
Connecting chloride solvation with hydration in deep eutectic systems†
Maria Enrica
Di Pietro‡
 a,
Oliver
Hammond‡
b,
Adriaan
van den Bruinhorst
b,
Alberto
Mannu
c,
Agilio
Padua
*b,
Andrea
Mele
*ad and
Margarida
Costa Gomes
b
a,
Oliver
Hammond‡
b,
Adriaan
van den Bruinhorst
b,
Alberto
Mannu
c,
Agilio
Padua
*b,
Andrea
Mele
*ad and
Margarida
Costa Gomes
b
aDepartment of Chemistry, Materials and Chemical Engineering ‘G. Natta’, Politecnico di Milano, Piazza L. da Vinci 32, 20133 Milano, Italy. E-mail: andrea.mele@polimi.it
bÉcole Normale Supérieure de Lyon and CNRS, Laboratoire de Chimie, 46 allée d’Italie, 69364 Lyon Cedex 07, France. E-mail: agilio.padua@ens-lyon.fr
cDepartment of Chemistry, University of Torino, Via P. Giuria 7, 10125 Torino, Italy
dCNR-SCITEC Istituto di Scienze e Tecnologie Chimiche, Via A. Corti 12, 20133 Milano, Italy
First published on 8th December 2020
Abstract
The Deep Eutectic Solvents/Systems (DESs) choline chloride:urea (xChCl = 0.33) and choline chloride:glycolic acid (xChCl = 0.5) were investigated using viscosity-corrected 35Cl NMR spectroscopy and molecular dynamics simulations to probe the role of chloride as a function of water content. Three Cl− solvation regimes are revealed, with high-symmetry environments for pure and highly dilute DES, and an unusual low-symmetry interstitial region where the primary coordination sphere is most disordered.
Eutectic mixtures show a melting temperature depression upon mixing, which increases the accessibility of their liquid window.1 Deep Eutectic Systems (DESs) are widely applied as a subclass of eutectic mixtures,2 defined by a eutectic temperature significantly below the ideal eutectic point.3 DES literature is mainly focused on extensively H-bonded mixtures,4 comprising a hydrogen bond acceptor (HBA) and hydrogen bond donor (HBD). Among other hygroscopic quaternary ammonium salts, choline chloride (ChCl), a non-toxic, widely available salt, is one of the most widely studied HBAs. The equilibrium water content of DESs ranges from ppm to significant mass fractions, making trace water unavoidable.5,6 This water acts as an additional H-bond donor and acceptor, modifying interactions between DES components,5 and strongly affecting crucial physicochemical properties and phase behaviour.
Developing fundamental understanding of intermolecular interactions in DESs will facilitate the design of task-specific systems. The structure of DESs is complex and disordered, with competing strong and weak H-bonds between the components,7 whose contributions are poorly understood due to heavy system-dependence. A recurrent finding is that the anion, usually chloride, plays a primary role by defining the H-bond network.7–14 In the archetypal DES ChCl:urea, the local structure sees chloride in constant H-bond flux with choline and urea, at the centre of a local cluster. The cation and HBD have different bonding affinities and short-range diffusion dynamics.9 The interaction between chloride and HBD also defines the structure of other DES such as ChCl:propylene glycol,15 ChCl:malic acid,16 and ChCl:oxalic acid.17 The anion therefore defines physicochemical properties of DESs,18,19 and affects the DES–solute interactions.20,21 Naturally, the distribution of species bound to chloride will vary as water content changes, but this is currently poorly understood.8,10,22
Various works have investigated the H-bond network of pure and hydrated DESs,16,23–32 but not specifically the environment of chloride, and how it changes with hydration. A single paper has applied chlorine NMR to some choline chloride:glycol DESs,33 which suggested weakened DES–DES H-bonds upon dilution with D2O, from trends in 35Cl linewidth. This study aims to directly explore how the key role of the anion is affected by the mixture's water content through connecting viscosity-corrected 35Cl NMR and MD simulations. Two model ChCl-based DESs were chosen (Fig. 1), choline chloride:urea (ChCl:U, xChCl = 0.33), and choline chloride:glycolic acid (ChCl:G, xChCl = 0.5); some critical discussion on DES nomenclature is included in the ESI.† We consider glycolic acid as a structural proxy for both carboxylic and alcoholic DES, which can overcome the experimental and computational challenges of the more common citric/malic acid based mixtures.
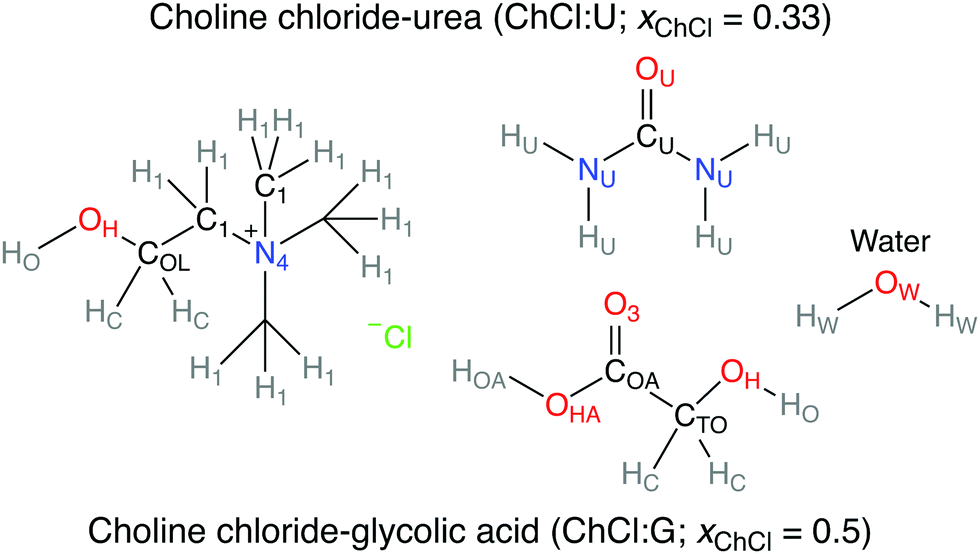 | ||
| Fig. 1 Chemical structure of the ChCl:U and ChCl:G DES studied in this work, and atom type labels for each molecule used in MD simulations. | ||
35Cl NMR spectroscopy has been used before to investigate H-bonding and Cl− binding in a multitude of systems, with Cl− chemical shift, linewidth and relaxation time characterising ion dynamics.35–43 Twenty samples of ChCl:U and ChCl:G were prepared at several dilutions, up to 98 wt% H2O (experimental details in the ESI†). Fig. 2a shows representative spectra of ChCl:U samples, with a large 35Cl upfield shift of ∼50 ppm for the most diluted sample relative to the pure DES (more details in Fig. S1–S3 and Table S5, ESI†). This shift indicates substantially greater shielding of Cl− upon dilution, consistent with the stronger Cl⋯H interaction with water molecules.44 The 35Cl linewidth is symptomatic of the Cl− bond character and local environment.45 A small quadrupolar coupling constant and thus narrow lineshapes are observed for highly symmetric scenarios, such as Cl− in isolation or with a homogeneous ligand population, due to a minimised electric field gradient.35 Decreasing the symmetry around Cl−, such as by adding a different HBD to the ligand shell, increases the quadrupolar coupling constant and broadens the NMR linewidth.42,46 Experimental linewidths at half height (FWHM) of the 35Cl peaks and corresponding relaxation times (T2) and rates (R2) were calculated and are shown in the ESI.† A marked drop in linewidth and relaxation rate are observed with increasing water content (Fig. 2b and Fig. S4, S6, ESI†). The log-scaled FWHM trend shows a steep initial decrease, a second region with lesser gradient, and a third regime with steeper gradient at high dilution; these findings are consistent with stronger aqueous H-bonding at high dilutions, where the asymmetrical network in the DES is replaced by spherically-symmetric hydrated Cl−.39,47 Pioneering studies of long-chain alkylammonium chlorides showed a marked effect of the cations on the relaxation rate of Cl−, due to asymmetric hydration of the anion in the aqueous cage around the hydrophobic cation tail.46
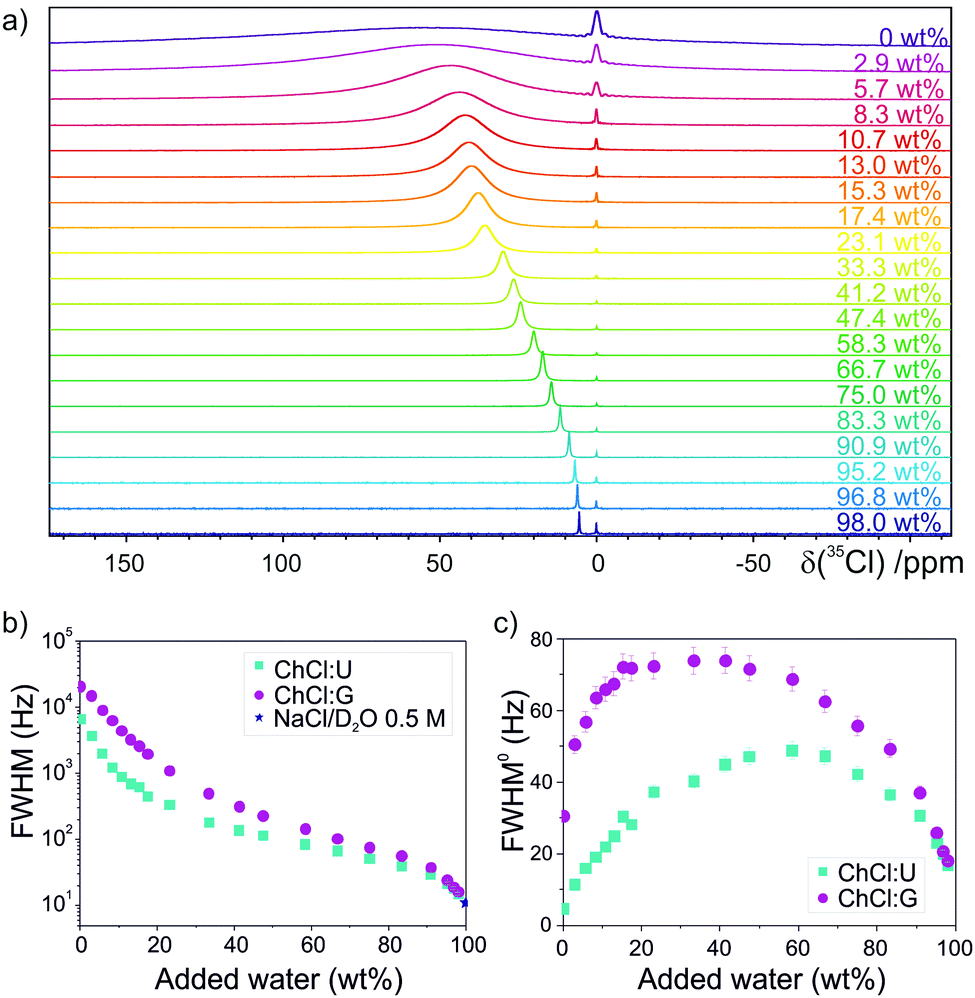 | ||
| Fig. 2 (a) Representative 35Cl NMR spectra of ChCl:U samples as a function of added water content (increasing from pure DES at the top to 98.0 wt% H2O at the bottom). The reference peak at 0 ppm corresponds to Cl− of a 0.5 M NaCl solution in D2O in the coaxial inset. (b) Experimental log-scaled linewidth (FWHM = full width at half maximum) and (c) viscosity-corrected linewidth (FWHM0) for the 35Cl signal of ChCl:U and ChCl:G samples as a function of the water content. FWHM of ChCl:U was corrected with viscosity data from Agieienko and Buchner.34 | ||
Because ion relaxation is closely related to the system viscosity,48 we applied a viscosity-correction to the linewidth for these viscous DESs, shown in Fig. 2c as a function of water content for both ChCl:U and ChCl:G, using experimental viscosity data:45
| FWHM0 = FWHM/(η/η0) | (1) |
ChCl:G partially (auto-)esterifies;51 the resulting water release and presence of esters can significantly affect the viscosity and other physicochemical properties.51,52 Dilution with water decreases this deviation, by shifting the reaction equilibrium towards the DES components. A small ester peak was distinguishable in 1H-NMR spectra, under the same conditions as the viscosity and 35Cl-NMR measurements, for ChCl:G at low water contents (esterification yield of 1.5–4%). The initial latent water content (0.983–1.029 wt% for ChCl:U, 1.022–1.314 wt% for ChCl:G) slightly shifts the viscosity corrections at low water contents. This was not considered for the 35Cl NMR experiments, but we show that this effect on the FWHM0 would be limited in Fig. S11 and S12 (ESI†).
The viscosity-corrected linewidths in Fig. 2c show a remarkable re-entrant behaviour as water content grows. The FWHM0 first increases up to a maximum value (ca. 40 wt% for ChCl:G and ca. 60 wt% for ChCl:U), followed by a decrease at higher dilutions. In the most diluted samples, the linewidth is ∼15 and 16 Hz for ChCl:U and ChCl:G, respectively (respective FWHM0 of 17 and 18 Hz). As Cl− approaches the Debye–Hückel limit of completely dissociated ions, i.e. predominantly hydrated Cl− at these higher dilutions,53 these FWHM match more closely the linewidths of ‘free ions’ (FWHM = 11 Hz for 0.5 M NaCl). Here, the 35Cl coordination shell is symmetrical, leading to the narrowest possible linewidths. An artefact of our correction is a slightly narrower FWHM0 for neat ChCl:U than for the most diluted aqueous samples (5 vs. 17 Hz). This is due to the proportionality between ionic relaxation rate and macroscopic viscosity. Future correction strategies for such viscous systems, which do not over-correct pure DES due to direct normalisation, constitute an interesting avenue.
Molecular dynamics (MD) trajectories of 10 ns of both DES, from 0–90.9 wt% H2O, were analysed to determine the Cl− coordination environment. Interactions between DES components (i.e. Ch+–Cl−, HBD–Cl−, and HBD–HBD) decrease, as expected, when water is added,54 until the DES–DES hydrogen bonding network is disrupted, with onset around a volume fraction of 50% H2O.8 Further MD details, including densities, diffusion coefficients and methodology, are given in the ESI.† The trends in diffusivity agree with previous computations.54
The models show a strong contraction in the Cl−–Cl− RDF as water content increases, which is most prominent in ChCl:G (Fig. 3a). The primary Cl−–Cl− interaction peak is diffuse in pure ChCl:G, spanning from ca. 4.8–10 Å. Once water is added, the Cl−–Cl− peak contracts to respective onset and first minima of ca. 4 and 6 Å. The closely-binned water contents studied here reveal this change even for the lowest water content of 2.9 wt% H2O, which is easily achieved if DES are prepared without control over water content.55 Other studies have recently noted similar phenomena in DES such as ChCl:U and ChCl:Ethylene Glycol,13,14,54 though this effect appears stronger in ChCl:G.
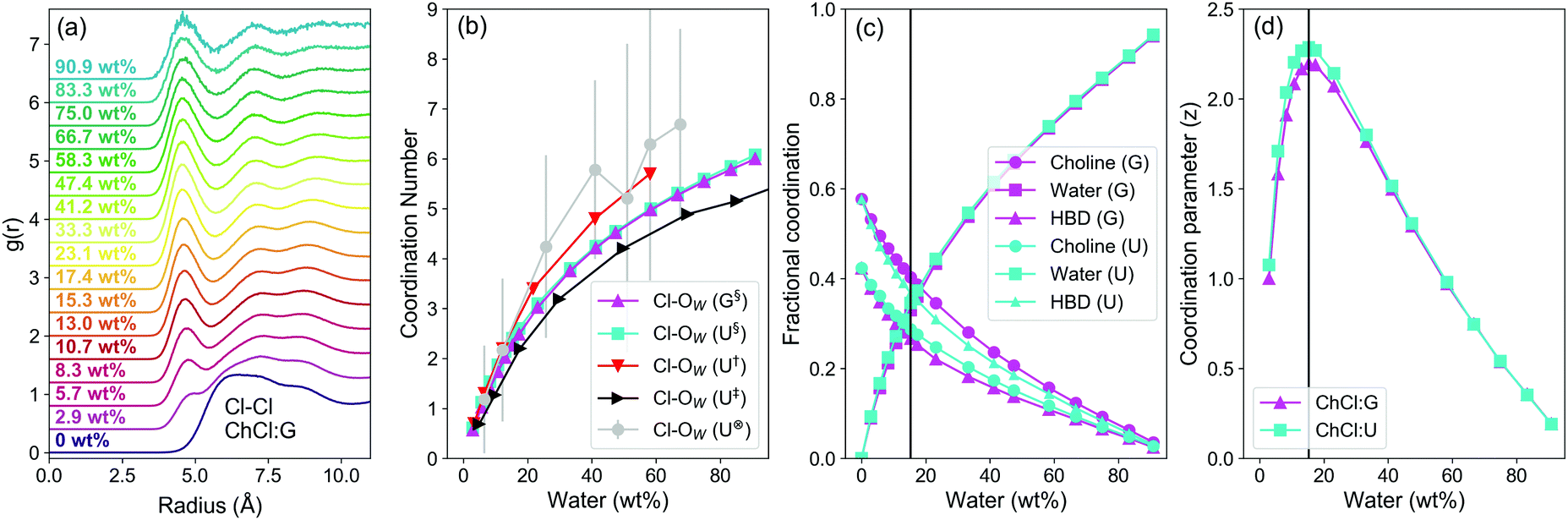 | ||
| Fig. 3 Data from MD simulations of ChCl:U and ChCl:G aqueous mixtures; (a) Cl–Cl RDFs for the ChCl:G DES as a function of water content (increasing from bottom to top); (b) Cl–OW (water oxygen) coordination number as a function of water concentration for both systems, and compared with 3 literature studies of ChCl:U as a function of water content: §Data from this study, †Data from Kumari et al.,13‡Data from Harries et al.,14⊗Data from Hammond et al.,8 (c) fractional coordination of each species around Cl− in both DES as a function of water content. Fractional coordination is the proportion of each species relative to the sum of all, omitting Cl−–Cl− because it does not form contact pairs and thus should have minimal effect on the NMR environment; (d) calculated coordination parameter (z) as a function of water content for both DES, where the maximum shows the point at which the ligand environment is most varied. Note that the lowest water content used for the z-parameter is 2.9 wt%. Coordination numbers were calculated by integrating RDFs of atom types proximal to the molecular geometric centres; N4 for cholinium, OW for water, CTO for glycolic acid. A tie line is drawn on both (c) and (d) for the concentration in both DES (15.3 wt%) where z, the coordination parameter, is maximised; lines serve otherwise as a guide to the eye. | ||
Even at very low water contents, water is a strong ligand for chloride, binding strongly and outcompeting DES species due to its H-bonding capability and small molecular volume. Water therefore occupies the space around Cl− anions, which helps to reduce Cl−–Cl− separation via bridging in solvent-separated pairs. At just 25 wt% H2O, the Cl−–H2O coordination number is ≈3, (Fig. 3b), so half-hydrated relative to [Cl(H2O)6]−.56 The Cl–OW coordination number follows the same trends for the two DESs, despite different structures and eutectic ratios. OPLS parameters with 0.8q scaling of ionic species gave good agreement with previous DES Cl–OW coordination from MD13,14 and neutron scattering studies.8
The strong hydration of Cl− is therefore key, but the contribution from other components is considered via the fractional coordination (Fig. 3c), i.e. the proportion of each species as a fraction of the sum of coordination numbers, excluding chloride, which is too distant to occupy the primary sphere. The linewidth of Cl− under dilute aqueous conditions was shown only to be affected by nearest neighbours, and two layers of water molecules screen this effect.45 ChCl:G has xChCl = 0.5 as compared to xChCl = 0.33 for ChCl:U. Hence, ChCl:G has significantly stronger Ch+–Cl− interaction than ChCl:U, whereas for the latter the HBD–Cl− interaction is more dominant. As a result, the fractional Cl− coordination of Ch+ and HBD are inverted (Fig. 3c). In both cases the Cl−–water fractional coordination follows the same trend, with the structure around Cl− rearranging to compensate for the systematic differences. In both systems, a crossover point is then reached where water begins to dominate. We therefore developed a simple coordination parameter, z, to describe the solvation asymmetry around Cl−, shown in eqn (2) for our 3-component system:
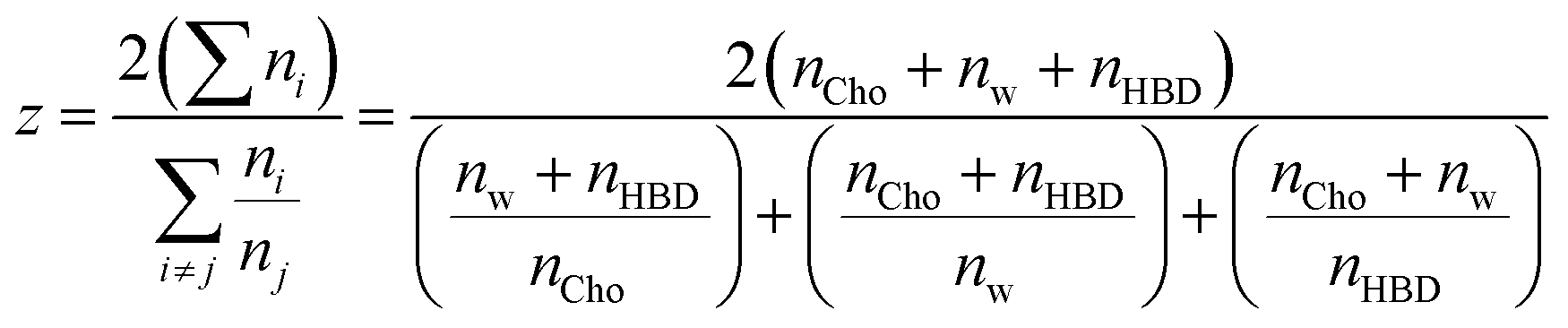 | (2) |
The calculated z (Fig. 3d) follows the same re-entrant trend shown by 35Cl NMR measurements. z peaks at 15.3 wt% H2O for both DES, or a water fractional coordination of 0.33 (Fig. 3c). The environment becomes more symmetrical as water becomes dominant. This ‘intermediate’ water content, with the highest asymmetry of the Cl− environment, corresponds with other observations of large water–chloride ‘pearl-on-string’ clusters in ChCl:U,14 and the minimum melting temperature of ChCl:U:water ternary systems.57 Though it is a bulk measure, the key role of Cl− is highlighted by the minimum in mixing enthalpy as a function of water content,58 which corresponds with maximum Cl− asymmetry. The mixing enthalpy then increases to a maximum at 60–70 wt% H2O. Initial added water is therefore energetically favourable, but this ceases when it has to directly compete with the HBD, Ch+, and Cl− interactions. At 25–35 wt%, ΔmixH = 0 coincides with a fractional water coordination around the chloride of about 0.5 (Fig. 3c). Finally, the competition between the water and DES components weakens after reaching maximum mixing enthalpy and conductivity.34
The unified NMR and MD analysis therefore highlights three distinct solvation regimes in DES/water mixtures:
1. In the pure/low-hydrated DES regime, the Cl− environment is mostly symmetrical, with a remarkably homogeneous solvation shell of the HBD and Ch+.
2. Increasing water content to maximum z/FWHM0 (ca. 40 wt% for ChCl:G and 60 wt% for ChCl:U), the asymmetry around Cl− is maximised, as water competes with the HBD and Ch+.
3. Above the maximum in z/FWHM0, water dominates the 35Cl local environment, maximising symmetry. This reduces the net field gradient at the Cl− nucleus, minimising linewidth.
In conclusion, viscosity-corrected 35Cl NMR spectroscopy and MD simulations elucidated three hydration regimes of Cl− in DES upon aqueous dilution. Adding water initiates asymmetric hydration around the Cl−, as it displaces DES components in the primary solvation shell, distorting the symmetry. This causes the initial rise in FWHM0, as the field gradient upon the nucleus increases. The asymmetry then decreases as water becomes the dominant ligand. Overall, these findings are in line with the debate suggesting structural heterogeneity on molecular scales in certain hydrated DESs,59,60 and the idea of different solvation regimes in the mixtures, where chloride (and the solvent as a whole) becomes partially and then fully hydrated.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. J. B. M. Kollau, M. Vis, A. Van Den Bruinhorst, A. C. C. Esteves and R. Tuinier, Chem. Commun., 2018, 54, 13351–13354 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- O. S. Hammond and K. J. Edler, Deep Eutectic Solvents: Synthesis, Properties, and Applications, 2019, pp. 25–42 Search PubMed.
- C. Ma, A. Laaksonen, X. Ji and X. Lu, Chem. Soc. Rev., 2018, 47, 8685–8720 RSC.
- Y. Chen, D. Yu, W. Chen, L. Fu and T. Mu, Phys. Chem. Chem. Phys., 2019, 21, 2601–2610 RSC.
- C. R. Ashworth, R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2016, 18, 18145–18160 RSC.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- E. O. Fetisov, D. B. Harwood, I.-F. W. Kuo, S. E. E. Warrag, M. C. Kroon, C. J. Peters and J. I. Siepmann, J. Phys. Chem. B, 2018, 122, 1245–1254 CrossRef CAS PubMed.
- V. Migliorati, F. Sessa and P. D’Angelo, Chem. Phys. Lett.: X, 2019, 2, 100001 CAS.
- R. Stefanovic, M. Ludwig, G. B. Webber, R. Atkin and A. J. Page, Phys. Chem. Chem. Phys., 2017, 19, 3297–3306 RSC.
- P. Kumari, Shobhna, S. Kaur and H. K. Kashyap, ACS Omega, 2018, 3, 15246–15255 CrossRef CAS PubMed.
- L. Sapir and D. Harries, J. Chem. Theory Comput., 2020, 16, 3335–3342 CrossRef CAS PubMed.
- E. S. C. Ferreira, I. V. Voroshylova, N. M. Figueiredo, C. M. Pereira and M. N. D. S. Cordeiro, J. Mol. Liq., 2020, 298, 111978 CrossRef CAS.
- O. S. Hammond, D. T. Bowron, A. J. Jackson, T. Arnold, A. Sanchez-Fernandez, N. Tsapatsaris, V. Garcia Sakai and K. J. Edler, J. Phys. Chem. B, 2017, 121, 7473–7483 CrossRef CAS PubMed.
- M. Gilmore, L. M. Moura, A. H. Turner, M. Swadźba-Kwaśny, S. K. Callear, J. A. McCune, O. A. Scherman and J. D. Holbrey, J. Chem. Phys., 2018, 148, 193823 CrossRef PubMed.
- A. Mannu, M. Ferro, G. Colombo, M. E. Di Pietro, S. Garroni and A. Mele, J. Mol. Liq., 2020, 301, 112441 CrossRef CAS.
- A. Mannu, M. E. Di Pietro and A. Mele, Molecules, 2020, 25, 1–11 CrossRef PubMed.
- D. Smink, A. Juan, B. Schuur and S. R. A. Kersten, Ind. Eng. Chem. Res., 2019, 58, 16348–16357 CrossRef CAS.
- A. Triolo, F. Lo Celso and O. Russina, J. Phys. Chem. B, 2020, 124, 2652–2660 CrossRef CAS PubMed.
- D. Shah and F. S. Mjalli, Phys. Chem. Chem. Phys., 2014, 16, 23900–23907 RSC.
- C. F. Araujo, J. A. P. Coutinho, M. M. Nolasco, S. F. Parker, P. J. A. Ribeiro-Claro, S. Rudic, B. I. G. Soares and P. D. Vaz, Phys. Chem. Chem. Phys., 2017, 19, 17998–18009 RSC.
- N. López-Salas, J. M. Vicent-Luna, S. Imberti, E. Posada, M. J. Roldán, J. A. Anta, S. R. G. Balestra, R. M. Madero Castro, S. Calero, R. J. Jiménez-Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. Del Monte, ACS Sustainable Chem. Eng., 2019, 7, 17565–17573 CrossRef.
- R. Ahmadi, B. Hemmateenejad, A. Safavi, Z. Shojaeifard, A. Shahsavar, A. Mohajeri, M. Heydari Dokoohaki and A. R. Zolghadr, Phys. Chem. Chem. Phys., 2018, 20, 18463–18473 RSC.
- A. Pandey and S. Pandey, J. Phys. Chem. B, 2014, 118, 14652–14661 CrossRef CAS PubMed.
- E. Posada, N. López-Salas, R. J. Jiménez Riobóo, M. L. Ferrer, M. C. Gutiérrez and F. Del Monte, Phys. Chem. Chem. Phys., 2017, 19, 17103–17110 RSC.
- C. D’Agostino, L. F. Gladden, M. D. Mantle, A. P. Abbott, E. I. Ahmed, A. Y. M. Al-Murshedi and R. C. Harris, Phys. Chem. Chem. Phys., 2015, 17, 15297–15304 RSC.
- E. Posada, M. J. Roldán-Ruiz, R. J. Jiménez Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, J. Mol. Liq., 2019, 276, 196–203 CrossRef CAS.
- M. J. Roldán-Ruiz, R. J. Jiménez-Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, J. Mol. Liq., 2019, 284, 175–181 CrossRef.
- P. L. Pisano, M. Espino, M. de los, Á. Fernández, M. F. Silva and A. C. Olivieri, Microchem. J., 2018, 143, 252–258 CrossRef CAS.
- A. H. Turner and J. D. Holbrey, Phys. Chem. Chem. Phys., 2019, 21, 21782–21789 RSC.
- F. Gabriele, M. Chiarini, R. Germani, M. Tiecco and N. Spreti, J. Mol. Liq., 2019, 291, 111301 CrossRef CAS.
- V. Agieienko and R. Buchner, J. Chem. Eng. Data, 2019, 64, 4763–4774 CrossRef CAS.
- D. L. Bryce, M. Gee and R. E. Wasylishen, J. Phys. Chem. A, 2001, 105, 10413–10421 CrossRef CAS.
- M. Silva, A. M. Figueiredo and E. J. Cabrita, Phys. Chem. Chem. Phys., 2014, 16, 23394–23403 RSC.
- H. Gustavsson and B. Lindman, J. Am. Chem. Soc., 1978, 100, 4647–4654 CrossRef CAS.
- N. Hedin, I. Furó and P. O. Eriksson, J. Phys. Chem. B, 2000, 104, 8544–8547 CrossRef CAS.
- R. C. Remsing, G. Hernandez, R. P. Swatloski, W. W. Massefski, R. D. Rogers and G. Moyna, J. Phys. Chem. B, 2008, 112, 11071–11078 CrossRef CAS PubMed.
- P. G. Gordon, D. H. Brouwer and J. A. Ripmeester, ChemPhysChem, 2010, 11, 260–268 CrossRef CAS.
- V. Klimavicius, Z. Gdaniec and V. Balevicius, Spectrochim. Acta, Part A, 2014, 132, 879–883 CrossRef CAS PubMed.
- R. P. Chapman, C. M. Widdifield and D. L. Bryce, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 55, 215–237 CrossRef CAS.
- P. M. J. Szell and D. L. Bryce, Recent advances in chlorine, bromine, and iodine solid-state NMR spectroscopy, Elsevier Ltd, 1st edn, 2015, vol. 84 Search PubMed.
- R. C. Remsing, J. L. Wildin, A. L. Rapp and G. Moyna, J. Phys. Chem. B, 2007, 111, 11619–11621 CrossRef CAS PubMed.
- M. Yudasaka, T. Sugawara, H. Iwamura and T. Fujiyama, Bull. Chem. Soc. Jpn., 1981, 54, 1933–1938 CrossRef CAS.
- J. W. Akitt, in Multinuclear NMR, ed. J. Mason, Plenum Press, New York, 1987, pp. 447–461 Search PubMed.
- R. C. Remsing, R. P. Swatloski, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 1271–1273 RSC.
- B. C. Hall, Q. Rev. Chem. Soc., 1971, 25, 87–109 RSC.
- H. Shekaari, M. T. Zafarani-moattar and B. Mohammadi, J. Mol. Liq., 2017, 243, 451–461 CrossRef CAS.
- Y. Xie, H. Dong, S. Zhang, X. Lu and X. Ji, J. Chem. Eng. Data, 2014, 59(11), 3344–3352 CrossRef CAS.
- N. Rodriguez Rodriguez, A. Van Den Bruinhorst, L. J. B. M. Kollau, M. C. Kroon and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 11521–11528 CrossRef CAS.
- A. Van Den Bruinhorst, T. Spyriouni, J. R. Hill and M. C. Kroon, J. Phys. Chem. B, 2018, 122, 369–379 CrossRef CAS PubMed.
- H. Wennerström, B. Lindman and S. Forsén, J. Phys. Chem., 1971, 75, 2936–2942 CrossRef.
- A. T. Celebi, T. J. H. Vlugt and O. A. Moultos, J. Phys. Chem. B, 2019, 123, 11014–11025 CrossRef CAS PubMed.
- X. Meng, K. Ballerat-Busserolles, P. Husson and J.-M. Andanson, New J. Chem., 2016, 40, 4492–4499 RSC.
- D. H. Powell, A. C. Barnes, J. E. Enderby, G. W. Neilson and P. S. Salmon, Faraday Discuss. Chem. Soc., 1988, 85, 137–146 RSC.
- P. J. Smith, C. B. Arroyo, F. Lopez Hernandez and J. C. Goeltz, J. Phys. Chem. B, 2019, 123, 5302–5306 CrossRef CAS PubMed.
- C. Ma, Y. Guo, D. Li, J. Zong, X. Ji, C. Liu and X. Lu, J. Chem. Eng. Data, 2016, 61, 4172–4177 CrossRef CAS.
- A. Abbott, L. Aldous, N. Borisenko, S. Coles, O. Fontaine, J. D. Gamarra Garcia, R. Gardas, O. Hammond, L. J. Hardwick, P. H. Haumesser, F. Hausen, C. Horwood, J. Jacquemin, R. Jones, E. Jónsson, A. Lahiri, D. Macfarlane, G. Marlair, B. May, H. Medhi, V. H. Paschoal, J. E. S. J. Reid, T. Schoetz, K. Tamura, M. L. Thomas, S. Tiwari, B. Uralcan, A. Van Den Bruinhorst, M. Watanabe and J. Wishart, Faraday Discuss., 2018, 206, 405–426 RSC.
- L. Percevault, A. Jani, T. Sohier, L. Noirez, L. Paquin, F. Gauffre and D. Morineau, J. Phys. Chem. B, 2020, 124, 9126–9135 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 35Cl NMR, viscosity and MD details. See DOI: 10.1039/d0cp05843b |
| ‡ These authors contributed equally as joint first author. |
| This journal is © the Owner Societies 2021 |