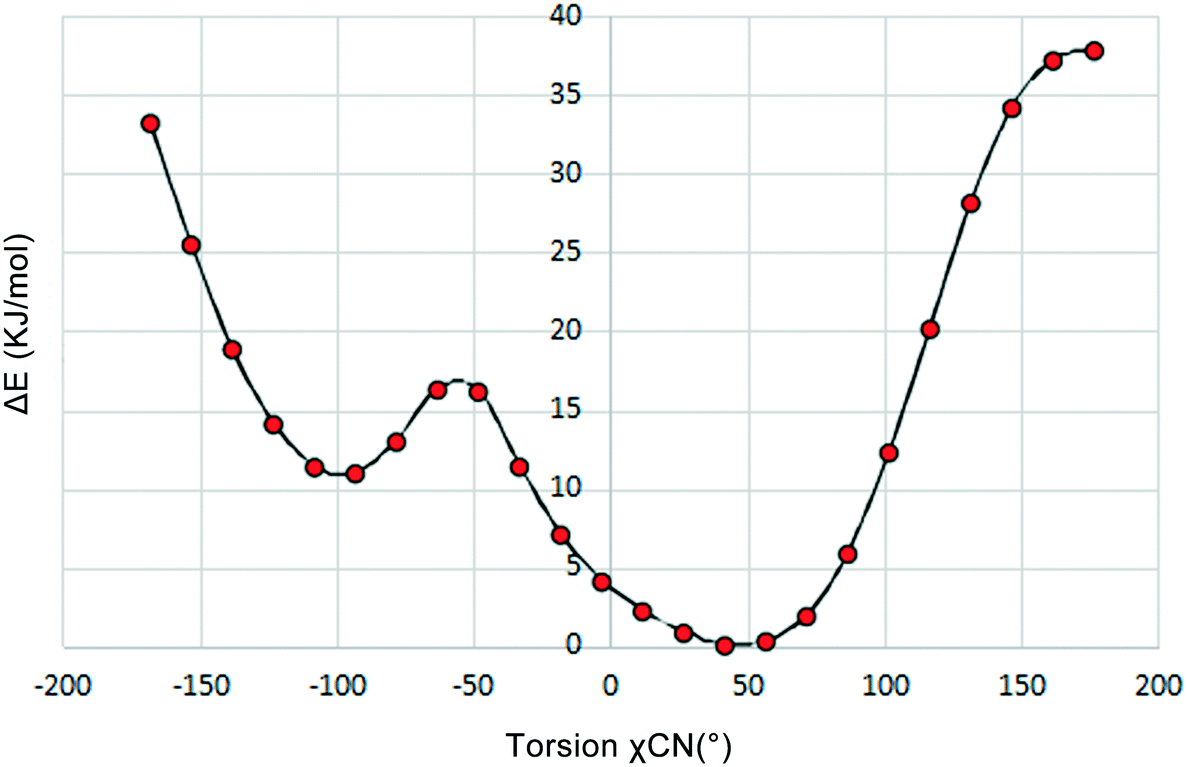 | ||
| Fig. 5 Diagram of the potential energy surface scan for the torsion χCN of cAMP−. | ||
The sodium coordination profiles for forms I–IV are shown in Fig. S3† and Table 1. For the dihydrate, the sodium ion in the asymmetric unit exhibits a five-fold coordination. It is coordinated with four different cAMP species by a base (N), furanose ring (O), and phosphate group (O) with coordination distances varying from 2.31 Å to 2.56 Å, making the structure compact. The sodium ions show a six-fold coordination in the crystal structures of the remaining hydrates, including methanol trihydrate. For the tetrahydrate, Na(2), three of the coordinating oxygen atoms belong to the water molecules and the remaining three are parts of the different cAMP molecules. For the Na(1) atom, only one coordinating oxygen atom is related to the host molecule, and the remaining five coordinating atoms are water oxygen atoms. For the pentahydrate and methanol trihydrate, only two atoms in the surrounding host molecules were involved in coordinating with sodium ion, and the remaining coordinating interaction occurred between the crystal water oxygen atoms and sodium ion.
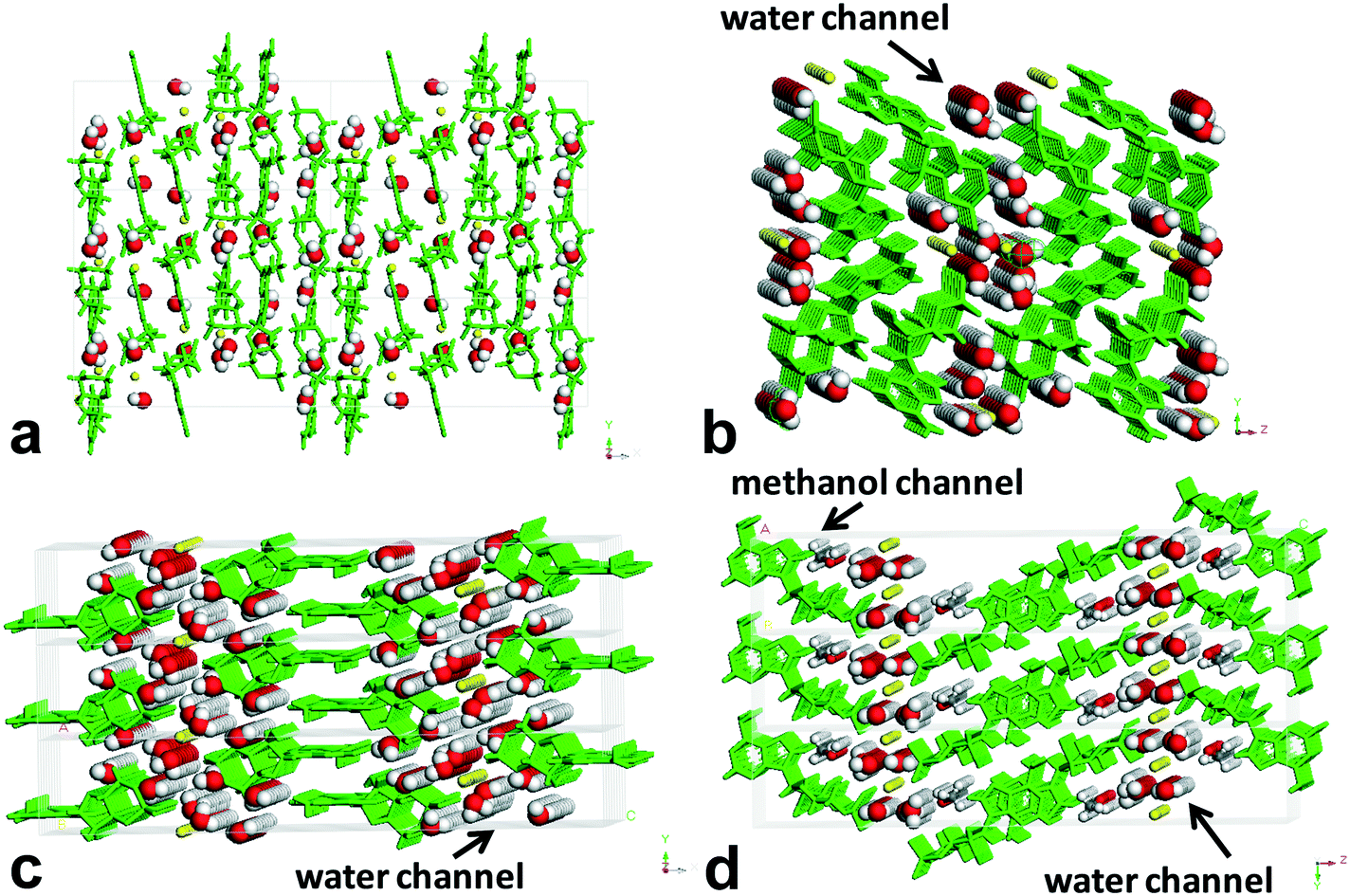 | ||
| Fig. 6 Environment profiles of guest molecules in the lattices of forms I–IV. (a) Form I, (b) form II, (c) form III, and (d) form IV. | ||
Obviously, form II, form III, and form IV belong to the channel-type hydrates,30 and form I is an isolated site-type hydrate.31 When viewed along the crystallographic b axis, the H-bond pattern of water molecules for form I shows a periodic discrete short chain that is composed of two water molecules, and the water molecules were surrounded and separated by the host molecules, existing as an isolated form, which resembles a guest in the “cage of clathrate”. For form II and form III, the H-bond patterns of water molecules show the discrete short chains along the b axis generated by two or three water molecules for the former, and the regular discrete 3-membered ring along the b axis generated by two water molecules and one O atom of P-oxide for the latter, both extending infinitely down the a axis to form the water tapes, which are separated by the sodium ion columns or host molecule columns. For form III, the water tape consisted of four water chains. As for form IV, the H-bond pattern along the b axis is similar to that of form III, but the difference is that the discrete 3-membered ring along the b axis was constructed by one methanol molecule, one water molecule, and one O atom of P-oxide, and the resulting water tape was composed of merely two water chains. One methanol molecule in the lattice of form IV was found to replace the original two water molecules in the lattice of form III, which led to a linear methanol chain down the a axis direction besides the water tape.
The arrangement profiles of the host molecule packing for forms I–IV are shown in Fig. S4.† It was found that the host molecule arrangement is the orientation of the adjacent two molecules in an antiparallel fashion, and the distances of the base plane vary from 3.26 Å to 3.58 Å. The water molecule was embedded in the crystal lattice, interacting with the surrounding host molecule or the sodium ion by either hydrogen bonding or coordination, thereby forming a complex framework. Detailed bond profiles for the water molecules in the different structures are summarized in Table S1 and S2.†
The H-bond donor/acceptor ratio in an organic molecule is related to the stability of the crystal structure according to L. Infantes's survey.32 As for the cAMP− species, the number of acceptor groups was far more than that of donor ones, leading to an imbalance between the number of acceptor and donor groups. Water molecules have two hydrogen atoms and two lone pairs enabling them to participate in four hydrogen bonds in a tetrahedral arrangement,33 yet unlike covalent bonds, the H-bond geometry is much more flexible. The inclusion of water allows the participation of some groups in H-bonding to some extent, and adjusts the donor/acceptor balance, which is important for establishing H-bonded contributions to the total lattice energy.32 The inclusion of water fills in the gaps in a close-packed structure of organic molecules. Water could be viewed as playing not only a space filling role, but also a “gluing factor” based on its properties with readiness to geometric adjustment and favorable bonding ability.34 The presence of water molecules in the cAMPNa structures can play a role in stabilizing the supramolecular species established by cAMP−–H2O–Na+, and it is significant for establishing the stability of the hydrated crystal structure. In contrast, compared to the hydrated form of cAMPNa, its anhydrous forms were proved to be hygroscopic and poorly stable in experiments (see section 3.4).
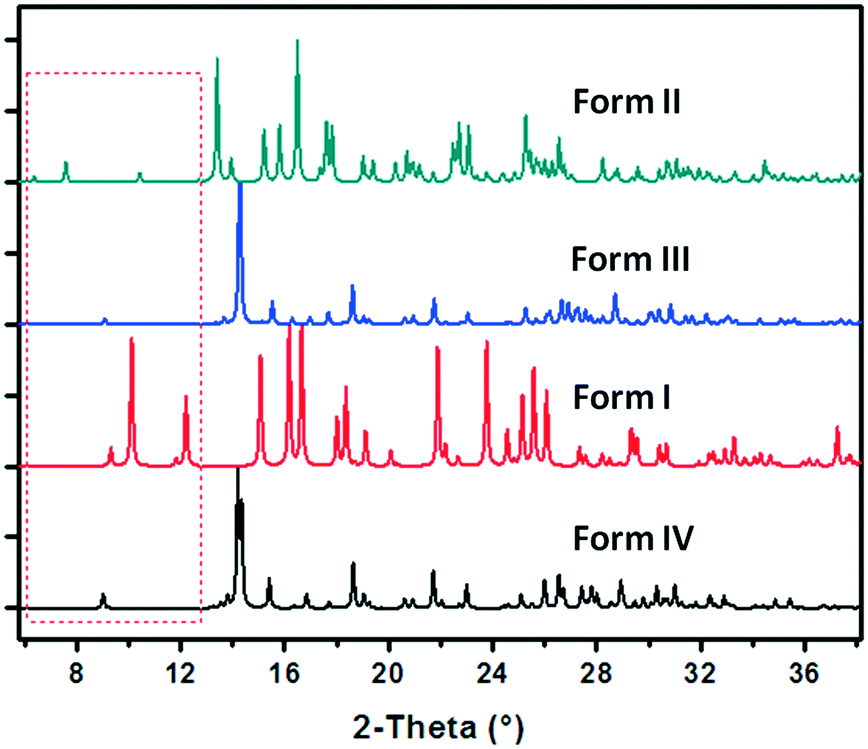 | ||
| Fig. 7 Comparison of the simulated PXRD patterns of forms I–IV. | ||
3.2. Sensibility of the cAMPNa crystal forms to humidity
Humidity sensitivity experiments of forms I–IV were carried out in several sealed glass chambers with different humidity environments created by the P2O5 solid powder (0% RH), or a saturated solution of K2SO4, NaCl, and MgCl2 with 98%, 76%, and 32% RH, respectively.35 Approximately 0.18 g of the four powders with different crystal forms after soft fine grinding were placed onto weighing dishes with a 5 cm diameter in chambers in a corresponding RH environment at room temperature for 13 weeks. The weights of the samples in the initial and final states were recorded, and the structures were analyzed by PXRD and composition analysis. It was found that for forms I–IV, no significant weight and structural changes were observed under 32% RH and 76% RH conditions, yet the majority of the crystal water or some of the solvent was lost in 0% RH, and they also exhibited serious moisture absorption in a high-humidity environment as presented in Table 2.| Sample | RH 0% | RH 32% | RH 76% | RH 98% |
|---|---|---|---|---|
| Note: the symbols “+” and “−” denote the weight increment and reduction, respectively. | ||||
| Form IV | −12.83 ± 0.56 | 0.11 ± 0.14 | 0.05 ± 0.12 | 19.85 ± 2.23 |
| Form II | −15.67 ± 0.33 | 0.30 ± 0.26 | 0.35 ± 0.15 | 33.17 ± 3.15 |
| Form III | −16.90 ± 0.31 | −0.20 ± 0.21 | 0.20 ± 0.10 | 38.60 ± 3.61 |
| Form I | 8.46 ± 0.24 | 0.25 ± 0.34 | −0.13 ± 0.24 | 44.43 ± 4.23 |
3.3. Desorption/uptake behaviors of crystal water and humidity-mediated reversible conversions
To investigate the crystal water desorption/uptake behaviors of the cAMPNa hydrates and the resulting structural transformation behaviors mediated by humidity, the experiments were designed and performed as follows. In the first stage, the four crystalline powders were placed in a 0% RH sealed chamber for several days at room temperature until their respective weight ceased to decrease. In the second stage, the dehydrated samples from the previous step were transferred to the 76% RH condition for a period until a new equilibrium was reached and the weight remained constant. The two stages were a cycle. Subsequently, the next cycle was restarted from the first stage for the samples at the end of the previous cycle. As such, three cycles were performed. During the experiments, the weight of the samples was tracked at two day intervals. The structure and composition of the subjected samples at the initial ①, intermediate ②, and final ③ stages in each cycle were analyzed by PXRD and composition analysis. For each of the solid forms, the experiments were repeated three times, and the data of the first cycle are shown in Fig. 8.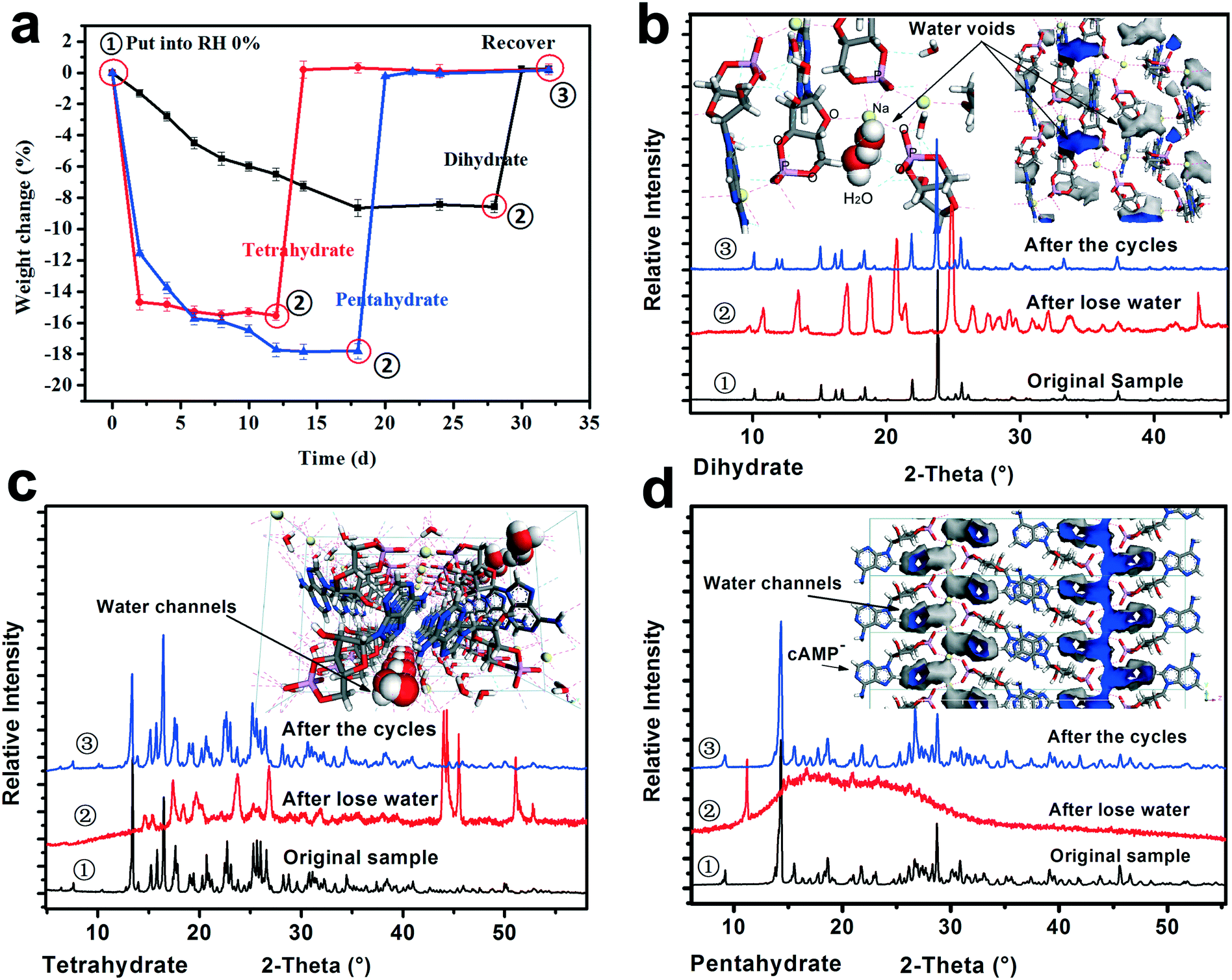 | ||
| Fig. 8 Desorption/uptake behaviors of the crystal water of the three hydrates mediated by humidity. (a) One cycle of desorption/adsorption dynamics of the crystal water of the cAMPNa hydrates. (b), (c), and (d) show the PXRD patterns of the three hydrates at ①/②/③ points and their respective crystal water environments, respectively. Theoretical water contents of dihydrate, tetrahydrate, and pentahydrate were 9.31%, 17.03%, and 20.42%, respectively. | ||
It was found that the water desorption rate of the dihydrate, tetrahydrate and pentahydrate of cAMPNa were strikingly different. The tetrahydrate and pentahydrate exhibited a rapid dehydration process compared with the dihydrate. For the dihydrate, two crystal water molecules were embedded into the lattices and surrounded by the host cAMP species, while for the tetrahydrate and pentahydrate, the water molecules are arranged linearly in a two-dimensional direction forming water channels and located among molecular columns formed by the host cAMP species. Based on the analysis of water distribution in the lattices and bonding profiles, such differences in dehydration dynamics were attributed mainly to their steric effects, despite the interaction difference of water molecules with the sodium ion coordination or the cAMP anion hydrogen bond. The desorption behavior of methanol trihydrate was similar to that of pentahydrate due to the similarity of structured packings and the water environment as seen in our previous report,27 but influenced by a larger steric hindrance of the methanol molecule than that of the water molecule.
In addition, the crystal water has a noticeable effect on the structure of the hydrates. For the dihydrate, the host molecules would undergo a well-ordered rearrangement with the loss of crystal water, while for the pentahydrate, the host structure collapses with the desorption of the water molecules, demonstrating a disordered state. For the tetrahydrate, an orderly reconstruction was observed as well although the crystallinity of the dehydrated solid somewhat declined to some extent. In addition to the respective packing characteristics, the structural variations may be related to the amount of crystal water loss. Seemingly, the more the loss of water, the more disordered the resulting structure becomes.
Interestingly, whether or not the structures of the already established solid powders at point ② were orderly or disorderly, the resulting respective powders generated by the dihydrate, tetrahydrate, and pentahydrate would, at the second stage, take up a certain amount of water from the surroundings, which is nearly equal to the amount of water loss at the first stage, and transform into the respective original states of the dihydrate, tetrahydrate and pentahydrate at the RH of 76%. This was confirmed by the PXRD pattern comparison combined with the composition analysis, as shown in Fig. 8. Furthermore, the test results of the three sampling points in three cycles were very consistent. The reversible transformation behaviors mediated by humidity can be described by the following Scheme 1:
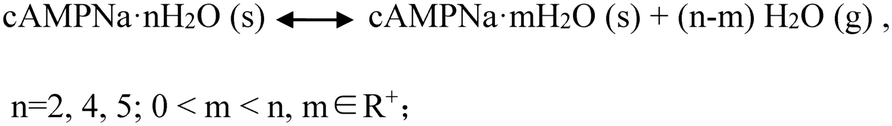 | ||
| Scheme 1 Reversible transformation behaviors mediated by humidity for the three hydrates. | ||
As for methanol trihydrate, the methanol in the crystal lattice was removed and replaced by gaseous water molecules from the surroundings, the solid was transformed into isostructural pentahydrate in the first cycle, and the subsequent desorption/uptake behavior of the crystal water coincided with that of the pentahydrate. The transition details can be seen in our previous report.21
The structures of the relatively low hydrates after the partial loss of crystal water for the dihydrate, tetrahydrate and pentahydrate of cAMPNa exhibited a potential that allowed the absorption of the quantitative gaseous water molecules from the environment, which integrated into respective lattices, recovering the respective initial states. It seemed that the gaseous water uptake process of these relatively low hydrates was similar to the adsorption of porous materials, yet the results of the typical N2 adsorption–desorption experiments show no detectable pores. Hence, classic gas sorption theories, such as the Langmuir isotherm model, Freundlich isotherm model, and Brunauer–Emmett–Teller isotherm model, were not available.36,37 Yet, it should be pointed out that such structure recovery capability might be disabled when the structures are destroyed owing to the excessive loss of crystal water brought by high-temperature treatment.
3.4. Variable-temperature powder XRD (VT-PXRD) analysis of forms I–IV
The VT-PXRD experiments from room temperature to 200 °C were performed to capture the structure variation of forms I–IV after complete dehydration/desolvation, and investigate the uptake capacity of the solid samples subjected to 200 °C in water as well as the structure recovery when they were transferred to RH 76% surroundings for 13 weeks at room temperature. The VT-PXRD profiles are shown in Fig. 9.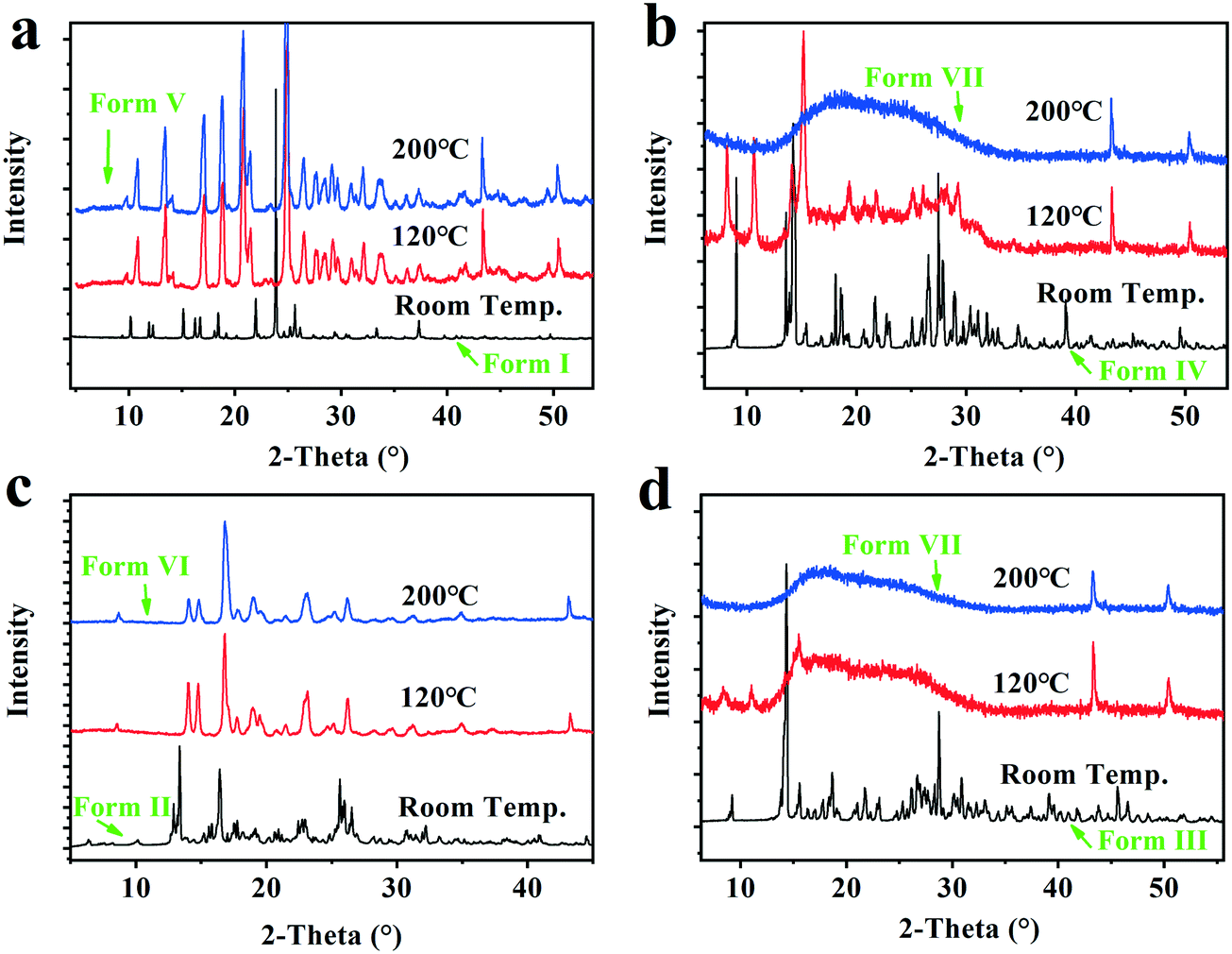 | ||
| Fig. 9 The VT-PXRD profiles of the Forms I–IV. a, Form I (dihydrate); b, Form IV (Methanol trihydrate); c, Form II (tetrahydrate); d, Form III (pentahydrate). | ||
It was found that the two solid powders after the dihydrate and tetrahydrate were completely dehydrated show new PXRD patterns with high crystallinity, herein called form V and form VI, respectively, which could take up 9.3 ± 0.2% and 17.1 ± 0.2% water and recover the original form I (dihydrate) and the original form II (tetrahydrate) when they were placed under the 76% RH condition or in an ambient atmosphere. However, the two solid powders, after form I (pentahydrate) and form IV (methanol trihydrate) were completely dehydrated/desolvated at 200 °C, show the same amorphous state from their PXRD patterns, herein called as form VII, which could only take up 8.5 ± 0.5% water and no longer transform into the pentahydrate when it was subjected to the 76% RH condition at room temperature. Hence, the new anhydrate crystals form V and form VI generated by the complete hydration of form I and form II could be converted back into the corresponding hydrates through absorbing water from the surroundings, but for the dehydrated form III, the structure recovery capability was disabled when its structure was destroyed with excessive loss of water.
3.5. Thermal analysis for the cAMPNa hydrates
To investigate the thermal behavior of forms I–IV subjected to heating, TG-DSC experiments were performed at a heating rate of 10 K min−1 from room temperature to 400 °C, and the results are shown in Fig. 10.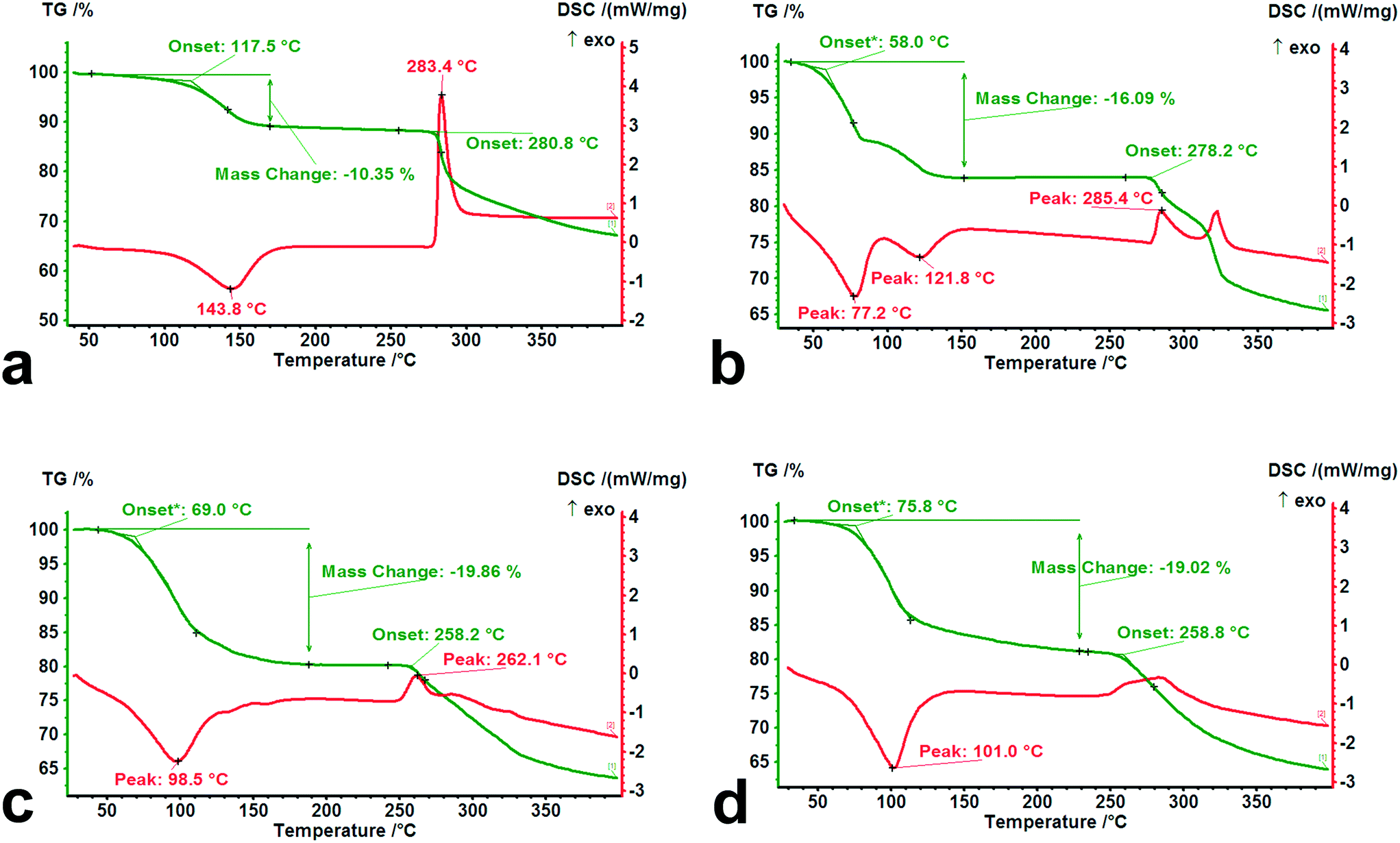 | ||
| Fig. 10 TG-DSC curves of forms I–IV. (a) Form I, (b) form II, (c) form III, and (d) form IV. | ||
It was observed that there are significant differences in the dehydration and decomposition behaviors for the different hydrates. The loss of crystal water occurs mainly within the temperature range from 30 °C to 150 °C, accompanied by apparent endothermic peaks. A two-step dehydration phenomenon was observed from the DSC curve of tetrahydrate, indicating that its adjusted structure brought about by the partial loss of crystal water influences the further desorption of the residual water molecules. From the onset temperature of dehydration in the TG curves for forms I–IV, the value of the isolated dihydrate (117.5 °C) was significantly higher than those of the channel tetrahydrate (58.0 °C) and pentahydrate (69.0 °C), which was in agreement with the results of the desorption experiment of the above-mentioned crystal water. In contrast to pentahydrate, a slightly high onset temperature (75.8 °C) and a relatively long tail of desorption may be ascribed to the relatively strong steric effect on the lattice caused by the size of the methanol molecule. In addition, from the decomposition behaviors of the four dehydrated/desolvated solid forms, the onset temperatures of the dihydrate (280.8 °C) and tetrahydrate (278.2 °C) were higher than those of the pentahydrate (258.2 °C) and methanol trihydrate (258.8 °C), which was attributed to the structural characteristics after the complete loss of water.
Despite having the same composition when completely losing water or methanol, the evolved structures of the dihydrate and tetrahydrate subjected to heating retained their relatively high crystallinity and regularity, while the resulting structures of the pentahydrate and methanol trihydrate were converted into the same amorphous forms with serious disorder.
4. Conclusion
It was found that there are seven solid forms for cAMPNa, forms I–VII. Forms I–III are hydrates, i.e. dihydrate, tetrahydrate, and pentahydrate, form IV is methanol trihydrate, forms V and VI are two anhydrous forms, and form VII is an amorphous state. The three hydrates and methanol trihydrate were stable under 32% RH and 76% RH conditions. Forms V and VI can be obtained by a complete dehydration process of form I and form II, respectively, and converted into corresponding hydrates by absorbing water from the surroundings at room temperature when exposed to the 76% RH condition. Form VII is the result of complete dehydration/desolvation of form III or form IV, proved to readily absorb moisture.The features of the crystal structures of forms I–IV were analyzed; the desorption/uptake behaviors of crystal water, the structure–property relationships, and thermal behavior were studied. The crystal water molecule was observed to have a linear arrangement, forming a water channel for the tetrahydrate, pentahydrate and methanol trihydrate, while it exists as an isolated state surrounded by cAMP− species for the dihydrate. The desorption dynamics of the crystal water were ascribed mainly to steric effects in crystal lattices, despite the interaction of water molecules with sodium ion coordination and the cAMP− species hydrogen bond. The crystal water has a noticeable effect on the structure of the hydrates. For the dihydrate and tetrahydrate, host molecules would undergo a well-ordered rearrangement with the loss of crystal water, while for the pentahydrate and methanol trihydrate, the host structure collapses with the desorption of water/solvent molecules in the lattices, demonstrating a disordered state. In addition, the structures of the relatively low hydrates after the partial loss of crystal water for the dihydrate, tetrahydrate and pentahydrate of cAMPNa exhibited a potential that allowed the absorption of the quantitative gaseous water molecules from the 76% RH environment, which integrated into respective lattices, recovering the respective initial states. However, for the pentahydrate, the reversible transformation capability mediated by humidity was disabled when the host structure was destroyed due to excessive loss of crystal water.
Conflicts of interest
All the authors have given approval for the final version of the manuscript. The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC 21908100), the Jiangsu Synergetic Innovation Center for Advanced Bio-Manufacture (XTE1847), the Key R&D Plan of Jiangsu Province (BE2019001), and the National Key Research and Development Program of China (2019YFD1101204 and 2019YFD1101202).References
- L. Wang, X. Wen, P. Li, J. Wang, P. Yang, H. Zhang and Z. Deng, CrystEngComm, 2014, 16, 8537–8545 RSC.
- Y. Sugawara, A. Nakamura, Y. Iimura, K. Kobayashi and H. Urabe, J. Phys. Chem. B, 2002, 106, 10363–10368 CrossRef CAS.
- Y. Sugawara, H. Urabe, K. Kobayashi, Y. Iimura and H. Iwasaki, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1996, 276, A255–A258 CrossRef.
- S. K. Katti, T. P. Seshadri and M. A. Viswamitra, Curr. Sci., 1980, 49, 533–535 CAS.
- T. P. Seshadri, M. A. Viswamitra and G. Kartha, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 925–927 CrossRef.
- F. A. Perras, I. Korobkov and D. L. Bryce, Phys. Chem. Chem. Phys., 2012, 14, 4677–4681 RSC.
- Y. Sugawara and H. Iwasaki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 68 CrossRef.
- M. Viswamitra, M. Post and O. Kennard, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1089–1094 CrossRef.
- N. Moreau, M. Buback, J. Garcia-Martinez and I. P. Committee, Pure Appl. Chem., 2014, 86, 1 CAS.
- R. K. Khankari and D. J. W. Grant, Thermochim. Acta, 1995, 248, 61–79 CrossRef CAS.
- Y. Wang, Y. Chi, W. Zhang, Q. Yang, S. Yang, C. Su, Z. Lin, J. Gu and C. Hu, Cryst. Growth Des., 2016, 16, 1492–1501 CrossRef CAS.
- C. Bilton, J. A. K. Howard, N. N. Laxmi Madhavi, A. Nangia, G. R. Desiraju, F. H. Allen and C. C. Wilson, Chem. Commun., 1999, 1675–1676 RSC.
- A. L. Gillon, N. Feeder, R. J. Davey and R. Storey, Cryst. Growth Des., 2003, 3, 663–673 CrossRef CAS.
- L. Infantes, L. Fabian and W. D. S. Motherwell, CrystEngComm, 2007, 9, 65–71 RSC.
- M. Dračínský, M. Šála and P. Hodgkinson, CrystEngComm, 2014, 16, 6756–6764 RSC.
- N. Mestdagh, B. Vandewalle, L. Hornez and J. P. Henichart, Biochem. Pharmacol., 1994, 48, 709–716 CrossRef CAS.
- R. R. Shankar, J. S. Zhu and A. D. Baron, Metab., Clin. Exp., 1998, 47, 573–577 CrossRef CAS.
- M. Pourcyrous, H. Parfenova, M. Shibata, H. S. Bada, S. B. Korones and C. W. Leffler, Pediatr. Res., 1997, 42, 305–310 CrossRef CAS.
- K. I. Varughese, C. T. Lu and G. Kartha, J. Am. Chem. Soc., 1982, 104, 3398–3401 CrossRef CAS.
- G. Kartha, K. I. Varughese and C. T. Lu, Summer Meeting of the American Crystallographic Association, 1979, vol. 52 Search PubMed.
- P. Yang, Q. Wen, J. Wu, W. Zhuang, Y. Zhang and H. Ying, Ind. Eng. Chem. Res., 2014, 53, 10803–10809 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- S. R. Vippagunta, H. G. Brittain and D. J. W. Grant, Adv. Drug Delivery Rev., 2001, 48, 3–26 CrossRef CAS.
- T. Srikrishnan, R. Parthasarathy, W. Pfleiderer, N. C. De and G. B. Chheda, Nucleosides Nucleotides, 1997, 16, 239–255 CrossRef CAS.
- M. E. Druyan and M. Sparagana, J. Cyclic Nucleotide Res., 1976, 2, 373–377 CAS.
- P. Yang, C. Lin, W. Zhuang, Q. Wen, F. Zou, J. Zhou, J. Wu and H. Ying, CrystEngComm, 2016, 18, 1699–1704 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, Gaussian 09, Revision A.02, Gaussian Inc, Wallingford CT, 2009 Search PubMed.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS.
- T. Steiner and W. Saenger, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 450–455 CrossRef.
- L. Rajput and K. Biradha, J. Mol. Struct., 2011, 991, 97–102 CrossRef CAS.
- L. Infantes and S. Motherwell, CrystEngComm, 2002, 4, 454–461 RSC.
- G. A. Jeffrey and H. Maluszynska, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 546–549 CrossRef.
- T. M. Krygowski, S. J. Grabowski and J. Konarski, Tetrahedron, 1998, 54, 11311–11316 CrossRef CAS.
- L. Greenspan, J. Res. Natl. Bur. Stand., 1977, 81, 81–89 Search PubMed.
- P. Kumar, W. L. Pei, S. Kale, S. Johnson, V. Pareek, R. Utikar and A. Lali, J. Chromatogr. A, 2014, 1356, 105–116 CrossRef CAS.
- A. B. Thompson, S. J. Cope, S. T. Dallas and J. M. Notestein, Langmuir, 2011, 27, 11990–11998 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details related to the asymmetric unit diagrams, crystal packing diagrams, arrangement profiles of the host molecule packing, sodium ion coordination, and hydrogen bond information. Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 976996 and 1030129. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01180k |
| This journal is © The Royal Society of Chemistry 2021 |