
Advances in polycyclization cascades in natural product synthesis
Yubo
Jiang†
 ab,
Ryan E.
McNamee†
a,
Philip J.
Smith†
a,
Ana
Sozanschi†
a,
Zixuan
Tong†
a and
Edward A.
Anderson
*a
ab,
Ryan E.
McNamee†
a,
Philip J.
Smith†
a,
Ana
Sozanschi†
a,
Zixuan
Tong†
a and
Edward A.
Anderson
*a
aChemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: edward.anderson@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 285000
bFaculty of Science, Kunming University of Science and Technology, Kunming 650500, China
First published on 23rd November 2020
Abstract
Cascade reactions (also known as domino reactions) are arguably the most powerful means to achieve the construction of multiple ring systems in a single step. In this Tutorial Review, highlights in cascade polycyclizations applied to natural product syntheses over the last five years are discussed, including pericyclic, ionic, metal-catalyzed, organocatalytic, and radical processes. Significant developments in each of these fields that have advanced the state-of-the-art are a particular focus, including photochemical and electrochemical methods, novel biomimetic routes, and enantioselective cascades.
 Yubo Jiang | Yubo Jiang completed his PhD in Chemistry from Tongji University in 2011 where he worked under the supervision of Prof. Chunxiang Kuang, including a year with Prof. Jianbo Wang at Peking University as a visiting fellow. He then joined the Kunming University of Science and Technology. He is currently a one-year visiting scholar working with Prof. Edward Anderson in University of Oxford. Professor Jiang's research interests revolve around the development of new coupling reactions through C–H functionalization, and ynamide chemistry. |
 Ryan E. McNamee | Ryan McNamee received his MSc in Medicinal Chemistry from Queen's University Belfast, having worked on the catalytic asymmetric synthesis of spirocyclic oxindoles with Dr Peter Knipe. He is currently a DPhil student with Professor Ed Anderson at the University of Oxford, where his research focuses on the synthesis and reactions of strained rings. |
 Philip J. Smith | Philip Smith completed his MChem degree at the University of Oxford. He is now part of the EPSRC Synthesis for Biology and Medicine CDT at the University of Oxford and is working with Professor Ed Anderson to develop novel transformations of yndiamides and ynamides, with a particular focus on their use in cycloisomerisation reactions to form substituted nitrogen heterocycles. |
 Ana Sozanschi | Ana Sozanschi received her BSc Honours in Medicinal Chemistry from University College Dublin in 2019. She is currently a DPhil student with Prof. Edward Anderson at the University of Oxford, where her research focuses on the synthesis and modification of antiparasitic natural products. |
 Zixuan Tong | Zixuan Tong gained his BSc in chemistry from City University of Hong Kong in 2019. After that, he joined Prof. Ed Anderson as a DPhil student in organic chemistry, where he is working on transition metal-catalyzed functionalizations of yndiamides. |
 Edward A. Anderson | Ed Anderson is Professor of Organic Chemistry at the University of Oxford. He began his independent career as an EPSRC Advanced Research Fellow in 2007, during which he was appointed as Associate Professor at Jesus College, Oxford in 2009, and then as Professor in 2016. His research interests encompass a wide range of synthetic organic chemistry, including natural product total synthesis, transition metal catalysis and mechanistic study, bicyclo[1.1.1]pentanes and related small rings, antiparasitic natural products, the chemistry of ynamides and yndiamides, and EPR spectroscopy in nucleic acids. |
Key learning points(1) Cascade (domino) polycyclizations are among the most efficient, ambitious, and elegant tools for the synthesis of polycyclic natural products.(2) A variety of reaction types (pericyclic, ionic, metal-catalyzed, organocatalytic and radical) are suitable for the design of these processes. (3) Contemporary methodologies, such as photocatalysis and synthetic electrochemistry, have emerged as new tools in cascade polycyclizations. (4) There is an increasing use of chiral catalysts to induce or control stereochemistry in the polycyclization. (5) Transition metal-catalyzed cascades continue to offer efficient and unique methods for bond formation to prepare polycyclic natural products. |
Introduction
The total synthesis of natural products remains a fascinating and fast-changing area of chemical research,1 not only providing access to bioactive compounds for applications in biology and medicine,2 but also stimulating the development of novel strategies and synthetic methods. The increasing pressure for sustainable technologies has touched many fields of chemical research, and natural product synthesis is no exception: contemporary total syntheses ideally not only attain the desired target, but do so with step-, atom- and redox economy.3,4 One of the most powerful ways to achieve this goal is to employ cascade (also known as domino) reactions, in which multiple bond formations occur sequentially, in a single reaction. Such processes typically lead to a rapid increase in molecular complexity while reducing overall step count, delivering efficiency in an elegant manner. This review highlights recent developments in cascade reactions that generate the polycyclic cores of natural products as a key step in their total synthesis. Within the constraints of this review, it is not possible to offer an exhaustive coverage of the field; the focus instead lies on new strategies and technologies in cascade polycyclization that have emerged over the last five years. In line with our previous coverage of this topic,5,6 these cyclizations are broadly classified into pericyclic, ionic, metal-catalyzed, organocatalytic and radical processes, although overlap is inevitable. Indeed, it is precisely the orthogonality between these various classes that is key to designing the most creative and efficient cascades.Pericyclic cascades
Pericyclic reactions are among the most elegant complexity-increasing transformations in organic synthesis. Chemists have long recognized Nature's use of cascade processes in biosynthetic pathways,7 and indeed often take inspiration from Nature in the design of pericyclic synthetic routes. One such example is the Gleason group's total synthesis of the isoxazolidine natural product (−)-virosaine A (Scheme 1).8 The dense cage polycycle structure of this natural product renders it a challenging yet attractive synthetic target, with a rare (3+2) nitrone-olefin cycloaddition proposed in its biosynthesis. The group envisaged that ring-opening of an epoxide (1) by a tethered oxime nucleophile could be used to form this nitrone, with (3+2) cycloaddition generating the carbocyclic portion of the core ring system. In the event, simple heating of epoxide 1 in acetic acid indeed effected regioselective epoxide ring-opening to nitrone 2, which spontaneously underwent the proposed biomimetic cycloaddition to give 3 in an excellent 92% yield. While formation of the virosaine butenolide ring was unsuccessful under various C–H insertion strategies, the relatively acidic nature of the bridgehead C–H bond enabled functionalization by carbamate-directed lithiation/bromination (4). (−)-Virosaine A was synthesized in an impressive 10 steps and an overall yield of 9%.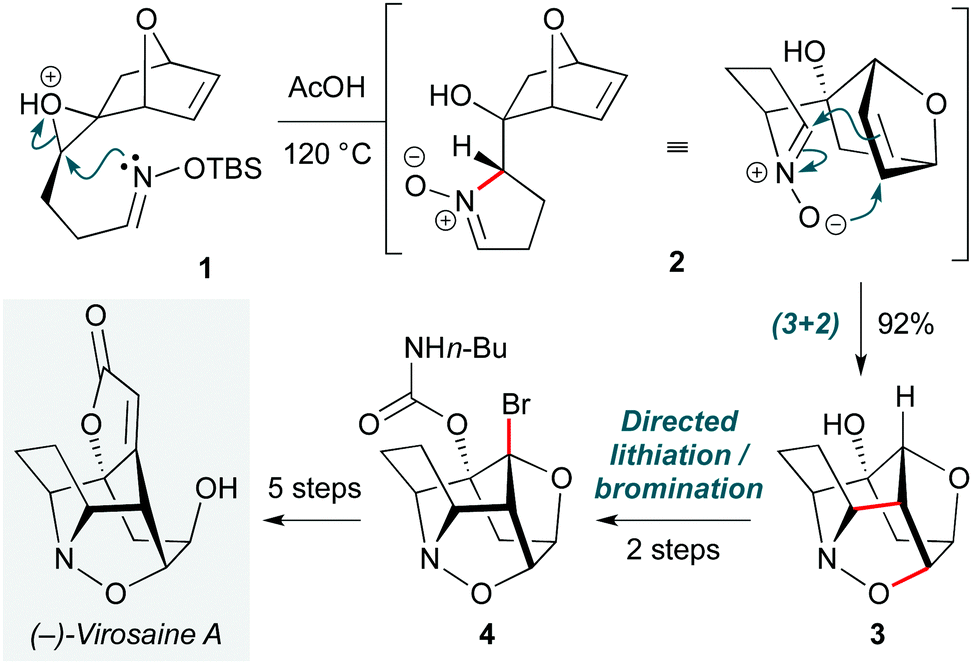 | ||
| Scheme 1 Cascade epoxide opening/1,3-dipolar nitrone-olefin cycloaddition in the synthesis of (−)-virosaine A (Gleason and co-workers).8 | ||
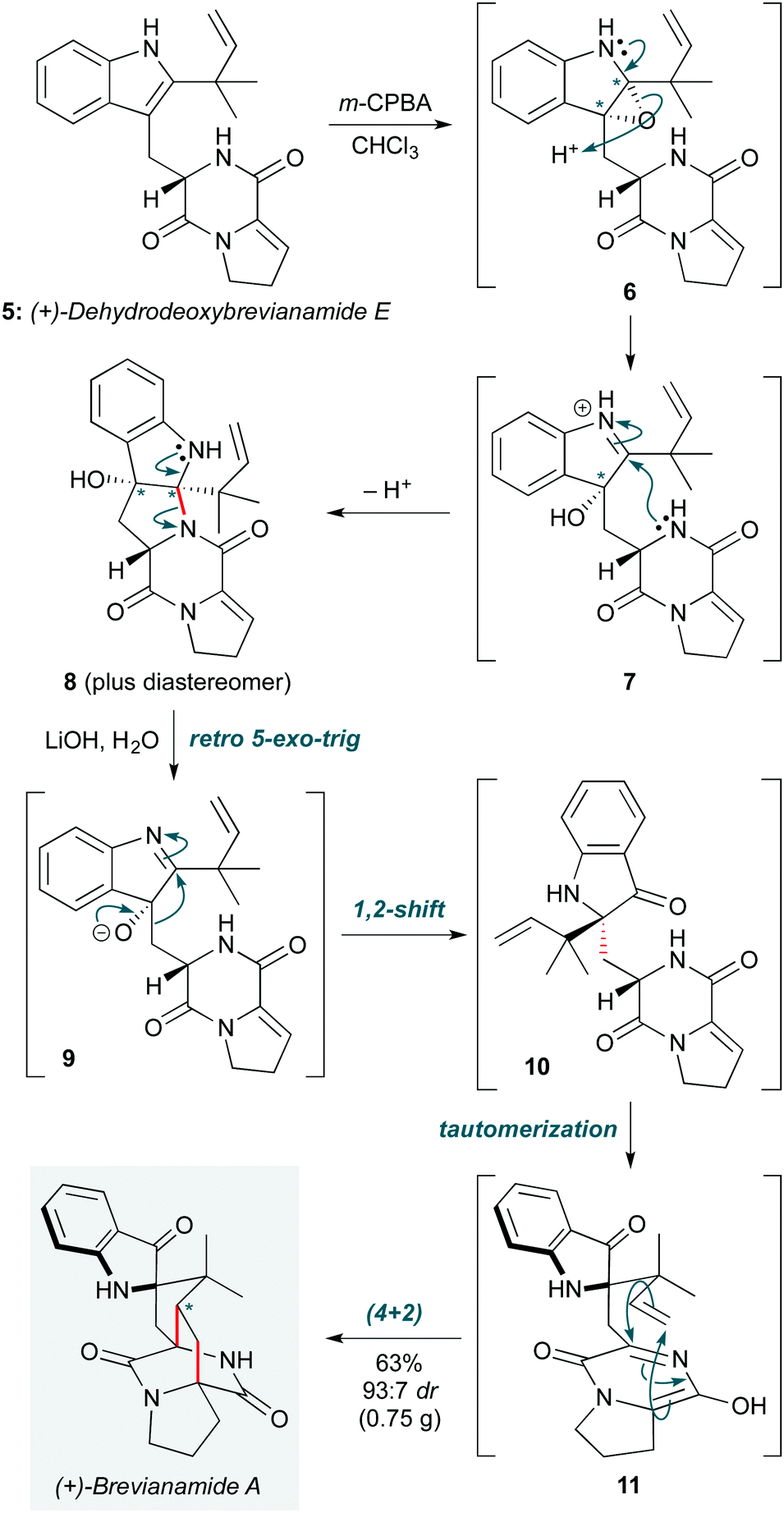
The theme of biosynthetic cascades also inspired Lawrence and co-workers to propose a modified biosynthesis of the diketopiperazine brevianamide A (Scheme 2), enabling the group to solve this 50 year challenge in natural product synthesis.9 Key to this work was the proposal of a different biosynthetic precursor to brevianamide A to that in previous biosynthetic hypotheses10 – specifically (+)-dehydrodeoxy brevianamide E (5). This natural product, which was prepared in five steps, was first treated with m-CPBA to effect a chemoselective oxidation of the indole. The likely diastereomeric epoxide intermediates resulting from this step underwent spontaneous ring opening/cyclization by the pendent diketopiperazine to afford two separable diastereomeric adducts (6→7→8). Treatment of the desired isomer 8 under basic conditions triggered a reverse of this cyclization to liberate putative intermediate 9, which underwent a 1,2-alkyl shift to give 10. Tautomerization of the diketopiperazine ring to azadiene 11 set up an aza-Diels–Alder cycloaddition to afford (+)-brevianamide A, and its diastereomer (+)-brevianamide B, in a 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 ratio (63%). This is similar to the ratio observed in the natural biosynthesis, supporting the revised biosynthetic hypothesis. This cascade completed the natural product synthesis in just 7 steps in total, with an overall yield of 7.2%.
:7 ratio (63%). This is similar to the ratio observed in the natural biosynthesis, supporting the revised biosynthetic hypothesis. This cascade completed the natural product synthesis in just 7 steps in total, with an overall yield of 7.2%.
 | ||
| Scheme 2 N,N-Acetal ring-opening, 1,2-alkyl shift, and aza-Diels–Alder reaction cascade in the biomimetic synthesis of brevianamide A (Lawrence and co-workers).9 Asterisks indicate the position of diastereomers (see text). | ||
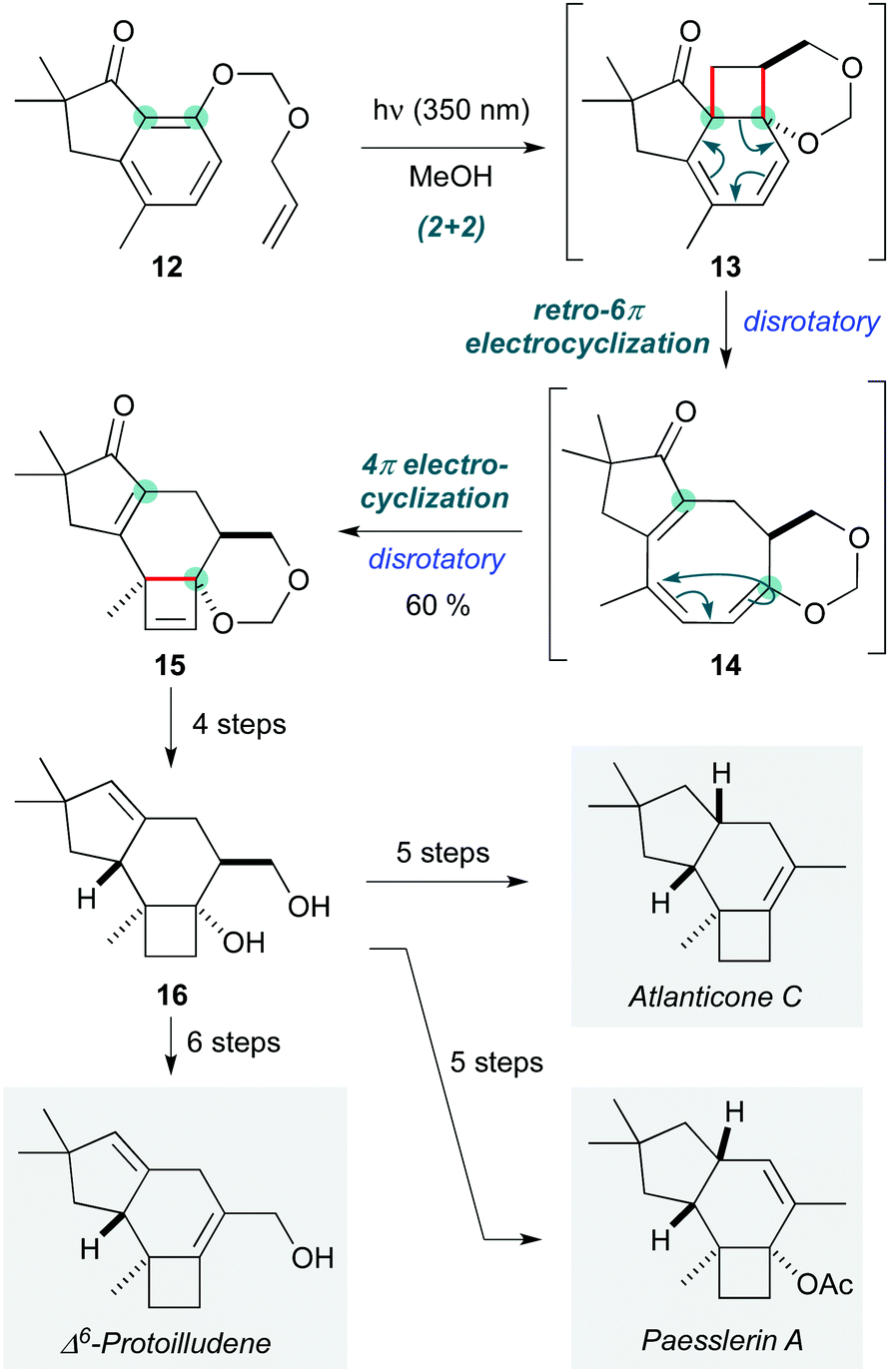
Chemists are by no means limited to following in Nature's footsteps when it comes to designing pericyclic cascades, and this is particularly the case for photochemical processes. Building on earlier explorations,11,12 the Bach group implemented an elegant pericyclic cascade to construct the tricyclic protoilludane skeleton found in the natural product atlanticone C (Scheme 3).13 This process proceeds by an initial (2+2) ortho-photocycloaddition of 12 (under irradiation at 350 nm in methanol) to afford a short-lived cyclohexadiene intermediate 13, which undergoes thermal disrotatory 6π-electrocyclic ring opening to give cyclooctatriene 14. Under the photochemical conditions, disrotatory 4π-electrocyclization of 14 afforded tetracycle 15 in 60% yield, an impressive outcome given that this sequence involves the formation of two four-membered rings. The remainder of the synthesis entailed a series of redox manipulations to adjust the oxidation state at various positions on the protoilludane skeleton; common intermediate 16 was thus converted to atlanticone C (14 steps longest linear sequence), and its relatives Δ6-protoilludene and paesslerin A.
 | ||
| Scheme 3 Photochemical pericyclic cascade resulting in the protoilludane skeleton, and application to the synthesis of three members of the family (Bach and co-workers).13 | ||
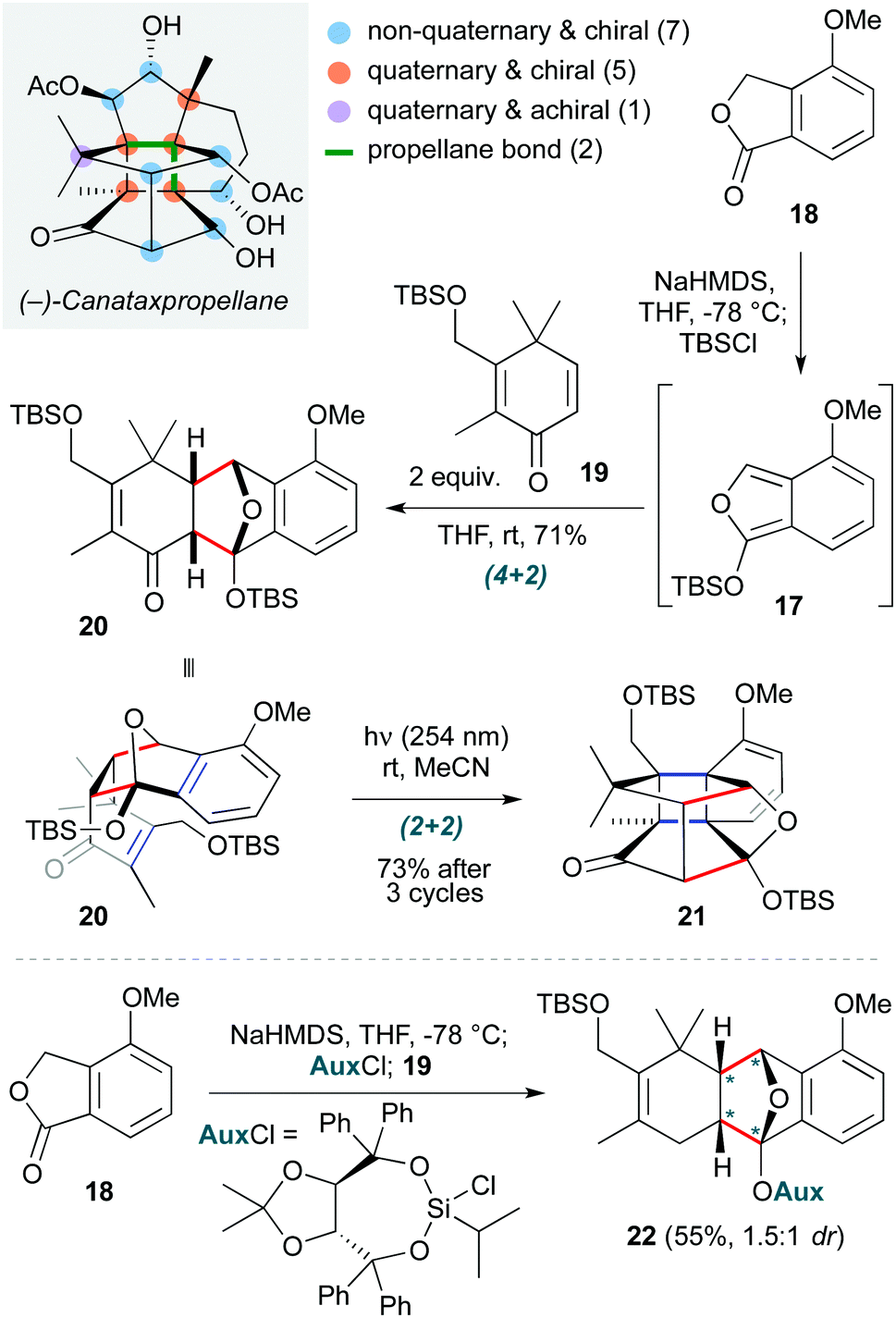
This ‘pericyclic’ section concludes with a total synthesis masterpiece: completion of the first synthesis of the complex taxane diterpene canataxpropellane by Gaich and co-workers (Scheme 4).14 This is a fascinating natural product, comprising the taxane skeleton with three additional transannular C–C bonds, which result in a double-propellane core featuring six contiguous quaternary carbons (four of which are on the cyclobutane ring at the heart of the molecule). While not a true cascade, two pericyclic steps are employed in direct succession early in the synthesis to establish the most challenging elements of this core. The first is a Diels–Alder cycloaddition of isobenzofuran 17 (formed by deprotonation/silylation of isobenzofuranone 18) with dienone 19, which afforded cycloadduct 20 in 71% yield with exceptional diastereoselectivity. The residual alkene in this endo product is in sufficiently proximity to the arene to then undergo a (2+2) ortho-photocycloaddition (irradiation at 254 nm in acetonitrile), affording propellane 21 in 73% yield after three irradiation cycles. The two cycloadditions were carried out on decagram scale, and a creative auxiliary approach was used to obtain enantioenriched material 22via formation of a chiral siloxy-isobenzofuran for the initial Diels–Alder cycloaddition. The overall yield of canataxpropellane was 0.5% (29 steps), with over half of the synthesis being carried out on decagram scale, highlighting its practicality and efficiency.
 | ||
| Scheme 4 Diels–Alder cycloaddition/(2+2) photocycloaddition yielding the core of canataxpropellane, and the asymmetric Diels–Alder version (Gaich and co-workers).14 Asterisks indicate the position of diastereomers, relative to the auxiliary. | ||
Ionic cascades
Cationic polyene cyclizations are commonplace in nature as a powerful means to construct complex architectures such as polycyclic terpenoid natural products.15 While many methods have been developed that mimic such pathways, the reactivity of heteroatom-substituted polyenes has received less attention, potentially since heteroatom functionalities might typically be introduced by ‘post-cyclase’ oxidations. Nonetheless, the realization of such cascades could facilitate access to a wide range of terpenoid natural products by enhancing the reactivity of the polyene. Building on seminal work in this field by Corey and Lin,16 this concept has been elegantly exploited in Magauer and co-workers’ total synthesis of (−)-cyclosmenospongine (Scheme 5),17 where a ‘non-biomimetic’ cationic polyene cyclization cascade of precursor 23, bearing two heteroatom substituents, was triggered by a Lewis acid-promoted epoxide ring-opening. On treatment with ethylaluminium dichloride, polyene 23 was transformed into a short-lived (but isolable) ketal 24via attack of the enol ether on the activated epoxide, then cyclization of the resultant aluminium alkoxide onto the oxocarbenium ion arising from the enol ether. This epoxide ring-opening likely proceeds via a concerted mechanism, with the stereochemical outcome dictated by the configurations of the enol ether and epoxide; however, formation of the bicyclic ketal is reversible under the reaction conditions, and regeneration of the oxocarbenium ion 25 triggers cyclization by the exocyclic thio enol ether to give sulfonium ion 26. The stereoselectivity of this step relies on the C8-stereocentre, but not on the geometry of the thio enol ether. 26 in turn is trapped by the adjacent axial p-methoxyphenyl (PMP) group in an intramolecular Friedel–Crafts reaction, affording the final meroterpenoid 27 as a single diastereomer in excellent yield (83%). A further eight steps completed the synthesis of (−)-cyclosmenospongine, which represents the first example of a cationic polyene cyclization that culminates in an exo-termination step. The Magauer group recently applied a similar approach to a synthesis of the natural product pimara-15-en-3α,8α-diol.18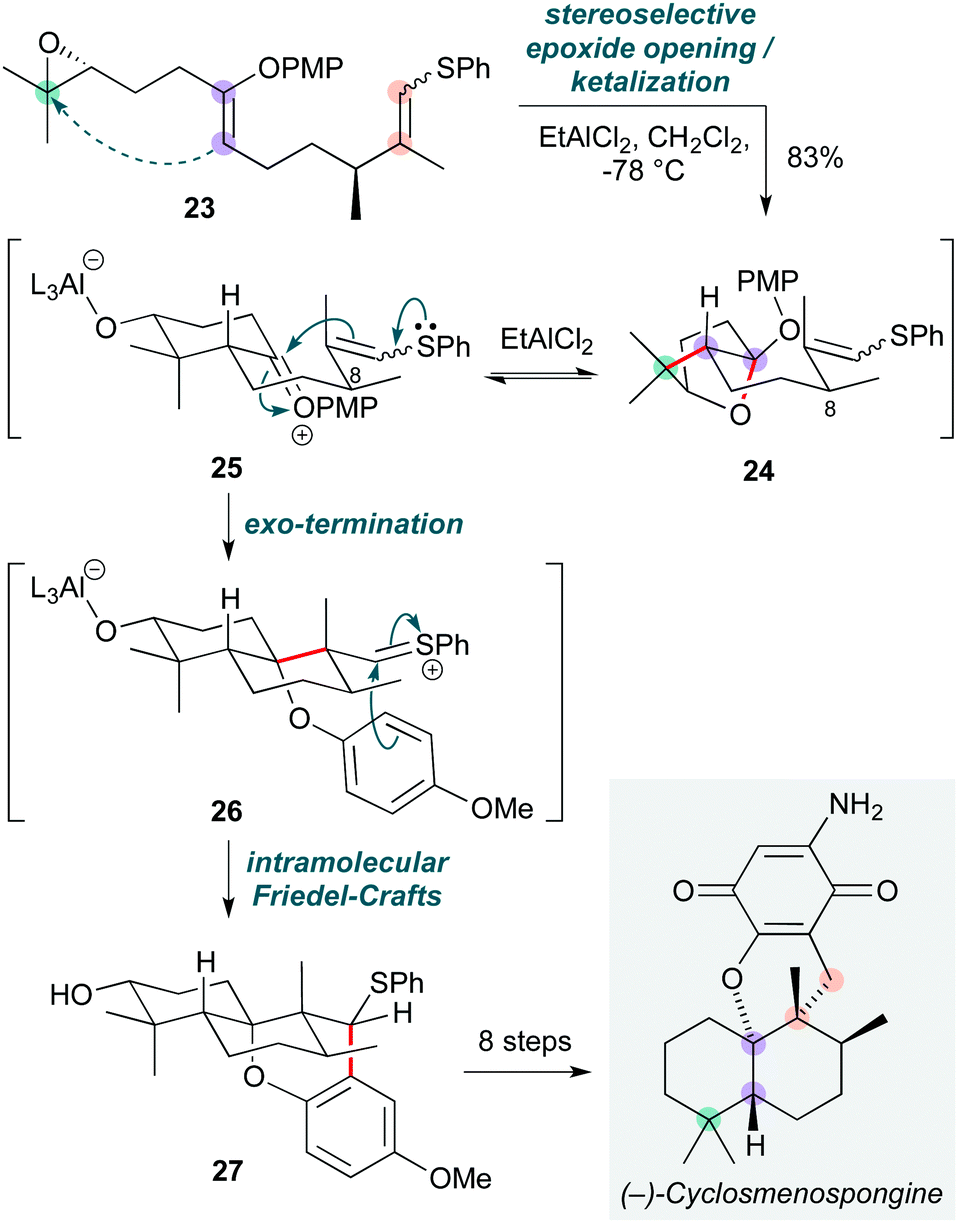 | ||
| Scheme 5 Non-biomimetic polyene cyclization towards (−)-cyclosmenospongine (Magauer and co-workers).18 | ||
Polycyclizations triggered by epoxide opening are by no means restricted to polyenes. Tong and co-workers used this concept to promote the cyclization of a vinylepoxy-δ-keto-alcohol (VEKA) 28 (Scheme 6) to forge the common tricyclic spiroketal core of several members of the alotaketal family.19 The cascade commenced with triene 29, desilylation of which with HF·Py, then chemoselective epoxidation of the most substituted alkene (on the less-hindered face) with dimethyldioxirane (DMDO), afforded the unstable VEKA 28 (10:1 dr). Treatment of 28 with triflic acid triggered the cyclization cascade via protonated epoxide 30, which underwent an SN2′ ring-opening by the proximal hemiketal, generating the tricyclic spiroketal 31 in good yield on multigram scale (56% over three steps). This spiroketal core ultimately led to (−)-alotaketals A–D and (−)-phorbaketal A – a unified asymmetric approach which offers a promising entry to other members of this natural product family.
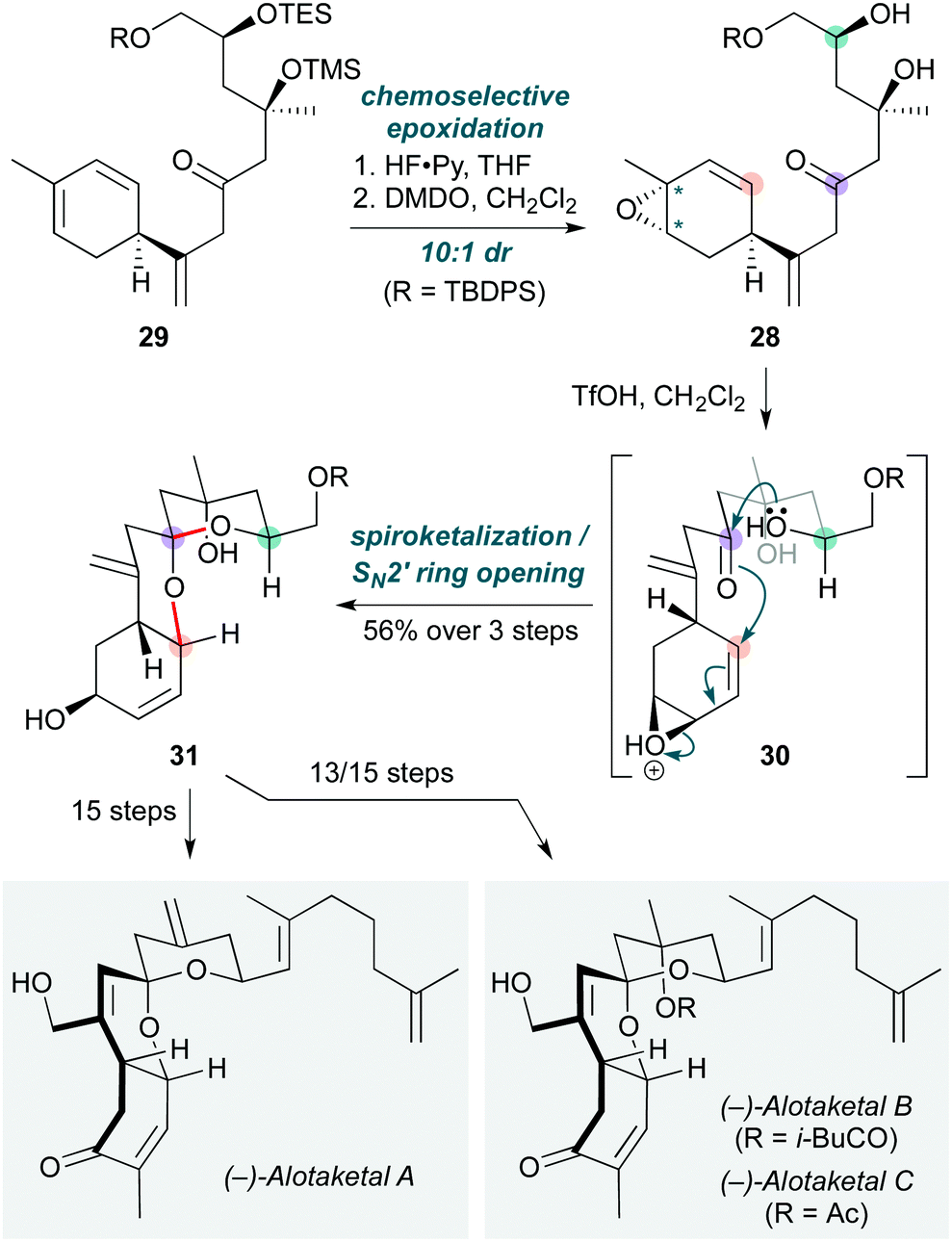 | ||
| Scheme 6 Cascade cyclization to the tricyclic spiroketal core of the alotaketal natural products (Tong and co-workers).19 Asterisks indicate the position of diastereomers. | ||
Epoxides, enol ethers and propellanes feature in a further oxidative cationic cascade in a synthesis of the monoterpene indole alkaloid cymoside (Scheme 7).20 The Vincent group directly accessed the cage hexacyclic core 32 of this natural product from strictosidine derivative 33. The transformation starts with a chemo- and stereoselective epoxidation of the indole ring with oxaziridine 34. Under the acidic reaction conditions, the resulting epoxide 35 undergoes epoxide ring opening, generating iminium ion 36, onto which 5-enolexo-exo-trig cyclization of the enol ether leads to oxocarbenium ion 37. The electrophilic carbon atom of this moiety is positioned in proximity to the hydroxyl group that resulted from the previous epoxide opening, and spontaneous ketalization takes place, producing the hexacyclic skeleton 32 as a single diastereomer in high yield (72%). Following further transformations, including late-stage β-glucosidation, 32 was converted to cymoside. The choice of a nosyl protecting group for the quinolizidine nitrogen proved important not only to protect the secondary amine from oxaziridine-mediated oxidation, but also to enhance the diastereoselectivity of the epoxidation through the proposed positioning of the nosyl aryl group under the indole ring.
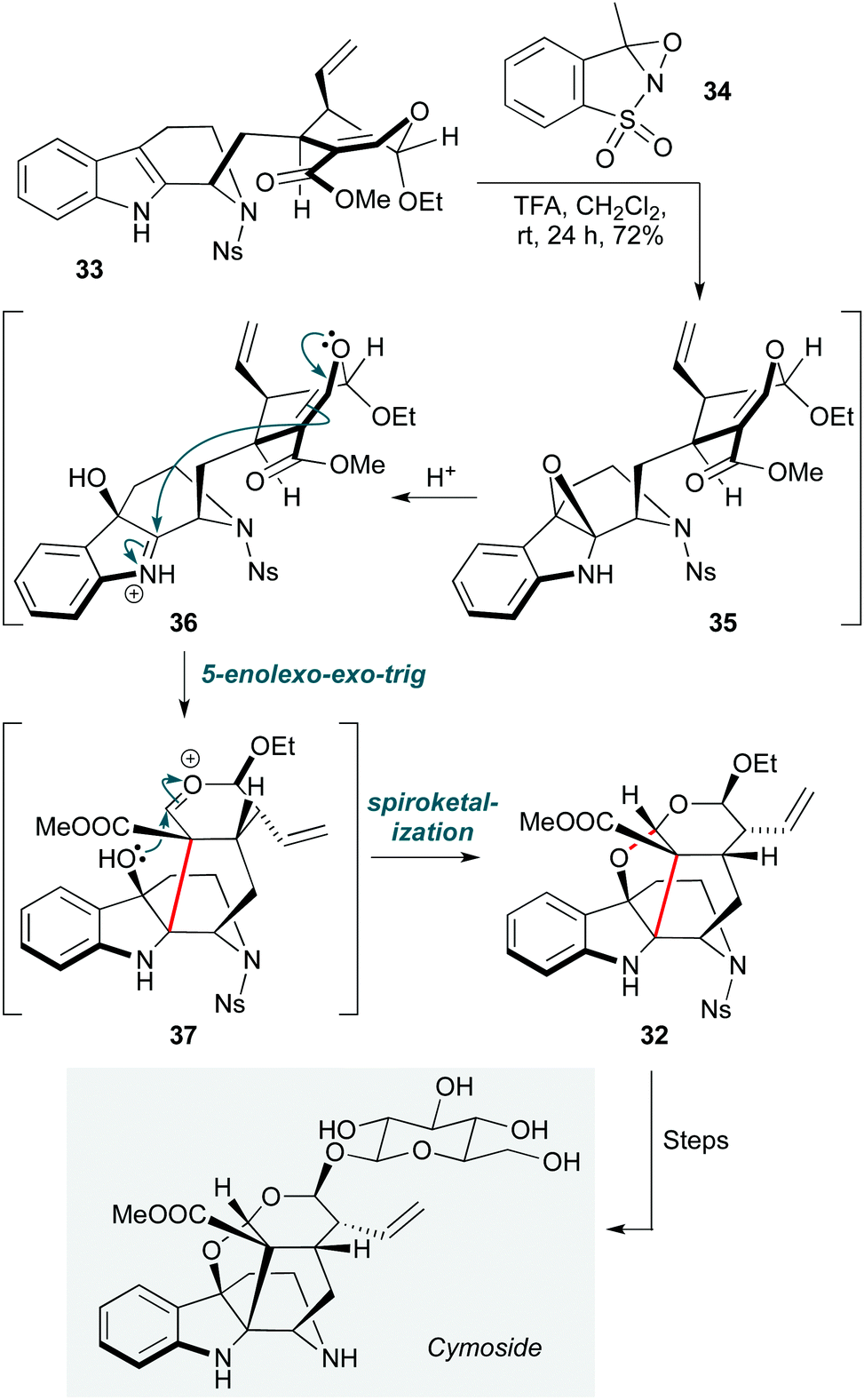 | ||
| Scheme 7 Oxidative cyclization towards cymoside (Vincent and co-workers).20 | ||
Biomimetic approaches can not only inspire the invention of cascades, but also provide solutions to key transformations towards natural products. In this vein, Trauner and co-workers employed a diastereoselective biomimetic transannular cyclization cascade to assemble the oxabicyclo[3.2.1]octane motif of (+)-aspergillin PZ in the final step of its synthesis (Scheme 8).21 Treatment of the bis-TBS-protected diol 38 with hydrogen fluoride in acetonitrile gave the natural product in 89% yield via a transannular Prins-Michael type cyclization, then capture of the intermediate carbocation 39 by the proximal oxygen nucleophile atom to form the oxabicyclic bridge. This transformation is particularly interesting since the lowest energy conformer of 40 is such that a different (trans-fused) product might be expected. The group proposed that the protonated enone that is presumably generated exists as an equilibrium mixture of conformational isomers, where the less-populated conformer 41 undergoes cyclization at a higher rate to install the observed (and required) cis-ring junction. While it is not clear at what stage the silyl ethers are removed, or whether the corresponding diol might exhibit different behaviour under acidic conditions, this manifestation of the Curtin–Hammett principle in a biosynthetic context is both appealing and essential to the success of the transformation.
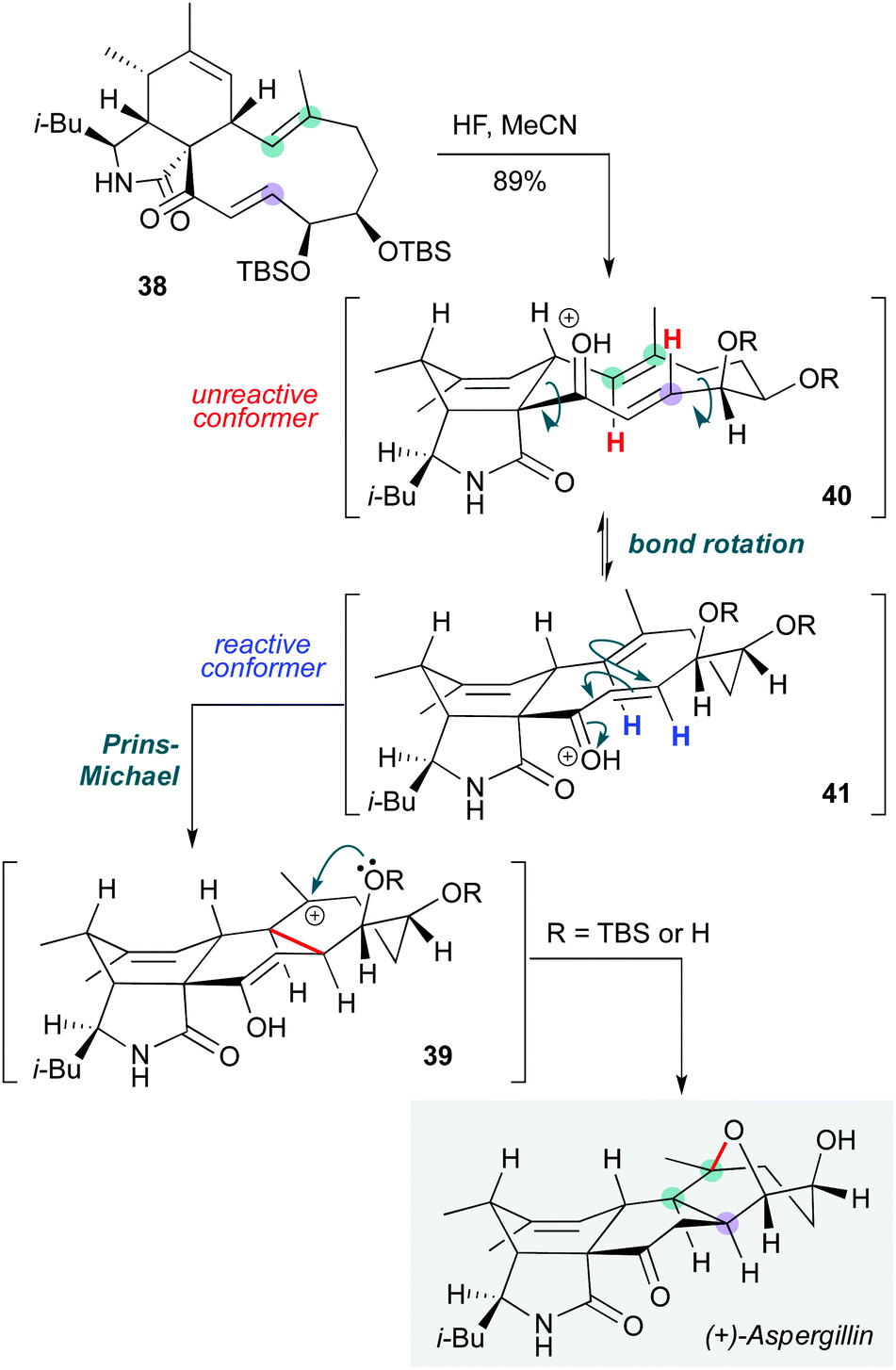 | ||
| Scheme 8 Biomimetic Prins-Michael cyclization cascade towards (+)-aspergillin (Trauner and co-workers).21 | ||
Intermolecular polycyclization cascades are significantly more challenging to engineer than intramolecular sequences, especially where stereochemistry must be introduced. An impressive example was reported by Nagorny and co-workers, who used a copper-catalyzed Michael/aldol cascade in a modular total synthesis of ouabagenin and a number of other cardiotonic steroids (Scheme 9).22 The cascade reaction was deployed early in the synthesis to access a common intermediate 42, from which many natural and non-natural steroids could be derived. The reaction itself begins with a diastereoselective copper-catalyzed Michael addition between 43 and 44, which is proposed to proceed via open transition state 45. The allylic benzoyloxy stereocentre is crucial for the diastereoselectivity of this addition, where stereoelectronically-favoured positioning of the C–O bond perpendicular to the enone π-system, with the smaller hydrogen atom on the ‘inside’ of the allyl unit, blocks the top face of the enone and favours approach of the copper-coordinated nucleophile from the bottom face with its cyclohexene ring in the least hindered quadrant. The enol in the resulting Michael adduct (which is formed as a single diastereomer) then undergoes a double acid-catalyzed aldol cyclization via intermediates 46, 47 and 48. Based on previous studies by the same group,23 it is likely that the first of these cyclizations occurs onto the cyclopentadione subunit 46, which then undergoes a second aldol cyclization to produce steroid 50. This was converted to 42, the common precursor to the cardenolides ouabagenin and its relatives, enabling anticancer structure–activity relationship studies.
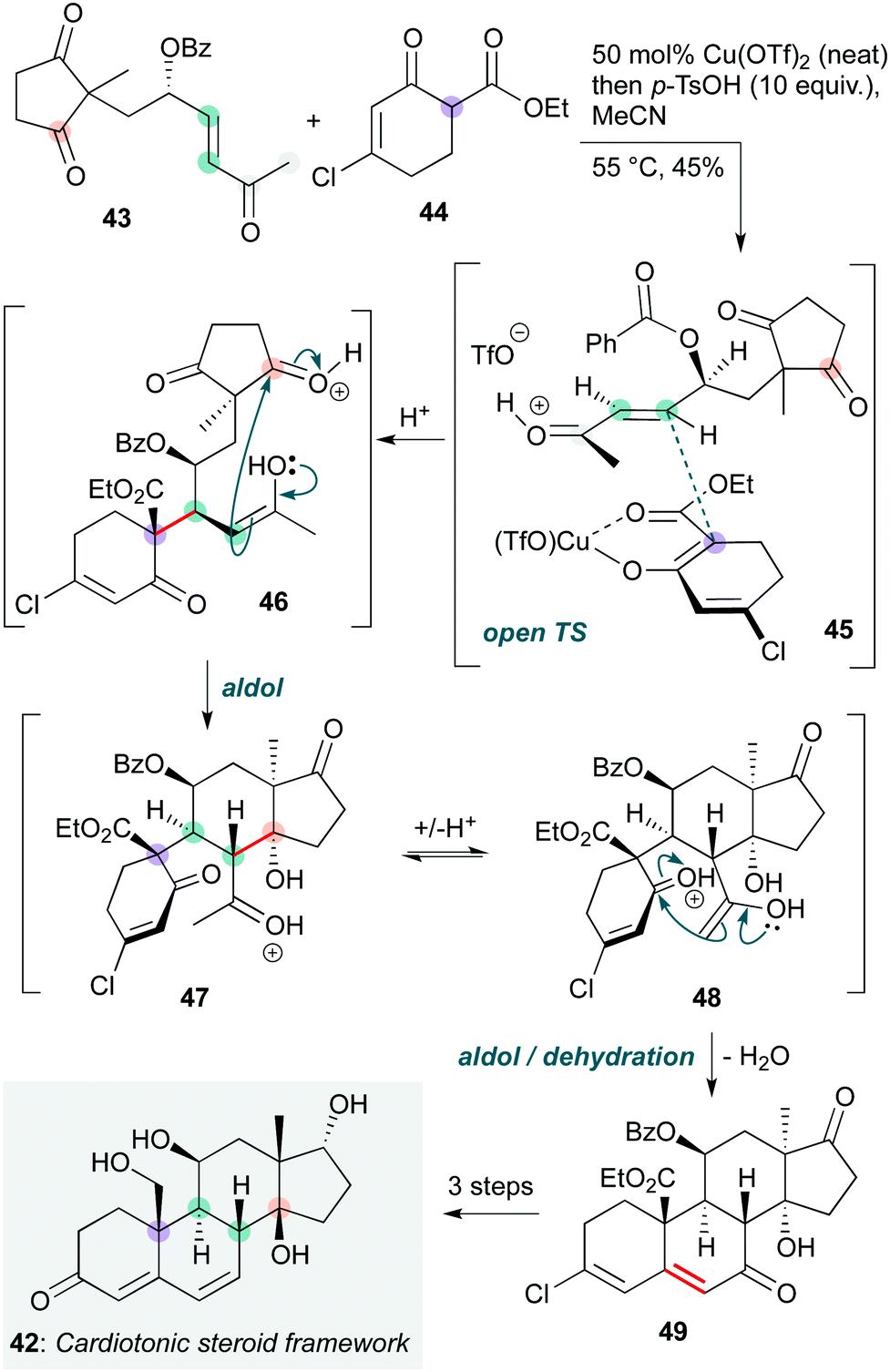 | ||
| Scheme 9 Copper-catalyzed Michael/aldol cascade towards the core framework of ouabagenin and other cardiotonic steroids (Nagorny and co-workers).22 | ||
Transition metal-catalyzed cascades
Transition metal-catalyzed reactions are indispensable in organic synthesis. In the context of cascade processes, they rank among the most prominent methods for accomplishing the construction of natural products with high selectivity and efficiency, not least due to their ability to access unique reaction pathways.A prime example is the position occupied by gold(I) catalysis, which shows particularly versatility in the electrophilic activation of C–C π-bonds.24 While applications in total synthesis are numerous,25 innovations continue to be described; three examples have been selected to highlight new modes of Au(I) activation in cascade processes. The first of these is the synthesis of curcusones I and J (Scheme 10) by Li and Dai,26 which is notable due to the use of a single gold catalyst to perform a twofold substrate activation. This cascade first involves formation of furan 50 through gold-catalyzed 5-endo-dig cyclization of enyne alcohol 51 followed by isomerization of 52. Complexation of the pendent allene to the gold catalyst then leads to the formal π-allyl cation 53, which undergoes (4+3) cycloaddition with the furan to construct the oxabicyclic 5,7-fused ring 54, the core of curcusones I and J. While this work is somewhat compromised by the lengthy sequence to convert 54 to the natural products, this does not detract from the efficiency of construction of this tricyclic core from a simple acyclic precursor. These syntheses revealed that the structures of the natural products require revision, as the NMR spectra of the synthetic samples did not match the reported data.
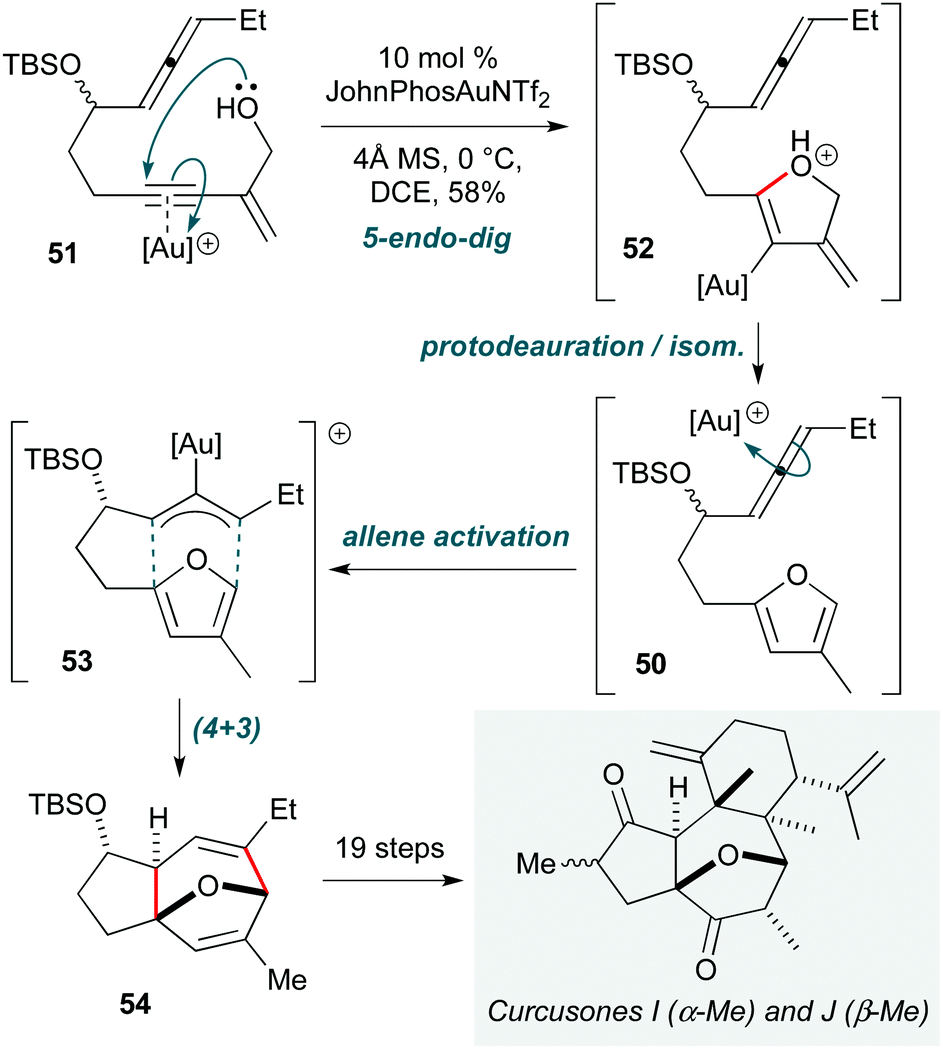 | ||
| Scheme 10 Au-Catalyzed cascade towards curcusones I and J (Li and Dai).26 | ||
Conjugated diynes are somewhat uncommon precursors to poly-heterocycles. The Ohno group achieved the total synthesis of dictyodendrins B, C, E, and F, with the key step involving a beautiful cascade of gold-catalyzed annulations on a 1,3-diyne 55 to form the natural product pyrroloindole core 56 (Scheme 11).27 This cascade initiates with nucleophilic attack of the azide group in diyne 55 onto the gold-activated diyne, producing gold carbene 57via elimination of N2 from 56.28 Subsequent Friedel–Crafts type reaction of this carbenoid at C3 of N-Boc pyrrole 59 delivered the pyrrole-substituted 2-alkynylindole intermediate 60, with the C2/C3 regioselectivity of this step depending critically on the pyrrole protecting group. This intermediate underwent a gold-promoted 6-endo-dig cyclization, generating pyrrolo-[2,3-c]carbazole derivative 56, which corresponds to the core of dictyodendrins B, C, E, and F.
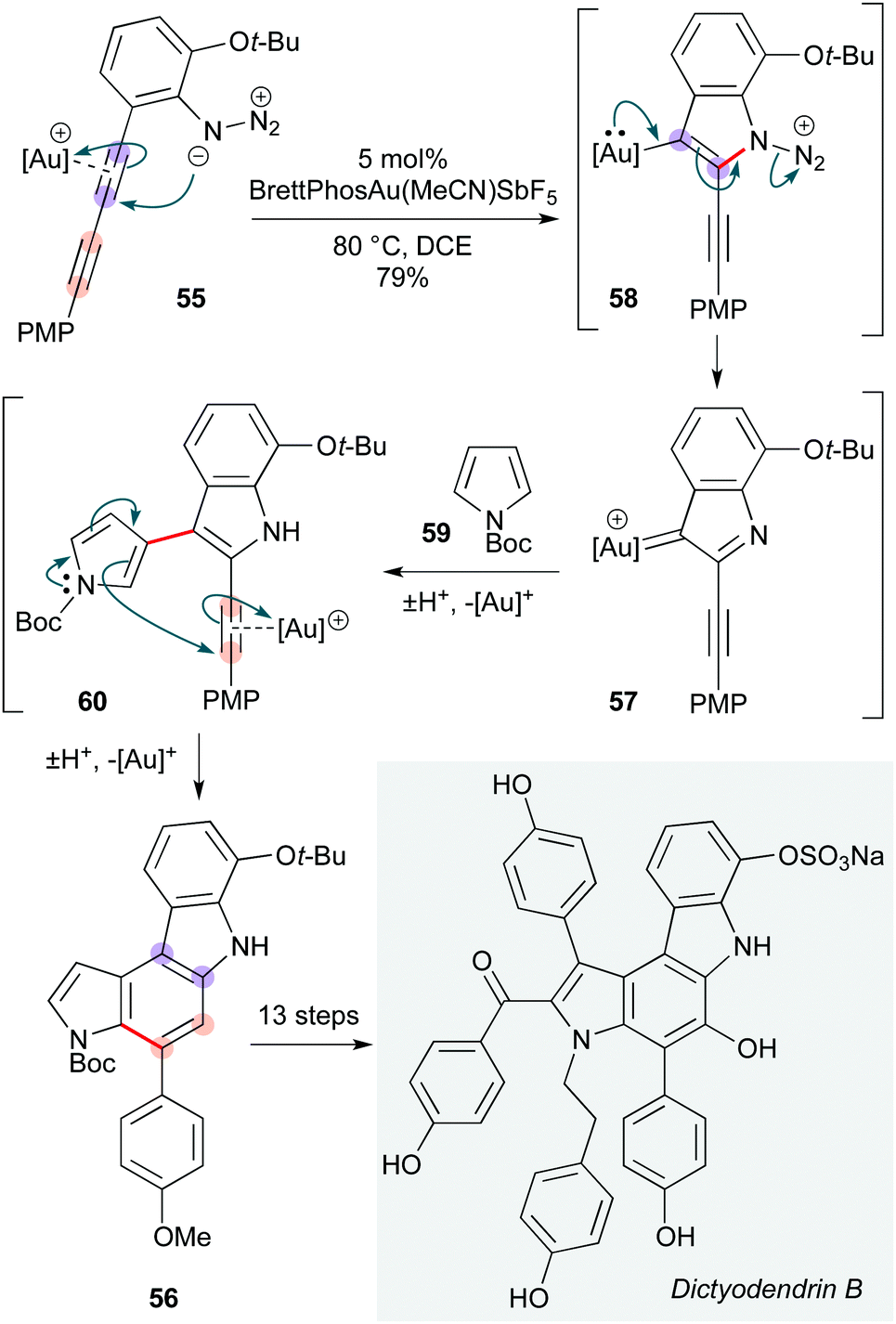 | ||
| Scheme 11 Au-Catalyzed route to dictyodendrins B, C, E and F (Ohno and co-workers).27 | ||
The Echavarren group recently developed a most impressive Au(I)-catalyzed bis-cyclopropanation of geranyl acetone 61 (Scheme 12), in which acetylene gas itself (generated in situ from calcium carbide) serves as the substrate.29 This cascade affords the tricyclo[5.1.0.02,4]octane natural product waitziacuminone in a single step, and in terms of atom and step economy constitutes a ‘near-ideal’ synthesis.3 Stereoselectively generating four C–C bonds and three rings, the reaction proceeds via initial cyclopropanation of 61 by the η2-acetylene gold(I) complex 62 to form cyclopropyl gold carbene 63; the site-selectivity and diastereoselectivity of this step was determined computationally to favour the terminal alkene by ∼1.6 kcal mol−1. 63 then effects a second intramolecular cyclopropanation to complete the natural product.
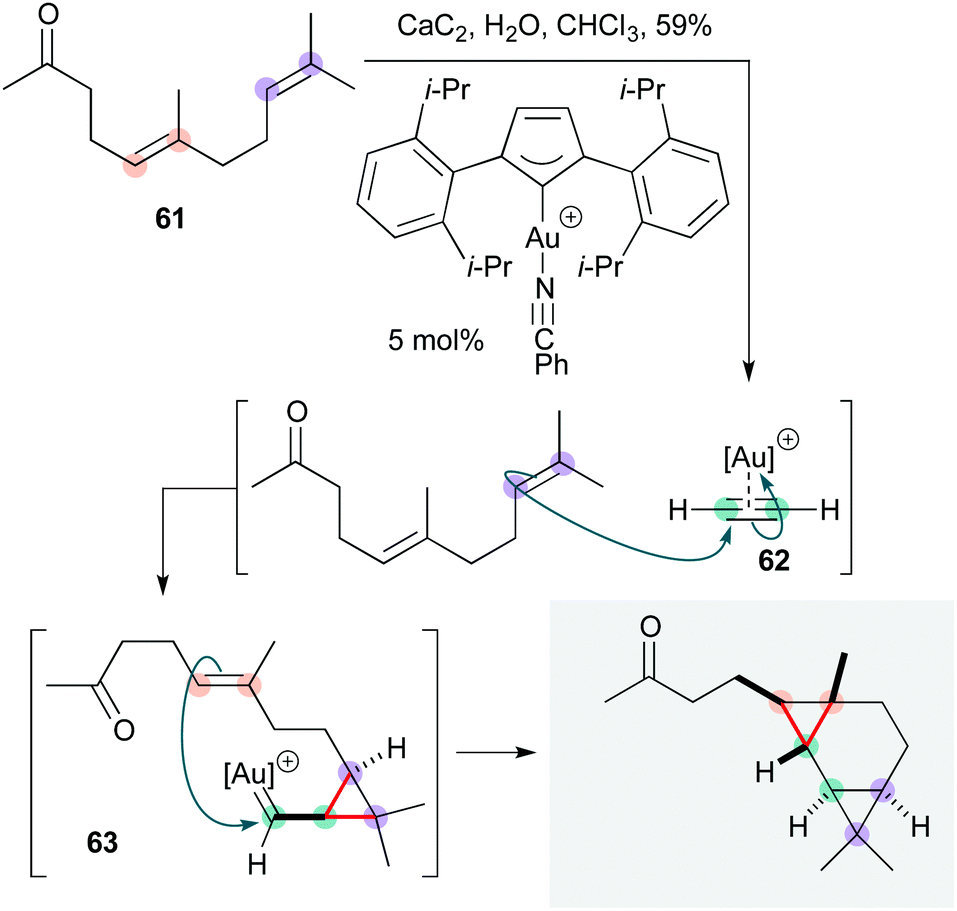 | ||
| Scheme 12 Au-Catalyzed cascade synthesis of waitziacuminone (Echavarren and co-workers).28 | ||
Another transition metal with a long history in cascade reactions is palladium, not least due to its position at the head of transition metal catalysis in organic chemistry. Nonetheless, as with gold catalysis, new applications of palladium-catalyzed cascades continue to be invented.30,31 An example is found in the first total synthesis of chaetophenol C by Li et al. (Scheme 13),32 where Pd(OAc)2 was used as a π-acid to activate the alkyne in o-alkynylbenzaldehyde 64 towards cyclization by the adjacent aldehyde. The resultant benzopyrilium ion 65 underwent a regio- and stereoselective (4+2) cycloaddition with acrylate enol ether 66, with the ensuing oxocarbenium ion 67 being trapped by the silyl ester to assemble the tetracyclic system 68 in a highly efficient manner. This cascade constructs four new bonds, three rings and four stereocenters (two of which are quaternary) in one step; 68 was converted to the natural product by simple desilylation.
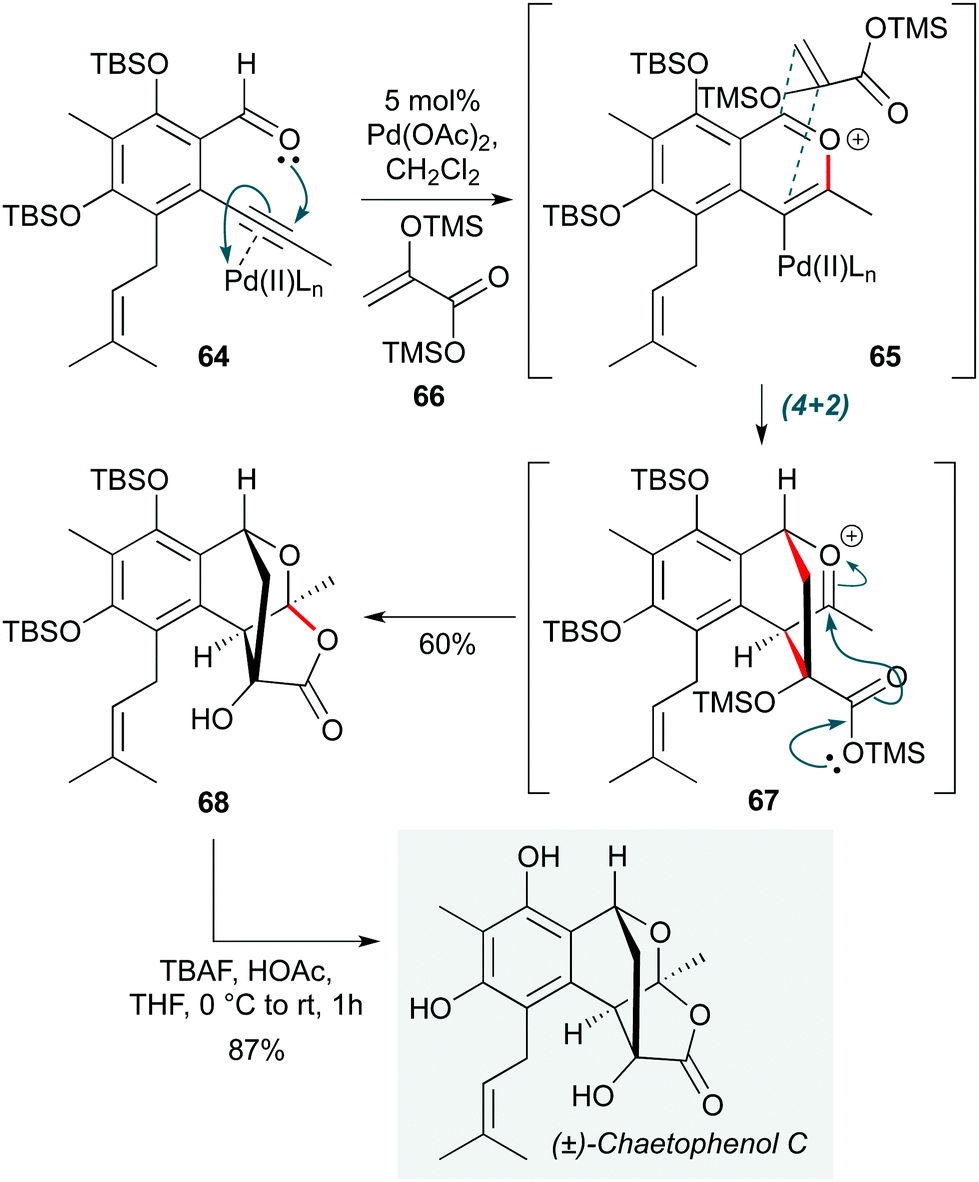 | ||
| Scheme 13 Pd-Catalyzed cascade towards chaetophenol C (Li and co-workers).32 | ||
Another novel use of a Pd-catalyzed cascade was reported by the Zhao group, who established the first total synthesis of (±)-cephanolides B and C (Scheme 14).33 This ingenious polycyclization is built around an ‘interrupted Catellani’ reaction:34 cyclization of substrate 70via oxidative addition and migratory insertion generates σ-alkylpalladium(II) species 71, which is incapable of undergoing β-hydride elimination due to Bredt's rule. Instead, CO insertion forms acylpalladium(II) complex 72, from which ortho C–H activation gives cyclopentanone 73 after reductive elimination of Pd(0). This was readily converted to the natural products cephanolides B and C by oxidation state adjustment. The presence of an acetal on the oxabicyclo[2.2.2]octene was crucial to achieve the desired diastereoselectivity in the initial migratory insertion, as use of the lactone led to the opposite stereochemical outcome.
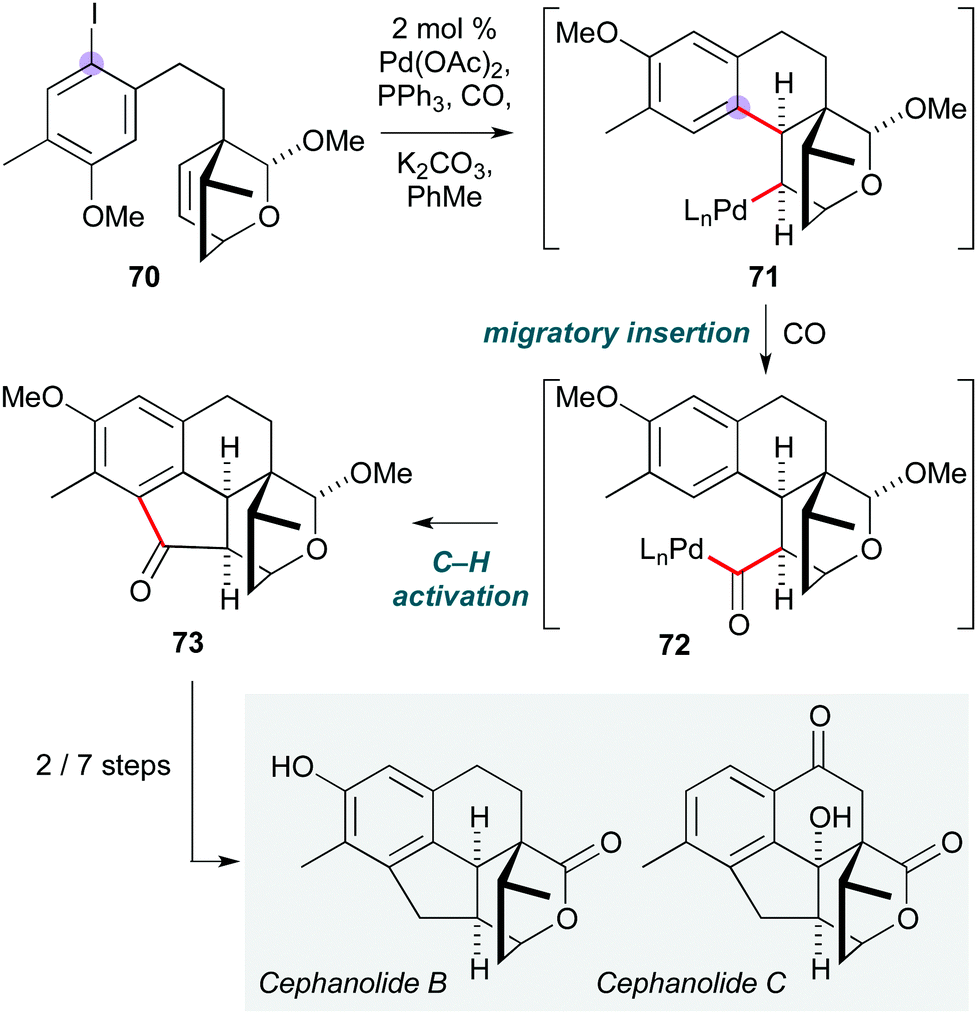 | ||
| Scheme 14 Pd-Catalyzed cascade to form cephanolides B and C (Zhao and co-workers).33 | ||
(2+2+2) Cyclotrimerizations are well established as a useful tactic in polycyclization processes. The ability of this reaction to operate in complex settings was recently reiterated by the Anderson group in highly convergent total syntheses of the natural products rubriflordilactone B and pseudo-rubriflordilactone B (Scheme 15).35 Rhodium-catalyzed cyclotrimerization of triyne 73 (assembled by chromium-mediated addition of iodoalkyne 74 to aldehyde 75) was achieved in the penultimate step of the synthesis using Wilkinson's catalyst. This reaction, which likely proceeds through rhodacyclopentadiene 76, afforded hexacycle 77, acidic dehydration of the benzylic alcohol of which gave the natural product. Application of equivalent chemistry to prepare the computationally predicted structure of pseudo-rubriflordilactone B confirmed its structure, thus resolving the stereochemical ambiguity of this intriguing natural product.
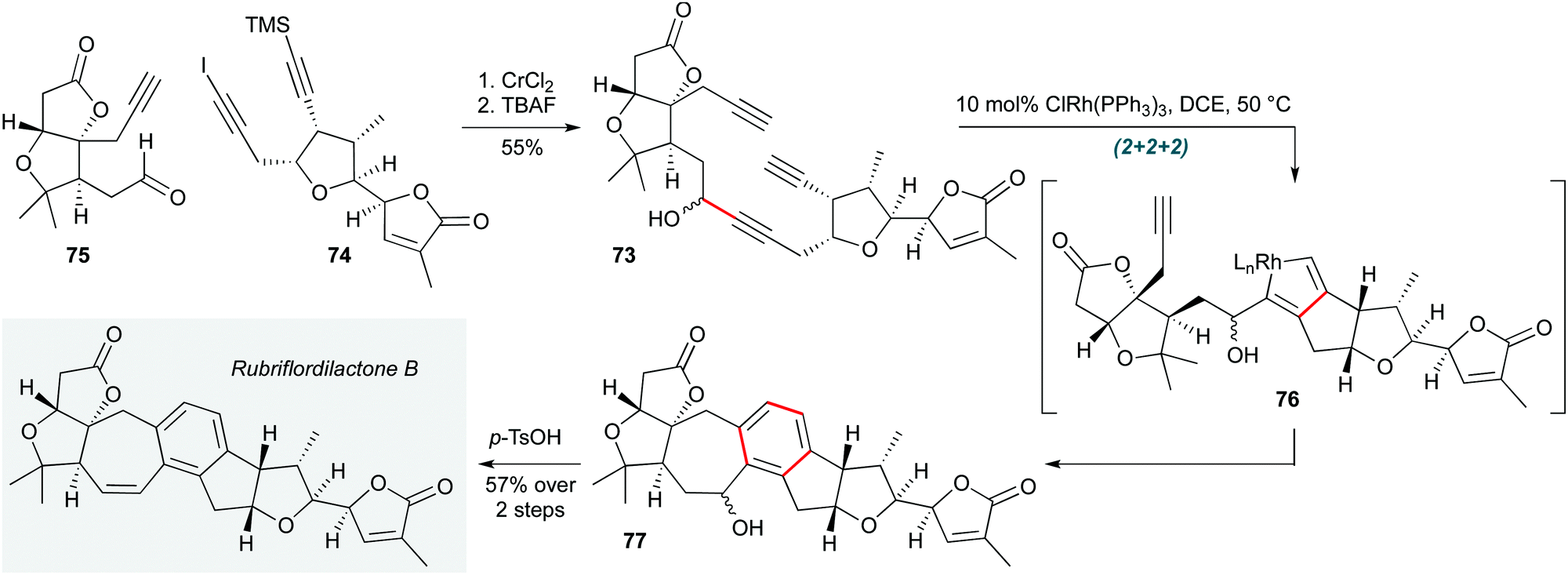 | ||
| Scheme 15 Rh-Catalyzed cyclotrimerization towards rubriflordilactone B (Anderson and co-workers).35 | ||
Organocatalytic cascades
In recent years, the forefront of organocatalysis has moved towards fields such as asymmetric bifunctional organocatalysis36 and asymmetric phase transfer catalysis.37 Applications of such methods in natural product polycyclizations are no exception, and two notable examples are presented that epitomise the former of these concepts.The use of chiral hydrogen bond donors enables a myriad of enantioselective transformations, and is appealing due to the easily tuned nature of the organocatalyst. In this vein, Zhu and co-workers achieved the first total synthesis of (+)-peganumine A (Scheme 16) by employing a remarkable organocatalytic cascade to assemble the octacyclic natural product core from indoles 78 and 79 in the final step of the synthesis.38 This process commences with condensation of 6-methoxytryptamine 78 with the ketone motif in 79 to form imine 80. This underwent an asymmetric Pictet–Spengler reaction, with the facial selectivity of the cyclization controlled by Jacobsen's thiourea catalyst 81 in combination with benzoic acid. While the exact pathway by which asymmetric induction is achieved is not clear, the resultant aza-Friedel–Crafts product 82 was then treated (without workup) with trifluoroacetic acid, promoting tautomerization of the enamide to an N-acyliminium ion intermediate 83. Transannular cyclization of the proximal piperidine amine afforded 84, and in situ N-Boc deprotection, yielded (+)-peganumine A in 92% ee and 69% yield. The efficiency of this convergent strategy is such that the natural product could be synthesized on gram scale in only 7 steps, with an impressive overall yield of 33%.
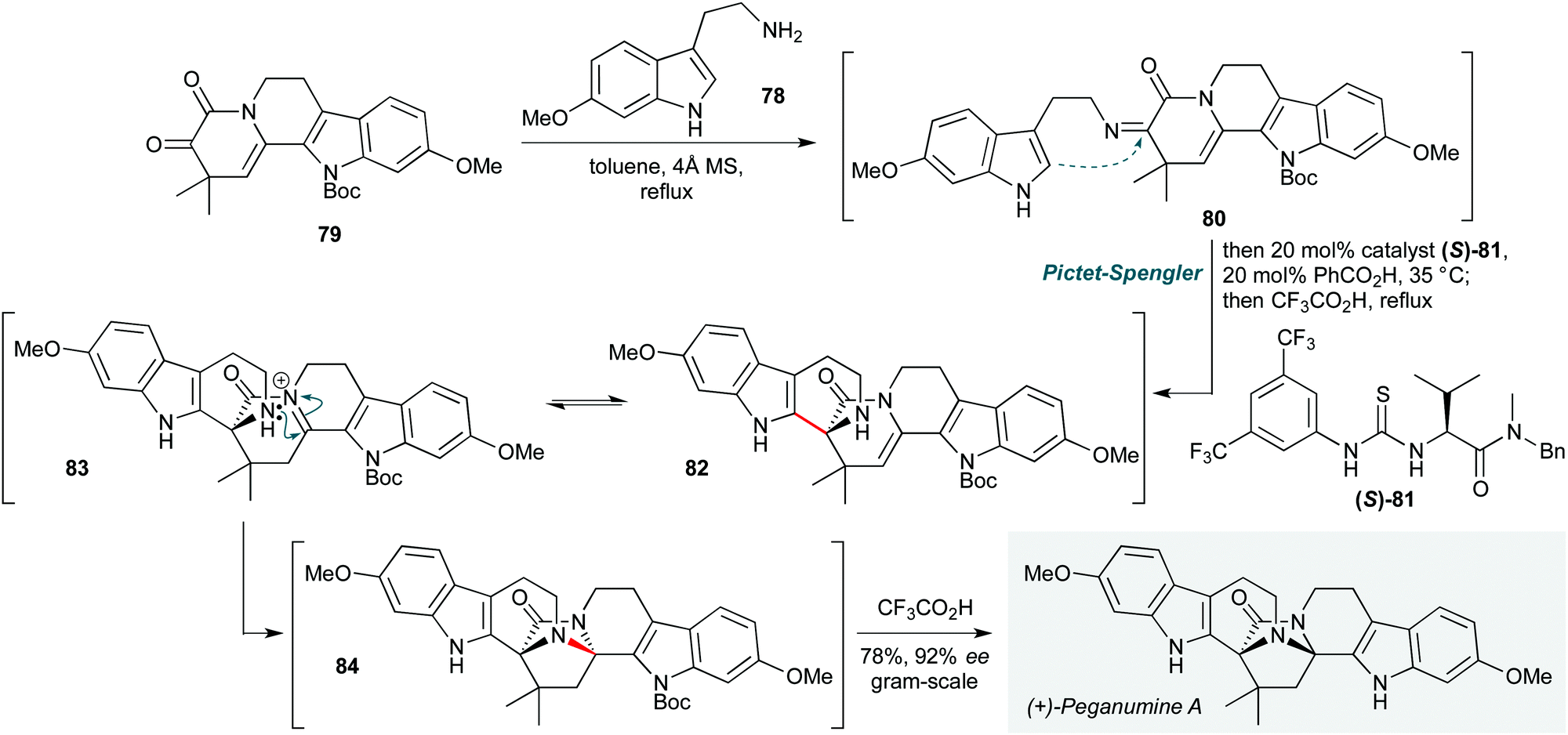 | ||
| Scheme 16 Total synthesis of (+)-peganumine A using a thiourea-catalyzed Pictet–Spengler reaction/acid-mediated transannular cyclization (Zhu and co-workers).38 | ||
Himalensine A (Scheme 17) has been the target of many research groups. Dixon and co-workers achieved the lowest step count to date (22 steps) and the first enantioselective total synthesis, with a cascade organocatalytic transformation playing a pivotal role in construction of the 5,6,7-tricyclic core of the natural product early in the route.39 This was achieved on treatment of achiral β,γ-unsaturated cycloheptenone 85 with the bifunctional iminophosphorane (BIMP) ‘superbase’ catalyst 86 (developed by the same group, and generated in situ by reaction of the corresponding azide and triphenylphosphine). This catalyst triggered the cascade via an enantioselective 1,3-prototropic shift to give α,β-unsaturated cycloheptenone 87, followed by an exo-selective intramolecular amidofuran Diels–Alder reaction 88, which afforded the tricyclic core 89 in 86% yield on multigram scale (92:8 dr, 90% ee). Importantly, the high basicity of 86 ensures that reprotonation of the intermediate dienolate in the isomerization of 87 is the rate-limiting and enantiodetermining step. DFT calculations suggest that a dual H-bonding interaction of the imide carbonyl with the amide N–H and the ortho aryl C–H of the catalyst orient the dienolate in such a way that the CF3 group is preferentially positioned proximal to a CH2 group of 87, rather than the methyl group. In the latter scenario, steric hindrance weakens hydrogen bonding. The product of this cascade was advanced to the natural product via steps including a lactam-directed exocyclic alkene reduction that is sensitive to both pressure and hydrogen/argon gas ratio.
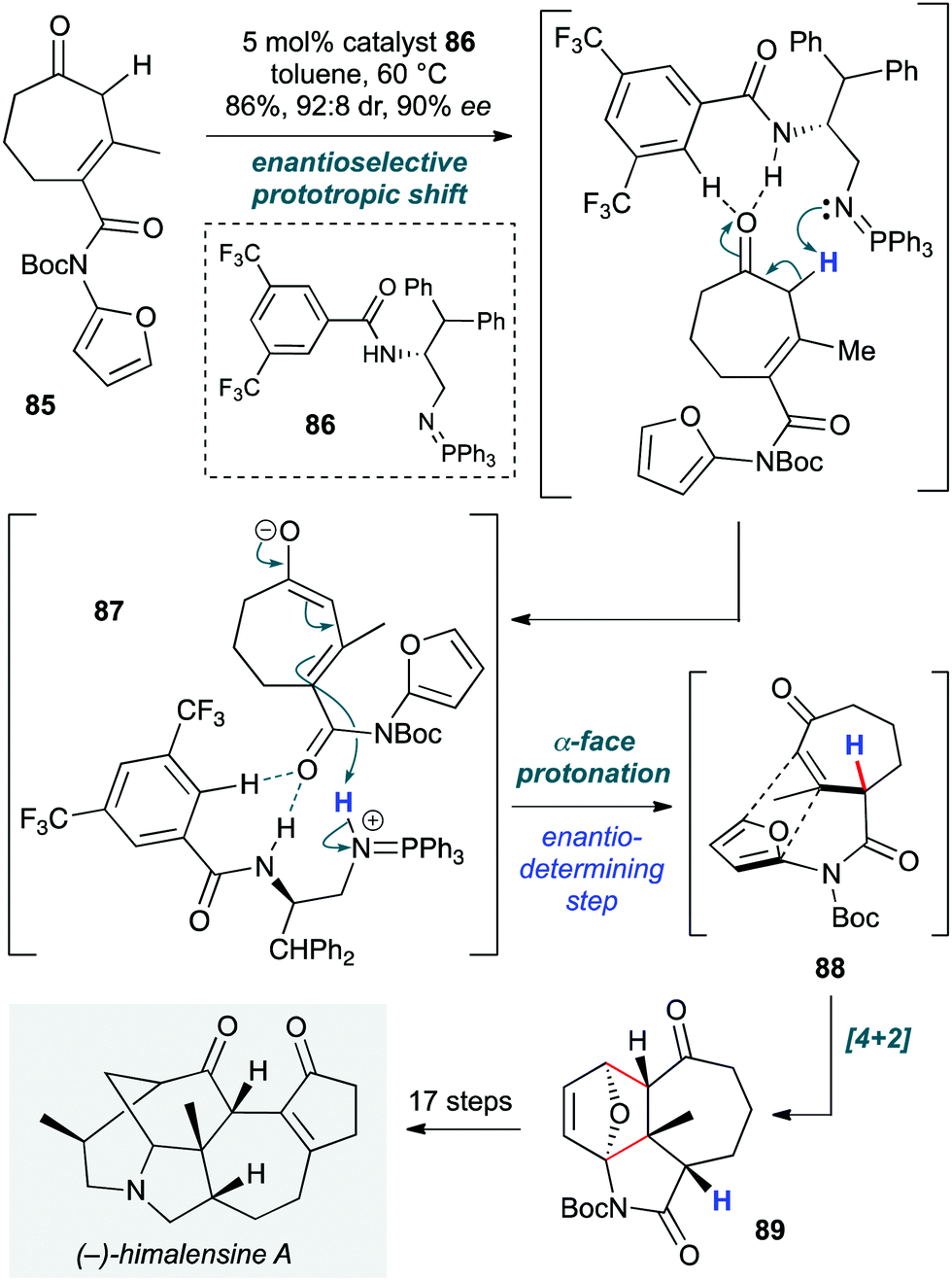 | ||
| Scheme 17 Enantioselective total synthesis of (−)-himalensine using an organocatalyzed prototropic shift/intramolecular Diels–Alder reaction (Dixon and co-workers).39 | ||
Radical cascades
While radical cascades are among the most classic methods to construct polycyclic ring systems, in recent years the wider field of radical chemistry has seen an explosion of new technologies such as photoredox catalysis40 (and other radical relays),41 and a renaissance of synthetic electrochemistry.42 This has inevitably stimulated the incorporation of such methods into natural product synthesis,43 with impressive results.The first such example is an elegant electrochemical polycyclization described by Baran and co-workers to prepare decalin 90 (Scheme 18), an intermediate in a divergent approach to the pyrone diterpene natural products subglutinols A and B, sesquicillin A, and higginsianin A.44 This cascade involves a manganese(III)-catalyzed oxidation of the β-keto ester in 91 to give tertiary radical 92, which undergoes a 6-endo-trig/6-exo-trig cyclization sequence, terminating with a further oxidation and loss of a proton to construct the exo-methylene group of 91. Since the pioneering work of Snider,45 Mn(III)-mediated cyclization cascades have been widely used in natural product synthesis, but the development of an electrochemical version offers significant advantages over previous methods in enabling the use of a sub-stoichiometric quantity of Mn(OAc)2, as opposed to the more expensive Mn(OAc)3 (which is typically used in excess in non-catalytic variants), and also by using a lower loading of copper(II) salt. Electrochemical methods are yet to be widely adopted by the synthetic community, in part due to the perceived inaccessibility of the methods and equipment needed;46 this first example of an electrochemical radical cascade in natural product synthesis provides a powerful demonstration of the potential of electrochemistry to facilitate the preparation of complex target molecules.
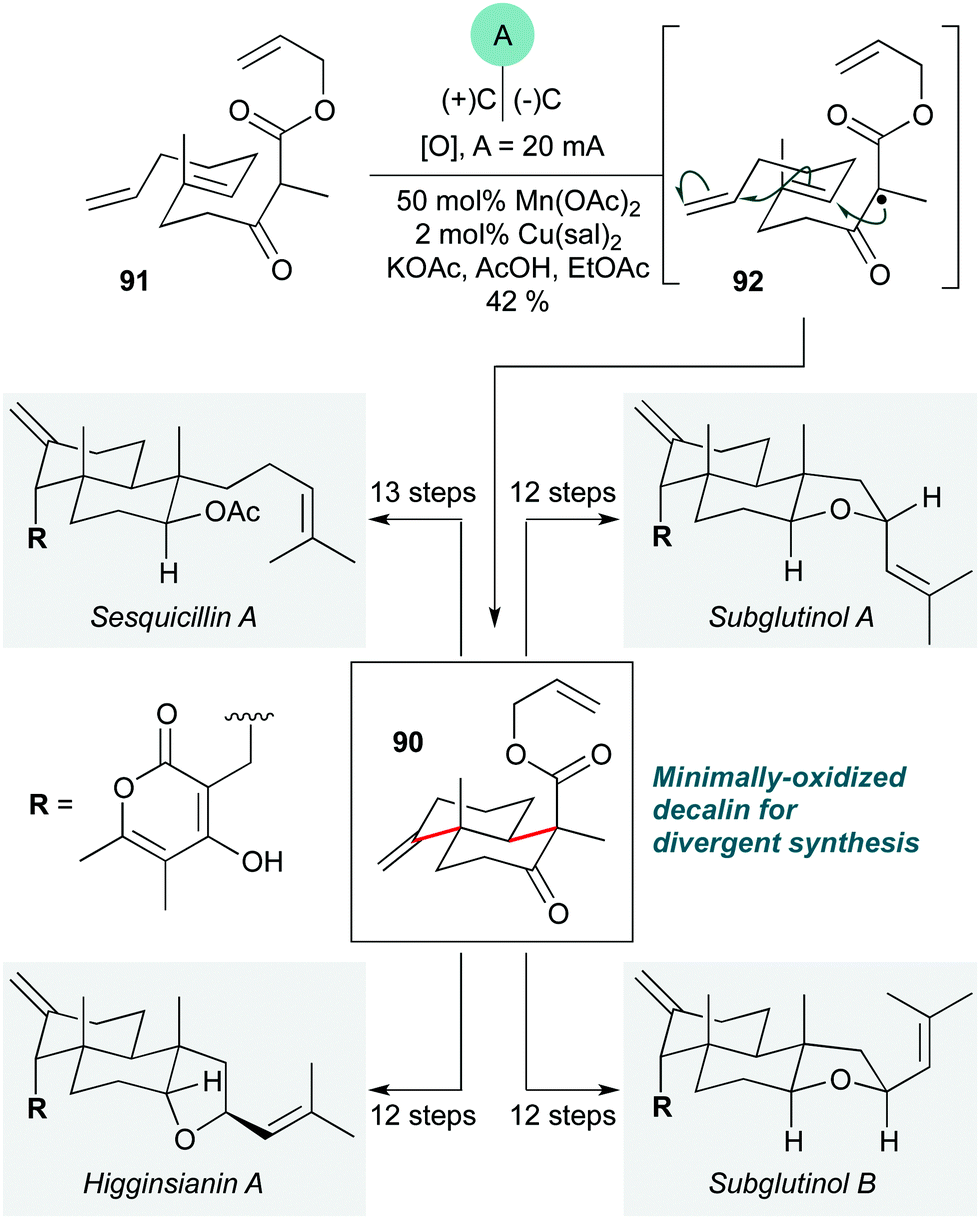 | ||
| Scheme 18 Electrochemical radical cascade for the preparation of decalin 90 in the divergent synthesis of pyrone diterpenes (Baran and co-workers).44 sal = 3,5-diisopropylsalicylate. | ||
The first use of photoredox-catalyzed cascade polycyclizations in natural product synthesis was reported by Qin and co-workers, in their preparation of 33 monoterpenoid alkaloids from four families of natural products (Scheme 19) – namely the eburnamine-vincamine, yohimbine, corynanthe, and heteroyohimbe families.47 The cascade initiates with deprotonation of a chiral aniline sulfonamide of general structure 93. Photochemical oxidation of the resulting anion gives a nitrogen-centred radical 94, which cyclizes onto the pendent enamine, forming a carbon centred radical 95. In the case of the eburnamine–vincamine alkaloids, the core skeleton is forged by stereoselective 6-exo-trig cyclization of 95 onto a tethered alkene, followed by quenching with a hydrogen atom source. In contrast, the yohimbine, corynanthe and heteroyohimbine alkaloids are accessed via radical 95′, which features a shorter tether between the lactam nitrogen atom and the C–C π–acceptor on the lactam sidechain. This first undergoes intermolecular attack onto an electron-deficient alkene or alkyne (96), followed by intramolecular attack onto the sidechain C–C multiple bond. This cascade was employed at an early stage of the divergent synthetic strategy, exploiting the high functional group tolerance of the reaction conditions to install synthetic handles for subsequent manipulations.
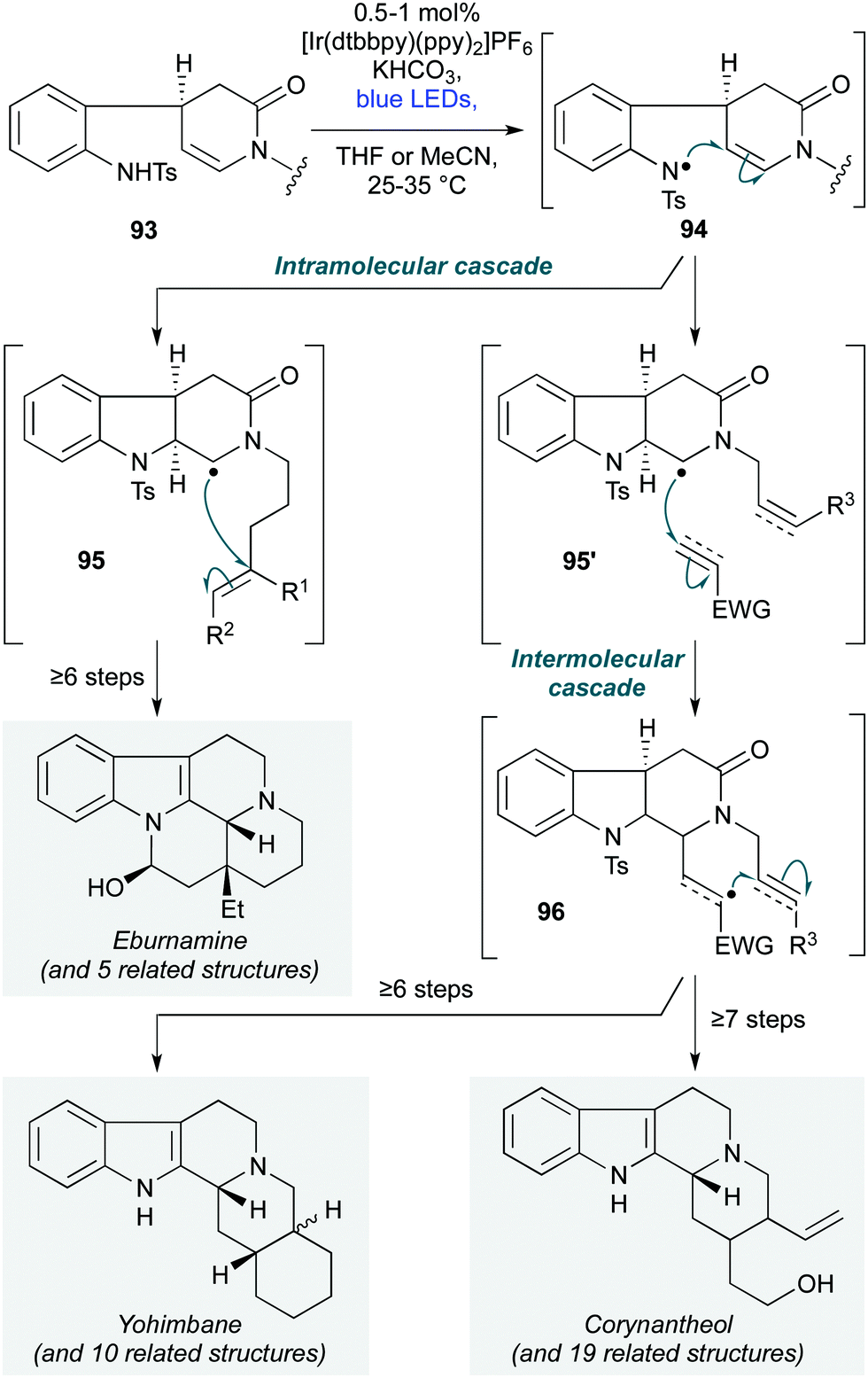 | ||
| Scheme 19 Divergent photochemical polycyclization cascade for the synthesis of multiple-ring-fused tetrahydrocarbolinones in the eburnamine–vincamine, yohimbine and corynanthe families (Qin and co-workers).47 | ||
Maimone and co-workers developed a triethylborane/air-initiated diastereoselective reductive radical cascade in their total synthesis of the sesterpene (−)-6-epi-ophiobolin N (Scheme 20).48 The notable features of this cascade are not only the rather impressive 8-endo-trig cyclization to form the 8-membered ring core of the natural product (97→98) from the triene 99, but also the stereoselective installation of three contiguous stereocentres in the ensuing 5-exo-trig/H-atom abstraction step (98→100→101). The latter is particularly notable in representing the first example of a reagent-controlled diastereoselective hydrogen atom transfer: the inherent substrate preference for formation of the undesired C15-(S) stereocentre could be reversed (to a moderate dr of 3.4:1 at C15) by using TADDOL-derived thiol catalyst 102. By controlling the stereochemical outcome of the reaction at the termination step, the authors demonstrated a complementary approach to the initiation-based stereocontrol typically observed in biomimetic cationic polycyclizations. The group has recently reported a related radical cascade in the synthesis of 6-epi-ophiobolin A.49
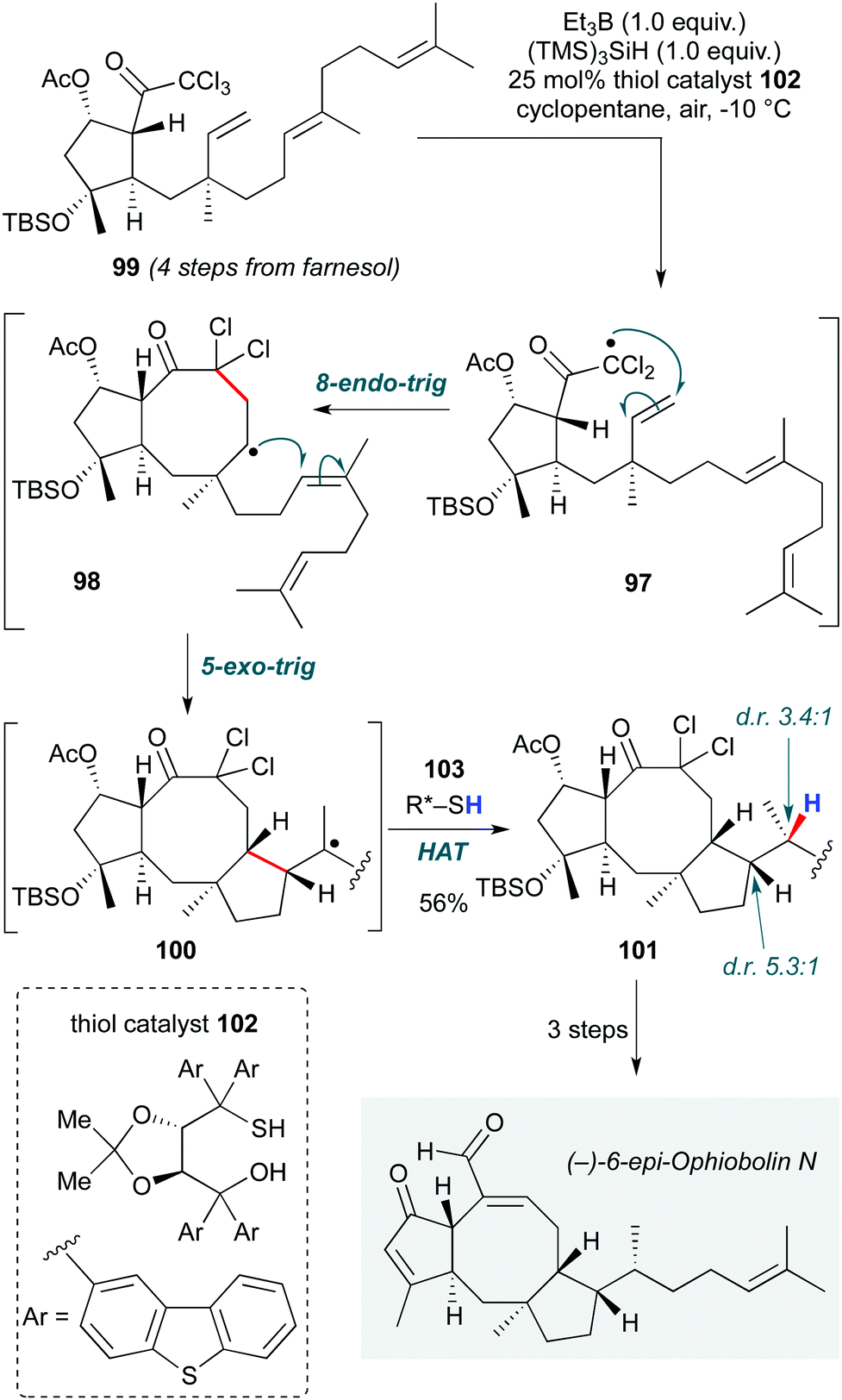 | ||
| Scheme 20 Stereoselective radical polycyclization cascade in total synthesis of (−)-6-epi-ophiobolin N (Maimone and co-workers).48 | ||
Finally, the recent asymmetric total synthesis of hispidanin A (Scheme 21) by Liu and co-workers50 features an Fe(acac)3/phenylsilane-mediated radical polycyclization to form the trans-decalin containing fragment 103 under substrate stereochemical control. The cascade is initiated by hydrogen atom transfer from an iron hydride species (generated in situ) to the more electron rich disubstituted alkene in 104. This generates a tertiary radical 105, which cyclizes onto the α,β-unsaturated ester to form an electron-deficient tertiary radical 106. Cyclization of this tertiary radical is slow due to the mismatched radical/acceptor philicity (the intramolecular acceptor butenolide is also electron-deficient) 107, which is proposed to improve selectivity for the trans decalin. This second cyclization is followed by HAT from the silane to give product 103 (after deprotection of the silyl ether). Alternative mechanisms involving reduction of the intermediate radical 107 followed by a protic quench, or indeed reduction/anionic cyclization of 106, are hard to exclude in the absence of further evidence. A similar Fe(acac)3/silane HAT cascade has been reported by Pronin and co-workers in their total synthesis of (−)-nodulisporic acid C.51
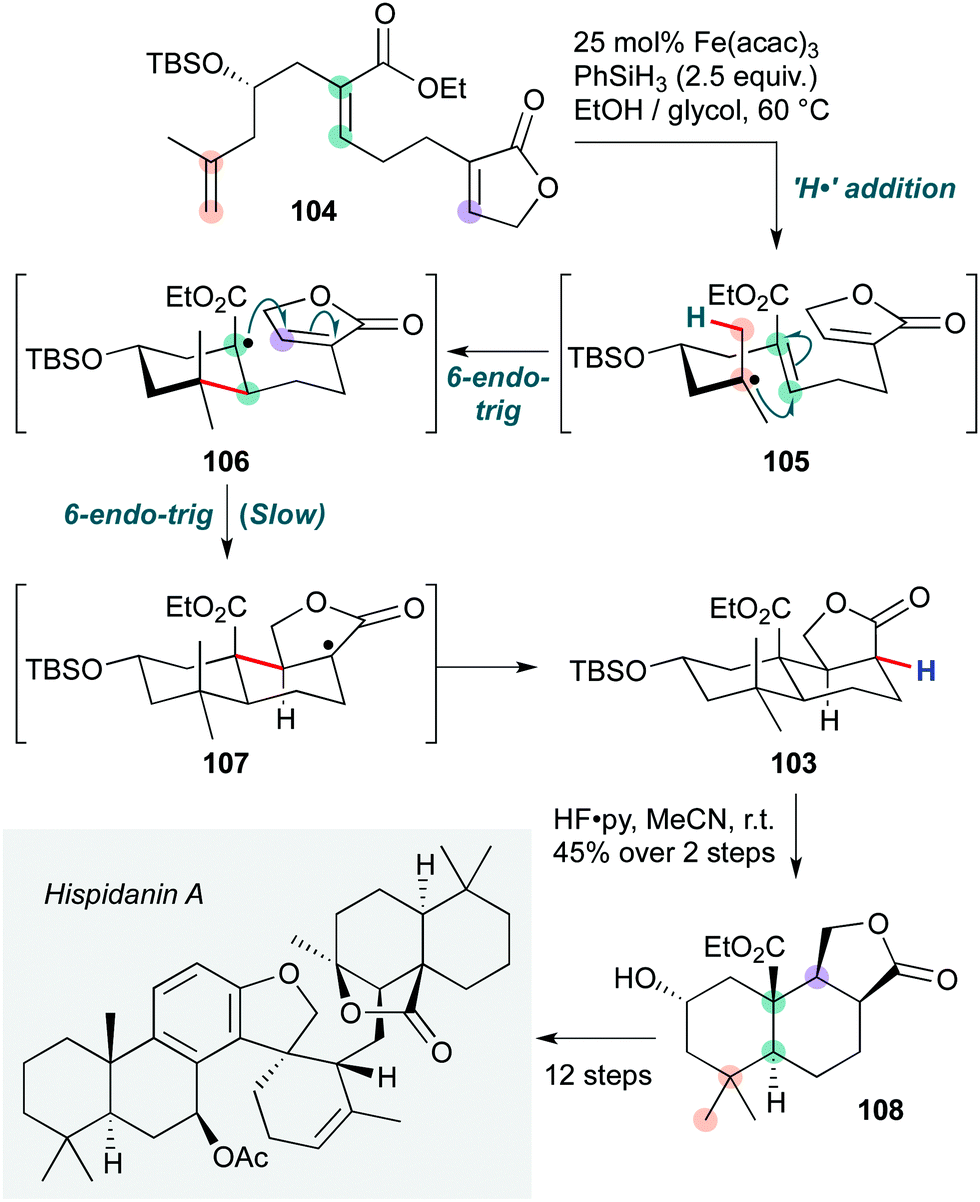 | ||
| Scheme 21 Hydrogen atom transfer (HAT) initiated radical polycyclization cascade in the total synthesis of hispidanin A (Liu and co-workers).50 | ||
Conclusions
Cascade polycyclizations continue to impart efficiency and elegance on total synthesis endeavours, while also pushing the frontiers of synthetic methodology. Alongside emerging areas such as electrochemistry and photoredox catalysis, new interpretations of classic polycyclization tactics, such as ‘non-biomimetic’ cyclization cascades, underline the unquenched ingenuity of the synthesis community. With the drive for ever more efficient and flexible synthetic routes, polycyclizations unarguably offer an indispensable means to enable biological applications of complex natural product targets. The question arises as to where the next developments in this field might be envisaged – areas such as combined synthetic/biological cascades, or the combination of emerging methods such as electrochemistry with more established tactics, are ripe for development. Whatever the future may bring, polycyclization cascades are sure to continue to push the frontiers of organic synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. J. thanks the China Scholarship Council (No. 201908535023) for financial support. R. E. M. and P. J. S. thank the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine for studentships (EP/L015838/1), generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. E. A. A. thanks the EPSRC for additional support (EP/S013172/1).Notes and references
- P. S. Baran, J. Am. Chem. Soc., 2018, 140, 4751–4755 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS.
- P. A. Wender, Nat. Prod. Rep., 2014, 31, 433–440 RSC.
- C. A. Kuttruff, M. D. Eastgate and P. S. Baran, Nat. Prod. Rep., 2014, 31, 419–432 RSC.
- R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557–1569 RSC.
- E. A. Anderson, Org. Biomol. Chem., 2011, 9, 3997–4006 RSC.
- C. T. Walsh and B. S. Moore, Angew. Chem., Int. Ed., 2019, 58, 6846–6879 CrossRef CAS.
- J. M. E. Hughes and J. L. Gleason, Angew. Chem., Int. Ed., 2017, 56, 10830–10834 CrossRef CAS.
- R. C. Godfrey, N. J. Green, G. S. Nichol and A. L. Lawrence, Nat. Chem., 2020, 12, 615–619 CrossRef CAS.
- K. R. Klas, H. Kato, J. C. Frisvad, F. Yu, S. A. Newmister, A. E. Fraley, D. H. Sherman, S. Tsukamoto and R. M. Williams, Nat. Prod. Rep., 2018, 35, 532–558 RSC.
- A. Zech and T. Bach, J. Org. Chem., 2018, 83, 3069–3077 CrossRef CAS.
- P. J. Wagner and R. P. Smart, Tetrahedron Lett., 1995, 36, 5135–5138 CrossRef CAS.
- A. Zech, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 14629–14632 CrossRef CAS.
- F. Schneider, K. Samarin, S. Zanella and T. Gaich, Science, 2020, 367, 676–681 CrossRef CAS.
- M. Baunach, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 2604–2626 CrossRef CAS.
- E. J. Corey and S. Lin, J. Am. Chem. Soc., 1996, 118, 8765–8766 CrossRef CAS.
- K. Speck, R. Wildermuth and T. Magauer, Angew. Chem., Int. Ed., 2016, 55, 14131–14135 CrossRef CAS.
- J. M. Feilner, K. Wurst and T. Magauer, Angew. Chem., Int. Ed., 2020, 59, 12436–12439 CrossRef CAS.
- H. Cheng, Z. Zhang, H. Yao, W. Zhang, J. Yu and R. Tong, Angew. Chem., Int. Ed., 2017, 56, 9096–9100 CrossRef CAS.
- Y. Dou, C. Kouklovsky, V. Gandon and G. Vincent, Angew. Chem., Int. Ed., 2020, 59, 1527–1531 CrossRef CAS.
- J. R. Reyes, N. Winter, L. Spessert and D. Trauner, Angew. Chem., Int. Ed., 2018, 57, 15587–15591 CrossRef CAS.
- H. R. Khatri, B. Bhattarai, W. Kaplan, Z. Li, M. J. Curtis Long, Y. Aye and P. Nagorny, J. Am. Chem. Soc., 2019, 141, 4849–4860 CrossRef CAS.
- W. Kaplan, H. R. Khatri and P. Nagorny, J. Am. Chem. Soc., 2016, 138, 7194–7198 CrossRef CAS.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS.
- D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC.
- Y. Li and M. Dai, Angew. Chem., Int. Ed., 2017, 56, 11624–11627 CrossRef CAS.
- J. Matsuoka, Y. Matsuda, Y. Kawada, S. Oishi and H. Ohno, Angew. Chem., Int. Ed., 2017, 56, 7444–7448 CrossRef CAS.
- D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260–11261 CrossRef CAS.
- D. Scharnagel, I. Escofet, H. Armengol-Relats, M. E. de Orbe, J. N. Korber and A. M. Echavarren, Angew. Chem., Int. Ed., 2020, 59, 4888–4891 CrossRef CAS.
- J. Biemolt and E. Ruijter, Adv. Synth. Catal., 2018, 360, 3821–3871 CrossRef CAS.
- Y. Ping, Y. Li, J. Zhu and W. Kong, Angew. Chem., Int. Ed., 2019, 58, 1562–1573 CrossRef CAS.
- Y. Li, Q. Zhang, H. Wang, B. Cheng and H. Zhai, Org. Lett., 2017, 19, 4387–4390 CrossRef CAS.
- L. Xu, C. Wang, Z. Gao and Y.-M. Zhao, J. Am. Chem. Soc., 2018, 140, 5653–5658 CrossRef CAS.
- J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS.
- M. Mohammad, V. Chintalapudi, J. M. Carney, S. J. Mansfield, P. Sanderson, K. E. Christensen and E. A. Anderson, Angew. Chem., Int. Ed., 2019, 58, 18177–18181 CrossRef CAS.
- M. Odagi and K. Nagasawa, Asian J. Org. Chem., 2019, 8, 1766–1774 CrossRef CAS.
- J. Tan and N. Yasuda, Org. Process Res. Dev., 2015, 19, 1731–1746 CrossRef CAS.
- C. Piemontesi, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2016, 138, 11148–11151 CrossRef CAS.
- H. Shi, I. N. Michaelides, B. Darses, P. Jakubec, Q. N. N. Nguyen, R. S. Paton and D. J. Dixon, J. Am. Chem. Soc., 2017, 139, 17755–17758 CrossRef CAS.
- C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible light photocatalysis in organic chemistry, Wiley-VCH, 2018 Search PubMed.
- H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Chem. Soc. Rev., 2019, 48, 4626–4638 RSC.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020 10.1039/D0SC01848A.
- J. B. Mateus-Ruiz and A. Cordero-Vargas, Synthesis, 2020, 3111–3128 CAS.
- R. R. Merchant, K. M. Oberg, Y. Lin, A. J. E. Novak, J. Felding and P. S. Baran, J. Am. Chem. Soc., 2018, 140, 7462–7465 CrossRef CAS.
- B. B. Snider, Chem. Rev., 1996, 96, 339–364 CrossRef CAS.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS.
- X. Wang, D. Xia, W. Qin, R. Zhou, X. Zhou, Q. Zhou, W. Liu, X. Dai, H. Wang, S. Wang, L. Tan, D. Zhang, H. Song, X. Y. Liu and Y. Qin, Chemistry, 2017, 2, 803–816 CrossRef CAS.
- Z. G. Brill, H. K. Grover and T. J. Maimone, Science, 2016, 352, 1078–1082 CrossRef CAS.
- D. Q. Thach, Z. G. Brill, H. K. Grover, K. V. Esguerra, J. K. Thompson and T. J. Maimone, Angew. Chem., Int. Ed., 2020, 59, 1532–1536 CrossRef CAS.
- H. Deng, W. Cao, R. Liu, Y. Zhang and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 5849–5852 CrossRef CAS.
- N. A. Godfrey, D. J. Schatz and S. V. Pronin, J. Am. Chem. Soc., 2018, 140, 12770–12774 CrossRef CAS.
Footnote |
| † These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2021 |