
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the formation of isolated cyclo-FF peptide clusters by far-infrared action spectroscopy†
Sjors
Bakels
a,
Iuliia
Stroganova
ba and
Anouk M.
Rijs
 *b
*b
aRadboud University, FELIX Laboratory, Institute for Molecules and Materials, Toernooiveld 7, 6525 ED Nijmegen, The Netherlands
bDivision of BioAnalytical Chemistry, AIMMS Amsterdam Institute of Molecular and Life Sciences, Vrije Universiteit Amsterdam, De Boelelaan 1108, 1081 HV, Amsterdam, The Netherlands. E-mail: a.m.rijs@vu.nl
First published on 15th September 2021
Abstract
Small cyclic peptides containing phenylalanine residues are prone to aggregate in the gas phase into highly hydrophobic chains. A combination of laser desorption, mass spectrometry and conformational selective IR-UV action spectroscopy allows us to obtain detailed structural insights into the formation processes of the cyclic L-phenylalanyl-L-phenylalanine dipeptide (named cyclo-FF) aggregates. The rigid properties of cyclo-FF result in highly resolved IR spectra for the smaller clusters (n ≤ 3) and corresponding conformational assignments. For the higher order clusters (n > 3) the spectra are less resolved, however the observed ratios, peak positions and trends in IR shifts are key to make predictions on their structural details. Whereas the mid-IR spectral region between 1000–1800 cm−1 turns out to be undiagnostic for these small aggregates and the 3 μm region only for specific calculated structures, the far-IR contains valuable information that allows for clear assignments.
Introduction
Peptides form a particularly interesting class of self-assembling molecules, as their resulting nanostructures are found to play an important role in neurodegenerative diseases, but they can also be directed to form functional nano-materials. An important example is the dipeptide diphenylalanine, FF or Phe–Phe, which is a key structural motif in Alzheimer's β-amyloid polypeptides.1 However, FF was also found to self-assemble in aqueous solution into highly structured nanotubes, and therefore consequently used as a template to produce silver nanowires.1 These results triggered in-depth research into FF containing nanostructures and their properties, showing their versatility by the formation of different nanostructures at different conditions.2–5 Adler-Abramovich et al. found that using a vapor deposition technique, so-called nanoforests could be produced consisting of nanotubes and nanorods.6 These nanorods actually consisted of the diketopiperazine cyclo-(Phe–Phe) or cyclo-FF, which was dehydrated and cyclized from the FF starting material as a result of the high temperature used in their experiment (250 degrees Celsius).7–9 Cyclo-FF nanorods or nanowires have very specific properties, as they are for example extremely hydrophobic, they can show a strong blue luminescence and they possess semi-conducting properties.7,10,11 Possible applications for these nanowires include water guiding in microfluidic devices by its hydrophobic properties,6 acting as triboelectric material for biocompatible nanogenerators,12 or as optical waveguide.11 X-Ray diffraction experiments on cyclo-FF nanorods showed that the peptides are ladder-like structured, where the peptides are connected to each other via double hydrogen bonds along the direction of the nanorods.10,13 The resulting interactions between the amino acid residues, such as intermolecular hydrogen bonding and π-stacking, were also observed using NMR14 and FT-IR.15 However, in these publications the nanostructures were obtained in solution (e.g., using methanol mixtures) and not under isolated conditions as is done in this work.A computational study by the group of Scott Shell brought insight into the early stages of the formation process of the nanorods in the vapor deposition method.16 They showed that the cyclo-FF peptides adopt ladder-like structures, in which the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H groups of the peptides are connected via double intermolecular hydrogen bonds. In this way ladders are formed of variable lengths (with an average of 3 cyclo-FF peptides per ladder), and are aligned to other ladders via electrostatic interactions, van der Waals forces and interactions between the phenyl groups, with the latter pointing outward. In this way very hydrophobic assemblies of associated ladders are formed, with similar packing interactions to the crystal structures, but in a more irregular way.
O and N–H groups of the peptides are connected via double intermolecular hydrogen bonds. In this way ladders are formed of variable lengths (with an average of 3 cyclo-FF peptides per ladder), and are aligned to other ladders via electrostatic interactions, van der Waals forces and interactions between the phenyl groups, with the latter pointing outward. In this way very hydrophobic assemblies of associated ladders are formed, with similar packing interactions to the crystal structures, but in a more irregular way.
To obtain experimental insight into the early self-assembly steps, peptide aggregation can be studied in the gas phase under controlled conditions combining laser desorption molecular beam technology with IR-UV ion dip spectroscopy. There are only a limited number of experimental studies on peptide aggregation in the gas phase since the pioneering work of Gerhards et al.,17 and these studies all focused solely on peptide dimers.18–20 Only recently, research from our group enabled us to investigate the behavior of higher order clusters in the gas phase (up to n = 9).21 The monomer of cyclo-FF was previously studied for chirality effects in the amide A region by Zehnacker et al., using conformer specific IR-UV laser techniques in the gas phase.22 They determined the presence of a single conformer in which a CH–π interaction was deemed important in the stabilizing process. In this work, we make use of infrared action spectroscopy techniques and focus thereby specifically on the far-IR region, which constitutes information on global motions, and is therefore ideal for research on aggregation.23,24
Methods
Experimental details
The experimental details of the molecular beam set-up used for the presented experiments have been discussed recently.23 Therefore, only a brief description is given of the experiment and its details. Cyclo-FF (mass 294.35 amu), was commercially acquired from Bachem (>99% purity), and used without any further purification. The molecule was mixed with carbon black in an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, and deposited on the surface of a graphite sample bar, which was placed in a high vacuum (>10−7 mbar) chamber. There, a low intense (1–1.5 mJ pulse−1) laser beam from a Nd:YAG laser (New Wave Research, Polaris II, 1064 nm) was focused on the surface of the sample bar. The sample bar is placed on a translational stage, which, by stepper motor control, provides new sample for every laser shot, and allows adjustment of the height of the sample bar. The nozzle of a pulsed valve (Jordan), with a modified linear orifice (0.5 mm Ø, operating at 10 Hz), is located directly perpendicular to the sample bar surface where the laser is focused. The desorbed molecules are entrained in the supersonic expansion created by the pulsed valve, and are cooled down as a result of collisions with the argon atoms (4 bar backing pressure). Downstream the coldest part of the beam is selected by a skimmer, which leads into a reflector time-of-flight mass spectrometer (Jordan). Here, the molecular beam is crossed perpendicularly by a UV laser (Nd:YAG pumped dye lasers, LIOP-TEC, around 2.5 mJ pulse−1), and counter propagated by the IR laser in order to perform the spectroscopic experiments. The infrared light is provided by the free electron laser FELIX in the wavenumber range between 90 and 1800 cm−1 (∼10 μs width), or in the 3 μm region by an OPO/OPA system (LaserVision).25 The UV wavelengths have been calibrated using a wave meter (HighFinesse, WS7).
:1 ratio, and deposited on the surface of a graphite sample bar, which was placed in a high vacuum (>10−7 mbar) chamber. There, a low intense (1–1.5 mJ pulse−1) laser beam from a Nd:YAG laser (New Wave Research, Polaris II, 1064 nm) was focused on the surface of the sample bar. The sample bar is placed on a translational stage, which, by stepper motor control, provides new sample for every laser shot, and allows adjustment of the height of the sample bar. The nozzle of a pulsed valve (Jordan), with a modified linear orifice (0.5 mm Ø, operating at 10 Hz), is located directly perpendicular to the sample bar surface where the laser is focused. The desorbed molecules are entrained in the supersonic expansion created by the pulsed valve, and are cooled down as a result of collisions with the argon atoms (4 bar backing pressure). Downstream the coldest part of the beam is selected by a skimmer, which leads into a reflector time-of-flight mass spectrometer (Jordan). Here, the molecular beam is crossed perpendicularly by a UV laser (Nd:YAG pumped dye lasers, LIOP-TEC, around 2.5 mJ pulse−1), and counter propagated by the IR laser in order to perform the spectroscopic experiments. The infrared light is provided by the free electron laser FELIX in the wavenumber range between 90 and 1800 cm−1 (∼10 μs width), or in the 3 μm region by an OPO/OPA system (LaserVision).25 The UV wavelengths have been calibrated using a wave meter (HighFinesse, WS7).
Theoretical approaches
Several theoretical approaches have been combined, namely for the monomer and dimer, a conformational search was performed using an amber force field.26 First, the starting structures were heated up to 1000 K (for the monomers), and 300 K (for the dimers), after which they were cooled down to 0 K in order to find minima in the conformational energy landscape. This process was repeated 500 times, and consequently provided 500 structures. Similar structures were grouped together, while structures above 70 kJ mol−1 were discarded, resulting in 6 structures to evaluate for the monomer and about 21 structures for the dimer. The difference in simulated heating temperatures for monomer and dimer is a result of how the bonds are treated by the force field: at larger temperatures (>300 K), all the intermolecular interactions that hold the two monomers in the dimer together are broken. This is solved by either performing more conformational searches, starting with different input structures to sample the entire conformational space, or by manually design the molecules and directly calculate them. This last approach was used for the dimer, in which all possible combinations of the six monomers with two hydrogen bonds were calculated: an additional 26 structures.The conformers obtained from the conformational searches (monomer and dimer) or by the design approach (dimer, trimer, tetramer) were optimized and their frequencies calculated at the B3LYP-D3/6-311+G(d,p) level of theory for the monomers and dimers, the B3LYP-D3/6-311+G(d) level of theory for the trimers and the B3LYP-D3/6-31+G(d) level of theory for the tetramers in the Gaussian 16 environment.27,28 In order to correct for anharmonicity, the calculated spectra between 0 and 1800 cm−1 were scaled by 0.976 for all molecules.20,21 For all molecules in the amide A region, between 3350 and 3450 cm−1, the spectra were scaled by 0.954, as determined by the overlap of the experiment and calculation of the monomer (see also Fig. S4, ESI†).
Results and discussion
Electronic spectra
The mass spectrum, presented in Fig. S1 (ESI†), obtained at UV = 37506 cm−1 shows that both the cyclo-FF monomer as higher-order clusters are formed together inside the molecular beam TOF set-up. Fig. 1 shows the REMPI spectra of cyclo-FF and its homogeneous clusters, which are obtained by scanning the UV laser in the region between 37100 and 38300 cm−1. The electronic excitation spectrum of the monomer (Fig. 1a) has been published before by Zehnacker et al.22 The monomer shows a well resolved spectrum with the origin located at 37588 cm−1. However, the origin measured in this article is shifted by −15 cm−1 with respect to the value reported by Zehnacker et al., possible resulting from a variance in laser calibration. For our work, the lasers are calibrated using a High Finesse wavemeter, verifying the position of the origin and vibrational progression in Fig. 1. Additional bands and different intensities can be observed in comparison to the spectrum obtained by Zehnacker and coworkers, possibly resulting from different experimental parameters.22 The spectra of the dimer and the trimer, in Fig. 1b and c, show their origins slightly red shifted (−10 cm−1) with respect to the monomer, and the peak is slightly broadened (7 cm−1 with respect to 1.5 cm−1 of the monomer). Intense features that appear in the monomer spectrum also show up in the UV spectra of the dimer and trimer spectra (n = 2 and 3), around +341 cm−1 and +532 cm−1. The peak at +645 cm−1 is however absent, and replaced by a peak at +614 cm−1 in the dimer/trimer spectrum respectively. The strong features arising to the red side of the origin (−18/−41 cm−1 for the dimer and trimer) can most likely be assigned to complexes of the molecule with argon, although the presence of hot bands cannot be excluded. The spectrum of the tetramer (Fig. 1d), shows no sharp resolved peaks as was observed in the spectra of the lower order clusters, however the broader features remain. The presence of a dominant conformation of the monomer22 was verified by recording an IR-UV hole-burning spectrum, although a small contribution of an additional conformer was observed (see Fig. S5, ESI†). The origin of both monomer conformers is expected to split, resulting from the electronic transition of each of the chromophores in the cyclo-FF molecule. However, as discussed in detail by Zehnacker et al.,22 this is likely smaller than the experimental resolution. For the cyclo-FF clusters, a broader origin peak is observed, which, in addition to reduced cooling, can indicate a small peak splitting of the multiple chromophores as a result of the clustering. Although very interesting, this lies outside the scope of this work.
 | ||
| Fig. 1 Normalized REMPI spectra of the measured molecules: The clusters of cyclo-FFn with n = 1–4 (a–d). The solid arrows indicate the position where the IR spectra presented in Fig. 2, 3, 5 and 6 are obtained (monomer, dimer, trimer and tetramer, respectively) and in Fig. S11 (ESI†). The dashed line in (a) indicates the second conformer named b, as shown in Fig. S5 (ESI†). The dashed arrows in (b) and (c) are assigned to complexes with argon. The inset in (a) depicts the molecular structure of cyclo-FF. | ||
All infrared spectra were obtained using the IR ion-dip method.23 The IR spectra for the monomer were taken at a wavenumber of 37588 cm−1, and 37612 cm−1 for the second conformer. IR spectra of the dimer and trimer were simultaneously recorded at a REMPI wavenumber of 37580 cm−1, which is for both aggregates the main band in the spectrum. Additional, coinciding IR spectra were taken for the peaks indicated in Fig. 1b and c with dashed arrows, which are assigned to possible complexes with argon. The IR spectra of the tetramer and higher order clusters were measured at a single wavenumber of 37506 cm−1. This is a result of optimization of the signal on wavelength at the day of IR measurement.
Infrared spectra
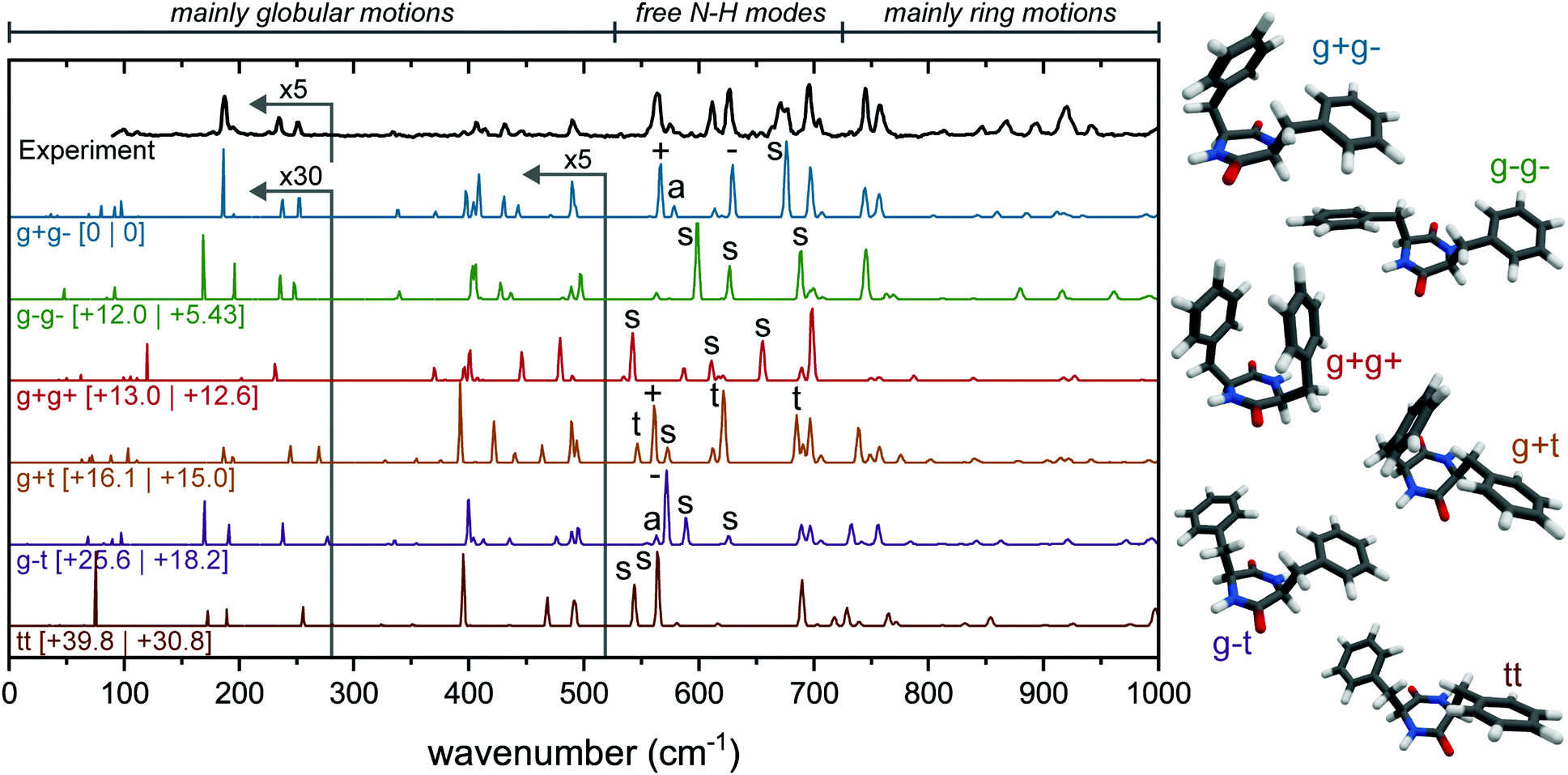 | ||
| Fig. 2 Experimental spectrum of the main conformer of the monomer (black, top) and calculated (colored) far-IR spectra of the six possible monomeric structures, arranged from lowest energy to highest (top to bottom). The corresponding structures are shown to the right of each spectrum. All calculated spectra are scaled by 0.976, calculations have been performed at the B3LYP-D3/6-311+G(d,p) level. Zero-point energies (left) and Gibbs free energies at 300 K are shown between square brackets. The region which exhibits free N–H bending modes is indicated above the figure, and indicated above each peak involved in such a vibration is the responsible N–H group (+, −, t for single N–H vibration, a and s for (anti-)symmetric N–H vibrations). For clarity, the region below 280 cm−1 has been multiplied by 30 (calculated spectra), and 5 (for the experimental spectrum), and between 280 and 520 cm−1 by 5 for the calculated spectra. | ||
The calculated far-IR spectra show that this region is very sensitive to subtle ring rotations; i.e., differences in the ring orientations result in completely different patterns throughout the whole far-IR region (Fig. 2, colored spectra). The region between 90 and 525 cm−1 contains mostly weak global motions, where rings, backbone and/or CH2 groups are involved.23 The region from 725 to 1000 cm−1 is dominated by ring motions. The larger peaks at 695, 745 and 755 cm−1 can be assigned to different ring vibrations, from which the latter two are typically seen in spectra containing phenylalanine residues. The most interesting region for this molecule, with also the most intensity (between 525 and 725 cm−1), is where free N–H bending modes are present.23,29,30 The different types of N–H out-of-plane motions are indicated by various signs; a and s for (anti-)symmetric motions with both N–H groups involved; +, − and t when only a single N–H group is involved (when located next to a ring in the g+, g− or t position, respectively). For example, in the g+g− spectra (blue, second trace from top), the peak at 567 cm−1 is indicated with a “+”. This means that only 1 N–H out of the two is involved in the vibration, and in this case the N–H that belongs to the amino acid of the ring in the g+ position.
The far-IR region below 400 cm−1 also comprises a number of interesting spectral features. At 334 cm−1 a very weak peak can be observed, which can be assigned to a CH2 rocking motion of the g− side group. Another peak is predicted at 371 cm−1, but this has not been observed as a result of experimental sensitivity limitations. This predicted peak belongs to the CH2 rocking motion of the g+ side group. The rocking motion of the CH2 peak of a t side group would show up around 353 cm−1, which makes this region particularly diagnostic to determine what ring orientations are present in the molecule.
Previously, the observed neutral cyclo-FF monomer has been assigned to the g+g− conformer, based on the IR spectrum recorded in the 3 μm region.22 Three peaks were observed in this IR region, while only two appeared in the calculated spectrum: the third peak was assigned to a combination band. To complete our IR study, we have included the mid-IR in both the 1000–1800 cm−1 and 3300–3500 cm−1 region of the monomer as well, see Fig. S3 and S4 (ESI†), obtained with the UV excitation of 37588 cm−1. However, the calculated IR spectra of all six conformers in the amide I and II region are very similar in this region, preventing an assignment based solely on this spectral region.
Based on the far-IR region, we can confirm the assignment of the main conformer of the monomer to the g+g− structure. Almost all calculated peaks are in excellent agreement with the experimental spectrum, in both wavenumber and intensity. Only the experimental peak at 672 cm−1 is broader than the calculation, and the calculated peaks around 900 cm−1 are slightly redshifted and show lower intensity compared to the experiment. The calculated spectra of the other conformers show a poor agreement in almost every region, which makes this a very confident assignment. The far-IR region proves to be very diagnostic in comparison to the other infrared regions. Specifically, the region between 525 and 725 cm−1 acts as a fingerprint for the ring orientation of the diketopiperazine molecule (i.e., g+, g−, t). We will use this information to characterize the structure of the aggregates of cyclo-FF. The assignment agrees with the work by Zehnacker et al.22 using the 3 μm region, although in our analysis of the 3 μm region we have an alternative peak assignment as discussed in S3 (ESI†).
An IR-UV hole-burning experiment, with the IR fixed at the 3402 cm−1 NH stretch peak, was performed to confirm the presence of a single conformer (Fig. S5, ESI†), however, this surprisingly yielded a second (minor) conformer at 37612 cm−1. The IR spectrum of this conformer was measured around 3400 cm−1, and is presented in Fig. S5b (ESI†). The second lowest energy structure (g−g−) shows good overlap with the experiment, and this structure is therefore assigned to the second conformer of the cyclo-FF monomer.
The experimental far-IR spectrum of the dominant conformer of the dimer (Fig. 3) was obtained at a UV wavenumber of 37580 cm−1 and shows a wealth of resolved peaks. The peaks below 280 cm−1 are slightly broadened with respect to the rest of the spectrum, as these are measured at different settings (and bandwidth) of the free electron laser. The large peak in the experimental spectrum around 900 cm−1 is predicted in all double hydrogen bonded dimers, while it is absent in all single or non-hydrogen bonded dimers (see Fig. S6, ESI† for the calculated far-IR infrared spectra of all structures below 30 kJ mol−1). Therefore, we only consider double hydrogen bonded dimers to be viable candidates. By comparing the overlap between experiment and calculations, and considering the energies, the three most promising conformations are presented in Fig. 3. Since all three conformers have very similar spectra between 1000 and 1800 cm−1 (see Fig. S7, ESI†), only the far-IR is used to make assignments, as was done with the monomer.
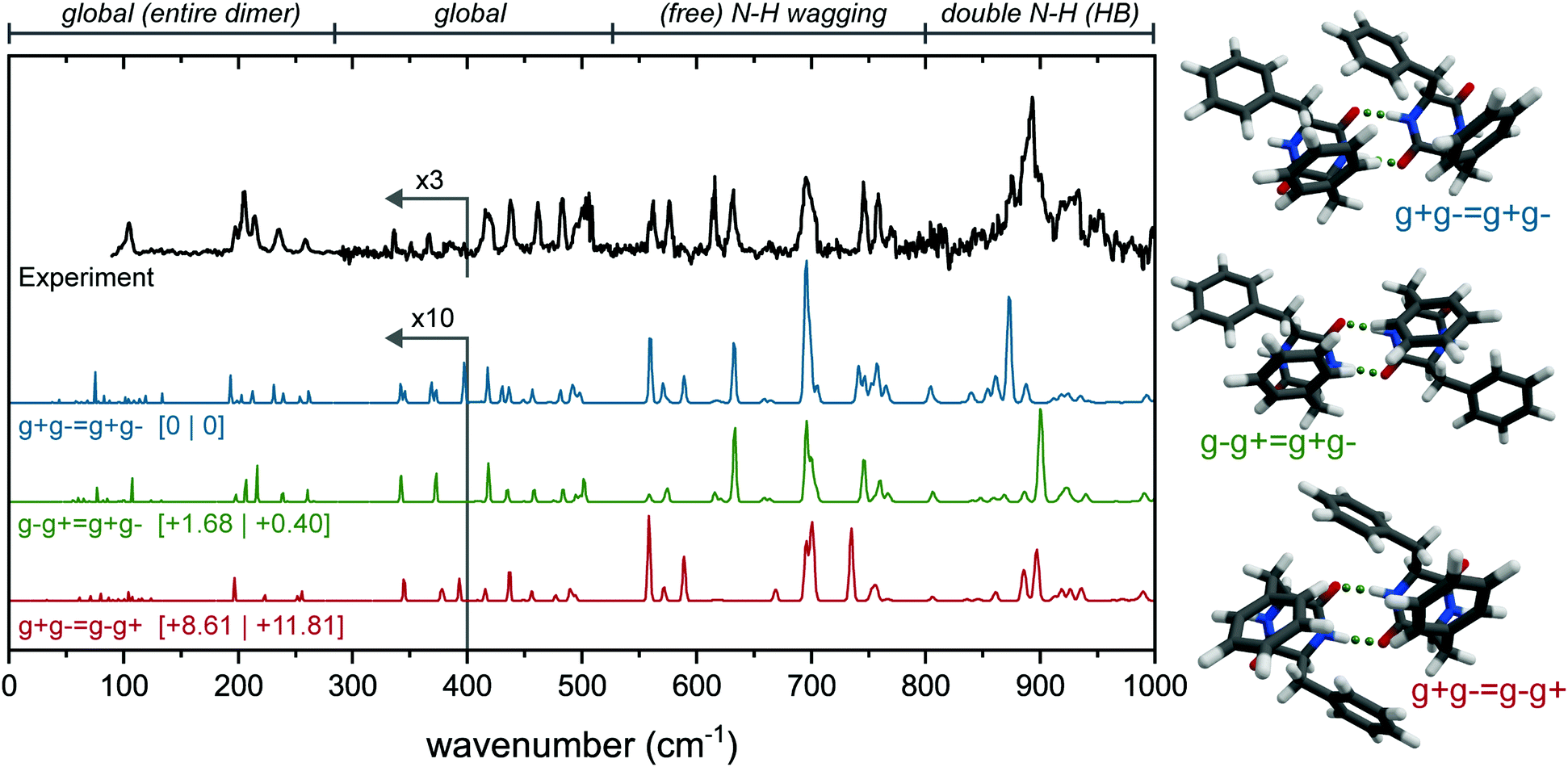 | ||
| Fig. 3 Experimental (black, top) of the main conformer of the cyclo-FF dimer and calculated (colored) far-IR spectra of three possible conformations, arranged from lowest energy to highest (top to bottom). The corresponding structures are shown to the right of each spectrum. All calculated spectra (B3LYP-D3/6-311+G(d,p) level) are scaled by 0.976. Zero-point energies (left) and Gibbs free energies at 300 K are shown between square brackets. Four regions of interest have been indicated above the spectrum: containing mostly global modes, free N–H vibrations, and N–H vibrations that are part of a double hydrogen bonded structure. The region below 380 cm−1 has been multiplied by 10 (calculated spectra), and 3 (for the experimental spectrum). | ||
Here, the calculated far-IR spectra of the g+g− = g+g−, g−g+ = g+g−, and g+g− = g−g+ conformers (see Fig. S2, ESI† for nomenclature) are presented, together with their structures and the experimental far-IR spectrum. Notice that these three structures are combinations of two g+g− subunits (the main monomer conformer), connected via double intermolecular hydrogen bonds. They differ in the relative orientation of the rings: g+g− = g+g− is the lowest in energy and has only C1 symmetry; g−g+ = g+g− has C2 symmetry and has the two g+ rings T-stacked and the g− subunits pointing to the outside, thereby adopting a more extended form; and g+g− = g−g+, slightly higher in energy (+8.6 kJ mol−1) and more packed, with all four rings T-stacking with each other.
The far-IR spectrum can roughly be divided into 4 regions: Below 280 cm−1; between 280 and 525 cm−1; between 525 and 800 cm−1 and between 800 and 1000 cm−1. The features in the region below 280 cm−1 result from large, global motions with involvement of the complete dimer (symmetrically between the two subunits). In this region, the experimental far-IR spectrum shows a peak with a shoulder to the red at 104 cm−1, followed by a triplet of peaks at 197, 205 and 215 cm−1, and two peaks at 236 and 258 cm−1. All these peaks are in near perfect agreement with the calculated IR spectrum of the g−g+ = g+g− dimer (green spectrum in Fig. 3). Only for the triplet around 200 cm−1 the intensities of the 205 and 215 cm−1 peaks are reversed. In contrast, the g+g− = g+g− (blue) dimer lacks the peak at 104 cm−1, shows additional activity between 110 and 150 cm−1, and has doublets instead of single peaks at the 236 and 258 cm−1 position. The g+g− = g−g+ conformer (red) misses a number of peaks around 200 cm−1, and is worse overall in this region.
In the second region from 280 to 550 cm−1, the g−g+ = g+g− dimer also shows the best agreement. First of all, the five features between 418 and 503 cm−1 are all explained by g−g+ = g+g−, while both other conformers either have doublets instead of singlets (g+g− = g+g−), or the peaks are shifted to different wavenumbers (g+g− = g−g+). Furthermore, the two distinct peaks at 336 and 366 cm−1 are only predicted correctly by the g−g+ = g+g− dimer.
The free N–H wagging region (525–800 cm−1), known to be sensitive to the phenyl ring orientation of the monomers, is diagnostic for the dimer conformation. This region can be used to identify the phenylalanine subunits within the monomers by their free N–H groups, such as two g− subunits in the g−g+ = g+g− dimer. This section of the experimental spectrum contains a set of peaks with similar intensities at 562, 576, 615, 631, 746 and 759 cm−1, a larger and broader feature at 695 cm−1 with a shoulder to the blue, and a smaller peak at 771 cm−1. In the same fashion as the previous regions, here g−g+ = g+g− shows the best agreement with the experiment. All peak positions are accounted for, and in particular the shoulder of the peak at 695 cm−1 and the three peaks around 750 cm−1 are predicted well.
The free N–H wagging motion of the g− subunit is located at 631 cm−1, which is absent in the g+g− = g−g+ dimer as here the g− subunits are involved in hydrogen bonding. Both g+g− = g+g− as g+g− = g−g+ have an additional peak at 589 cm−1 resulting from the free N–H wagging vibrations of the g+ subunit, which is not observed in the experimental spectrum. In g−g+ = g+g−, only the g− subunits have free N–H groups which explains the absence of this peak in the experimental spectrum. The g−g+ = g+g− conformer shows excellent agreement based on the absorption frequencies, however, the intensities of the N–H out-of-plane vibrations are not well predicted in this region, which has been observed before for this N–H wagging region when using static DFT calculations rather than dynamic DFT.29,30
The region between 800 and 1000 cm−1 is dominated by hydrogen bonded symmetric N–H wagging/out-of-plane vibrations. The experimental spectrum shows a strong and broad peak centered around 891 cm−1. Interestingly, from all the calculated IR spectra, only the dimers with two (double) hydrogen bonds have significant activity in this IR region. The single and non-hydrogen bonded dimers all lack intense peaks here, see Fig. S6 (ESI†). Although all three highlighted conformations in Fig. 3 are not overlapping perfectly, the g−g+ = g+g− conformer shows the most convincing match.
The far-IR spectrum clearly indicates that the g−g+ = g+g− conformer is the dimer present in our experiment. The 3 μm spectrum of the free N–H bending is also measured and compared with the three possible conformers in Fig. S7 (ESI†) using the obtained scaling factor for the monomer (Fig. S4, ESI†). The 3 μm region confirms our far-IR findings: the g−g+ = g+g− structure provides the best overlap with experiment in the amide A region.
It is expected that the higher-order clusters of cyclo-FF show the same behavior as was reported on the aggregation of Ac–Ala–Ala–OBn and Z–Ala–Ala–OMe, where also the monomeric subunits were represented in the dimer and so on.20 Therefore, we have used a more focused approach for the conformation search of the trimer: trimer conformers have been built out of g+g− monomers and for the assignment, diagnostic peaks and features from the monomer and dimer assignment have been used. The size of the trimer and the consequent increase in the degrees of freedom prevented us to perform large conformational searches in order to sample the entire conformational space.
The experimental far-IR spectrum of the trimer is presented in the top of Fig. 4a (black trace) and is compared with the calculated IR spectra. The trimer IR spectrum is recorded at the same REMPI wavelength as the dimer and simultaneously acquired. The experimental far-IR spectrum shows a set of well-resolved and diagnostic peaks and has a similar pattern as the dimer far-IR spectrum, namely activity in the 90–280 cm−1 region; two small peaks between 300 and 400 cm−1; a group of peaks between 400 and 525 cm−1 and between 525 and 800 cm−1, and a large and broad feature around 890 cm−1, however with an additional broadened peak around 860 cm−1.
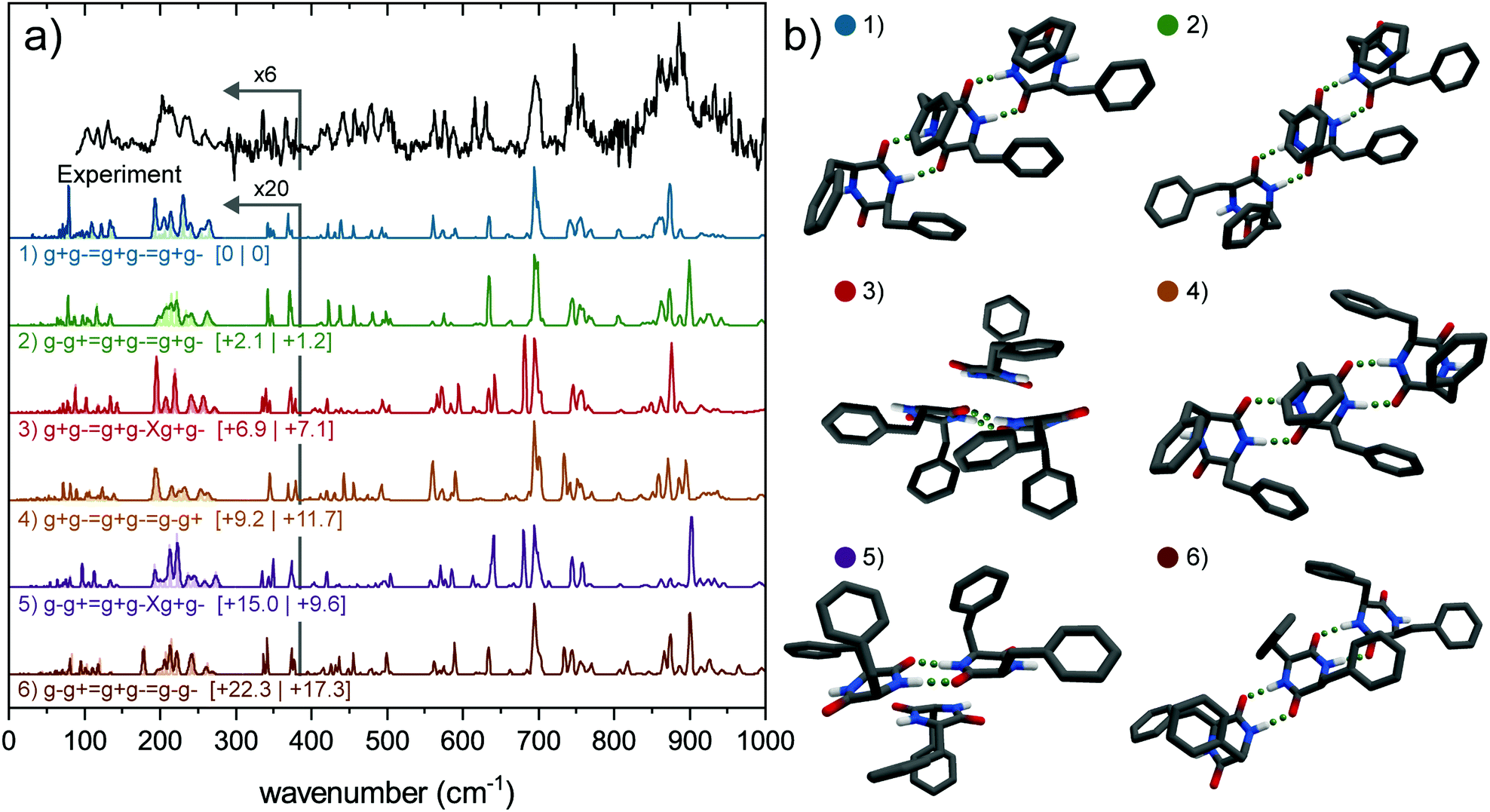 | ||
| Fig. 4 (a) Experimental spectrum of the trimer of cyclo-FF (black, top) and the calculated (colored) far-IR spectra of six possible conformers, arranged from lowest energy to highest (top to bottom). Relative zero-point energies (left) and Gibbs free energies at 300 K are in kJ mol−1 between square brackets. All calculated spectra are scaled by 0.976, calculations have been performed at the B3LYP-D3/6-311+G(d) level. The region below 380 cm−1 has been multiplied by 20 (calculated spectra), and 6 (experimental spectrum) for reasons of clarity. The region below 280 cm−1 is convoluted by 2.5% of the wavenumber and 1% (light colors), to match the experimental conditions. (b) Corresponding 3D structures of (a). | ||
Nine different trimer conformations have been calculated, all but one including only the g+g− subunit. The ninth structure contains a single g−g− conformer. The calculated IR spectra of the six lowest energy conformations are presented in Fig. 4a (colored spectra), together with their structures in Fig. 4b. Mid-IR spectra and relative energies can be found in Fig. S8 and Table S1 (ESI†). Four conformers include the assigned dimer conformation (g−g+ = g+g−) and five conformers contain the lowest energy dimer (g+g− = g+g−) conformer. The calculated spectra of the four structures that are all connected via intermolecular double hydrogen bonds forming a so-called ladder show a much better agreement with the mid-IR experimental spectrum than the conformers where one monomeric subunit is only connected through π–π interactions. The calculated IR spectra of the latter structures show an additional peak around 1410 cm−1, which is completely absent in the experiment. Moreover, the intensities of the two peaks in the amide I region are reversed (see Fig. S8, ESI†).
The experimental far-IR spectra of the monomer, dimer and trimer are plotted together in Fig. 5 to directly observe changes or similarities in peak positions upon aggregation, to identify conserved structural monomeric or dimeric signatures. The green and red lines indicate similarities and differences between the spectra, respectively. Starting at the red side of the far-IR region (90–280 cm−1), a new set of peaks appeared in the trimer spectrum, which were not observed in the dimer or monomer spectrum. The peaks above 180 cm−1 in the dimer spectrum are preserved in the trimer spectrum, but broadened. Importantly, the two small peaks that originate from CH2 rocking motions specific to the ring orientation (i.e., g+, g− or t), are found at the same position as in the dimer and monomer (336 cm−1 for g−, 366 cm−1 for g+). This substantiates the assumption that only g+g− subunits are present in the trimer. The dimer spectrum in the region between 400 and 525 cm−1 is completely different to the monomer, where only a few low intense peaks are observed. In turn, the trimer spectrum comprises here the same peak pattern as the dimer, with again a broadening or splitting of the peaks. Since this part of the far-IR region is, in addition to the region below 300 cm−1, very sensitive to the overall structure of the molecule, the trimer appears to contain the assigned dimer as a building block.
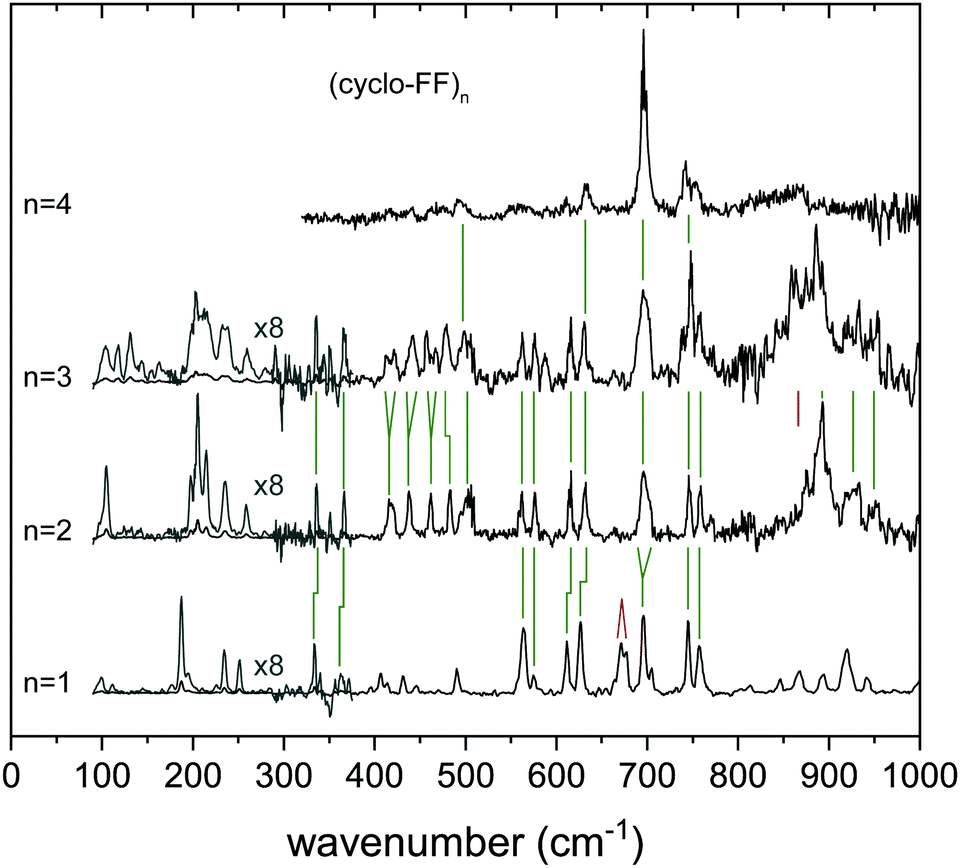 | ||
| Fig. 5 Experimental far-IR spectra of the aggregates of (cyclo-FF)n, with n = 1–4. In some regions green or red lines have been added to show the similarities or differences between the different spectra, respectively. The region between 90 and 280 cm−1 has been multiplied by 8 for clarity (grey spectrum). | ||
The free N–H wagging region between 525 and 800 cm−1 shows distinctive peaks for free N–H groups that are connected to a g+, g− or t side. This makes this region diagnostic for the type of functional groups that are oriented on the outside of the molecule. Upon dimerization, most peaks are retained in the dimer spectrum when compared with the monomer, although some peaks are slightly blue shifted or broadened (e.g., at 696 cm−1) due to intermolecular hydrogen bond interactions. The peak at 673 cm−1, resulting from a symmetric out-of-plane bending motion by both (g+ and g−) N–H groups in the monomer, is absent in the dimer spectrum (red line in Fig. 5); a signature that the molecule is hydrogen bonded in the dimer. The trimer spectrum shows a very similar pattern to the assigned dimer conformer. Here, all the peaks (with IR wavenumbers 562, 572, 615, 631, 714, 746, 759 and 771 cm−1) are preserved in the trimer spectrum, although more intense. Two new peaks are present, one at 589 cm−1 and a small shoulder at 740 cm−1. Since these are not related to the monomer, they must result from hydrogen bonded N–H moieties. This, together with other dissimilarities with the IR spectrum of the monomer, also suggests that the third subunit is hydrogen bonded to the dimer and not connected via π-interactions (thereby excluding the g−g+ = g+g−Xg+g− and g+g− = g+g−Xg+g− conformers).
The region above 800 cm−1 constitutes broad and intense peaks in both dimer and trimer spectra, therefore likely to originate from hydrogen bonded groups. The trimer constitutes a slightly redshifted peak at 886 cm−1 (−6 cm−1) with respect to the dimer, and a large additional feature at 863 cm−1. Both are assigned to vibrations of hydrogen bonded N–H groups. The presence of two separate peaks in this region points to two different double hydrogen bonds (e.g., in one g+ and g− involved, in the other g+ and g+). Therefore, we conclude that the trimer should consist of the assigned dimer (g−g+ = g+g−), with another g+g− monomeric subunit connected via double hydrogen bond with either the g+ or the g− group pointing to the outside, probably in a way that the lowest energy dimer (g+g− = g+g−) is also part of the trimer.
By comparing the calculated far-IR spectra of the six conformers to the experiment (Fig. 4), we can discard the two (2, 1) conformers (red and purple traces), which constitute a dimer and monomer attached only via backbone interactions. They show the poorest overlap with the experiment overall, in both the far-IR as the mid-IR region. The remaining four conformers all show a decent overlap with the experiment. Amongst them, the g−g+ = g+g− = g+g− conformer shows the best overlap, predicting the set of peaks between 200–260 cm−1 and the feature around 500 cm−1 very well. The other conformers all fail to predict certain diagnostic features or predict peaks at wrong frequencies. We can therefore confirm the findings in the previous paragraphs. The trimer observed in our experiment can most likely be assigned to the g−g+ = g+g− = g+g− conformer (green) as it shows the best overlap in both the far-IR and mid-IR (Fig. S8, ESI†) region. Moreover, this assignment is in agreement with the evolution of the IR spectra upon aggregating from monomer to trimer (see Fig. 5).
506 cm−1, and compared with calculated IR spectra (in color) of a selection of predicted conformers (corresponding structures in Fig. 6b). Three main groups can be distinguished within these calculations (Table S2, ESI†), i.e., consisting of a single ladder with length n = 4 (named 4, 0), consisting of two ladders with length n = 2 (2, 2) or of two ladders with lengths n = 3 and n = 1 (3, 1). These structures contain either one or two of the dimers g+g− = g+g− and g−g+ = g+g−, the assigned monomer g+g− or the assigned trimer g−g+ = g+g− = g+g−. Energetically, a clear division is visible between these structural families. The two ladder structures with a loose monomer (3, 1) are highest in energy (above 50 kJ mol−1), the single ladder species (4, 0) are lower in energy (>27 kJ mol−1), and the double ladder structures with 2 dimers (2, 2) are found to be energetically favorable. These (2, 2) dimers comprise the 5 lowest energy structures.
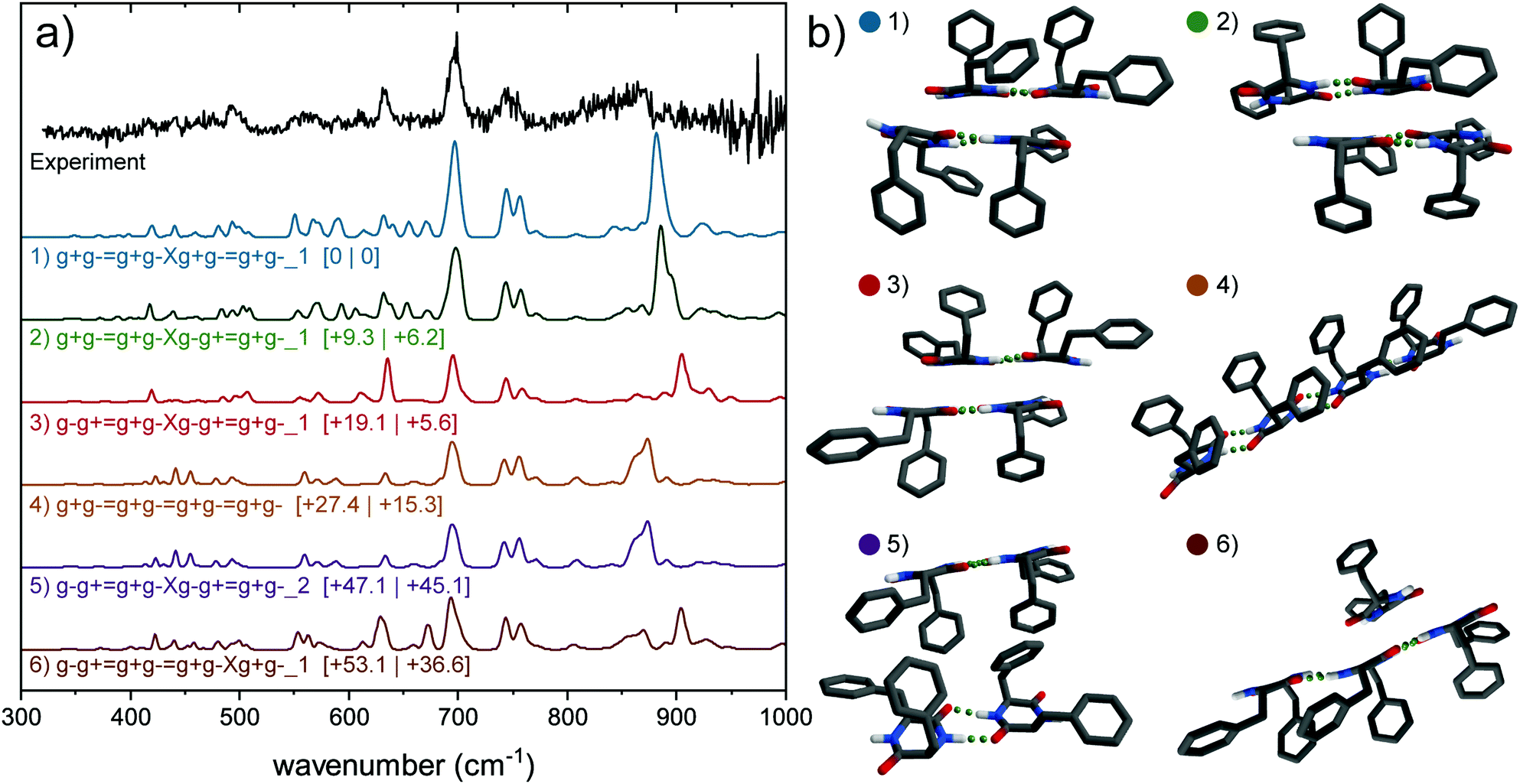 | ||
| Fig. 6 (a) Experimental spectrum of the tetramer of cyclo-FF (black, top) and the calculated (colored) far-IR spectra of five possible conformers containing only g+g− subunits, arranged from lowest energy to highest (top to bottom). Relative zero-point energies (left) and Gibbs free energies at 300 K are in kJ mol−1 between square brackets. All calculated spectra are scaled by 0.976, calculations have been performed at the B3LYP-D3/6-31+G(d) level. (b) Corresponding 3D structures of (a). Hydrogen atoms attached to carbon atoms are not showed for clarity. The lowest three energy structures and the purple (5) structure are of the form (2, 2), the orange structure (4, 0) and the brown structure (3, 1). | ||
The mid-IR region of the IR spectrum shows a double peak in the amide I region, which can be best explained by the (2, 2) structures, although the other structural families cannot fully be discarded here. The rest of the mid-IR does not provide diagnostic structural information, as a result of similarities between all calculations and the scarce amount of resolved features in the experimental spectrum. The far-IR spectrum of the tetramer shows less resolved signatures than observed in the spectrum of the trimer, but similar general features are present. Specific peaks are preserved from the trimer to the tetramer, such as the peak at 632 cm−1 originating from free N–H out-of-plane wagging. The region between 540 and 685 cm−1 is very sensitive to free N–H groups. A comparison with calculations of the monomer, dimer and trimer shows that the peak attributed to free g− N–H groups (633 cm−1, Fig. S10, ESI†) is retained in the tetramer spectrum, whereas the free g+ N–H wagging motions (605 cm−1) seem to have lost a considerable amount of intensity. Combining these conclusions, we cannot assign the tetramer to a specific conformation, however, we conclude that the tetramer most likely forms a (2, 2) dimeric structure made of the g+g− = g+g− and/or g−g+ = g+g− dimers, with the backbones of both ladders faced toward each other and the rings pointing away. The presence of a g−g− conformer in the tetramer cannot be excluded.
506 cm−1. Obtaining the IR spectra of these higher order clusters is experimentally challenging, which is reflected in the quality (signal-to-noise ratio) of the spectra of the highest order clusters. Overall, the IR spectra show less resolved features than the clusters with n = 1–3, although the most intense peaks are still prominently present, such as the amide I and II peaks and the ring vibration modes around 700 cm−1. Specific structural assignments on clusters larger than the tetramer are not possible at this moment, since their spectra are too broad, and lack diagnostic bands.
Discussion
In this paper, we have experimentally studied the aggregation process of cyclo-FF molecules under isolated conditions. To compare our work to computational studies of vapor-depositing experiments,16 the variations in aggregation conditions should be kept in mind. In our experiments, cyclo-FF self-assembles on the fly, starting from desorbing cyclo-FF powder. By collisions with each other and with argon atoms the molecules are cooled down and aggregate during the travel through the vacuum chamber, where they are probed in flight.21 In the vapor depositing process,6 the starting material, lyophilized cyclo-FF, is heated using thermal evaporation at temperatures around 220 degrees Celsius, after which they are deposited onto a substrate.12The first conclusion is that the double hydrogen-bond “ladder” structures that have been found in previous experiments and calculations,16 such as X-ray and NMR studies,13,14 are also clearly observed in our experiments. Very convincing assignments to these double hydrogen-bonded structures can be made for the clusters of cyclo-FF. In particular, our work showed that the region between 800 and 1000 cm−1 provides strong evidence with high intense peaks. These peaks only appear in the calculated IR spectra with double hydrogen bonds, while almost no intensity is found in the single or non-hydrogen-bonded calculations. In molecular simulations performed by Jeon and Scott Shell on 50 cyclo-FF molecules, two states were found, separated by a free energy barrier.16 One state (98%) with a small average length (n < 3.5) and a large average angle between the ladders (>30°), and a minor, second state with a larger average length (>3.5) and smaller angles between the ladders (<30°). This second state is likely responsible for forming the nanowires and rods. It shows a temperature dependence, with its maximum at 80 °C, which is precisely the temperature the vapor deposition experiment was conducted. A sharp decrease was observed for lower temperatures (up to 27 °C). Although no simulations were performed at even lower temperatures (towards temperatures in our experiment), it seems probable that this state with its larger average ladder length, would be non-existent. This would be in line with our conclusions that the largest observed ladder is n = 3, while the tetramer consists of two ladders of n = 2.
Scott Shell et al. also concluded that in vacuum electrostatic interactions between the backbones of the ladders are the most significant driving forces, which ensure that these backbones are oriented towards each other, leaving the aromatic side-chains facing outward. This behavior is what could explain the hydrophobic properties of the cyclo-FF nanorods. In our work, we assigned the structures of the monomer, dimer and trimer to single ladder species, and only the tetramer and larger clusters to double ladder species. One of the calculations of the tetramer, the g−g+ = g+g−Xg−g+ = g+g−_2 conformer, has its aromatic rings oriented to each other, and the backbones to the outside. Compared to the same structure with the backbones oriented towards each other (g−g+ = g+g− Xg−g+ = g+g−_1), no clear differences can be observed in the far-IR, and only minor differences in the mid-IR (small shift of the 1450 cm−1 peak and bands in amide I region). While spectrally nearly indistinguishable, energetically a large difference is observed in favor of the structure with the rings pointing outward. Our work shows that indeed this type of structure is the preferred assignment for the tetramer, however, the energetically unfavorable structure cannot completely be disregarded.
The amide I (CO stretch vibration) peaks are shown in Fig. 7a for the clusters n = 1–8, with two lines to guide the eye. The orange line indicates the position of the hydrogen bonded CO stretch peak of the 8-mer, while the blue line is centered at the non-hydrogen bonded CO stretch vibration of the monomer. The percentage of the area occupied by the hydrogen bonded CO stretch vibration compared to the total amide I region is presented in Fig. 7b in the red columns. In the dimer spectrum, both hydrogen bonded and free CO stretch peaks have the same intensity, resulting in a percentage of 50% hydrogen bonded amide I (see Fig. 7b). This percentage grows steadily with the cluster size, and ultimately more than 80% of the intensity in the amide I region is a result of the hydrogen bonded CO stretch peak. The black line in Fig. 7b indicates the peak position of the hydrogen bonded CO stretch peak, and shows a clear redshift with growing cluster size (−14 cm−1 from n = 2 to n = 8). This behavior was also observed before for other model peptides, where the hydrogen bonding CO stretch peak redshifted as a result of clustering, toward the obtained FT-IR value of parallel beta-sheet CO mode for the bulk material.21
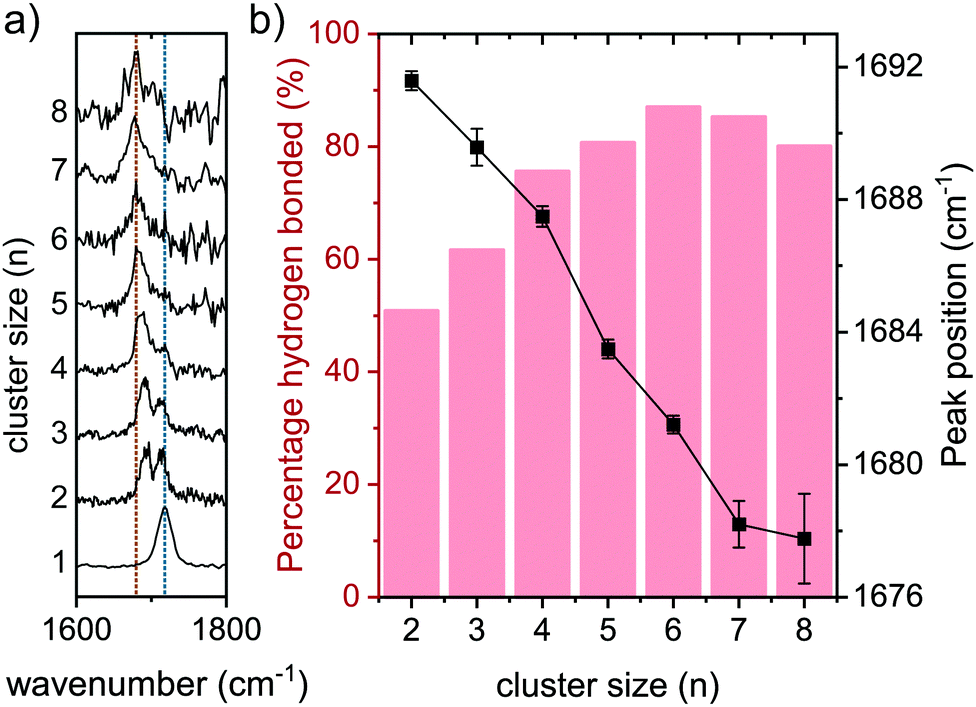 | ||
| Fig. 7 (a) Amide I spectral region for aggregates of (cyclo-FF)n with n = 1–8. Blue and orange lines indicate positions of the top of the peak for the aggregates with n = 1 and n = 8, respectively; (b) black line: peak position of the hydrogen bonded CO stretch peak (orange line) from (a) per cluster; Red columns: the part of the total intensity in (a) that is allocated to the hydrogen bonded CO stretch. The remaining percentage is a result of the free CO stretch peak (blue line in (a)). | ||
Two conformations were found for the monomer: One in the lowest energy g+g− configuration, with an important CH–π interaction that stabilizes the molecule, and a minor second g−g− configuration, with both phenyl groups pointing away from the central ring. Previously, we studied dimers of multiple model peptides, from which the monomer showed the presence of two conformers.20 There, the observed dimers showed to be consisting of a combination of both conformers, whereas the assigned dimer of cyclo-FF in this work solely consist of the main conformer. There is no indication of the presence of a second dimer conformer which includes the g−g− conformer. However, the UV excitation wavelength of a possible second dimer conformer could be different (and hence not probed by us), or its IR or UV signals are too weak to be detected, and hence the signatures are not observed in the IR spectrum. For the previous examples where both monomeric conformers contributed to the dimer, both conformers were found to have relatively similar UV intensities in the REMPI spectrum, while in this work the signal of the second conformer is almost negligible compared to the main conformer.
From the tetramer onwards the REMPI spectrum is broader and lacks the sharp peaks of the smaller clusters. It is remarkable that there seems to be a sharp transition between the trimer and tetramer in this respect. The broadening could be explained by a larger conformational diversity, or insufficient cooling. The large difference between the trimer and tetramer could be fueled by the double ladder structure of the tetramer, which is floppier than the trimer, and allows also for more similar conformations.
Conclusions
We have investigated the aggregation of the cyclic dipeptide cyclo-FF in the gas phase in detail using a combination of laser desorption, mass spectrometry and IR action spectroscopy. The hyphenation of IR-UV spectroscopy with molecular beam time-of-flight detection allowed us to form neutral cyclo-FF clusters on the fly, and to obtain mass- and conformer selective mid-IR and far-IR spectra, resulting in a detailed structural elucidation. Overall, our results show that the clusters are grown from the smaller clusters. The monomer adopts two conformers, the lowest energy g+g− conformer (major contribution), and a minor g−g− conformation. One conformer was identified for the dimer, with double hydrogen bonds and built up of g+g− monomers, thereby aligning in a so-called ladder configuration. In this conformer (g−g+ = g+g−), they are lined up such that both phenyl rings (g−) that stick out of the molecule are pointing outward, while the 2 g+ rings are T-stacked at the inside of the “ladder”, at the hydrogen bonding site. For the trimer also one conformer was identified, with a single ladder (n = 3), namely g−g+ = g+g− = g+g−. This can originate from the assigned dimer configuration and again consists solely of the g+g− monomers. Again, on both outsides of the molecule the rings are pointing away from the ladder. The tetramer and other higher order clusters lose the highly resolved spectra as seen for the smaller molecules, and are likely subject to a larger conformational diversity. Nevertheless, using the knowledge obtained from the monomer, dimer and trimer on peak positions and ratios, predictions on the structure of the larger clusters could be made. Whereas the mid-IR between 1000 and 1800 cm−1 is not sufficiently diagnostic for these molecules, the N–H stretch region (to a certain extent) and especially the far-IR region allowed us to retrieve these unprecedented structural details.Author contributions
Sjors Bakels: conceptualization, data curation, formal analysis, investigation, project administration, visualization, writing–original draft, writing–review & editing. Iuliia Stroganova: investigation, writing–review and editing. Anouk M. Rijs: conceptualization, funding acquisition, project administration, resources, supervision, validation, writing–review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. We thank the FELIX team for their experimental support and helpful discussions. This work was sponsored by NWO Exact and Natural Sciences for the use of supercomputer facilities (Grant no. 2019.062) at the SurfSARA Cartesius cluster. Furthermore, Alexander Lemmens is particularly acknowledged for his help on the OPO measurements, and helpful discussions in general.Notes and references
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- N. Amdursky, P. Beker, I. Koren, B. Bank-Srour, E. Mishina, S. Semin, T. Rasing, Y. Rosenberg, Z. Barkay and E. Gazit, Biomacromolecules, 2011, 12, 1349–1354 CrossRef CAS PubMed.
- L. Adler-Abramovich, P. Marco, Z. A. Arnon, R. C. Creasey, T. C. Michaels, A. Levin, D. J. Scurr, C. J. Roberts, T. P. Knowles and S. J. Tendler, ACS Nano, 2016, 10, 7436–7442 CrossRef CAS PubMed.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- J. Kim, T. H. Han, Y. I. Kim, J. S. Park, J. Choi, D. G. Churchill, S. O. Kim and H. Ihee, Adv. Mater., 2010, 22, 583–587 CrossRef CAS PubMed.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS PubMed.
- P. Beker and G. Rosenman, J. Mater. Res., 2010, 25, 1661–1666 CrossRef CAS.
- M. Jaworska, A. Jeziorna, E. Drabik and M. J. Potrzebowski, J. Phys. Chem. C, 2012, 116, 12330–12338 CrossRef CAS.
- M. A. Ziganshin, A. V. Gerasimov, S. A. Ziganshina, N. S. Gubina, G. R. Abdullina, A. E. Klimovitskii, V. V. Gorbatchuk and A. A. Bukharaev, J. Therm. Anal. Calorim., 2016, 125, 905–912 CrossRef CAS.
- J. S. Lee, I. Yoon, J. Kim, H. Ihee, B. Kim and C. B. Park, Angew. Chem., Int. Ed., 2011, 50, 1164–1167 CrossRef CAS PubMed.
- X. H. Yan, Y. Su, J. B. Li, J. Fruh and H. Mohwald, Angew. Chem., Int. Ed., 2011, 50, 11186–11191 CrossRef CAS PubMed.
- I. W. Park, J. Choi, K. Y. Kim, J. Jeong, D. Gwak, Y. Lee, Y. H. Ahn, Y. J. Choi, Y. J. Hong and W.-J. Chung, Nano Energy, 2019, 57, 737–745 CrossRef CAS.
- M. Gdaniec and B. Liberek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1343–1345 CrossRef.
- H. Takahashi, B. Viverge, D. Lee, P. Rannou and G. De Paëpe, Angew. Chem., Int. Ed., 2013, 52, 6979–6982 CrossRef CAS PubMed.
- K. Joshi and S. Verma, Tetrahedron Lett., 2008, 49, 4231–4234 CrossRef CAS.
- J. Jeon and M. S. Shell, J. Phys. Chem. B, 2014, 118, 6644–6652 CrossRef CAS PubMed.
- M. Gerhards and C. Unterberg, Phys. Chem. Chem. Phys., 2002, 4, 1760–1765 RSC.
- M. Gerhards, C. Unterberg, A. Gerlach and A. Jansen, Phys. Chem. Chem. Phys., 2004, 6, 2682–2690 RSC.
- T. D. Vaden, S. A. N. Gowers and L. C. Snoek, J. Am. Chem. Soc., 2009, 131, 2472–2474 CrossRef CAS PubMed.
- S. Bakels, E. M. Meijer, M. Greuell, S. B. A. Porskamp, G. Rouwhorst, J. Mahé, M. P. Gaigeot and A. M. Rijs, Faraday Discuss., 2019, 217, 322–342 RSC.
- S. Bakels, S. B. A. Porskamp and A. M. Rijs, Angew. Chem., Int. Ed., 2019, 58, 10537–10541 CrossRef CAS PubMed.
- A. Perez-Mellor, I. Alata, V. Lepere and A. Zehnacker, J. Mol. Spectrosc., 2018, 349, 71–84 CrossRef CAS.
- S. Bakels, M.-P. Gaigeot and A. M. Rijs, Chem. Rev., 2020, 120, 3233–3260 CrossRef CAS PubMed.
- D. R. Galimberti, S. Bougueroua, J. Mahe, M. Tommasini, A. M. Rijs and M. P. Gaigeot, Faraday Discuss., 2019, 217, 67–97 RSC.
- D. Oepts, A. F. G. Vandermeer and P. W. Vanamersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CrossRef CAS.
- D. A. Case, T. A. Darden, T. E. I. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, K. F. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M. J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, 2012 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, 2016 Search PubMed.
- S. Jaeqx, J. Oomens, A. Cimas, M. P. Gaigeot and A. M. Rijs, Angew. Chem., Int. Ed., 2014, 53, 3663–3666 CrossRef CAS PubMed.
- J. Mahe, S. Jaeqx, A. M. Rijs and M. P. Gaigeot, Phys. Chem. Chem. Phys., 2015, 17, 25905–25914 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03237b |
| This journal is © the Owner Societies 2021 |