DOI:
10.1039/D0CS00666A
(Review Article)
Chem. Soc. Rev., 2021,
50, 658-666
Asymmetric desymmetrization of alkene-, alkyne- and allene-tethered cyclohexadienones using transition metal catalysis†
Received
3rd July 2020
First published on 7th December 2020
Abstract
This review is covering the recent development of catalytic asymmetric domino reactions for the desymmetrization of alkene-, alkyne- and allene-tethered cyclohexadienones using transition metals and chiral ligands. This desymmetrization has emerged as an important strategy for the rapid construction of complex molecular skeletons, such as fused-polycycles or spirocyclic compounds in controlling multiple stereogenic centers.

Tao Shu
| Tao Shu was born in Hubei, China. He got his bachelor and Master degree from Dalian University of Technology, China in 2010 and 2013 respectively. Then he moved to Germany, and obtained his doctoral degree from RWTH Aachen University, under the supervision of Prof. Dieter Enders in the field of asymmetric domino reactions. In 2018, he came to ESPCI Paris as a postdoctoral fellow, in Prof. Janine Cossy's group, focusing on the synthesis of a bioactive compound. |

Janine Cossy
| Janine Cossy was born in Reims (France). After her PhD in Reims, she spent two years in Prof. Trost's group as a postdoc. After an ealier career at the CNRS, she was appointed Professor of Organic Chemistry at the ESPCI in 1990. Janine Cossy's research interests focus on the synthesis of bioactive molecules and on the development of synthetic methods. Her research efforts have resulted in more than 530 publications and 17 patents. Among the awards, she received the CNRS Silver Medal (1996), she was elected at the French Academy of sciences (2017) and nominated Fellow of the ACS (2018). |
1. Introduction
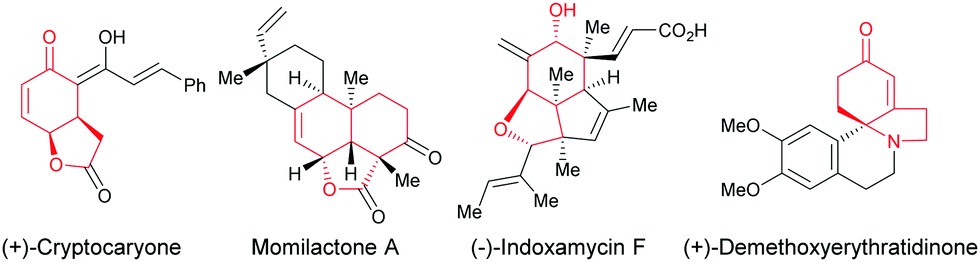
Enantioselective desymmetrization of simple prochiral molecules to complex scaffolds, by breaking one or more elements of symmetry, in an attractive process for organic chemists to access optically active compounds. In this context, C4-substituted cyclohexadienones are highly appealing molecules to investigate an enantioselective desymmetrization as they have a C2 element of symmetry. Among them, the alkene-, alkyne-, and allene-tethered cyclohexadienones are especially popular and have been extensively involved in desymmetrization processes as prochiral cyclohexadienones are easily prepared in one step by oxidative dearomatization of para-substituted phenols using commercially available PhI(CH3CO2)2 or PhI(CF3CO2)2 and alcohols. In addition, alkenes, alkynes, and allenes are functional groups that can be easily transformed to other functionalities using various transition metals. In addition, the two double bonds present in cyclohexadienones can easily be involved in conjugate additions or cycloadditions, thus providing bicyclic or polycyclic scaffolds featuring multiple stereocenters. These scaffolds are ubiquitous moieties present in natural products such as, for example, (+)-cryptocaryone, momilactone A, (−)-indoxamycin F, (+)-demethoxy-erythratidinone (Fig. 1).
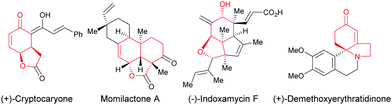 |
| | Fig. 1 Bioactive natural products containing hydrobenzofurans and hydroindoles. | |
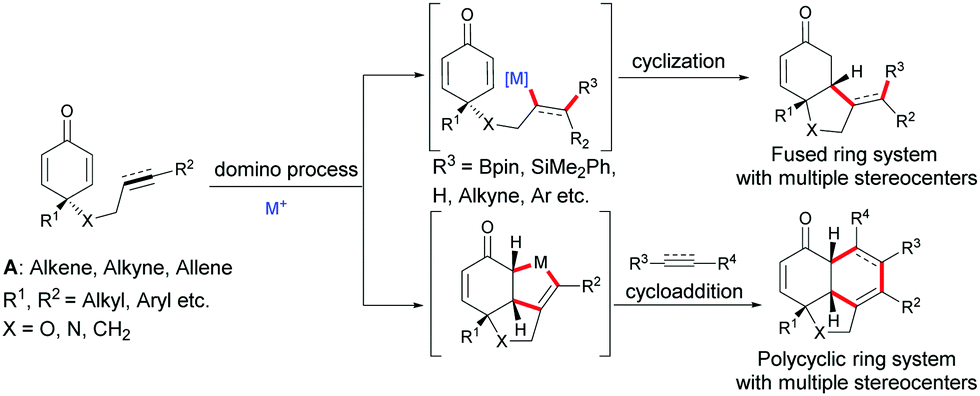
Due to the number of chemical transformations that alkenes, alkynes, allenes and cyclohexadienones can undergo, the alkene-, alkyne-, allene-tethered cyclohexadienones A have opened up many possibilities for domino transformations catalyzed by transition metals. Transition metal/chiral ligand complexes create a chiral environment which provides the shielding of one face of symmetric cyclohexadienones thereby creating a facial differentiation during a 1,4-addition or a cycloaddition (Scheme 1).
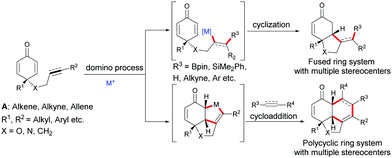 |
| | Scheme 1 General domino reactions involving alkene-, alkyne-, and allene-tethered cyclohexadienones. | |
Although there has been tremendous progress in the field of enantioselective desymmetrization of prochiral cyclohexadienones using transition-metal catalysis, organocatalysis or enzyme catalysis,1 the catalytic asymmetric desymmetrization of alkene-, alkyne-, and allene-tethered cyclohexadienones has not been reviewed specifically. The aim of this review is to give an overview of the recent progress in transition metal-catalyzed enantioselective desymmetrization of cyclohexadienones. This review is divided in three parts according to the cyclohexadienone unsaturated substituent present at C4 e.g. an alkene, an alkyne, or an allene substituent.
2. Alkene-tethered cyclohexadienones
2.1 Rh-Catalyzed enantioselective desymmetrization
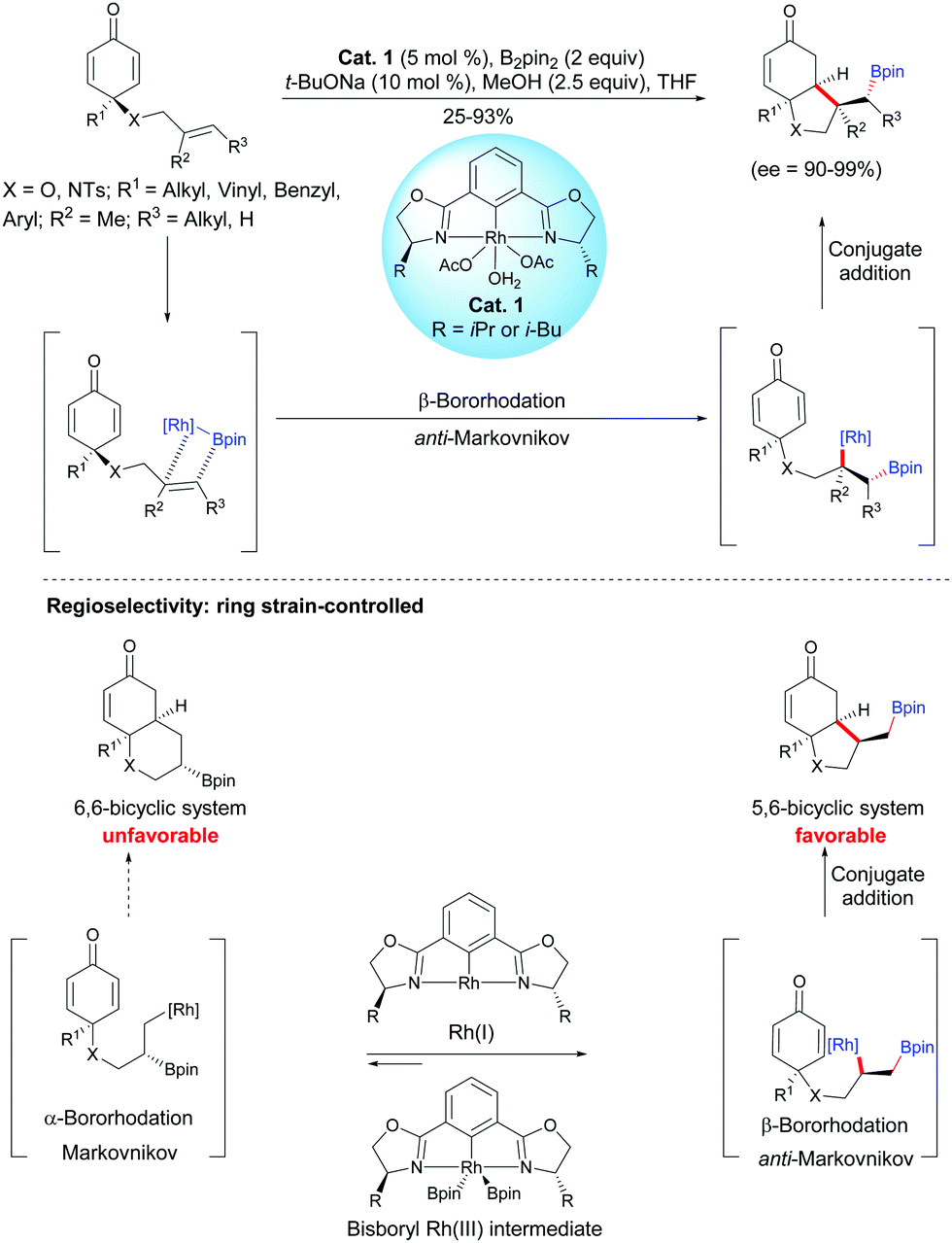
As unactivated alkenes are less reactive than alkenes activated by electron-withdrawing groups towards nucleophiles, the development of methods to differentiate and overcome the inherent properties of these two type of alkenes is of great value. In 2019, Tian, Hong, Lin et al. developed a Rh-catalyzed asymmetric boration/cyclization domino reaction of alkene-tethered cyclohexadienones. By using the chiral catalyst Cat. 1, enantio-enriched hydrobenzofuran scaffolds were produced and three or four contiguous stereocenters were controlled.2 The process works well with various substituted alkenes, e.g. monosubstituted terminal alkenes (65–93%, ee = 90–96%), 1,1-disubstituted terminal alkenes (25%, ee = 94%), and (E)-1,2-disubstituted alkenes (37–66%, ee = 97–99%) affording substituted fused bicyclic compounds with good enantiomeric excess (ee = 90–99%) (Scheme 2).
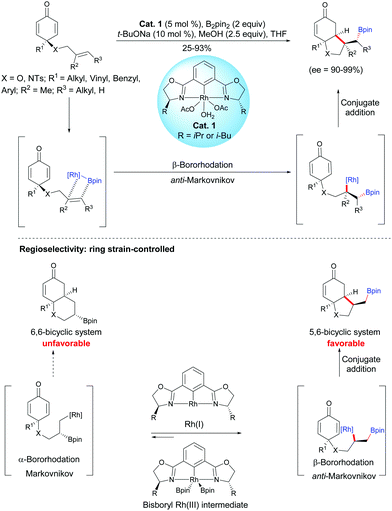 |
| | Scheme 2 Rh-Catalyzed asymmetric boration/cyclization domino reaction of alkene-tethered cyclohexadienones. | |
Based on a SAESI-MS experiment and density functional theory calculations (DFT), the authors have proposed a Rh(I)/Rh(III) catalytic cycle for this domino process, in which a Rh(I) species is the active species (oxidative addition of B2pin2, olefin insertion, 1,4-addition). According to the DFT calculations, the authors found that the insertion of bisboryl-Rh(III) intermediate in the double bond of cyclohexadienone is significantly less favored than its insertion in the double bond of the tethered-alkene, which accounts for the chemoselectivity. However, the addition of the bisboryl-Rh species on the double bond of the tethered-alkene is not regioselective and two regioisomeric alkylrhodium(III) intermediates are in equilibrium but, by a subsequent irreversible 1,4-addition cyclization, the formation of the fused 5,6-compound is favored over the fused 6,6-bicyclic compound due to a difference of ring strain. The power of ring strain on the control of the alkene-tethered cyclohexadienone cyclization was supported by DFT calculations which have shown that a fused 6,6-bicyclic compound is more strained than a fused 5,6-bicyclic one. It is worth mentioning that the enantioselectivity of the domino process is arising from the chiral NCN-pincer rhodium(III) catalyst Cat. 1.
2.2 Cu-Catalyzed enantioselective desymmetrization
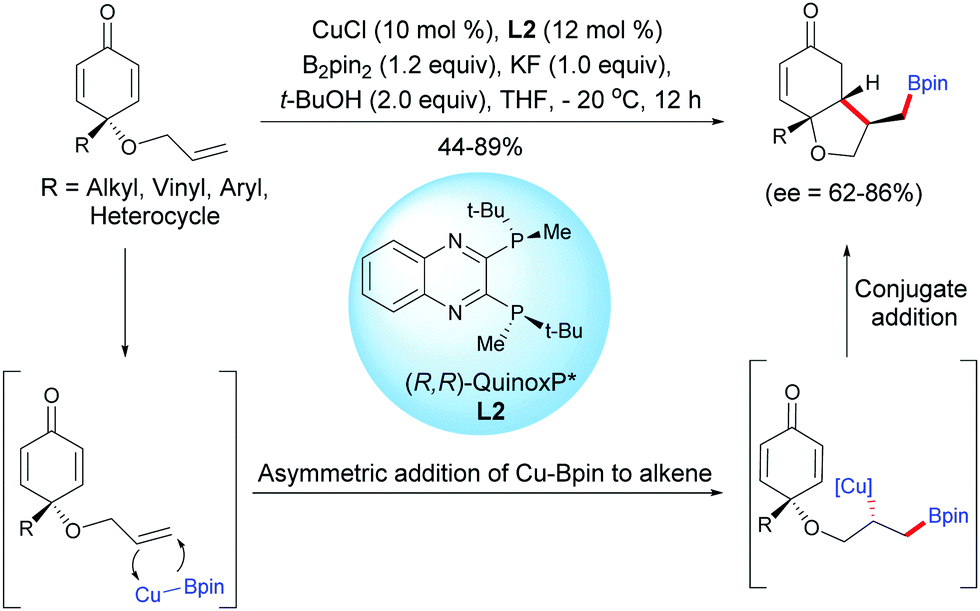
Tian, Lin et al. have also developed a process by employing a copper catalyst. A Cu-catalyzed asymmetric boration/cyclization domino reaction, applied to monosubstituted terminal alkene-tethered cyclohexadienones, using CuCl (10 mol%) and the chiral ligand L2, afforded fused bicyclic products in moderate to high yields (44–89%) and with good enantiomeric excess (ee = 62–86%).3
The fused bicyclic compounds are the result of a regioselective anti-Markovnikov cuproboration of tethered electron-rich terminal alkenes followed by a 1,4-addition of a stereospecific alkyl copper intermediate onto the cyclohexadienone. The authors suggested that the 1,4-borylation of the electron-deficient double bonds of the cyclohexadienone is suppressed by the presence of two substituents at the C4 position which induces steric hindrance and thus, the tethered alkene is preferentially borylated. Interestingly, in the Rh-catalyzed process the fused bicyclic products were obtained with a trans relationship between the substituent on the 5-membered ring (–CHRBpin) and the substituent at the ring junction while the Cu-catalyzed process afforded the fused bicyclic product with a cis relationship between the substituent on the 5-membered ring (–CH2Bpin) and the substituent at the ring junction (Scheme 3). This difference of stereoselectivity is probably related to the substitution pattern of the alkene.
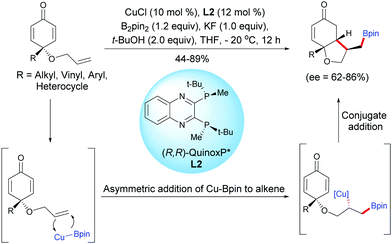 |
| | Scheme 3 Cu-Catalyzed asymmetric boration/cyclization of alkene-tethered cyclohexadienones. | |
3. Alkyne-tethered cyclohexadienones
As many examples of asymmetric domino reactions, involving alkyne-tethered cyclohexadienones have been reported, this section will be classified according to the different transition metals used to achieve these reactions e.g. Rh, Pd, Cu, Ni, and Au catalysts.
3.1 Rh-Catalyzed enantioselective desymmetrization
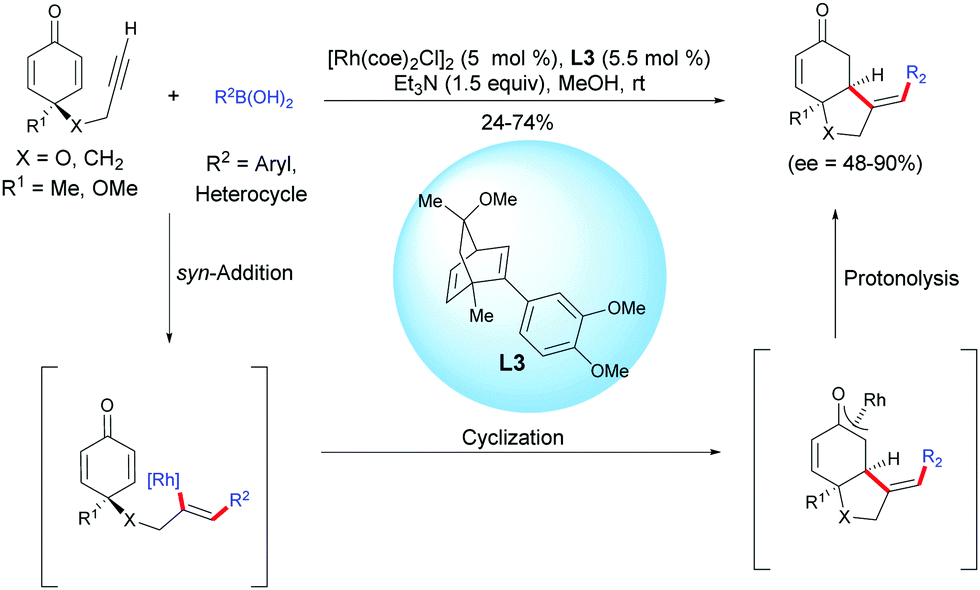
In 2013, Lautens et al. reported an enantioselective Rh-catalyzed hydroarylation of alkyne-tethered cyclohexadienones with arylboronic acids.4 In the presence of [Rh(coe)2Cl]2 and the chiral dienic ligand L3. A variety of enantio-enriched bicyclo[4.3.0]nonane scaffolds, possessing a five-membered heterocyclic ring or an all-carbon five-membered ring were obtained in moderate to good yields (24–74%) and with good to excellent enantiomeric excess (ee = 48–90%). The authors suggested that a syn-addition of the rhodium-aryl species onto the alkyne moiety is favored over the addition of this species on the activated double bond (Scheme 4).
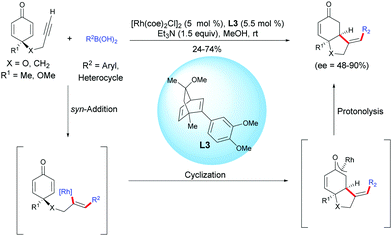 |
| | Scheme 4 Rh-Catalyzed hydroarylation/cyclization sequence of alkyne-tethered cyclohexadienones using arylboronic acids. | |
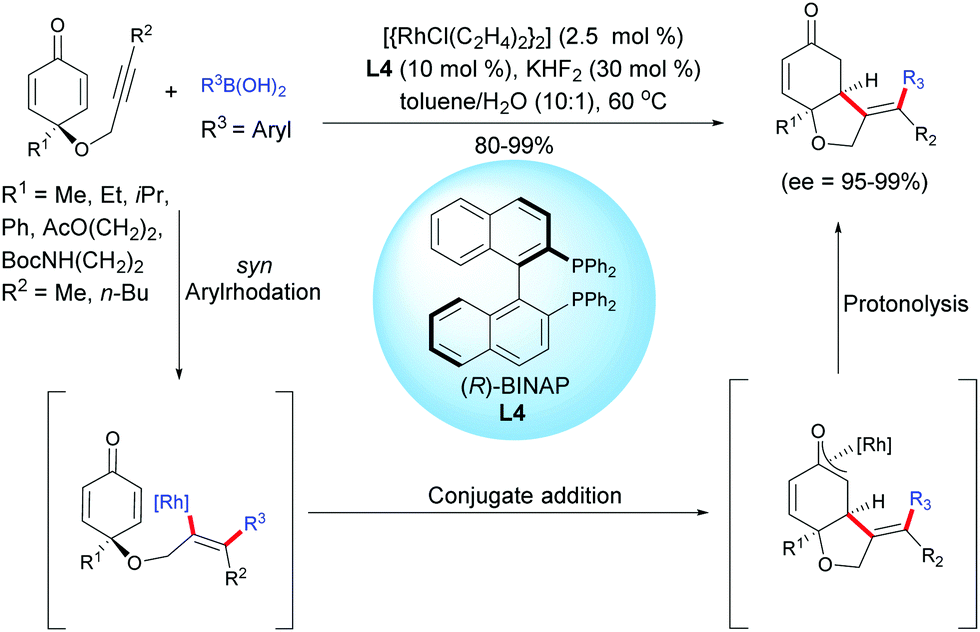
Another example of desymmetrization using a Rh-catalyzed enantioselective hydroarylation of alkyne-tethered cyclohexadienones, involving arylboronic acids, was reported by Tian, Lin et al.5 by using a different catalytic system than the one utilized by Lautens et al. In the presence of [{RhCl(C2H4)2}2] (2.5 mol%) and the (R)-BINAP ligand, L4 (10 mol%), a variety of enantio-enriched cis-hydrobenzofurans were obtained with excellent yields (80–99%) and excellent enantiomeric excess (ee = 95–99%). The aryl rhodation/conjugate addition domino process tolerates a variety of arylboronic acids in which the aryl group can be substituted either by electron-donating or electron-withdrawing groups (Scheme 5).
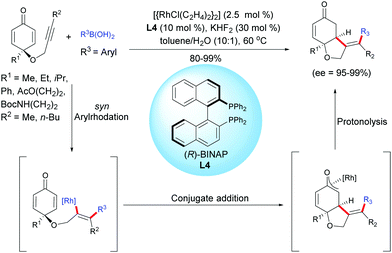 |
| | Scheme 5 Rh-Catalyzed hydroarylation/cyclization of alkyne-tethered cyclohexadienones using arylboronic acids. | |
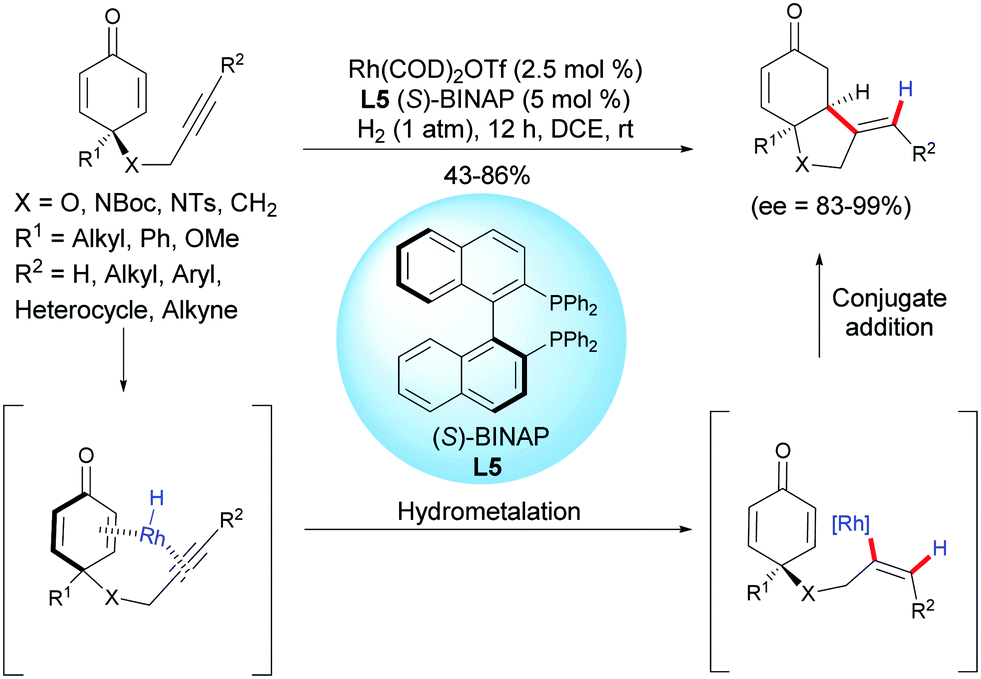
In 2018, Chegondi et al. reported a rhodium-catalyzed highly regio- and enantioselective reductive cyclization of alkyne-tethered cyclohexadienones.6 In the presence of Rh(cod)2OTf (2.5 mol%) and (S)-BINAP L5 (5 mol%), under 1 atm of H2, a variety of cis-hydrobenzofurans and cis-hydroindoles were obtained in good yields (43–86%) and excellent enantioselectivities (ee = 83–99%) (Scheme 6). In addition, the authors also demonstrated that the desymmetrization process works well when applied to 1,3-diyne-tethered cyclohexadienones. To further understand the mechanism, the authors conducted deuterium-labeling experiments and studied the kinetic isotope effects. They found that the hydrogen activation by the catalyst is the rate-determining step.
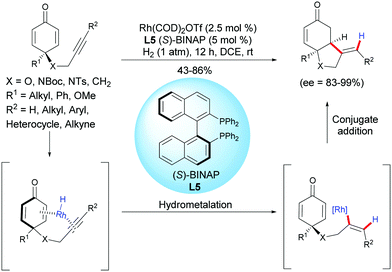 |
| | Scheme 6 Rh-Catalyzed reductive cyclization of alkyne-tethered cyclohexadienones. | |
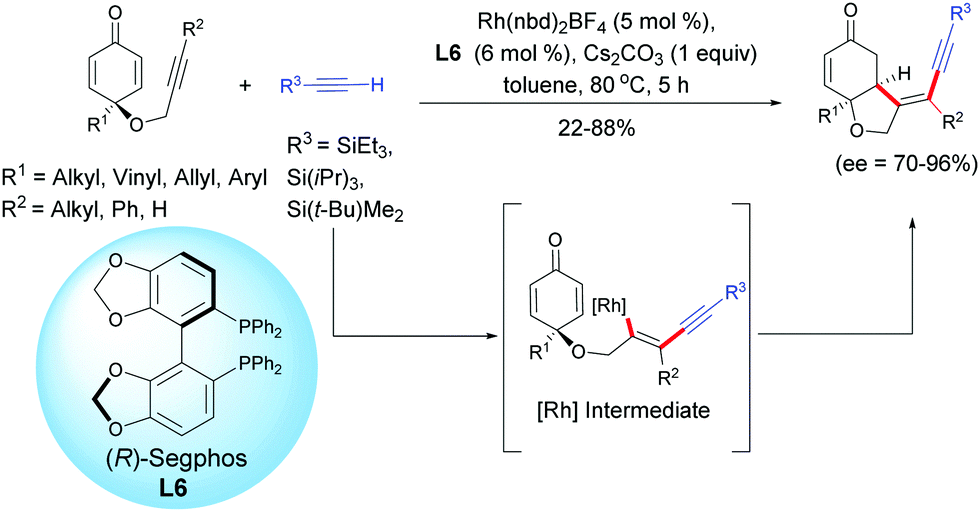
A recent example using a rhodium catalyst, Rh(nbd)2BF4, in the presence of (R)-Segphos L6 was reported by Tian, Lin et al. in 2019.7 These authors developed a highly enantioselective rhodium-catalyzed cross-addition of trialkylsilyl acetylenic derivatives [R3 = SiEt3, Si(iPr)3, Si(t-Bu)Me2] with the internal alkynyl-tethered cyclohexadienones. Through a regioselective alkynylation of the alkynyl-tethered and a subsequent intramolecular 1,4-addition domino reaction, cis-hydrobenzofurans were isolated with moderate to high yields (22–88%) and excellent enantioselectivities (ee = 70–96%). The reaction works well with internal alkyne-tethered cyclohexadienones however, when a terminal alkyne-tethered cyclohexadienone was tested under the optimal conditions, the desired cyclized product was obtained in only 22% yield but with a good enantiomeric excess (ee = 70%). It is worth mentioning that under the conditions developed by Tian, Lin et al., if the R3 substituent of the acetylenic derivative is a SiMe3, t-Bu, or CO2Me group, the domino reaction failed and the desired cyclization product was not delivered. In these cases, the homodimerization of the less bulky terminal alkyne was observed as the main side reaction (Scheme 7).
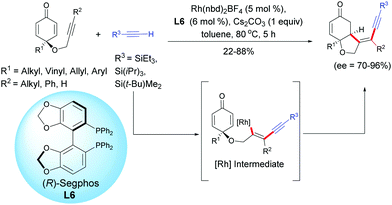 |
| | Scheme 7 Rh-Catalyzed hydroalkynylation/cyclization sequence of alkyne-tethered cyclohexadienones and external terminal alkynes. | |
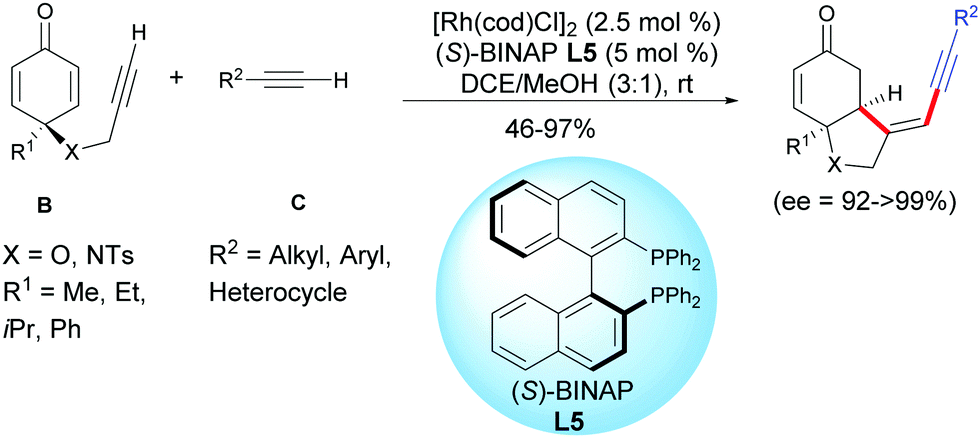
Xu et al. found that external terminal alkynes C were tolerated and a cross-dimerization process was taking place with the terminal alkynyl of the cyclohexadienones B using [Rh(cod)Cl]2 as the catalyst and (S)-BINAP L5 as the ligand. Head-to-head regioisomers were isolated in moderate to excellent yields (46–97%) and excellent enantiomeric excess (ee = 92 to >99%)8 (Scheme 8).
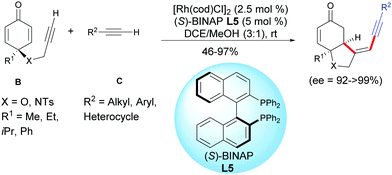 |
| | Scheme 8 Rh-Catalyzed cross-hydroalkynylation/cyclization of alkyne-tethered cyclohexadienones and external terminal alkynes. | |
Good results were obtained with various terminal acetylenic derivatives C substituted by aromatics bearing electron-withdrawing or electron-donating groups whatever the para, ortho, or meta position of the substituent. Alkynes C, substituted by a heterocycles such as an indole, a thiophene, a ferrocene, or substituted by a cyclopropyl- or a cyclohexenyl group can also be involved in domino reactions with alkyne-tethered cyclohexadienones B. Simple acetylenic derivatives C, substituted by an alkyl group also afforded the desired products with excellent enantiomeric excess, albeit in moderate yields (R2 = C6H5CH2CH2CH2, 55%; R2 = C6H5OCH2, 46%) (Scheme 8).
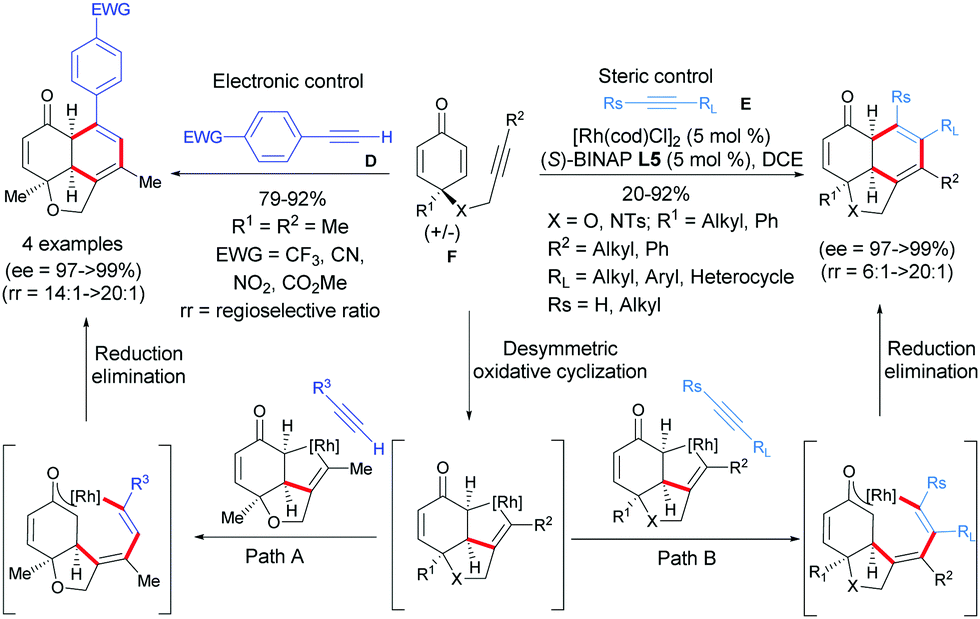
With the success of terminal acetylenic derivatives C, acetylenic derivatives of type D or E, which are more challenging, were tested. In 2019, Xu et al. established a Rh-catalyzed [2+2+2]-cycloaddition of internal alkyne-tethered cyclohexadienones F with acetylenic derivatives D or E, which afforded diverse fused tricyclic hydronaphthofuran scaffolds in one step (Scheme 8).9 Excellent regio-, diastereo-, and enantioselectivities were obtained and three contiguous stereocenters were controlled (ee = 97 to >99%) with 100% atom economy. A plausible catalytic cycle was then postulated by the authors involving an oxidative cyclization/cycloaddition/reductive elimination domino sequence for the desymmetrization. The domino process was initiated by a desymmetric oxidative cyclization of alkyne-tethered cyclohexadienones induced by a Rh(I) catalyst and the chiral (S)-BINAP ligand. A chiral rhodacyclopentene intermediate is formed and then this latter reacts with an external alkyne to afford, after reductive elimination, the [2+2+2]-cycloaddition product and the Rh catalyst is regenerated (Scheme 9). It is worth mentioning that the steric and electronic properties of the alkynes D and E have a significant influence on the regioselectivity of the cycloaddition. According to the authors, the steric hindrance between the RL substituent of the alkyne E and the ketone moiety is responsible for the regioselectivity and the less congested products are formed. In the case of alkynes D, substituted by an aryl group possessing a strong electron-withdrawing group in the para position, an electronic effect is responsible of the observed regioselectivity. It is hypothesized that the presence of an electron-withdrawing group is beneficial to the stabilization of the rhodacyclopentene intermediate, facilating the reductive elimination step and allowing the exclusive formation of one regioisomer.
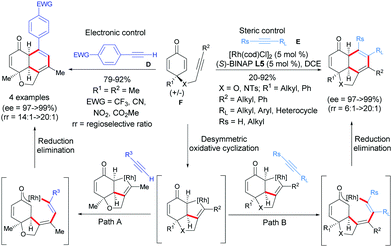 |
| | Scheme 9 Rh(I)-Catalyzed desymmetric [2+2+2]-cycloaddition of alkyne-tethered cyclohexadienones using external terminal alkynes. | |
3.2 Cu-Catalyzed enantioselective desymmetrization
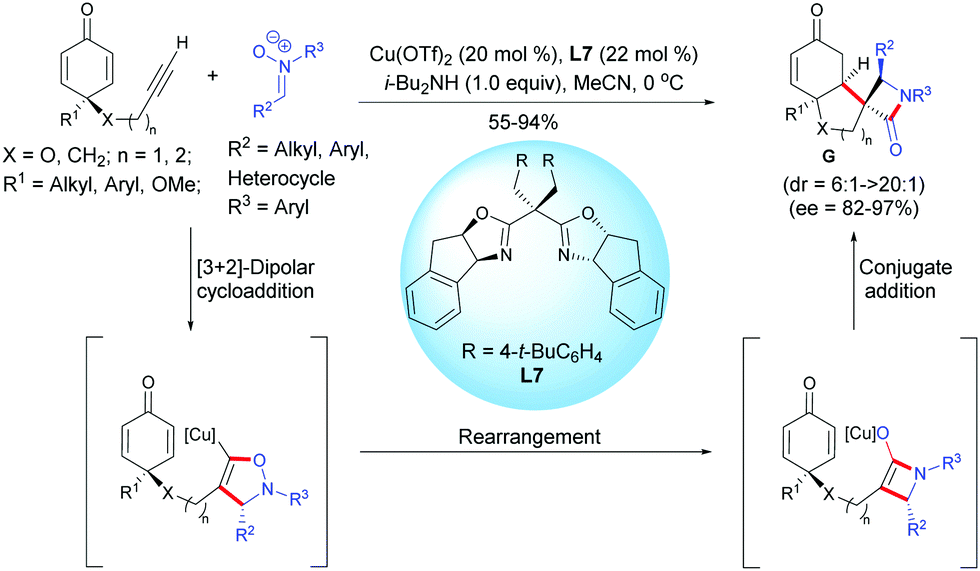
In 2018, Enders et al. reported a copper-catalyzed Kinugasa/1,4-addition domino reaction for the desymmetrization of prochiral cyclohexadienones, affording spirocyclic β-lactams.10 In the presence of Cu(OTf)2 (20 mol%) and the chiral Box ligand, L7 (22 mol%), the alkyne-tethered cyclohexadienones underwent a classical Kinugasa reaction in the presence of a nitrone. The in situ-formed copper enolate was trapped by the cyclohexadienone to afford the enantio-enriched spirocyclic β-lactams in high to excellent yields (55–94%), diastereoselectivities (dr = 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to >20:1), and enantiomeric excess (ee = 82–97%). The process tolerated a broad range of cyclohexadienones substituted at C4 (substituent R1) by an alkyl group or by an aryl substituted by electron-donating or electron-withdrawing groups. Cyclohexadienones were smoothly transformed to the desired spirocyclic compounds G in good yields (X = CH2, n = 1, 72%; X = O, n = 2, 55%) with moderate to excellent diastereoselectivity and good enantiomeric excess (X = CH2, n = 1, ee = 88%; X = O, n = 2, ee = 82%) (Scheme 10).
:1 to >20:1), and enantiomeric excess (ee = 82–97%). The process tolerated a broad range of cyclohexadienones substituted at C4 (substituent R1) by an alkyl group or by an aryl substituted by electron-donating or electron-withdrawing groups. Cyclohexadienones were smoothly transformed to the desired spirocyclic compounds G in good yields (X = CH2, n = 1, 72%; X = O, n = 2, 55%) with moderate to excellent diastereoselectivity and good enantiomeric excess (X = CH2, n = 1, ee = 88%; X = O, n = 2, ee = 82%) (Scheme 10).
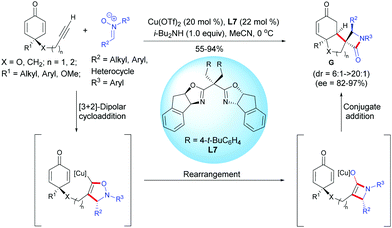 |
| | Scheme 10 Cu-Catalyzed Kinugasa/1,4-addition domino reaction of alkyne-tethered cyclohexadienones involving nitrones. | |
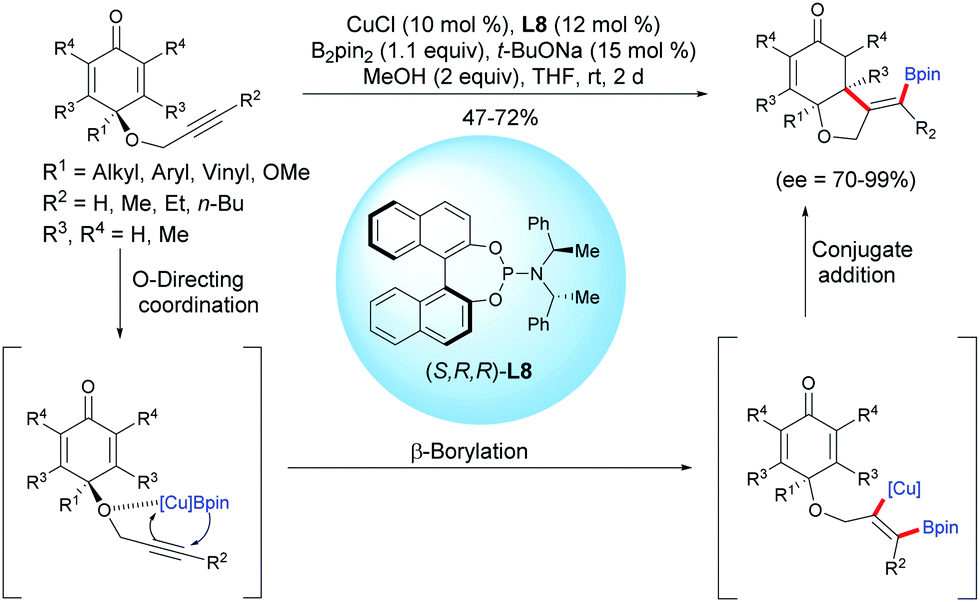
Tian and Lin et al. reported a highly efficient copper-catalyzed asymmetric boration/cyclization sequence of alkyne-tethered cyclohexadienones using CuCl and the chiral ligand L8.11 A selective β-boration of the pendant alkyne took place, followed by a 1,4-addition on the cyclohexadienone. A variety of enantio-enriched cis-hydrobenzofurans were obtained in moderate to high yields (47–72%) and with high to excellent enantioselectivities (ee = 70–99%). It seems that the propargylic ether is crucial for the β-boration of the alkyne moiety through an O-directing coordination, and then the formed alkenyl-Cu intermediate reacts with the cyclohexadienone according to an enantioselective 1,4-addition (Scheme 11).
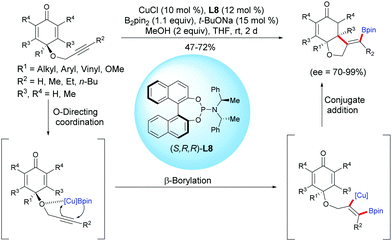 |
| | Scheme 11 Cu-Catalyzed enantioselective boration/cyclization of alkyne-tethered cyclohexadienones using B2pin2. | |
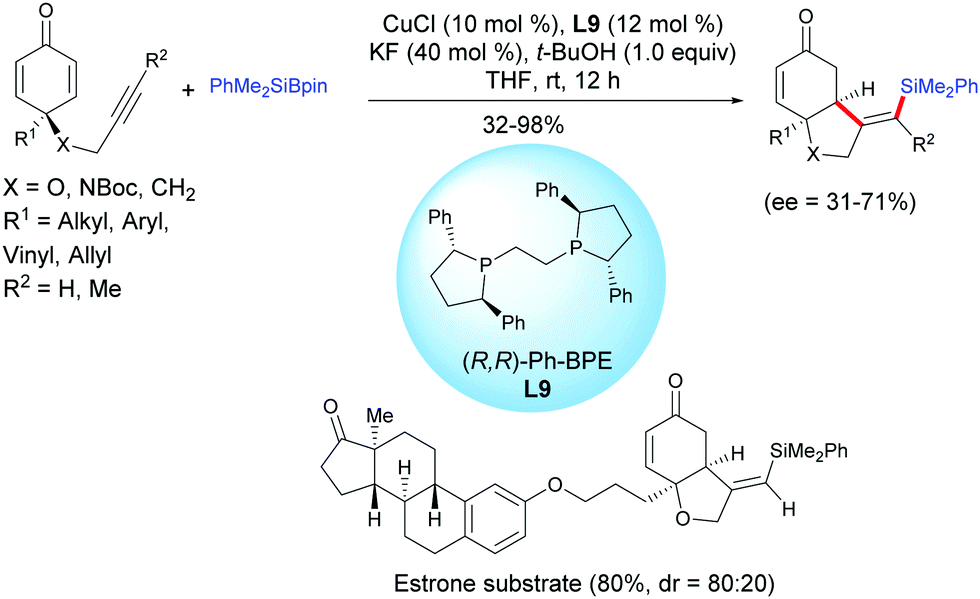
The same group reported a protocol for the desymmetrization of prochiral alkyne-tethered cyclohexadienones via a copper-catalyzed asymmetric silylation/cyclization domino reaction in the presence of the chiral ligand (R,R)-Ph-BPE, L9.12 A regioselective silylcupration of the alkyne-tethered cyclohexadienone took place when PhMe2Si–Bpin was used and subsequently the alkenylsilyl copper intermediate reacted with cyclohexadienones according to an enantioselective 1,4-addition. In the presence of CuCl (10 mol%) and (R,R)-Ph-BPE, L9 (12 mol%), the cis-hydrobenzofuran and cis-hydroindole scaffolds, bearing functionalized alkenyl silane motifs, were obtained in moderate to excellent yields (32–98%) and with moderate enantiomeric excess (ee = 31–71%) (Scheme 11). It is worth mentioning that alkyl, aryl, benzyl, vinyl and allyl substituents on the cyclohexadienone were well tolerated under the developed optimized conditions. For the alkyne-tethered cyclohexadienone, with an internal alkyne, the process is also efficient, however the reaction failed with 4-butynyl cyclohexadienones. The authors have applied this method to the synthesis of an estrone-tethered 1,6-enyne which was obtained in good yield (80%) but with a modest diastereoselectivity (dr = 4:1) (Scheme 12).
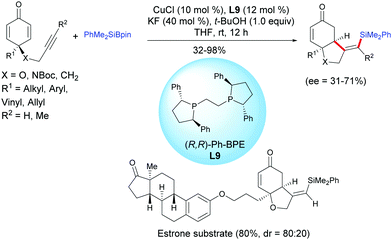 |
| | Scheme 12 Cu-Catalyzed asymmetric silylative cyclization of alkyne-tethered cyclohexadienones and PhMe2Bpin. | |
3.3 Ni-Catalyzed enantioselective desymmetrization
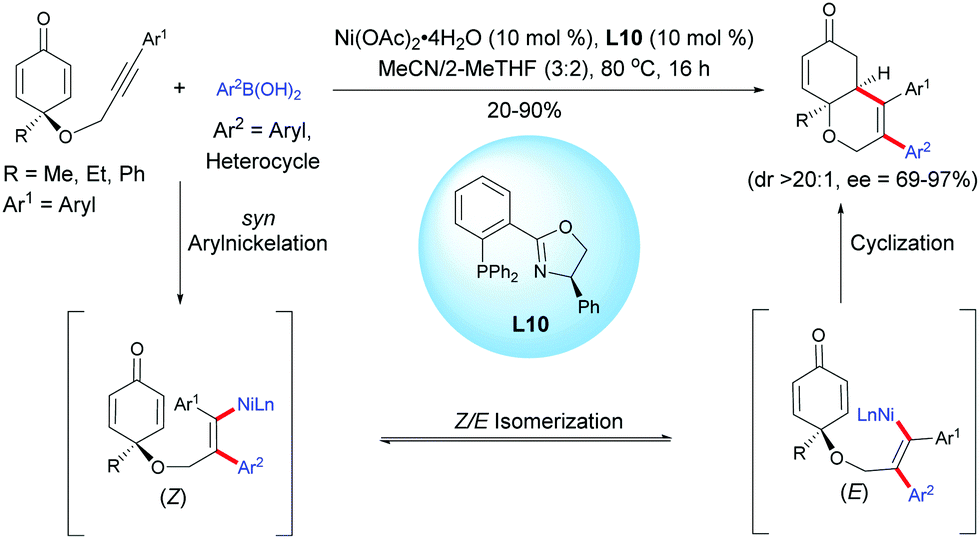
Lam et al. reported an enantioselective nickel-catalyzed anti-carbometalation/cyclization domino reaction of alkyne-tethered cyclohexadienones using arylboronic acids and Ni(OAc)2·4H2O as the catalyst, in the presence of the chiral ligand L10.13 The reaction proceeds through a transmetalation of ArB(OH)2 with the nickel catalyst to give an aryl nickel species and then a syn-aryl metalation followed by a Z/E isomerization lead to the (E)-alkenyl nickel species. The reaction of the alkenyl nickel species with the cyclohexadienone moiety produced a cyclized product which, after protonation of the resulting nickel enolate intermediate, releases the desired fused bicyclic product and the nickel catalyst is regenerated for the next catalytic cycle. It is worth mentioning that the mechanism involving a nickel species is different from the one involving a rhodium species. These two distinct transition metal-catalyzed reactions proceed via different reaction mechanisms, thus providing different fused bicyclic products. With a rhodium catalyst, a stereospecific syn-addition of the rhodium-aryl species onto the alkyne leads to an alkenyl rhodium intermediate while with a nickel catalyst, the nickel-aryl species insertion proceeds in a syn-stereospecific manner, leading to a (Z) configuration. A Z/E isomerization provides a (E) configuration alkenyl nickel intermediate which has the right configuration for a 1,4-addition. In both cases, the resulting alkenyl rhodium and nickel intermediates were trapped by the cyclohexadienone in an enantioselective manner, affording the corresponding fused 5,6- and 6,6-bicyclic products respectively (Scheme 13).
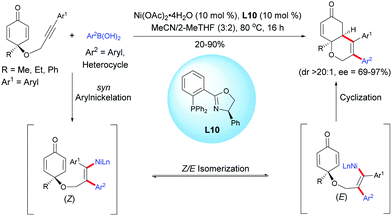 |
| | Scheme 13 Nickel-catalyzed anti-carbometalation/cyclization of alkyne-tethered cyclohexadienones using arylboronic acids. | |
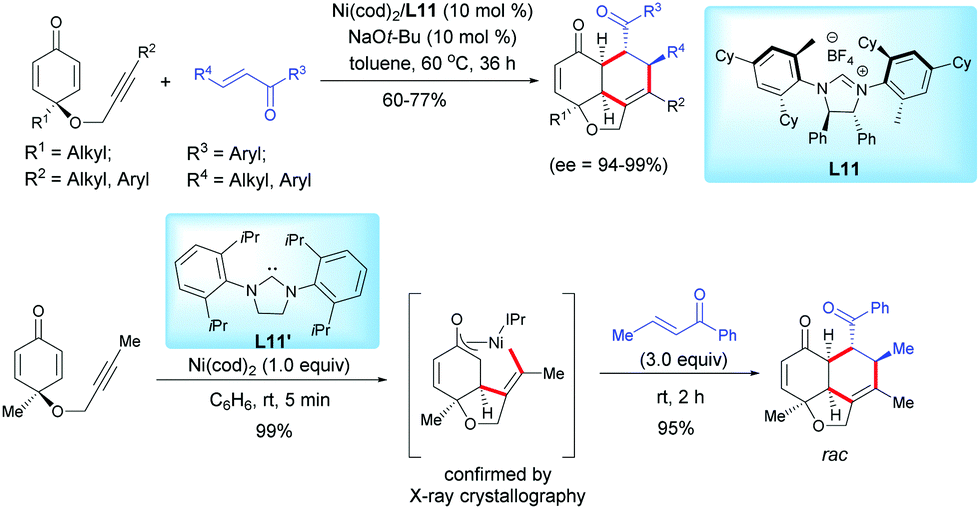
In 2017, Ogoshi et al. reported an elegant and highly enantioselective nickel-catalyzed synthesis of tricyclic hydronaphthofurans via an oxidative cyclization and a formal [4+2]-cycloaddition of alkyne-tethered cyclohexadienones with chalcones using Ni(OAc)2 and the chiral ligand L11 (Scheme 14).14 A range of complex densely functionalized chiral fused tricyclic hydronaphtho[1,8-b,c]-furan scaffolds were synthesized in high yields (60–77%) and excellent enantioselectivities (ee = 94–99%). A η3-oxaallyl nickelacycle intermediate was isolated when the reaction was performed with an alkyne-tethered cyclohexadienone in the presence of a stoichiometric amount of Ni(cod)2 and the IPr ligand L11′, which is in favor of an oxidative cyclization process (Scheme 14).
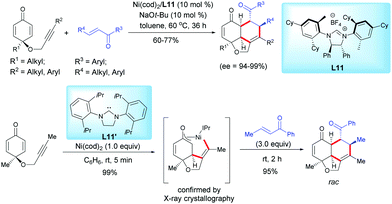 |
| | Scheme 14 Nickel-catalyzed oxidative cyclization and formal [4+2]-cycloaddition of alkyne-tethered cyclohexadienones and chalcones. | |
3.4 Pd-Catalyzed enantioselective desymmetrization
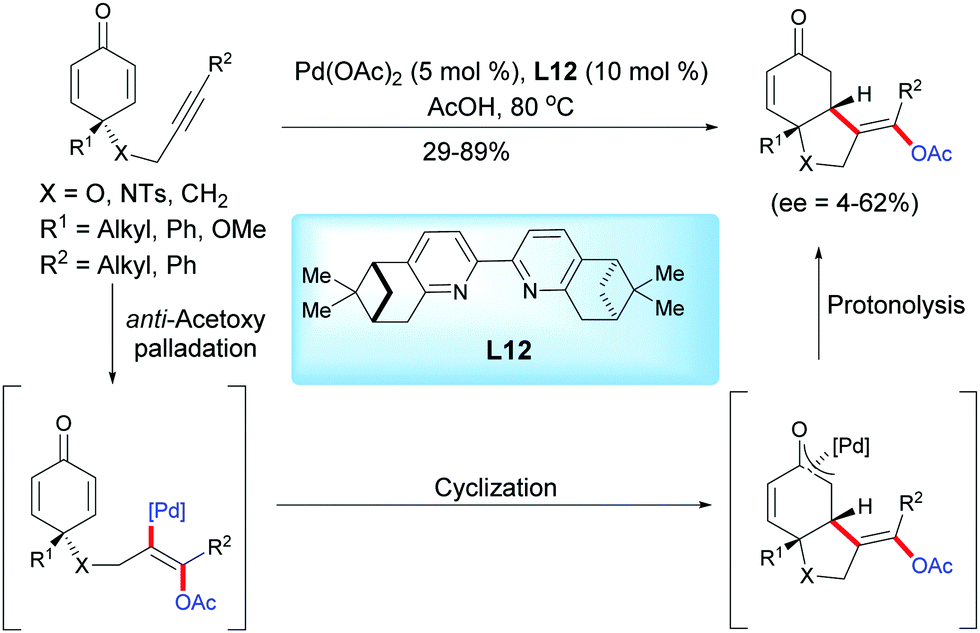
In 2013, Harned et al. reported a palladium-catalyzed enantioselective acetoxylation/cyclization domino reaction of alkyne-tethered cyclohexadienones in acetic acid using the ligand L12.15 In the presence of Pd(OAc)2 (5 mol%) and the pinene-derived bipyridine ligand L12 (10 mol%), the fused bicyclic products were obtained in moderate to high yields (29–89%) and with low to modest enantiomeric excess (ee = 4–62%) (Scheme 15).
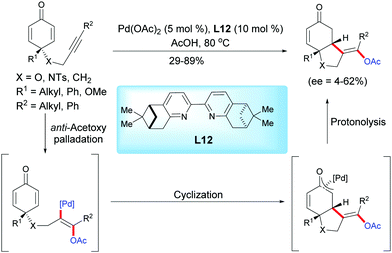 |
| | Scheme 15 Palladium-catalyzed acetoxylation/cyclization domino reaction of alkyne-tethered cyclohexadienones. | |
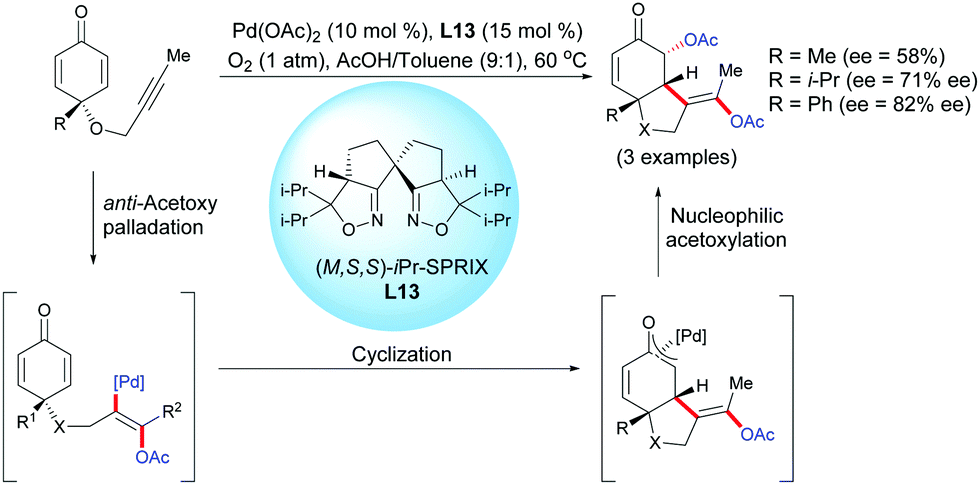
In 2014, Sasai et al. reported a novel enantioselective palladium-catalyzed diacetoxylation-induced cyclization of alkyne-tethered cyclohexadienones using ligand L13 and O2 as a green oxidant.16 The process involving an alkyne acetoxylation/intramolecular cyclization/umpolung acetoxylation domino process afforded diacetoxylated benzofuranones. A preliminary screening achieved on three compounds, using the chiral (M,S)-iPr-SPRIX ligand, L13, showed promising results, as the desired products were obtained in moderate to high enantioselectivities (ee = 58–82%) (Scheme 16).
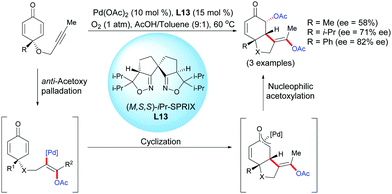 |
| | Scheme 16 Palladium-catalyzed acetoxylation/cyclization/umpolung acetoxylation domino reaction of alkyne-tethered cyclohexadienones. | |
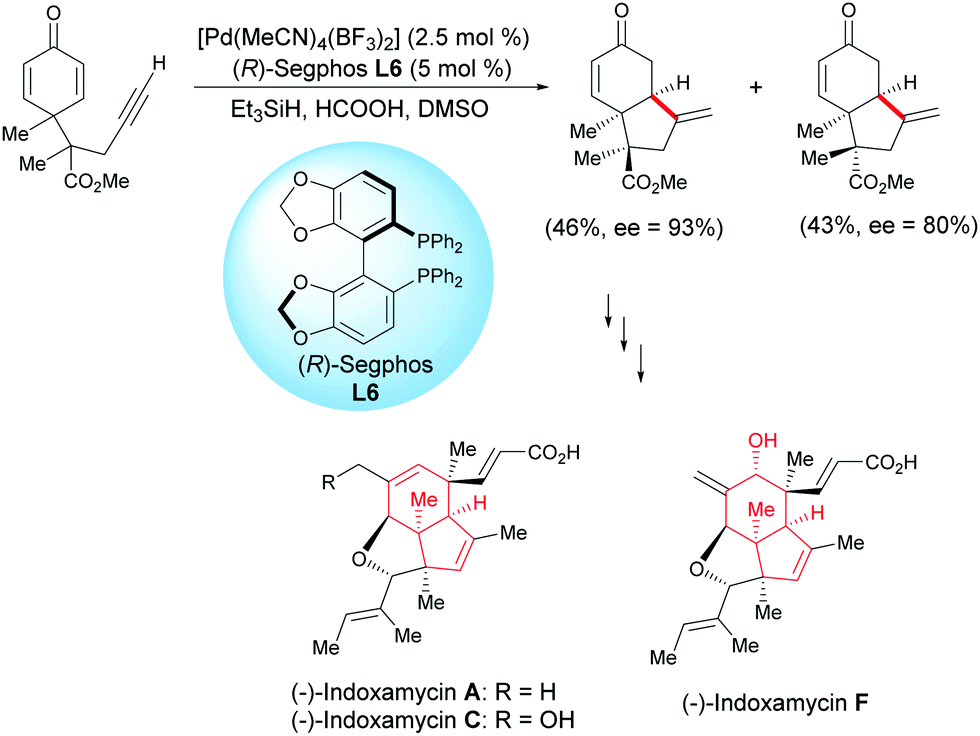
Ding et al. developed a Pd-catalyzed asymmetric cyclization for the enantioselective synthesis of fused bicyclic scaffolds.17 In the presence of Pd(MeCN)4(BF3)2 catalyst (2.5 mol%) and the (R)-Segphos ligand, L6 (5 mol%), the racemic alkyne-tethered cyclohexadienone underwent an asymmetric cyclization and two epimeric bicyclic compounds were formed with an enantiomeric excess of 93% and 80%, respectively. One fused bicyclic compound was used to synthesize indoxamycins A, C, and F (Scheme 17).
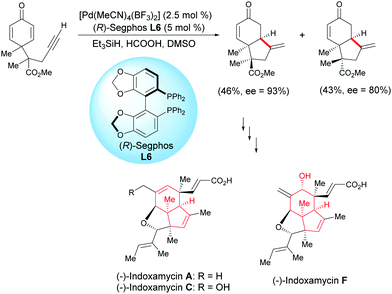 |
| | Scheme 17 Pd-Catalyzed asymmetric cyclization of alkyne-tethered cyclohexadienones. | |
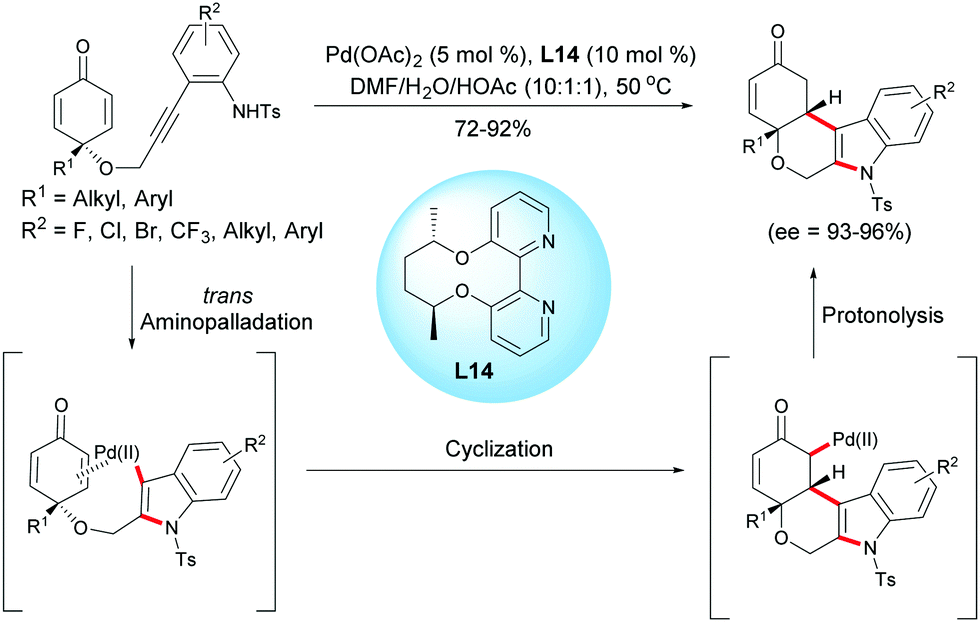
In 2017, Han, Lu et al. reported a palladium(II)-catalyzed aminopalladation/1,4-addition domino reaction.18 In the presence of Pd(OAc)2 (5 mol%) and the chiral bipyridine ligand, L14 (10 mol%), a variety of enantio-enriched cyclohexanone-fused tetrahydropyrano[3,4-b]indoles were obtained in high yields (72–92%) and excellent enantioselectivities (ee = 93–96%). The authors suggested that the domino process was initiated by an intramolecular aminopalladation of the alkyne which is followed by a conjugate addition of the vinylpalladium intermediate to the cyclohexadienone to form a palladium enolate. Protonation of the enolate leads to tetrahydropyrano[3,4-b]indoles and the palladium catalyst is regenerated for the next catalytic cycle (Scheme 18).18
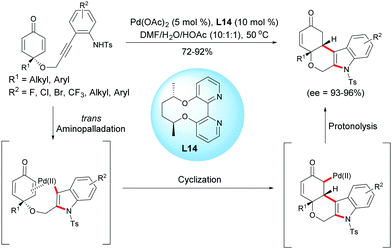 |
| | Scheme 18 Palladium-catalyzed aminopalladation/1,4-addition of alkyne-tethered cyclohexadienones. | |
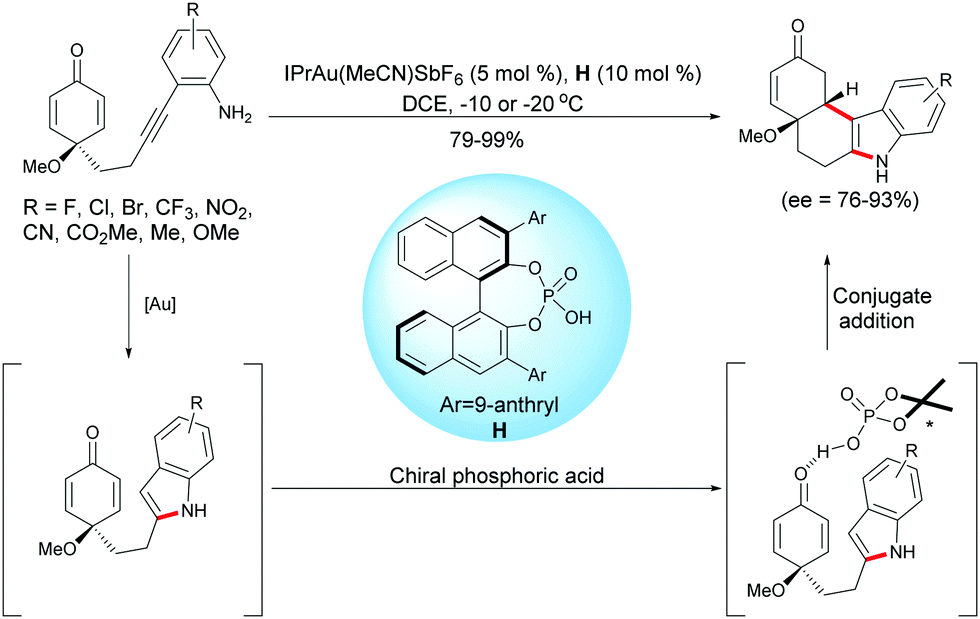
3.5 Au-Catalyzed enantioselective desymmetrization
As described above, Han, Lu et al. developed a palladium-catalyzed domino reaction for the synthesis of enantio-enriched tetrahydrocarbazoles. Han and coworkers have also developed an intramolecular hydroamination/1,4-addition domino reaction using a gold complex, IPrAu(MeCN)SbF6, in the presence of a chiral Brønsted acid such as the chiral phosphoric acid H.19 Under these conditions, diverse functionalized tetrahydrocarbazoles were obtained in excellent yields (79–99%) and with high enantiomeric excess (ee = 76–93%) (Scheme 19).
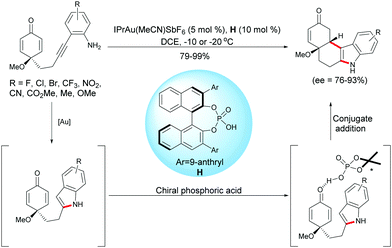 |
| | Scheme 19 Gold and chiral phosphoric acid relay catalysis for the desymmetrization of alkyne-tethered cyclohexadienones. | |
To gain insight into the reaction mechanism, the authors conducted the 1H NMR experiments to monitor the reaction, and these experiments supported the formation of an indole intermediate obtained by a gold-catalyzed intramolecular hydroamination of a tethered 2-ethynylaniline. As they were able to successfully isolated the indole intermediate substituted by a methyl ester on the aryl group (R = CO2Me), they treated this intermediate with the chiral phosphoric acid H. They found that the asymmetric Friedel–Crafts reaction took place smoothly as anticipated, which further demonstrated that the reaction proceeded through a relay gold-catalyzed hydroamination, followed by a chiral phosphoric acid-catalyzed intramolecular Friedel–Crafts type 1,4-addition of the in situ formed indole onto the cyclohexadienone.
4. Allene-tethered cyclohexadienones
Allene-tethered cyclohexadienones have also been involved in the enantioselective desymmetrization using transition metal catalysts, mainly copper and nickel, to produce functionalized fused bicyclic compounds.
4.1 Cu-Catalyzed enantioselective desymmetrization
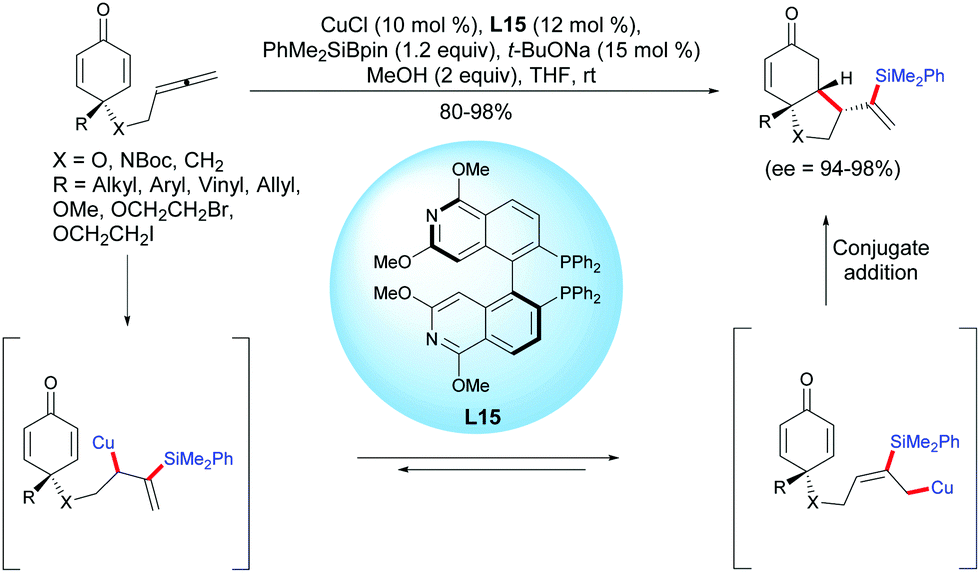
The unique physical and chemical properties of allenes make them attractive building blocks for organic synthesis. Compared to the well-established methods of alkene- and alkyne-tethered cyclohexadienones, the allene-tethered cyclohexadienones are relatively less studied. In 2015, Tian et al. reported a highly efficient copper-catalyzed asymmetric silylation/cyclization of allene-tethered cyclohexadienones.20 By using CuCl and (S)-P-Phos L15, a regioselective β-silylation of the pendant allene took place followed by an asymmetric 1,4-addition of the β-allylcopper intermediate to the cyclohexadienone moiety. A variety of enantio-enriched cis-hydrobenzofuran, cis-hydroindole, and cis-hydroindene scaffolds were obtained in high to excellent yields (80–98%) and excellent enantioselectivities when the reaction was performed with CuCl in the presence of the ligand L15 (ee = 94–98%) (Scheme 20).
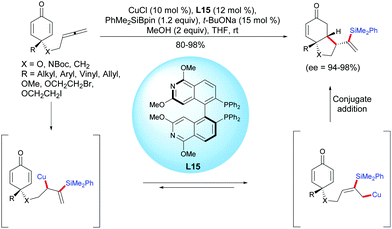 |
| | Scheme 20 Cu-Catalyzed asymmetric silylation/cyclization sequence of allene-tethered cyclohexadienones. | |
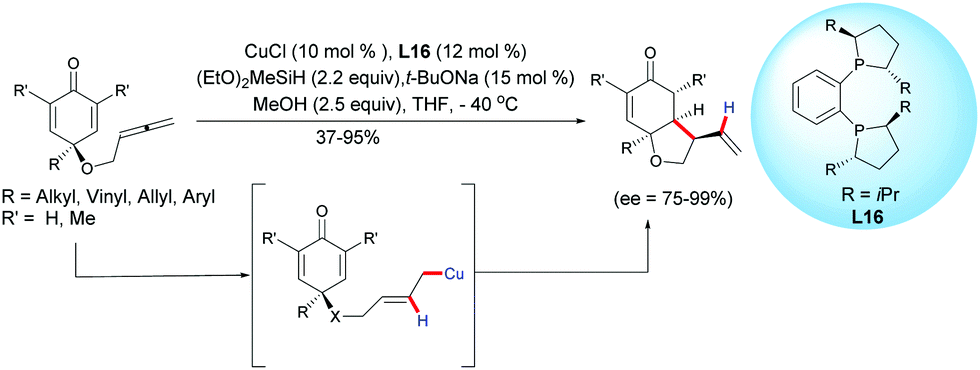
In 2018, Chen, Tian, Lin et al. extended the process to the challenging CuH-catalyzed asymmetric reductive coupling of allene-tethered cyclohexadienones.21 The process proceeded with CuCl, in the presence of diethoxy methylsilane and in the presence of the chiral ligand L16. A regioselective insertion of CuH to the pendant allene followed by an intramolecular 1,4-addition of the in situ generated alkylcopper intermediate led to cis-hydrobenzofurans with moderate to high yields (37–95%) and high to excellent enantioselectivities (ee = 75–99%) (Scheme 21).
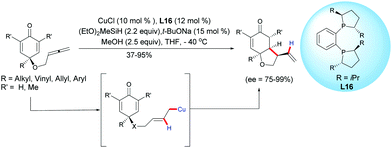 |
| | Scheme 21 CuH-Catalyzed asymmetric reductive coupling of allene-tethered cyclohexadienones. | |
4.2 Ni-Catalyzed enantioselective desymmetrization
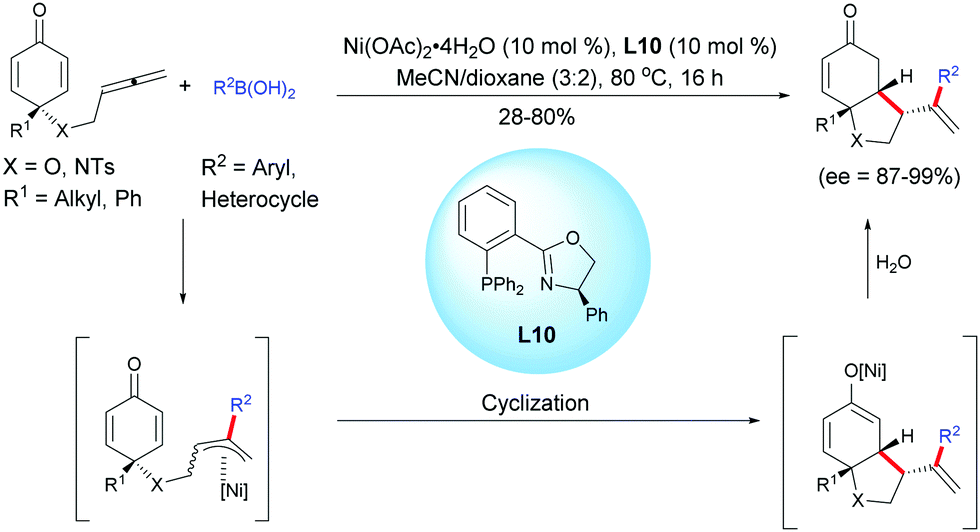
In 2018, Lam et al. described a nickel-catalyzed asymmetric arylation/cyclization of allene-tethered cyclohexadienones using arylboronic acids, Ni(OAc)2 and L10.22
The desymmetrization process underwent a nickel-catalyzed arylation and the resulting allylnickel intermediate underwent a 1,4-allylation, affording hexahydroindol-5-ones and hexahydrobenzofuran-5-ones in moderate to high yields (28–80%) and with high to excellent enantiomeric excess (ee = 87–99%). However, if the linker has an additional carbon, only traces of the desired six-membered ring products were detected (Scheme 22).
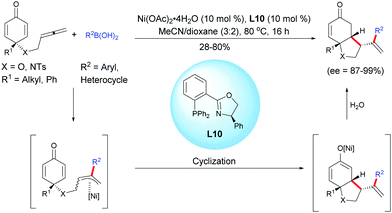 |
| | Scheme 22 Nickel-catalyzed asymmetric arylation/cyclization of allene-tethered cyclohexadienones. | |
5. Conclusion
The examples summarized in this review demonstrate the power of the asymmetric desymmetrization of C4-substituted cyclohexadienones in the construction of chiral complex scaffolds. The addition of the transition metals on the unsaturated tethers are very regio- and stereoselective, and due to the chelation of the transition metal by a chiral ligand, highly face selective and enantioselective reactions are taking place. Moreover, a wide range of functional groups on both the cyclohexadienone and on the unsaturated tethers are tolerated. Despite the progress made in the desymmetrization of alkene-, alkyne-, allene-tethered cyclohexadienones, and due to the diverse reactivities of the alkene, alkyne, allene and cyclohexadienone functionalities, more novel asymmetric domino reactions can be developed. For example, very recently 1,3-enynes-tethered cyclohexadienones have been involved in the asymmetric desymmetrization.23 In the future, it will be important to use cheap and non-toxic transition metals and to find an “universal” ligand to achieve the asymmetric desymmetrization of cyclohexadienones, and also to apply this efficient method to the synthesis of natural products and/or bioactive compounds.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- Selected reviews:
(a) G. Maertens, M.-A. Menard and S. Canesi, Synthesis, 2014, 1573 Search PubMed;
(b) K. A. Kalstabakken and A. M. Harned, Tetrahedron, 2014, 70, 9571 CrossRef CAS;
(c) A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474 RSC.
- Y.-X. Tan, F. Zhang, P.-P. Xie, S.-Q. Zhang, Y.-F. Wang, Q.-H. Li, P. Tian, X. Hong and G.-Q. Lin, J. Am. Chem. Soc., 2019, 141, 12770 CrossRef CAS.
- C.-Y. He, Q.-H. Li, X. Wang, F. Wang, P. Tian and G.-Q. Lin, Adv. Synth. Catal., 2020, 362, 765 CrossRef CAS.
- J. Keilitz, S. G. Newman and M. Lautens, Org. Lett., 2013, 15, 1148 CrossRef CAS.
- Z.-T. He, B. Tian, Y. Fukui, X. Tong, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2013, 52, 5314 CrossRef CAS.
- K. K. Gollapelli, S. Donikela, N. Manjula and R. Chegondi, ACS Catal., 2018, 8, 1440 CrossRef CAS.
- C.-L. Duan, Y.-X. Tan, J.-L. Zhang, S. Yang, H.-Q. Dong, P. Tian and G.-Q. Lin, Org. Lett., 2019, 21, 1690 CrossRef CAS.
- Q. Teng, N. Thirupathi, C.-H. Tung and Z. Xu, Chem. Sci., 2019, 10, 6863 RSC.
- Q. Teng, W. Mao, D. Chen, Z. Wang, C.-H. Tung and Z. Xu, Angew. Chem., Int. Ed., 2020, 59, 2220 CrossRef CAS.
- T. Shu, L. Zhao, S. Li, X.-Y. Chen, C. von Essen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 10985 CrossRef CAS.
- P. Liu, Y. Fukui, P. Tian, Z.-T. He, C.-Y. Sun, N.-Y. Wu and G.-Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700 CrossRef CAS.
- C.-Y. He, L.-B. Xie, R. Ding, P. Tian and G.-Q. Lin, Tetrahedron, 2019, 75, 1682 CrossRef CAS.
- C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068 CrossRef CAS.
- R. Kumar, Y. Hoshimoto, E. Tamai, M. Ohashi and S. Ogoshi, Nat. Commun., 2017, 8(32), 1 Search PubMed.
- R. Tello-Aburto, K. A. Kalstabakken and A. M. Harned, Org. Biomol. Chem., 2013, 11, 5596 RSC.
- K. Takenaka, S. C. Mohanta and H. Sasai, Angew. Chem., Int. Ed., 2014, 53, 4675 CrossRef CAS.
- C. He, C. Zhu, Z. Dai, C.-C. Tseng and H. Ding, Angew. Chem., Int. Ed., 2013, 52, 13256 CrossRef CAS.
- J. Chen, X. Han and X. Lu, Angew. Chem., Int. Ed., 2017, 56, 14698 CrossRef CAS.
- F. Zhao, N. Li, Y.-F. Zhu and Z.-Y. Han, Org. Lett., 2016, 18, 1506 CrossRef CAS.
- Z.-T. He, X.-Q. Tang, L.-B. Xie, M. Cheng, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2015, 54, 14815 CrossRef CAS.
- Y.-X. Tan, X.-Q. Tang, P. Liu, D.-S. Kong, Y.-L. Chen, P. Tian and G.-Q. Lin, Org. Lett., 2018, 20, 248 CrossRef CAS.
- T. L. N. Nguyen, C. A. Incerti-Pradillos, W. Lewis and H. W. Lam, Chem. Commun., 2018, 54, 5622 RSC.
- During the revision of the manuscript, Gao, Tian, Lin and co-workers reported an enantioselective copper-catalyzed reductive coupling desymmetrization of 1,3-enyne-tethered cyclohexadienones for the synthesis of chiral exocyclic allenes. C.-Y. He, Y.-X. Tan, X. Wang, R. Ding, Y.-F. Wang, F. Wang, D. Gao, P. Tian and G.-Q. Lin, Nat. Commun., 2020, 11, 4293 CrossRef CAS.
Footnote |
| † This review is dedicated to the memory of Prof. Dieter Enders. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Janine
Cossy
and
Janine
Cossy