
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental measurement and prediction of ionic liquid ionisation energies†
Jake M.
Seymour
 a,
Ekaterina
Gousseva
a,
Alexander I.
Large
ad,
Coby J.
Clarke
b,
Peter
Licence
b,
Richard M.
Fogarty
c,
David A.
Duncan
d,
Pilar
Ferrer
d,
Federica
Venturini
d,
Roger A.
Bennett
a,
Robert G.
Palgrave
e and
Kevin R. J.
Lovelock
*a
a,
Ekaterina
Gousseva
a,
Alexander I.
Large
ad,
Coby J.
Clarke
b,
Peter
Licence
b,
Richard M.
Fogarty
c,
David A.
Duncan
d,
Pilar
Ferrer
d,
Federica
Venturini
d,
Roger A.
Bennett
a,
Robert G.
Palgrave
e and
Kevin R. J.
Lovelock
*a
aDepartment of Chemistry, University of Reading, Reading, RG6 6AD, UK. E-mail: k.r.j.lovelock@reading.ac.uk
bSchool of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK
cDepartment of Materials, Imperial College London, SW7 2AZ, UK
dDiamond Light Source, Didcot, Oxfordshire OX11 0DE, UK
eDepartment of Chemistry, University College London, UK
First published on 3rd September 2021
Abstract
Ionic liquid (IL) valence electronic structure provides key descriptors for understanding and predicting IL properties. The ionisation energies of 60 ILs are measured and the most readily ionised valence state of each IL (the highest occupied molecular orbital, HOMO) is identified using a combination of X-ray photoelectron spectroscopy (XPS) and synchrotron resonant XPS. A structurally diverse range of cations and anions were studied. The cation gave rise to the HOMO for nine of the 60 ILs presented here, meaning it is energetically more favourable to remove an electron from the cation than the anion. The influence of the cation on the anion electronic structure (and vice versa) were established; the electrostatic effects are well understood and demonstrated to be consistently predictable. We used this knowledge to make predictions of both ionisation energy and HOMO identity for a further 516 ILs, providing a very valuable dataset for benchmarking electronic structure calculations and enabling the development of models linking experimental valence electronic structure descriptors to other IL properties, e.g. electrochemical stability. Furthermore, we provide design rules for the prediction of the electronic structure of ILs.
 Kevin R. J. Lovelock | Kevin Lovelock has been selected as a PCCP Emerging Investigator. He held an Imperial College London Junior Research Fellowship from 2012 to 2015, followed by a year as a stay-at-home dad. In 2016 he was awarded a Royal Society University Research Fellowship, hosted by the University of Oxford; in 2017 he switched his fellowship to the University of Reading. His current research interests include studying ions in solution and at interfaces for energy applications using both laboratory and synchrotron X-ray spectroscopy. |
1. Introduction
Ionisation energy, Ei, is a key descriptor for chemical, photochemical and electrochemical reactivity,1–5 especially any application that involves exchange of electrons, particularly formal donation of an electron (ionisation) or donation of electron density (partial ionisation). For ionic liquids (ILs), these potential applications include: electrochemical energy storage; gas capture/separation/storage; as solvents for catalysis and metal extraction/separation.6–11 The identity of the most readily ionised valence state, often called the highest occupied molecular orbital (HOMO),12 is also a reactivity descriptor, particularly for ILs given the HOMO could come from the anion or from the cation. Furthermore, given their importance, Ei and the HOMO identity can be used for quantitative validation of calculations of ILs.13Ei can be used to validate methods, e.g. choice of functional/basis set in density functional theory (DFT) can be benchmarked.13 HOMO identity can be used to validate the ability of calculations to capture the solvation effects of ions in liquid phase. However, for ILs there is limited experimental data on electronic structure, including Ei and HOMO identity.Most measurements of Ei have been made using non-resonant X-ray photoelectron spectroscopy (XPS) and ultraviolet photoelectron spectroscopy (UPS).14 Almost all Ei values for ILs, Ei(IL), have been measured on vaporised gas phase neutral ion pairs.15–26 Whilst measuring Ei is relatively facile in the gas phase, a major problem is that most ILs are very tricky to vaporise without significant thermal decomposition occurring/dominating, meaning many IL ion pairs cannot be easily studied in the vapour phase;27–32 furthermore, vapour phase ion pairs do not have the complete solvation environment of the liquid phase. A major hurdle for measuring reliable, reproducible, and comparable binding energies (EB)14 and Ei for liquid phase ILs is dealing with sample charging during XPS measurements, which is not understood.33,34 A widely-used, very robust method of charge referencing XP spectra for ILs is using EB(Calkyl 1s) = 285 eV for ILs with relatively long alkyl chains, usually –C8H17.33,35–48EB values of valence states using this charge referencing method have been reported, e.g. EB(HOMO), EB(cation HOFO) and EB(anion HOFO), where the ion HOFOs are the highest occupied fragment orbital of each ion (one of which is the IL HOMO).48 However, all of these EB values were effectively charge referenced to an apparent Fermi level for the alkyl chains, not the vacuum level. Reporting of experimental Ei(IL) values (by definition, charge referenced to the vacuum level) for liquid phase ILs have been very limited, with little or no mention of charge referencing given;49–51 these studies were published before IL sample charging was an acknowledged problem. Recently, Ei(CC–C/C–H 1s) = 289.58 ± 0.14 eV was given as a reference to the vacuum level for C–C/C–H carbon adsorbed on conducting metal surfaces,52–56 although this value has not been applied to IL XPS data to date.
A key challenge is to identify the valence states for ILs. Most importantly, which of the cation and the anion gives rise to the HOMO? For traditional salts such as NaCl, the anion is very clearly the HOMO, with the Na 2p cation HOFO valence state ∼26 eV larger EB than the Cl 3p anion HOFO.57 However, for ILs EB of the cation HOFO and anion HOFO are far more similar. Furthermore, ILs have a relatively large number of valence electrons per the cation–anion molecular unit. Water and NaCl both have eight valence electrons and therefore the valence states are relatively easy to identify using XPS.57–60 In contrast, common ILs can have between 50 and 300 valence electrons.61 Therefore, ILs have many valence states at similar EB. Compounding this problem, the large range of ion solvation environments in the liquid phase is expected to give a significant range of EB for the nominally the same valence state, as demonstrated for Cl− ion solvated in water.62 Consequently, valence XP spectra of ILs often have many overlapping contributions, making the separation of cation and anion contributions very difficult. The most common laboratory-based XPS apparatus employs Al Kα radiation at hν = 1486.6 eV, giving non-resonant XPS. Valence XPS data from hν = 1486.6 eV has been analysed using a visual fingerprint method and subtraction,48 although this approach is difficult when using results measured on different apparatus. Furthermore, for XPS measured at hν = 1486.6 eV the most important contributors to cation-based valence states, C 2p and N 2p, have very low photoionisation cross-sections relative to many of the common anion-based valence states, e.g. Cl 3p, S 3p, making identification of cationic-based valence states very challenging in particular. Measuring photoelectron spectroscopy with a second hν, e.g. hν = 21.2 eV from He(I) giving non-resonant UPS, helps valence state identification due to variation in photoionisation cross-sections with varying hν.48,63–68 However, most valence state identification for ILs to date has relied on comparisons to calculations; this situation is less than ideal when trying to use experimental data to validate calculations. Valence state identification for ILs has mostly been limited to ILs comprised of [CnC1Im]+ (1-alkyl-3-methylimidazolium), with a small selection of commonly studied anions, particularly cyano-based anions and [NTf2]− (bis[(trifluoromethane)sulfonyl]imide).48,63–73 A recent development has been the use of resonant Auger electron spectroscopy (RAES, also known as resonant XPS, RXPS), which allows identification of valence states, particularly those states with strong π-bonding contributions, e.g. imidazolium ring, cyano-based anions.48 Using an approach combining variable hν XPS and RXPS, key values for 37 ILs were determined: EB(HOMO), EB(cation HOFO) and EB(anion HOFO).48 ΔEB(ion HOFO) = EB(cation HOFO) − EB(anion HOFO) has been used as the key variable for judging IL HOMO identity.48 For [CnC1Im][A] ILs (where [A]− = anion), XPS data charge referenced to EB(Calkyl 1s) = 285.0 eV gave EB(cation HOFO) = 4.8 ± 0.4 eV (independent of [A]−) and the [CnC1Im]+ HOFO (at least for n ≤ 8) was composed of N 2p and C 2p contributions from the imidazolium ring.48 For the ILs [CnC1Im][BF4], [C4C1Im][PF6] and [C6C1Im][B(CN)4] the cation (rather than the anion) has been identified as giving rise to the HOMO.48,49,74–76
The possibility, out of the potentially vast number of ILs, that an ideal IL exists for a particular application is an appealing prospect. The challenge of synthesising, characterising and testing a large number of potential ILs for an application is daunting and makes screening using predictions hugely advantageous. One important question for understanding and predicting IL properties is: how independent is the electronic structure of the cation from the anion and vice versa? Using XPS it has been demonstrated implicitly, i.e. by studying core state EB for elements located specifically in the cation, that the anion influenced the valence electronic structure of the cation (for the cations imidazolium,35,37 pyridinium,41,45 ammonium (linear39 and cyclic36,44) and phosphonium39). Anions with larger Kamlet–Taft β solvent parameters gave lower EB, e.g. EB(Ncation 1s) and EB(Cring 1s) for [C8C1Im][A], and vice versa.35,37,77 Furthermore, using XPS it has been demonstrated that the cation influenced the electronic structure of the anion; [C8C1Im][A] gave larger core state EB for elements located specifically in the anion (denoted here as EB(elementanion core)) than [P6,6,6,14][A] when [A]− was the same.78 However, to date no experimental studies have been published to explicitly demonstrate the influence of cation–anion interactions on IL valence states.
Quantitative comparisons of IL electronic structure experimental descriptors to other experimentally measured physical properties are very limited, principally due to a lack of IL electronic structure experimental descriptors. A positive correlation was found between EB(HOMO) from XPS and anodic stability for [C2C1Im][A] where [A]− = [N(CN)2]−, [C(CN)3]− and [B(CN)4]−; both EB(HOMO) and the anodic stability were largest when [A]− = [B(CN)4]− and smallest when [A]− = [C(CN)3]−.69 Gas phase Ei(IL) have been compared to electrochemical stability for a number of ILs (the IL choice being limited to those ILs that can be vaporised).25
Comparisons exist between liquid phase experimental XP/UP spectra for ILs and calculated data, but the structural range of ILs studied is limited.24,49,64–70,72–76,79 Calculations showed the HOMO came from the cation for [CnC1Im][BF4] (n = 1, 2, 4),24,80–82 [C4C1Im][PF6],24,81,82 [C2C1Im][B(CN)4],73,83 which matched well to experimental HOMO identification,48,49,74–76 demonstrating that for this limited range of ILs the calculations captured the solvation of the cation and anion acceptably. Ei(IL) values have been calculated, e.g. ref. 84–87. Absolute comparisons of calculated Ei(IL) and experimental EB values are unsatisfactory due to the calculated data being charge referenced to the vacuum level and the experimental data being charge referenced to a Fermi level (of e.g. metal substrate or alkyl carbon). Therefore, a lack of experimental Ei(IL) data means validation of calculated IL electronic structure, e.g. functional/basis set choices, has not been possible to date.
Calculated IL electronic structure descriptors, e.g. Ei(IL), have been used to predict other properties, e.g. electrochemical stability82–84,86–91 and thermal stability.92,93 For a very small number of ILs a linear correlation between anodic stability and calculated Ei was identified, showing promise for such comparisons;88 such comparisons should be made against experimental data measured on inert electrodes to minimise the importance of specific chemical reactivity with the electrode materials.84 For quantitative structure–property relationships (QSPR), IL electronic structure descriptors such as Ei have been used to understand and predict IL properties.94 Importantly, the accuracy of these calculated electronic structure descriptors is not routinely validated against IL experimental electronic structure data, most likely due to a lack of available experimental data.
In this article, we investigate IL valence electronic structure using experimental methods, without the aid of calculations. Key ions studied are given in Fig. 1; all 60 ILs studied in this paper are given in ESI,† Table S1. All 60 ILs were liquid at room temperature, making XPS experiments relatively straightforward, as no heating was required for any IL studied here. Compared to the ILs studied in ref. 48, two new cation cores were studied here, [CnC1Pyrr]+ and [CnPy]+. Furthermore, 20 new anions were studied, with a variety of properties/reasons to study; how the new anions were chosen is given in ESI,† Section S1. We have studied a total of 60 ILs using laboratory-based XPS; 37 ILs from ref. 48 and 23 ILs newly published here. 18 of these ILs were studied using synchrotron resonant XPS (also known as RAES); 7 ILs from ref. 48 and 11 ILs newly published here. We have measured Ei for all 60 ILs using a combination of our EB(HOMO) values and applying a charge referencing method that is new to ILs. Furthermore, we have identified the HOMO for all 60 ILs and have quantified the electronic effect of the cation on the anion, and vice versa. Finally, we have predicted Ei and the HOMO identity for a further 516 ILs.
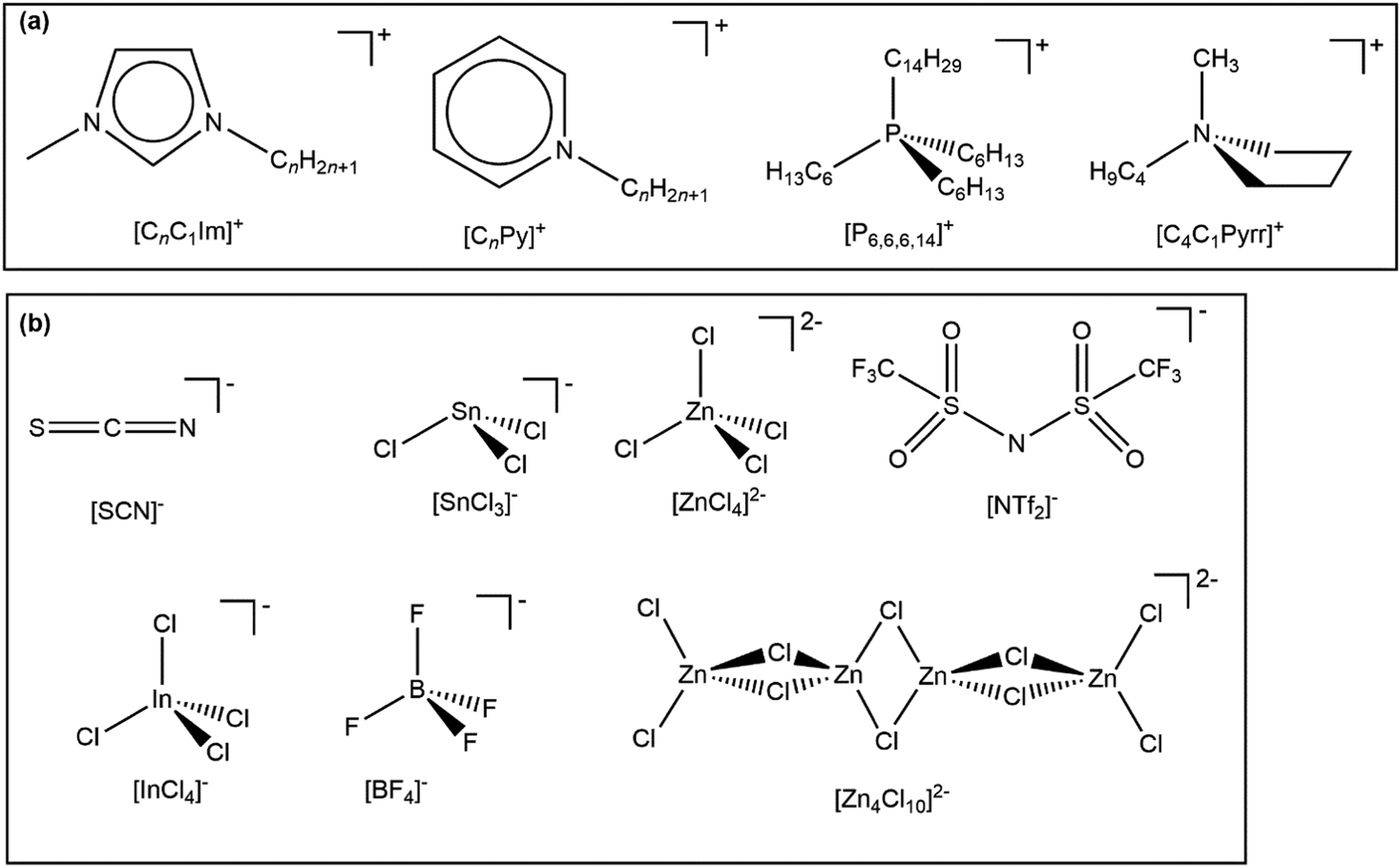 | ||
| Fig. 1 Key ions studied in this paper. A full list of ILs studied is given in the ESI,† Table S1. | ||
2. Experimental
2.1. IL synthesis
Details of IL synthesis are given in the ESI,† Section S1.2.2. Laboratory XPS
Laboratory-based XPS was carried out using four separate XP spectrometers for the 23 ILs studied here. In general, a drop of IL was placed directly onto a stainless steel sample plate (one IL was studied on a glass substrate). This sample was placed in a loadlock and the pressure reduced to 10−7 mbar by pumping down for >6 hours. After attaining the required pressure, the IL was transferred to the analysis chamber. Etching (where necessary) was carried out using a 500 eV Ar+ ion gun (∼10 minutes per sample). Acquisition parameters were matched where possible to give comparable energy resolution; generally, a pass energy of 20 eV was used for core states and ∼40 eV for valence states.(i) Non-resonant XPS of 16 ILs were recorded at University College London on a Thermo Scientific K-alpha monochromated Al Kα source (hν = 1486.6 eV) spectrometer. Charge compensation was achieved using a dual beam flood gun which applied both electrons and low energy Ar+ ions to the sample.
(ii) Non-resonant XPS of 16 ILs were recorded at University College London on a Thermo Scientific Theta Probe monochromated Al Kα source (hν = 1486.6 eV) spectrometer. Charge compensation was achieved using a dual beam flood gun which applied both electrons and low energy Ar+ ions to the sample.
(iii) Non-resonant XPS of four ILs were recorded at the University of Nottingham on a Kratos Axis Ultra equipped with a monochromatic Al Kα source (hν = 1486.6 eV). The core states were published already in ref. 36 and 41–43. Charge compensation was achieved using a flood gun which applied low energy Ar+ ions to the sample.
(iv) Non-resonant XPS of one IL was recorded at Harwell XPS using a Kratos Axis Ultra DLD equipped with a monochromatic Al Kα source (hν = 1486.6 eV). The X-ray source was operated at 150 W (10 mA × 15 kV). Charge compensation was achieved using a flood gun which applied low energy Ar+ ions to the sample.
2.3. Synchrotron XPS and resonant XPS
Synchrotron XPS and resonant XPS were carried out using two separate beamlines, I09 and B07 at Diamond Light Source. In both cases a thin IL film was spread from less than 0.1 ml droplet on a tantalum sample holder so no drop could be observed by eye.The soft synchrotron XPS for [C8C1Im][SnCl3] was performed on the I09 beamline at Diamond Light Source (UK).95 The XP spectra and RXP/RAE spectra were acquired using a VG Scienta EW4000 HAXPES analyser, which had an angular acceptance of ±30°. The analyser was mounted with its lens axis approximately 90° away from the direction of the incident X-ray light in a horizontal plane; the analyser slits (and thus the angular acceptance direction) were also in the horizontal plane. Due to significant observable beam damage/sample charging (ESI,† Section S2), the flux of the synchrotron light was decreased by first defocussing the incident light ∼20 fold and by detuning the undulator (i.e. offsetting the undulator gap) away from the maximum intensity so as to detune the flux a further 100-fold. Prior to XPS measurements the sample was Ar+ sputtered for 30 minutes at a voltage of 500 V.
The soft synchrotron XPS for 10 ILs was performed on the B07 beamline at Diamond Light Source (UK).96 A thin film (less than 0.1 ml, essentially so no drop could be observed) of the IL sample was placed on a tantalum sample holder. For the T-cup apparatus, nine ILs were measured at the N 1s edge. Due to significant observable beam damage, the flux was reduced by using the 1200 l mm−1 grating (T-cup apparatus only). For the T-pot apparatus, one IL was measured at the C 1s edge. Due to significant observable beam damage, the sample was rastered continually perpendicular to the analyser entrance nozzle during X-ray irradiation (T-pot apparatus only); this rastering allowed a higher flux (400 l mm−1 grating) to be used than for the T-cup apparatus.
The RXPS/RAES data were acquired across the N 1s edge (hν ∼ 402 eV) or the C 1s edge (hν ∼ 285 eV); at each hν a RXP spectrum was acquired. Partial electron yield near edge X-ray absorption fine structure (NEXAFS) spectra for the N 1s and C 1s edges were recorded by summing the recorded RAE/RXPS intensity at each hν.
2.4. Analysing XP spectra
All non-resonant XP spectra were fitted using the CASAXPS™ software. Fitting was carried out using a Shirley background and GL30 line shapes (70% Gaussian, 30% Lorentzian). The peak constraints used for core XP spectra are outlined ESI,† Section S3 and peak constraints used for valence XP spectra are outlined ESI,† Section S4. The purity of the ILs studied here is demonstrated in the ESI,† Section S7.2.5. Charge referencing methods for XP spectra
All XP spectra for ILs were effectively charge referenced to C–C/C–H carbon for long alkyl chains. Two different values of EB(Calkyl 1s) were used.(a) EB(Calkyl 1s) = 285.00 eV, which is equivalent to charge referencing to the Fermi level for long alkyl chains. This EB value is standard in the IL literature.33,35–48
(b) Ei(Calkyl 1s) = 289.58 eV,52–56 which is equivalent to charge referencing to the vacuum level for long alkyl chains. Adding the work function97 for long alkyl chains would convert EB to Ei.14 The C–C/C–H carbon contribution to adventitious carbon has been found to match the vacuum level when setting Ei(Calkyl 1s) = 289.58 ± 0.14 eV.52–56 For our measurements, this value effectively means the work function was 289.58 − 285.00 = 4.58 eV. This value for the effective work function of alkyl carbon matches to expected work functions, which often range from 4 eV to 5 eV.98 Therefore, to produce Ei values referenced to the vacuum level from our EB values charge referenced to EB(Calkyl 1s) = 285.0 eV, we added 4.58 eV (Table 1). This charge referencing approach has not been used for ILs to date.
| Descriptors referenced to Fermi level | How determined | Descriptors referenced to vacuum level | How determined | Descriptors for which the reference level does not matter | How determined |
|---|---|---|---|---|---|
| E B(anion HOFO) | Peak fitting and/or estimation | E i(anion) | E B(anion HOFO) + 4.58 eV | ||
| E B(cation HOFO) | Peak fitting and/or estimation | E i(cation) | E B(cation HOFO) + 4.58 eV | ||
| E B(HOMO) | Lowest value of EB(anion HOFO) and EB(cation HOFO) | E i(IL) | E B(HOMO) + 4.58 eV | ΔEB(ion HOFO) | E B(cation HOFO) − EB(anion HOFO) |
| E B(anion onset) | Onset method and/or estimation | E th(anion) | E B(anion onset) + 4.58 eV | ||
| E B(cation onset) | Onset method and/or estimation | E th(cation) | E B(cation onset) + 4.58 eV | ||
| E B(IL onset) | Lowest value of EB(anion onset) and EB(cation onset) | E th(IL) | E B(IL onset) + 4.58 eV | ΔEB(ion onset) | E B(cation onset) − EB(anion onset) |
| E B(cation HOFO,pred.) for [CnC1Im][A] | E B(Ncation 1s) − 397.1 eV | E i(cation,pred.) for [CnC1Im][A] | E B(cation HOFO,pred.) + 4.58 eV | ||
| E B(cation,pred.) for [CnC1Im][A] | E B(cation onset,pred.) − 1.1 eV | E th(cation,pred.) for [CnC1Im][A] | E i(cation,pred.) − 1.1 eV | ||
| HOMO identity | ΔEB(ion HOFO), ΔEB(ion onset) and visual assessment |
For the 60 ILs studied here, six different charge referencing methods were used to charge reference to EB(Calkyl 1s); all charge referencing was carried out after the measurements were completed.
(i) 36 ILs had a sufficiently long alkyl chain that a fitted component for EB(Calkyl 1s) for long alkyl chains was used for charge referencing all XP spectra. This approach to charge reference to EB(Calkyl 1s) has a very high confidence, with an error of less than ±0.1 eV.
(ii) 16 [CnCmIm][A] (where n ≤ 4 and m = 1 or 0) ILs where [C8C1Im][A] IL with the same anion had already been studied, so EB(Ncation 1s) or EB(elementanion core) was used for charge referencing all XP spectra, effectively charge referenced to EB(Calkyl 1s) for long alkyl chains. This approach has a very high confidence, with an error of less than ±0.1 eV.
(iii) 1 IL, [C4C1Im][MeSO4], EB(Ncation 1s) for [C4C1Im][OcSO4] was used to charge reference all XP spectra. As both anions are alkylsulfate, the same functional group was interacting with the countercations; hence, this approach to effectively charge reference to EB(Calkyl 1s) has a very high confidence, with an estimated error of less than ±0.1 eV.
(iv) 1 IL, [C4C0Im][HSO4] with a protic cation, EB(Calkyl 1s) for [C4C1Im][HSO4] was used to charge reference all XP spectra. As n = 4, based on data in ref. 33, this approach to effectively charge reference to EB(Calkyl 1s) has an estimated error of ±0.2 eV.
(v) 4 ILs with relatively short alkyl chains, where an IL with the same anion and a different cation with a long alkyl chain had already been studied (i.e. ILs from charge referencing method i) but the cation was new for XPS studies, so EB(elementanion core) from [C8C1Im][A] was used for charge referencing all XP spectra. Based on data for [C][A] (where [C]+ = cation) where the cation was varied (all with sufficiently long alkyl chains) and the anion kept constant, EB(elementanion core) varied by a maximum of 0.4 eV.78 Therefore, this approach to effectively charge reference to EB(Calkyl 1s) has an error of ±0.2 eV.
(vi) 2 [CnC1Im][A] (where n ≤ 4) ILs where the anion had not been studied for [C8C1Im][A] previously, so EB(Ncation 1s) = 401.9 eV was used for charge referencing all XP spectra as an average EB(Ncation 1s) value. Almost all [C8C1Im][A] ILs gave 401.7 eV < EB(Ncation 1s) < 402.1 eV;33,35 therefore, this approach to effectively charge reference to EB(Calkyl 1s) has an error of ±0.2 eV.
Details of the charge referencing method applied to the synchrotron XP spectra are given in ESI,† Section S5.
2.6. Determining key valence electronic structure descriptors
The onset method used to determine EB(anion onset), EB(cation onset) and EB(IL onset) is explained in ref. 48. Threshold energies, Eth(IL), were obtained by adding 4.58 eV to Ei(IL onset), the IL onset energy charge referenced to the vacuum level); these Eth(IL) values are compared to literature values.The valence electronic structure descriptors charge referenced to the Fermi level are given in column 1 of Table 1, and how they were determined in column 2. Values for valence electronic structure descriptors charge referenced to the Fermi level are given: EB(HOMO) (ESI,† Table S5), EB(anion HOFO) and EB(anion onset) (ESI,† Tables S5 and S6), EB(cation HOFO) and EB(cation onset) (ESI,† Tables S5 and S7), EB(cation HOFO,pred.) (Table 4 and ESI,† Table S8).
The valence electronic structure descriptors charge referenced to the vacuum level are given in column 3 of Table 1, and how they were determined in column 4. Values for valence electronic structure descriptors charge referenced to the vacuum level are given: Ei(IL) values (Table 3), Ei(anion) and Ei(cation) (ESI,† Tables S6 and S7 respectively) and Ei(cation,pred.) (Table 4 and ESI,† Table S8).
The valence electronic structure descriptors for which the reference level does not matter are given in column 5 of Table 1, and how they were determined in column 6. Values for valence electronic structure descriptors for which the reference level does not matter are given: ΔEB(ion HOFO), ΔEB(ion onset) and HOMO identity in Table 3 and ESI,† Table S5.
3. Experimental results
3.1. Determining EB and Ei
For five ILs studied here ([C8C1Im][BF4], [C4C1Im][PF6], [C2C1Im][FAP], [C8C1Im][SbF6] and [C8Py][BF4]) the lowest EB feature due to cationic valence states in the non-resonant valence XP spectra was observable at 4 eV < EB < 7 eV (Fig. 2 and ESI,† Fig. S9f, S12f, S13f, S14f, S15f). However, for 18 ILs studied here, features due to cationic valence states were not readily observed in non-resonant valence XP spectra recorded at hν = 1486.6 eV due to features from the anion valence states dominating (ESI,† Fig. S10–S32); the photoionisation cross-sections of N 2p and C 2p atomic orbitals (AOs) are much lower than many of the anion-based AOs, e.g. Cl 3p.99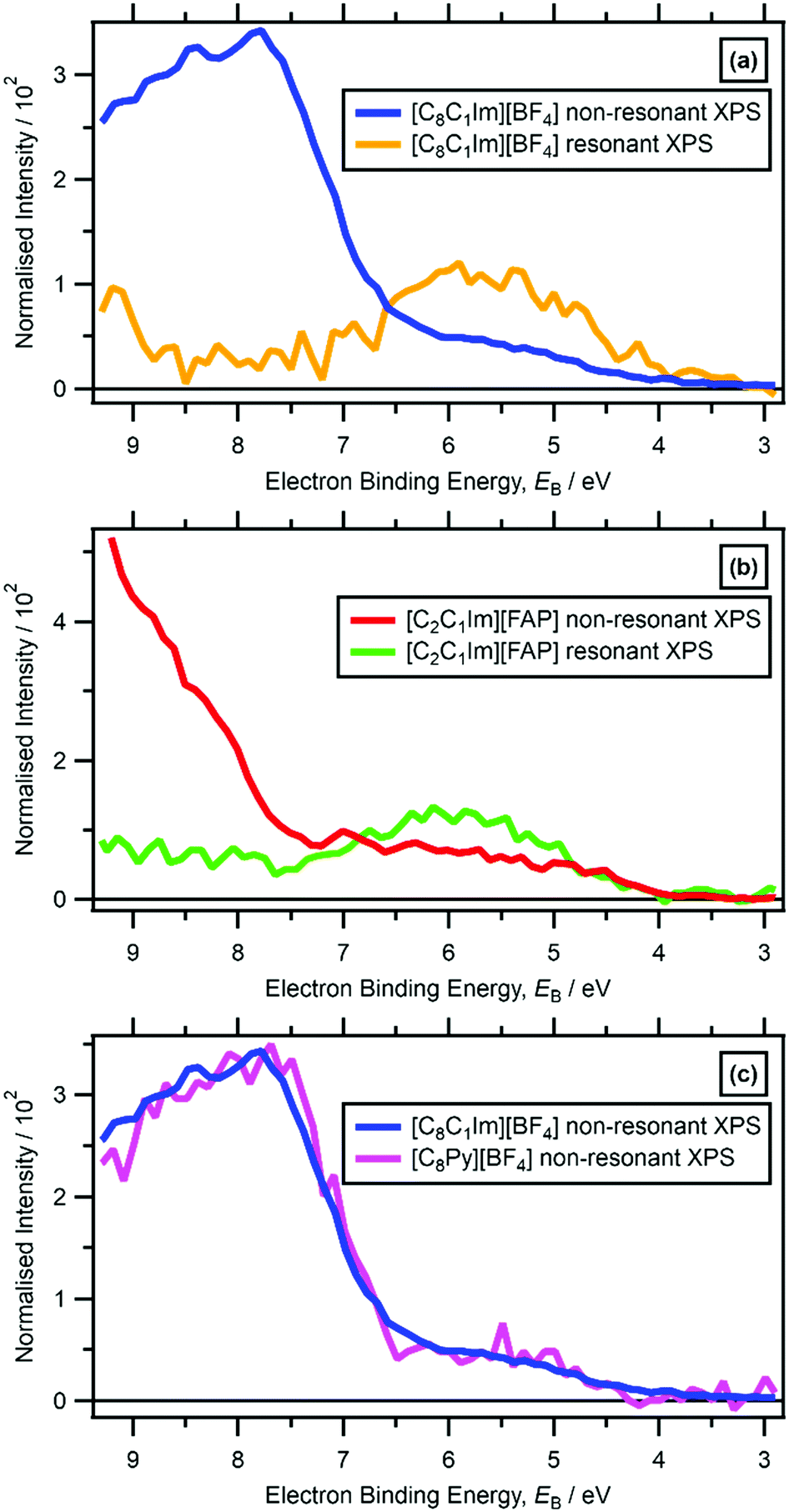 | ||
| Fig. 2 Subtracted Ncation RXP spectrum and non-resonant laboratory valence XPS at hν = 1486.6 eV for: (a) [C8C1Im][BF4], (b) [C2C1Im][FAP]. (c) Non-resonant laboratory valence XPS at hν = 1486.6 eV for [C8C1Im][BF4] and [C8Py][BF4]. These RXPS traces were produced by subtraction of resonant XP spectrum minus non-resonant XP spectrum using the procedure outlined in ref. 48. All electron spectra were charge referenced using methods outlined in Section 2.4. | ||
For RXPS of the 15 [CnC1Im]+-based ILs reported here and in ref. 48, including for [C8C1Im][SnCl3] and [C8C1Im]2[ZnCl4], a broad feature due to resonant enhancement was observed at hν ≈ 402 eV and 3.5 eV < EB(Ncation RXPS) < 7.5 eV (Fig. 3a, c, 4 and ESI,† Fig. S44). hν ≈ 402 eV corresponded to X-ray absorption from the Ncation 1s core state to imidazolium ring π* unoccupied valence state(s) (Fig. 3b and d).48,100 The feature at 3.5 eV < EB(Ncation RXPS) < 7.5 eV was from participator Auger transitions involving valence states with good overlap with the Ncation 1s core hole. Consequently, for [CnC1Im]+-based ILs, valence states at 3.5 eV < EB(Ncation RXPS) < 7.5 eV had strong contributions from Ncation in the imidazolium ring, i.e. from Ncation 2p-based AOs. The anion charge ([A]− or [A]2−) did not have a strong effect on EB(Ncation RXPS) (Fig. 4). The average EB(Ncation 2p) for [C8C1Im][A] was estimated as EB ∼ 5.7 eV (Fig. 4). Based on results presented in ESI,† Fig. S45 and ref. 48, the lowest EB(Ccation 2p) for [C8C1Im][A] was at ∼0.9 eV lower than the feature at EB ∼ 5.7 eV for EB(Ncation 2p) and represents the cation HOFO, as summarised in ESI,† Fig. S47 and the accompanying text. Therefore, for [C8C1Im][A] and [C8C1Im]2[A] charge referenced to EB(Calkyl 1s) = 285.0 eV, EB(cation HOFO) = 4.8 ± 0.4 eV, independent of the identity of the anion [A]− or [A]2−. For two of the [CnC1Im]+-based ILs studied here, [C8C1Im][BF4] and [C2C1Im][FAP], the nitrogen and carbon RXPS features matched very well to the lowest EB feature in the non-resonant valence XP spectra (Fig. 2a and b). This observation demonstrated that our use of RXPS to identify EB(cation HOFO) was very reliable. A similar analysis can be made for [CnPy][A] (see ESI,† Section S8 for more details). Overall, when charge referenced to EB(Calkyl 1s) = 285.0 eV, EB(cation HOFO) = 5.3 ± 0.6 eV for [CnPy][A].
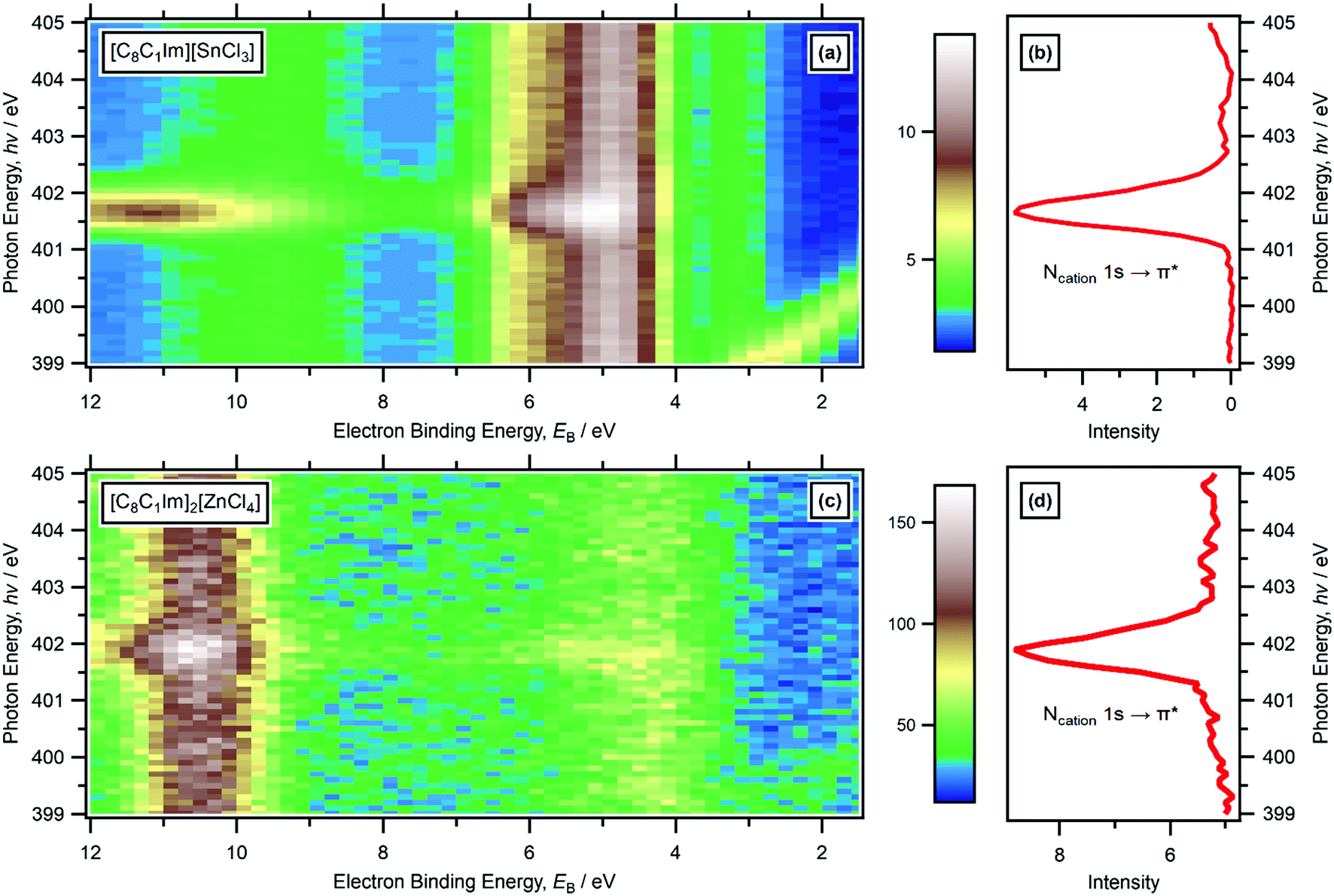 | ||
| Fig. 3 RXPS N 1s edge data. [C8C1Im][SnCl3]: (a) heat map of hν against EB for the N 1s edge; (b) partial electron yield NEXAFS spectrum for the N 1s edge. [C8C1Im]2[ZnCl4]: (c) heat map of hν against EB for the N 1s edge; (d) partial electron yield NEXAFS spectrum for the N 1s edge. These RXPS plots represent the raw data, with no subtraction performed. All electron spectra were charge referenced using methods outlined in Section 2.4. The features at 399 eV < hν < 401 eV and 0 eV < EB < 3 eV were due to N 1s photoemission from 2nd order light. | ||
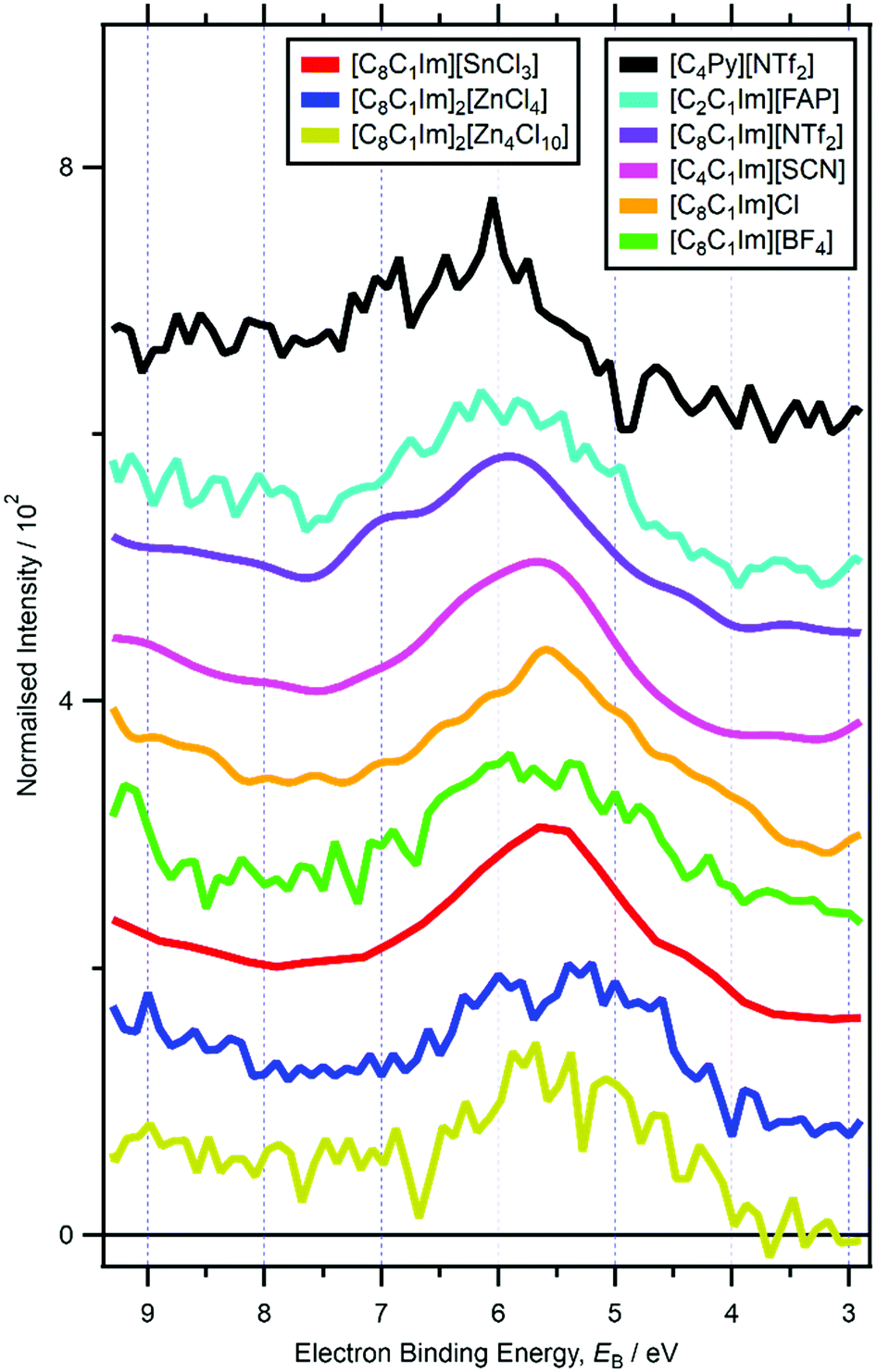 | ||
| Fig. 4 Subtracted RXP spectra for Ncation for nine ionic liquids (hν ∼ 402 eV). These RXPS traces were produced by subtraction of resonant XP spectrum minus non-resonant XP spectrum using the procedure outlined in ref. 48. All electron spectra were charge referenced using methods outlined in Section 2.4. | ||
For ILs with quaternary cations (e.g. [N4,1,1,0][HSO4], [N8,8,8,1][NTf2], [P6,6,6,14][A]), the number of alkyl carbons had a dramatic effect on EB(cation HOFO). For [N4,1,1,0][HSO4] EB(cation HOFO) = 6.5 eV and for [N8,8,8,1][NTf2] and [P6,6,6,14][A] EB(cation HOFO) = 5.0 eV (ESI,† Table S5), demonstrating that longer alkyl chain lengths gave smaller EB(cation HOFO). At this stage, the relationship between the number alkyl carbons/length of alkyl chain and EB(cation HOFO) is unclear. For imidazolium-based ILs where a feature from cationic valence states were readily observed in non-resonant XPS (i.e. [C8C1Im][BF4], [C8C1Im][SbF6], [C6C1Im][B(CN)4], [C4C1Im][PF6], [C2C1Im][FAP]), there was no clear effect of the alkyl chain length on EB(cation HOFO). This observation is most likely because the cation HOFO was from the imidazolium ring and not from the alkyl chain. EB(cation HOFO) was much lower for [N2OH,2OH,2OH,1][TfO] compared to [N4,1,1,0][HSO4] (1.7 eV lower), certainly more than any effect due to the different counteranions or number of CH2 groups; adding OH groups will lower EB(cation HOFO) versus alkyl groups. In ref. 48, EB(cation HOFO) for [N8,1,1,0]+ was estimated to match that of [N4,1,1,0]+. However, based on this effect of the alkyl chain length on EB(cation HOFO), the EB(cation HOFO) value for [N8,1,1,0]+ was re-evaluated as 6.0 eV.
3.2. Quantifying the effect of counterions on EB and Ei
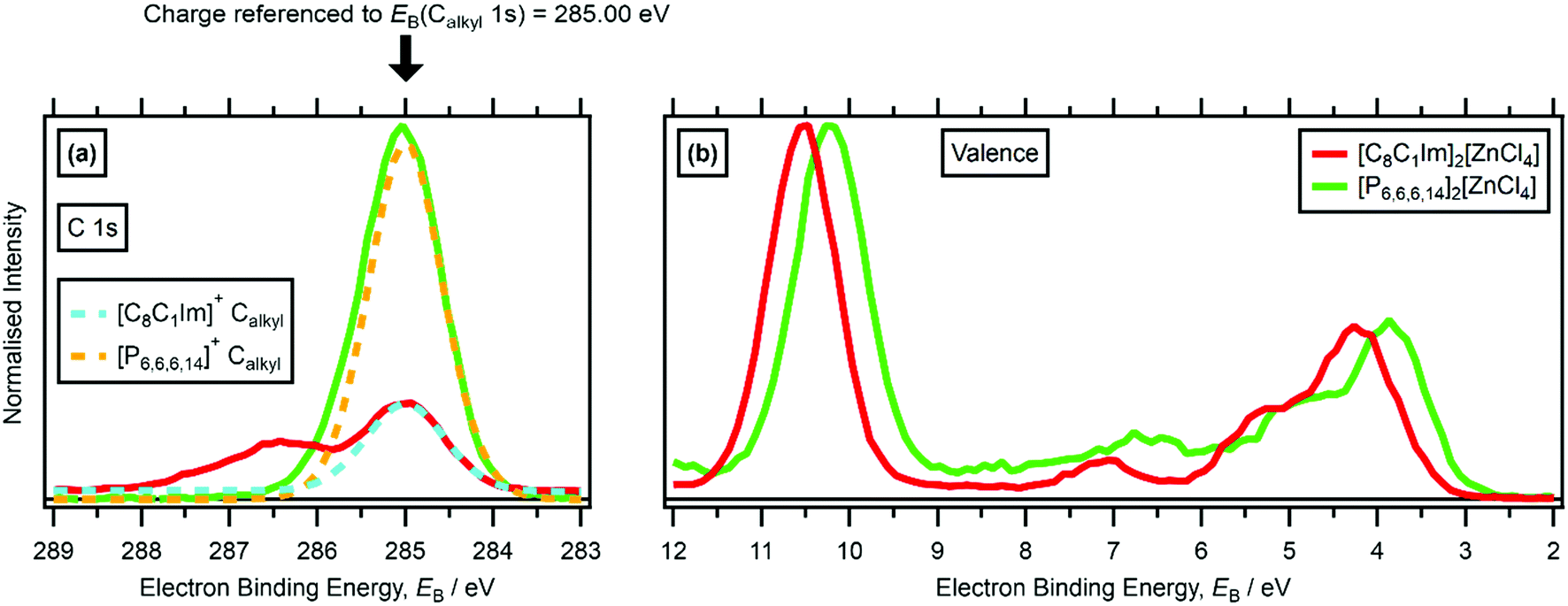 | ||
| Fig. 5 Non-resonant laboratory XPS at hν = 1486.6 eV for [C8C1Im]2[ZnCl4] and [P6,6,6,14]2[ZnCl4]: (a) C 1s, (b) valence. XP spectra were charge referenced to EB(Calkyl 1s) = 285.00 eV (i.e. using method 1 outlined in Section 2.5). | ||
| IL | E B(elementanion core) charge referenced to EB(Calkyl 1s) = 285.0 eV/eV | E B(anion HOFO) charge referenced to EB(Calkyl 1s) = 285.0 eV/eV | E B(anion HOFO) for [C8C1Im][A] − EB(anion HOFO) for [P6,6,6,14][A]/eV |
|---|---|---|---|
| All values were recorded to two decimal places, but the values are reported here to one decimal place; hence, the subtracted values do not appear to match the original values for some ILs. | |||
| [C8C1Im]2[ZnCl4] | 198.2 (Cl 2p3/2) | 4.1 | 0.3 |
| [P6,6,6,14]2[ZnCl4] | 197.8 (Cl 2p3/2) | 3.8 | |
| [C8C1Im]Cl | 197.0 (Cl 2p3/2) | 3.5 | 0.4 |
| [P6,6,6,14]Cl | 196.6 (Cl 2p3/2) | 3.1 | |
| [C8C1Im]Br | 67.4 (Br 3d5/2) | 3.1 | 0.2 |
| [P6,6,6,14]Br | 67.1 (Br 3d5/2) | 2.9 | |
| [C4C1Im][N(CN)2] | 398.4 (Nterminal 1s) | 3.5 | 0.3 |
| [P6,6,6,14][N(CN)2] | 398.0 (Nterminal 1s) | 3.2 | |
| [C8C1Im][NTf2] | 398.5 (Nanion 1s) | 5.0 | 0.2 |
| [P6,6,6,14][NTf2] | 398.3 (Nanion 1s) | 4.8 | |
| Ionic liquid | E i(IL)/eV | HOMO identity |
|---|---|---|
| [C8C1Im]2[FeCl4] | 6.0 ± 0.3 | Anion |
| [C6C1Im]I | 7.2 ± 0.2 | Anion |
| [C8C1Im]2[CoBr4] | 7.3 ± 0.3 | Anion |
| [C8C1Im][C(CN)3] | 7.4 ± 0.2 | Anion |
| [P6,6,6,14]Br | 7.5 ± 0.2 | Anion |
| [C8C1Im]2[CoCl4] | 7.5 ± 0.3 | Anion |
| [C4C1Im][SCN] | 7.5 ± 0.2 | Anion |
| [C8C1Im][SCN] | 7.5 ± 0.2 | Anion |
| [C8C1Im][I3] | 7.5 ± 0.3 | Anion |
| [P6,6,6,14]Cl | 7.7 ± 0.2 | Anion |
| [C8C1Im][SnCl3] | 7.7 ± 0.2 | Anion |
| [C8C1Im]Br | 7.7 ± 0.2 | Anion |
| [P6,6,6,14][N(CN)2] | 7.8 ± 0.4 | Anion |
| [C8C1Im]2[NiCl4] | 7.9 ± 0.3 | Anion |
| [P6,6,6,14][NO3] | 8.1 ± 0.4 | Anion |
| [C8C1Im]2[ZnBr4] | 8.1 ± 0.3 | Anion |
| [C4C1Im][N(CN)2] | 8.1 ± 0.4 | Anion |
| [C8C1Im]Cl | 8.1 ± 0.2 | Anion |
| [C8C1Im]2[ZnCl2Br2] | 8.2 ± 0.3 | Anion |
| [P6,6,6,14]2[ZnCl4] | 8.4 ± 0.3 | Anion |
| [C8C1Im]2[Zn2Br6] | 8.4 ± 0.3 | Anion |
| [C8C1Im]2[ZnCl4] | 8.7 ± 0.3 | Anion |
| [C8C1Im]2[Zn3Br8] | 8.7 ± 0.3 | Anion |
| [C8C1Im]2[Zn4Br10] | 8.8 ± 0.3 | Anion |
| [C8C1Im]2[Zn2Cl6] | 9.0 ± 0.3 | Anion |
| [C4C1Im][HSO4] | 9.1 ± 0.4 | Anion |
| [C8C1Im][HSO4] | 9.1 ± 0.4 | Anion |
| [C4C0Im][HSO4] | 9.1 ± 0.4 | Anion |
| [C4C1Im][MeSO4] | 9.1 ± 0.4 | Anion |
| [C4C1Im][OcSO4] | 9.1 ± 0.4 | Anion |
| [C2C1Im][MeSO3] | 9.1 ± 0.4 | Anion |
| [C4C1Im][Me2PO4] | 9.1 ± 0.4 | Anion |
| [N4,1,1,0][HSO4] | 9.1 ± 0.4 | Anion |
| [N8,1,1,0][HSO4] | 9.1 ± 0.4 | Anion |
| [C4C1Im][TfO] | 9.4 ± 0.4 | Cation/anion |
| [C8C1Im][TfO] | 9.4 ± 0.4 | Cation/anion |
| [C4C1Im][NTf2] | 9.4 ± 0.4 | Cation/anion |
| [C6C1Im][NTf2] | 9.4 ± 0.4 | Cation/anion |
| [C8C1Im][NTf2] | 9.4 ± 0.4 | Cation/anion |
| [C2C0Im][NTf2] | 9.4 ± 0.4 | Cation/anion |
| [C4C0Im][NTf2] | 9.4 ± 0.4 | Cation/anion |
| [C8C1Im]2[Co(NTf2)4] | 9.4 ± 0.4 | Cation/anion |
| [N2OH,2OH,2OH,1][TfO] | 9.4 ± 0.4 | Cation/anion |
| [C8C1Im]2[Zn4Cl10] | 9.4 ± 0.4 | Cation/anion |
| [C8C1Im][InCl4] | 9.4 ± 0.4 | Cation |
| [C6C1Im][B(CN)4] | 9.4 ± 0.4 | Cation |
| [C8C1Im][BF4] | 9.4 ± 0.4 | Cation |
| [C4C1Im][PF6] | 9.4 ± 0.4 | Cation |
| [C2C1Im][FAP] | 9.4 ± 0.4 | Cation |
| [C8C1Im][SbF6] | 9.4 ± 0.4 | Cation |
| [C4Py][NTf2] | 9.6 ± 0.4 | Cation/anion |
| [P6,6,6,14][NTf2] | 9.6 ± 0.4 | Cation/anion |
| [N8,8,8,1][NTf2] | 9.6 ± 0.4 | Cation/anion |
| [N2,2,1,0][TfO] | 9.6 ± 0.4 | Anion |
| [C4C1Pyrr][NTf2] | 9.6 ± 0.4 | Anion |
| [N4,1,1,1][NTf2] | 9.6 ± 0.4 | Anion |
| [N3,2,1,1][NTf2] | 9.6 ± 0.4 | Anion |
| [S2,2,2][NTf2] | 9.6 ± 0.4 | Anion |
| [S2,2,1][NTf2] | 9.6 ± 0.4 | Anion |
| [C8Py][BF4] | 9.9 ± 0.6 | Cation |
| Ionic liquid | E B(Ncation 1s)/eV | E B(cation HOFO,pred.)/eV | E i(cation,pred.)/eV |
|---|---|---|---|
| [C8C1Im]Cl | 401.7 | 4.6 | 9.2 |
| [C8C1Im]2[ZnCl4] | 401.8 | 4.7 | 9.3 |
| [C4C1Im][SCN] | 401.9 | 4.8 | 9.4 |
| [C8C1Im][BF4] | 402.0 | 4.9 | 9.5 |
| [C8C1Im][SnCl3] | 402.0 | 4.9 | 9.5 |
| [C8C1Im]2[Zn4Cl10] | 402.1 | 5.0 | 9.5 |
| [C8C1Im][NTf2] | 402.1 | 5.0 | 9.6 |
| [C8C1Im][InCl4] | 402.2 | 5.1 | 9.7 |
| [C2C1Im][FAP] | 402.3 | 5.2 | 9.8 |
No countercation effect on EB(anion HOFO) was observed for [C8C1Im][NTf2] versus [C8Py][NTf2] versus [N8,8,8,1][NTf2] or for [C4Py][BF4] versus [C8Py][BF4] (ESI,† Table S5), when effectively charge referenced to EB(Calkyl 1s) = 285.0 eV.
For features at EB > 12 eV (ESI,† Fig. S48), the dominant contributions were from spectator Auger transitions (i.e. not from participator Auger transitions). When charge referenced to EB(Ncation 1s) (ESI,† Fig. S48), the subtracted Ncation traces (which include peaks due to both participator and spectator Auger transitions) for [CnC1Im][A] where the anion was varied were the same (ESI,† Fig. S48). The main features appeared at the same EB for all [CnC1Im][A] ILs. These findings demonstrate that cationic EB differences were the same (within experimental error) for both valence and core levels, irrespective of the identity of the anion.
For [P6,6,6,14][A], linear/cyclic ammoniums ([C8Pyrr][A], [N6,6,6,14][A], [C8C1Pip][A]) and [C8Py][A], the central group 15 N (or P) atom showed EB(Ncation 1s) (or EB(Pcation 2p3/2)) differences due to the counteranion when charge referenced to EB(Calkyl 1s) = 285.0 eV.36,39,41,44 At present, it is not clear if this counteranion effect on the cation core state translates to any counteranion effect on the cation valence states, i.e. EB(cation HOFO). Given the lack of strong cation participator features for many of these ILs48 and the significant impact of the alkyl chain length on EB(cation HOFO) for these ILs, observing any counteranion effect on the cation valence states appears very challenging.
3.3. HOMO identification
The HOMO identity was judged mainly using ΔEB(ion HOFO) = EB(cation HOFO) − EB(anion HOFO) and ΔEB(ion onset) = EB(cation onset) − EB(anion onset), in combination with a visual assessment of the both resonant and non-resonant valence XP spectra (Table 1). For example, for [C8C1Im][SnCl3] ΔEB(ion onset) = 1.6 ± 0.5 eV and ΔEB(ion HOFO) = 1.7 ± 0.6 eV; no peak due to resonant enhancement was observed at EB(anion HOFO) = 3.1 eV (i.e. only the same non-resonant XPS contribution can be observed at all hν values, Fig. 3a), demonstrating that the peak at lowest EB for [C8C1Im][SnCl3] was from the [SnCl3]− anion. Therefore, for [C8C1Im][SnCl3] the HOMO was from the [SnCl3]− anion (Table 3).For the 60 ILs studied here and in ref. 48, 39 ILs had the anion as the HOMO, 7 ILs had the cation as the HOMO, and for 14 ILs the HOMO was either the cation or the anion as it was too close to judge (Table 3 and ESI,† Table S5). Unambiguously, a significant number of ILs had the cation as the HOMO.
4. Predictions
The consistent EB shift of all valence states when varying the counterion demonstrates that IL valence electronic structure can be predicted, as the non-specific, electrostatic-based EB shift can be applied.For [CnC1Im][A], [CnPy][A], and [ammonium][A] the effect of the countercation on the anion was relatively small, so no changes to EB(anion HOFO) were needed for these cations. For [CnC1Im][A] versus [P6,6,6,14][A] the effect of the countercation on EB(anion HOFO) was significant, with EB(anion HOFO) for [P6,6,6,14][A] ∼0.3 eV lower on average than EB(anion HOFO) for [CnC1Im][A] (Table 2). Therefore, for predictions of ΔEB(ion HOFO) and Ei(IL), EB(anion HOFO) was set 0.3 eV lower for [P6,6,6,14][A] compared to EB(anion HOFO) for [CnC1Im][A]. For example, for [P6,6,6,14][SCN] EB(anion HOFO) = 2.6 eV whereas for EB(anion HOFO) = 2.9 eV for [C8C1Im][SCN].
ΔEB(Ncorrelation) ≈ 396.2 eV was used to predict EB(cation HOFO,pred.), Ei(cation,pred.) and Eth(cation,pred.) for 36 [CnC1Im][A] ILs (Table 4 for select ILs, ESI,† Table S8 for all 36 ILs). However, the effect of the counteranion on EB(cation HOFO) for [P6,6,6,14]+, [CnPy]+, ammonium or [S2,2,n]+ has not been determined. Therefore, for predictions given in Fig. 6–8, no effects of the counteranion on EB(cation HOFO) were included, i.e. EB(cation HOFO) for each cation was kept constant whatever the identity of the anion, e.g. for all imidazolium-based ILs EB(cation HOFO) = 4.8 ± 0.4 eV was used for the predictions presented in Fig. 6–8.
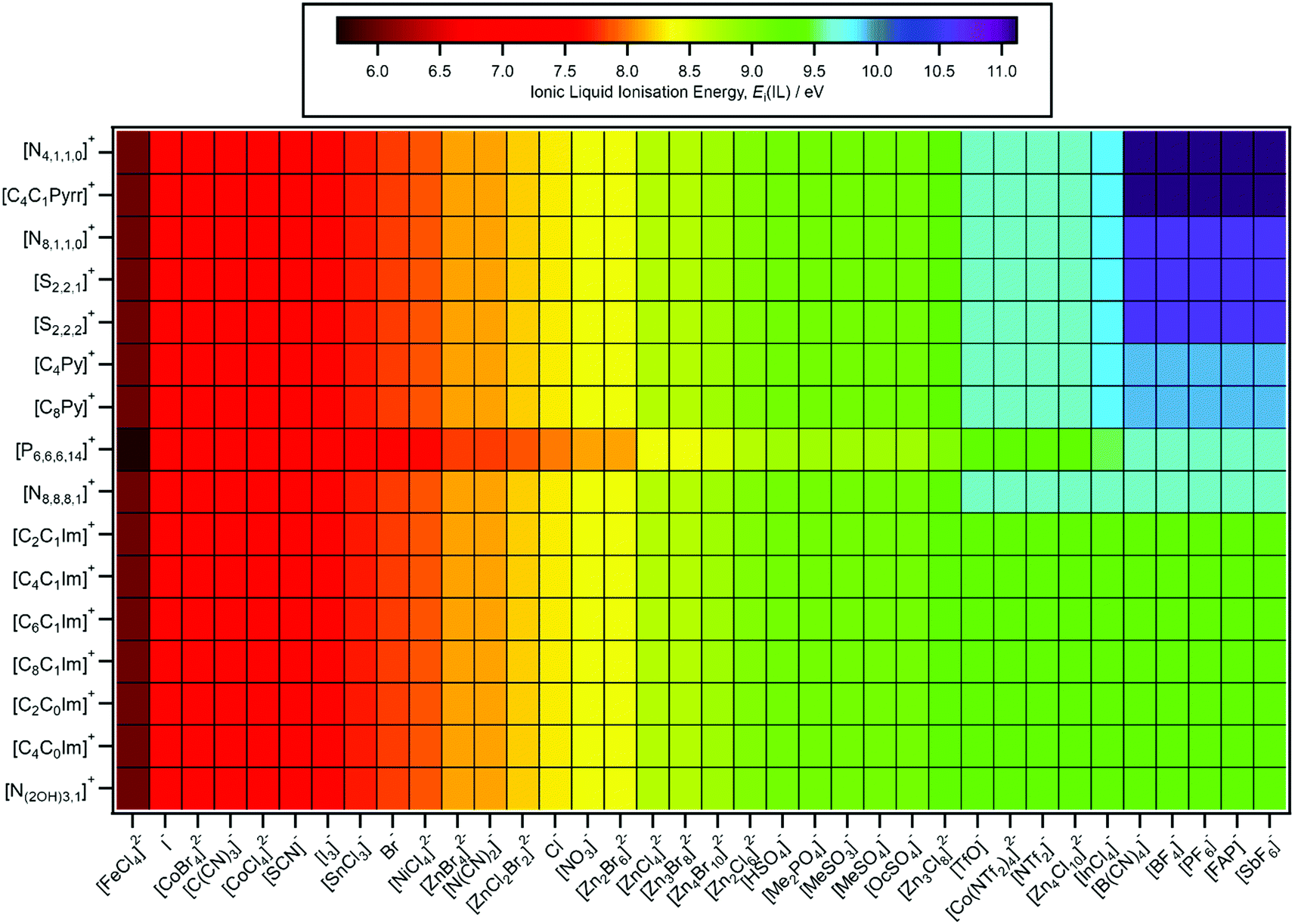 | ||
| Fig. 6 Predicted IL ionisation energies, Ei(IL,pred.). The errors in Ei(IL,pred.) generally range from ±0.2 eV to ±0.4 eV. | ||
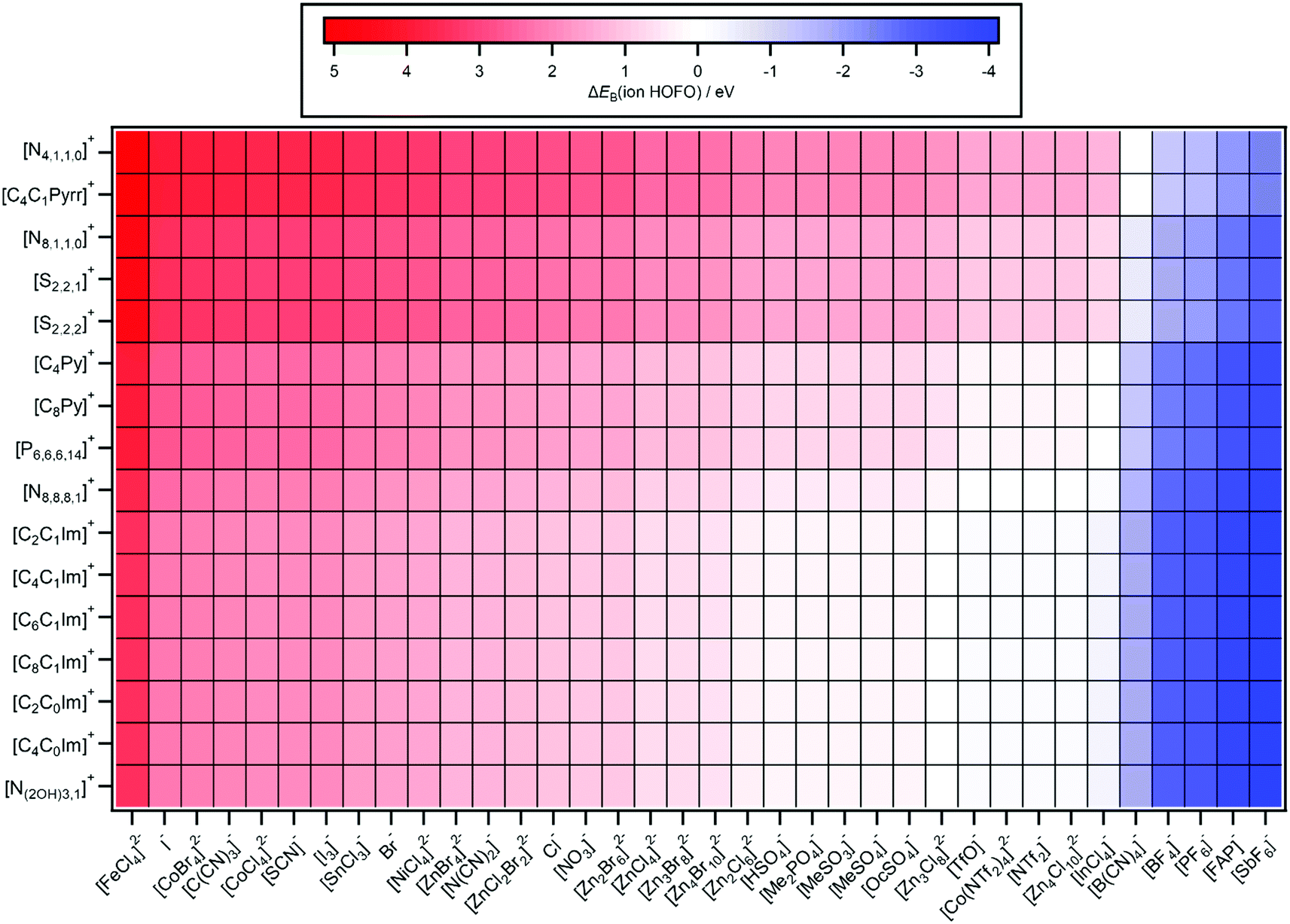 | ||
| Fig. 7 Predicted ΔEB(ion HOFO,pred.) = EB(cation HOFO,pred.) − EB(anion HOFO,pred.) = Ei(cation,pred.) − Ei(anion,pred.). The errors in ΔEB(ion HOFO,pred.) generally range from ±0.4 eV to ±0.6 eV. | ||
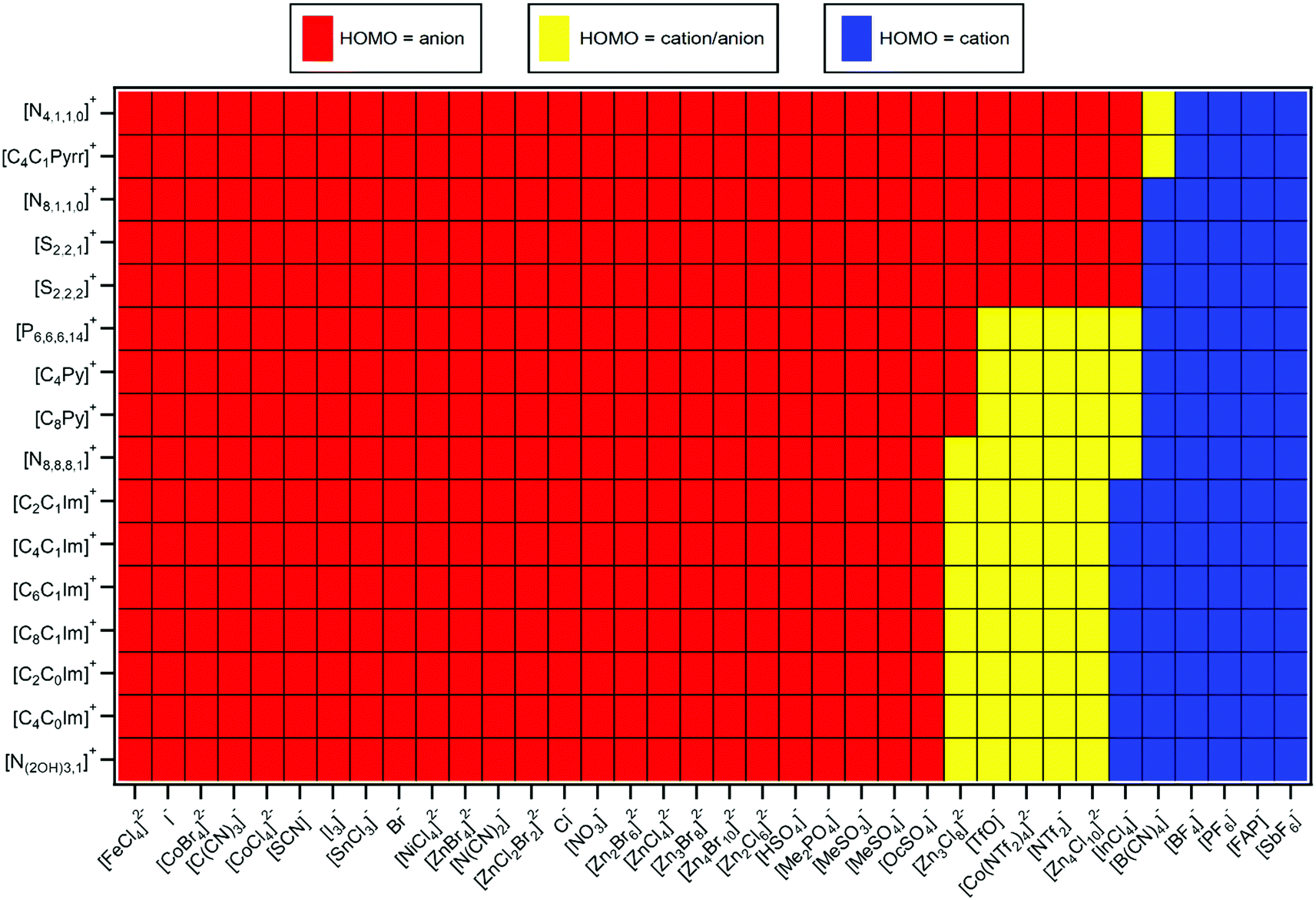 | ||
| Fig. 8 Predicted HOMO identity, judged mainly using ΔEB(ion HOFO,pred.). | ||
Predictions of Ei(IL), ΔEB(ion HOFO) and HOMO identity for 576 ILs are presented in Fig. 6–8 respectively; 60 ILs for which experimental data exists and 516 ILs for which experimental data has not been measured. For these predictions, the IL might not be liquid at room temperature, unlike the 60 ILs studied experimentally here. Furthermore, for some cation–anion combinations, the speciation of the metal complex may be affected by the cation identity.
4.1. E i(IL) predictions
The predicted EB(cation HOFO) and EB(cation HOFO) values were used to obtain Ei(cation), Ei(anion) and Ei(IL) (Fig. 6). As with the experimentally determined values of Ei(IL), the lowest value of Ei(cation) and Ei(anion) for each IL represents Ei(IL).4.2. HOMO identity predictions
ΔEB(ion HOFO) = EB(cation HOFO) − EB(anion HOFO) = Ei(cation) − Ei(anion) was calculated for 576 ILs to produce Fig. 7. Positive ΔEB(ion HOFO) values (red in Fig. 7) represent the anion as the HOMO, whereas negative ΔEB(ion HOFO) values (blue in Fig. 7) represent the cation as the HOMO; the ILs represented by near white have the cation/anion as the HOMO. The decision over which category (HOMO = anion, cation/anion or cation) each IL was placed into to produce Fig. 8 was based mainly on the predicted ΔEB(ion HOFO) value for that IL, although the experimental data was also taken into account for e.g. the alkylsulfate-based ILs. For most ILs, the choice was easy, but for a few ILs the judgement was trickier. This area is expanded upon in the discussion section. Overall, of the 576 ILs, 431 were predicted to have an anion HOMO, 59 were predicted to have a cation/anion HOMO, and 86 were predicted to have a cation HOMO.5. Discussion
5.1. Electrostatic effects of counterions on EB and Ei
The electronic structure of the cation was not independent from the identity of the counteranion, and vice versa; non-specific, electrostatic interactions dominated and specific, directional ion–ion interactions were not important. In comparison, for the NaI dissolved in water, solvation effects on the water caused changes to some valence states of the water but not to other valence states of the water, i.e. the solvation effects were due to specific, directional ion–water interactions between individual water valence states and iodide anion valence states.1025.2. Design rules for tuning Ei and HOMO identity
Given the structural diversity of 36 anions and 16 cations studied here, gathering the anions and cations into groups is very challenging. All anions with Ei(anion) larger than Cl− (i.e. Ei(anion) > 8.1 eV) are defined as superhalogen anions, demonstrating the relative stability of IL anions in general with respect to ionisation.81,103A design rule for producing (closed shell) anions with relatively small Ei(anion) values is the central atom having a formal lone pair, i.e. the anion HOFO is formed from ligand AOs only; conversely, for relatively large Ei(anion) values the central atom should have no formal lone pair. For example, the two cyano-containing ILs with the central atom having a lone pair (using Lewis structures), [N(CN)2]− and [C(CN)3]−, both gave relatively small Ei(anion) values, whereas [B(CN)4]−, for which the central B atom does not have a formal lone pair, gave a relatively large Ei(anion), i.e. the anion HOFO for [B(CN)4]− was formed from cyano ligand AOs only. Furthermore, the [SnCl3]− anion also contains a formal lone pair.79 For [SCN]−, the central atom is carbon, but for this design rule the key atom is sulfur, which contains two formal lone pairs in the most favoured Lewis structure. One outlier to this design rule was [NTf2]−, which has two formal lone pairs (or one lone pair, depending on the resonance structure drawn104) on the central N atom but gave a relatively large Ei(anion) value. This design rule has also been observed for anions with F and O ligands.105 A number of the anions with the smaller Ei(anion) values with a formal lone pair on the key atom also contained the soft and polarisable elements S and I; I−, [I3]−, [SCN]−.
A design rule for producing (closed shell) anions with relatively large Ei(anion) values is the presence of fluorine, e.g. [SbF6]−, [PF6]−, [BF4]−, [FAP]−, [NTf2]− and [TfO]−. All ILs containing a fluorinated anion gave a relatively large Ei(anion). Atomic fluorine has a relatively large EB(F 2p) = 18.7 eV (compared to EB(N 2p) = 13.2 eV to EB(C 2p) = 10.7 eV),106 showing the influence of atomic EB values on EB(anion HOFO) and Ei(anion). Only three anions with Ei(anion) > 9.5 eV contained no F atoms: [B(CN)4]−, [InCl4]− and [Zn4Cl10]2−.
The dianions where the central atom was a transition metal (i.e. open shell) gave relatively small Ei(anion) values; indeed, the anion that gave the smallest Ei(anion) value was [FeCl4]2−. However, having a dianion does not guarantee a small Ei(anion); [Zn4Cl10]2− gave relatively a large Ei(anion) value.
A design rule for producing relatively small Ei(cation) values is either: (a) aromatic cations with readily ionised π systems (e.g. imidazolium and pyridinium) or (b) very long alkyl chains (e.g. [P6,6,6,14]+). Conversely, a design rule for producing relatively large Ei(cation) values is a non-aromatic cation with short alkyl chains (e.g. [N4,1,1,0]+, [S2,2,2]+ and [S2,2,1]+). The identity of the central atom does not appear to be hugely impactful on the Ei(cation) value; the relatively soft, polarisable S atom in the sulfonium cations has a formal lone pair, but this lone pair still does not lead to a low Ei(cation) value. Therefore, the design rule developed for anions based on the central atom does not hold for cations.
A design rule for obtaining large or small Ei(IL) is to use the design rules for cations and anions. For example, [C8Py][BF4] gave the largest Ei(IL) measured here, Ei(IL) = 9.9 eV, larger than Ei(IL) for [C8C1Im][BF4], demonstrating that Ei(IL) can be tuned by selection of both the cation and the anion. The largest Ei(IL,pred.) = 11.1 eV for five [N4,1,1,0][A] and five [C4C1Pyrr][A], where [A]− = [B(CN)4]− and four of the fluorinated anions. The smallest Ei(IL,pred.) = 5.7 eV for [P6,6,6,14]2[FeCl4]. A further design rule is that the [P6,6,6,14][A] ILs give smaller Ei(IL,pred.) than ILs with other cations, i.e. any anion put with a [P6,6,6,14]+ cation would give a lower Ei value than with other cations such as [CnC1Im]+.
5.3. Ionisation: competition between cation and anion
For two thirds of the anions studied here and in ref. 48, the anion was comfortably the HOMO. An electron was more readily removed from the anion than the cation, i.e. easier to remove an electron from the already negatively charged anion to form a neutral species, [A]− → A˙, rather than remove an electron from the positively charged cation to form a dicationic ion, [C]+ → [C]2+.∼10% of the ILs studied here gave the cation as the HOMO. Using the design rules laid out in Section 5.2, a combination of a cation with π-bonding/long alkyl chains and fluorinated anion/anion with no lone pair on central atom will likely give the cation as the HOMO, e.g. [C8C1Im][SbF6], [C2C1Im][FAP] and [C8Py][BF4]. For [C8C1Im][InCl4] this result was a surprise, given that the anion is a metal complex; it was easier to remove an electron from the +1 cation, [C]+ → [C]2+, than it was from the −1 anion, [A]− → [A]˙. Furthermore, [C4C1Pyrr][PF6] was predicted to have the cation as the HOMO, validating calculations for [C3C1Pyrr][PF6].82
∼20% of ILs studied here the HOMO could be from the cation or the anion; it was too close to tell, given both the experimental errors involved and the contribution from the IL sample to the variability in EB(anion HOFO) and EB(cation HOFO). There will be a significant variation in the valence state energies due to the large range of ion solvation environments in the liquid phase, as observed for high quality calculations of the Cl− ion solvated in water62 and demonstrated here. In this work we identify the average EB and Ei values, but it is important to consider the variability in these EB and Ei values when attempting to identify the HOMO. This variability in EB and Ei values is important when considering rare events such as chemical reactions, as ions in the liquid phase with extremes of EB and Ei values are the ions that are likely to undergo reactions. One of the ILs for which the origin of the HOMO was uncertain was [C8C1Im]2[Zn4Cl10], a stunning result; it required similar energy to remove an electron from the +1 cation, [C]+ → [C]2+, and from the −2 dianion, [A]2− → [A]−.
5.4. Comparisons of Ei with other data sources
From UPS measurements of a microscopically thick but macroscopically thin film of [C8C1Im][BF4] produced using physical vapour deposition, the work function was 4.2 eV and the first peak came at approximately 5.5 eV from visually judging the UP spectra.107 Therefore, Ei(IL) using this approach was Ei(IL) = 4.2 eV + 5.5 eV = 9.7 eV.107Ei(IL) = 9.4 eV from this work for [C8C1Im][BF4], an excellent match to the Ei(IL) value derived from data in ref. 107.For [C4C1Im][BF4], [C4C1Im][PF6] and [C4C1Im][NTf2] using liquid phase external photoelectron emission spectroscopy, Eth = 7.8 eV, 7.9 eV and 8.1 eV respectively;50 for [C8C1Im][BF4], [C4C1Im][PF6] and [C4C1Im][NTf2], Eth(IL) = 8.3 eV from this work, a good match. Furthermore, for [C4C1Im]I Eth(IL) = 6.2 eV;50Eth(IL) = 6.3 eV from this work for [C6C1Im]I, as excellent match. Lastly, for [N3,1,1,1][NTf2] Eth(IL) > 9.2 eV;50Eth(IL) = 8.6 eV for the very similar IL [C4C1Pyrr][NTf2] for our work gave a reasonable match. For [C4C1Im][BF4], [C4C1Im][PF6] and [C4C1Im][NTf2] using liquid phase UPS, Eth(IL) = 6.6 eV, 7.8 eV and 7.8 eV respectively.49 The Eth(IL) value for [C4C1Im][BF4] from ref. 49 was significantly lower than Eth(IL) = 7.8 eV for [C4C1Im][BF4] from ref. 50 and Eth(IL) = 8.3 eV for [C8C1Im][BF4] from our work, strongly suggesting a problem with the charge referencing for the data in ref. 49.
Using gas phase UPS of neutral ion pairs, Eth(ion pair) values were measured by two groups, Leone and co-workers15–18 and Kuusik and co-workers.19–25 Most of these Eth(ion pair) values were for a combination of imidazolium cations and an imide anion, e.g. [NTf2]−; Eth(ion pair) ∼ 8.5 eV for these ILs, which matches well to our Eth(IL,pred.) = 8.5 eV. Kuusik and co-workers have also published Eth(ion pair) values for a small selection of other ILs. For [C4C1Pyrr][PF6] Eth(ion pair) = 10.5 eV, a good match to Eth(IL,pred.) = 11.2 eV; these values are very large, highlighting the very large Ei(IL) values for ILs with short alkyl chain ammonium cations and fluorinated anions.
Lastly, for comparisons to a molecular liquid, Ei = 11.16 eV for liquid phase water.108 This value is significantly larger than any of the Ei(IL,exp.) values presented here, although a small selection of Ei(IL,pred.) values were similar, e.g. for [N4,1,1,0][BF4] Ei(IL,pred.) = 11.1 eV.
For optical spectroscopies of [CnC1Im][A] ILs, the lowest absorption energy was between ∼3.4 eV and 6.0 eV.109–113 The smaller values were observed for [A]− = halide ion, likely due to anion–cation charge transfer transitions not observed with [A]− other than the halide ions.113 For [tetraalkylammonium][NTf2] ILs the lowest absorption energy was ∼8.3 eV.111,113 From N 1s resonant X-ray emission spectroscopy for [C2C1Im]Br and [C2C1Im][NTf2], the energy transfer between N 2p-based occupied valence state and N 2p-based unoccupied valence state for the [C2C1Im]+ cation for both ILs was 6.9 eV (401.9 − 395.0 eV).114 By comparison, Ei(cation,pred.) = 9.2 eV for [C8C1Im]Br and Ei(cation,pred.) = 9.6 eV for [C8C1Im][NTf2] (Table 4). Ei(IL) was always larger than these absorption energies (and transfer energies) from absorption spectroscopy (and resonant X-ray emission spectroscopy), demonstrating that the transitions in both absorption and resonant X-ray emission spectroscopies were to bound states; in the case of [CnC1Im][A] ILs the bound states were likely from the cationic ring. From N 1s resonant X-ray emission spectroscopy for [C2C1Im][NTf2], N 2p-based occupied valence state to N 2p-based unoccupied valence state for the [NTf2]+ anion was 9.5 eV (403.5 − 394.0 eV).114Ei(IL) = 9.4 eV for [CnC1Im][NTf2], suggesting that the transition measured using N 1s resonant X-ray emission spectroscopy was to an unbound state.
These measurements of valence XPS for liquid phase halometallate anions and dianions serve as an excellent complement to gas phase measurements of halometallate anions and dianions; many of the anions and dianions studied here would not be stable enough to be studied in the gas phase, e.g. [FeCl4]2−.115
5.5. Relationships of Ei with other IL properties
Comparing our data to electrochemical stability, our ILs with very large Ei(IL,pred), e.g. [C4C1Pyrr][PF6], would be predicted to have excellent electrochemical stability. This prediction matches to experience, where tetraalkylammonium cations and highly fluorinated anions, e.g. [N4,4,4,4][PF6] or [N4,4,4,4][BF4], are used as supporting electrolytes, which need to be very electrochemically stable.116 Further comparisons to electrochemical stability data are challenging at this stage, given the tricky task of finding an experimental electrochemical dataset to test against, as the IL selection needs to be sufficiently diverse as a test set, but also a relatively inert electrode must have been used. We believe we have produced an excellent experimental dataset of IL valence electronic structures for which comparisons can be made in the future. It is a similar story with respect to comparisons of experimental electronic structure and thermal stability; is there an experimental thermal stability dataset of sufficient IL diversity to provide a high-quality test of our electronic structure data? One significant challenge is quantifying thermal stability; there are a number of different metrics, e.g. onset temperature at a certain % of mass loss, activation energy.117,118A pyridinium-based cation in an IL can act as an electron donor to a neutral dye solute.119 This study demonstrates that the cation has been considered as an electron donor in ILs, but the full potential and importance has not yet been considered.
6. Conclusions and future work
We have successfully measured valence electronic structure descriptors for 60 ILs, most importantly, Ei and the HOMO identity. Measuring Ei for such a structurally diverse set of ILs represents a significant step forward in the understanding of the valence electronic structure of ILs. The structurally diverse range of cations and anions studied allow us to provide qualitative design rules linking ion structure to valence electronic structure. The electronic influence of the countercation on the anion valence electronic structure (and vice versa) was demonstrated to be dominated by non-specific, electrostatic interactions; the largest effect was 0.6 eV, although most effects were much smaller than that. Given that the cation–anion effects were relatively predictable, we were able to make predictions of both Ei and the HOMO identity for a further 516 ILs.∼10% of the ILs have the cation rather than the anion as the HOMO. The cation must be considered as a possible electron donor (or partial electron donor, when donating electron density rather than a formal electron pair) in such ILs in particular, especially for neutral solutes where electrostatic ion-solute interactions are expected to be less dominant.
Adding new anions to the dataset should be relatively facile if studied on a standard lab apparatus, given most anions dominate non-resonant XP spectra recorded at hν = 1486.6 eV; suitable charge referencing is achievable for any new IL. Adding new cations to the dataset will prove far more of a challenge, given the multiple experimental difficulties, especially those caused by the normally dominant anionic contributions to non-resonant XP spectra recorded at hν = 1486.6 eV.
Given our significant experimental and predicted data of valence electronic structure descriptors, the development of models linking experimental valence electronic structure descriptors to other IL properties, e.g. electrochemical stability and thermal stability, is now possible. Furthermore, our dataset will provide a very valuable benchmark for validation of electronic structure calculations.
Both qualitative comparisons (e.g. visual) and quantitative comparisons (e.g. peak EB separation) of liquid phase and gas phase photoelectron spectra have great potential to provide insight into the effect of solvation on electronic structure. In the gas phase, a standard [C][A] IL has only one counterion, whereas in the liquid phase each ion is fully solvated. ILs that gave the cation as the HOMO, e.g. [CnC1Im][FAP], would be ideal candidates, given the cationic contributions to the valence electronic structure can be readily identified along with the anionic contributions, allowing any phase-related EB shifts to be observed.
Author contributions
CJC synthesised the samples (where necessary). JMS, EG, AL, CJC, PL, DAD, PF, FV, RAB, RGP and KRJL performed the XPS/RXPS experiments. JMS, RMF and KRJL analysed the results. The manuscript was written by JMS and KRJL with the help from other authors. KRJL proposed and initiated the project, and supervised the investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KRJL acknowledges support from a Royal Society University Research Fellowship (URF\R\150353). JMS acknowledges support from a Royal Society University Research Fellowship Enhancement Award (RGF\EA\180089). EG acknowledges support from a Royal Society Research Grant for Research Fellows (RGF\R1\180053). AL acknowledges part funding for a studentship from Johnson Matthey plc. PL acknowledges support from the EPSRC (EP/K005138/1 and EP/P013341/1). We acknowledge Diamond Light Source for time on Beamline I09 under Proposal SI20463 and Beamline B07 (VERSOX) under Proposals SI24304 and SI25929. Georg Held and Matthijs Van Spronsen are acknowledged for beamtime support on Proposal SI25929. XP spectra for [C2C1Im][FAP] were recorded by the EPSRC National Facility for Photoelectron Spectroscopy (HarwellXPS), operated by Cardiff University and UCL under contract number PR16195. Mark Isaacs is acknowledged for recording the HarwellXPS results.References
- I. Fleming, Molecular Orbitals and Organic Chemical Reactions, Wiley, Chichester, 2010 Search PubMed.
- K. Fukui, Angew. Chem., Int. Ed. Engl., 1982, 21, 801–809 CrossRef.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 CrossRef.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- R. G. Pearson, J. Org. Chem., 1989, 54, 1423–1430 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. J. Zheng, S. G. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- E. W. Castner and J. F. Wishart, J. Chem. Phys., 2010, 132, 120901 CrossRef PubMed.
- E. W. Castner, C. J. Margulis, M. Maroncelli and J. F. Wishart, Annu. Rev. Phys. Chem., 2011, 62, 85–105 CrossRef CAS PubMed.
- B. J. Mincher and J. F. Wishart, Solvent Extr. Ion Exch., 2014, 32, 563–583 CrossRef CAS.
- In the calculation community, the HOMO usually refers to the lowest energy valence state in the ground state of the molecule or ion. In the experimental XPS community, the HOMO often represents the most readily ionised valence state of the molecule or ion ( A. Kahn, Mater. Horizons, 2016, 3, 7–10 RSC ). In this article, we use the latter convention.
- E. I. Izgorodina, Z. L. Seeger, D. L. A. Scarborough and S. Y. S. Tan, Chem. Rev., 2017, 117, 6696–6754 CrossRef CAS PubMed.
- In this article, ionisation energies, Ei, are charge referenced to the vacuum level; binding energies, EB, are charge referenced to the alkyl carbon Fermi level.
- D. Strasser, F. Goulay, M. S. Kelkar, E. J. Maginn and S. R. Leone, J. Phys. Chem. A, 2007, 111, 3191–3195 CrossRef CAS PubMed.
- D. Strasser, F. Goulay, L. Belau, O. Kostko, C. Koh, S. D. Chambreau, G. L. Vaghjiani, M. Ahmed and S. R. Leone, J. Phys. Chem. A, 2010, 114, 879–883 CrossRef CAS PubMed.
- S. D. Chambreau, G. L. Vaghjiani, A. To, C. Koh, D. Strasser, O. Kostko and S. R. Leone, J. Phys. Chem. B, 2010, 114, 1361–1367 CrossRef CAS PubMed.
- S. D. Chambreau, G. L. Vaghjiani, C. J. Koh, A. Golan and S. R. Leone, J. Phys. Chem. Lett., 2012, 3, 2910–2914 CrossRef CAS.
- I. Kuusik, M. Tarkanovskaja, J. Kruusma, V. Reedo, R. Valbe, A. Lohmus, V. Kisand, E. Lust, E. Kukk and E. Nommiste, RSC Adv., 2015, 5, 6834–6842 RSC.
- I. Kuusik, M. Tarkanovskaja, J. Kruusma, V. Kisand, A. Tonisoo, E. Lust and E. Nommiste, J. Mol. Liq., 2016, 223, 939–942 CrossRef CAS.
- I. Kuusik, M. Berholts, J. Kruusma, V. Kisand, A. Tonisoo, E. Lust and E. Nommiste, RSC Adv., 2018, 8, 30298–30304 RSC.
- I. Kuusik, M. Kook, R. Parna, A. Kivimaki, T. Kaambre, L. Reisberg, A. Kikas and V. Kisand, J. Mol. Liq., 2019, 294, 111580 CrossRef CAS.
- I. Kuusik, M. Berholts, J. Kruusma, A. Tonisoo, E. Lust, E. Nommiste and V. Kisand, RSC Adv., 2019, 9, 33140–33146 RSC.
- J. M. Kahk, I. Kuusik, V. Kisand, K. R. J. Lovelock and J. Lischner, npj Comput. Mater., 2020, 6, 148 CrossRef CAS.
- I. Kuusik, M. Kook, R. Parna and V. Kisand, ACS Omega, 2021, 6, 5255–5265 CrossRef CAS PubMed.
- A. Deyko, K. R. J. Lovelock, P. Licence and R. G. Jones, Phys. Chem. Chem. Phys., 2011, 13, 16841–16850 RSC.
- K. R. J. Lovelock, J. P. Armstrong, P. Licence and R. G. Jones, Phys. Chem. Chem. Phys., 2014, 16, 1339–1353 RSC.
- C. J. Clarke, S. Puttick, T. J. Sanderson, A. W. Taylor, R. A. Bourne, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2018, 20, 16786–16800 RSC.
- S. D. Chambreau, J. A. Boatz, G. L. Vaghjiani, C. Koh, O. Kostko, A. Golan and S. R. Leone, J. Phys. Chem. A, 2012, 116, 5867–5876 CrossRef CAS PubMed.
- S. D. Chambreau, A. C. Schenk, A. J. Sheppard, G. R. Yandek, G. L. Vaghjiani, J. Maciejewski, C. J. Koh, A. Golan and S. R. Leone, J. Phys. Chem. A, 2014, 118, 11119–11132 CrossRef CAS PubMed.
- V. Volpe, B. Brunetti, G. Gigli, A. Lapi, S. V. Ciprioti and A. Ciccioli, J. Phys. Chem. B, 2017, 121, 10382–10393 CrossRef CAS PubMed.
- C. J. Clarke, N. D. Richardson, A. E. J. Firth, J. P. Hallett and P. Licence, ACS Sustainable Chem. Eng., 2020, 8, 16386–16390 CrossRef CAS.
- I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. L. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797–2808 RSC.
- A. Foelske-Schmitz and M. Sauer, J. Electron Spectrosc. Relat. Phenom., 2018, 224, 51–58 CrossRef CAS.
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wölfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H. P. Steinrück, Chem. – Eur. J., 2010, 16, 9018–9033 CrossRef CAS PubMed.
- S. Men, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 15244–15255 RSC.
- B. B. Hurisso, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 17737–17748 RSC.
- A. W. Taylor, S. Men, C. J. Clarke and P. Licence, RSC Adv., 2013, 3, 9436–9445 RSC.
- R. K. Blundell and P. Licence, Phys. Chem. Chem. Phys., 2014, 16, 15278–15288 RSC.
- I. J. Villar-Garcia, K. R. J. Lovelock, S. Men and P. Licence, Chem. Sci., 2014, 5, 2573–2579 RSC.
- S. Men, D. S. Mitchell, K. R. J. Lovelock and P. Licence, ChemPhysChem, 2015, 16, 2211–2218 CrossRef CAS PubMed.
- L. S. Longo, E. F. Smith and P. Licence, ACS Sustainable Chem. Eng., 2016, 4, 5953–5962 CrossRef CAS.
- S. A. Men, K. R. J. Lovelock and P. Licence, Chem. Phys. Lett., 2017, 679, 207–211 CrossRef CAS.
- S. Men, P. Licence, C. L. Do-Thanh, H. M. Luo and S. Dai, Phys. Chem. Chem. Phys., 2020, 22, 11976–11983 RSC.
- S. Men, P. Licence, H. M. Luo and S. Dai, J. Phys. Chem. B, 2020, 124, 6657–6663 CrossRef CAS PubMed.
- R. M. Fogarty, R. P. Matthews, C. R. Ashworth, A. Brandt-Talbot, R. G. Palgrave, R. A. Bourne, T. V. Hoogerstraete, P. A. Hunt and K. R. J. Lovelock, J. Chem. Phys., 2018, 148, 193817 CrossRef PubMed.
- R. M. Fogarty, R. Rowe, R. P. Matthews, M. T. Clough, C. R. Ashworth, A. Brandt, P. J. Corbett, R. G. Palgrave, E. F. Smith, R. A. Bourne, T. W. Chamberlain, P. B. J. Thompson, P. A. Hunt and K. R. J. Lovelock, Faraday Discuss., 2018, 206, 183–201 RSC.
- R. M. Fogarty, R. G. Palgrave, R. A. Bourne, K. Handrup, I. J. Villar-Garcia, D. J. Payne, P. A. Hunt and K. R. J. Lovelock, Phys. Chem. Chem. Phys., 2019, 21, 18893–18910 RSC.
- D. Yoshimura, T. Yokoyama, T. Nishi, H. Ishii, R. Ozawa, H. Hamaguchi and K. Seki, J. Electron Spectrosc. Relat. Phenom., 2005, 144, 319–322 CrossRef.
- R. Katoh, J. Phys. Chem. B, 2008, 112, 14971–14975 CrossRef CAS PubMed.
- R. Katoh, Chem. Phys. Lett., 2009, 482, 259–262 CrossRef CAS.
- G. Greczynski and L. Hultman, ChemPhysChem, 2017, 18, 1507–1512 CrossRef CAS PubMed.
- G. Greczynski and L. Hultman, Appl. Surf. Sci., 2018, 451, 99–103 CrossRef CAS.
- D. R. Baer, K. Artyushkova, H. Cohen, C. D. Easton, M. Engelhard, T. R. Gengenbach, G. Greczynski, P. Mack, D. J. Morgan and A. Roberts, J. Vac. Sci. Technol., A, 2020, 38, 031204 CrossRef CAS.
- G. Greczynski and L. Hultman, Angew. Chem., Int. Ed., 2020, 59, 5002–5006 CrossRef CAS PubMed.
- G. Greczynski and L. Hultman, Prog. Mater. Sci., 2020, 107, 100591 CrossRef CAS.
- J. M. Thomas, I. Adams and M. Barber, Solid State Commun., 1971, 9, 1571–1573 CrossRef CAS.
- B. Winter and M. Faubel, Chem. Rev., 2006, 106, 1176–1211 CrossRef CAS PubMed.
- R. Seidel, S. Thürmer and B. Winter, J. Phys. Chem. Lett., 2011, 2, 633–641 CrossRef CAS.
- B. Winter, R. Weber, I. V. Hertel, M. Faubel, P. Jungwirth, E. C. Brown and S. E. Bradforth, J. Am. Chem. Soc., 2005, 127, 7203–7214 CrossRef CAS PubMed.
- [C4C1Im][SCN] has 72 valence electrons (1-butyl-3-methylimidazolium, [C4C1Im]+), [C8C1Im]Cl has 88 valence electrons (1-octyl-3-methylimidazolium, [C8C1Im]+), [C8C1Im][NTf2] has 172 valence electrons ([NTf2]− = [(CF3SO2)2N]−, bis[(trifluoromethane)sulfonyl]imide) and [P6,6,6,14][NTf2] has 292 valence electrons ([P6,6,6,14]+ = tetradecyl(trihexyl)phosphonium).
- L. Ge, L. Bernasconi and P. Hunt, Phys. Chem. Chem. Phys., 2013, 15, 13169–13183 RSC.
- O. Höfft, S. Bahr, M. Himmerlich, S. Krischok, J. A. Schaefer and V. Kempter, Langmuir, 2006, 22, 7120–7123 CrossRef PubMed.
- S. Krischok, R. Öttking, W. J. D. Beenken, M. Himmerlich, P. Lorenz, O. Höfft, S. Bahr, V. Kempter and J. A. Schaefer, Z. Phys. Chem.-Int. J. Res. Phys. Chem. Chem. Phys., 2006, 220, 1407–1416 CAS.
- T. Ikari, A. Keppler, M. Reinmöller, W. J. D. Beenken, S. Krischok, M. Marschewski, W. Maus-Friedrichs, O. Höfft and F. Endres, e-J. Surf. Sci. Nanotechnol., 2010, 8, 241–245 CrossRef CAS.
- M. Reinmöller, A. Ulbrich, T. Ikari, J. Preiss, O. Höfft, F. Endres, S. Krischok and W. J. D. Beenken, Phys. Chem. Chem. Phys., 2011, 13, 19526–19533 RSC.
- A. Ulbrich, M. Reinmöller, W. J. D. Beenken and S. Krischok, ChemPhysChem, 2012, 13, 1718–1724 CrossRef CAS PubMed.
- A. Ulbrich, M. Reinmöller, W. J. D. Beenken and S. Krischok, J. Mol. Liq., 2014, 192, 77–86 CrossRef CAS.
- D. Weingarth, I. Czekaj, Z. F. Fei, A. Foelske-Schmitz, P. J. Dyson, A. Wokaun and R. Kötz, J. Electrochem. Soc., 2012, 159, H611–H615 CrossRef CAS.
- S. Krischok, M. Eremtchenko, M. Himmerlich, P. Lorenz, J. Uhlig, A. Neumann, R. Öttking, W. J. D. Beenken, O. Höfft, S. Bahr, V. Kempter and J. A. Schaefer, J. Phys. Chem. B, 2007, 111, 4801–4806 CrossRef CAS PubMed.
- S. Krischok, A. Ulbrich, T. Ikari, V. Kempter, M. Marschewski and O. Höfft, Nucl. Instrum. Methods Phys. Res. Sect. B-Beam Interact. Mater. Atoms, 2014, 340, 51–57 CrossRef CAS.
- A. B. Biedron, E. L. Garfunkel, E. W. Castner and S. Rangan, J. Chem. Phys., 2017, 146, 054704 CrossRef PubMed.
- S. Rangan, J. Viereck and R. A. Bartynski, J. Phys. Chem. B, 2020, 124, 7909–7917 CrossRef CAS PubMed.
- K. Kanai, T. Nishi, T. Iwahashi, Y. Ouchi, K. Seki, Y. Harada and S. Shin, J. Chem. Phys., 2008, 129, 224507 CrossRef CAS PubMed.
- T. Nishi, T. Iwahashi, H. Yamane, Y. Ouchi, K. Kanai and K. Seki, Chem. Phys. Lett., 2008, 455, 213–217 CrossRef CAS.
- K. Kanai, T. Nishi, T. Iwahashi, Y. Ouchi, K. Seki, Y. Harada and S. Shin, J. Electron Spectrosc. Relat. Phenom., 2009, 174, 110–115 CrossRef CAS.
- J. M. Seymour, C. J. Clarke, R. G. Palgrave, N. A. Besley and K. R. J. Lovelock, 2021, in preparation.
- S. Men and P. Licence, Chem. Phys. Lett., 2017, 681, 40–43 CrossRef CAS.
- J. M. Seymour, E. Gousseva, R. M. Fogarty, L. Frankemoelle, R. Rowe, C. J. Clarke, D. A. Duncan, R. A. Bennett, R. G. Palgrave, P. A. Hunt and K. R. J. Lovelock, 2021, in preparation.
- R. P. Matthews, C. Ashworth, T. Welton and P. A. Hunt, J. Phys.: Condens. Matter, 2014, 26, 284112 CrossRef PubMed.
- A. K. Srivastava, A. Kumar and N. Misra, J. Phys. Chem. A, 2021, 125, 2146–2153 CrossRef CAS PubMed.
- S. P. Ong, O. Andreussi, Y. B. Wu, N. Marzari and G. Ceder, Chem. Mater., 2011, 23, 2979–2986 CrossRef CAS.
- K. B. Dhungana, L. F. O. Faria, B. N. Wu, M. Liang, M. C. C. Ribeiro, C. J. Margulis and E. W. Castner, J. Chem. Phys., 2016, 145, 024503 CrossRef PubMed.
- S. P. Ong and G. Ceder, Electrochim. Acta, 2010, 55, 3804–3811 CrossRef CAS.
- J. B. Haskins, C. W. Bauschlicher and J. W. Lawson, J. Phys. Chem. B, 2015, 119, 14705–14719 CrossRef CAS PubMed.
- S. Kazemiabnavi, Z. C. Zhang, K. Thornton and S. Banerjee, J. Phys. Chem. B, 2016, 120, 5691–5702 CrossRef CAS PubMed.
- N. V. Ilawe, J. Fu, S. Ramanathan, B. M. Wong and J. Z. Wu, J. Phys. Chem. C, 2016, 120, 27757–27767 CrossRef CAS.
- V. R. Koch, L. A. Dominey, C. Nanjundiah and M. J. Ondrechen, J. Electrochem. Soc., 1996, 143, 798–803 CrossRef CAS.
- P. Ballone and R. Cortes-Huerto, Faraday Discuss., 2012, 154, 373–389 RSC.
- Y. Zhang, C. J. Shi, J. F. Brennecke and E. J. Maginn, J. Phys. Chem. B, 2014, 118, 6250–6255 CrossRef CAS PubMed.
- C. Lian, H. L. Liu, C. Z. Li and J. Z. Wu, AIChE J., 2019, 65, 804–810 CrossRef CAS.
- E. Thomas, D. Thomas, K. P. Vijayalakshmi and B. K. George, RSC Adv., 2016, 6, 9421–9428 RSC.
- E. Thomas, K. P. Vijayalakshmi and B. K. George, J. Mol. Liq., 2019, 276, 721–727 CrossRef CAS.
- S. Koutsoukos, F. Philippi, F. Malaret and T. Welton, Chem. Sci., 2021, 12, 6820–6843 RSC.
- T. L. Lee and D. A. Duncan, Synchrotron Radiat. News, 2018, 31, 16–22 CrossRef.
- G. Held, F. Venturini, D. C. Grinter, P. Ferrer, R. Arrigo, L. Deacon, W. Q. Garzon, K. Roy, A. Large, C. Stephens, A. Watts, P. Larkin, M. Hand, H. C. Wang, L. Pratt, J. J. Mudd, T. Richardson, S. Patel, M. Hillman and S. Scott, J. Synchrotron Radiat., 2020, 27, 1153–1166 CrossRef CAS PubMed.
- K. W. Kolasinski, Surface Science: Foundations of Catalysis and Nanoscience, Wiley, Chichester, 4th edn, 2020 Search PubMed.
- Surface Analysis: The Principal Techniques, ed. J. C. Vickerman and I. S. Gilmore, John Wiley & Sons, Chichester, 2009 Search PubMed.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- R. M. Fogarty, R. P. Matthews, M. T. Clough, C. R. Ashworth, A. Brandt-Talbot, P. J. Corbett, R. G. Palgrave, R. A. Bourne, T. W. Chamberlain, T. Vander Hoogerstraete, P. B. J. Thompson, P. A. Hunt, N. A. Besley and K. R. J. Lovelock, Phys. Chem. Chem. Phys., 2017, 19, 31156–31167 RSC.
- J. M. Seymour, E. Gousseva, R. M. Fogarty, J. Werner, G. Öhrwall, O. Björneholm, D. Hein, G. Wartner, C. Kolbeck, B. Winter, R. Seidel and K. R. J. Lovelock, 2021, in preparation.
- M. N. Pohl, E. Muchová, R. Seidel, H. Ali, S. Sršeň, I. Wilkinson, B. Winter and P. Slavíček, Chem. Sci., 2019, 10, 848–865 RSC.
- J. Zhang, P. Yang, Z. R. Sun and X. B. Wang, J. Phys. Chem. A, 2014, 118, 8074–8080 CrossRef CAS PubMed.
- L. Péter and J. Arai, J. Appl. Electrochem., 1999, 29, 1053–1061 CrossRef.
- G. L. Gutsev and A. I. Boldyrev, Chem. Phys., 1981, 56, 277–283 CrossRef CAS.
- J. B. Mann, T. L. Meek and L. C. Allen, J. Am. Chem. Soc., 2000, 122, 2780–2783 CrossRef CAS.
- K. L. Syres and R. G. Jones, Langmuir, 2015, 31, 9799–9808 CrossRef CAS PubMed.
- B. Winter, R. Weber, W. Widdra, M. Dittmar, M. Faubel and I. V. Hertel, J. Phys. Chem. A, 2004, 108, 2625–2632 CrossRef CAS.
- R. Katoh, Chem. Lett., 2007, 36, 1256–1257 CrossRef CAS.
- T. Miyata, M. Fukuyama, A. Hibara, E. Okunishi, M. Mukai and T. Mizoguchi, Microscopy, 2014, 63, 377–382 CrossRef CAS PubMed.
- K. Regeta, C. Bannwarth, S. Grimme and M. Allan, Phys. Chem. Chem. Phys., 2015, 17, 15771–15780 RSC.
- I. Tanabe, Y. Kurawaki, Y. Morisawa and Y. Ozaki, Phys. Chem. Chem. Phys., 2016, 18, 22526–22530 RSC.
- I. Tanabe, A. Suyama, T. Sato and K. Fukui, Analyst, 2018, 143, 2539–2545 RSC.
- Y. Horikawa, T. Tokushima, O. Takahashi, H. Hoke and T. Takamuku, J. Phys. Chem. B, 2016, 120, 7480–7487 CrossRef CAS PubMed.
- T. Waters, X. B. Wang and L. S. Wang, Coord. Chem. Rev., 2007, 251, 474–491 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- F. Heym, B. J. M. Etzold, C. Kern and A. Jess, Phys. Chem. Chem. Phys., 2010, 12, 12089–12100 RSC.
- F. Heym, B. J. M. Etzold, C. Kern and A. Jess, Green Chem., 2011, 13, 1453–1466 RSC.
- B. N. Wu, M. Liang, N. Zmich, J. Hatcher, S. I. Lall-Ramnarine, J. F. Wishart, M. Maroncelli and E. W. Castner, J. Phys. Chem. B, 2018, 122, 2379–2388 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02441h |
| This journal is © the Owner Societies 2021 |