
Two-dimensional materials in biomedical, biosensing and sensing applications
Nasuha
Rohaizad†
ab,
Carmen C.
Mayorga-Martinez†
 c,
Michaela
Fojtů
c,
Naziah M.
Latiff
b and
Martin
Pumera
*cdef
c,
Michaela
Fojtů
c,
Naziah M.
Latiff
b and
Martin
Pumera
*cdef
aNTU Institute for Health Technologies, Interdisciplinary Graduate School, Nanyang Technological University, Singapore
bDivision of Chemistry & Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore
cCenter of Advanced Functional Nanorobots, Department of Inorganic Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic. E-mail: pumera.research@gmail.com
dFuture Energy and Innovation Laboratory, Central European Institute of Technology, Brno University of Technology, Purkyňova 656/123, Brno, CZ-616 00, Czech Republic
eDepartment of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea
fDepartment of Medical Research, China Medical University Hospital, China Medical University, No. 91 Hsueh-Shih Road, Taichung, Taiwan
First published on 18th November 2020
Abstract
Two-dimensional (2D) materials are at the forefront of materials research. Here we overview their applications beyond graphene, such as transition metal dichalcogenides, monoelemental Xenes (including phosphorene and bismuthene), carbon nitrides, boron nitrides along with transition metal carbides and nitrides (MXenes). We discuss their usage in various biomedical and environmental monitoring applications, from biosensors to therapeutic treatment agents, their toxicity and their utility in chemical sensing. We highlight how a specific chemical, physical and optical property of 2D materials can influence the performance of bio/sensing, improve drug delivery and photo/thermal therapy as well as affect their toxicity. Such properties are determined by crystal phases electrical conductivity, degree of exfoliation, surface functionalization, strong photoluminescence, strong optical absorption in the near-infrared range and high photothermal conversion efficiency. This review conveys the great future of all the families of 2D materials, especially with the expanding 2D materials’ landscape as new materials emerge such as germanene and silicene.
 Nasuha Rohaizad | Nasuha Rohaizad obtained her BSc (Hons) degree in Chemistry and Biological Chemistry from Nanyang Technological University (NTU), Singapore, in 2016. She continues to pursue her PhD under the guidance of Prof. Martin Pumera. Her research interest is centred on the electrochemistry of layered nanomaterials, motivated by the development of biosensors for healthcare applications. |
 Carmen C. Mayorga-Martinez | Carmen Mayorga is currently Kralupy unit Leader and senior scientist from the Center for Advanced Functional Nanorobots, UCT-Prague. She was a research fellow in the nanobioelectronics and biosensors group/ICN2, Barcelona-Spain and in Nanyang Technological University, Singapore. She completed her PhD degree at the National University of Tucuman, Argentina, in 2009. Currently, her main research fields include development of bio/sensors based on 2D-materials and nanoparticle platforms functionalized with bioreceptors (enzymes, DNA, and antibodies) as well as micro/nano motors for biomedical applications and environmental monitoring. Moreover, she is also interested in 2D-materials’ catalysis for energy application. |
 Michaela Fojtů | Michaela Fojtů received her MSc in Biochemistry in 2014 and in 2018 she received a PhD in Physiology and Pathological Physiology from the Masaryk University in Brno, Czech Republic. During her PhD studies, she spent several months at Nanyang Technological University (NTU) in Singapore in the group of Prof. Martin Pumera. Since 2018 she has been working as a postdoctoral fellow in the Center for Advanced Functional Nanorobots led by Prof. Pumera in Prague, Czech Republic. In 2019 she received the Werner von Siemens Award and in 2020 she received a Fulbright Fellowship and will join the group of Prof. Sengupta at Harvard Medical School in Boston, USA. Her research is focused on cancer biology, testing of novel anticancer compounds, drug resistance, nanocarrier-mediated targeted drug delivery, nanomaterial toxicity and novel biomedical applications of nanomaterials and nanorobots. |
 Naziah M. Latiff | Naziah M. Latiff obtained her BSc and PhD degrees in Chemistry and Biological Chemistry from Nanyang Technological University (NTU), Singapore, in 2014 and 2019, respectively. Her PhD was under the supervision of Prof. Martin Pumera. Cytotoxicity of nanomaterials as well as their electrochemical properties was her main research interest during her graduate studies, which range from graphene, to transition metal chalcogenides and black phosphorus. |
 Martin Pumera | Martin Pumera is Director of the Center for Advanced Functional Nanorobots and a Distinguished Professor of Chemistry at the University of Chemistry and Technology, Prague, and Chief Investigator of Future Energy & Innovation Lab at CEITEC, Brno, Czech Republic. He in 2001 received a PhD from Charles University, Czech. After two postdoctoral stays, in 2006 he became a tenured group leader at the National Institute for Materials Science (NIMS), Japan. In 2010 he joined Nanyang Technological University, Singapore, as a tenured associate professor for nearly a decade. He has broad interest in nanomaterials and microsystems, in specific areas of electrochemistry and synthetic chemistry of 2D nanomaterials, nanotoxicity, micro and nanomachines, and 3D printing. He is ‘‘2017, 2018, 2019 and 2020 Highly Cited Researcher’’ by Clarivate Analytics. |
1. Introduction
Two-dimensional (2D) materials represent a novel class of nanomaterials which are used in numerous applications and in different fields of research such as biomedicine, biosensing and chemical sensing as well as energy storage and generation, electronics, etc. 2D materials exist as sheet-like structures of a single atom or a few atoms thick with a lateral size from a few nanometers to hundreds of nanometers and above.1–4 Such structures have shown enhanced electronic, optical, chemical, and physical properties that are mainly related to their signature characteristics such as phase, crystallinity, degree of exfoliation, stability and size. These characteristics may depend on the synthesis method used for obtaining them.1,5,6 Recently, these particular properties of 2D materials have been reported to have remarkably improved the performance indexes of sensing and biosensing systems in electrochemical and optical transduction modes. Apart from application as sensors, these properties make them promising candidates in biomedical applications notably in areas of drug delivery and drug potentiation as well as photothermal and photodynamic therapies. Moreover, these properties of 2D materials can impact and influence their toxicity.After the great initial fervor around graphene, attention turned to other 2D structures such as transition metal dichalcogenides (TMDs),7,8 boron nitrides (BN),9,10 carbon nitrides (CN),11 monoelemental Xenes (phosphorene, antimonene, arsenene, bismuthene, silicene, germanene, etc.),12–14 transition metal carbides and/or nitrides (MXenes),15,16 and more recently, metal phosphorus chalcogenides (MPX3).17–19 Strikingly, TMDs representing the main 2D material family have been exhaustively used in developing electrochemical and optical diagnostic devices owing to their sizeable bandgap, good conductivity, fast electron transfer as well as strong fluorescence, electrochemiluminescence and photoelectrochemical activity. The other 2D material families engaged for similar purposes are MXenes, carbon nitrides followed by Xenes and boron nitrides.
The annotations in this review are envisioned to promote how the distinctive properties of each 2D material are utilized: (i) enhancing the performance of biosensing and sensing (chemical and gas sensing) systems, (ii) improving photothermal and photodynamic therapies as well as drug's potentiation and delivery and (iii) correlating to the material's intrinsic toxicity. This review will focus on five classes of 2D materials: TMDs, MXenes, Xenes, boron nitrides and carbon nitrides. They are shortlisted due to their important role in biomedical applications (diagnostic and therapy) that have been reported in our pioneering works, in addition to sensing and biosensing applications that leverage on specific chemical or physical properties of TMDs and Xenes. Likewise, there is enthusiasm for boron nitrides, carbon nitrides and MXenes by other research groups,20 all of which are outstanding among 2D materials as will be put forth in this review. Such studies have also been extended to therapeutic applications and in-depth insights on how these properties affect the toxicity of 2D materials.
2. 2D materials for biosensing application
The superior optical, electrical and electrochemical properties of 2D materials render them attractive as platforms and/or probes for developing sensitive electrochemical and optical biosensors, towards the detection of environmental pollutants and biologically active molecules. These properties are governed by phase, crystallinity, metallicity, degree of exfoliation, stability, sheets’ size and decoration with metal particles. In this section, we will describe the latest advances of electrochemical and optical biosensors that harness the unique properties of 2D materials reported for biological metabolite or toxic compound detection. The materials are used as platforms for enzymatic biosensors and a platform or label for genosensors, aptasensors and immunosensors.2.1 Electrochemical biosensors
Electrochemical biosensors have been touted as a promising class of analytical devices. They rely on biological recognition elements, offering high selectivity towards the target analyte. Simultaneously, electrochemical transduction demonstrates advantages superior to conventional analytical tools.21 The instrumentation is low-cost and simple, yet capable of robust and quantitative measurements. Portability and miniaturization are also possible, an encouraging progress towards the ideal biosensor.22 The basic principle of this class of biosensors is that the recognition event between immobilized biomolecule and target analyte leads to changes in the electrical properties of the sensing material or solution such as electric current, conductance, potential, and ionic strength.23Potentiometry, amperometry, voltammetry, and impedance are detection techniques selected to measure those changes through their respective transduction mechanisms.24,25 At a glance, potentiometry generates electric potential based on the conversion of charge carriers from the detected ions to electrons. Impedance probes the interfacial properties of an electrode after imposing a small-amplitude, sinusoidal AC potential. On the other hand, the dominant voltammetry and amperometry techniques rely on an applied potential to drive electron transfer, yielding a measurable change in current which is proportional to the target analyte. Inherently, there are advantages and disadvantages of each detection technique. Cyclic voltammetry (CV) is known as a “fingerprint” technique to obtain qualitative information regarding electrochemical processes. A quantifiable and sensitive response, however, can be achieved with different excitation profiles such as differential pulse voltammetry (DPV) and square-wave voltammetry (SWV). Recognizing that detection techniques influence the transduction mechanism and analytical parameters, they are included in the summary of developed electrochemical biosensors (Tables 1–3).
| Analyte | 2D material class | Material | Technique | Linear range | LOD | Response intensity | Ref. |
|---|---|---|---|---|---|---|---|
| Glucose | g-C3N4 | g-C3N4 | CV | 1 to 12 mM | 11 μM | 0–28 μA | 112 |
| H2O2 | Xene | BP-PLL | CV | 10 to 700 μM | 3–18 μA | 63 | |
| H2O2 | Xene | BP | Amp | 5 to 275 μM | 0.14 μM | 0–2 μA | 64 |
| Nitrite (NO2−) | Xene | BP-PDDA-IL | Amp | 80 μM to 3.8 mM | 3.65 μM | 0–9 μA | 113 |
| Phenol | Xene | Antimonene | Amp | 0.5 to 2.5 μM; 7.5 to 27.5 μM | 0.255 μM | 0–0.3 μA | 65 |
| Glucose | MXene | Ti3C2-HF/TBA | Amp | 50 to 250 μM; 27 750 μM | 23.0 μM | 0–50 μA | 71 |
| Glucose | MXene | Ti3C2Tx–Au | Amp | 0.1 to 18 mM | 5.9 μM | 0–12 μA | 72 |
| Glucose | MXene | Ti3C2Tx–graphene | CV | 0.2 to 5.5 mM | 0.10 mM | 0–120 μA | 75 |
| Glucose; lactate | MXene | Ti3C2Tx-CNT | Amp | 10 μM to 1.5 mM; 0 to 22 mM | 0.33 μM; 0.67 μM | 0–60 μA cm−2; 0–300 μA cm−2 | 77 |
| H2O2 | MXene | Ti3C2 | Amp | 0.1 to 260 μM | 20 nM | 0–6 μA | 68 |
| H2O2 | MXene | Ti3C2–TiO2 | Amp | 0.1 to 380 μM | 14 nM | 0–14 μA | 74 |
| H2O2 | MXene | Ti3C2Tx–GO | DPV | 2 μM to 1 mM | 1.95 μM | 5–25 μA | 78 |
| Nitrite (NO2−) | MXene | Ti3C2 | Amp | 0.5 to 11800 μM | 0.12 μM | 0–40 μA | 69 |
| OPs (malathion) | MXene | Ti3C2Tx–Ag | DPV | 10 fM to 10 nM | 3.27 fM | 0.2–2.3 μA | 73 |
| OPs (malathion) | MXene | Ti3C2Tx–Chi | DPV | 10 fM to 10 nM | 3 fM | 0.3–3.0 μA | 114 |
| OPs (methamidophos) | MXene | Ti3C2–Au/MnO2–Mn3O4 | DPV | 1 pM to 1 μM | 0.134 pM | 0.5–4.5 μA | 76 |
| Phenol | MXene | Ti3C2Tx–Chi | Amp | 0.05 to 15.5 μmol L−1 | 12 nmol L−1 | 0–7 μA | 70 |
| Cholesterol | TMDs | MoS2–Au NPs | Amp | 0.5 to 48 μM | 0.26 μM | 5–20 μA | 45 |
| Glucose | TMDs | TiS2 (Nb-doped) | Amp | 74.6 to 272.9 μM 0.767 to 12.6 mM 17.6 to 27.3 mM | 25.7 μM | 0–20 μA | 53 |
| Glucose | TMDs | WS2; WSe2 (1T phase) | Amp | 176 to 766 μM & 1.3 to 22.3 mM; 77 to 274 μM & 0.77 to 22.3 mM | 82.6 μM; 52.0 μM | 0–30 μA; 0–25 μA | 40 |
| Glucose | TMDs | MoS2–Au NPs | Amp | 0.25 to 13.2 mM | 0.042 μM | 0–65 μA | 44 |
| Glucose | TMDs | MoS2–Au NPs | CV | 10 to 300 μM | 2.8 μM | 2–14 μA | 46 |
| Glucose | TMDs | rMoS2 | CV | 3 to 20 mM | 7–10 μA | 52 | |
| H2O2 | TMDs | WS2 (1T phase) | Amp | 2 to 38 μM, 48 to 1728 μM | 36 nM | 0–16 μA | 34 |
| H2O2 | TMDs | MoS2 | DPV | 1 to 950 μM | 0.26 μM | 0–12 μA | 42 |
| H2O2 | TMDs | MoS2 | CV | 20 to 180 μM | 6.7 μM | 0–5 μA | 43 |
| H2O2; NO | TMDs | MoS2–Au NPs | CV | 10 to 300 μM; 10 to 1100 μM | 4 μM; 5 μM | 0–3.5 μA; 0–28 μA | 47 |
| H2O2 | TMDs | MoS2–Au | Amp | 0.5 to 200 μM | 0.1 μM | 0–1.4 μA | 48 |
| H2O2 | TMDs | MoS2–Gr | Amp | 0.2 μM to 1.103 mM | 0.049 μM | 0–80 μA | 51 |
| H2O2 | TMDs | WS2–TiO2 | Amp | 0.5 to 30 μM, 50 to 300 μM, 0.5 to 3 mM | 8.7 μM | 0–16 μA | 115 |
| Lactate | TMDs | MoS2 | CV | 0.056 to 0.77 mM | 17 μM | 0–5 μA | 33 |
| OPs (chlorpyrifos; monocrotophos) | TMDs | MoS2 (N, F-doped) | DPV | 0.15 nM to 3 μM; 0.4 pM to 4 nM | 3 pM; 0.2 pM | 1–6 μA; 1–5 μA | 116 |
| OPs (fenitrothion) | TMDs | WS2 (1T phase) | Amp | 1 to 1000 nM | 2.86 nM | 38 | |
| OPs (omethoate) | TMDs | MoS2–PdNi NW | DPV | 0.1 pM to 0.1 μM | 0.05 pM | 0–1.2 μA | 50 |
| OPs (paraoxon) | TMDs | MoS2 (1T phase) | Amp | 1.0 to 1000 μg L−1 | 0.013 μg L−1 | 0–1.5 μA | 117 |
| Polyphenolic compounds (caffeic acid; chlorogenic acid; epicatechin) | TMDs | MoS2–Gr QDs | Amp | 0.38 to 100.00 μM; 0.38 to 100.00 μM; 2.86 to 100 μM | 0.32 μM; 0.19 μM; 2.04 μM | 0–0.9 μA | 49 |
| Analyte | 2D material class | Material | Technique | Linear range | LOD | Response intensity | Ref. |
|---|---|---|---|---|---|---|---|
| Mb | Xene | BP-PLL | CV | 1 pg mL−1 to 16 μg mL−1 | 0.524 pg mL−1 | 1.3–1.8 mA | 90 |
| PCB77 | Xene | BP-Au NPs | DPV | 100 pg L−1 to 10 μg L−1 | 33 pg L−1 | 40–110 μA | 118 |
| Gliotoxin | MXene | Ti3C2 | Amp | 5 pM to 20 nM | 5 pM | 0–1.25 μA | 79 |
| 17β-Estradiol | TMDs | VS2-Au NPs | DPV | 0.01 pM to 10 nM | 1.0 pM | 42–50 μA | 119 |
| AIV (H5N1) | TMDs | VS2-Gr/Au NPs | DPV | 0.5 pM to 0.5 nM | 0.052 pM | 15–25 μA | 120 |
| ATP; Hg2+ | TMDs | WS2 | EIS | 0.1 μM to 5 mM; 0.1 to 500 nM | 1.5 nM; 0.5 pM | 0.6–2 KΩ; 0.8–1.6 KΩ | 95 |
| ATP; thrombin | TMDs | MoS2–Au NPs | SWV | 1 nM to 10 mM; 0.01 nM to 10 μM | 0.74 nM; 0.0012 nM | 0–1.2 μA; 0.05–0.5 μA | 121 |
| dsDNA | TMDs | MoS2–Thi | SWV | 0.09 to 1.9 ng mL−1 | 2.7–3.3 μA | 87 | |
| DNA | TMDs | MoS2–Gr/Au NPs | DPV | 50 fM to 5 nM | 2.2 fM | 3–9 μA | 80 |
| DNA | TMDs | WS2-Gr/Au-NPs | DPV | 0.01 to 500 pM | 2.3 fM | 60–140 μA | 81 |
| DNA | TMDs | MoS2–MWCNT/Au NPs | CV | 10 fM to 10 nM | 0.79 fM | 18–30 μA | 84 |
| DNA | TMDs | WS2-MWCNT/Au NPs | DPV | 10 fM to 0.1 nM | 2.5 fM | 16–32 μA | 85 |
| DNA | TMDs | WS2-AB/Au NPs | DPV | 0.001 to 100 pM | 0.12 fM | 5–35 μA | 86 |
| DNA | TMDs | MoS2–ZnO | DPV | 1 fM to 1 μM | 0.66 fM | 0–6 μA | 89 |
| DNA | TMDs | MoS2 | DPV | 0.1 fM to 0.1 nM | 0.019 fM | 0.3–2.8 μA | 93 |
| DNA | TMDs | MoS2–Gr | DPV | 0.1 fM to 0.1 pM | 0.01 fM | 45–80 μA | 94 |
| DNA | TMDs | MoS2 | DPV | 0.03 to 300 nM | 0.4–0.65 μA | 96 | |
| DNA | TMDs | MoSe2-Au NPs | DPV | 10 fM to 0.1 nM | 4 fM | 2–18 μA | 122 |
| DNA | TMDs | MoSe2-SiO2/Au NPs | DPV | 0.1 fM to 100 pM | 0.068 fM | 15–45 μA | 123 |
| IgE | TMDs | WS2-Gr/Au NPs | DPV | 1 pM to 10 nM | 0.12 pM | 50–80 μA | 124 |
| miR-21 | TMDs | MoS2–Thi/Au NPs | SWV | 1.0 pM to 10.0 nM | 0.26 pM | 0.2–1.2 μA | 88 |
| miR-21 | TMDs | MoS2–Au NPs | DPV | 0.1 fM to 0.1 pM | 0.086 fM | 0–25 μA | 125 |
| OTA | TMDs | MoSe2-Au NPs | 0.0001 to 1 nM | 0.08 pM | 5–12 μA | 126 | |
| PDGF-BB | TMDs | MoSe2-Gr/Au NPs | DPV | 0.0001 to 1 nM | 20 fM | 20–40 μA | 83 |
| PDGF-BB | TMDs | MoS2–CA/Au NPs | DPV | 0.001 to 10 nM | 0.3 pM | 3–8 μA | 127 |
| Analyte | 2D material class | Material | Technique | Linear range | LOD | Response intensity | Ref. |
|---|---|---|---|---|---|---|---|
| Hp | Xene | BP-PLL | DPV, Amp | 0.01 to 10 mg mL−1 | 0.011 mg ml−1 | 4–10 μA | 100 |
| IgG | Xene | BP | Amp | 2 to 100 ng mL−1 | 0.98 ng mL−1 | 50–200 spikes | 111 |
| CEA | MXene | Ti3C2-APTES | CV | 0.1 pg mL−1 to 2 μg mL−1 | 18 fg mL−1 | 1.5–1.9 mA cm−2 | 101 |
| BSA | TMDs | MoS2 | CV | 0.01 to 10 ng mL−1 | 6 pg mL−1 | 4.0–4.1 μA | 99 |
| CEA | TMDs | MoS2–Au NPs | DPV | 1 pg mL−1 to 50 ng mL−1 | 0.27 pg mL−1 | 0–100 μA | 102 |
| CEA | TMDs | MoS2–Thi/Au NPs | SWV | 1 pg mL−1 to 10 ng mL−1 | 0.52 pg mL−1 | 0.03–0.24 μA | 103 |
| CEA | TMDs | MoS2–PB | DPV | 0.005 to 10 ng mL−1 | 0.54 pg mL−1 | 0–20 μA | 104 |
| IgG | TMDs | WS2 | LSV, EIS | 2 to 500 ng mL−1 | 2 ng mL−1 | 1–5 KΩ | 107 |
| IgG | TMDs | MoSe2 | Amp | 2 to 500 ng mL−1 | 1.23 ng mL−1 | 0.3–0.8 mA | 108 |
| IgG | TMDs | WSe2 | Amp | 5 to 500 ng mL−1 | 1.01 ng mL−1 | 0.5–0.9 mA | 109 |
| IgG | TMDs | MoS2 | Amp | 2 to 6 pg mL−1 | 1.94 pg mL−1 | 0.2–0.7 mA | 110 |
| PTH | TMDs | MoS2–Gr | CV | 1 to 50 pg mL−1 | 4–14 μA | 105 |
Nanomaterials have been garnering attention and studied intensively towards improving the performance of biosensors. In general, their attributes of small size effect, quantum size effect as well as surface and interface effect remarkably improve the important performance indexes of biosensors.26 Modification of conventional electrodes with nanomaterials addresses the size mismatch between electronic transducer and biological elements, which significantly enhances biocompatibility and sensitivity.27 Meanwhile, the quantum size effects unearth catalytic activities not found in bulk structures, due to shifts in the valence band as well as oxidation potentials.
2D materials not only possess favorable size characteristics, but also dimensionality and signature properties that will enhance the performance of biosensors. These planar materials flaunt a large surface-to-volume ratio thereby providing a platform for straightforward and easy functionalization with biological elements or chemical moieties.28 The optimal exposure would increase efficiency as well as sensitivity.29 Attributes of high conductivity and chemical stability, depending on the degree of exfoliation, further result in desirable electrochemical properties like enhanced heterogeneous electron transfer (HET) and water electrolysis reactions. Owing to these qualities along with their biocompatibility, 2D materials are sought after for the development of high-performance electrochemical biosensors.22,30
It is imperative to foremostly discuss the preparation methods of 2D materials since their properties will be greatly influenced, especially electrical conductivity, stability and high surface area. Sonication-assisted liquid exfoliation is one of the top-down methods which scales down bulk materials to single or few layers dispersed in solvents. N-Methyl pyrrolidone (NMP), dimethylformamide (DMF), and isopropyl alcohol (IPA)/water are amongst the solvents that have been suggested for effective exfoliation.32 Including ethanol (EtOH)/water, these four solvents are studied in the preparation of MoS2 nanosheets towards a lactate electrochemical biosensor.33 The lactate oxidase enzyme (LOx) is drop cast following the layer of exfoliated materials on GC, to selectively detect lactate with hydroxymethylferrocene as the mediator. In the comparison of lactate response in cyclic voltammograms, MoS2 exfoliated in NMP solvent is asserted to have superior conductivity based on the highest electrochemical signal generated. An apparent explanation for this is that since the surface energy of NMP is close to that of MoS2, high quality MoS2 nanosheets with enhanced conductivity are obtained.
Another preparation method worth mentioning in the context of electrochemical biosensors is lithium intercalation-assisted liquid exfoliation. It is recognized as a strategy for phase engineering, granting access to tune the properties of TMDs. Lithium ions intercalate in between layers of the bulk material, at the same time inducing an effective change in the d electron count of the metal. As a result, there is a phase transformation from the natural semiconducting 2H to metallic 1T. WS2, prepared by this method, has been coupled with hemoglobin (Hb) for a hydrogen peroxide biosensor.34 Characterization by transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS) revealed a conspicuous 1T-phase, although the 2H-phase was still present. In comparison to the bulk, the exfoliated material displayed an improved HET rate as well as sensitivity towards H2O2. Size, dimensionality along with metallicity bring about fast electron transfer kinetics, good conductivity and increased catalytic sites at both edges and basal planes.35–37
The remarkable improvement in conductivity and electrochemical properties owing to phase transformation has continued the pursuit of biosensors based on solely TMDs without any noble metal nanoparticles. Metallic 1T-WS2 successfully enhances signal transduction for the detection of fenitrothion, an organophosphorus pesticide (Fig. 1A).38 The sensing mechanism relies on the irreversible inhibition of acetylcholinesterase by fenitrothion, consequently preventing the breakdown of the acetylcholine substrate to electroactive choline. With less choline present for oxidation, the measurable decrease in current response is expressed as inhibition percentage. Practicality in real samples is assessed using store-bought apple juice. After sample treatment by blending and filtration, the developed WS2-based biosensor recovers 80% of the spiked concentration of fenitrothion. Compared with other group 6 TMDs (MoS2, MoSe2, WSe2), the outstanding performance of WS2 for electrochemical biosensors is anticipated from characterization data. WS2 undergoes the greatest extent of exfoliation and conversion to the metallic 1T phase, in agreement with the literature.39
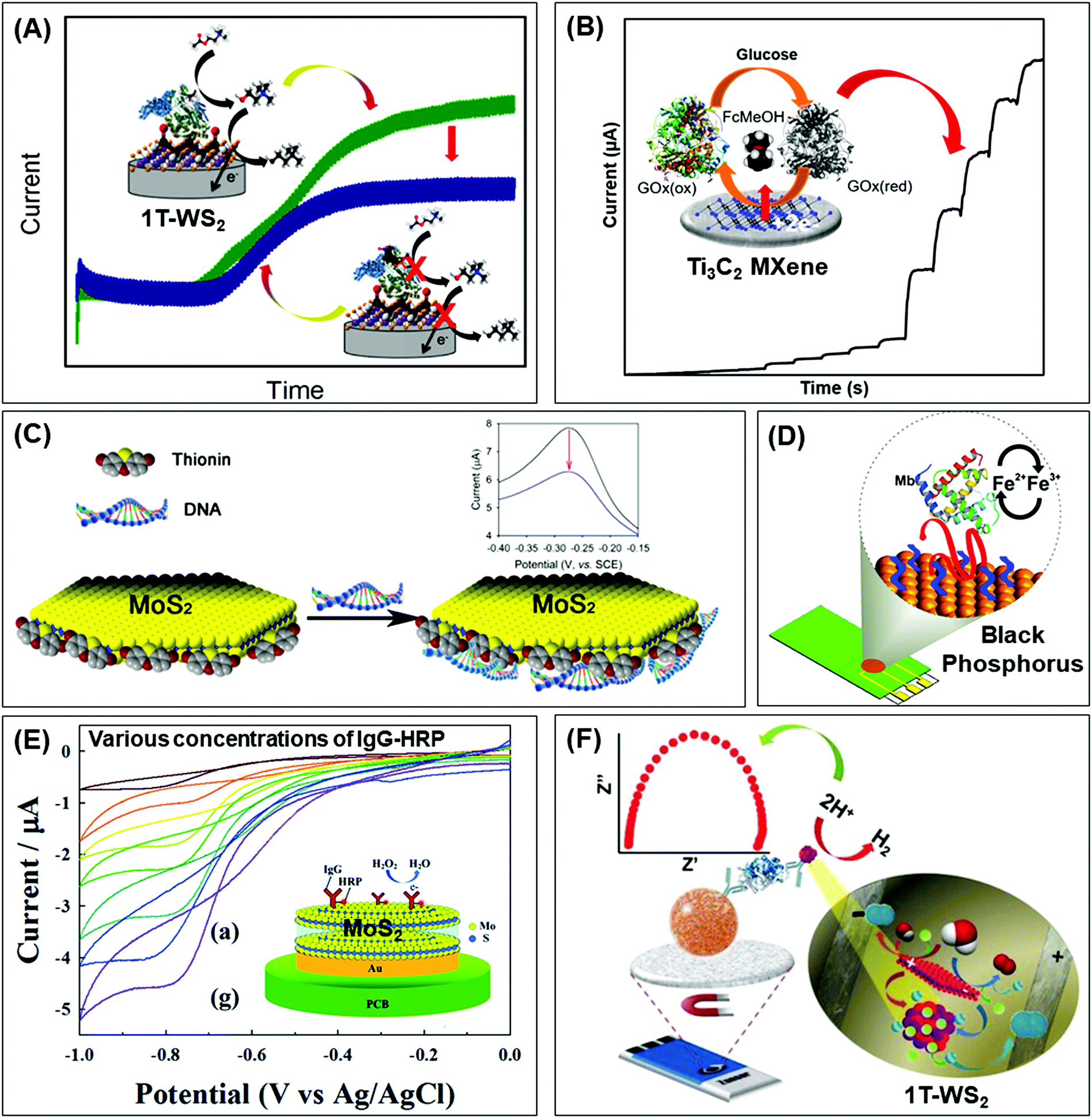 | ||
| Fig. 1 Electrochemical biosensors integrated with 2D materials, of varying elemental compositions, mostly as electrode platforms to detect diverse target biomarkers. Their properties and strategies attractive for biosensors are depicted according to different biorecognition elements: enzymes (top row), DNA or aptamers (middle row) and antibodies (lowest row). Conductivity can be enhanced via (A) phase engineering of TMDs from semiconducting 2H to metallic 1T, and (B) etched-then-delaminated Ti3C2 MXene to optimize surface functional groups for the development of sensitive enzymatic electrochemical biosensors to detect fenitrothion and glucose respectively. The large surface area as well as surface charges of 2D materials facilitates functionalization of positively charged compounds, (C) thionin onto MoS2, (D) PLL onto BP, for binding with the negatively charged phosphate backbone of DNA or aptamers. Advanced preparation methods of TMDs are utilized for immunosensors, namely (E) plasma enhanced chemical vapor deposition to directly integrate MoS2 on polymeric substrates and (F) bipolar electrochemistry to downsize and match the sizes of proteins and magnetic beads in magneto-immunoassays. Strikingly, 1T-WS2 prepared via bipolar electrochemistry is employed as a label which catalyzes hydrogen evolution reaction to generate a measurable response. Figures adapted with permission from: (A) ref. 38, Copyright 2017 American Chemical Society; (B) ref. 71, Copyright 2020 American Chemical Society; (C) ref. 87, Copyright 2014 American Chemical Society; (D) ref. 90, Copyright 2016 American Chemical Society; (E) ref. 106, Copyright 2015 The Royal Society of Chemistry; (F) ref. 107, Copyright 2016 Wiley-VCH. | ||
A coherent relationship between metallic 1T-phase and electrochemical performance has been reported in a second generation glucose biosensor.40 Characterization across group 6 TMDs reiterates that exfoliation together with phase transformation is more effective for tungsten dichalcogenides than molybdenum dichalcogenides. TEM and HR-TEM images together with X-ray diffractograms prove their well-exfoliated structures compared to MoS2 and MoSe2. The dominance as well as stability of the 1T phase in both WS2 and WSe2 is supported by XPS spectra which translate to smallest peak-to-peak separation in HET studies involving the ferrocenemethanol mediator. Subsequently, they achieve the highest sensitivity in glucose detection compared to MoS2 and MoSe2. Their superior sensing capabilities are ascribed to the metallic 1T-phase, with emphasis on exfoliation efficiency and phase transformation. Concurrently, this study highlights the significant role of the transition metal component, besides intercalating agents,41 in influencing the outcomes of lithium intercalation-assisted exfoliation. However, the developed biosensor is less successful in its applicability in real sample analysis. The concentration of glucose detected falls short, albeit close to that, of the specified range in human serum samples. There are possible effects of interference which calls for an improvement in selectivity, currently obtained by tuning the operating potential.
Moving on from the preparation of 2D materials to their immobilization with enzymes, MoS2 nanosheets are revealed to be negatively charged through zeta potential measurements. Such a property proves to be desirable in the construction of biosensors as positively charged enzymes can be immobilized onto the electrode surface. This is demonstrated for the detection of H2O2 where electrostatic interactions between MoS2 and HRP stabilize the electrode system.42 The biocompatibility of MoS2 nanosheets is also assured by Fourier-transform infrared (FTIR) spectroscopy as well as differential pulse voltammograms which indicates that the assembled HRP maintains its native activity as it effectively reduces H2O2 with the aid of the hydroquinone mediator. Enhanced by dimensionality, the few-layered MoS2 (3 to 4 monolayers) achieved better analytical parameters than 3D flower-like MoS2 based biosensors (Table 1).43
The negative charges of MoS2 can further be relied on to form nanocomposites with noble metal particles, namely gold nanoparticles (AuNPs). Above pH 7.4, the surface charges of AuNPs are positive; therefore spontaneous as well as homogeneous self-assembly can occur onto MoS2via electrostatic interactions.44 The elemental presence of sulfur in MoS2 also allows bonding with Au NPs.45 While MoS2 nanosheets intrinsically demonstrate fast electron transfer kinetics, MoS2/Au NPs improve the dynamic range as well as sensitivity for glucose or cholesterol. Forming nanocomposites with Au NPs46,47 or Au nanorods48 is a prevalent approach due to its established contribution in electrochemical biosensors, especially increasing electroactive surface area and conductivity. A strong synergistic effect of higher enzyme loading and facilitated electron transfer is consequently achieved. There are also other MoS2 nanocomposites produced with materials of varying dimensionality – 0D graphene quantum dots (QDs),49 1D bimetallic alloy nanowires50 and 2D graphene.51
The inherent electrochemistry of materials is an important characteristic of consideration prior to their application in electrochemical biosensors. Intrinsic oxidation or reduction of the material may pose either an interference or advantage to the target signal. Wu et al. reported the first MoS2 based biosensor, who put forth an irreversible electrochemical reduction of the nanomaterial in NaCl aqueous solution saturated with N2.52 When comparing MoS2 and the electrochemically reduced MoS2, signals arise for the latter when probed with redox couples such as [Fe(CN)6]3−/4− and [Ru(NH3)6]2+/3+, implying efficient electron transfer and good conductivity. Its sensing capabilities were then tested for glucose using glucose oxidase (GOx) as the biorecognition element. Recognizing the inherent electrochemistry of the material, electrochemical pretreatment presents a straightforward strategy for enhancing the conductivity of the material which in turn improves the sensitivity of the biosensor.
TMDs do not constitute only group 6 transition metals like Mo and W, which have overshadowed many other layered TMDs. To close the gap, we have reported group 4 TiS2 for the detection of glucose. Additionally, doping with Nb was employed as a strategy to enhance the biosensor's sensitivity.53 Identifying that Ti and Nb are a group apart in the periodic table, their sizes, valencies and polymorphs are discussed to understand the doping effects. More importantly, Nb is more electron rich and would partially fill the originally empty conduction band of TiS2. The increase in electrical conductivity substantially improves the defining electrochemical properties for biosensing, namely fast HET, low resistance and large electroactive surface area. However, too high a dopant concentration results in the formation of TiS3 nanobelts that impede electrochemical performance. Subsequently developed into a 2nd generation glucose biosensor, group 4 TiS2 optimally doped with Nb demonstrates competitive advantages such as a wider linear range and higher sensitivity than group 6 WS2 (Table 1).
Exploring various chemical compositions under the umbrella of 2D materials set the stage for newer properties and strategies. The attention is turned to black phosphorus (BP), a layered material based on puckered sheets of only phosphorus atoms.54 Single- or even few-layered black phosphorus is alternatively referred to as phosphorene,55 just like graphene, or phosphane following the type of hybridization and IUPAC nomenclature.56 Each sheet is composed of sp3-hybridized P4 units in an orthorhombic crystal structure.57–59 Every P atom, along with its lone pairs, covalently bonds to two P atoms in the same plane and another P atom in the parallel adjacent plane. This results in a puckered and bilayer configuration along the armchair (x axis) and zigzag (y axis) directions respectively. Anisotropic properties are reported, in addition to a direct and tunable band gap, sufficiently large carrier mobility as well as good biocompatibility. The combination of properties balances between those of popular graphene and MoS2, favoring BP for device applications.60
A pressing challenge for BP is its chemical instability in air, water and light. Oxygen is identified to play the leading role in degradation, rapidly oxidizing BP to PxOy species.61,62 To develop a biosensor for H2O2 detection, BP was henceforth subjected to liquid exfoliation in deoxygenated water to obtain stable nanoflakes.63 Moreover, our group has evaluated phosphorene as a sensing platform in two different enzymatic systems where their detection is based on either reduction or oxidation of a mediator (ferrocene methanol). Interestingly, enhanced activity is observed only in the reductive system unlike the oxidative counterpart. This phenomenon is attributed to the reductive environment keeping the structure of phosphorene intact whereas in an oxidative environment, phosphorene is easily oxidized. Since phosphorene possesses relatively poor stability in an ambient atmosphere, the electroactivity of phosphorene can be controlled by the mediator-based enzymatic system. These findings of the binary nature of phosphorene give rise to a highly sensitive biosensor for the detection of H2O2 and have high importance in the construction of enzyme logic systems.64
Besides phosphorene, there are other monoelemental layered compounds in group VA (group 15), namely arsenene, antimonene, and bismuthene. We have reported for the first time a highly sensitive and selective phenol biosensor based on these 2D pnictogens.65 Due to their thiophilic characteristics, they are relevant to the development of biosensors as they are capable of binding to the cysteine groups of enzymes. All four materials were exfoliated using the shear-force method, prior to their integration with tyrosinase. In the comparison of their performance to detect phenol, antimonene ranked first followed by phosphorene, bismuthene and lastly arsenene. Antimonene is characterized by the lowest oxidation-to-bulk ratio and the highest degree of exfoliation, resulting in enhanced electron transfer capability. Arsenene, in contrast, is recognized for enzyme inhibition.
Another prominent class of 2D materials reported for enzyme-based biosensors is MXenes. They are represented by the general formula Mn+1Xn (n = 1, 2, 3), where M is an early transition metal while X is carbon, nitrogen or both. Understandably, they are also referred to as transition metal carbides, nitrides or carbonitrides. Twenty MXenes of varying elemental compositions have been successfully synthesized,66 yet only Ti3C2 is reported for electrochemical biosensing. This is owing to its established top-down synthesis route together with attractive properties especially unique morphology, hydrophilicity, and metallic conductivity.
Ti3C2 is derived from its parent MAX phase, Ti3AlC2, with Al atoms between sheets composed of Ti in the hexagonal lattice and C occupying octahedral sites. In other words, these sheets are held by Ti–Al metallic bonds instead of the classic van der Waals forces in layered compounds. The increased bond strength forbids simple mechanical exfoliation, rather demands an optimized temperature and an etchant to selectively remove Al from Ti3AlC2.67 Upon the loss of Al atoms together with interlayer metallic bonds, Ti3C2 sheets are readily delaminated with Ti atoms exposed on the surface. Functional groups subsequently terminate on the surface enabling stability in air, which simultaneously expands the lattice.15
Delamination as well as lattice expansion results in the unique morphology of Ti3C2, often described as organ or accordion-like. The supposedly horizontal sheets are tilted and stacked on top of each other in a vertical zig-zag pattern. Structural advantages for electrochemical biosensors are exemplified in Nafion/Hb/Ti3C2/GC electrodes for H2O2 and nitrite ion detection.68,69 Hb enzymes are depicted to be adsorbed at the opening end of Ti3C2 sheets, sloped towards the other closing end then retained.68 The enzyme immobilization and entrapment are further facilitated by the biocompatibility of Ti3C2. Both UV-vis and FTIR spectroscopy ascertain that the essential conformational features of Hb are kept intact after immobilization. Combining the unique morphology and biocompatibility of Ti3C2, together with its large surface area, a high enzyme loading concentration is achieved.69
Another exclusive aspect of MXenes is their abundant surface groups, largely determined by etchant composition. Most of the Ti3C2 developed for electrochemical biosensors are etched using hydrofluoric acid, thereby generating fluorine and hydroxyl surface terminations. Although there may be a mixture of –F and –OH functional groups, the latter seems to be prevalent due to the conventional preparation in aqueous conditions.15 The surface is rendered hydrophilic which facilitates uniform dispersion in aqueous medium and provides a favorable microenvironment for enzyme immobilization, as demonstrated by Ti3C2 coupled with tyrosinase enzymes to detect phenols in water samples.70 In real sample analysis, the developed biosensor could detect 87 to 107% of phenol spiked in filtered tap water. Hydrophilicity offered by the surface groups while preserving metallic conductivity boosts the popularity of Ti3C2 for electrochemical biosensors.
Besides surface interactions, functional groups are crucial for modulating conductivity. Our group took a step further with the already HF etched Ti3C2 by delaminating it using tetrabutylammonium hydroxide.71 Comprehensive characterization of the compound reveals a substantial percentage decrease of fluorine terminating groups upon exfoliation. Higher electron transfer capability is realized, evident from the enhanced HET. Alongside exfoliation outcomes of thinness and high surface to volume ratio, the material is developed into a 2nd generation glucose biosensor with high sensitivity and selectivity (Fig. 1B). Food analysis is a prospective application for the developed biosensor as 92.5% recovery is attained for glucose spiked in commercialized, sugar-free drinks.
The aforementioned biosensors based on solely Ti3C2 exhibit satisfactory electrochemical properties and detection parameters. Nevertheless, building composites is a strategy indifferently applied to MXenes. Higher conductivity is sought to overcome the electrode surface resistance upon enzyme immobilization. Synergistic effects in amplifying electrochemical signals while maximizing electroactive surface area are achieved between Ti3C2 and nanoparticles, namely Au, Ag and TiO2.72–74 Au NPs impart added biocompatibility by retaining the biological activity of GOx enzymes as well as reducing the insulating effect of the protein shell.72 As such, direct electron transfer is optimized between enzymes and electrode surface. Meanwhile, composites with TiO2 slightly improve the detection limit, linear range and long-term stability of the developed biosensor for H2O2.68,74
Taking advantage of the layered structure of Ti3C2, composites with other 2D materials can be constructed. Graphene, a representative of 2D materials, is superior in terms of conductivity and mechanical strength but suffers from hydrophobicity. Due to the hydrophobicity of graphene, low enzyme affinity results which poses a challenge for biosensors. Conversely, Ti3C2 is reiterated herein as hydrophilic; therefore a composite of graphene and Ti3C2 would bestow superior properties.75 The hydrophilicity of Ti3C2 is pronounced from experimental low water contact angles as well as good dispersion in aqueous solutions without surfactant. During the assembly of 3D Ti3C2-graphene, abundant internal pores are introduced which are advantageous in improving hydrophilicity as well as immobilization and entrapment of GOx enzymes. Another multilayered composite of transition metal oxide MnO2–Mn3O4 and MXene Ti3C2–Au has been developed, this time for electrochemical pesticide detection.76 The latter mainly contributes to the large surface area and high conductivity, nonetheless the composite synergistically enhances these properties.
MXene Ti3C2 continues to gain traction for electrochemical biosensors towards wearable and printing technology, driven by its favorable properties of hydrophilicity and conductivity.77,78 A wearable multifunctional biosensing patch is developed to measure glucose and lactate from sweat uptake.77 The requirement of stretchability is fulfilled by the mechanical strength of Ti3C2 enforced with carbon nanotubes (CNTs). To investigate the feasibility of the device for real applications, real-time monitoring of lactate is conducted in an intense cycling session. After cycling, the subject consumes a banana to analyze sweat glucose levels. Both lactate and glucose values obtained are acceptable with other reports. On the other hand, a composite of Ti3C2 and graphene oxide (GO) is formulated for inkjet printing to prepare a H2O2 biosensor.78
Overall, enzyme-based electrochemical biosensors incorporated with 2D materials are compiled in Table 1 to showcase their progress as well as facilitate comparison. They are sorted according to material class to highlight rising materials, then analyte. Key analytical parameters are listed including linear range, limit of detection (LOD) and response intensity. The prominence of TMDs is apparent, granted their earlier foothold upon outperforming the revolutionary graphene with a direct band gap. They are well-established in terms of synthesis methods and strategies for improving conductivity, hence are successfully incorporated into biosensors to detect a spectrum of analytes. Nonetheless, MXenes are following closely with increasing biosensors developed. Using the table to pick out MXene- and TMD-based biosensors for lactate detection, the former achieves a lower LOD as well as a linear range corresponding to the human lactate concentration in arm sweat.33,77 Apart from differences in the biosensor construction and electrochemical technique, MXenes prove to be promising with their favorable properties of hydrophilicity and metallic conductivity.
Mono-elemental Xenes, on the other hand, have yet to enjoy similar success. Chemical instability remains the main hurdle, evident from the following comparison of H2O2 biosensors constructed with BP (Xene), Ti3C2 (MXene), MoS2 and WS2 (TMDs). The developed WS2-based biosensor could only retain 81.5% of its initial current response after 5 days.34 A postulation is that the engineered 1T phase is less stable than the natural 2H. The BP-based biosensor retains 93% of its initial current response after 7 days but, as anticipated, the percentage dropped to 69% after 15 days (∼2 weeks).64 MoS2- and Ti3C2-based biosensors have superior chemical stability, with a retention rate of 95% after 2 weeks and 93% after 3 weeks respectively.43,68 In terms of analytical parameters, the WS2-based biosensor demonstrates the widest linear range, from μM to mM, and LOD in the nM range together with the Ti3C2-based biosensor, attributed to high conductivity. Although BP has a lower LOD (0.14 μM) than MoS2 (6.7 μM), it is the least sensitive among all four biosensors, implied from the response intensity. There is much to explore for BP or generally Xenes, in terms of modulating essential properties prior to their development as biosensors.
The ease of functionalization coupled with large surface area of 2D materials provides access to various compounds including thionin, PLL, ZnO, etc. Their positive charges could interact with the abundant negative charges of S edges in MoS2 (Fig. 1C).87,88 Such electrostatic effects are demonstrated between MoS2 and ZnO nanosheets, as the adsorption of Zn2+ initiated nucleation followed by growth in a vertical direction.89 A positively charged interface is intended to immobilize aptamer receptors with negatively charged phosphate backbone via electrostatic interactions. In parallel, a BP based biosensor was constructed for the detection of myoglobin (Mb) where the material was foremostly exfoliated in aqueous medium with surfactants, allowing direct application in biological systems.90 It was subsequently functionalized with PLL so that its positively charged amide groups would bind to an anti-Mb aptamer. When the target Mb is captured, an oxidation signal arises from its own iron(II) center (Fig. 1D). The signal is further amplified by the favorable electrochemical properties of BP, including high conductivity. With the biosensor designed to be selective towards only Mb, there is negligible response from the interference of structurally similar Hb. The biosensor could retain 95% of the initial current response up to 21 days, implying high stability. Although TMDs have always been in the limelight (Table 2), works on Xenes and MXenes are notable as they convey a wider perspective of 2D materials for nucleic acid-based biosensors.
Apart from enhancing electrochemical signal transduction, 2D materials continue to fascinate us with another of their attribute in relation to nucleic acid-based biosensors. The physisorption of aromatic and conjugated compounds on the basal planes of MoS2 inspired and led to the realization of contrasting affinity towards single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA).91,92 Higher affinity, therefore higher adsorption, is observed for ssDNA due to van der Waals’ forces between the nucleobases and 2D materials. On the other hand, the nucleobases are buried in the densely negatively charged helical phosphate backbone for dsDNA, which accounts for the low affinity and weak adsorption.
The differential affinity of 2D materials towards ssDNA and dsDNA translates to a direct involvement in biological recognition events, specifically hybridization of capture probes with their desired targets. Following this unique principle, a label-free electrochemical biosensor has been developed based on MoS2 nanosheets prepared by ultrasound exfoliation.93 The material is drop cast onto bare glassy carbon electrodes prior to immobilization of the ssDNA probe. A strong interaction exists between guanine bases of the ssDNA probe and methylene blue, the selected electroactive indicator that produces a dramatically high current. Upon hybridization with target DNA, the resulting dsDNA desorbs from the electrode surface and diminishes the current response. The current difference is measured against concentration of target DNA. A similar biosensor is developed based on the MoS2–graphene composite where intermolecular π–π stacking interactions between nucleobases of ssDNA and graphene facilitate the differential affinity.94 Combining the large surface area, biocompatibility, and high conductivity of 2D materials, both the developed biosensors are both sensitive and selective. The specific affinity towards ssDNA is validated by introducing complementary, mismatched bases as well as non-complementary target DNA. Furthermore, comparison with bulk MoS2 emphasizes the advantageous properties of 2D materials.
In an attempt to improve the stability of biosensors, an Au/SH-DNA/WS2 platform was constructed by linking SH-DNA to a Au electrode and WS2via Au–S bonds and van der Waals’ forces respectively.95 Stability was indeed achieved when compared to a control biosensor, WS2 drop-cast onto a Au electrode without the DNA linker. WS2 then continued to serve as a platform for probe DNA. The charge transfer resistance is large due to the repulsion between negative charges of both probe DNA and redox probe [Fe(CN)6]3−/4−. In the presence of complementary targets, specifically ATP and heavy metal ion Hg+, a duplex structure will be formed, desorbed from the surface of WS2, causing a decrease in the charge transfer resistance. Herein, the affinity of WS2 towards ssDNA is utilized for the immobilization of the material to the electrode as well as a platform for DNA probes.
Another interesting biosensor design employs MoS2 as an electrochemical label (label-based).96 The signal arises from the inherent electrochemistry of MoS2 itself, as it undergoes chemically irreversible oxidation. The ssDNA probe is firstly immobilized onto a bare disposable electrical printed (DEP) carbon chip before introducing the target DNA. MoS2 is then incubated in the resulting solution to be attached as a label where it will have a higher adsorption onto ssDNA and otherwise for dsDNA. Consequently, there will be a lower voltammetric signal upon hybridization.
A correlation between chemical compositions of TMDs and their differential affinity towards DNA was investigated within group 6 TMDs (MoS2, MoSe2, WS, WSe2).97 Based on their interactions with hairpin DNA, greater physisorption occurs for Mo than W, and Se than S components. The transition metal superiority is supported by the comparison of MoS2 against WS2. Acknowledging that chemical composition plays a significant role in influencing a biosensor's performance, 2D materials offer a myriad of compounds including MXenes and Xenes (Table 2). Their properties can be exploited and they can be further fabricated into electrochemical biosensors, as discussed above. It is therefore promising to explore other 2D materials apart from group 6 TMDs (MoS2, WS2). Doping with metal atoms can also be considered to strengthen the adsorption of nucleobases onto the materials, as a DFT study puts forth the charge transfer and chemisorption of Li atoms onto MoS2.98
The large surface area, coupled with abundant surface groups, spurs the usage of MXene Ti3C2 towards an immunosensor for carcinoembryonic antigen (CEA) detection.101 Ti3C2 easily functionalizes with (3-aminopropyl)triethoxysilane (APTES) to create amino groups on the surface which subsequently bond covalently with carboxyl groups of the anti-CEA receptor. Upon capture of the target CEA, the electron transfer by the [Ru(NH3)6]2+/3+ redox probe is hindered and decreases the current response. The analytical parameters are impressive, especially when compared to other 2D-based immunosensors (Table 3). It flaunts a wider linear range and lower LOD despite employing the less sensitive CV as a quantification technique. The high density of amino functional groups has facilitated an effective immobilization of anti-CEA thereby achieving detection of CEA at low concentrations. However, there is no interference study conducted and the immunosensor lost 20% of its initial current response after just 7 days. Contrastingly, the biosensor based on the MoS2 and Au NP nanocomposite could selectively detect CEA amidst interferents and was stable with a current retention rate of 91% after 30 days.102 More composites of MoS2 have been reported with thionine,103 Prussian blue (PB)104 and graphene105 for synergistic enhancement of both biomolecule immobilization and electrical conductivity.
Moving on to a fundamental aspect of biosensors, the preparation or synthesis of 2D materials is equally intriguing. There are various methods explored from the conventional ones, such that the materials will be optimized in terms of biocompatibility and sensitivity. MoS2 was successfully synthesized in situ at low temperature via plasma enhanced chemical vapor deposition (PECVD).106 The Mo metal was foremostly deposited by e-beam evaporation on the electrode surface and it may even penetrate to form Au–Mo composites. Subsequently, H2S source gas was introduced whose dissociation was facilitated by the presence of radicals as well as dense sheaths in plasma conditions, enabling the reaction with Mo to obtain MoS2 at low temperature. The fabricated electrode is biocompatible as it adsorbs HRP-conjugated IgG for subsequent detection (Fig. 1E). More notably, this synthesis method eliminates the need to transfer the nanosheets, therefore suggesting the direct integration on polymeric substrates for flexible devices.
The preparation of materials can also be achieved with electrochemical techniques, namely bipolar electrochemistry as exemplified in group 6 TMDs (WS2, WSe2, MoS2, MoSe2).107–110 These layered materials are suspended in Na2SO4 solution while a constant DC potential is applied across two Pt driving electrodes. At the extremities of the materials, electrolysis of water occurs which is relatively violent enough to cause fragmentation of sheets into NPs. Recognized as promising electrocatalysts for hydrogen evolution reaction (HER), the group 6 TMD NPs are incorporated as labels for magneto-immunoassays. Downsizing is thus desirable to achieve smaller or similar sizes to those of proteins and paramagnetic beads.
Magnetic beads are sequentially incubated with anti-rabbit IgG, target IgG and lastly secondary anti-rabbit IgG conjugated with WS2.107 Washing and blocking are performed between each incubation step to ensure an accurate immunosandwich assembly. With increasing concentrations of IgG, more conjugated WS2 labels are present to catalyze HER in H2SO4 solution. This translates to a decrease in the charge transfer resistance, which is inversely proportional to the rate of electron transfer, measured by electrochemical impedance spectroscopy (EIS) (Fig. 1F). Selectivity of the biosensing system is tested by replacing the IgG protein with non-specific human Hb. The immunosandwich fails to assemble since anti-IgG would not capture Hb, implying the absence of WS2 labels in the final architecture. Without HER occurring on the surface, the charge transfer resistance is significantly higher.
There are slight variations in immunosensors developed based on MoS2, MoSe2 and WSe2.108–110 These NPs are conjugated directly to the target protein IgG which is captured by anti-rabbit IgG immobilized on magnetic beads. Chronoamperometry is an electrochemical technique used to quantify IgG concentration via the current generated from HER, electrocatalyzed by TMD NPs. Across group 6 TMDs of varying elemental compositions, the immunosensor based on MoS2 has the lowest LOD for IgG detection (Table 3). These works nonetheless highlight that electrochemistry is not only limited as a transduction mode in the context of biosensors but is a key to open more doors, including nanoparticle preparation as well as sensing labels instead of electrode materials.
Meanwhile, contemporary studies of impact electrochemistry may lead to a more sensitive approach for detection. BP particles are foremostly treated with bipolar electrochemistry, resulting in a decrease of size as well as electron transfer resistance.111 They are subsequently used as labels for magneto-immunosandwich assays, like the materials mentioned earlier. H2SO4 is added to the solution upon conjugation with the target. This is necessary for the denaturation of the protein complex to release BP NPs, and the detection signal is produced via nanoimpacts of the BP NPs followed by HER catalysis. The levels of IgG are quantified by the number of spikes which correlate to the attached BP NPs. The following work beautifully conveys the superiority of electrochemistry for biosensors, from the preparation of materials to detection of biomolecules and signal transduction.
2.2 Optical biosensors
Beyond graphene, graphitic carbon nitride (g-C3N4) nanosheets and some TMD quantum dots exhibit strong fluorescence. g-C3N4 nanosheets have a direct bandgap of 2.7 eV and emit blue PL around 450 nm when excited using UV light irradiation, due to the transition of the s-triazine ring.128 On the other hand, TMD QDs are obtained by breaking intraplane X–M–X bonds. The bandgaps of TMD QDs open as a result of quantum confining effects, and the strong absorption peak observed at 277 nm is an outcome of the optical transition between the density of state peaks in the valence and conduction bands.129 Such properties enable them to function as fluorophore labels for protein, immunoassay and biomolecule detection. Additionally, 2D materials have been used as nanoquenchers because their fluorescence or Förster resonance energy transfer (FRET) effect is independent of the emission spectra of the donor. This is commendable since the distance-dependent fluorescence quenching is closely coupled with recognition events.130 Shifting from fluorescence to electrochemiluminescence (ECL), this transduction mode involves a light emission process as a result of redox reactions electrogenerated by redox species present on the electrode surface. In other words, ECL is a combination of electrochemical and chemiluminescence methods.131 2D materials play two roles in ECL bio/sensors: (i) as a robust substrate for platform signal amplification that is generated through another material like QDs, and (ii) having ECL activity that is generated in the presence of reductive–oxidative co-reactants. MoS2 and g-C3N4 have been widely implemented in each role respectively.131 The third optical type is photoelectrochemical (PEC) bio/sensors, relying on a novel technology that measures light interaction with electrochemical reaction at the electrode surface. Because of their semiconductor nature and photoactive properties, 2D materials are nominated for the development of sensitive PEC sensors.132 Lastly, the colorimetric system is based on 2D materials’ peroxidase-mimic activity and their ability to oxidize 3,3′,5,5′-tetramethylbenzidine (TMB) in the presence of H2O2. The color response is observed by the naked-eye and/or quantified using a UV-VIS spectrometer.133The optical biosensors based on 2D materials have been developed using enzymes, antibodies and ssDNA (or aptamers) as bioreceptors. The enzyme-based optical biosensors mainly involve TMDs (MoS2 and WS2) as well as g-C3N4. For these approaches, the peroxidase-like catalytic activity of these materials is used to obtain glucose,134–140 xanthine141 and cholesterol142 colorimetric biosensors with high sensitivity. The mechanism is based on the production of H2O2 from the oxidation of glucose, xanthine and cholesterol by their respective enzymes: glucose oxidase (GOx), xanthine oxidase (XOD) and cholesterol oxidase (ChOx). H2O2 together with 2D materials will oxidize TMB, yielding a quantifiable colored product. Pristine WS2,135 MoS2 (Fig. 2A)138 and g-C3N4 (Fig. 2B)140 have been studied for the detection of glucose, while xanthine and cholesterol are detected using Se doped g-C3N4141 and MoS2 decorated with AuNPs142 respectively. Furthermore, a MnSe-loaded g-C3N4 nanocomposite,136 Fe-doped g-C3N4137 and MoS2 decorated with AuNPs139 have been used to improve the sensitivity of the colorimetric glucose biosensor. In parallel, Wang et al. implemented a fluorescent glucose biosensing system based on the fluorescence quenching of MoS2 QDs by H2O2 produced from the oxidation of glucose by GOx (see Fig. 2C).143 Peng and Weng demonstrated that visible light irradiation could enhance peroxidase-like activity, resulting in an improved sensitivity of the biosensing system based on a GO/MoS2 hybrid material.134
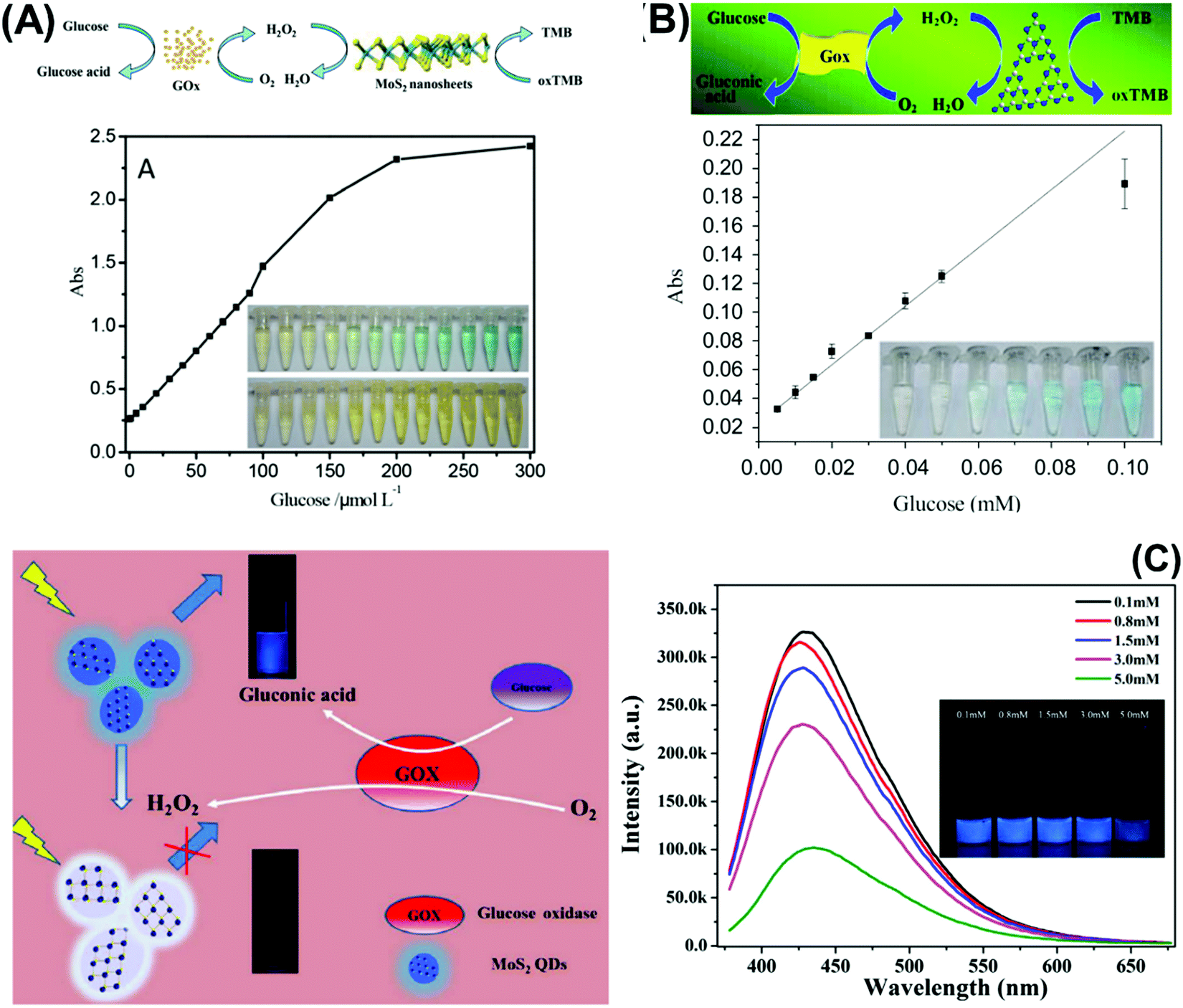 | ||
| Fig. 2 Colorimetric enzymatic biosensor based on 2D materials’ peroxidase-mimic activity and their ability to oxidize 3,3′,5,5′-tetramethylbenzidine (TMB) in the presence of H2O2 using (A) MoS2 nanosheets and (B) g-C3N4 nanosheets. (C) Enzymatic glucose biosensor based on a fluorescence glucose biosensing system based on the fluorescence quenching of MoS2 QDs by H2O2 produced from the oxidation of glucose by glucose oxidase. Figures adapted with permission from: (A) ref. 138, Copyright 2014 The Royal Society of Chemistry; (B) ref. 140, Copyright 2014 Elsevier; (C) ref. 143, Copyright 2017 Elsevier. | ||
Table 4 summarizes analytical performances of the described optical enzyme-based biosensors. In the case of glucose detection, a lower limit of detection (LOD) is observed when 2D materials are decorated with Fe, graphene oxide or AuNPs (below 1 μM) which is expected due to the synergy of peroxidase activity of these materials. However, the LOD of biosensors based on pure 2D materials is found between 1 and 5 μM. This demonstrates the role of pure 2D materials in developing enzyme-based colorimetric biosensors. Interestingly, the TMDs (MoS2 and WS2) consist of a few layers (1–8 layers) with lateral size below 200 nm. The importance of the exfoliation level and size of TMDs for this implementation is emphasized. Moreover, it is claimed that both MoS2 and WS2 nanosheets would facilitate electron transfer between TMB and H2O2 through decomposition of the latter in acidic media, giving rise to a blue product. On the other hand, bulk g-C3N4 is reported as a colorimetric enzymatic biosensor which achieves enhanced catalytic capabilities. Nevertheless, an enhanced linear range is observed in TMD nanosheets compared to bulk g-C3N4. Moreover, the response intensities in terms of absorbance (a. u.) of TMDs are higher than those of g-C3N4. Subsequently, the authors developed a highly sensitive portable test kit using TMDs for instrument-free visual detection of glucose in real serum samples, made possible owing to the superior response of TMDs in mimicking enzyme activity.
| Analyte | 2D material class | Material | Technique | Linear range | LOD | Response intensity | Ref. |
|---|---|---|---|---|---|---|---|
| Glucose | g-C3N4 | g-C3N4 | Colorimetric | 5–100 μM | 1 μM | 0.04–0.22 a.u. (Abs) | 140 |
| Glucose | g-C3N4 | MnSe-g-C3N4 | Colorimetric | 0.16–1.6 mM | 8 μM | 0.24–0.32 a.u. (Abs) | 136 |
| Glucose | g-C3N4 | g-C3N4/Fe | Colorimetric | 0.5–10 μM | 0.5 μM | 0.2–0.5 a.u. (Abs) | 137 |
| Xantine | g-C3N4 | g-C3N4 | Colorimetric | 0.16–40 μM | 0.016 μM | 0.07–0.35 a.u. (Abs) | 141 |
| Cholesterol | TMDs | MoS2 nanoribbons/Au NPs | Colorimetric | 0.04–1 mM | 15 μM | 0.1–0.4 a.u. (Abs) | 142 |
| Glucose | TMDs | MoS2/GO | Colorimetric | 1–50 μM | 86 nM | 0–1 a.u. (Abs) | 134 |
| 0–1.1 a.u. (Abs) | |||||||
| Glucose | TMDs | MoS2–QDs/AuNPs | Colorimetric | 1 to 400 μM | 0.068 μM | 0.48–0.66 a.u. (Abs) | 139 |
| Glucose | TMDs | MoS2 | Colorimetric | 5–150 μM | 1.2 μM | 0.2–2 a.u. (Abs) | 138 |
| Glucose | TMDs | WS2 | Colorimetric | 5–300 μM | 2.9 μM | 0.2–1.2 a.u. (Abs) | 135 |
| Glucose | TMDs | MoS2 QDs | Fluorescent | 10 to 1500 μM | 5.16 μM | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 a.u. (FL) 000–30000 a.u. (FL) |
143 |
In addition, MoS2 QDs obtained by a hydrothermal method show blue emission with a high quantum yield (−10.3%) as well as robust dispersibility and storage stability whose optical property is quenched in the presence of H2O2 (product of glucose decomposition through GOx). The system is successful for glucose detection in fetal bovine serum samples, demonstrating viability of the proposed method for blood glucose monitoring in real samples. This is another endorsement for how synthesis methods of TMDs influence their size and optical properties, which are key considerations for fluorescence enzymatic biosensor systems. However, lower sensitivity of glucose detection is observed using fluorescence quenching of TMD QDs.
The fluorescence quenching capabilities of 2D materials beyond graphene are well optimized for DNA detection, especially the extensively studied TMDs and g-C3N4. In 2013, Zhu et al. demonstrated for the first time that a single-layered MoS2 nanosheet possesses high fluorescence quenching efficiency as well as different affinity towards ssDNA and dsDNA.144 Later, Ge et al. designed a novel fluorescence sensor to monitor the activity of polynucleotide kinase using T4 polynucleotide kinase as a model target. The sensor is based on phosphorylation-specific exonuclease reaction together with efficient fluorescence quenching of ssDNA by WS2 nanosheets. Owing to the high quenching efficiency of WS2 nanosheets, this system offers excellent analytical performance.145 In order to reveal the effect of elemental composition, our group compared the quenching ability of MoS2 and WS2 nanoflakes for DNA corresponding to Alzheimer's disease detection. MoS2 demonstrated superior performance than its group 6 counterpart.146
Moreover, Kong et al., Deng et al., and Huang et al. respectively used the MoS2 quencher for sensitive detection of prostate specific antigen (Fig. 3A),147 DNA methyltransferase activity148 and DNA detection via hybridization chain reactions (HCRs).149 The detection of DNA methyltransferase (MTase) activity was materialized by designing a substrate DNA with affinity to MoS2.148 It was composed of both ssDNA and dsDNA. The ssDNA allows the substrate DNA to adsorb on MoS2 nanosheets via van der Waals interactions. There would consequently be fluorescence quenching of the fluorophore attached at the end of the dsDNA counterpart. Simultaneously, the dsDNA embodies a sequence specific to DNA adenosine methyltransferase (Dam). When methylated by Dam, the methylation sensitive restriction endonuclease DpnI cleaves and releases the fluorophore. Fluorescence is recovered and quantified to reflect the methylation level. Meanwhile, detection of DNA via hybridization chain reactions (HCRs) is achieved with MoS2 nanosheets.149 The 2D material is responsible for suppressing the background signal as well as controlling the fluorescence emission of the detection system. The “on” and “off” switch corresponds to the presence or absence of target DNA respectively. Target-triggered HCRs between two hairpin probes amplify the signal generated, resulting in high sensitivity of DNA detection. Finally, Huang et al. developed a novel fluorescence microfluidic biosensor for ultrasensitive detection of DNA using the efficient fluorescence quenching capability of MoS2 to distinguish ssDNA and dsDNA in a very short time, within minutes. Moreover, this system shows high sensitivity for DNA detection at fmol levels and in a very low sample volume.150
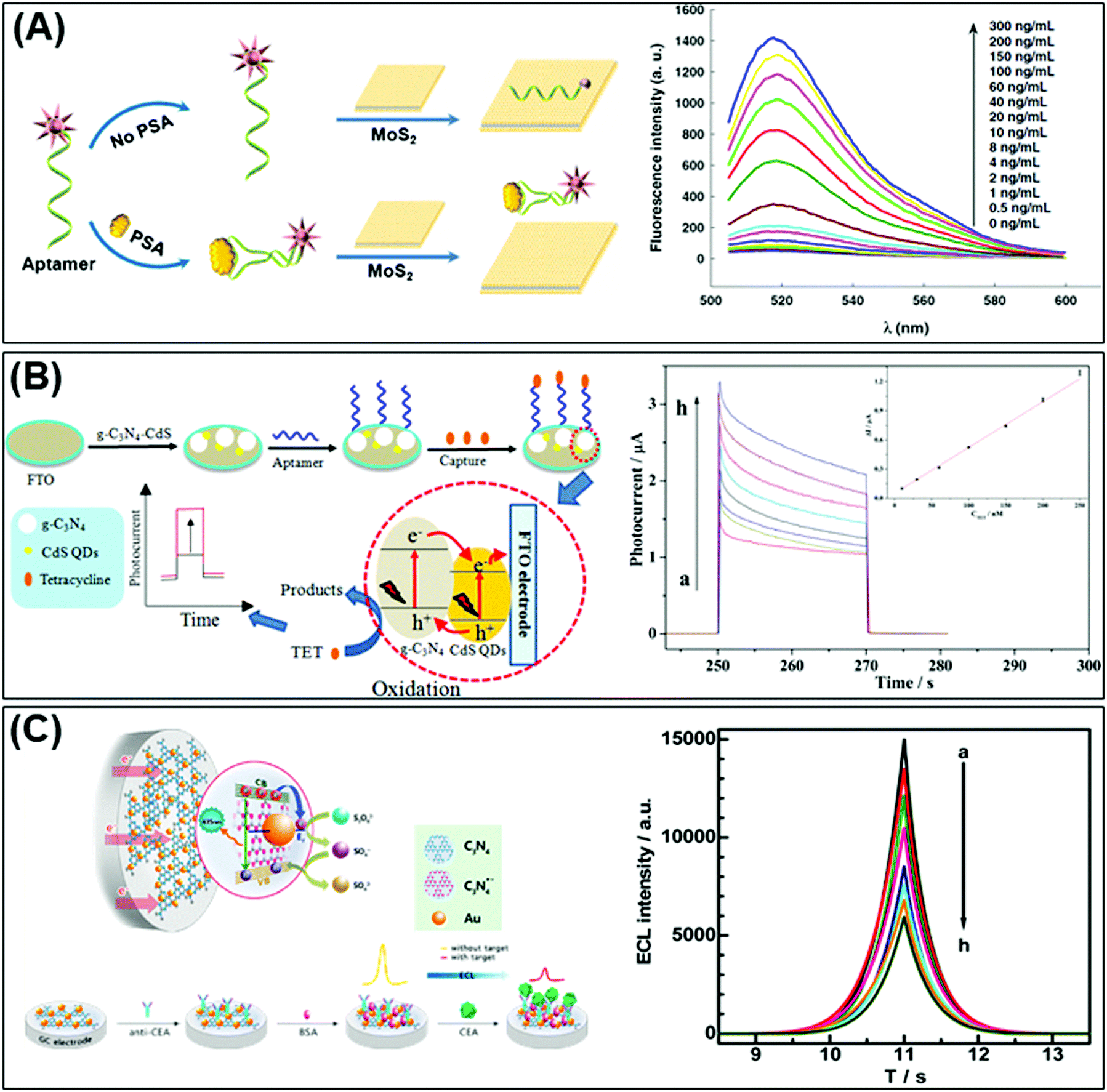 | ||
| Fig. 3 (A) Fluorescent PSA aptasensor based on the fluorescence-quenching ability of MoS2 nanosheets when applied to a dye-labeled single-stranded DNA probe. (B) g-C3N4 coupled with CdS quantum dots served as a highly efficient photoactive species in a photoelectrochemical aptasensor for tetracycline detection; UV-visible diffuse reflection spectra (DRS) indicated that the absorption of g-C3N4 in the visible region was enhanced by CdS QDs. (C) Au NP-functionalized g-C3N4 NS nanohybrids exhibit strong and stable cathodic electrochemiluminescence (ECL) activity; these nanohybrids are used as a novel ECL immunosensor to detect a carcinoembryonic antigen. Figures are adapted with permission from: (A) ref. 147, Copyright 2014 Springer Nature; (B) ref. 154, Copyright 2015 American Chemical Society; (C) ref. 155, Copyright 2014 American Chemical Society. | ||
Moving away from TMDs, another prominent 2D material used to develop fluorescence DNA sensors is g-C3N4. Wang et al. in 2013 put forth the fluorescence quenching effect of g-C3N4 nanosheets caused by their affinity change to DNA probes. The concept is implemented in several fluorescence detection methods, including ratiometric fluorescence and coupling with Exo III mediated target recycling. A short assay time and high sensitivity are reaped for DNA and Hg2+ detection.151 Furthermore, g-C3N4 is used for simultaneous sensing of intracellular microRNAs; in this case g-C3N4 nanosheets were assembled with two different dye-labeled ssDNAs (dye-ssDNAs) and folate (FA)-Poly, via π–π interactions. The fluorescence of dye-ssDNAs is quenched by g-C3N4 and the presence of FA on g-C3N4 achieved cell-target-specific delivery. After g-C3N4 loaded with dye-ssDNAs is transfected into the cells, the hybridization of the assembled dye-ssDNAs with complementary targets weakened the π–π interaction between bases and g-C3N4. As a result, the release of dye-ssDNAs recovers fluorescence leading to simultaneous detection of multiple miRNAs in a living cell.152
Table 5 compiles the analytical performances of DNA/aptamer-based optical biosensors using 2D materials. For DNA detection, a very thin layer of MoS2 with thickness below 1 nm attained a very low LOD in the pM level by the fluorescence quenching approach.149,150 The material is prepared by two methodologies, (i) expansion using hydrated hydrazine via hydrothermal technique, followed by physical treatment using an ultrasound bath and, (ii) lithium intercalation using the butyllithium intercalator. For the fluorescence quencher, it is demonstrated that single-layered TMD nanosheets can spontaneously adsorb single-stranded DNA (ssDNA) by van der Waals force between nucleobases and basal plane of TMDs nanosheets, generating a high fluorescence quenching ability. This reaffirms how the exfoliation level of TMDs is critical in developing highly sensitive fluorescence biosensing systems. On the other hand, the exfoliation extent of commercial MoS2 and WS2 is not guaranteed given the lower sensitivity.146 It is seen that the quenching process by TMDs is completed within a very short time, ∼1 s, much faster than other nanomaterials used for that aim. The explanation lies in the large planar surface area and superb adsorption property of MoS2.149
| Analyte | 2D material class | Material | Technique | Linear range | LOD | Response intensity | Ref. |
|---|---|---|---|---|---|---|---|
| Tetracycline | g-C3N4 | g-C3N4/CdS QDs | Photoelectrochemical | 10 to 250 nM | 5.3 nM | 0–3 μA (PC) | 154 |
| Kanamycin | g-C3N4 | g-C3N4 | Photoelectrochemical | 1 nM to 230 nM | 0.2 nM | 0.01–0.06 μA (FC) | 163 |
| MCF-7 cells | g-C3N4 | g-C3N4-Au NPs/MoS2 | Chemiluminescence | 102 to 106 cells mL−1 | 2000–800 a.u. (ECL) | 153 | |
| DNA | g-C3N4 | g-C3N4 | Fluorescent | 3.0 to 30 nM | 2.1 nM | 40–160 a.u. (FI) | 151 |
| DNA | TMDs | MoS2 | Fluorescent | 9.60–366 nM | 146 | ||
| WS2 | 13.3–143 nM | ||||||
| PSA | TMDs | MoS2 | Fluorescent | 0.5 to 300 ng mL−1 | 0.2 ng mL−1 | 0–1400 a.u. (FI) | 147 |
| DNA methyl transferase | TMDs | MoS2 | Fluorescent | 0.2–20 U mL−1 | 0.14 U mL−1 | 40–340 a.u. (FI) | 148 |
| DNA | TMDs | MoS2 | Fluorescent | 0–50 nM | 500 pM | 25–45 a.u. (FL) | 150 |
| DNA (hairpin probes) | TMDs | MoS2 | Fluorescent | 0 to 200 pM | 15 pM | 250–450 a.u. (FI) | 149 |
| T4 polynucleotide kinase | TMDs | WS2 | Fluorescent | 0.01 to 10 U mL−1 | 0.01 U mL−1 | 5000–1000 a.u. (FI) | 145 |
Again relying on the optical properties of 2D materials, a reusable and dual-potential electrogenerated chemiluminescence (ECL) biosensor is developed. It is simultaneously responsive to both MCF-7 cancer cells and in situ evaluation of cell surface glycan expression. By measuring the ratio of ECL intensity between negative and positive potentials, the cytosensing and cell surface N-glycan evaluation could therefore be synchronously determined. In addition to the meritable selectivity and sensitivity of the biosensor, traditional cell counting procedures can be avoided. Electrochemically reduced MoS2 nanosheets are used for the immobilization of capture DNA whereas a conjugated gold NP modified g-C3N4 serves as a negative ECL nanoprobe and is applied for cell surface N-glycan evaluation.153 On the other hand, Liu et al. fabricated a highly efficient photoactive species from g-C3N4 coupled with CdS QDs as a transducer for PEC aptasensors for tetracycline detection. The developed sensor shows a linear PEC response to tetracycline concentrations (see Fig. 3B).154
In the optical immunosensor design, Chen et al. implemented a system using g-C3N4 nanosheets decorated with Au NPs referred to as the “Au-g-C3N4 nanohybrid” and constructed an electrochemiluminescence (ECL) immunosensor for highly sensitive and selective carcinoembryonic antigen (CEA) detection. The Au-g-C3N4 nanohybrid exhibits strong and stable cathodic ECL activity in comparison to pristine g-C3N4. That cathodic ECL activity of Au-g-C3N4 decreased in the presence of CEA (Fig. 3C).155
In this section we have discussed several approaches based on the optical properties of emerging 2D materials. We hope to provide an avenue to understand how these devices work and what is the role of these materials in improving both sensitivity and selectivity of optical bio/sensors.
2.3 Future factors to explore
We have elucidated 2D materials from the aspects of structure as well as advantageous properties and limitations pertaining to biosensors. Bulk layered materials are either top-down exfoliated or bottom-up synthesized, ultimately achieving a high surface to volume ratio desirable for immobilization of biorecognition elements. Surface charges or bonding interactions within the materials, depending on the elemental composition, further facilitate immobilization. As a result of high concentration of biorecognition elements, the developed biosensors fulfil the fundamental requirements of sensitivity and selectivity. There are also other properties relevant to biosensors that are discussed, including biocompatibility and stability of the materials.A concrete understanding of materials, especially their limitations, paves the way for innovative strategies towards developing successful electrochemical and optical biosensors. Taking MoS2 as an example, phase engineering via lithium intercalation and exfoliation transforms the natural semiconducting 2H phase to metallic 1T. A satisfactory conductive material with increased electrocatalytic sites results, without the application of noble metal nanoparticles for electrochemical biosensor development. This is made possible owing to the established correlation between conductivity of TMDs and their d-electron count along with the coordination environment of transition metals.
Among 2D materials beyond graphene, TMDs are the most prominent across biosensors regardless of biorecognition elements or transduction modes. Nonetheless, MXenes are rising in popularity for electrochemical biosensors due to their commendable conductivity and hydrophilicity while g-C3N4 with its intrinsic peroxidase activity is increasingly sought for optical biosensors. Monoelemental Xenes such as phosphorene, on the other hand, are less visible in the biosensing landscape due to their main drawback of chemical stability.
Since 2D materials of different elemental compositions exhibit diverse properties, one enticing strategy is to precisely select and sequence the materials layer by layer. These designer compounds are known as van der Waals’ heterostructures which put together desired properties best for an application.156 Furthermore, neighbouring crystals can induce structural changes and charge redistribution, leading to improved or novel performances. Research on van der Waals’ heterostructures is gathering momentum with the development of preparation methods157 and sophisticated devices.158 Specific to biosensors, monolayer graphene has been manually stacked on top of monolayer MoS2.159 The latter is responsible for the change in photoluminescence intensity corresponding to the label-free detection of target DNA. Concurrently, graphene protects MoS2 from moisture and oxygen in the air as well as provides a biocompatible interface layer to host DNA. Strikingly when stacked, electron concentration of the initially n-doped MoS2 reduces as electrons are transferred to graphene. The effect is laudable as high sensitivity of the biosensor is achieved.
New breakthroughs and rapid advancements of 2D materials will continue to propel the progress of biosensors. With increasing publications, a standard methodology or guideline in reporting fundamental parameters would greatly facilitate comparison while communicating significant advantages of any developed biosensor.160 Because 2D materials are commonly incorporated as the electrode to enhance signal transduction, determining the electroactive surface area is imperative. A standardized calculation of the electroactive surface area, different from the geometrical area, can then be relied on to compute other key parameters.161 In parallel, developed biosensors should evolve from proof-of concept to actualization. Rigorous criteria such as stability for a specified duration and comparison with current technology are put forth, in addition to sensitivity and selectivity.162 Other essential analytical parameters namely linear range and limit of detection have been consistently reported.
3. 2D materials for sensing application
3.1 Electrochemical sensors
Nowadays, the enhanced electrical and electrochemical properties of 2D materials equip them as excellent alternatives in developing electrochemical sensors for detection of toxic compounds and biological markers for biomedical, environmental as well as safety and security applications. In this section, we will describe the latest achievements of 2D materials in the field of electrochemical sensing. There will be two sub-sections, the first for pollutant detection while the second for biological biomarker detection.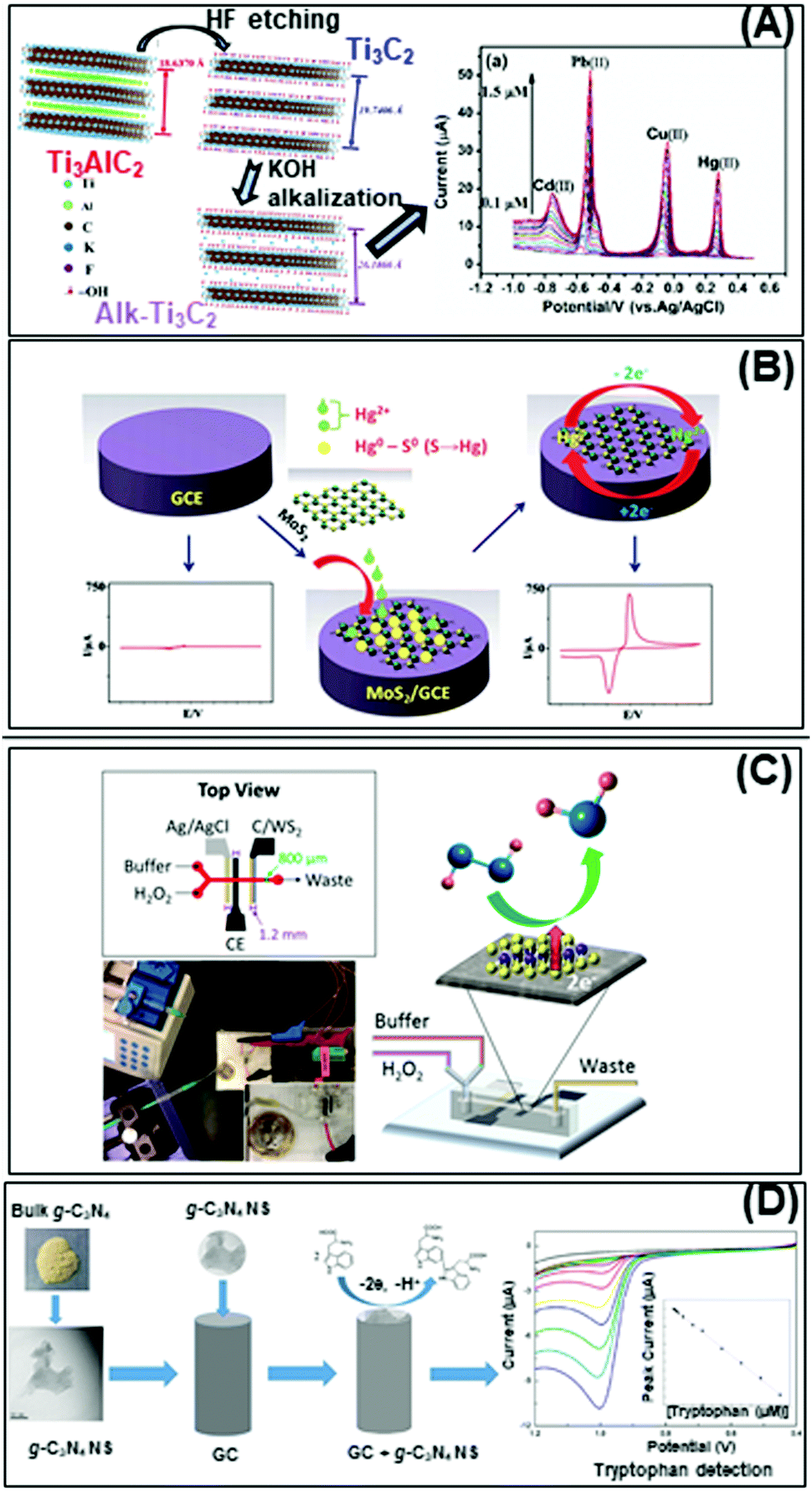 | ||
| Fig. 4 Electrochemical sensors for heavy metal detection: (A) alkaline intercalated Ti3C2 for simultaneous detection of multiple heavy metal ions (Cd2+, Pb2+, Cu2+ and Hg2+) and (B) MoS2, the high selectivity and sensitivity are due to the interactions between S2− groups of MoS2 and Hg2+. (C) Highly sensitive H2O2 detection using 1T-WS2 nanosheets as a platform integrated into a microfluidic channel and (D) sensitive tryptophan detection using g-C3N4 nanosheets (NS). Figures adapted with permission from: (A) ref. 170, Copyright 2017 Elsevier; (B) ref. 171, Copyright 2018 The Royal Society of Chemistry; (C) ref. 180, Copyright 2017 American Chemical Society; (D) ref. 182, Copyright 2017 Elsevier. | ||
On the other hand, the more representative TMD, MoS2, is used for electrochemical detection of Hg2+ ions in 1 M HCl solution, and tap and sea water samples (Fig. 4B). The remarkable selectivity and sensitivity are assigned to the possible interactions between S2− groups of MoS2 and Hg2+. S2−, being a natural reducer, would donate electrons from MoS2 to the adsorbed Hg2+, producing Hg0.171 Another TMD, TiS2, is reported for heavy metal detection (Cu2+). Gan et al. prepared a nanocomposite of hydric titanium disulfide (HxTiS2) ultrathin nanosheets and polyaniline. The obtained nanocomposite was used as a selective electrochemical platform for Cu2+ detection. They claim that the incorporation of HxTiS2 in the nanocomposite regulates the growth of PANI, enhances electrode stability, and improves the rate of interfacial electron transfer. Furthermore, high selectivity is observed due to the coordination bond between Cu2+ and the N atoms of the imine groups of PANI. Finally, the enhanced sensitivity is derived from the synergic conductivity between both HxTiS2 nanosheets and PANI.
The bottom-up fabricated two-dimensional carbon nitride is another 2D material successfully employed for selective detection of Hg2+ ions. 2D g-C3N4 nanosheets are produced by evaporation induced self-assembly and condensation of carbon nitride dots, previously formed by microwave irradiation of formamide. The simultaneous electrochemical detection of Pb2+, Cu2+ and Hg2+ performed demonstrates the enhanced capability of C3N4 for Hg2+ ion detection, plausibly based on the formation of amalgam with the reduced mercury on the carbon nitride sheet surface. There is also a report of the MoS2-poly(m-aminobenzenesulfonic acid) nanocomposite used for 2,4,6-trinitrotoluene (TNT) detection.172 TNT is an explosive commonly used for military, industrial, and mining applications. The authors attribute the high sensitivity of this system to the large surface area of the nanocomposite, especially upon introducing MoS2 nanosheets (produced by physical ultrasound exfoliation). At the same time, the absorption efficiency could be increased through the π–π stacking effect of the TNT aromatic ring and the MoS2-based composite.173
Moreover, WS2 nanospheres supported in carbon nanofibers show high sensitivity and selectivity for dopamine detection with a reasonable limit of detection.177 The reasons are the enhanced surface area of carbon nanofibers by WS2 nanospheres, the abundant active sites coupled with high electric conductivity of the resultant materials. WS2 nanospheres are grown on carbon nanofibers by hydrothermal synthesis and the resultant hexagonal phase is indexed by XRD analysis. On the other hand, our group has proven that shear exfoliated antimonene is able to oxidize ascorbic acid efficiently compared to other pnictogens, namely arsenene and bismuthene, exfoliated by the same method.178 This is due to the low oxidation ratio and high level of exfoliation of antimonene after shear exfoliation.
Developing highly sensitive enzymatic-free devices for H2O2 detection is tremendously sought for in chemical and food industries as well as for diagnostic and environmental control.179 Pristine MXene Ti3C2Tx exhibits excellent capability in reducing H2O2 with a low limit of detection, comparable to a microfluidic sensing system based on 1T-phase WS2 implemented by our group (Fig. 4C).180 We have also published that pristine PtTe2 shows enhanced peroxidase-like activity for H2O2 detection.181 Interestingly, Ti3C2Tx, WS2 and PtTe2 possess a metallic nature which contributes to enhanced sensing activity. In addition, g-C3N4 nanosheets obtained by the ultrasound exfoliation method provided a large surface area for the interaction with tryptophan and an ultrathin g-C3N4 structure for fast carrier transport. The obtained g-C3N4-based tryptophan sensor shows enhanced analytical performance that allows its application in real samples (Fig. 4D).182 The high sensitivity of this system could be explained firstly by the functional groups on g-C3N4 nanosheets that enhance their interaction with tryptophan and secondly, the π–π stacking interaction of the aromatic structure of tryptophan and g-C3N4. Moreover, the enhanced surface area of g-C3N4 nanosheets provides a high density of active sites and consequently fast carrier transport. Such discoveries are evident of the high capabilities of pristine 2D materials in developing bioreceptor-free sensors for the detection of several biological molecules.
3.2 Optical sensor
There are interesting approaches developed for heavy metal detection through optical sensors, mainly employing g-C3N4 due to its fluorescence and ECL properties. The heavy metal fluorescence sensors reported are based on the fluorescence quenching of g-C3N4 by Cu2+,183–186 Ag+,186 Fe3+187 and Cr4+188 ions. Lee et al. reported for the first time a very sensitive Cu2+ sensor using high surface area 3D cubic mesoporous g-C3N4 with –NH2, –NH–, and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– functional groups.183 These groups function as ligands and show high adsorption capacity for metal ions through chelation or redox reactions. A broad peak at wavelength 455 nm with an excitation wavelength of 375 nm is observed in the PL spectrum of pure 3D cubic mesoporous g-C3N4 that is selectively quenched in the presence of Cu2+ over other metal ions such Zn2+, Hg2+, Pb2+, Co2+, Mg2+, Fe3+, Mn2+, Cd2+, and Ni2+. The PL quenching is elucidated by the redox potential of Cu2+/Cu+ ranging between the conduction band and the valence band of 3D cubic mesoporous g-C3N4, where a photoinduced electron transfer from the conduction band to the complexed Cu2+ leads to fluorescence quenching.
N– functional groups.183 These groups function as ligands and show high adsorption capacity for metal ions through chelation or redox reactions. A broad peak at wavelength 455 nm with an excitation wavelength of 375 nm is observed in the PL spectrum of pure 3D cubic mesoporous g-C3N4 that is selectively quenched in the presence of Cu2+ over other metal ions such Zn2+, Hg2+, Pb2+, Co2+, Mg2+, Fe3+, Mn2+, Cd2+, and Ni2+. The PL quenching is elucidated by the redox potential of Cu2+/Cu+ ranging between the conduction band and the valence band of 3D cubic mesoporous g-C3N4, where a photoinduced electron transfer from the conduction band to the complexed Cu2+ leads to fluorescence quenching.
Tian et al. produced ultrathin g-C3N4 nanosheets via ultrasonication-assisted liquid exfoliation of bulk g-C3N4, aimed to use these nanosheets for Cu2+ detection by PL quenching (Fig. 5A).184 It is demonstrated that nanostructuring of g-C3N4 is indeed essential for the fluorescence detection of metal ions. In addition, Huang and coworkers combined g-C3N4 and Mg2Al–Cl–LDH NPs for the detection of Cu2+ and Ag+.186
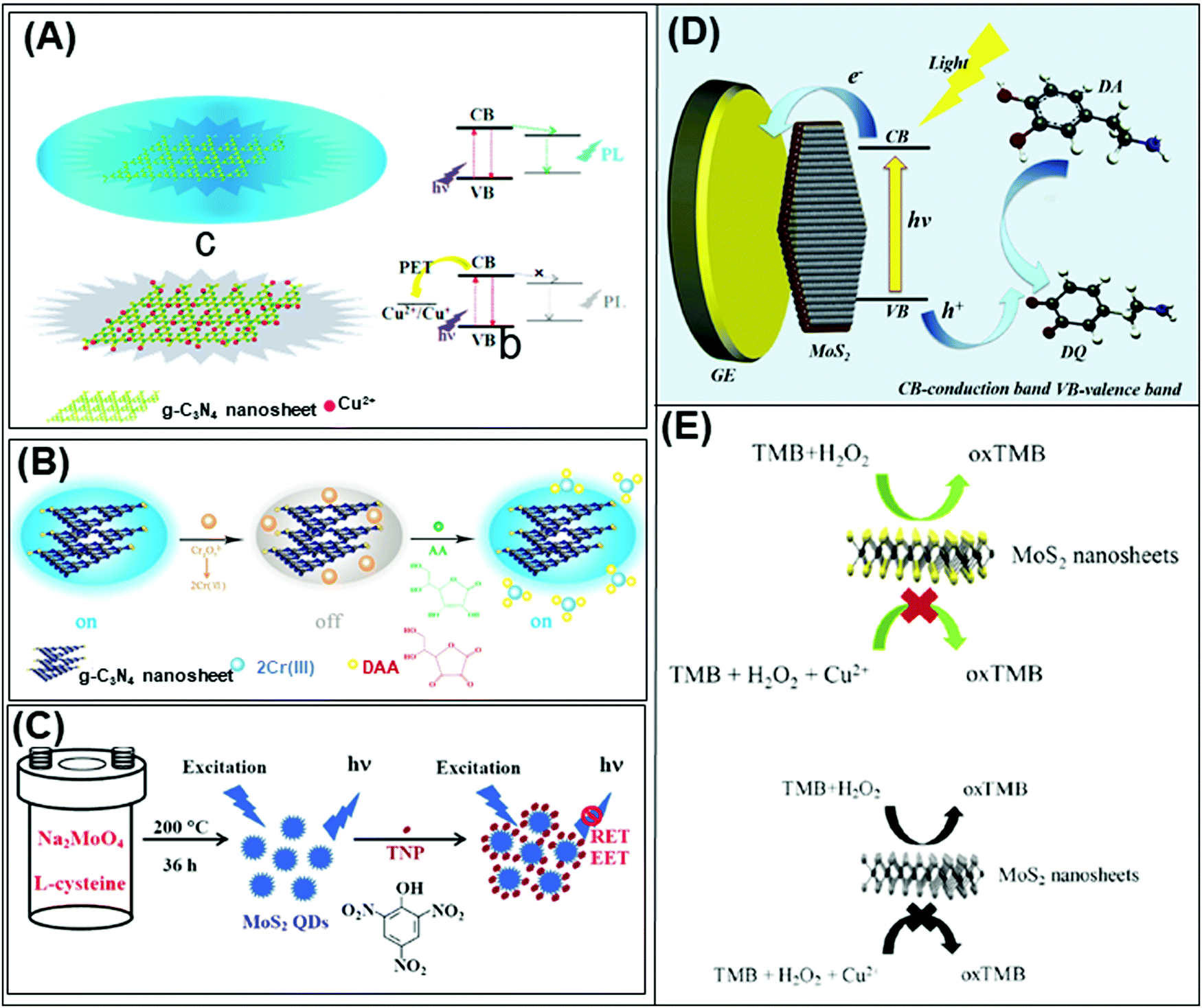 | ||
| Fig. 5 Schematic representation of optical sensors: fluorosensors based on g-C3N4 nanosheets for (A) Cu2+ along with (B) simultaneous detection of Cr3+ and ascorbic acid as a fluorescent “switch”. (C) MoS2 QDs as a photoluminescence sensing system for 2,4,6-trinitrophenol quantification. (D) Photoelectrochemical sensor for ultrasensitive dopamine detection using single-layer nano MoS2. (E) Colorimetric sensing system for Cu2+ detection based on the peroxidase-like activity of MoS2 nanosheets. Figures adapted with permission from: (A) ref. 184, Copyright 2013 American Chemical Society; (B) ref. 188, Copyright 2015 Elsevier; (C) ref. 189, Copyright 2014 American Chemical Society; (D) ref. 132, Copyright 2017 Elsevier; (E) ref. 133, Copyright 2017 Elsevier. CB = conduction band, VB = valence band, PET= photoinduced electron transfer, hν = photon energy, h+ photon, GE = glassy carbon, DA = dopamine, DQ = dopaminequinone, TMB = 3,3′,5,5′-tetramethylbenzidine. | ||
Later, Rong et al. implemented a dual fluorescence sensing system for Cr4+ and ascorbic acid detection,188 as the Cr4+ ions effectively quench the fluorescence of g-C3N4 nanosheets, via the redox reaction between Cr4+ and ascorbic acid (Fig. 5B). Guo and coworkers extended this approach to develop a fluorescence sensor for Fe3+ ions instead of Cr4+ ions.187 In the meantime, Cheng et al. leveraged on the ECL activity of g-C3N4 to develop a sensitive Cu2+ sensor using Na2S2O8 as the co-reactant. A linear relationship was established between the increasing concentrations of Cu2+ and decreasing ECL intensity of g-C3N4, giving rise to a sensitive Cu2+ sensor.185 This system is highly selective due to the redox potential of Cu2+/Cu+ (0.159 V vs. NHE) that lies between the valence (1.83 V) and conduction (−0.83 V) bands, allowing efficient electron transfer from the negatively charged g-C3N4 nanosheets to Cu2+ thereby resulting in ECL quenching. Moreover, the more positive redox potential of Cu2+ favors the electron transfer between Cu2+ and g-C3N4. In the case of the other metals, the electron transfer is not so efficient since their redox potentials are less positive.
Moving on, Wang et al. used MoS2 QDs as photoluminescent probes to construct a sensitive and selective photoluminescence quenching sensor for the detection of 2,4,6-trinitrophenol (TNP) (see Fig. 5C).189 The high selectivity of this sensor is attributed to the favorable PL resonance energy transfer and electronic energy transfer between MoS2 QDs and TNP. This is supported by the analysis of the Stern–Volmer quenching constant obtained, as well as electrostatic interactions.189 On the other hand, Hun et al. developed an ultrasensitive dopamine photoelectrochemical (PEC) sensor based on a single-layered nano MoS2 modified gold electrode.132 Interestingly, in this sensor the photocurrent of nano MoS2 improved in the presence of dopamine, which is attributed to the life improvement of the electron–hole pairs (Fig. 5D).
As for colorimetric detection method, Chen et al. took advantage of the peroxidase-like activity of MoS2 nanosheets and established a colorimetric assay for Cu2+ (Fig. 5E).133 The peroxidase-like activity of MoS2 was validated by the oxidation of TMB in the presence of H2O2 producing a colorimetric product. The detection mechanism was based on the inhibition of the peroxidase-like catalytic activity of MoS2 by Cu2+.133
3.3 Future factors to explore
In this section we have described the important role of 2D materials in developing different free bioreceptor sensing systems for the detection of different biomolecules and pollutants. Moreover, it has been demonstrated that 2D materials are able to mimic the peroxidase activity and can be used for optical and electrochemical sensing systems. All these findings have a great implication for the development of multitasking smart sensing devices with long durability and high stability, since they avoid the low stability of bioreceptors. However, systematic studies to evaluate the scalability and miniaturisation of developed portable point of care devices are still missing, as well as their mass production.4. 2D materials for gas sensing application
Recently, 2D materials have attracted research interest in the development of highly sensitive and selective gas sensing devices to detect hazardous gases, organic vapors, and humidity. Such devices are widely used for diagnostics, environmental monitoring, and emission control as well as for safety and security. A wide variety of materials have been explored, ranging from carbon materials (CNTs and graphene) to metal oxides and polymers. Not to be missed are the emerging 2D materials, including TMDs, black phosphorus, layered metal oxides, MXenes, pnictogens, and h-boron nitride, as they boast impressive capabilities for gas sensing due to their large surface area and high adsorption properties. These 2D materials are incorporated into the gas sensing devices based on two configurations: (i) field effect transistors (FETs)190 (Fig. 6A) and (ii) chemiresistors (Fig. 6B).191 Gas sensors of 2D materials will be discussed according to their working principle. | ||
| Fig. 6 (A) Field effect transistor based on monolayer MoS2 for chemical vapor sensing. (B) Layered PtSe2 films in chemiresistor configuration. Figures adapted with permission from: (A) ref. 190, Copyright 2013 American Chemical Society; (B) ref. 191, Copyright 2016 American Chemical Society. | ||
4.1 Field effect transistor-based gas sensors
FET-based gas sensors take advantage of the band gap modulation in 2D materials. When the surfaces of these materials interact with gas molecules, the drain current change causes changes in their conductivity.192 Among the 2D materials (excluding graphene), TMDs are widely used and studied to develop FET gas sensor devices of high sensitivity. Showcasing diversity in elemental compositions, TMDs that have been reported are MoS2, MoTe2, WSe2, and SnS2. The developed systems possess very high sensitivity and selectivity in detecting compounds that are imperative for environmental monitoring as well as for safety and security. Interestingly, MoS2 exhibits a special predilection to nitrogen-containing compounds such as NO2,192–195 NO,196 NH3,193,194,197 N2,195 and triethylamine (TEA–N(CH2–CH2)3).198 Triethylamine is a decomposition product of the V-series nerve gas agents.198 Understanding the relationship between the features of 2D materials and gas sensing performance is fundamental in paving the way toward optimization and realization. A correlation with the number of layers is investigated by Li et al.196 and Late et al.192 Based on MoS2 prepared by micromechanical exfoliation, both authors posit that single-layered gas sensing devices are unstable unlike few-layered MoS2 of enhanced sensitivity (Fig. 7A). | ||
| Fig. 7 Field effect transistor-based gas sensors. (A) NH3 and NO2 gas sensor prepared using single-layer or few layer MoS2. (B) MoTe2 chemical sensor and its performance evaluation through gate biasing. (C) Nitrogen dioxide (NO2) gas sensor based on black phosphorus field effect transistors. Figures adapted with permission from: (A) ref. 192, Copyright 2013 American Chemical Society; (B) ref. 193, Copyright 2017 Institute of Physics; (C) ref. 199, Copyright 2015 American Chemical Society. | ||
Meanwhile, Liu et al.194 and Cho et al. have developed FETs based on an MoS2 single layer prepared by chemical vapor deposition (CVD). Liu et al. demonstrated that CVD-grown monolayer MoS2 transistors using Schottky contacts are able to detect few ppb of NO2 and down to 1 ppm of NH3. The observed sensing capability of this device is due to (i) the charge transfer between gaseous species and MoS2 monolayers and (ii) Schottky barrier modulation at the MoS2–metal electrode junctions. Moreover, the enhanced sensitivity is mainly by the Schottky barrier modulation.194 As for the alignment direction, Cho et al. observed that the use of a vertically aligned MoS2 film in the FET increases the sensitivity of NO2 gas sensing by about five fold and provides superior resistance variation when compared with its counterpart (horizontally aligned MoS2 film) under the same experimental conditions.195
The other reported TMDs are used to detect NH3 (SnS2199 and MoTe2193,197) and NO2 (MoTe2193). In addition, a device based on SnO2–SnS2 hybrids, synthesized by the high-temperature oxidation of SnS2, shows enhanced sensitivity for NH3 sensing. The authors demonstrated that the device response depends on the amount of interfacial Sn bonds (O–Sn–S) generated on the hybrid's surface. High amounts of interfacial chemical bonds give rise to lower interface state density and increase the charge density of SnO2; this fact allows the improvement of oxygen chemisorption, which increases the NH3 sensing response.199 Another NH3 gas sensor, developed and improved by Feng et al., is based on MoTe2 through continuous light illumination.197 This group later demonstrated for the same MoTe2-based gas sensor that the recovery kinetics can be effectively adjusted by biasing and enhancing the recovery rate for both NO2 and NH3 sensing (Fig. 7B).193 The other reported TMDs are used to detect NH3 (MoTe2193,197 and SnS2198) and NO2 using MoTe2.193
Finally, an FET based on multilayer BP was reported for NO2 detection (Fig. 7C). This gas sensor shows enhanced response and excellent sensitivity and can detect NO2 at 5 ppb. Additionally, the relative stability of BP before and after sensing is satisfactory, with no apparent change observed via Raman spectroscopy.199
4.2 Chemiresistor gas sensors
In the chemiresistor configuration, the nanosheets of 2D materials are connected by two electrodes and the change in the resistance, capacitance, or impedance is quantified after the gas or vapor molecules are adsorbed onto the surfaces. The 2D materials used to develop chemiresistor gas sensors are MoS2,200 WS2,201 boron nitride,202 black phosphorus,203 and MXenes.204–206 These devices are used mainly for the detection of volatile organic compounds (VOCs).Kim et al. developed a high-performance chemiresistor from primitive MoS2 conjugated with mercaptoundecanoic acid (MUA) to attain a highly sensitive and tunable sensor for VOCs. The pristine MoS2 showed a positive response to oxygen-functionalized VOCs and the MUA-conjugated MoS2 gave negative responses in the presence of the same VOCs. This phenomenon is due to the strong interaction between VOCs and the sulfur vacancies of the pristine MoS2, whereas in the MoS2 conjugated with MUA, the oxygen-functionalized VOCs bond with hydrogen present in MUA molecules, thus enhancing the charge carrier density on the MoS2 chemiresistor.200 Moreover, our group has reported a selective VOC sensor for methanol and humidity based on 1T-phase WS2 (Fig. 8A). The impedance phase spectra showed two different resonant frequencies in the presence of methanol vapor (1 Hz) and water (1 kHz) (Fig. 8B and C). With these well-distinguished signals, this system is able to detect selectively and simultaneously methanol and water molecules even when they are present in a mixture.201 On the other hand, boron nitride (Fig. 9)202 and black phosphorus (Fig. 9B)203 are used as selective chemiresistor gas sensors for the respective sensing of methane and methanol.
 | ||
| Fig. 8 (A) Selective gas sensor using metallic 1T-phase WS2 as a gas sensing platform and impedance phase as a selective transduction method; this device can discriminate methanol and water vapour within their mixture. (B) Methanol is detected at low resonance frequency and (C) humidity detection is observed at high resonance frequency. Figure reproduced with permission from ref. 201, Copyright 2015 Wiley-VCH. | ||
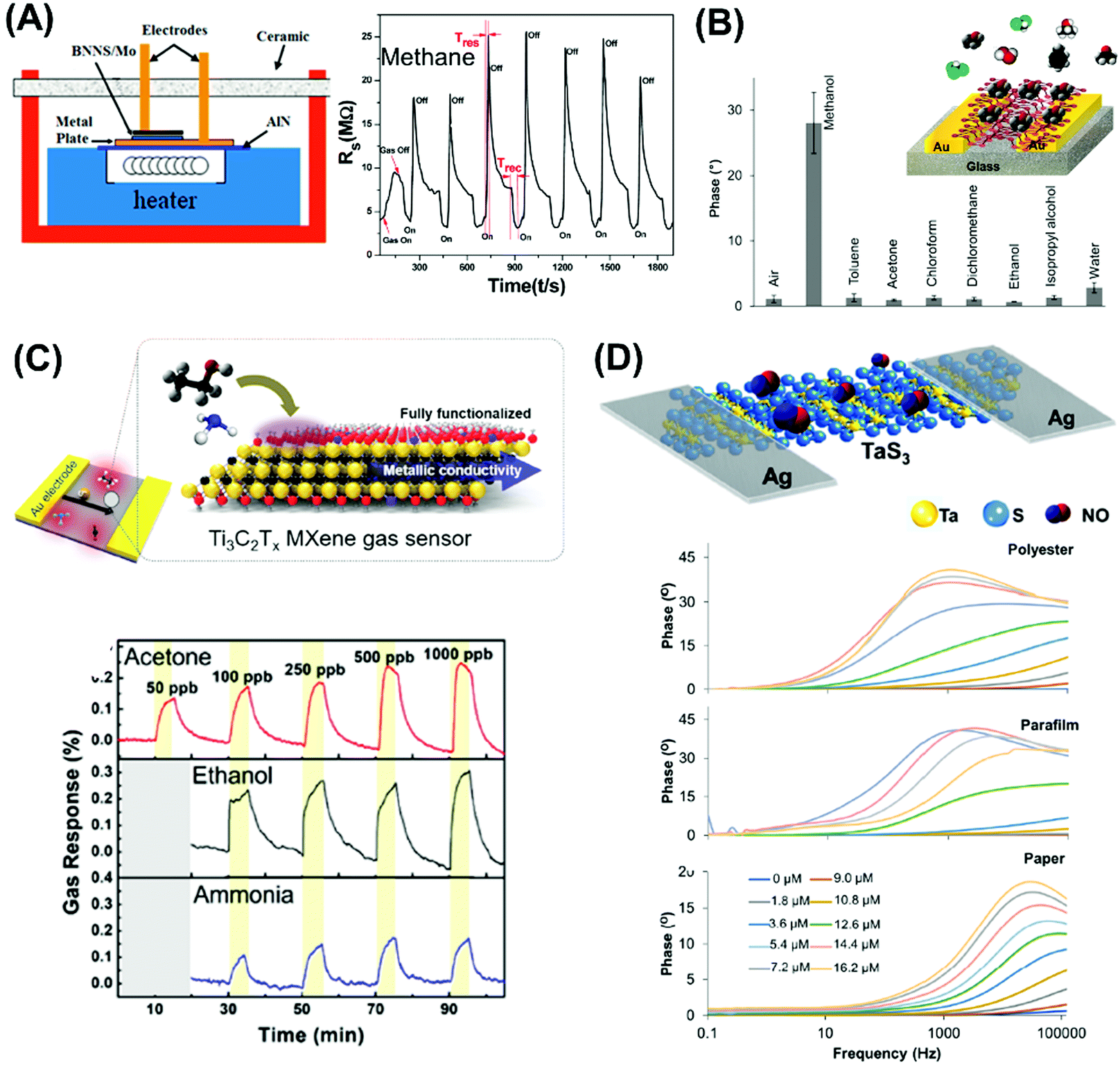 | ||
| Fig. 9 Chemiresistor gas sensors: (A) boron nitride nanosheets for methane gas sensors. (B) Impedimetric vapor sensor based on black phosphorus. (C) Metallic Ti3C2Tx MXene for VOC gas sensors. (D) Metallic TaS3 nanofibers for NO flexible gas sensors. Figures adapted with permission from: (A) ref. 202, Copyright 2013 American Chemical Society; (B) ref. 203, Copyright 2015 Wiley-VCH; (C) ref. 204, Copyright 2018 American Chemical Society; (D) ref. 211, Copyright 2017 American Chemical Society. | ||
Both gas sensing systems show enhanced analytical performance. Recently, MXene (Ti3C2Tx) prepared by Al etching of MAX phase (Ti3AlC2) was used to prepare VOC sensors. The sensors were fabricated using SiO2 wafers (Fig. 9C),204 a flexible polyimide substrate,205 and a 3D Mxene framework prepared through electrospinning technology.206 These devices were used for the detection of a group of VOCs (acetone, ethanol, ammonia, propanol, and methanol) and the first two designs showed similar performances.204,205 Nevertheless, the 3D Mxene framework sensor showed enhanced performance for VOC sensing and also for the detection of inorganic gases (e.g., NO2, NH3, H2O) but with lower sensitivity.206
MoS2207,208 and SnS2209 chemiresistors fabricated by CVD showed highly sensitive and selective gas sensor response for NO2. Cho et al. performed in situ photoluminescence (PL) characterization and additional theoretical studies that helped to elucidate the mechanism of charge transfer between NO2 and NH3 molecules and the MoS2 surface.208 The different responses observed are due to NO2 molecules’ actions as electron acceptors (p-type dopants) and NH3 molecules’ actions as electron donors (n-type dopants) onto the surface of MoS2.208 Meanwhile, Yan et al. demonstrated enhanced response of the SnS2 gas sensor for NO2 detection. They showed that S vacancies are produced during chemical vapor deposition. These S vacancies work as adsorption sites and give rise to an NO2 gas sensor with fast response times, enhanced adsorption energy, and higher resistance change compared with primitive SnS2.209 In addition, PtSe2191 can be produced by the thermally assisted conversion synthesis method, consisting of two steps: first, sputter-coating of the Pt layer onto the SiO2/Si substrates followed by selenization in a quartz tube furnace (Fig. 6B). This device can detect NO2 from 0.1 to 1 ppm with a fast response of 10 s.
Uniformly stacked TaS2 films made by filtration of a homogeneous suspension of TaS2 nanosheets prepared by electrochemical lithium intercalation and exfoliation210 and layered metallic TaS3 nanofibers211 have been used to prepare high-sensitivity NO-flexible gas sensors (Fig. 9D). The NO gas sensor based on metallic TaS3 nanofibers presents high analytical performance and a low limit of detection of 0.48 ppb compared to the TaS2 congener.210
In 2008, Wang et al. reported an NO2 gas sensor based on conductive C-rich carbon nitride. This sensor showed enhanced sensitivity under exposure to NO2 and this response was selective toward NH3, ethanol, and CO. The authors claim that the detection mechanism is based on the high content of pyridine N that can adsorb NO2. As a result, the negative charge of pyridine N can transfer to the NO2 molecule, resulting in a resistivity change.212 Moreover, carbon nitride was used for humidity and ammonia sensing; in both cases, the detection mechanism is also based on the nitrogen-rich content of carbon nitride.213,214
4.3 Future factors to explore
In this section, we have described gas sensors based on pristine 2D materials. We have focused on the more novel 2D materials beyond graphene such as TMDs, MXenes, h-BN, BP, as well as carbon nitride, and described the sensing mechanism of these reported devices. Summarizing, 2D materials show promising performance for developing highly sensitive and selective gas sensing devices that can be applied to the environmental monitoring of humidity and hazardous gases in order to mitigate environmental hazards. However, comprehensive studies related to their portability and mass production are still necessary to realize the potential of these devices for real applications.5. 2D materials for therapeutic applications
The application of 2D materials in the biomedical field is attracting growing attention, especially in the area of drug delivery and cancer therapy.215,216The application of nanotechnology to drug delivery became a blockbuster of biomedical research gradually revolutionizing the clinical settings. In the biomedical field, nanomaterials are used in a broad spectrum of applications ranging from enhanced drug delivery even of poorly water-soluble compounds, tissue-specific targeting of therapeutics, to photothermal (PTT) or photodynamic therapy (PDT).217–220 The employment of 2D materials has been reported especially for the therapy of malignant diseases.216 Graphene and graphene-based materials were the first 2D materials investigated for such a purpose. After that, research focus shifted also to the TMDs and lately also to black phosphorus. The use of arsenene, antimonene and bismuthene in the treatment of malignant diseases is so far in its infancy, however showing great potential. To date there is a negligible number of studies investigating the behavior of these nanomaterials under physiological conditions.221,222
5.1 Drug delivery
Nanocarriers bearing therapeutic agents proved their efficiency predominantly in cancer treatment.223,224 Nanocarrier-mediated targeted delivery of therapeutic agents into the tumour tissue simultaneously mitigates the side effects on healthy cells.225 Besides therapeutics, other molecules can be attached on the surface of nanoparticles and thus impact their physicochemical properties and pharmacokinetics. For example, the broadly used attachment of polyethylene glycol (PEG) improves particle stabilization and prolongs their circulation time in the bloodstream and thus influences the therapeutic efficiency of the payload.226To date, the first and one of the most detailed studies concerning the intracellular fate and internalization of 2D materials was published by Zhu et al.227 They chose MoS2 as their model representative of 2D nanomaterials and found out that it can be internalized through three different pathways: clathrin- and caveolae-dependent endocytosis and macropinosis. MoS2 was several times investigated for the loading effectivity of various compounds.228–230 The PLGA/MoS2/DOX nanocomposite may be used for the synergistic tumour photothermal and chemotherapy. Near-infrared (NIR) laser irradiation not only induces tumour necrosis, but also further facilitates the release of cytostatic doxorubicin (DOX) and by that enhances its therapeutic efficiency. In this way tumours may be completely destroyed without any reoccurrence.228 Further, a synergistic therapeutic effect of chemotherapy and photothermal therapy was observed in vivo after NIR irradiation of MoS2 nanosheets modified with chitosan (CS) and DOX.229 CS was introduced during the exfoliation process to increase the stability of the whole delivery system under physiological conditions as well as to enhance its biocompatibility. Heat-induced tumour killing mediated by MoS2-CS-DOX under NIR laser irradiation was proven in nude mice bearing pancreatic tumours. The local temperature in the tumour increased to about 22.5 °C (Fig. 10A). In both MoS2-CS and especially MoS2-CS-DOX groups a great reduction of tumour volume was observed after applying an 808 nm laser (Fig. 10B). The representative photographs of surgically removed tumours in each group (Fig. 10C) nicely illustrate the superior antitumour effect of MoS2-CS-DOX after irradiation mediated by the synergistic effect of photothermal therapy and chemotherapy and/or enhanced on-demand release of DOX. The anticancer effect was observable even macroscopically (Fig. 10D). In the irradiated MoS2-CS-DOX group one of three tumours on three mice even fell off without any recurrence observed. Histological examination revealed that unlike control groups (Fig. 10E and F) majority of the tumour tissue in the irradiated MoS2-CS-DOX group died from necrosis (Fig. 10G).
 | ||
| Fig. 10 Comparison of tumour growth inhibiting efficiency in vivo. (A) Infrared thermal images of Panc-1 tumour-bearing mice injected with saline, MoS2-CS + NIR laser and MoS2-CS-DOX + NIR laser. (B) Tumour growth curves of tumours after various treatments for five groups. (C) Photograph of tumours from the control group, MoS2-CS group, DOX group, and MoS2-CS + NIR group, and MoS2-CS-DOX + NIR group. (D) Photographs of the typical mice of tested groups corresponding to (i) saline + NIR, (ii) MoS2-CS, (iii) DOX, (iv) MoS2-CS + NIR, and (v) MoS2-CS-DOX after observation for 24 days. (E–G) Histological images of tumours collected from the group of (E) saline, (F) saline + NIR, and (G) MoS2-CS-DOX + NIR. Figure reproduced with permission from ref. 229, Copyright 2014 American Chemical Society. | ||
In another study, DOX was also loaded on the surface of silica-coated PEGylated MoS2 nanoplates in order to construct a photo-responsive drug delivery system.230 Here, the system was capable of 30-times more effective liver and colon cancer cell growth inhibition in vitro in comparison with free DOX. Interestingly, the drug release was found to be facilitated by the acidic environment characteristic for the tumour microenvironment and to be further enhanced by NIR irradiation. Additionally, higher DOX effectivity was also attributed to facilitated endosomal escape of the carrier. Besides other nanomaterials, zero-dimensional MoS2 QDs were also described to be applicable in the multiphoton imaging of malignant diseases. After surface modification with prostate-specific membrane antigen (PSMA), MoS2 QDs were used for the diagnosis of prostate cancer.231
To our knowledge, one of the first papers using BP nanosheets as a drug carrier was published by Tao et al.232 PEGylated BP nanosheets were individually modified with two agents: doxorubicin (DOX) for cancer therapy and cyanine7 (Cy7) for in vivo NIR imaging. BP nanosheets proved to have high drug loading capacity. Further, folic acid (FA) was attached to the surface of the particles for higher accumulation in the tumour tissue in vivo. Here, FA-BP NPs modified with therapeutic agents DOX and chloroquinone (CQ) and irradiated with 808 nm NIR light were identified as the most efficient therapeutic strategy tested within this study, extensively reducing the tumour size. CQ prevented endosomal acidification by blocking the fusion of autophagosomes with lysosomes.233 Simultaneously, no signs of acute toxicity were found, as well as no damage of major organs was observed. Endocytosis was identified to be the intake pathway prevalent for the internalization of BP NPs, more specifically caveolae-dependent endocytosis and macropinosis.232,234 After that, more studies confirmed BP's high loading capacity235 of various cytostatics,232,236 negligible induction of hemolysis,235 high stability under physiological conditions237 and even facilitated drug release of therapeutics by the acidic environment within the tumour microenvironment235 that can be even potentiated by the high photothermal conversion efficiency of BP nanosheets.238 This controlled drug release could be especially beneficial for BP utilization in clinical practice due to the precisely controllable drug release rate achievable by altering the external and internal parameters, e.g. drug concentration, laser light intensity or irradiation duration. This together with low toxicity for the major organs and biodegradability make BP a suitable material for the bench-to-bedside transition with high therapeutic efficiency and reduced side effects.
5.2 Photothermal and photodynamic therapy
In the management of malignant diseases, photothermal (PTT) and photodynamic therapy (PDT) are among intensively investigated experimental therapeutic approaches. PTT and PDT represent an especially promising alternative in the management of tumours that are untreatable by the conventional therapy (surgery and radio-, or chemotherapy), or in the case that implementation of these conventional treatment modalities could induce severe side effects.239 PTT uses agents capable of light energy conversion (preferably near-infrared light, NIR) in the form of photons to heat while creating hyperthermia.240 Such an increased temperature induced by a laser then damages the tissues in the affected area as a result of protein denaturation and cell organelle damage.241,242 Irreversible hyperthermia-induced tissue damage occurs from up to 41 °C and increases with increasing temperature and time at which the tissue is held at this given temperature.243 Besides, tumour cells are known to be more temperature-sensitive than the healthy ones.244 Two-dimensional nanomaterials are capable of strong interaction with NIR light and therefore proved to be especially suitable for biophotonic nanomedicine.245 These are used as so-called photothermal agents (PTAs) in order to target the malignantly transformed tissue more specifically and to spare the surrounding healthy cells. One of the key factors characterizing the effectivity of PTAs is photothermal conversion efficiency (PTCE) summarized in Table 6. PTCE can be calculated using the formulawhere η represents the photothermal conversion efficiency (PTCE), h is the coefficient of heat transfer, A is the surface area of the container, ΔTmax is the maximum temperature change of the particle solution, Q stands for the heat associated with the light absorbance of water, I stands for the power density of the NIR laser and Ax is the absorbance of the particle solution at the laser wavelength.221,246 Photodynamic therapy (PDT) uses non-toxic photosensitizers capable of reactive oxygen species (ROS) generation. These are created after irradiation with a harmless laser light of a specific wavelength by transferring the accumulated energy to the surrounding molecular oxygen. ROS then induce cellular death, predominantly by necrotic or apoptotic pathways. When using photosensitisers, the PTT or PDT effects are often combined.
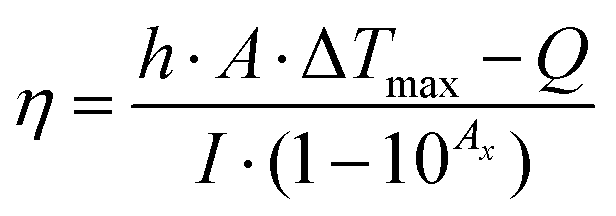
| 2D Materials | Mice used | Tumour | Cells injected | In vitro cell lines | λ (nm) | I (W cm−2) | t (min) | η (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| BPQDs | — | — | — | HSC, 293T, C6, MCF7 | 808 | 1 | 10 | 28.4 | 257 |
| PEG-coated BP NPs | Balb/c | Breast | 4T1 | 4T1 | 808 | 2 | 5 | 36.8 | 256 |
| BP nanosheets (BP NSs) | Nude | Breast | MDA-MB-231 | MDA-MB-231 | 660 | 0.5 | 20 | — | 260 |
| PEG-coated BP nanosheets | Nude | Cervix | HeLa | HeLa | 808 | 1 | 10 | 29.8 | 232 |
| PLGA-loaded BP QDs (BPQDs/PLGA) | Balb/c | Breast | MCF-7 | HSF, MCG7, B16 | 808 | 1 | 10 | — | 261 |
| BP NSs, BP-DOX NSs | Balb/c | Breast | 4T1 | 4T1 | 808 | 1 | 5 | — | 235 |
| Dye-modified BP NSs | Balb/c | Breast | MCF-7 | MCF7, LO2 | 808 | 1.5 | 10 | — | 258 |
| BP QDs | Balb/c | Breast | 4T1 | Hep G2 | 635; 808 | 80; 1 | 20; 2 | — | 259 |
| BP NSs | Nude | Breast | MDA-MB-231 | MDA-MB-231, A549, HeLa, B16 | 808 | 1 | 5 | 38.8 | 238 |
| WS2-PEG NSs | Balb/c | Breast | 4T1 | 4T1, HeLa, 293T | 808 | 0.8 | 5 | — | 292 |
| MoS2–PEG nanoflakes | Balb/c | Breast | 4T1 | 4T1, HeLa | 808 | 1 | 15 | 27.6 | 247 |
| PEGylated MoS2 NSs | Balb/c | Breast | 4T1 | L929, 4T1 | 808 | 1 | 5 | — | 248 |
| 1T-WS2 ultrathin NSs | NOD/SCID | Cervix | HeLa | HeLa, MDA-MB-231, HepG2 | 808 | 0.6 | 10 | — | 253 |
| WS2 with thiol-modified PEGylation | — | — | — | HeLa | 808 | 0.4 | 10 | ∼35.0 | 250 |
| MoS2 nanodots modified with glutathion | Balb/c, nude | Breast | 4T1 | 4T1 | 808 | 1 | 5 | — | 249 |
| DOX- and CS-loaded MoS2 NSs | Nude | pancreatic | Panc-1 | KB, Panc-1 | 808 | 0.5–0.9 | 7 | 24.37 | 229 |
| PEG-coated antimonene QDs (AMQDs) | Nude | Breast | MCF-7 | MCF7, HeLa | 808 | 1 | 10 | 45.5 | 221 |
| PEG-coated antimonene NPs (SbNPs) | Nude | Breast | 4T1 | 4T1 | 808 | 1 | 5 | <41.0 | 222 |
Out of the TMDs, MoS2247–249 and WS2250 (frequently modified with PEG) represent the most intensively investigated materials in PTT and PDT. MoS2 NPs were several times reported to have negligible cytotoxicity and not to induce haemolysis and blood coagulation.231,247,248,251,252 Under physiological conditions, particles prepared by Chen et al. exhibited natural degradability in vivo to water-soluble MoO42− ions followed by gradual clearance from the body.251 The particles induced in vitro cancer cell growth inhibition and in vivo tumour growth suppression or even ablation without any damage of vital organs.247,248,251 MoS2 NPs evinced great photothermal stability upon NIR irradiation and photothermal conversion efficiency.231,247,248,252 Even though MoS2 exhibited low cytotoxicity under physiological conditions, it showed high cancer cell killing ability after NIR irradiation by damaging the integrity of the lysosomal membrane and the cytoskeleton.247 The high photothermal performance of MoS2–PEG nanosheets was found to be modifiable via modulation of the particle size and their surface chemistry. Nanosheets with a smaller diameter evinced the highest photothermal conversion efficiency than the larger ones. This effect was found to be potentiated by the surface modification of particles with PEG.248 Further, the WS2 nanomaterial was also found to be non-toxic against a wide scale of cell lines with simultaneous strong optical absorption in the NIR range associated with high photothermal conversion efficiency and inducing no systemic toxicity while having a great tumour growth suppression potential.253–255
The low cytotoxicity and high photothermal conversion efficiency of BP were reported several times.232,256–259 PTCE higher than 38% was proved by Sun et al.256 They found out that BP toxicity in vitro rapidly increases with NIR light irradiation and aggravates with increased laser power density. The photoacoustic (PA) images of the vital organs confirmed preferential accumulation and retention of BP NPs in the tumour lesion. A lower PA signal was also observed in the liver and kidneys; however, the BP particles were gradually eliminated from the body. Further, the high efficiency of PTT was confirmed since BP treated and subsequently irradiated mice tumours reduced their size in just 3 days and the animals survived for more than one month after this therapy. No signs of metastasis formation, nor the damage of vital organs was observed. Further, BP nanosheets were found to meet the needs required for an ideal nanocarrier – biocompatibility, biodegradability, low systemic toxicity, and capability of ROS generation.235,258–261 Irradiation with a 660 nm laser was capable of complete degradation of BP nanosheets to non-toxic PxOy species.260 Further, BP was shown to generate singlet oxygen under light irradiation (λ ≥ 600 nm) which was attributed to the ultrathin structure of the nanosheets. This was confirmed in in vivo experiments proving tumour growth suppression after i.t. injection of BP nanosheets followed by light irradiation.
The first study on the photothermal properties of antimony NPs and their application as a photothermal agent was published by Li et al.222 They synthesized PEG-modified Sb NPs (PEG Sb NPs) and used them as a photothermal agent driven by an 808 nm laser. Compared to other PTT agents (Table 6), Sb NPs evinced particularly high photothermal efficiency reaching up to 41%. Nanoparticles showed low cytotoxicity in vitro up to the concentration 200 μg mL−1. Further, in vivo application followed by 808 nm laser irradiation resulted in not only tumour elimination, but also in animal survival for the whole time of the observation (30 days). Besides proving the PEGylated Sb NPs to be a highly efficient photothermal agent with excellent photothermal conversion performance and without any signs of side effects, they also demonstrated their potential as a NIR photoacoustic agent for tumour imaging. Recently Tao et al. developed a novel liquid exfoliation method for the preparation of antimonene quantum dots (AMQDs). Their PTCE was found to be superior over the traditional PTAs reaching nearly 46%. The cytotoxicity of particles was evaluated in vitro up to the concentration of 200 μg mL−1 on a panel of cancerous and non-cancerous cell lines. At the highest AMQD concentration (200 μg mL−1) the cellular viability fluctuated around 80% regardless of the cell line. After short laser irradiation, it steeply dropped to <10%. The irradiated AMQDs were capable of destroying the tumours also in vivo without any signs of tumour growth reoccurrence or surrounding tissue damage. Further, laser-induced degradation of AMQDs was investigated. The NIR-induced material degradation occurred within only a few minutes highlighting the potential of antimonene in biomedical applications and greatly increasing its applicability in the clinical practice. Altogether, this study proved the biocompatibility and biodegradability of AMQDs and proved their potential in cancer treatment.221
5.3 Future factors to explore
Currently, the vast majority of the therapeutic applications of 2D nanomaterials beyond graphene is finding their place in the area of cancer therapy. More potential therapeutic areas should be explored as in the case of graphene-based nanomaterials and their use in bone tissue engineering or diabetic wound healing.262,263 New anticancer compounds and strategies are still under development. The search for more efficient approaches to combat cancer is attracting tremendous efforts of the scientific community. The conventional administration of anticancer drugs is known to be frequently accompanied with serious side effects and problems with the induction of drug resistance in cancer cells. Both could be prevented or at least significantly reduced by refining the delivery mechanisms of these compounds. This is where 2D nanomaterials meet oncology. Novel types of 2D nanomaterials are being intensively synthesized and their employment in cancer therapy can be expected. Because of the large surface area, more or less efficient drug binding might be expected across the wide spectrum of 2D nanomaterials. However, in addition to the high binding efficiency of 2D nanomaterials usually mentioned, the release mechanisms should also be tuned.2D nanomaterials should be capable of delivering a therapeutically efficient amount of drug including the on-demand drug release triggered by external (e.g. laser irradiation, ultrasound) or internal (e.g. pH) stimuli. Furthermore, detailed in vitro studies are needed as they can unravel interesting biological phenomena. For example in our recent study we showed an increased efficiency of DOX therapy in a DOX-resistant ovarian cancer cell line mediated by a novel type of 2D nanomaterial 4-carboxybutylgermanane.264 Here, the anticancer effect of DOX was increased for up to 63% as the material presumably blocked the P-glycoprotein pumps. These pumps under standard conditions pump the chemotherapeutics out of the cells and therefore reduce the efficacy of cancer therapy. In this work we illustrated that nanomaterials might besides targeted drug delivery exhibit also other interesting properties working with the anticancer effect of drugs in synergy. In addition to unravelling the mechanisms implicated in the increased toxicity of therapeutics mediated by 2D nanomaterials, other therapeutic strategies should also be explored.
One of the most promising directions where the attention should be focused is the employment of 2D nanomaterials in immunotherapy.265 Immunotherapy is a highly attractive treatment opportunity providing a tool to aid the body's own immune system not only to recognize cancer cells, but also to fight them while sparing the healthy tissues. Under physiological conditions, the immune system defends the body against infection and disease. However, one of the hallmarks of cancer cells is their ability to escape recognition by the immune system. Contrarily, cancer cells may even convince the components of the innate as well as adaptive immune system to promote tumour progression. As an example one of the several mechanisms cancer cells use is that they create a tumour microenvironment enriched with signals causing polarization of tumour-associated macrophages (TAMs) from the tumour-inhibiting M1 phenotype towards an M2-like lineage promoting tumour progression and especially metastatic spreading. Therapeutic strategies reprogramming the pro-tumour M2 phenotype into the tumoricidal M1 phenotype and thus inhibiting TAMs’ supportive role in tumours are needed.266,267 Recently iron oxide as well as GO NPs were found to induce significant inhibition of tumour growth by inducing M1 macrophage polarization.265,268 The effect of 2D nanomaterials beyond graphene and GO on the components of the immune system and their function should be thoroughly explored as a prospective synergic effect with chemotherapy might be expected. Further, the effect of 2D nanomaterial-mediated immunotherapy can be combined with their photothermal therapy. However, reports employing 2D nanomaterials beyond graphene in the area of photoimmunotherapy are again only seldom.
Finally, detailed studies investigating the behaviour of nanomaterials in vivo are needed to thoroughly outline their prospective therapeutic potential. More attention should be paid though to the deeper appraisal of their systemic toxicity. For example GO was several times reported to induce acute lung injury or chronic pulmonary fibrosis and similar effects might be expected also in some other 2D nanomaterials.269–271 This is of particular importance not only because of their use in biomedicine. The overall impact of novel nanomaterials' usage on human and animal health as well as their environmental impact should be studied in detail prior to their implementation in the products of everyday use.
6. Toxicity of 2D materials
This section of the review details the toxicity aspect of 2D materials. This field is important to anticipate possible toxicological effects of 2D materials to humans and the environment when they are manufactured in large scale industries. Without proper knowledge of their toxicological effects, they may be mishandled and exposed to humans and the environment at dangerous levels and result in chronic health problems or even death.272,273 Within an individual class of 2D materials (i.e. graphene, TMDs, BP, pnictogens), differences in toxicity have been reported. For example, MoS2 and WS2 have shown low toxicity to human cell lines in several studies.30,274 Interestingly, these materials have also been reported to have antibacterial activity.275–277 Similar is the case for graphene.278,279 It appears that the toxicity profile of 2D materials can range over a wide spectrum. Several reviews have already discussed on factors that can influence the toxicity of 2D materials. This includes dosage, duration of exposure, particle dimensions, surface charge, functionalization, structure, aggregation/sedimentation, impurities and protein corona effect. However, the reviews are mostly in relation with graphene or nanoparticles.280–285Looking at the current literature, we find many reports on toxicity of TMDs, BP and pnictogens corroborating the factors influencing the toxicity of graphene mentioned above. This suggests that these factors are likely to be generalized and extended to other families of 2D materials. Here, we attempt to provide an update on the matter to include recent findings of factors influencing the toxicity of reported 2D materials, such as surface functionalization, size, protein corona effect, chemical composition, solubility as well as impurity content, morphology and polymorphs (Fig. 11).
 | ||
| Fig. 11 Possible factors that can influence the toxicity of 2D materials include (A) surface functionalization, (B) size effect, (C) protein corona effect and (D) chemical dissolution. Figures adapted with permission from: (A) ref. 286, Copyright 2017 American Chemical Society; (B) ref. 304, Copyright 2017 Wiley-VCH; (C) ref. 313, Copyright 2018 Springer Nature; (D) ref. 318, Copyright 2016 American Chemical Society. | ||
6.1 Surface functionalization
Surface functionalization is a useful strategy to tune or reduce the toxicity of 2D materials. For MoS2 functionalized with thiobarbituric acid (TBA), not only does its electrocatalytic activity towards proton reduction improve after this functionalization,286 but its toxicity response to human lung cancer cells (A549) is also reduced (Fig. 12).287 Another study by Qu et al. displayed improved biocompatibility of BP nanosheets modified by a titanium sulfonate ligand (TiL4) through in vitro and in vivo experiments.288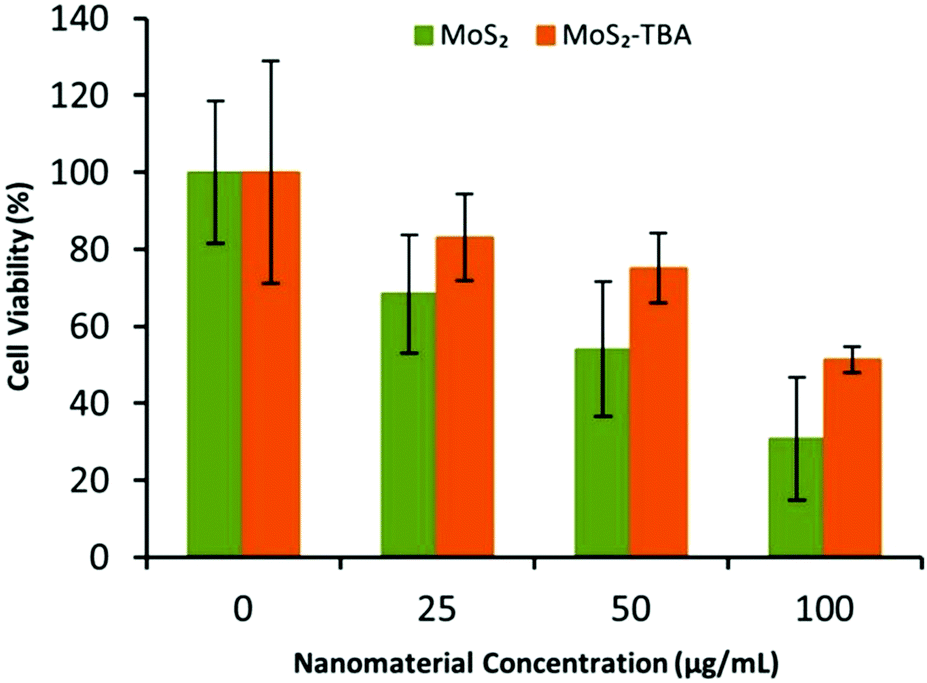 | ||
| Fig. 12 Cell viability profile obtained using MTT assay after 24 h incubation of A549 cells with MoS2 and its functionalized counterpart (MoS2–TBA). Figure adapted with permission from ref. 287, Copyright 2018 Elsevier. | ||
Surface functionalization is commonly seen in drug delivery and drug photothermal therapies to improve the biocompatibility of 2D materials.289–291 Often, we find polyethylene glycol (PEG) favoured for this purpose. This strategy has been proven effective in lowering the toxicity of graphene, TMDs, BP as well as antimonene.292–298 A possible reason for this observation is the alteration in characteristics that accompanies surface functionalization such as changes in hydrophilicity/hydrophobicity, surface charge and size.299 This consequently affects the nanomaterial's solubility and cellular uptake. It has been reported that nanomaterials including MoS2 and BP can sediment or form aggregates, while surface functionalization can improve their stability in solutions.280,300,301 Without functionalization, the effects of aggregation and sedimentation can greatly inhibit nutrient availability for adherent cells as reported for graphene and GO.302
6.2 Size effects
Besides surface functionalization, size effects can play a significant role in influencing the toxicity of 2D materials. This can be in the form of lateral dimension, thickness, diameter, etc. For instance, MoS2 exfoliated by different lithium intercalating agents (i.e. Me-butyl, n-butyl and t-butyl lithium) shows different thickness as well as different toxicity profiles.303 XPS was used to infer the thickness of MoS2 nanosheets upon exfoliation. The result indicates that various exfoliating agents can result in different extents of exfoliation which in turn affects the material's toxicity.In the case of BP, a systematic study investigating its size effect was conducted on various cell lines by Zhang et al.304 The authors found that BP particles of larger size show higher toxicity compared to the smaller ones (Fig. 13). The different sizes and thickness were determined using atomic force microscopy (AFM) and dynamic light scattering (DLS) measurements. This finding further develops on a previous study on cytotoxicity investigation of layered BP which showed intermediate toxicity between graphene and TMD but did not consider size effects.305 However, after consideration of size effects, this observation is possibly not true for all sizes of BP particles. This illustrates the importance of carefully characterizing a material's size before reporting on its toxicity as it could lead to a bias conclusion.
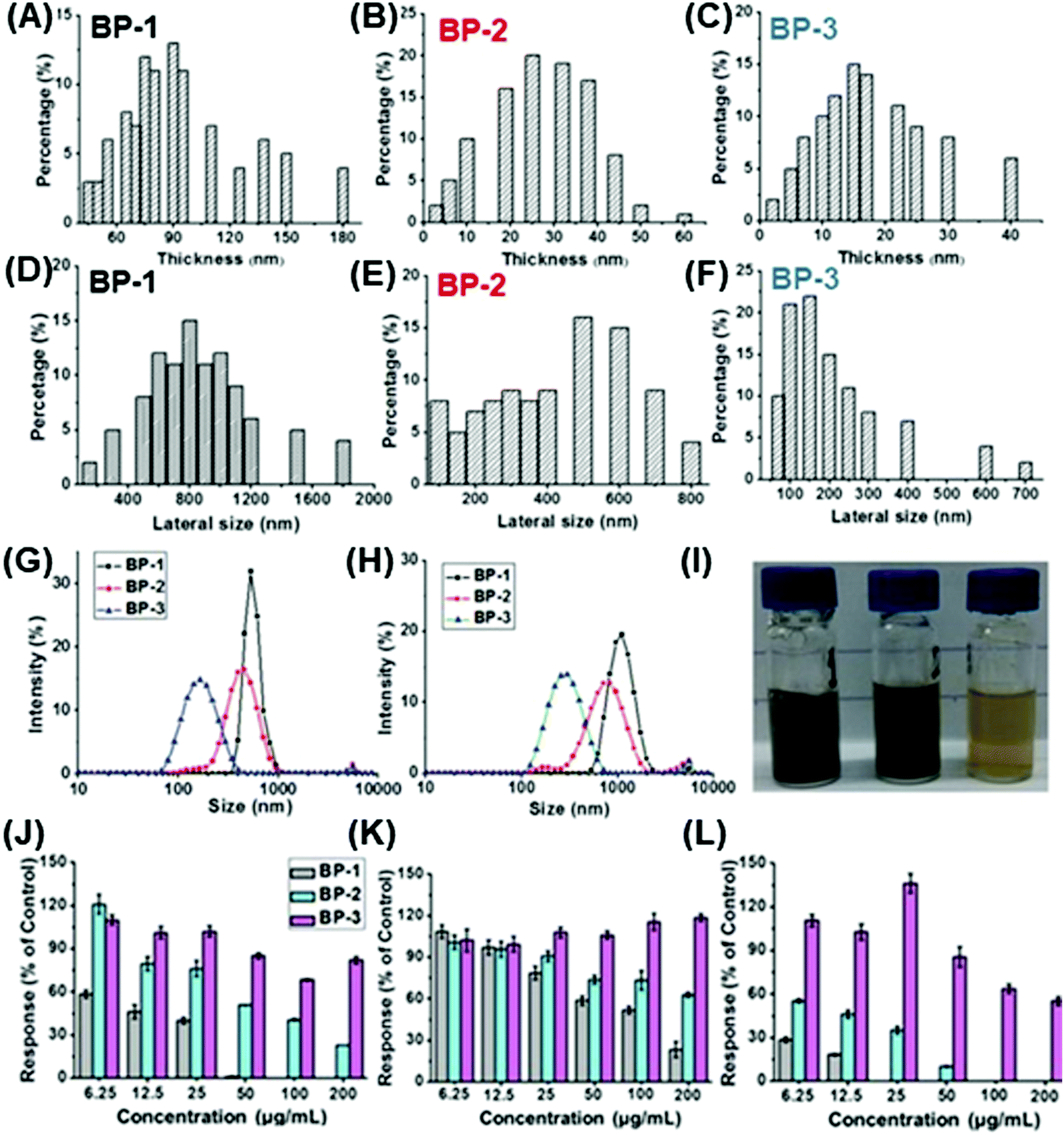 | ||
| Fig. 13 Size effects on the toxicity of BP nanosheets and their size characterization by atomic force microscopy in terms of (A–C) thickness and (D–F) lateral sizes, as well as (G and H) by dynamic light scattering (DLS). (I) Photographs of the prepared BP dispersions. (J–L) Cytotoxicity was evaluated after 12 h incubation across three different cell lines: (J) mouse fibroblast cells (NIH 3T3), (K) human colonic epithelial cells (HCoEpiC), and (L) human embryonic kidney cells (293T). Figure adapted with permission from ref. 304, Copyright 2017 Wiley-VCH. | ||
Interestingly, the observation that smaller BP particles show lower toxicity is opposite to that observed for graphene. GO of smaller lateral sizes were found to have higher toxicity compared to smaller ones in in vitro and in vivo studies, possibly due to higher cellular uptake of smaller sized particles.280,306 In contrast, larger BP sizes were found to produce greater cell membrane damage relative to smaller ones. Besides that, all BP sizes were observed to induce intracellular reactive oxygen species.303
6.3 Protein corona effect
The protein corona effect is another important parameter to consider for understanding the toxicity of nanomaterials in protein-containing solutions. Upon contact with biological fluids such as blood or cell culture media, a layer of proteins would instantaneously envelop the nanomaterial, producing a new interface known as “protein corona”.307 Cells would then respond to the interaction with this protein-adsorbed nanomaterial. Therefore, protein corona can regulate nanomaterial–cell recognition and has significant impact on modulating a nanomaterial's toxicity.308–311 It has been reported to help reduce the toxicity of GO and it differs with different biological media used.312 This protein corona effect shows that the toxicity of a material is not determined by the nature of nanomaterial alone, but is also linked to the physiological system it is in. There is a recent in-depth investigation by Mo et al. on the protein corona associated with different sizes of BP nanomaterials (nanosheets and QDs) and their effect on in vitro cytotoxicity and cellular uptake.313 The authors found that there are significant changes in surface charge and size after protein corona formation for the BP materials (Fig. 14). Also, they observed that the size of BP nanomaterials greatly influences the type of proteins defined in the BP–corona complexes. In terms of the cytotoxic effect, it appears to be reduced when the BP nanomaterials were coated with protein corona. Cellular uptake was determined by ICP-MS. They found that the presence of protein corona facilitated the uptake of BP nanomaterials. Between the different sizes, BP QDs showed higher cellular uptake compared to BP nanosheets. This observation is a reverse trend from that seen for GO.314 Overall, this study demonstrates the influence of size effects on protein corona formation for BP materials, suggesting that the different factors affecting the toxicity of 2D materials can be inter-related.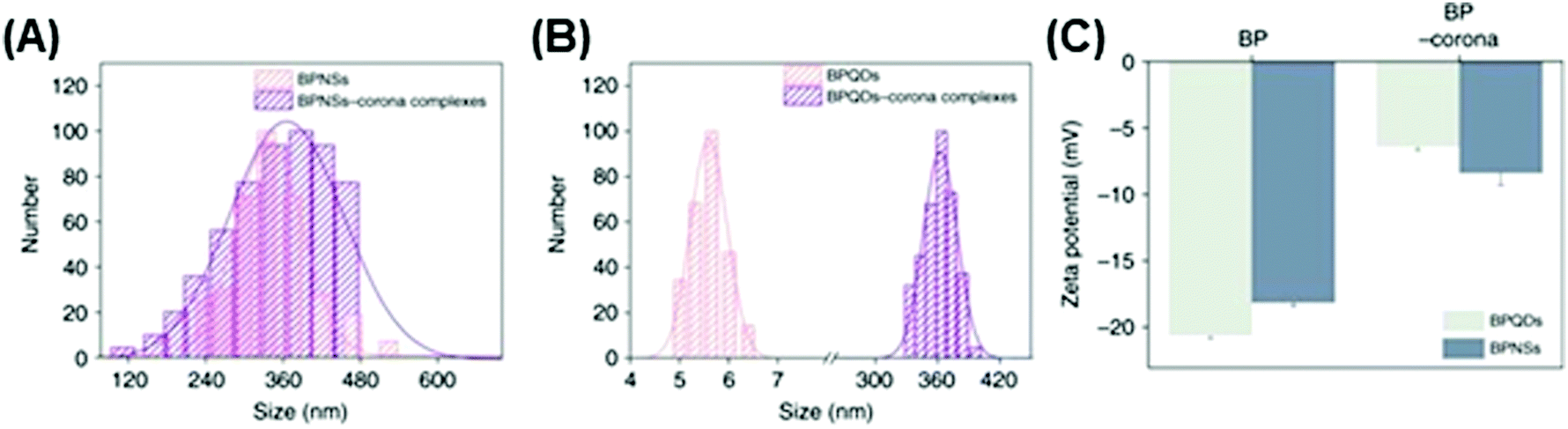 | ||
| Fig. 14 Changes in terms of (A) size and (B) surface charge observed for (C) black phosphorus nanosheets (BPNSs) and black phosphorus quantum dots (BP QDs) upon protein corona formation in biological media. Figure adapted with permission from ref. 313, Copyright 2018 Springer Nature. | ||
6.4 Chemical composition
Apart from surface functionalization, size and protein corona effects, chemical composition also plays a role in influencing the toxicity of 2D materials. This is illustrated by our cytotoxicity assessment of vanadium dichalcogenides (i.e. VS2, VSe2, VTe2).315 The presence of elements mentioned was verified by energy dispersive X-ray spectroscopy (EDS). We found that VTe2 shows high toxicity while VS2 and VSe2 show moderate toxicity. The toxicity of TMDs with the same metal element appears to increase down the chalcogen group. In another study, Chia et al. studied the cytotoxicity of Group 5 ditellurides, namely VTe2, NbTe2 and TaTe2.316 The authors found that VTe2 exhibits high toxicity while NbTe2 and TaTe2 show mild toxicity. Down Group 5 metals, we observed decreasing toxicity of TMDs sharing the same chalcogen element. These findings highlight the role different elements (or even trace elements) can have in the toxicity of a layered material. Interestingly, there may also be synergistic effects between elements present. This can be seen in a work by Rosli et al., who reported low toxicity of layered PtTe2 despite Pt/C showing inherent toxic behavior.317 Here, the authors also found that toxicity increases down the chalcogen group (i.e. tellurides > selenides > sulfides) for platinum dichalcogenides (Fig. 15).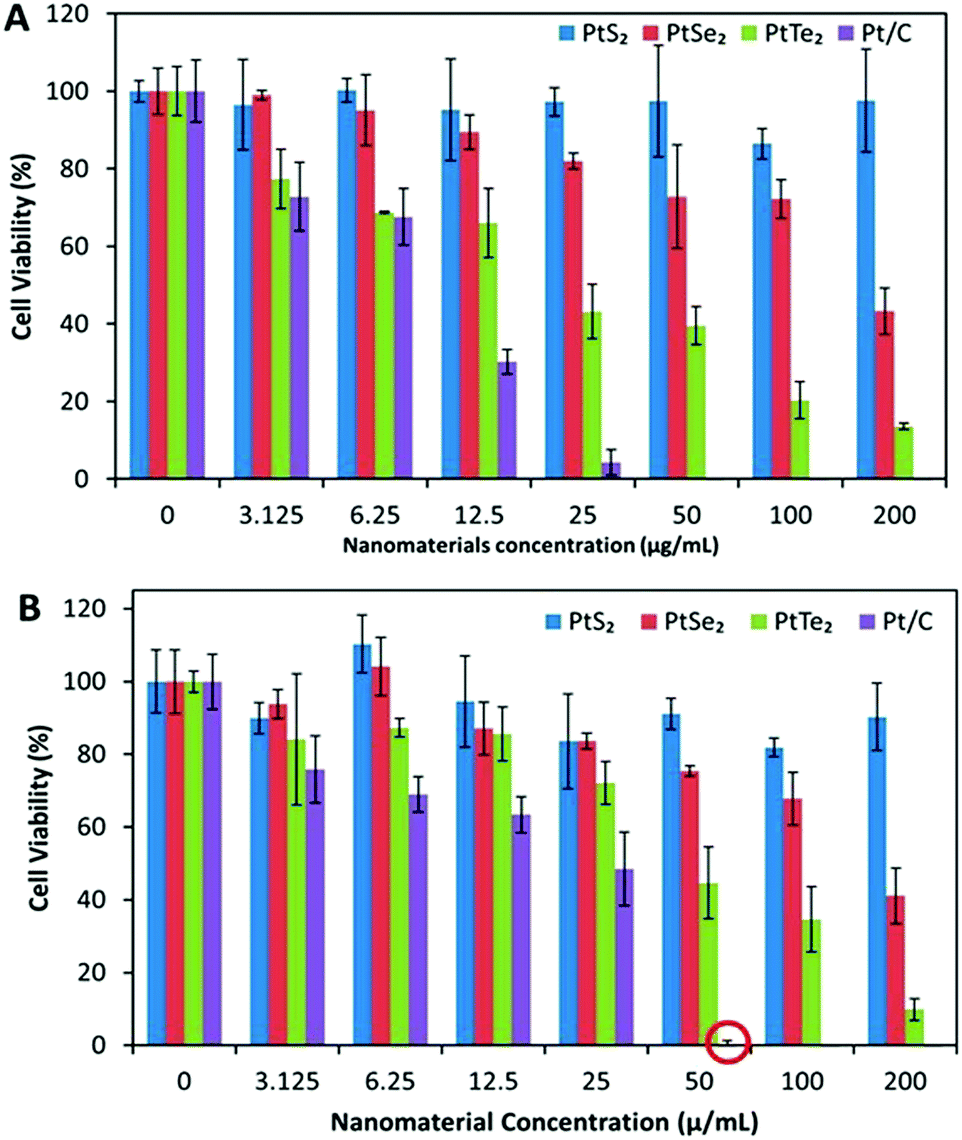 | ||
| Fig. 15 Cytotoxicity of platinum dichalcogenides after 24 h incubation with (A) human lung carcinoma cells and (B) human bronchial epithelial cells (BEAS-2B). Figure reproduced with permission from ref. 317, Copyright 2018 American Chemical Society. | ||
6.5 Chemical dissolution
Linked to chemical composition, the solubility or rate of dissolution of chemicals present in the nanomaterial can also influence its toxicity. Recently, Wang et al. investigated the dissolution pathways of MoS2 nanosheets and found them to be susceptible to oxidation under ambient conditions in aqueous media.318 Moreover, this oxidation process results in degradation of the MoS2 nanosheets, thereby producing soluble Mo and S species as well as releasing protons which can destabilize the other nanosheets. This process was determined to be pH-dependent and was found to occur more rapidly for MoS2 nanosheets dispersed in air-saturated water than in deoxygenated water. Also, the authors observed that the dissolution of MoS2 nanosheets occurred faster when they are in mixed phases (1T and 2H) and slower for MoS2 nanosheets in pure 2H phase. In the case of BP, it has also been found to be unstable in water due to its high reactivity with oxygen.319,320 However, the degradation products are phosphate and phosphonate which are present in and tolerated by the human body.3006.6 Future factors to explore
Through our literature search, we have identified two factors which can contribute to the toxicity of graphene materials but are currently less explored or less established in the case of the more recent families of 2D materials (i.e. TMDs, BP and pnictogens). They are effect of impurities and different morphology or polymorphs.There have been several reports on traces of metallic impurities present in graphene, carbon nanotubes (CNT) and BP that remained in the materials after their synthesis.321–323 For instance, manganese ions in GO have been found to increase its cellular toxicity (Fig. 16).324 Therefore, it would be useful to explore this possibility for other 2D materials too. It could explain the inconsistent conclusions observed in nanotoxicity reports.325
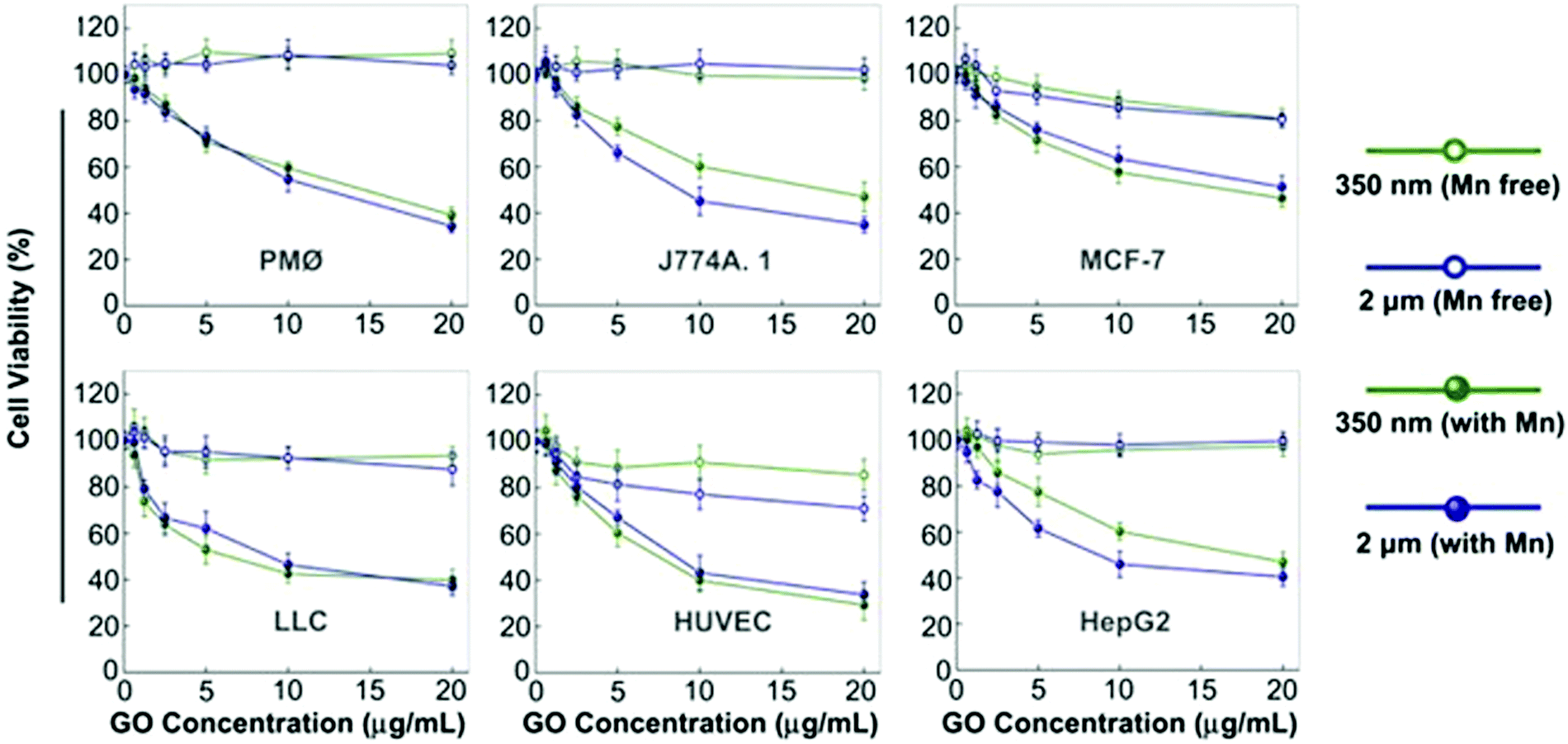 | ||
| Fig. 16 Comparison of cytotoxicity of graphene oxide (GO) with and without Mn impurities for two different sizes after 48 h incubation across six different cell lines: peritoneal macrophages (PMØ), murine macrophages (J774A), human breast cancer cells (MCF-7), human hepatocarcinoma cells (HepG2), human umbilical vein endothelial cells (HUVEC) and murine Lewis lung carcinoma cells (LLC). Figure reproduced with permission from ref. 324, Copyright 2012 Elsevier. | ||
As seen with graphene, there can be various possible forms of morphology associated with layered materials such as bulk form, fullerene-like, single-wall nanotubes (SWNTs), multi-wall nanotubes (MWNTs), few-layer nanosheets, single layer material, etc. Jia et al. had reported that different geometric structures of carbon nanomaterials exhibit unique cytotoxicity with the following trend: SWNTs > MWNTs > fullerenes.326 Furthermore, when exploring the toxicity of nanotubes, differences in tube diameter should be considered as it has been observed to be an important parameter in influencing the toxicity of multiwalled carbon nanotubes. Thin MWCNTs were found to have higher toxicity relative to thicker ones in both in vitro and in vivo experiments.327 Additionally, there has been report of graphene nanoribbons and nanoplatelets displaying different toxicity effects.328 Similarly, we can expect to see differences in toxicity when varying the morphology for other 2D materials beyond graphene. Earlier on, we have seen a report comparing the toxicological effect of BP nanosheets and BP QDs. It would be interesting to extend this scope to other BP morphologies as well (i.e. nanotubes, nanoribbons).
While different morphologies may be more associated with mono-elemental 2D materials (i.e. graphene and BP), TMDs possess different polymorphs (e.g. metallic 1T and semi-conducting 2H and 3R phases).7 It would be interesting to study the implications of different phases on the toxicological behavior of TMDs.
In studying the toxicity of 2D materials, it is important to recognize the complexity of the field with many different parameters that come in play (Table 7). These factors arise through a diverse variety of synthetic routes possible for 2D materials. With many exciting advances in computational toxicity studies,329–332 further expansion into composite 2D materials (graphene/MoS2) for improved biocompatibility,333 and emerging pnictogens (i.e. antimonene) gaining visibility in nanomaterials research, we can expect this field of nanotoxicology to continue developing rapidly in the future.
| Factors | 2D material | Cell line tested | Cell viability assessment method | Finding reported | Ref. |
|---|---|---|---|---|---|
| Surface functionalization | MoS2 functionalized with thiobarbituric acid (TBA) | A549 | MTT and WST-8 assays | Functionalization with TBA can lower the toxicity of MoS2 by about 20% at 100 μg mL−1 concentration tested | 287 |
| BP QDs functionalized with titanium sulfonate ligand (TiL4) | J774A.1, and raw 264.7 | ATP assay | Functionalized BP QDs showed a reduced toxic effect for raw 264.7 cells | 288 | |
| Size effects | BP nanosheets of different size and thickness | NIH 3T3, HCoEpiC, and 293T | Real-time cell analysis (RTCA) | Larger BP nanosheets have higher toxicity than smaller ones as they can cause more severe damage to cell membranes. Different cell lines show different sensitivity | 304 |
| Protein corona effect | BP nanosheets and BP QDs | H1299, L0-2, 293T, dTHP-1 and SC | MTT assay | Presence of protein corona surrounding nanomaterials can significantly alter the size and surface charge of 2D materials. The type and amount of protein in the corona is influenced by the size of the material. Cytotoxicity effect appears to be reduced with the formation of protein corona, even though its presence can facilitate the cell uptake of BP nanomaterials | 313 |
| Chemical composition | Vanadium dichalcogenides (VS2, VSe2, VTe2) | A549 | MTT and WST-8 assays | Toxicity of vanadium dichalcogenides appears to increase down the chalcogen group (VTe2 > VSe2 > VS2) | 315 |
| Group 5 ditellurides, namely VTe2, NbTe2 and TaTe2 | A549 | WST-8 assay | Toxicity of TMDs decreases down Group 5 metals (i.e. VTe2 > NbTe2 ∼ TaTe2) | 316 | |
| Platinum dichalcogenides | A549 | MTT and WST-8 assays | Toxicity of Pt dichalcogenides increases down the chalcogen group (i.e. PtTe2 > PtSe2 > PtS2) | 317 | |
| Chemical dissolution | MoS2 | TIB-67, HTB-177 | WST-8 assay | MoS2 nanosheets are susceptible to oxidation under ambient conditions in aqueous media to produce Mo and S species. The process is pH dependent and is slower for pure 2H phase than mixed 1T and 2H phases | 318 |
| Impurities | GO with and without Mn impurities | PMØ, J774A.1, LLC, MCF-7, HepG2, HUVEC | WST-8 assay | Presence of Mn ions in GO is found to increase its cellular toxicity | 324 |
| Morphology | SWNTs, MWNTs and fullerenes | Alveolar macrophage (AM) | MTT assay | Cytotoxicity was observed to follow the order similar to that of mass basis: SWNTs > MWNT > C60 |
328 |
7. Conclusions
Two-dimensional materials beyond graphene have taken off dramatically in the past few years. These materials offer new opportunities in the whole plethora of biomedical and environmental monitoring applications, from bio/sensors to drug delivery devices. The discoveries of new 2D materials, such as phosphorene, germanene or silicene, bring new opportunities since many of these materials are biocompatible and/or biodegradable. Moreover, the signature physical, chemical and optical properties of 2D materials demonstrate improvements in the capabilities of the developed devices and biomedical applications discussed. These properties included phase engineering, metallicity, electrical conductivity, degree of exfoliation, surface functionalization, strong photoluminescence, strong optical absorption in the NIR range and high photothermal conversion efficiency. There is still a lot of space for discovering new 2D materials; therefore there will be even more opportunities for their application in biomedical and biochemical sciences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. P. acknowledges the financial support of Grant Agency of the Czech Republic (EXPRO: 19-26896X).References
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS
.
- P. Miró, M. Audiffred and T. Heine, Chem. Soc. Rev., 2014, 43, 6537–6554 RSC
- X. Chia and M. Pumera, Nat. Catal., 2018, 1, 909–921 CrossRef
- S. Zhai, L. Wei, H. E. Karahan, X. Chen, C. Wang, X. Zhang, J. Chen, X. Wang and Y. Chen, Energy Storage Mater., 2019, 19, 102–123 CrossRef
- J. H. Han, M. Kwak, Y. Kim and J. Cheon, Chem. Rev., 2018, 118, 6151–6188 CrossRef CAS
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Bøggild, S. Borini, F. H. L. Koppens, V. Palermo, N. Pugno, J. A. Garrido, R. Sordan, A. Bianco, L. Ballerini, M. Prato, E. Lidorikis, J. Kivioja, C. Marinelli, T. Ryhänen, A. Morpurgo, J. N. Coleman, V. Nicolosi, L. Colombo, A. Fert, M. Garcia-Hernandez, A. Bachtold, G. F. Schneider, F. Guinea, C. Dekker, M. Barbone, Z. Sun, C. Galiotis, A. N. Grigorenko, G. Konstantatos, A. Kis, M. Katsnelson, L. Vandersypen, A. Loiseau, V. Morandi, D. Neumaier, E. Treossi, V. Pellegrini, M. Polini, A. Tredicucci, G. M. Williams, B. H. Hong, J.-H. Ahn, J. M. Kim, H. Zirath, B. J. V. Wees, H. V. D. Zant, L. Occhipinti, A. D. Matteo, I. A. Kinloch, T. Seyller, E. Quesnel, X. Feng, K. Teo, N. Rupesinghe, P. Hakonen, S. R. T. Neil, Q. Tannock, T. Löfwander and J. Kinaret, Nanoscale, 2015, 7, 4598–4810 RSC
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef
- X. Chia and M. Pumera, Chem. Soc. Rev., 2018, 47, 5602–5613 RSC
- K. Zhang, Y. Feng, F. Wang, Z. Yang and J. Wang, J. Mater. Chem. C, 2017, 5, 11992–12022 RSC
- M. Yankowitz, Q. Ma, P. Jarillo-Herrero and B. J. Leroy, Nat. Rev. Phys., 2019, 1, 112–125 CrossRef CAS
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS
- M. Pumera and Z. Sofer, Adv. Mater., 2017, 29, 1605299 CrossRef
- D. Akinwande, N. Petrone and J. Hone, Nat. Commun., 2014, 5, 5678 CrossRef CAS
- V. Lloret, M. Á. Rivero-Crespo, J. A. Vidal-Moya, S. Wild, A. Doménech-Carbó, B. S. J. Heller, S. Shin, H.-P. Steinrück, F. Maier, F. Hauke, M. Varela, A. Hirsch, A. Leyva-Pérez and G. Abellán, Nat. Commun., 2019, 10, 509 CrossRef
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS
- J. L. Hart, K. Hantanasirisakul, A. C. Lang, B. Anasori, D. Pinto, Y. Pivak, J. T. V. Omme, S. J. May, Y. Gogotsi and M. L. Taheri, Nat. Commun., 2019, 10, 522 CrossRef CAS
- F. Wang, T. A. Shifa, P. Yu, P. He, Y. Liu, F. Wang, Z. Wang, X. Zhan, X. Lou, F. Xia and J. He, Adv. Funct. Mater., 2018, 28, 1802151 CrossRef
- C. C. Mayorga-Martinez, Z. Sofer, D. Sedmidubský, Š. Huber, A. Y. S. Eng and M. Pumera, ACS Appl. Mater. Interfaces, 2017, 9, 12563–12573 CrossRef CAS
- R. Gusmão, Z. Sofer and M. Pumera, Angew. Chem. Int. Ed., 2019, 58, 9326–9337 CrossRef
- K. Huang, Z. Li, J. Lin, G. Han and P. Huang, Chem. Soc. Rev., 2018, 47, 5109–5124 RSC
- I. A. Gorodetskaya and A. A. Gorodetsky, Nat. Chem., 2015, 7, 541–542 CrossRef CAS
- L. Wang, Q. Xiong, F. Xiao and H. Duan, Biosens. Bioelectron., 2017, 89, 136–151 CrossRef CAS
- R. Monošík, M. Stredanský and E. Šturdík, J. Clin. Lab. Anal., 2012, 26, 22–34 CrossRef
- Z. Meng, R. M. Stolz, L. Mendecki and K. A. Mirica, Chem. Rev., 2019, 119, 478–598 CrossRef CAS
- A. Bonanni and M. D. Valle, Anal. Chim. Acta, 2010, 678, 7–17 CrossRef CAS
- Y. Zhang and Q. Wei, J. Electroanal. Chem., 2016, 781, 401–409 CrossRef CAS
- A. Zhang and C. M. Lieber, Chem. Rev., 2015, 116, 215–257 CrossRef
- T. Wang, H. Zhu, J. Zhuo, Z. Zhu, P. Papakonstantinou, G. Lubarsky, J. Lin and M. Li, Anal. Chem., 2013, 85, 10289–10295 CrossRef CAS
- Z. Li and S. L. Wong, Mater. Sci. Eng., C, 2017, 70, 1095–1106 CrossRef CAS
- W. Z. Teo, E. L. K. Chng, Z. Sofer and M. Pumera, Chem. – Eur. J., 2014, 20, 9627–9632 CrossRef CAS
- L. C. Clark and C. Lyons, Ann. N. Y. Acad. Sci., 2006, 102, 29–45 CrossRef
- J. N. Coleman, M. Lotya, A. Oneill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. Mccomb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS
- A. M. Parra-Alfambra, E. Casero, L. Vázquez, C. Quintana, M. D. Pozo and M. D. Petit-Domínguez, Sens. Actuators, B, 2018, 274, 310–317 CrossRef CAS
- R. J. Toh, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, Adv. Funct. Mater., 2017, 27, 1604923 CrossRef
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS
- K. Chang, X. Hai, H. Pang, H. Zhang, L. Shi, G. Liu, H. Liu, G. Zhao, M. Li and J. Ye, Adv. Mater., 2016, 28, 10033–10041 CrossRef CAS
- M. Z. M. Nasir, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, ACS Nano, 2017, 11, 5774–5784 CrossRef CAS
- A. Ambrosi, Z. Sofer and M. Pumera, Chem. Commun., 2015, 51, 8450–8453 RSC
- N. Rohaizad, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, ACS Appl. Mater. Interfaces, 2017, 9, 40697–40706 CrossRef CAS
- C. C. Mayorga-Martinez, A. Ambrosi, A. Y. S. Eng, Z. Sofer and M. Pumera, Electrochem. Commun., 2015, 56, 24–28 CrossRef CAS
- G.-X. Wang, W.-J. Bao, J. Wang, Q.-Q. Lu and X.-H. Xia, Electrochem. Commun., 2013, 35, 146–148 CrossRef
- H. Liu, X. Su, C. Duan, X. Dong and Z. Zhu, Mater. Lett., 2014, 122, 182–185 CrossRef CAS
- O. Parlak, A. Incel, L. Uzun, A. P. Turner and A. Tiwari, Biosens. Bioelectron., 2017, 89, 545–550 CrossRef CAS
- X. Lin, Y. Ni and S. Kokot, Sens. Actuators, B, 2016, 233, 100–106 CrossRef CAS
- S. Su, H. Sun, F. Xu, L. Yuwen, C. Fan and L. Wang, Microchim. Acta, 2014, 181, 1497–1503 CrossRef CAS
- J. Chao, M. Zou, C. Zhang, H. Sun, D. Pan, H. Pei, S. Su, L. Yuwen, C. Fan and L. Wang, Nanotechnology, 2015, 26, 274005 CrossRef
- Y. Shu, J. Chen, Q. Xu, Z. Wei, F. Liu, R. Lu, S. Xu and X. Hu, J. Mater. Chem. B, 2017, 5, 1446–1453 RSC
- I. Vasilescu, S. A. Eremia, M. Kusko, A. Radoi, E. Vasile and G.-L. Radu, Biosens. Bioelectron., 2016, 75, 232–237 CrossRef CAS
- D. Song, Q. Li, X. Lu, Y. Li, Y. Li, Y. Wang and F. Gao, J. Hazard. Mater., 2018, 357, 466–474 CrossRef CAS
- H. Song, Y. Ni and S. Kokot, Biosens. Bioelectron., 2014, 56, 137–143 CrossRef CAS
- S. Wu, Z. Zeng, Q. He, Z. Wang, S. J. Wang, Y. Du, Z. Yin, X. Sun, W. Chen and H. Zhang, Small, 2012, 8, 2264–2270 CrossRef CAS
- N. Rohaizad, C. C. Mayorga-Martinez, Z. Sofer, R. D. Webster and M. Pumera, Biosens. Bioelectron., 2020, 155, 112114 CrossRef CAS
- P. W. Bridgman, J. Am. Chem. Soc., 1914, 36, 1344–1363 CrossRef CAS
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS
- A. Favron, E. Gaufrès, F. Fossard, A.-L. Phaneuf-L’Heureux, N. Y.-W. Tang, P. L. Lévesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS
- R. Hultgren, N. S. Gingrich and B. E. Warren, J. Chem. Phys., 1935, 3, 351–355 CrossRef CAS
- A. Brown and S. Rundqvist, Acta Crystallogr., 1965, 19, 684–685 CrossRef CAS
- D.-K. Seo and R. Hoffmann, J. Solid State Chem., 1999, 147, 26–37 CrossRef CAS
- L. Kou, C. Chen and S. C. Smith, J. Phys. Chem. Lett., 2015, 6, 2794–2805 CrossRef CAS
- Y. Huang, J. Qiao, K. He, S. Bliznakov, E. Sutter, X. Chen, D. Luo, F. Meng, D. Su, J. Decker, W. Ji, R. S. Ruoff and P. Sutter, Chem. Mater., 2016, 28, 8330–8339 CrossRef CAS
- T. Zhang, Y. Wan, H. Xie, Y. Mu, P. Du, D. Wang, X. Wu, H. Ji and L. Wan, J. Am. Chem. Soc., 2018, 140, 7561–7567 CrossRef CAS
- Y. Zhao, Y.-H. Zhang, Z. Zhuge, Y.-H. Tang, J.-W. Tao and Y. Chen, Anal. Chem., 2018, 90, 3149–3155 CrossRef CAS
- C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, ACS Nano, 2019, 13, 13217–13224 CrossRef CAS
- C. C. Mayorga-Martinez, R. Gusmão, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2018, 58, 134–138 CrossRef
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS
- F. Wang, C. Yang, C. Duan, D. Xiao, Y. Tang and J. Zhu, J. Electrochem. Soc., 2015, 162, B16–B21 CrossRef CAS
- H. Liu, C. Duan, C. Yang, W. Shen, F. Wang and Z. Zhu, Sens. Actuators, B, 2015, 218, 60–66 CrossRef CAS
- L. Wu, X. Lu, Dhanjai, Z.-S. Wu, Y. Dong, X. Wang, S. Zheng and J. Chen, Biosens. Bioelectron., 2018, 107, 69–75 CrossRef CAS
- H. L. Chia, C. C. Mayorga-Martinez, N. Antonatos, Z. Sofer, J. J. Gonzalez-Julian, R. D. Webster and M. Pumera, Anal. Chem., 2020, 92, 2452–2459 CrossRef CAS
- R. B. Rakhi, P. Nayak, C. Xia and H. N. Alshareef, Sci. Rep., 2016, 6, 36422 CrossRef CAS
- Y. Jiang, X. Zhang, L. Pei, S. Yue, L. Ma, L. Zhou, Z. Huang, Y. He and J. Gao, Chem. Eng. J., 2018, 339, 547–556 CrossRef CAS
- F. Wang, C. Yang, M. Duan, Y. Tang and J. Zhu, Biosens. Bioelectron., 2015, 74, 1022–1028 CrossRef CAS
- H. Gu, Y. Xing, P. Xiong, H. Tang, C. Li, S. Chen, R. Zeng, K. Han and G. Shi, ACS Appl. Nano Mater., 2019, 2, 6537–6545 CrossRef CAS
- D. Song, X. Jiang, Y. Li, X. Lu, S. Luan, Y. Wang, Y. Li and F. Gao, J. Hazard. Mater., 2019, 373, 367–376 CrossRef CAS
- Y. Lei, W. Zhao, Y. Zhang, Q. Jiang, J. H. He, A. J. Baeumner, O. S. Wolfbeis, Z. L. Wang, K. N. Salama and H. N. Alshareef, Small, 2019, 15, 1901190 CrossRef
- J. Zheng, J. Diao, Y. Jin, A. Ding, B. Wang, L. Wu, B. Weng and J. Chen, J. Electrochem. Soc., 2018, 165, B227–B231 CrossRef CAS
- H. Wang, H. Li, Y. Huang, M. Xiong, F. Wang and C. Li, Biosens. Bioelectron., 2019, 142, 111531 CrossRef CAS
- X. Cao, Microchim. Acta, 2014, 181, 1133–1141 CrossRef CAS
- K.-J. Huang, Y.-J. Liu, H.-B. Wang, T. Gan, Y.-M. Liu and L.-L. Wang, Sens. Actuators, B, 2014, 191, 828–836 CrossRef CAS
- K.-J. Huang, Y.-J. Liu and Q.-F. Zhai, J. Mater. Chem. B, 2015, 3, 8180–8187 RSC
- K.-J. Huang, H.-L. Shuai and J.-Z. Zhang, Biosens. Bioelectron., 2016, 77, 69–75 CrossRef CAS
- K.-J. Huang, Y.-J. Liu, H.-B. Wang, Y.-Y. Wang and Y.-M. Liu, Biosens. Bioelectron., 2014, 55, 195–202 CrossRef CAS
- X. Liu, H.-L. Shuai, Y.-J. Liu and K.-J. Huang, Sens. Actuators, B, 2016, 235, 603–613 CrossRef CAS
- H.-L. Shuai, K.-J. Huang and Y.-X. Chen, J. Mater. Chem. B, 2016, 4, 1186–1196 RSC
- T. Wang, R. Zhu, J. Zhuo, Z. Zhu, Y. Shao and M. Li, Anal. Chem., 2014, 86, 12064–12069 CrossRef CAS
- D. Zhu, W. Liu, D. Zhao, Q. Hao, J. Li, J. Huang, J. Shi, J. Chao, S. Su and L. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 35597–35603 CrossRef CAS
- T. Yang, M. Chen, Q. Kong, X. Luo and K. Jiao, Biosens. Bioelectron., 2017, 89, 538–544 CrossRef CAS
- V. Kumar, J. R. Brent, M. Shorie, H. Kaur, G. Chadha, A. G. Thomas, E. A. Lewis, A. P. Rooney, L. Nguyen, X. L. Zhong, M. G. Burke, S. J. Haigh, A. Walton, P. D. Mcnaughter, A. A. Tedstone, N. Savjani, C. A. Muryn, P. O’Brien, A. K. Ganguli, D. J. Lewis and P. Sabherwal, ACS Appl. Mater. Interfaces, 2016, 8, 22860–22868 CrossRef CAS
- C. Zhu, Z. Zeng, H. Li, F. Li, C. Fan and H. Zhang, J. Am. Chem. Soc., 2013, 135, 5998–6001 CrossRef CAS
- Y. Yuan, R. Li and Z. Liu, Anal. Chem., 2014, 86, 3610–3615 CrossRef CAS
- X. Wang, F. Nan, J. Zhao, T. Yang, T. Ge and K. Jiao, Biosens. Bioelectron., 2015, 64, 386–391 CrossRef CAS
- Y. Chu, B. Cai, Y. Ma, M. Zhao, Z. Ye and J. Huang, RSC Adv., 2016, 6, 22673–22678 RSC
- A. Li, J. Zhang, J. Qiu, Z. Zhao, C. Wang, C. Zhao and H. Liu, Talanta, 2017, 163, 78–84 CrossRef CAS
- A. H. Loo, A. Bonanni, A. Ambrosi and M. Pumera, Nanoscale, 2014, 6, 11971–11975 RSC
- A. H. Loo, A. Bonanni, Z. Sofer and M. Pumera, ChemPhysChem, 2015, 16, 2304–2306 CrossRef CAS
- M. Sadeghi, M. Jahanshahi, M. Ghorbanzadeh and G. Najafpour, Appl. Surf. Sci., 2018, 434, 176–187 CrossRef CAS
- M. Kukkar, A. Sharma, P. Kumar, K.-H. Kim and A. Deep, Anal. Chim. Acta, 2016, 939, 101–107 CrossRef CAS
- S. K. Tuteja and S. Neethirajan, Nanotechnology, 2018, 29, 135101 CrossRef
- S. Kumar, Y. Lei, N. H. Alshareef, M. Quevedo-Lopez and K. N. Salama, Biosens. Bioelectron., 2018, 121, 243–249 CrossRef CAS
- X. Wang, C. Chu, L. Shen, W. Deng, M. Yan, S. Ge, J. Yu and X. Song, Sens. Actuators, B, 2015, 206, 30–36 CrossRef CAS
- S. Su, M. Zou, H. Zhao, C. Yuan, Y. Xu, C. Zhang, L. Wang, C. Fan and L. Wang, Nanoscale, 2015, 7, 19129–19135 RSC
- S. Su, X. Han, Z. Lu, W. Liu, D. Zhu, J. Chao, C. Fan, L. Wang, S. Song, L. Weng and L. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 12773–12781 CrossRef CAS
- H.-U. Kim, H. Y. Kim, A. Kulkarni, C. Ahn, Y. Jin, Y. Kim, K.-N. Lee, M.-H. Lee and T. Kim, Sci. Rep., 2016, 6, 34587 CrossRef CAS
- H.-U. Kim, H. Kim, C. Ahn, A. Kulkarni, M. Jeon, G. Y. Yeom, M.-H. Lee and T. Kim, RSC Adv., 2015, 5, 10134–10138 RSC
- C. C. Mayorga-Martinez, B. Khezri, A. Y. S. Eng, Z. Sofer, P. Ulbrich and M. Pumera, Adv. Funct. Mater., 2016, 26, 4094–4098 CrossRef CAS
- R. J. Toh, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, Anal. Chem., 2016, 88, 12204–12209 CrossRef CAS
- V. Mazánek, C. C. Mayorga-Martinez, D. Bouša, Z. Sofer and M. Pumera, Nanoscale, 2018, 10, 23149–23156 RSC
- D. Bouša, C. C. Mayorga-Martinez, V. Mazánek, Z. Sofer, K. Boušová and M. Pumera, ACS Appl. Mater. Interfaces, 2018, 10, 16861–16866 CrossRef
- C. C. Mayorga-Martinez, N. M. Latiff, A. Y. S. Eng, Z. Sofer and M. Pumera, Anal. Chem., 2016, 88, 10074–10079 CrossRef CAS
- J. Tian, Q. Liu, C. Ge, Z. Xing, A. M. Asiri, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 8921–8924 RSC
- Z. Zhuge, Y. H. Tang, J. W. Tao and Y. Zhao, ChemElectroChem, 2019, 6, 1129–1133 CrossRef CAS
- L. Zhou, X. Zhang, L. Ma, J. Gao and Y. Jiang, Biochem. Eng. J., 2017, 128, 243–249 CrossRef CAS
- E. Rahmanian, C. C. Mayorga-Martinez, R. Malekfar, J. Luxa, Z. Sofer and M. Pumera, ACS Appl. Nano Mater., 2018, 1, 7006–7015 CrossRef CAS
- D. Song, Y. Wang, X. Lu, Y. Gao, Y. Li and F. Gao, Sens. Actuators, B, 2018, 267, 5–13 CrossRef CAS
- F. Zhao, Y. Yao, X. Li, L. Lan, C. Jiang and J. Ping, Anal. Chem., 2018, 90, 11658–11664 CrossRef CAS
- S. Liang, L. Wu, H. Liu, J. Li, M. Chen and M. Zhang, Biosens. Bioelectron., 2019, 126, 30–35 CrossRef CAS
- K.-J. Huang, Y.-J. Liu, G.-W. Shi, X.-R. Yang and Y.-M. Liu, Sens. Actuators, B, 2014, 201, 579–585 CrossRef CAS
- L.-X. Fang, J.-T. Cao and K.-J. Huang, J. Electroanal. Chem., 2015, 746, 1–8 CrossRef CAS
- S. Su, H. Sun, W. Cao, J. Chao, H. Peng, X. Zuo, L. Yuwen, C. Fan and L. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 6826–6833 CrossRef CAS
- L. Yang and Y. Li, Anal. Methods, 2016, 8, 5234–5241 RSC
- H.-L. Shuai, X. Wu, K.-J. Huang and Z.-B. Zhai, Biosens. Bioelectron., 2017, 94, 616–625 CrossRef CAS
- K.-J. Huang, Y.-J. Liu, J.-T. Cao and H.-B. Wang, RSC Adv., 2014, 4, 36742 RSC
- H.-L. Shuai, K.-J. Huang, Y.-X. Chen, L.-X. Fang and M.-P. Jia, Biosens. Bioelectron., 2017, 89, 989–997 CrossRef CAS
- K.-J. Huang, H.-L. Shuai and Y.-X. Chen, Sens. Actuators, B, 2016, 225, 391–397 CrossRef CAS
- L.-X. Fang, K.-J. Huang and Y. Liu, Biosens. Bioelectron., 2015, 71, 171–178 CrossRef CAS
- A. Wang, C. Wang, L. Fu, W. Wong-Ng and Y. Lan, Nano-Micro Lett., 2017, 9, 47 CrossRef
- A. Manikandan, Y.-Z. Chen, C.-C. Shen, C.-W. Sher, H.-C. Kuo and Y.-L. Chueh, Prog. Quantum Electron., 2019, 68, 100226 CrossRef
- J. Tian, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2013, 85, 5595–5599 CrossRef CAS
- N. Cheng, P. Jiang, Q. Liu, J. Tian, A. M. Asiri and X. Sun, Analyst, 2014, 139, 5065–5068 RSC
- X. Hun, S. Wang, S. Wang, J. Zhao and X. Luo, Sens. Actuators, B, 2017, 249, 83–89 CrossRef
- H. Chen, Z. Li, X. Liu, J. Zhong, T. Lin, L. Guo and F. Fu, Spectrochim. Acta, Part A, 2017, 185, 271–275 CrossRef CAS
- J. Peng and J. Weng, Biosens. Bioelectron., 2017, 89, 652–658 CrossRef CAS
- T. Lin, L. Zhong, Z. Song, L. Guo, H. Wu, Q. Guo, Y. Chen, F. Fu and G. Chen, Biosens. Bioelectron., 2014, 62, 302–307 CrossRef CAS
- F. Qiao, Q. Qi, Z. Wang, K. Xu and S. Ai, Sens. Actuators, B, 2016, 229, 379–386 CrossRef CAS
- J. Tian, Q. Liu, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 11604 RSC
- T. Lin, L. Zhong, L. Guo, F. Fu and G. Chen, Nanoscale, 2014, 6, 11856–11862 RSC
- Vinita, N. R. Nirala and R. Prakash, Sens. Actuators, B, 2018, 263, 109–119 CrossRef CAS
- T. Lin, L. Zhong, J. Wang, L. Guo, H. Wu, Q. Guo, F. Fu and G. Chen, Biosens. Bioelectron., 2014, 59, 89–93 CrossRef CAS
- F. Qiao, J. Wang, S. Ai and L. Li, Sens. Actuators, B, 2015, 216, 418–427 CrossRef CAS
- N. R. Nirala, S. Pandey, A. Bansal, V. K. Singh, B. Mukherjee, P. S. Saxena and A. Srivastava, Biosens. Bioelectron., 2015, 74, 207–213 CrossRef CAS
- X. Wang, Q. Wu, K. Jiang, C. Wang and C. Zhang, Sens. Actuators, B, 2017, 252, 183–190 CrossRef CAS
- C. Zhu, Z. Zeng, H. Li, F. Li, C. Fan and H. Zhang, J. Am. Chem. Soc., 2013, 135, 5998–6001 CrossRef CAS
- J. Ge, L.-J. Tang, Q. Xi, X.-P. Li, R.-Q. Yu, J.-H. Jiang and X. Chu, Nanoscale, 2014, 6, 6866–6872 RSC
- A. H. Loo, A. Bonanni and M. Pumera, Analyst, 2016, 141, 4654–4658 RSC
- R.-M. Kong, L. Ding, Z. Wang, J. You and F. Qu, Anal. Bioanal. Chem., 2014, 407, 369–377 CrossRef
- H. Deng, X. Yang and Z. Gao, Analyst, 2015, 140, 3210–3215 RSC
- J. Huang, L. Ye, X. Gao, H. Li, J. Xu and Z. Li, J. Mater. Chem. B, 2015, 3, 2395–2401 RSC
- Y. Huang, Y. Shi, H. Y. Yang and Y. Ai, Nanoscale, 2015, 7, 2245–2249 RSC
- Q. Wang, W. Wang, J. Lei, N. Xu, F. Gao and H. Ju, Anal. Chem., 2013, 85, 12182–12188 CrossRef CAS
- X. Liao, Q. Wang and H. Ju, Chem. Commun., 2014, 50, 13604–13607 RSC
- Y. He, J. Li and Y. Liu, Anal. Chem., 2015, 87, 9777–9785 CrossRef CAS
- Y. Liu, K. Yan and J. Zhang, ACS Appl. Mater. Interfaces, 2015, 8, 28255–28264 CrossRef
- L. Chen, X. Zeng, P. Si, Y. Chen, Y. Chi, D.-H. Kim and G. Chen, Anal. Chem., 2014, 86, 4188–4195 CrossRef CAS
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS
- Z. Cai, B. Liu, X. Zou and H.-M. Cheng, Chem. Rev., 2018, 118, 6091–6133 CrossRef CAS
- K. S. Novoselov, A. Mishchenko, A. Carvalho and A. H. C. Neto, Science, 2016, 353, aac9439 CrossRef CAS
- P. T. K. Loan, W. Zhang, C.-T. Lin, K.-H. Wei, L.-J. Li and C.-H. Chen, Adv. Mater., 2014, 26, 4838–4844 CrossRef CAS
- J. M. Buriak, C. W. Jones, P. V. Kamat, K. S. Schanze, G. C. Schatz, G. D. Scholes and P. S. Weiss, Chem. Mater., 2016, 28, 3525–3526 CrossRef CAS
- D. Voiry, M. Chhowalla, Y. Gogotsi, N. A. Kotov, Y. Li, R. M. Penner, R. E. Schaak and P. S. Weiss, ACS Nano, 2018, 12, 9635–9638 CrossRef CAS
- P. T. Kissinger, Biosens. Bioelectron., 2005, 20, 2512–2516 CrossRef CAS
- R. Li, Y. Liu, L. Cheng, C. Yang and J. Zhang, Anal. Chem., 2014, 86, 9372–9375 CrossRef CAS
- M. Zhang, F. Jia, M. Dai and S. Song, Appl. Surf. Sci., 2018, 455, 258–266 CrossRef CAS
- K. Ai, C. Ruan, M. Shen and L. Lu, Adv. Funct. Mater., 2016, 26, 5542–5549 CrossRef CAS
- J. Wang, W. Zhang, X. Yue, Q. Yang, F. Liu, Y. Wang, D. Zhang, Z. Li and J. Wang, J. Mater. Chem. A, 2016, 4, 3893–3900 RSC
- F. Jia, Q. Wang, J. Wu, Y. Li and S. Song, ACS Sustainable Chem. Eng., 2017, 5, 7410–7419 CrossRef CAS
- C. Liu, F. Jia, Q. Wang, B. Yang and S. Song, Appl. Mater. Today, 2017, 9, 220–228 CrossRef
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef CAS
- X. Zhu, B. Liu, H. Hou, Z. Huang, K. M. Zeinu, L. Huang, X. Yuan, D. Guo, J. Hu and J. Yang, Electrochim. Acta, 2017, 248, 46–57 CrossRef CAS
- R. Aswathi and K. Y. Sandhya, J. Mater. Chem. A, 2018, 6, 14602–14613 RSC
- M. Sadhukhan and S. Barman, J. Mater. Chem. A, 2013, 1, 2752 RSC
- T. Yang, R. Yu, H. Chen, R. Yang, S. Wang, X. Luo and K. Jiao, J. Electroanal. Chem., 2016, 781, 70–75 CrossRef CAS
- R. Sakthivel, Int. J. Electrochem. Sci., 2017, 9288–9300 CrossRef CAS
- A. Yin, X. Wei, Y. Cao and H. Li, Appl. Surf. Sci., 2016, 385, 63–71 CrossRef CAS
- K.-J. Huang, J.-Z. Zhang, Y.-J. Liu and L.-L. Wang, Sens. Actuators, B, 2014, 194, 303–310 CrossRef CAS
- H. Y. Yue, P. F. Wu, S. Huang, X. Gao, S. S. Song, W. Q. Wang, H. J. Zhang and X. R. Guo, J. Electroanal. Chem., 2019, 833, 427–432 CrossRef CAS
- R. Gusmão, Z. Sofer, D. Bouša and M. Pumera, Angew. Chem., Int. Ed., 2017, 129, 14609–14614 CrossRef
- L. Lorencova, T. Bertok, E. Dosekova, A. Holazova, D. Paprckova, A. Vikartovska, V. Sasinkova, J. Filip, P. Kasak, M. Jerigova, D. Velic, K. A. Mahmoud and J. Tkac, Electrochim. Acta, 2017, 235, 471–479 CrossRef CAS
- R. J. Toh, C. C. Mayorga-Martinez, J. Han, Z. Sofer and M. Pumera, Anal. Chem., 2017, 89, 4978–4985 CrossRef CAS
- N. Rohaizad, C. C. Mayorga-Martinez, Z. Sofer, R. D. Webster and M. Pumera, Appl. Mater. Today, 2020, 19, 100606 CrossRef
- X. Liu, J. Zhang, J. Di, Y. Long, W. Li and Y. Tu, J. Colloid Sci., 2017, 505, 964–972 CrossRef CAS
- E. Z. Lee, Y.-S. Jun, W. H. Hong, A. Thomas and M. M. Jin, Angew. Chem., Int. Ed., 2010, 49, 9706–9710 CrossRef CAS
- J. Tian, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2013, 85, 5595–5599 CrossRef CAS
- N. Cheng, P. Jiang, Q. Liu, J. Tian, A. M. Asiri and X. Sun, Analyst, 2014, 139, 5065–5068 RSC
- H. Huang, R. Chen, J. Ma, L. Yan, Y. Zhao, Y. Wang, W. Zhang, J. Fan and X. Chen, Chem. Commun., 2014, 50, 15415–15418 RSC
- X. Guo, G. Yue, J. Huang, C. Liu, Q. Zeng and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 26118–26127 CrossRef CAS
- M. Rong, L. Lin, X. Song, Y. Wang, Y. Zhong, J. Yan, Y. Feng, X. Zeng and X. Chen, Biosens. Bioelectron., 2015, 68, 210–217 CrossRef CAS
- Y. Wang and Y. Ni, Anal. Chem., 2014, 86, 7463–7470 CrossRef CAS
- F. K. Perkins, A. L. Friedman, E. Cobas, P. M. Campbell, G. G. Jernigan and B. T. Jonker, Nano Lett., 2013, 13, 668–673 CrossRef CAS
- C. Yim, K. Lee, N. Mcevoy, M. O’Brien, S. Riazimehr, N. C. Berner, C. P. Cullen, J. Kotakoski, J. C. Meyer, M. C. Lemme and G. S. Duesberg, ACS Nano, 2016, 10, 9550–9558 CrossRef CAS
- D. J. Late, Y.-K. Huang, B. Liu, J. Acharya, S. N. Shirodkar, J. Luo, A. Yan, D. Charles, U. V. Waghmare, V. P. Dravid and C. N. R. Rao, ACS Nano, 2013, 7, 4879–4891 CrossRef CAS
- Z. Feng, Y. Xie, J. Chen, Y. Yu, S. Zheng, R. Zhang, Q. Li, X. Chen, C. Sun, H. Zhang, W. Pang, J. Liu and D. Zhang, 2D Mater., 2017, 4, 025018 CrossRef
- B. Liu, L. Chen, G. Liu, A. N. Abbas, M. Fathi and C. Zhou, ACS Nano, 2014, 8, 5304–5314 CrossRef CAS
- S.-Y. Cho, S. J. Kim, Y. Lee, J.-S. Kim, W.-B. Jung, H.-W. Yoo, J. Kim and H.-T. Jung, ACS Nano, 2015, 9, 9314–9321 CrossRef
- H. Li, Z. Yin, Q. He, H. Li, X. Huang, G. Lu, D. W. H. Fam, A. I. Y. Tok, Q. Zhang and H. Zhang, Small, 2011, 8, 63–67 CrossRef
- Z. Feng, Y. Xie, E. Wu, Y. Yu, S. Zheng, R. Zhang, X. Chen, C. Sun, H. Zhang, W. Pang, J. Liu and D. Zhang, Micromachines, 2017, 8, 155 CrossRef
- K. Xu, N. Li, D. Zeng, S. Tian, S. Zhang, D. Hu and C. Xie, ACS Appl. Mater. Interfaces, 2015, 7, 11359–11368 CrossRef CAS
- A. N. Abbas, B. Liu, L. Chen, Y. Ma, S. Cong, N. Aroonyadet, M. Köpf, T. Nilges and C. Zhou, ACS Nano, 2015, 9, 5618–5624 CrossRef CAS
- J.-S. Kim, H.-W. Yoo, H. O. Choi and H.-T. Jung, Nano Lett., 2014, 14, 5941–5947 CrossRef CAS
- C. C. Mayorga-Martinez, A. Ambrosi, A. Y. S. Eng, Z. Sofer and M. Pumera, Adv. Funct. Mater., 2015, 25, 5611–5616 CrossRef CAS
- M. Sajjad, G. Morell and P. Feng, ACS Appl. Mater. Interfaces, 2013, 5, 5051–5056 CrossRef CAS
- C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2015, 54, 14317–14320 CrossRef CAS
- S. J. Kim, H.-J. Koh, C. E. Ren, O. Kwon, K. Maleski, S.-Y. Cho, B. Anasori, C.-K. Kim, Y.-K. Choi, J. Kim, Y. Gogotsi and H.-T. Jung, ACS Nano, 2018, 12, 986–993 CrossRef CAS
- E. Lee, A. Vahidmohammadi, B. C. Prorok, Y. S. Yoon, M. Beidaghi and D.-J. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 37184–37190 CrossRef CAS
- W. Yuan, K. Yang, H. Peng, F. Li and F. Yin, J. Mater. Chem. A, 2018, 6, 18116–18124 RSC
- B. Cho, A. R. Kim, Y. Park, J. Yoon, Y.-J. Lee, S. Lee, T. J. Yoo, C. G. Kang, B. H. Lee, H. C. Ko, D.-H. Kim and M. G. Hahm, ACS Appl. Mater. Interfaces, 2015, 7, 2952–2959 CrossRef CAS
- B. Cho, M. G. Hahm, M. Choi, J. Yoon, A. R. Kim, Y.-J. Lee, S.-G. Park, J.-D. Kwon, C. S. Kim, M. Song, Y. Jeong, K.-S. Nam, S. Lee, T. J. Yoo, C. G. Kang, B. H. Lee, H. C. Ko, P. M. Ajayan and D.-H. Kim, Sci. Rep., 2015, 5, 8052 CrossRef CAS
- W.-J. Yan, D.-Y. Chen, H.-R. Fuh, Y.-L. Li, D. Zhang, H. Liu, G. Wu, L. Zhang, X. Ren, J. Cho, M. Choi, B. S. Chun, C. Ó. Coileáin, H.-J. Xu, Z. Wang, Z. Jiang, C.-R. Chang and H.-C. Wu, RSC Adv., 2019, 9, 626–635 RSC
- Q. He, Q. Ma, B. Chen, Z. Yin, Z. Zeng, S. Wu, X. Cao, X. Kong and H. Zhang, APL Mater., 2014, 2, 092506 CrossRef
- C. C. Mayorga-Martinez, Z. Sofer, J. Luxa, Š. Huber, D. Sedmidubský, P. Brázda, L. Palatinus, M. Mikulics, P. Lazar, R. Medlín and M. Pumera, ACS Nano, 2017, 12, 464–473 CrossRef
- D. Wang, W. Gu, Y. Zhang, Y. Hu, T. Zhang, X. Tao and W. Chen, RSC Adv., 2014, 4, 18003–18006 RSC
- L. M. Zambov, C. Popov, N. Abedinov, M. F. Plass, W. Kulisch, T. Gotszalk, P. Grabiec, I. W. Rangelow and R. Kassing, Adv. Mater., 2000, 12, 656–660 CrossRef CAS
- S. Lee, Sensors, 2008, 8, 1508–1518 CrossRef CAS
- L. Wang, Q. Xiong, F. Xiao and H. Duan, Biosens. Bioelectron., 2017, 89, 136–151 CrossRef CAS
- Y. Chen, L. Wang and J. Shi, Nano Today, 2016, 11, 292–308 CrossRef CAS
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS
- H. Chen, T. Liu, Z. Su, L. Shang and G. Wei, Nanoscale Horiz., 2018, 3, 74–89 RSC
- D. Chimene, D. L. Alge and A. K. Gaharwar, Adv. Mater., 2015, 27, 7261–7284 CrossRef CAS
- Z. Wang, W. Zhu, Y. Qiu, X. Yi, A. V. D. Bussche, A. Kane, H. Gao, K. Koski and R. Hurt, Chem. Soc. Rev., 2016, 45, 1750–1780 RSC
- W. Tao, X. Ji, X. Xu, M. A. Islam, Z. Li, S. Chen, P. E. Saw, H. Zhang, Z. Bharwani, Z. Guo, J. Shi and O. C. Farokhzad, Angew. Chem., Int. Ed., 2017, 56, 11896–11900 CrossRef CAS
- W. Li, P. Rong, K. Yang, P. Huang, K. Sun and X. Chen, Biomaterials, 2015, 45, 18–26 CrossRef CAS
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS
- V. P. Torchilin, AAPS J., 2007, 9, E128–E147 CrossRef CAS
-
A. Z. Wang, R. Langer and O. C. Farokhzad, Nanoparticle Delivery of Cancer Drugs, in Annual Review of Medicine, ed. C. T. Caskey, C. P. Austin and J. A. Hoxie, Annual Reviews, Palo Alto, 2012, vol. 63, pp. 185–198 Search PubMed
- S. Rivankar, J. Cancer Res. Ther., 2014, 10, 853–858 CrossRef
- X. Zhu, X. Ji, N. Kong, Y. Chen, M. Mahmoudi, X. Xu, L. Ding, W. Tao, T. Cai, Y. Li, T. Gan, A. Barrett, Z. Bharwani, H. Chen and O. C. Farokhzad, ACS Nano, 2018, 12, 2922–2938 CrossRef CAS
- S. Wang, Y. Chen, X. Li, W. Gao, L. Zhang, J. Liu, Y. Zheng, H. Chen and J. Shi, Adv. Mater., 2015, 27, 7117–7122 CrossRef CAS
- W. Yin, L. Yan, J. Yu, G. Tian, L. Zhou, X. Zheng, X. Zhang, Y. Yong, J. Li, Z. Gu and Y. Zhao, ACS Nano, 2014, 8, 6922–6933 CrossRef CAS
- J. Lee, J. Kim and W. J. Kim, Chem. Mater., 2016, 28, 6417–6424 CrossRef CAS
- C. Sweet, A. Pramanik, S. Jones and P. C. Ray, ACS Omega, 2017, 2, 1826–1835 CrossRef CAS
- W. Tao, X. Zhu, X. Yu, X. Zeng, Q. Xiao, X. Zhang, X. Ji, X. Wang, J. Shi, H. Zhang and L. Mei, Adv. Mater., 2017, 29, 1603276 CrossRef
- X. Zhang, Y. Dong, X. Zeng, X. Liang, X. Li, W. Tao, H. Chen, Y. Jiang, L. Mei and S.-S. Feng, Biomaterials, 2014, 35, 1932–1943 CrossRef CAS
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37–44 CrossRef CAS
- W. Chen, J. Ouyang, H. Liu, M. Chen, K. Zeng, J. Sheng, Z. Liu, Y. Han, L. Wang, J. Li, L. Deng, Y.-N. Liu and S. Guo, Adv. Mater., 2017, 29, 1603864 CrossRef
- M. Fojtů, X. Chia, Z. Sofer, M. Masařík and M. Pumera, Adv. Funct. Mater., 2017, 27, 1701955 CrossRef
- D. Yang, G. Yang, P. Yang, R. Lv, S. Gai, C. Li, F. He and J. Lin, Adv. Funct. Mater., 2017, 27, 1700371 CrossRef
- M. Qiu, D. Wang, W. Liang, L. Liu, Y. Zhang, X. Chen, D. K. Sang, C. Xing, Z. Li, B. Dong, F. Xing, D. Fan, S. Bao, H. Zhang and Y. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 501–506 CrossRef CAS
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS
- S. Wang, A. Riedinger, H. Li, C. Fu, H. Liu, L. Li, T. Liu, L. Tan, M. J. Barthel, G. Pugliese, F. D. Donato, M. S. D’Abbusco, X. Meng, L. Manna, H. Meng and T. Pellegrino, ACS Nano, 2015, 9, 1788–1800 CrossRef CAS
- Y. Chen, Y. Wu, B. Sun, S. Liu and H. Liu, Small, 2017, 13, 1603446 CrossRef
- E. S. Shibu, M. Hamada, N. Murase and V. Biju, J. Photochem. Photobiol., C, 2013, 15, 53–72 CrossRef CAS
- G. P. Raaphorst, D. P. Heller, A. Bussey and C. E. Ng, Int. J. Hyperthermia, 1994, 10, 263–270 CrossRef CAS
- C. Christophi, A. Winkworth, V. Muralihdaran and P. Evans, Surg. Oncol., 1998, 7, 83–90 CrossRef CAS
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS
- D. K. Roper, W. Ahn and M. Hoepfner, J. Phys. Chem. C, 2007, 111, 3636–3641 CrossRef CAS
- W. Feng, L. Chen, M. Qin, X. Zhou, Q. Zhang, Y. Miao, K. Qiu, Y. Zhang and C. He, Sci. Rep., 2015, 5, 17422 CrossRef CAS
- S. Wang, K. Li, Y. Chen, H. Chen, M. Ma, J. Feng, Q. Zhao and J. Shi, Biomaterials, 2015, 39, 206–217 CrossRef CAS
- T. Liu, Y. Chao, M. Gao, C. Liang, Q. Chen, G. Song, L. Cheng and Z. Liu, Nano Res., 2016, 9, 3003–3017 CrossRef CAS
- X.-Z. Cui, Z.-G. Zhou, Y. Yang, J. Wei, J. Wang, M.-W. Wang, H. Yang, Y.-J. Zhang and S.-P. Yang, Chin. Chem. Lett., 2015, 26, 749–754 CrossRef CAS
- L. Chen, Y. Feng, X. Zhou, Q. Zhang, W. Nie, W. Wang, Y. Zhang and C. He, ACS Appl. Mater. Interfaces, 2017, 9, 17347–17358 CrossRef CAS
- S. Wang, X. Li, Y. Chen, X. Cai, H. Yao, W. Gao, Y. Zheng, X. An, J. Shi and H. Chen, Adv. Mater., 2015, 27, 2775–2782 CrossRef CAS
- Q. Liu, C. Sun, Q. He, A. Khalil, T. Xiang, D. Liu, Y. Zhou, J. Wang and L. Song, Nano Res., 2015, 8, 3982–3991 CrossRef CAS
- Y. Yong, L. Zhou, Z. Gu, L. Yan, G. Tian, X. Zheng, X. Liu, X. Zhang, J. Shi, W. Cong, W. Yin and Y. Zhao, Nanoscale, 2014, 6, 10394–10403 RSC
- L. Cheng, J. Liu, X. Gu, H. Gong, X. Shi, T. Liu, C. Wang, X. Wang, G. Liu, H. Xing, W. Bu, B. Sun and Z. Liu, Adv. Mater., 2014, 26, 1886–1893 CrossRef CAS
- C. Sun, L. Wen, J. Zeng, Y. Wang, Q. Sun, L. Deng, C. Zhao and Z. Li, Biomaterials, 2016, 91, 81–89 CrossRef CAS
- Z. Sun, H. Xie, S. Tang, X.-F. Yu, Z. Guo, J. Shao, H. Zhang, H. Huang, H. Wang and P. K. Chu, Angew. Chem., Int. Ed., 2015, 54, 11526–11530 CrossRef CAS
- Y. Zhao, L. Tong, Z. Li, N. Yang, H. Fu, L. Wu, H. Cui, W. Zhou, J. Wang, H. Wang, P. K. Chu and X.-F. Yu, Chem. Mater., 2017, 29, 7131–7139 CrossRef CAS
- Y. Li, Z. Liu, Y. Hou, G. Yang, X. Fei, H. Zhao, Y. Guo, C. Su, Z. Wang, H. Zhong, Z. Zhuang and Z. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 25098–25106 CrossRef CAS
- H. Wang, X. Yang, W. Shao, S. Chen, J. Xie, X. Zhang, J. Wang and Y. Xie, J. Am. Chem. Soc., 2015, 137, 11376–11382 CrossRef CAS
- J. Shao, H. Xie, H. Huang, Z. Li, Z. Sun, Y. Xu, Q. Xiao, X.-F. Yu, Y. Zhao, H. Zhang, H. Wang and P. K. Chu, Nat. Commun., 2016, 7, 12967 CrossRef CAS
- C. C. Zhou, S. K. Liu, J. L. Li, K. Guo, Q. Yuan, A. M. Zhong, J. Yang, J. C. Wang, J. M. Sun and Z. X. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 44080–44091 CrossRef CAS
- J. Chu, P. P. Shi, W. X. Yan, J. P. Fu, Z. Yang, C. M. He, X. Y. Deng and H. P. Liu, Nanoscale, 2018, 10, 9547–9560 RSC
- M. Fojtů, J. Balvan, M. Raudenská, T. Vičar, J. Šturala, Z. Sofer, J. Luxa, J. Plutnar, M. Masařík and M. Pumera, Appl. Mater. Today, 2020, 20, 100697 CrossRef
- J. Ma, R. Liu, X. Wang, Q. Liu, Y. N. Chen, R. P. Valle, Y. Y. Zuo, T. Xia and S. J. Liu, ACS Nano, 2015, 9, 10498–10515 CrossRef CAS
- Y. X. Lin, J. X. Xu and H. Y. Lan, J. Hematol. Oncol., 2019, 12, 16 CrossRef
- G. L. Beatty, E. G. Chiorean, M. P. Fishman, B. Saboury, U. R. Teitelbaum, W. J. Sun, R. D. Huhn, W. R. Song, D. G. Li, L. L. Sharp, D. A. Torigian, P. J. O'Dwyer and R. H. Vonderheide, Science, 2011, 331, 1612–1616 CrossRef CAS
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS
- S. Sengupta, Trends Cancer, 2017, 3, 551–560 CrossRef CAS
- B. Li, J. Z. Yang, Q. Huang, Y. Zhang, C. Peng, Y. J. Zhang, Y. He, J. Y. Shi, W. X. Li, J. Hu and C. H. Fan, NPG Asia Mater., 2013, 5, e44 CrossRef CAS
- R. B. Li, L. M. Guiney, C. H. Chang, N. D. Mansukhani, Z. X. Ji, X. Wang, Y. P. Liao, W. Jiang, B. B. Sun, M. C. Hersam, A. E. Nel and T. Xia, ACS Nano, 2018, 12, 1390–1402 CrossRef CAS
- K. Donaldson and A. Seaton, Part. Fibre Toxicol., 2012, 9, 13 CrossRef
- C. R. Thomas and T. R. Kelley, Environ. Health Insights, 2010, 4, 21–26 CAS
- J. H. Appel, D. O. Li, J. D. Podlevsky, A. Debnath, A. A. Green, Q. H. Wang and J. Chae, ACS Biomater. Sci. Eng., 2016, 2, 361–367 CrossRef CAS
- S. Pandit, S. Karunakaran, S. K. Boda, B. Basu and M. De, ACS Appl. Mater. Interfaces, 2016, 8, 31567–31573 CrossRef CAS
- X. Yang, J. Li, T. Liang, C. Ma, Y. Zhang, H. Chen, N. Hanagata, H. Su and M. Xu, Nanoscale, 2014, 6, 10126–10133 RSC
- A. Pal, T. K. Jana, T. Roy, A. Pradhan, R. Maiti, S. M. Choudhury and K. Chatterjee, ChemistrySelect, 2018, 3, 81–90 CrossRef CAS
- A. Bianco, Angew. Chem., Int. Ed., 2013, 52, 4986–4997 CrossRef CAS
- I. E. M. Carpio, C. M. Santos, X. Wei and D. F. Rodrigues, Nanoscale, 2012, 4, 4746 RSC
- L. Ou, B. Song, H. Liang, J. Liu, X. Feng, B. Deng, T. Sun and L. Shao, Part. Fibre Toxicol., 2016, 13, 57 CrossRef
- P. Ganguly, A. Breen and S. C. Pillai, ACS Biomater. Sci. Eng., 2018, 4, 2237–2275 CrossRef CAS
- E. L. K. Chng and M. Pumera, RSC Adv., 2015, 5, 3074–3080 RSC
- X. Li, W. Liu, L. Sun, K. E. Aifantis, B. Yu, Y. Fan, Q. Feng, F. Cui and F. Watari, J. Biomed. Mater. Res., Part A, 2014, 103, 2499–2507 CrossRef
- K. Luyts, D. Napierska, B. Nemery and P. H. M. Hoet, Environ. Sci.: Processes Impacts, 2013, 15, 23–38 RSC
- M. V. Park, E. A. Bleeker, W. Brand, F. R. Cassee, M. V. Elk, I. Gosens, W. H. D. Jong, J. A. Meesters, W. J. Peijnenburg, J. T. Quik, R. J. Vandebriel and A. J. Sips, ACS Nano, 2017, 11, 9574–9593 CrossRef CAS
- S. Presolski, L. Wang, A. H. Loo, A. Ambrosi, P. Lazar, V. Ranc, M. Otyepka, R. Zboril, O. Tomanec, J. Ugolotti, Z. Sofer and M. Pumera, Chem. Mater., 2017, 29, 2066–2073 CrossRef CAS
- N. F. Rosli, N. M. Latiff, Z. Sofer, A. C. Fisher and M. Pumera, Appl. Mater. Today, 2018, 11, 200–206 CrossRef
- G. Qu, W. Liu, Y. Zhao, J. Gao, T. Xia, J. Shi, L. Hu, W. Zhou, J. Gao, H. Wang, Q. Luo, Q. Zhou, S. Liu, X.-F. Yu and G. Jiang, Angew. Chem., Int. Ed., 2017, 56, 14488–14493 CrossRef CAS
- K. Kalantar-Zadeh, J. Z. Ou, T. Daeneke, M. S. Strano, M. Pumera and S. L. Gras, Adv. Funct. Mater., 2015, 25, 5086–5099 CrossRef CAS
- L. Yan, F. Zhao, S. Li, Z. Hu and Y. Zhao, Nanoscale, 2011, 3, 362–382 RSC
- Y. Zhang, T. R. Nayak, H. Hong and W. Cai, Nanoscale, 2012, 4, 3833 RSC
- L. Cheng, J. Liu, X. Gu, H. Gong, X. Shi, T. Liu, C. Wang, X. Wang, G. Liu, H. Xing, W. Bu, B. Sun and Z. Liu, Adv. Mater., 2013, 26, 1886–1893 CrossRef
- T. Liu, C. Wang, W. Cui, H. Gong, C. Liang, X. Shi, Z. Li, B. Sun and Z. Liu, Nanoscale, 2014, 6, 11219–11225 RSC
- X. Yang, D. Wang, Y. Shi, J. Zou, Q. Zhao, Q. Zhang, W. Huang, J. Shao, X. Xie and X. Dong, ACS Appl. Mater. Interfaces, 2018, 10, 12431–12440 CrossRef CAS
- J. R. Choi, K. W. Yong, J. Y. Choi, A. Nilghaz, Y. Lin, J. Xu and X. Lu, Theranostics, 2018, 8, 1005–1026 CrossRef CAS
- T. Liu, C. Wang, X. Gu, H. Gong, L. Cheng, X. Shi, L. Feng, B. Sun and Z. Liu, Adv. Mater., 2014, 26, 3433–3440 CrossRef CAS
- W. Tao, X. Ji, X. Zhu, L. Li, J. Wang, Y. Zhang, P. E. Saw, W. Li, N. Kong, M. A. Islam, T. Gan, X. Zeng, H. Zhang, M. Mahmoudi, G. J. Tearney and O. C. Farokhzad, Adv. Mater., 2018, 30, 1802061 CrossRef
- W. Tao, X. Ji, X. Xu, M. A. Islam, Z. Li, S. Chen, P. E. Saw, H. Zhang, Z. Bharwani, Z. Guo, J. Shi and O. C. Farokhzad, Angew. Chem., Int. Ed., 2017, 56, 11896–11900 CrossRef CAS
- R. Thiruppathi, S. Mishra, M. Ganapathy, P. Padmanabhan and B. Gulyás, Adv. Sci., 2016, 4, 1600279 CrossRef
- J. Shao, H. Xie, H. Huang, Z. Li, Z. Sun, Y. Xu, Q. Xiao, X.-F. Yu, Y. Zhao, H. Zhang, H. Wang and P. K. Chu, Nat. Commun., 2016, 7, 12967 CrossRef CAS
- X. Li, J. Shan, W. Zhang, S. Su, L. Yuwen and L. Wang, Small, 2016, 13, 1602660 CrossRef
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CrossRef CAS
- E. L. K. Chng, Z. Sofer and M. Pumera, Nanoscale, 2014, 6, 14412–14418 RSC
- X. Zhang, Z. Zhang, S. Zhang, D. Li, W. Ma, C. Ma, F. Wu, Q. Zhao, Q. Yan and B. Xing, Small, 2017, 13, 1701210 CrossRef
- N. M. Latiff, W. Z. Teo, Z. Sofer, A. C. Fisher and M. Pumera, Chem. – Eur. J., 2015, 21, 13991–13995 CrossRef CAS
- H. Zhang, C. Peng, J. Yang, M. Lv, R. Liu, D. He, C. Fan and Q. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 1761–1767 CrossRef CAS
- S. Wan, P. M. Kelly, E. Mahon, H. Stöckmann, P. M. Rudd, F. Caruso, K. A. Dawson, Y. Yan and M. P. Monopoli, ACS Nano, 2015, 9, 2157–2166 CrossRef CAS
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503–513 CrossRef CAS
- G. Maiorano, S. Sabella, B. Sorce, V. Brunetti, M. A. Malvindi, R. Cingolani and P. P. Pompa, ACS Nano, 2010, 4, 7481–7491 CrossRef CAS
- P. Aggarwal, J. B. Hall, C. B. Mcleland, M. A. Dobrovolskaia and S. E. Mcneil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS
- W. Hu, C. Peng, M. Lv, X. Li, Y. Zhang, N. Chen, C. Fan and Q. Huang, ACS Nano, 2011, 5, 3693–3700 CrossRef CAS
- J. Mo, Q. Xie, W. Wei and J. Zhao, Nat. Commun., 2018, 9, 2480 CrossRef
- Q. Mu, G. Su, L. Li, B. O. Gilbertson, L. H. Yu, Q. Zhang, Y.-P. Sun and B. Yan, ACS Appl. Mater. Interfaces, 2012, 4, 2259–2266 CrossRef CAS
- N. M. Latiff, Z. Sofer, A. C. Fisher and M. Pumera, Chem. – Eur. J., 2016, 23, 684–690 CrossRef
- H. L. Chia, N. M. Latiff, Z. Sofer and M. Pumera, Chem. – Eur. J., 2017, 24, 206–211 CrossRef
- N. F. Rosli, C. C. Mayorga-Martinez, N. M. Latiff, N. Rohaizad, Z. Sofer, A. C. Fisher and M. Pumera, ACS Sustainable Chem. Eng., 2018, 6, 7432–7441 CrossRef CAS
- Z. Wang, A. V. D. Bussche, Y. Qiu, T. M. Valentin, K. Gion, A. B. Kane and R. H. Hurt, Environ. Sci. Technol., 2016, 50, 7208–7217 CrossRef CAS
- T. Zhang, Y. Wan, H. Xie, Y. Mu, P. Du, D. Wang, X. Wu, H. Ji and L. Wan, J. Am. Chem. Soc., 2018, 140, 7561–7567 CrossRef CAS
- Y. Huang, J. Qiao, K. He, S. Bliznakov, E. Sutter, X. Chen, D. Luo, F. Meng, D. Su, J. Decker, W. Ji, R. S. Ruoff and P. Sutter, Chem. Mater., 2016, 28, 8330–8339 CrossRef CAS
- C. H. A. Wong, Z. Sofer, M. Kube Ova, J. Ku Era, S. Mat Jkova and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13774–13779 CrossRef CAS
- E. J. E. Stuart and M. Pumera, J. Phys. Chem. C, 2010, 114, 21296–21298 CrossRef CAS
- C. C. Mayorga-Martinez, Z. Sofer, D. Sedmidubský, J. Luxa, B. Kherzi and M. Pumera, Nanoscale, 2018, 10, 1540–1546 RSC
- H. Yue, W. Wei, Z. Yue, B. Wang, N. Luo, Y. Gao, D. Ma, G. Ma and Z. Su, Biomaterials, 2012, 33, 4013–4021 CrossRef CAS
- A. B. Seabra, A. J. Paula, R. D. Lima, O. L. Alves and N. Durán, Chem. Res. Toxicol., 2014, 27, 159–168 Search PubMed
- G. Jia, H. Wang, L. Yan, X. Wang, R. Pei, T. Yan, Y. Zhao and X. Guo, Environ. Sci. Technol., 2005, 39, 1378–1383 CrossRef CAS
- I. Fenoglio, E. Aldieri, E. Gazzano, F. Cesano, M. Colonna, D. Scarano, G. Mazzucco, A. Attanasio, Y. Yakoub, D. Lison and B. Fubini, Chem. Res. Toxicol., 2012, 25, 74–82 Search PubMed
- E. L. K. Chng, C. K. Chua and M. Pumera, Nanoscale, 2014, 6, 10792–10797 RSC
- S. George, T. Xia, R. Rallo, Y. Zhao, Z. Ji, S. Lin, X. Wang, H. Zhang, B. France, D. Schoenfeld, R. Damoiseaux, R. Liu, S. Lin, K. A. Bradley, Y. Cohen and A. E. Nel, ACS Nano, 2011, 5, 1805–1817 CrossRef CAS
- T. Laomettachit, I. Puri and M. Liangruksa, Toxicol. Appl. Pharmacol., 2017, 334, 47–54 CrossRef CAS
- Y. Gao, J. Feng, L. Kang, X. Xu and L. Zhu, Sci. Total Environ, 2018, 610–611, 442–450 CrossRef CAS
- D. R. Bell, S.-G. Kang, T. Huynh and R. Zhou, Nanoscale, 2018, 10, 351–358 RSC
- Y. Liu, J. Peng, S. Wang, M. Xu, M. Gao, T. Xia, J. Weng, A. Xu and S. Liu, NPG Asia Mater., 2018, 10, e458 CrossRef
Footnote |
| † These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |