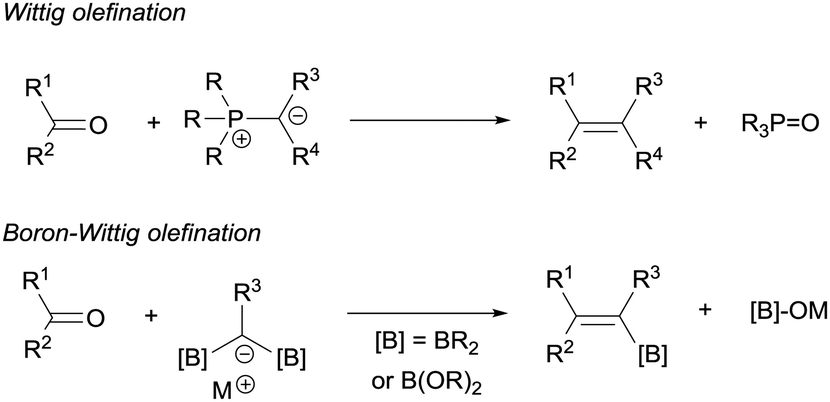
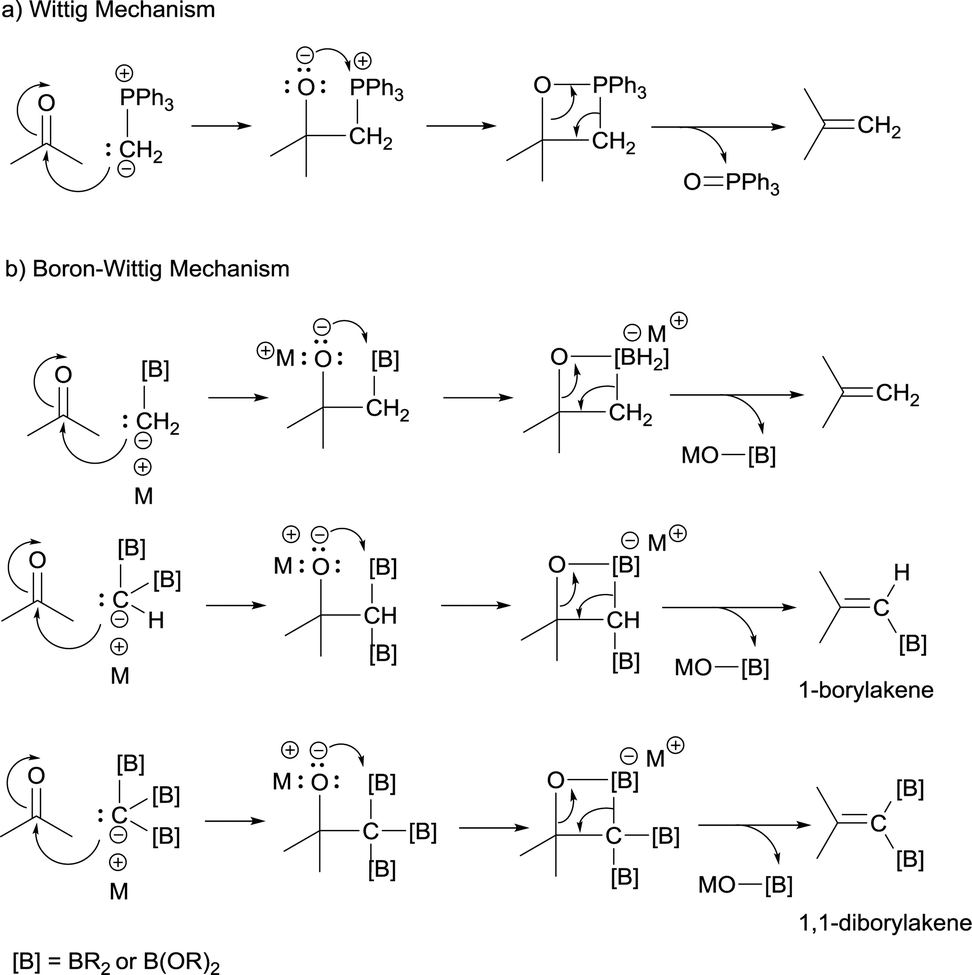
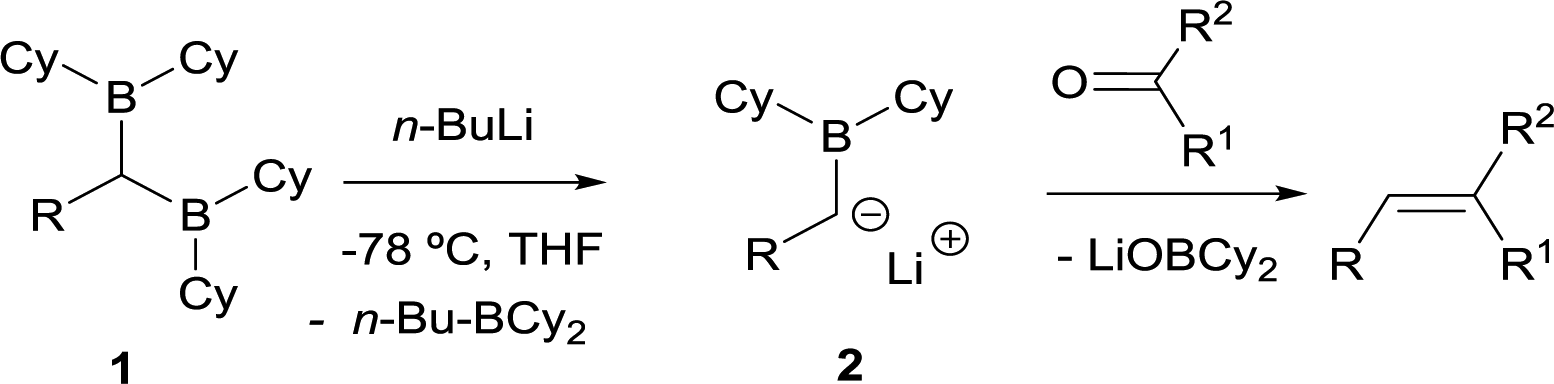
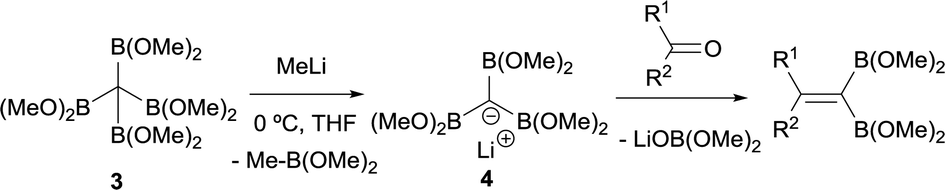
DOI:
10.1039/D0CS00953A
(Tutorial Review)
Chem. Soc. Rev., 2021,
50, 72-86
Boron-Wittig olefination with gem-bis(boryl)alkanes
Received
4th September 2020
First published on 15th December 2020
Abstract
The condensation of easy manageable lithium α-bis(boryl)carbanions with carbonyl derivatives, the so-called boron-Wittig reaction, allows for the straightforward and often stereoselective formation of synthetically highly versatile metalloid-substituted alkenes, which are key building blocks on route to all-carbon substituted olefins. In this Tutorial review the concept behind this olefination reaction and its application to ketones, aldehydes and other carbonyl derivatives, such amides, ester and carboxylic acids, are presented in a systematic manner. A special emphasis has been placed on parameters controlling the stereochemical outcome of these transformations. To illustrate the great synthetic potential of this new methodological tool, a section is also included covering a selection of applications of the boron-Wittig reaction to target compounds via subsequent C–C bond-forming process.

Ana B. Cuenca
| Ana B. Cuenca joined the group of Prof. G. Asensio (U. Valencia) for a PhD in enantioselective protonation. Later on, she completed two postdoctoral stays, first at IRCOF-CNRS (Rouen) and after at MIT under the supervision of Prof. S. L. Buchwald (2004–06). From 2007–13 she became assistant professor at the U. Valencia and then she moved to the U. Rovira I Virgili (Tarragona) to work with Prof. E. Fernández. Since Sept-2016 Cuenca is associate professor at IQS-School of Engineering (U. Ramon Llull). Her research interest focuses on the development of new methodologies involving main group elements, specifically I, B, Si. |

Elena Fernández
| Elena Fernández received her degree in chemistry at the University of Barcelona in 1991. She did PhD studies in catalytic hydroformylation of sugars with Prof. S. Castillón (1991–1995) and she moved to Oxford University (UK) (1995–1997) for a postdoctoral position with Prof. John M. Brown where her studies culminated with an approach towards the first catalytic asymmetric hydroboration-amination reaction. Elena accepted in 1997 a lecturer position at the University Rovira i Virgili, becoming part of the permanent staff in 2000 and Full Professor in 2019. She received the Award on Excellence of Research in Organometallic Chemistry 2014 and the Award on Excellence of Research in Chemistry 2017, both from the Spanish Royal Society of Chemistry. She is Distinguished Professor at the URV from 2018. Her current scientific campaign is aimed to generate knowledge and awareness about activation modes of borane reagents to be used in selective synthesis of multifunctional compounds. |
Key learning points
(1) Concept of boron-Wittig olefination and comparison with Wittig transformation.
(2) Mechanistic insights and application to the use of gem-bis(boryl)alkanes.
(3) Condensation with aldehydes and ketones and primary amides, with control of the stereoselectivity.
(4) Nucleophilic attack of α-bis(boryl)carbanions to tertiary amides, esters and acids via enolate formation.
(5) Boron-Wittig reaction as current synthetic tool for construction of polysubstituted alkenes via cross-coupling transformations.
|
1. Introduction
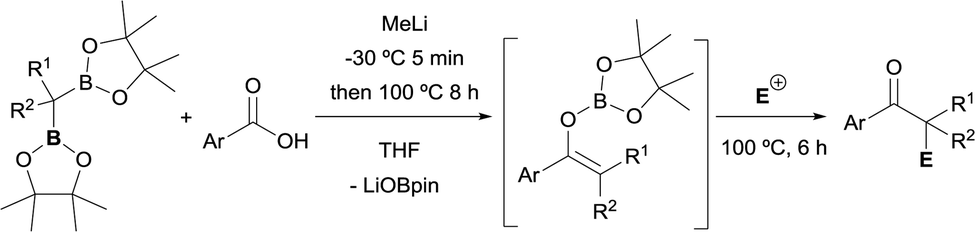
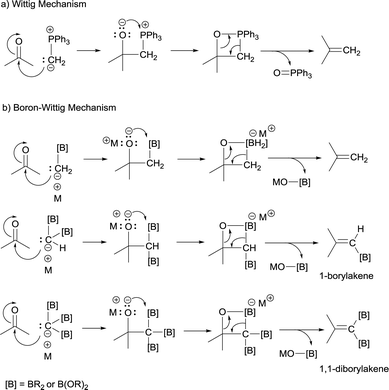
The boron-Wittig reaction with gem-bis(boryl)alkanes allows the preparation of alkenes by the nucleophilic attack of α-bis(boryl)carbanions to carbonyl compounds followed by a B–O elimination step. Formally, this reaction resembles the original Wittig olefination that is based on the reactivity of aldehydes or ketones with ylides generated from phosphonium salts (Scheme 1). It has been established that the driving force of the Wittig reaction is the formation of a very stable phosphine oxide, whereas in the boron-Wittig reaction the boronate by-products formed becomes the driving force due to the stability associated to B–O bonds.
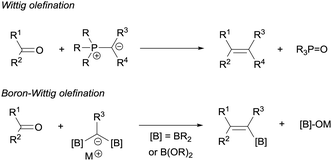 |
| | Scheme 1 Comparison between Wittig and boron-Wittig olefinations. | |
From a mechanistic point of view, both type of condensations shares the same elementary steps. For the stepwise Wittig reaction: (1) attack of the ylide carbon to the carbonyl group to form the betaine intermediate, (2) attack of oxygen on phosphorous towards oxaphosphetane and (3) reverse [2+2] cycloaddition to deliver the phosphine oxide and the alkene (Scheme 2a). For boron-Wittig reaction: (1) nucleophilic attack of the α-(boryl)carbanion to the carbonyl derivative, (2) attack of oxygen to Lewis acid B to form the corresponding tetrasubstituted borylated intermediate and (3) B–O elimination and formation of the corresponding alkene (Scheme 2b). However, depending on the use of α-mono(boryl)carbanions, α-bis(boryl)carbanions or α-tris(boryl)carbanions, the olefination produces alkenes, 1-borylalkenes or 1,1-diborylalkenes, respectively. The synthesis of vinylboranes1 through the boron-Wittig reaction has gained great interest within the last decade, as a synthetic tool, with a significant level of stereochemical control and applications in target compounds.
 |
| | Scheme 2 Mechanistic trends of Wittig and boron-Wittig olefinations. | |
The first condensation of gem-bis(boryl)alkanes with aldehydes and ketones to yield olefins was observed by Zubiani and co-workers in 1966.2 These authors observed that benzaldehyde and a series of aliphatic, aromatic and alicyclic ketones could efficiently be converted to the corresponding olefins by reaction with 1,1-bis(dicyclohexylboryl)alkanes (1). The activation of the gem-bis(boryl)alkanes with n-BuLi favoured the deborylation and therefore the in situ formation of the α-(boryl)carbanion lithium salts (2), that condensed with carbonyl compounds (Scheme 3). In this approach, both boryl moieties were eliminated from the starting compound, via deborylation with n-BuLi and subsequent B–O elimination. Consequently, the resulting olefin did not contain any boryl moiety. The authors found a strong dependence of the olefin stereochemistry as a function of the reaction conditions.
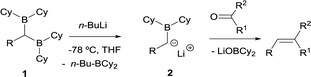 |
| | Scheme 3 First observed condensation of gem-bis(boryl)alkanes with aldehydes and ketones via B–O elimination by Zubiani and co-workers. | |
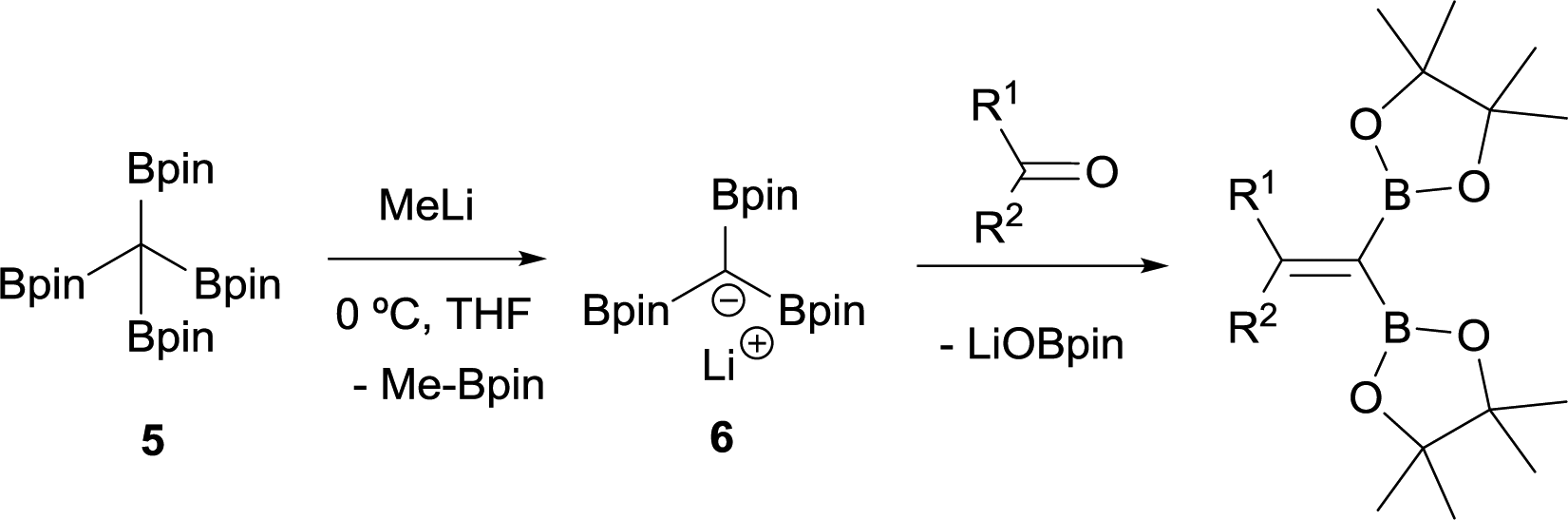
Matteson and co-workers extended this condensation to polyboronic esters,3 and conducted the olefination starting from tetra(dimethoxyboryl)methane (3)4 in the presence of MeLi. The in situ generated tris(dimethoxyboryl)methide ion (4) is able to condense with aldehydes and ketones to produce 1,1-bis(boryl)alkenes (Scheme 4). The same authors extended the concept to the more stable pinacol-based boronic esters (Bpin) (Scheme 5).5 This fact became the foundation of the synthesis of stable 1,1-diborylalkene compounds which can next undergo double cross-coupling reactivity with different electrophiles, thus allowing stereoselective synthesis of non-symmetrically substituted alkenes.6
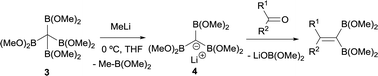 |
| | Scheme 4 Condensation of tris(dimethoxyboryl)methide ion with aldehydes and ketones via B–O elimination. | |
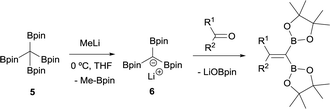 |
| | Scheme 5 Condensation of tris(pinacolboryl)methide ion with aldehydes and ketones via B–O elimination. | |
The same authors highlighted the chemoselectivity of the α-tris(boryl)carbanion's nucleophilic attack when reacted with chloroacetone which occurred preferentially at the carbonyl group rather than the alkyl halide function. They also explored the in situ transformations of alkene-1,1-diboronic esters into α-bromoalkeneboronic esters when react with bromine.5
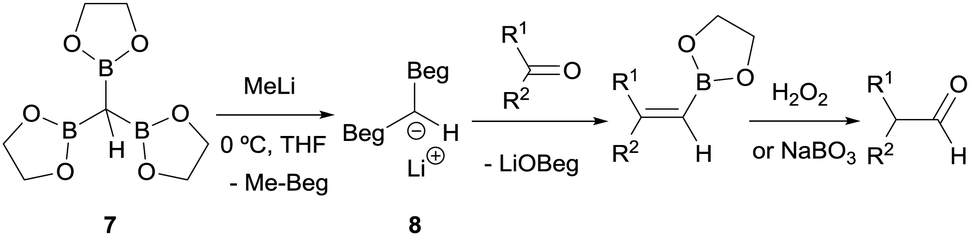
Matteson and co-workers launched the convenient homologation of aldehydes and ketones through condensation with α-bis(boryl)carbanions followed by oxidative work up with hydrogen peroxide (Scheme 6).7 Specifically, tris(ethylenedioxyboryl)methane (7) reacted with MeLi to yield a bis(boryl)carbanion 8 which in turn reacts with a series of aldehydes or ketones to give the corresponding alkene boronic esters. These intermediates were subsequently converted into their homologated aldehydes through oxidation with buffered aqueous hydrogen peroxide or alkaline sodium perborate (Scheme 6).
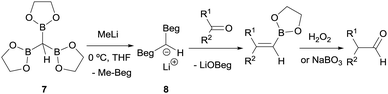 |
| | Scheme 6 Condensation of lithium bis(ethylenedioxyboryl)methide with aldehydes and ketones followed by in situ oxidation. | |
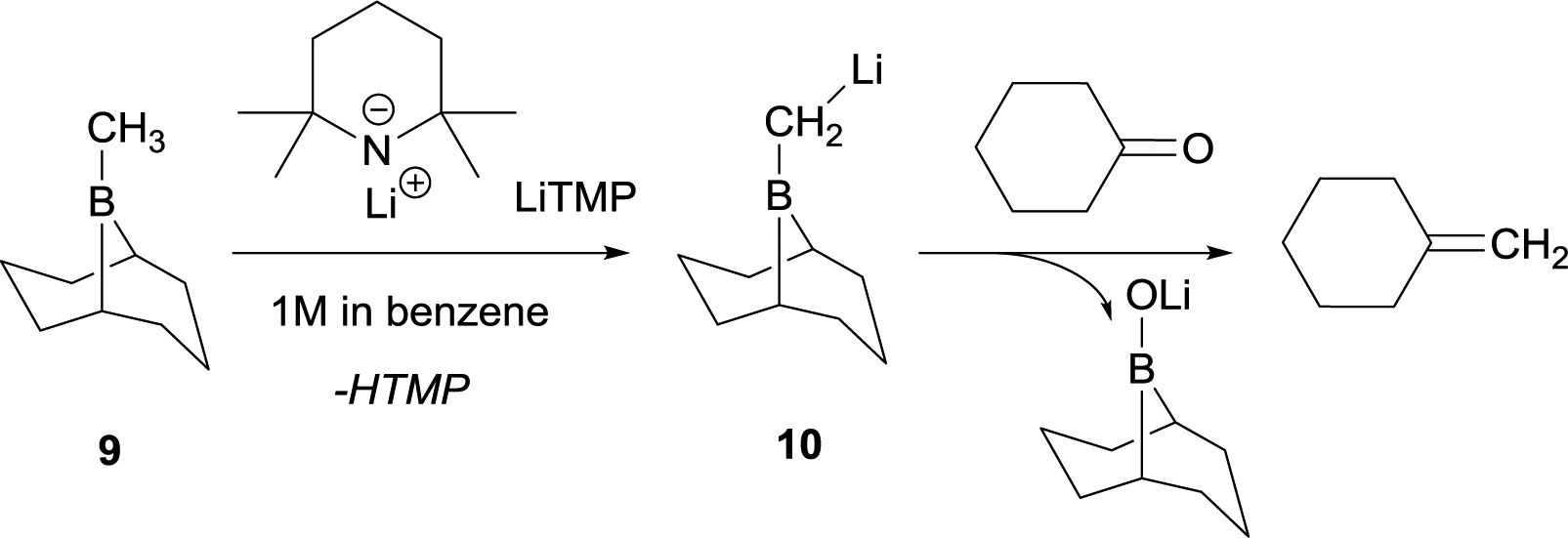
The synthetic utility of aldehyde or ketone condensation reactions with methane tetraboronic and methane triboronic esters was formerly limited to the requirement that one of the boryl moieties had to be sacrificed by the aid of MeLi or n-BuLi in order to generate the corresponding α-bis(boryl)carbanions.7–11 However, Rathke and Kow12 reported a highly efficient deprotonation of B-methyl-9-borabicyclo[3.3.1]nonane (B-methyl-9BBN-H) (9) with lithium 2,2,6,6-tetramethylpiperidide (LiTMP) and made the first approach to condense the lithium borylmethide salt 10 with cyclohexanone to produce methylenecyclohexane (Scheme 7).
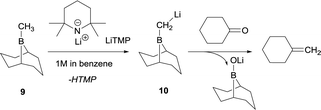 |
| | Scheme 7 Deprotonation of B-methyl-9BBN-H with LiTMP and further condensation with cyclohexanone. | |
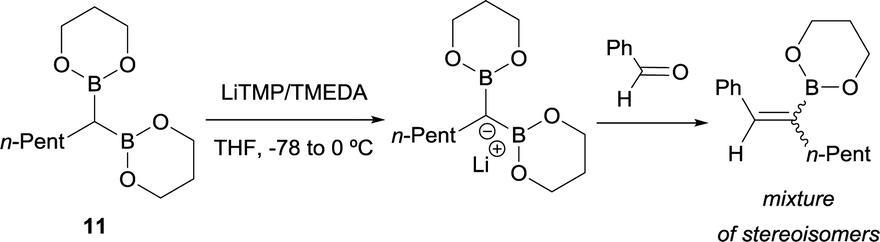
Inspired by this precedent, Matteson and co-workers extended the application of LiTMP to deprotonate methanediboronate reagents13 to form the corresponding lithium diborylmethide salts in the presence of tetramethylethylenediamine (TMEDA) in tetrahydrofuran.14 These authors proved the efficient condensation of 1-lithio-1,1-bis(l,3,2-dioxaborin-2-yl)hexane (11) with aldehydes but not stereochemical issues were addressed at this point (Scheme 8).
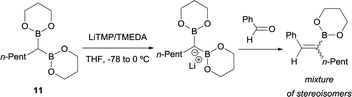 |
| | Scheme 8 Condensation of 1-lithio-1,1-bis(l,3,2-dioxaborin-2-yl)hexane with ketones via B–O elimination. | |
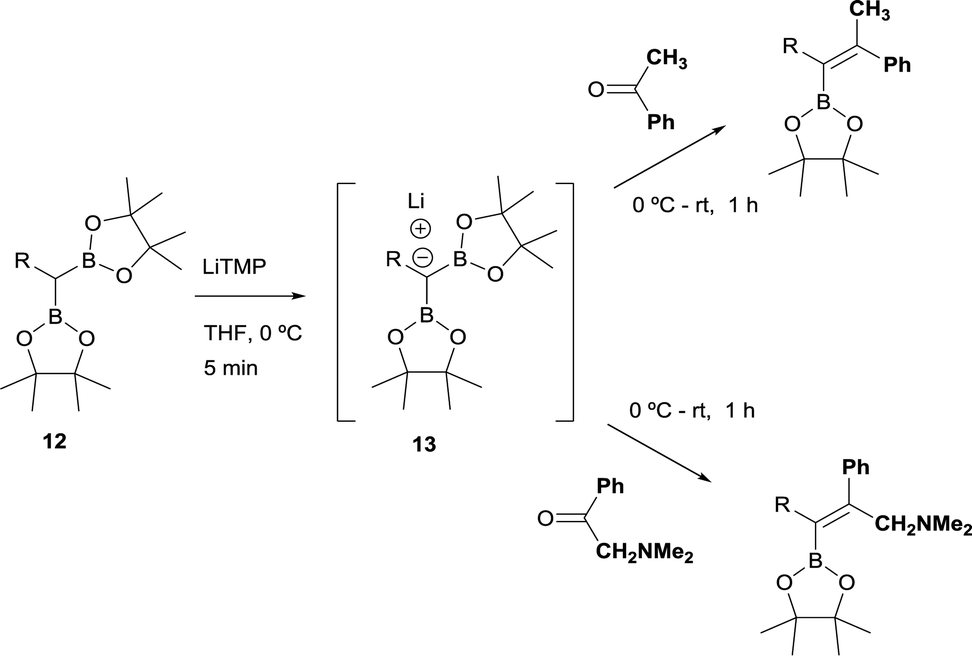
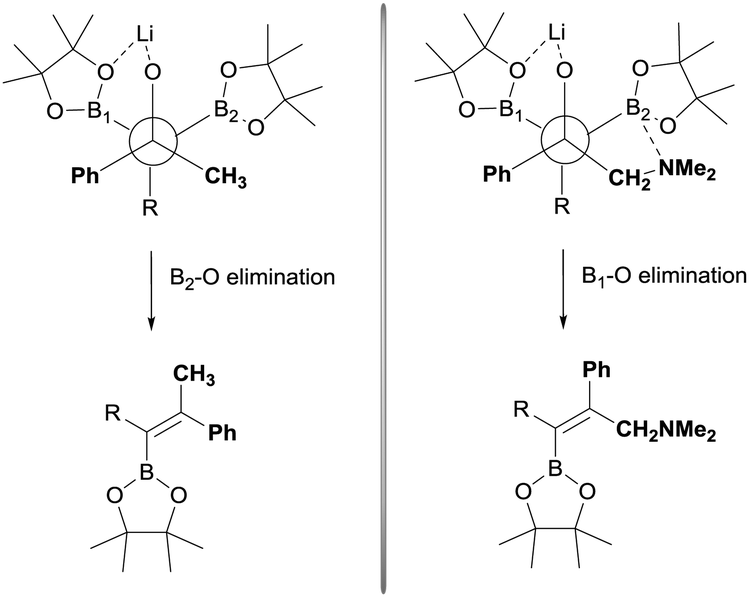
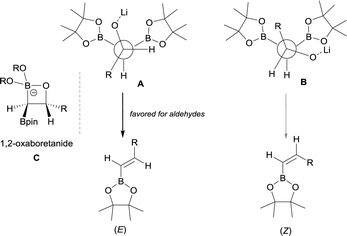
The stereoselective control on the synthesis of tetrasubstituted alkenylboronates was first established by Endo and Shibata in 201015via lithiation of 1,1-organodiboronates and subsequent stereoselective nucleophilic addition to carbonyl compounds. Hence, they explored the in situ deprotonation of 1,1-bis(pinacolboryl)alkane (12) with LiTMP to obtain the stable lithium α-bis(pinacolboryl)carbanion 13 that underwent subsequent nucleophilic addition to a series of ketones affording the favored tetrasubstituted alkenylboronate with the Bpin moiety positioned syn with respect to the aryl groups from the ketone (Scheme 9, top). However, the introduction of coordinating groups in the ketone favored the inversion on the stereoselectivity. Thus, in this case the main product is a tetrasubstituted alkenylboronate with Bpin moiety anti to the aryl groups from ketone (Scheme 9, bottom). Endo and Shibata rationalised these observations suggesting that the stereoselectivity of the syn-B–O elimination might be controlled by the lithium interaction to one of the oxygens on the Bpin fragment. Fig. 1 shows the models that have been used to justify the observed stereoselectivity in the boron-Wittig reactions based on the preliminary observations of intramolecular coordination of heteroatom to lithium center.16
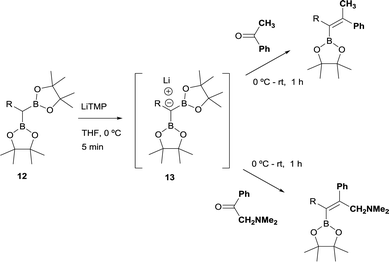 |
| | Scheme 9 Stereoselective trends on boron-Wittig reaction with aryl ketones. | |
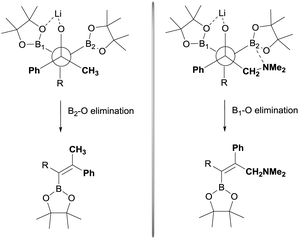 |
| | Fig. 1 Hypothesis on the origin of the stereoselective for boron-Wittig reaction with aryl ketones. | |
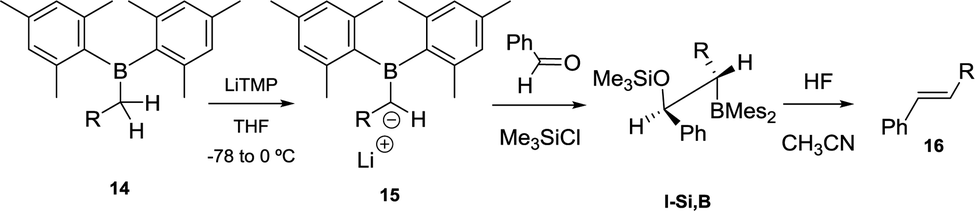
When lithium dimesitylboron stabilised carbanions 15 reacted with aromatic ketones and aldehydes the formation of intermediates I-Si,B could be trapped with chlorotrimethylsilane (Scheme 10). Further treatment of I-Si,B with aq. HF/CH3CN, allows the B–O elimination and subsequent isolation of the corresponding alkenes 16 with E configuration.17
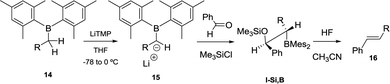 |
| | Scheme 10 Condensation of lithium dimesitylboryl methide with aldehydes, trapping with Me3SiCl and subsequent fluoride-assisted B–O elimination. | |
2. Boron-Wittig olefination of ketones with gem-bis(boryl)alkanes
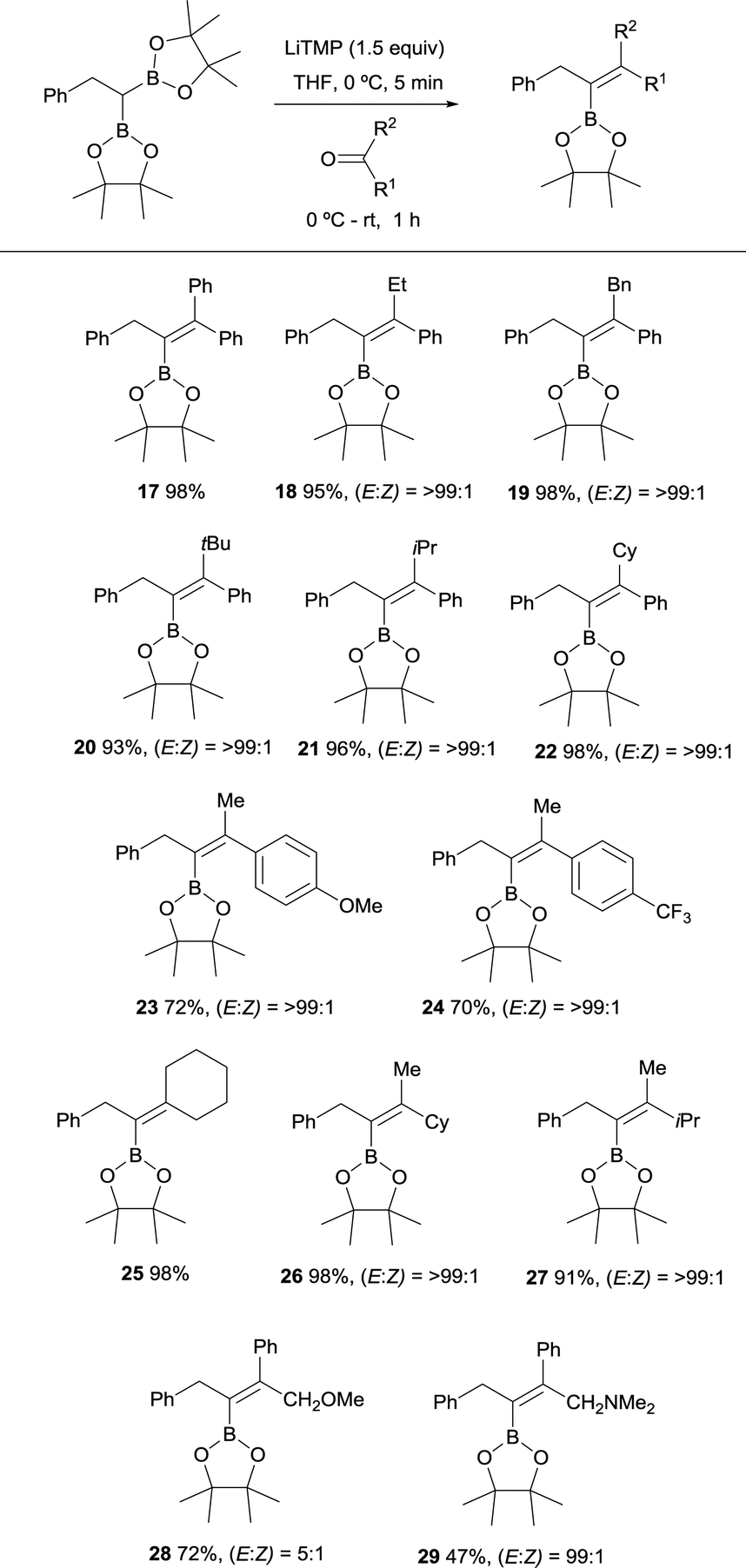
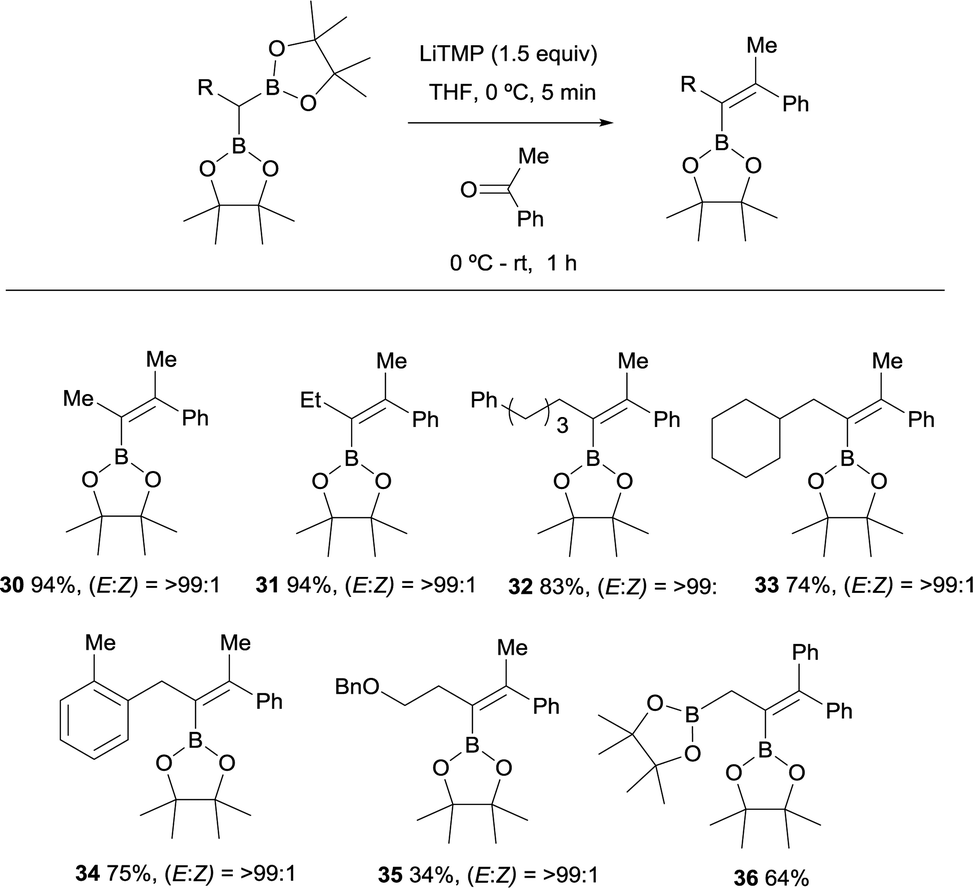
Endo and Shibata established in 2010 the stereoselective synthesis of tetrasubstituted alkenyl boronates via deprotonation of the α-C–H bond adjacent to the gem-diboryl fragments, followed by subsequent nucleophilic addition to a various type of symmetrical ketones such as benzophenone and cyclohexanone (Scheme 11).15 LiTMP was added in 1.5 equivalents to a 0 °C solution of the diboryl reagent in THF. After 5 min the ketone was added and reacted up to room temperature for 1 h. The reaction of non-symmetric aryl ketones provided the vinylboronates 17–22 with the Bpin moiety placed syn with respect to the aryl group. Electron-withdrawing or electron-donating substituents did not influence the stereochemical outcome of the alkenes 23 and 24. Aliphatic ketones also afforded the corresponding vinylboranes 25–27 in high yields and excellent stereoselectivities as it is reported for compounds 26 and 27 with the bulkiest substituent syn to the Bpin moiety.
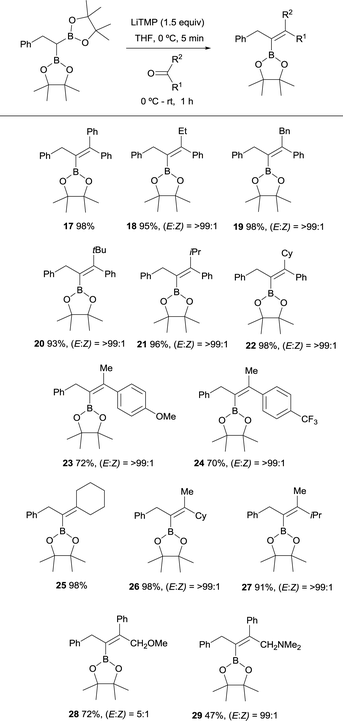 |
| | Scheme 11 Boron-Wittig olefination between 2,2′-(2-phenylethane-1,1-diyl)bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) with symmetric and non-symmetric ketones using LiTMP at rt. | |
However, the presence of coordinating groups in ketones drastically switched the stereoselectivity of products, that is the case of 2-methoxy-1-phenylethan-1-one and 2-(dimethylamino)-1-phenylethan-1-one that positioned the Ph group anti with respect to the Bpin moiety in alkenes 28 and 29 (Scheme 11).
A variety of gem-bis(boryl)alkanes were reacted with acetophenone under the same reaction conditions, demonstrating the compatibility of aliphatic and benzylic substituents on the 1,1-organodiboronates (Scheme 12).15 The benzyloxy group diminished the yield of the expected product albeit the final vinylboronate 35 was obtained with excellent stereoselectivity. The boron-Wittig reaction between ethane-1,1,2-triboronate and benzophenone allowed the formation of the corresponding allylborane 36 (Scheme 12).18 This is considered a practical approach towards the synthesis of tetrasubstituted alkenylboronates.19
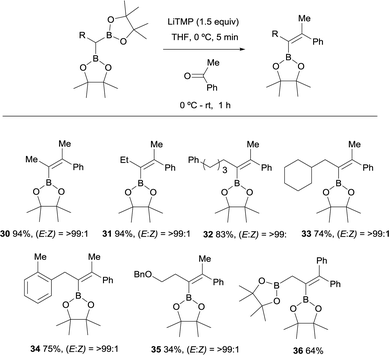 |
| | Scheme 12 Boron-Wittig olefination between substituted gem-bis(boryl)alkanes and acetophenone with LiTMP at rt. | |
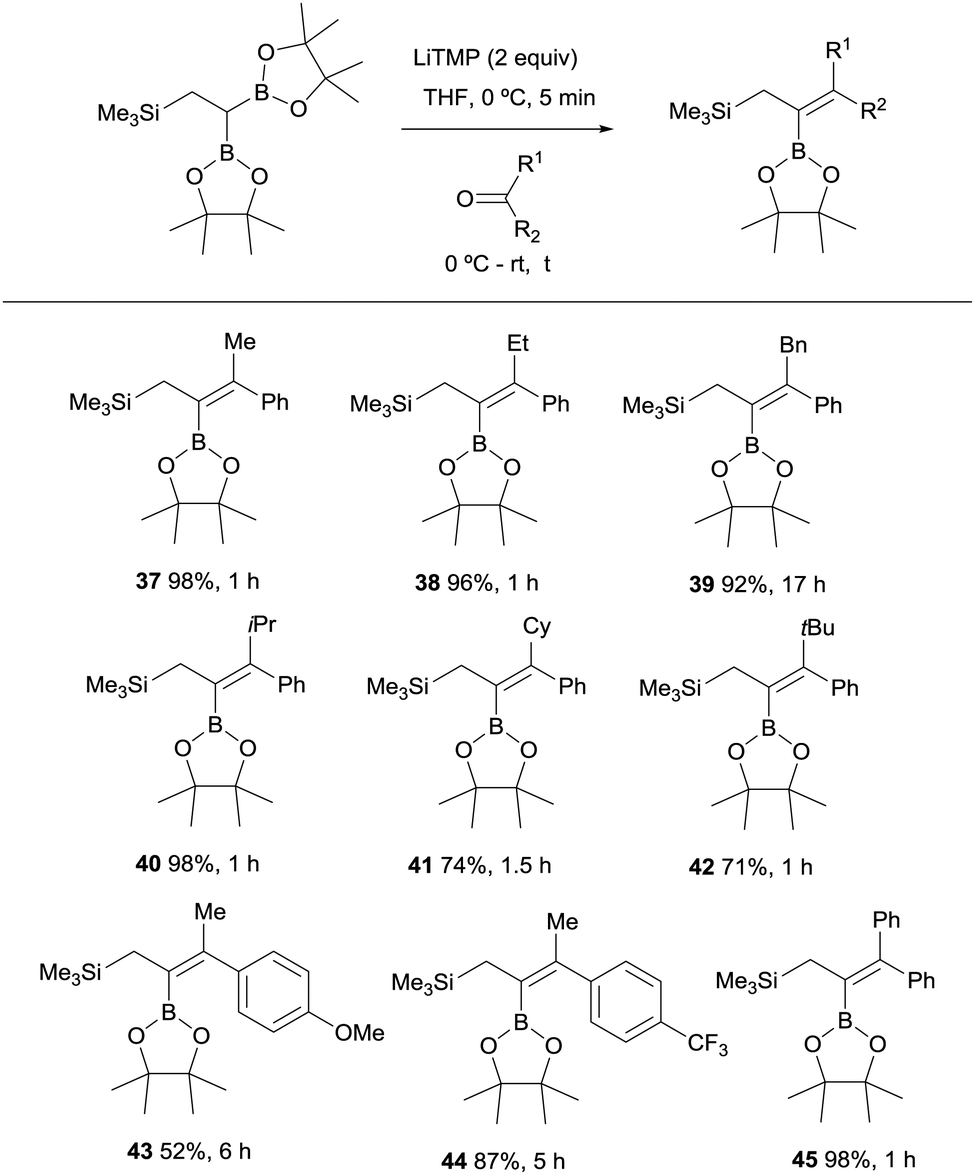
Endo, Shibata co-workers explored next the deprotonation of 2,2-diborylethylsilane with 2 equivalents of LiTMP and subsequent nucleophilic addition of the anion generated to ketones in order to obtain stereoselective synthesis of allylsilane compounds bearing a tetrasubstituted alkenylboronate group.18 The use of aryl ketones allowed the formation of the 2-borylallylsilane 37–45 with excellent (E)-stereoselectivity positioning the aromatic group syn with respect to the Bpin moiety (Scheme 13). However, the reaction with aliphatic ketones resulted in a mixture of unidentified by-products.
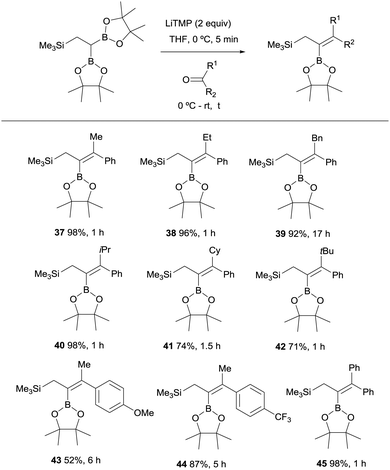 |
| | Scheme 13 Boron-Wittig between 2,2-diborylethylsilane and aromatic ketones with LiTMP at rt. | |
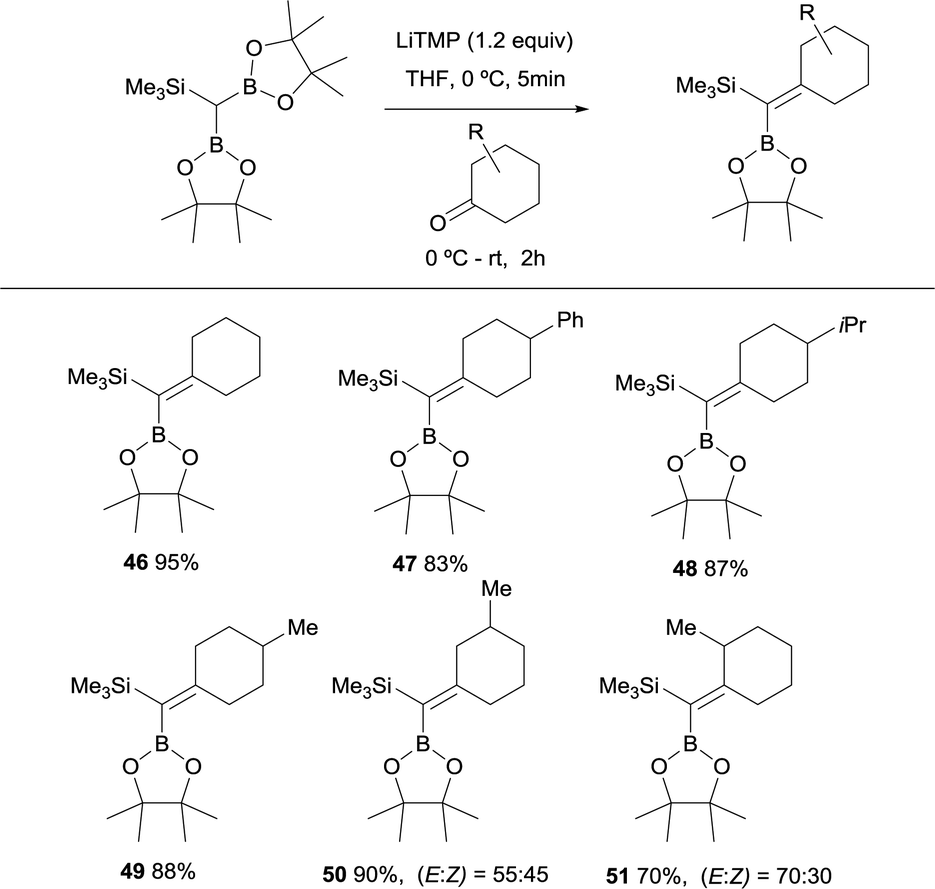
Fernández, Cuenca and co-workers performed a new boron-Wittig olefination with the reagent HC(Bpin)2(SiMe3) that was easily prepared by insertion of (trimethylsilyl)diazomethane into bis(pinacolato)diboron (B2pin2).20 The trimetalloid HC(Bpin)2(SiMe3) could be deprotonated in the presence of 1.2 equivalents of LiTMP, in THF at 0 °C, to form a diboryl and silyl stabilised carbanion. Its subsequent addition to a series of cyclic ketones and warming up to room temperature for 2 h, allowed the formation of the gem-silylborylated alkenes 46–49 in high yields (Scheme 14). Similarly, excellent reactivity was exhibited by a series of 3- and 2-substituted cyclohexanones, affording the corresponding gem-silylborylated alkene products 50 and 51 with high yield but moderate stereoselectivity with the preferred position of the 3- or 2-methyl group syn with respect to the SiMe3 group (Scheme 14). Interestingly, this protocol confirms that the B–O elimination is faster than the analogous Si–O Peterson-type elimination.21
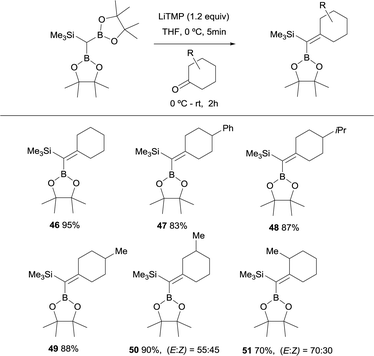 |
| | Scheme 14 Boron-Wittig between HC(Bpin)2(SiMe3) and cyclohexanone derivatives with LiTMP at rt. | |
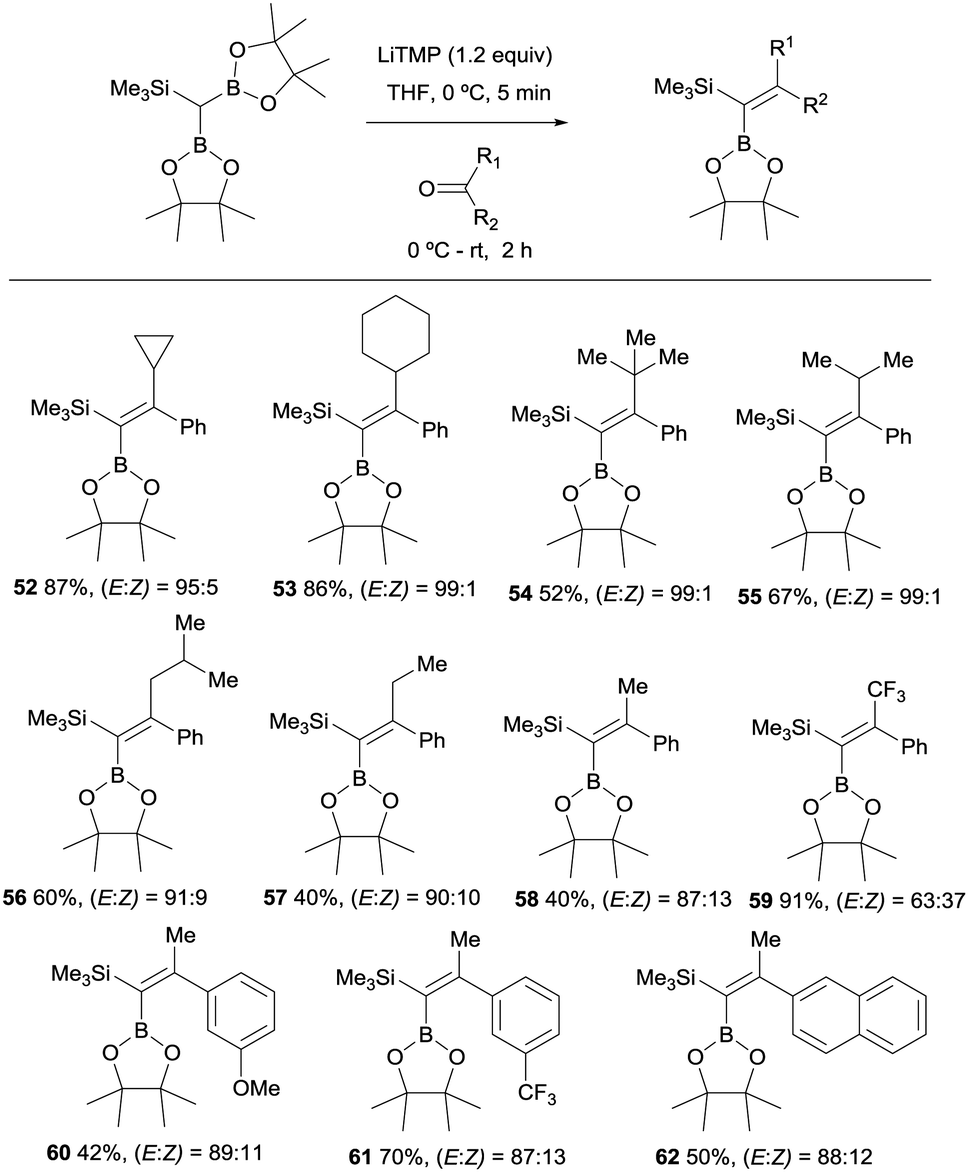
These authors also report the boron-Wittig olefination of acyclic aromatic ketones with HC(Bpin)2(SiMe3) in the presence of LiTMP. This reaction gave rise to the formation of a series of gem-silylborylated tetrasubstituted alkenes 52–62 with high degree of stereocontrol (Scheme 15). Interestingly, the major isomer places the Bpin moiety syn with respect to the aryl group.20 The reasons for such stereoselectivity might be justified by an unfavorable steric interaction between the aliphatic substituents on the ketone and a hindered Bpin moiety in the olefination transition state. Also, a plausible interaction between the aryl group and the empty p-orbital on B might contribute to the high stereoselectivity observed.20
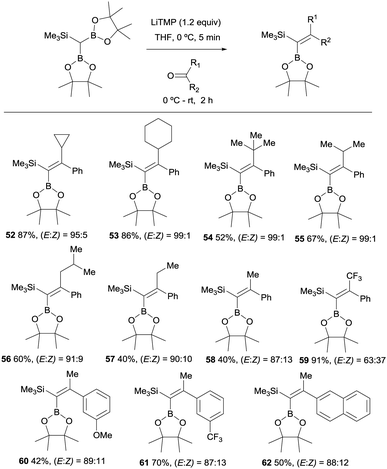 |
| | Scheme 15 Boron-Wittig olefination between HC(Bpin)2(SiMe3) and aromatic ketones with LiTPM at rt. | |
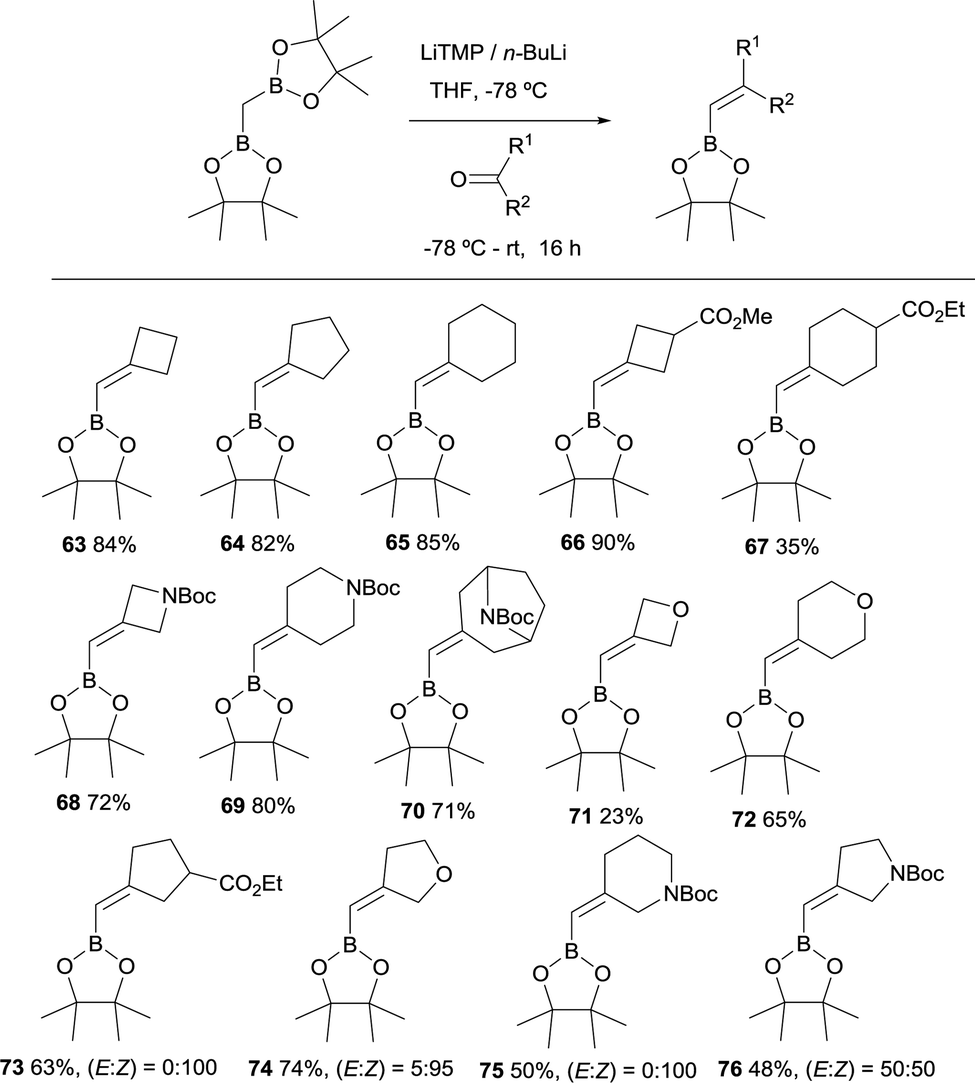
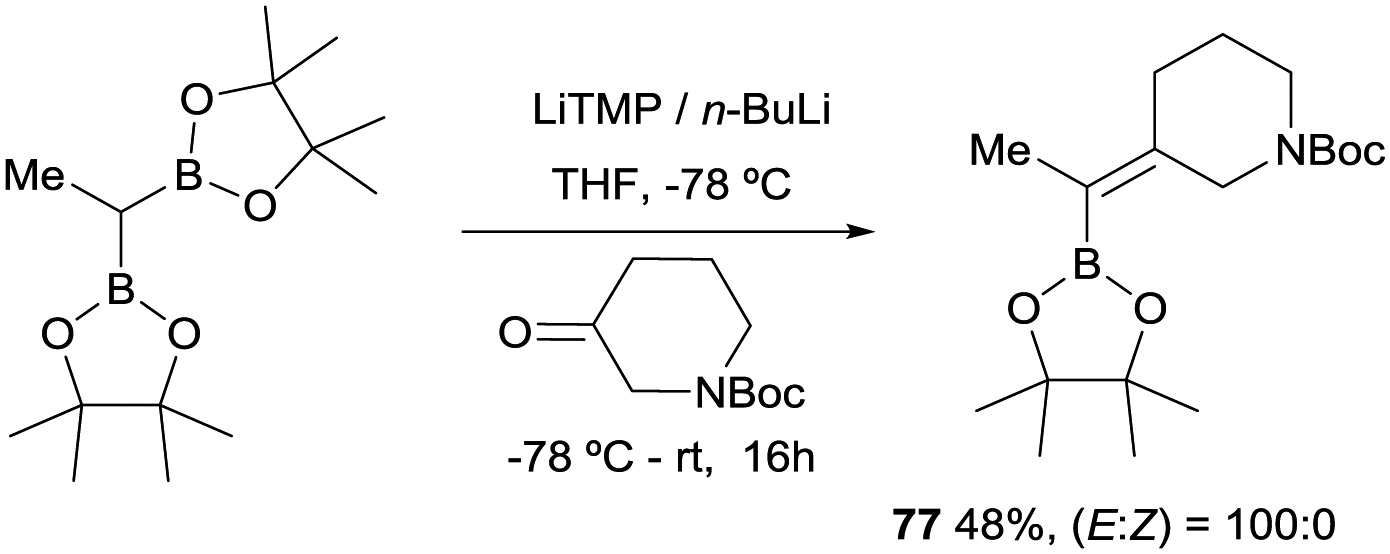
In 2019, Grygorenko22 and Morken23 have independently launched a systematic study of the boron-Wittig olefination of ketones with bis(pinacolboryl)methane in order to obtain trisubstituted alkenes with a significant control of the stereoselectivity. In this way Grygorenko and co-workers have efficiently synthesised a series of (hetero)cycloalkylidenemethyl boronates 63–76 from the corresponding cyclic ketones in the presence of the lithium bis(pinacolboryl)methide, generated from a mixture of LiTMP and nBuLi, at −78 °C (Scheme 16).22 The stereochemical control of the boron-Wittig reaction with non-symmetrical cyclic ketones favoured the (Z)-isomer in most of the cases (see products 73–75). The authors suggested that this marked stereocontrol could presumably be ascribed to a plausible interaction between the Bpin moiety and the oxygen or nitrogen atoms present in the substrate. Noteworthy, both (E) and (Z) stereoisomers were obtained in equal amounts for product 76 (Scheme 16). The authors rationalised this result based on the observation that geometric constrains might prevent a productive interaction between the Bpin fragment and the remote carbamate group. Interestingly, the same authors demonstrated that the boron-Wittig olefination with the most substituted substrate 2,2′-(ethane-1,1-diyl)bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) produced exclusively the (E)-alkene 77 with the carbamate group positioned syn with respect to the Bpin moiety (Scheme 17).
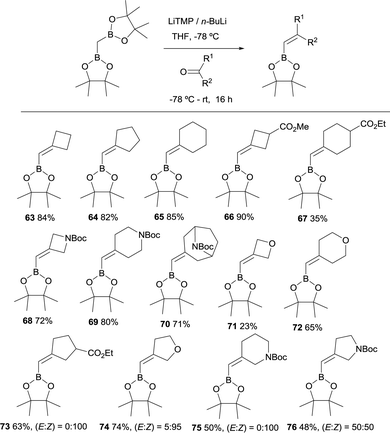 |
| | Scheme 16 Boron-Wittig olefination between CH2(Bpin)2 and cyclic ketones with LiTMP and nBuLi at −78 °C. | |
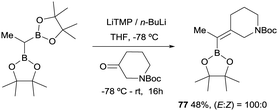 |
| | Scheme 17 Boron-Wittig olefination towards complementary (E)-stereoselectivity. | |
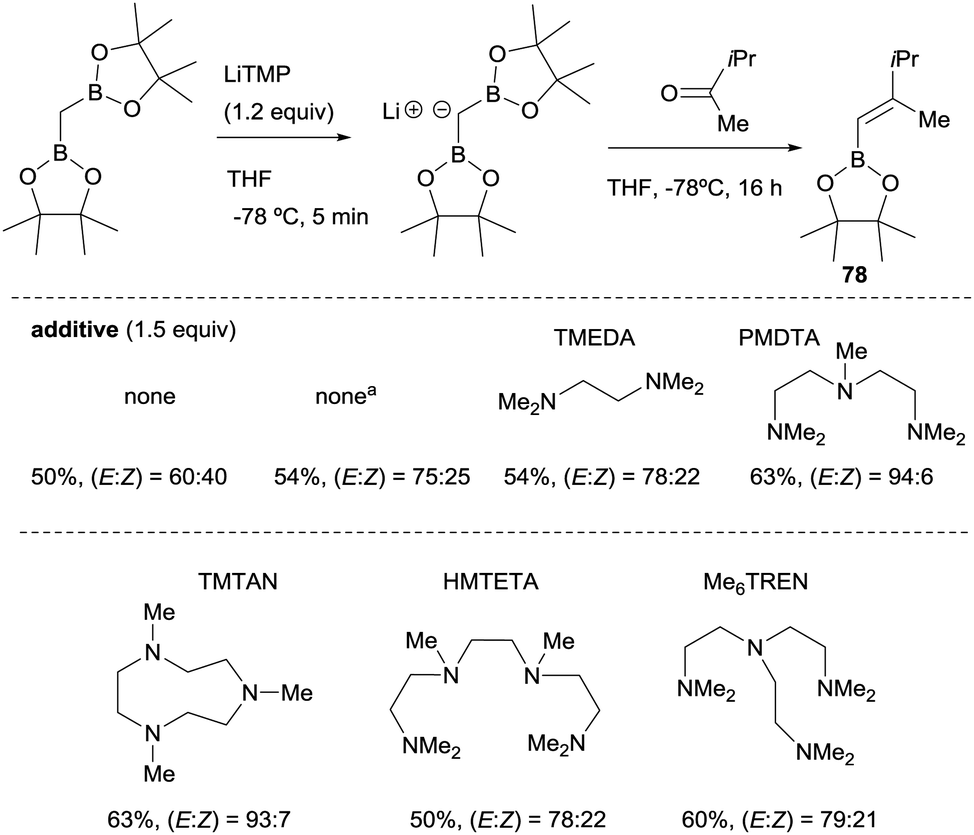
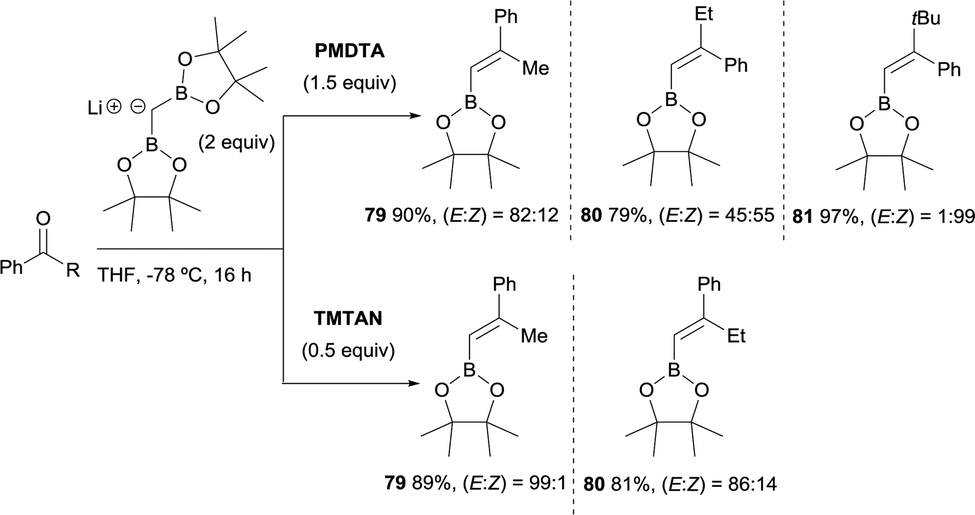
Morken and co-workers23 compared the use of lithium bis(pinacolboryl)methide previously isolated versus the one prepared in situ from a mixture of CH2(Bpin)2 and LiTMP at −78 °C, along the boron-Wittig reaction with methyl isopropyl ketone (Scheme 18). These authors realised that although the yield of the product 78 was comparable under both conditions, the olefination occurred with improved stereoselectivity in the case of in situ prepared lithium bis(pinacolboryl)methide. They explored therefore, the possible influence of amine as additive in the stereoselective outcome of the reaction, since Matteson and co-workers observed previously14 that the use of TMEDA (1.5 equiv.) as additive favored a slight increase in stereocontrol towards the (E)-isomer 78. The use of triamine PMDTA (1.5 equiv.) increased the stereoselectivity to (E)/(Z) = 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 as well as other triamine additives that offered comparable values of yield and stereoselectivity, whereas tetraamine additives provided only moderate results (Scheme 18).23
:6 as well as other triamine additives that offered comparable values of yield and stereoselectivity, whereas tetraamine additives provided only moderate results (Scheme 18).23
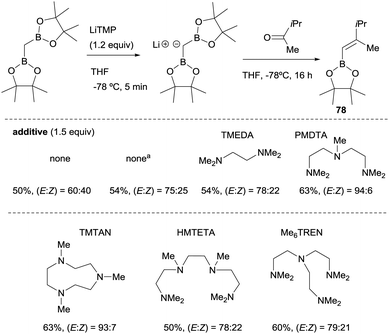 |
| | Scheme 18 Influence of polydentated amines as additives on boron-Wittig olefination between CH2(Bpin)2 and methyl isopropyl ketone at −78 °C. aLiCH(Bpin)2 used as isolated salt. | |
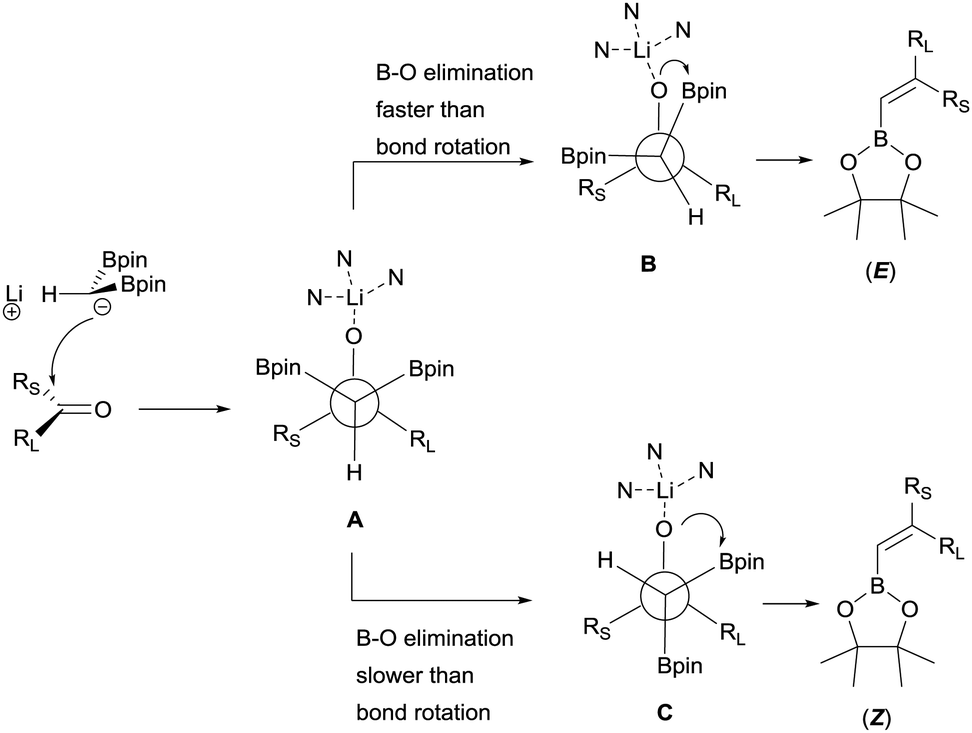
Morken and co-workers justified the origin of the stereoselectivity based on the Bassindale and Taylor model24 to predict the stereochemical outcome of reactions between carbanions and carbonyl compounds. The addition of the lithium bis(pinacolboryl)methide to the ketone might proceed lying the smallest group on the nucleophile (H in this case) between the carbonyl substituents thus providing intermediate A in Scheme 19. Considering that B–O elimination is faster than bond rotation, the evolution to intermediate B that minimise the steric interactions with the resting Bpin fragment should be favoured and the (E)-alkene might be obtained. Alternatively, when B–O elimination is slow, bond rotation may allow conversion of A into C where H is sited proximal to the stabilised Li with bulky triamine additive. From intermediate C, B–O elimination would give the (Z)-alkene. The authors assumed that the use of triamines such as PMDTA and TMTAN might favour the formation of monomeric species B, in agreement with previous studies about higher lithiate complexes.25,26 Morken and co-workers also found differences in the stereoselectivity control depending of the base used. Because of the intrinsic value of alkenylboronates derived from the reaction of aryl methyl ketones, the reaction with acetophenone was deeply explored. Hence, in this study it was found that the boron-Wittig reaction on acetophenone furnished the corresponding alkenylboronate 79 in a E/Z ratio = 82:18 when 1.5 equiv. of PMDTA was used, becoming less stereoselective the olefination of propiophenone with a E/Z ratio = 45:55 on product 80 (Scheme 20). However, the use of TMTAN in only 0.5 equiv. favoured a high stereoselectivity on the (E)-alkene for both products (Scheme 20). Interestingly, the boron-Wittig reaction with 2,2-dimethyl-1-phenylpropan-1-one in the presence of 1.5 equiv. PMDTA resulted highly stereoselective on the (Z)-isomer 81 (Scheme 20). The preferred formation of the (Z)-alkene was general for alkyl moieties with α-branch groups such as iPr, Cy and cyclopropyl.23
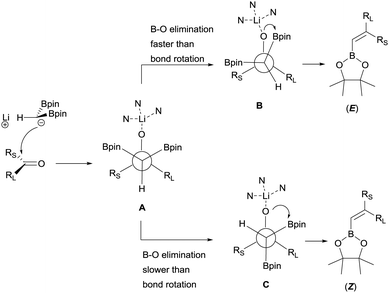 |
| | Scheme 19 Proposed origin of stereoselectivity in the boron-Wittig reaction of ketones using triamines as additives. | |
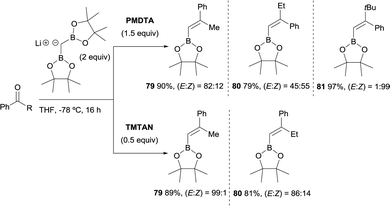 |
| | Scheme 20 Differences of boron-Wittig reaction of aryl ketones in the presence of triamine additives PMDTA and TMTAN. | |
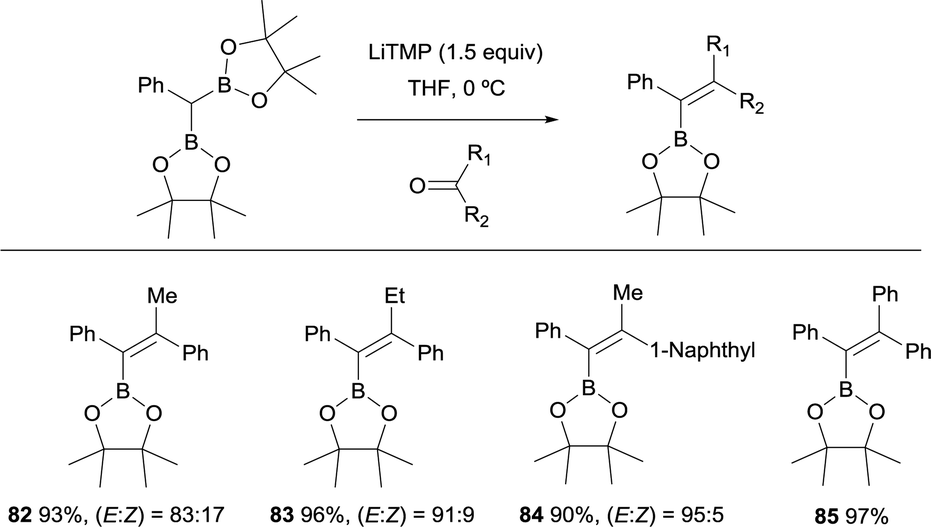
Hartwig and co-workers explored the convenient synthesis of tetrasubstituted alkenes from lithiation of CHPh(Bpin)2 with 1.5 equiv. of LiTMP and concomitant nucleophilic attack to aryl ketones.27 The reaction was conducted at 0 °C in THF and favoured the formation of (E)-alkenes 82–85 with high levels of stereoselectivity (Scheme 21).
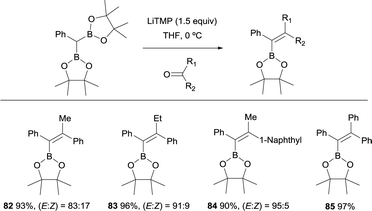 |
| | Scheme 21 Boron-Wittig olefination of aryl ketones with CHPh(Bpin)2 and LiTMP at 0 °C. | |
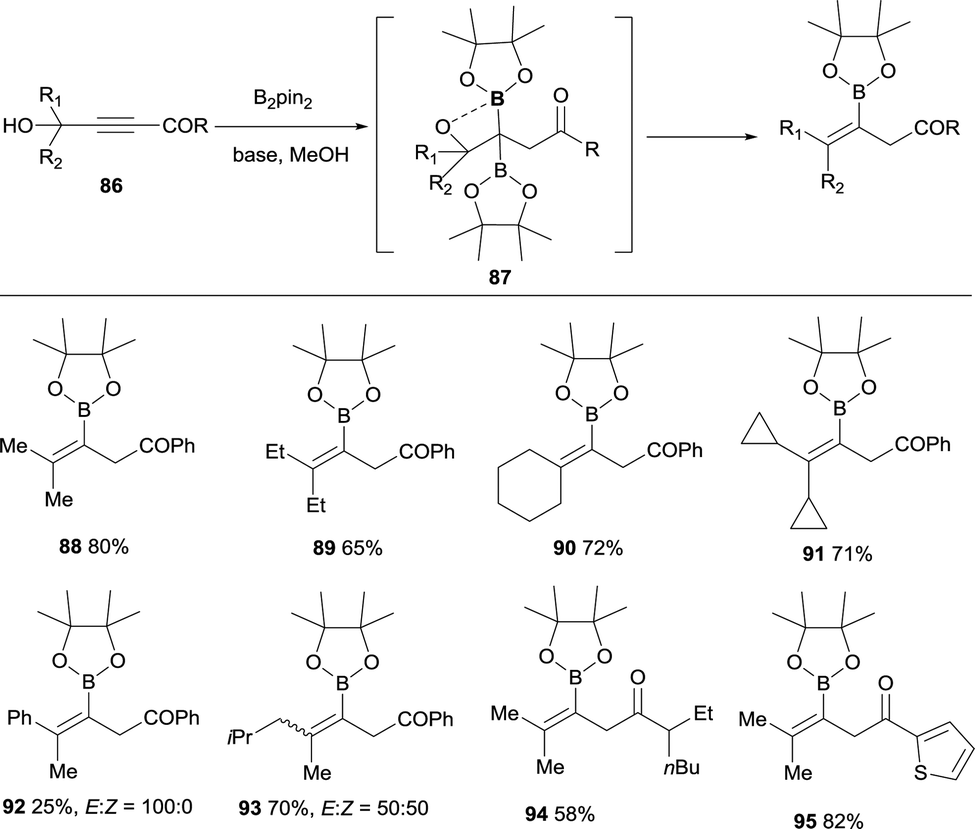
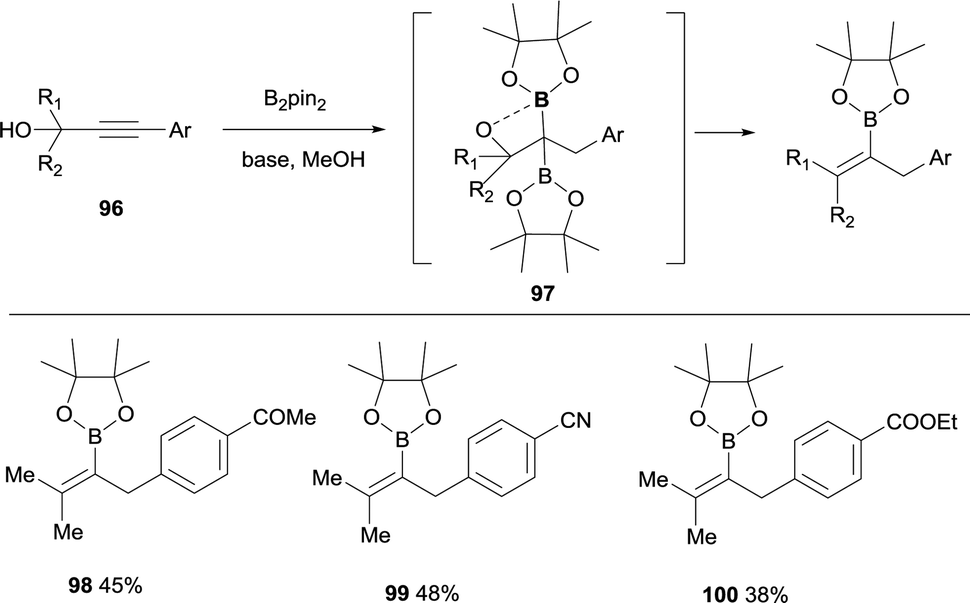
Lan, Song and co-workers have demonstrated an elegant intramolecular reaction through a cascade borylation/B–O elimination of propynols, in the absence of LiTMP.28 Although this reaction is not conducted through a boron-Wittig pathway, the intermediates are similar and we introduce here this example for comparison. The authors envisaged that ynones 86 could react in a transition-metal free manner with B2pin2, MeOH and base,29 to provide gem-diboronates 87 that further reacted intramolecularly with the hydroxyl group adjacent to the two Bpin moieties. Eventually, B–O elimination took place resulting in the synthesis of valuable tetrasubstituted alkenylboronates 88–95 (Scheme 22). When the groups adjacent to the hydroxyl group were not identical, a mixture of stereoisomers was obtained (see for example product 93), except for the Ph/Me groups that favoured the exclusive formation of the (E)-tetrasubstituted alkenylboronate 92 (Scheme 22). Interestingly, alkyne with strong electron-withdrawing groups instead the carbonyl groups could also convert under the standard conditions, towards the tetrasubstituted alkenylboronates 96–98 in moderate yields (Scheme 23).
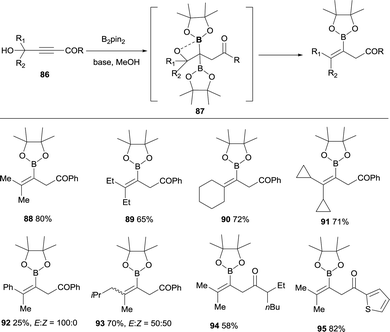 |
| | Scheme 22 Access to vinylboronates via gem-borylation of ynones and subsequent B–O elimination. | |
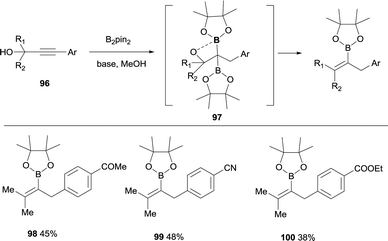 |
| | Scheme 23 Synthesis of alkenyl boronates through diborylation of propynols and subsequent B–O elimination. | |
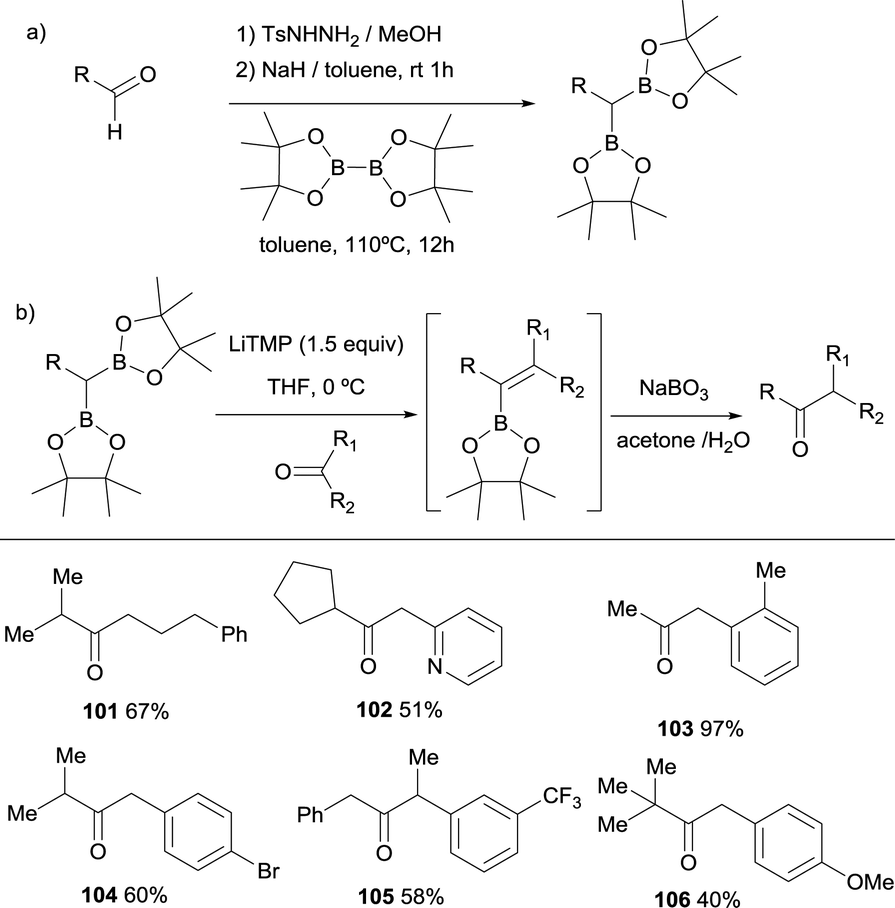
Pattison and co-workers reported an interesting application of the boron-Wittig reaction with a series of alkyl-substituted gem-bis(boronate) compounds and ketones by performing the in situ oxidation of the generated alkenyl boronates towards a new ketone functionality (Scheme 24b).30 In fact, given that the gem-bis(boryl)alkanes can be synthesised from the tosylhydrazone salts of the corresponding aldehydes (Scheme 24a), this transformation becomes a versatile homologative coupling understood formally as a umpolung coupling of two carbonyl compounds that give rise access to ketones 101–106. It was important to find that the aqueous sodium perborate dissolved in acetone gave the most consistent and cleanest results, by solubilizing both the oxidant and the vinyl boronate. Therefore, the process can be performed in a one pot fashion with only a switch in solvent at the intermediate stage. After completion of the boron-Wittig reaction of the carbonyl compound with lithiated geminal bis(pinacolboryl) reagent in THF, the solvent can be removed and replaced with acetone followed by oxidation with aqueous sodium perborate.
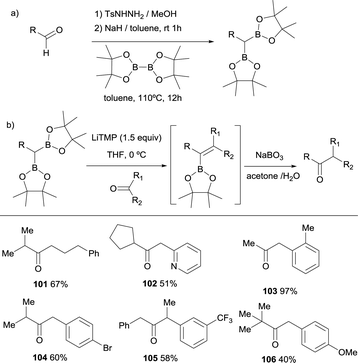 |
| | Scheme 24 Homologation of ketones by boron-Wittig/oxidation sequence. | |
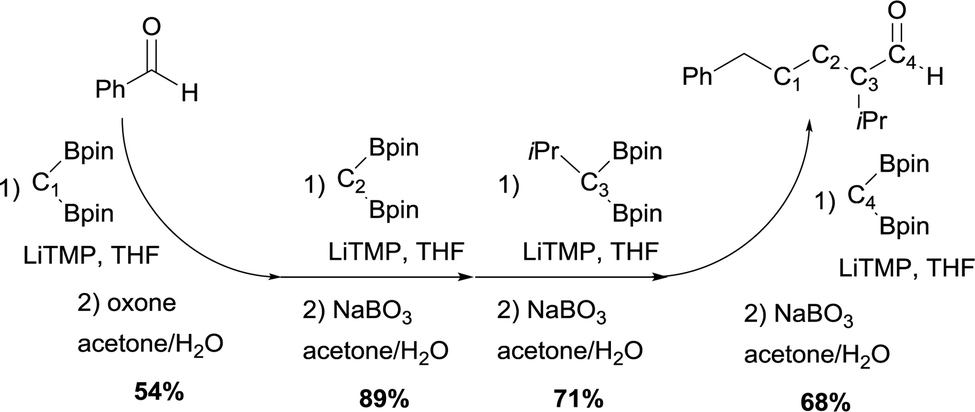
The authors also demonstrated that this strategy can be used in iterative sequences, though boron-Wittig/oxidation.30Scheme 25 shows a 4-carbon extension of benzaldehyde with the introduction of isopropyl group at a defined point of the chain. This is an elegant example of iterative synthesis of a large molecule constructed stepwise by addition of one subunit in sequential fashion through boron-Wittig/oxidation.
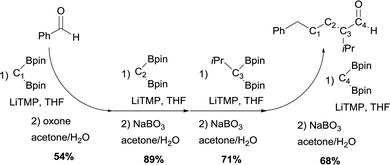 |
| | Scheme 25 Iterative homologation through boron-Wittig reaction. | |
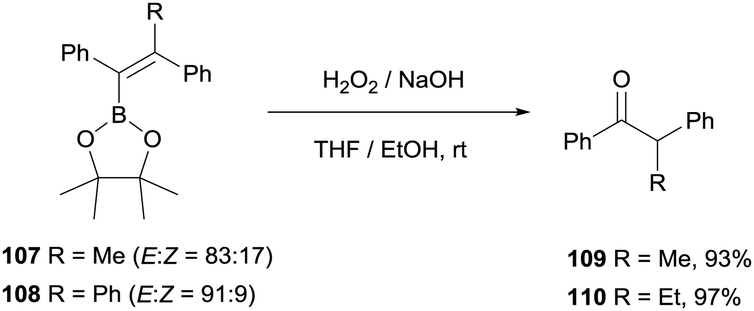
Hartwig and co-workers27 adapted the methodology for the oxidation of tetrasubstituted alkenylboronate by using an excess amount of NaOH and H2O2 to gain access to aryl substituted ketones 109 and 110 (Scheme 26).
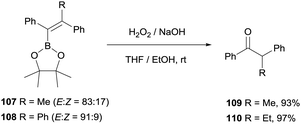 |
| | Scheme 26 Oxidation of tetrasubstituted alkenylboronates with H2O2 in basic media. | |
3. Boron-Wittig olefination of aldehydes with gem-bis(boryl)alkanes
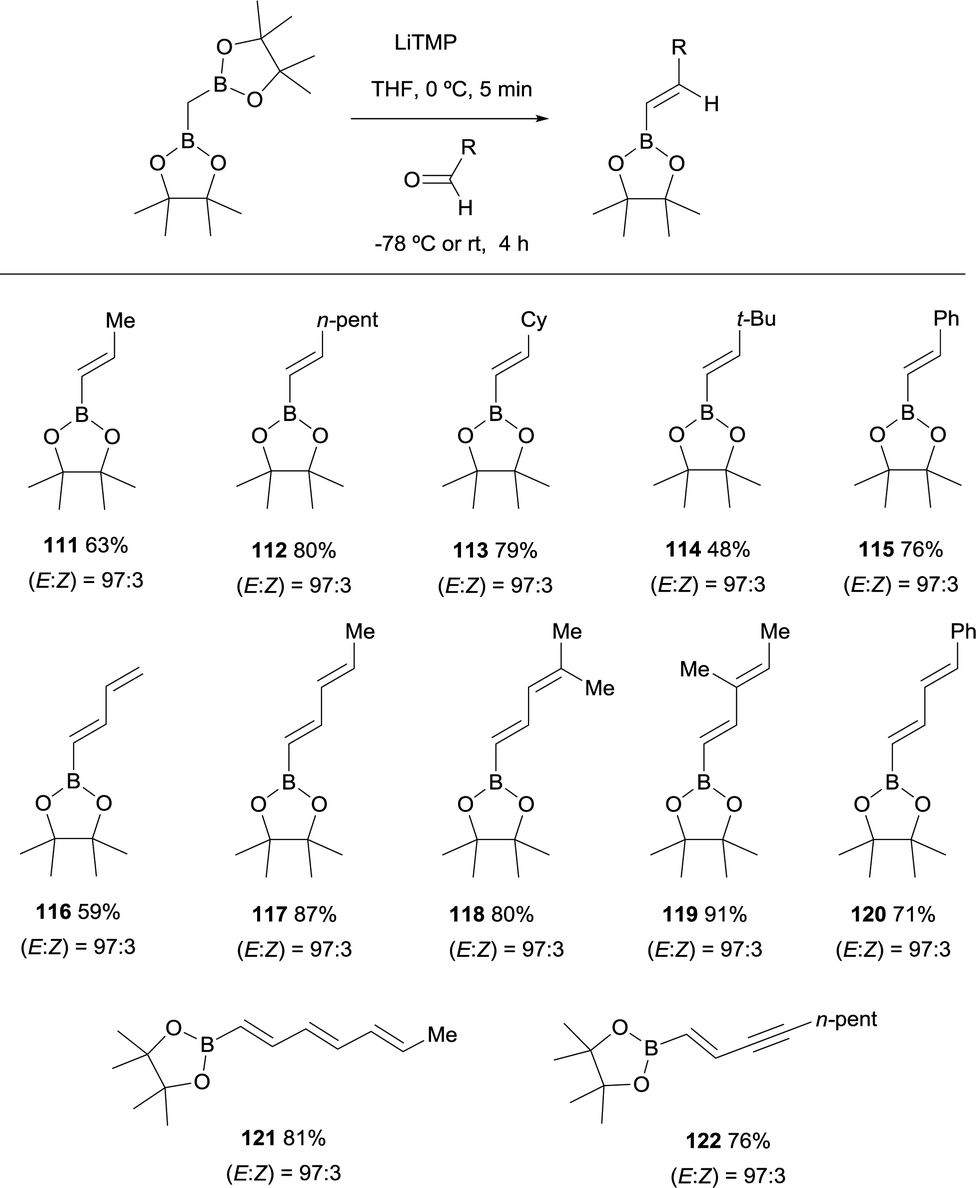
Morken and co-workers established in 2015, a stereoselective boron-Wittig olefination between bis(pinacolboryl)methane and aldehydes to furnish a variety of synthetically useful trans-vinylboronate esters 111–122 (Scheme 27).31 The base of choice was LiTMP that was added to CH2(Bpin)2 at 0 °C, followed by a decrease of temperature to −78 °C when the aldehyde was added. The reaction was stirred for 4 h before being warmed to room temperature for work up. It is worthy to say that authors also observed that similar reaction outcome was observed when the reaction took place at room temperature (E/Z ratio 97/3). Synthetically useful dienyl (116–120), trienyl (121) and enynyl boronates (122) were efficiently prepared from the corresponding unsaturated aldehydes (Scheme 27).31
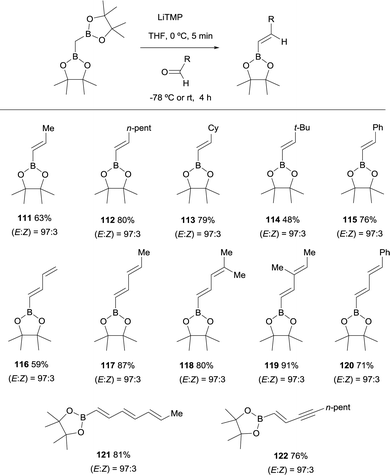 |
| | Scheme 27 Stereoselective boron-Wittig reactions for trans-vinylboronates. | |
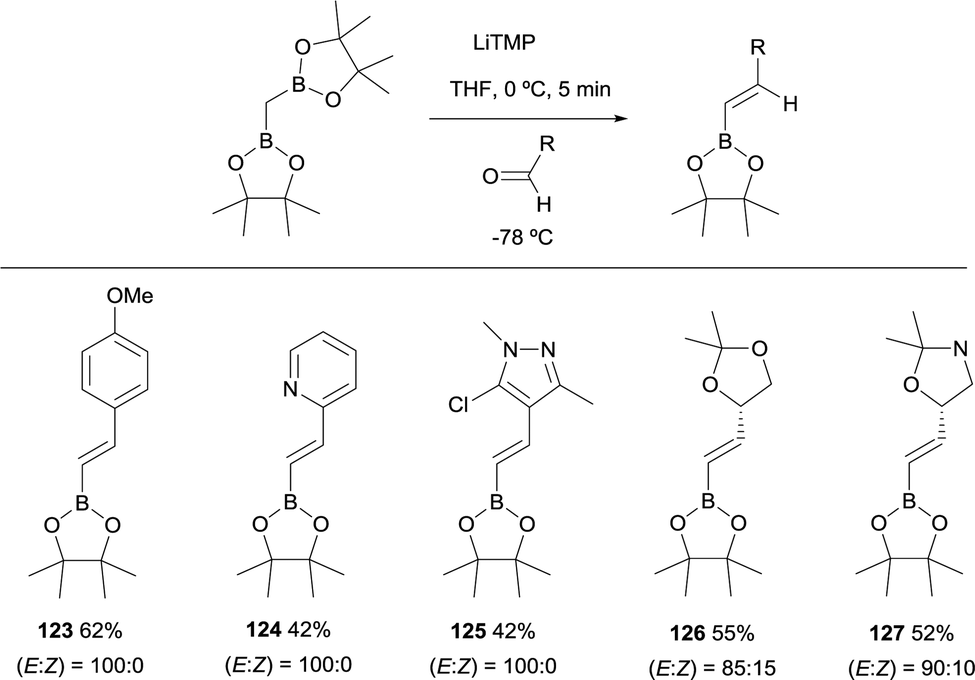
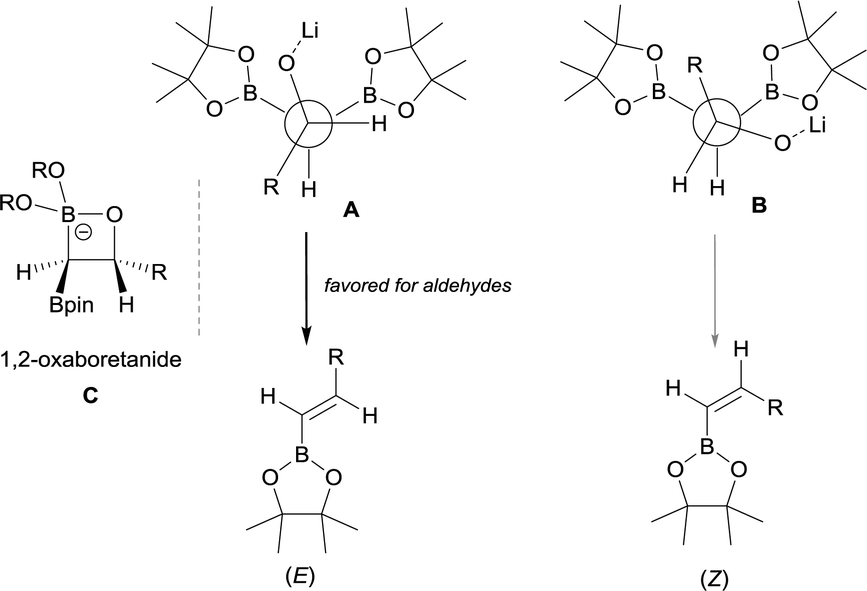
Remarkably, an exclusive formation of the (E)-vinyl boronates 123–127 was observed by Grygorenko and co-workers for the boron-Wittig reaction between aryl aldehydes and CH2(Bpin)2 in the presence of LiTMP at −78 °C (Scheme 28).22 The same authors justified the total control on the stereoselectivity in favor of the (E)-stereoisomer trough the models shown in Scheme 29,22 where the conformation A might be completely preferred versusB, in the case of aldehydes. However, the involvement of a four-membered intermediate 1,2-oxaboretanide of type C cannot be ruled out.32–34
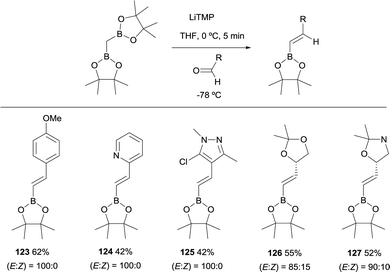 |
| | Scheme 28 High stereoselective control on boron-Wittig reactions with aldehydes. | |
 |
| | Scheme 29 Origin of the complete stereoselectivity in boron-Wittig olefination with aldehydes. | |
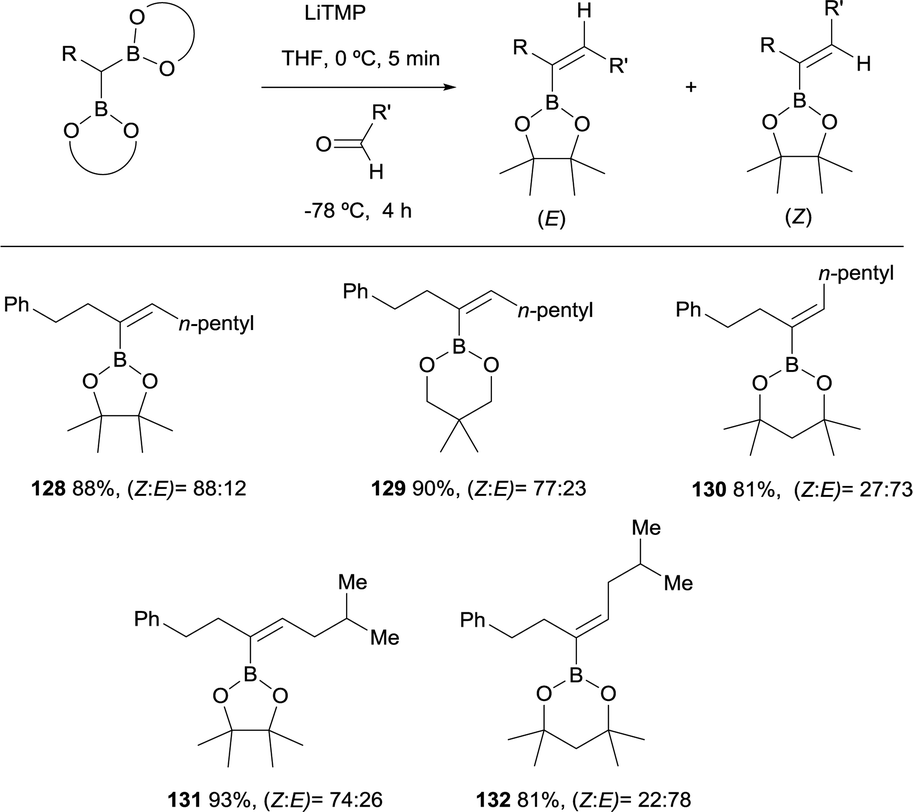
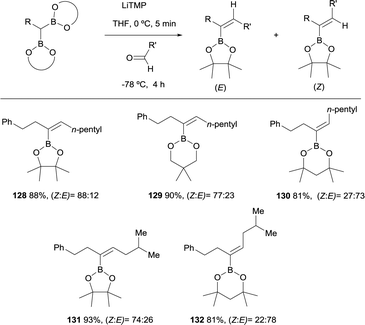
Morken and co-workers considered the application of the boron-Wittig reaction to the synthesis of trisubstituted vinylboronates.31 However, depending on the nature of the boryl moiety, the stereoselectivity switches from E to Z stereoisomer. The boron-Wittig of phenyl propane derived pinacolboryl reagent with hexanal, in the presence of LiTMP, favored the condensation towards the Z-isomer 128 in a ratio Z/E = 88:12 (Scheme 30). Similar behavior was observed with the related reagent neopentylglycolato prioritizing the formation of the Z-isomer 129 in a ratio Z/E = 77:23, while the same reaction with dimethylpentanediolato boryl derivative favored the E-isomer 130 in a Z/E = 27:73. Scheme 30 illustrates this observation, where both cis- or trans-isomers of the trisubstituted vinyl boronates can be afforded in moderate stereoselectivity by simply varying the boryl moiety. The authors did not justify the diverse stereoselectivity observed.31
 |
| | Scheme 30 Complementary moderate stereoselective boron-Wittig reactions for trisubstituted vinylboronates. | |
Since formaldehyde did not efficiently react with substituted geminal bis(boronates) in the presence of LiTMP, Morken and co-workers studied the nucleophilic substitution of the lithium bis(pinacolboryl)methide salts with CH2I2 and subsequent B–I elimination, resulting in the formation of 1,1-disubstituted vinyl boronates.31 This alkylation procedure was already developed by Matteson and co-workers with significant success.8
4. Boron-Wittig olefination of primary amides with gem-bis(boryl)alkanes
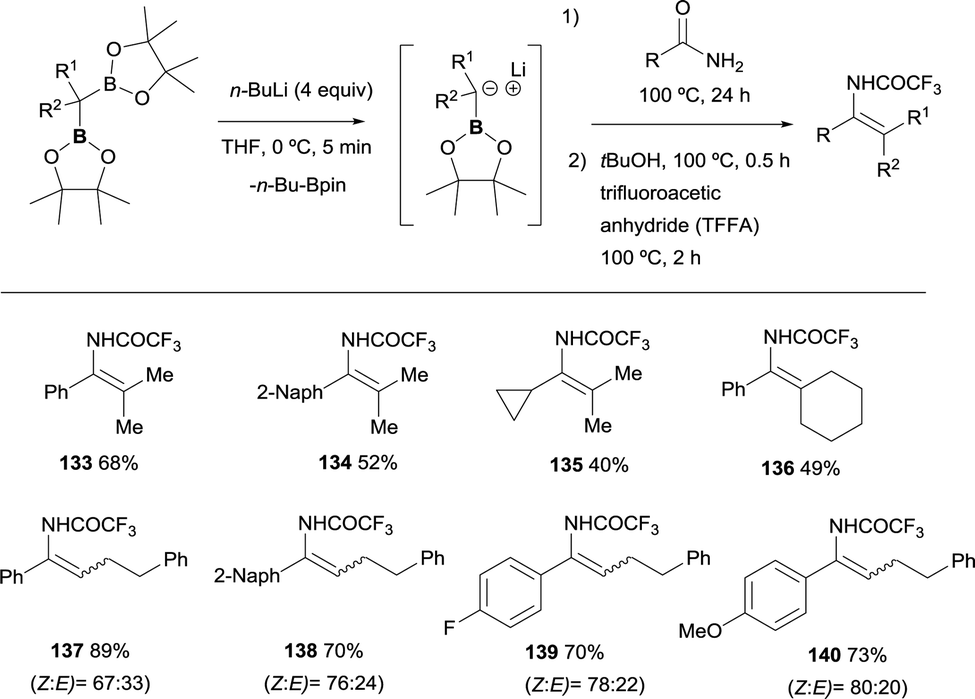
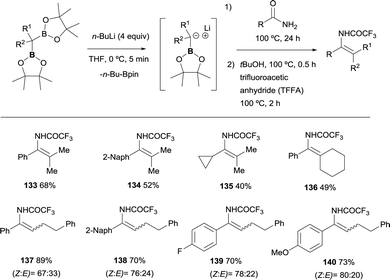
Liu and co-workers have disclosed a chemodivergent transformation of amides by using 1,1-diborylalkanes as pro-nucleophiles.35 They found that in general, among the two possible B–X removal, selective B–O elimination occurs for primary amides to generate an enamine intermediate. The generation of the α-boryl carbanion was carried out in the presence of nBuLi, that performed the deborylation of the gem-bis(pinacolboryl)alkanes instead of deprotonation associated to the use of LiTMP. The nucleophilic attack on primary amides, followed by B–O elimination generates enamine intermediates that were in situ transformed into isolable enamides by the addition of tertiary butanol and trifluoroacetic anhydride (TFAA) (Scheme 31). For non-symmetric gem-bis(pinacolboryl)alkanes, only moderate stereoselectivities was obtained.35
 |
| | Scheme 31 Boron-Wittig olefination between gem-bis(pinacolboryl)alkanes and primary amides. | |
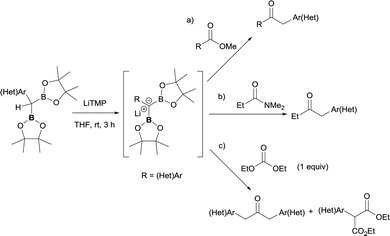
5. Nucleophilic attack of α-bis(boryl)carbanions to tertiary amides, esters and acids
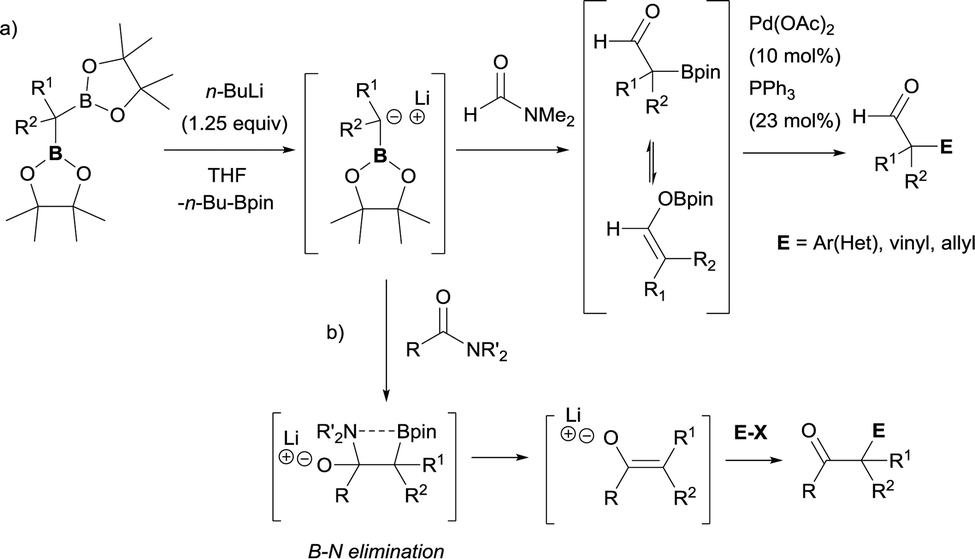
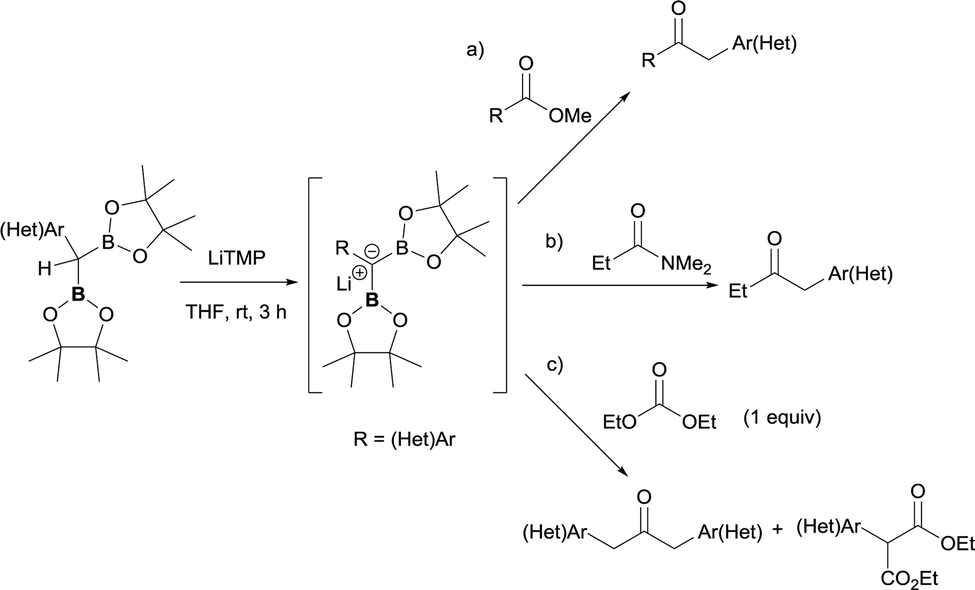
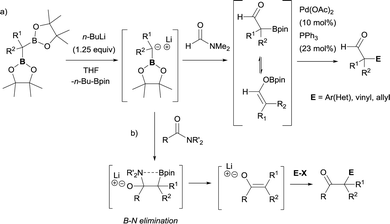
Due to the possible reactivity via addition/elimination sequence, the addition of α-(boryl)carbanions to amides may give carbonyl intermediates that bears a β-boryl group, which may result in the in situ generation of boron enolates. This enolization trend provides advantages in blocking a plausible second nucleophilic attack and their further electrophilic functionalization provides highly functionalized carbonylic compounds from widely accessible amides. In that context, Xu and Zhao reported the transformation of a particular tertiary amide (DMF, that was used as solvent) to generate α-boryl aldehydes as stable intermediates that efficiently reacted with electrophilic reagents via Pd-catalyzed cross-coupling reactions (Scheme 32a).36,37 Liu and co-workers developed a strategic pathway to transform tertiary amides into valuable enolates via B–N elimination. The subsequent electrophilic trapping with electrophiles allows the isolation of a large number of α-functionalized ketone (Scheme 32b).35
 |
| | Scheme 32 Reactivity of α-monoboryl carbanions with tertiary amides followed by enolization/electrophilic trapping protocols. | |
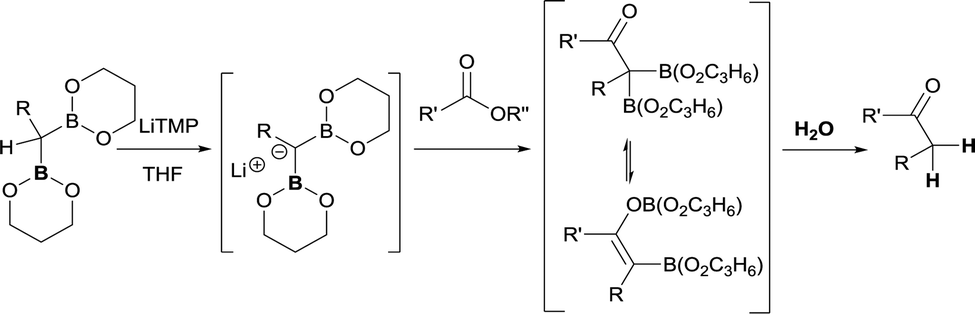
As far as ester groups is concerned, preliminary work by Matteson and co-workers about deprotonation of bis(trimethylenedioxyboryl)alkanes with LiTMP yielded the corresponding diborylcarbanion that reacted with methyl benzoate and methyl butyrate to generate the enolate intermediate that eventually was hydrolysed towards the corresponding ketone (Scheme 33).13 Mukaiyama et al. also developed a similar reactivity but using 9-borabicyclo-[3,3,1] nonane (9-BBN) as the boryl moiety.38
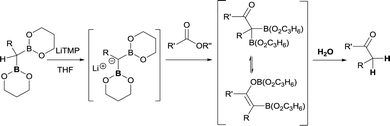 |
| | Scheme 33 Ketone formation from nucleophilic attack of diborylcarbanions to esters. | |
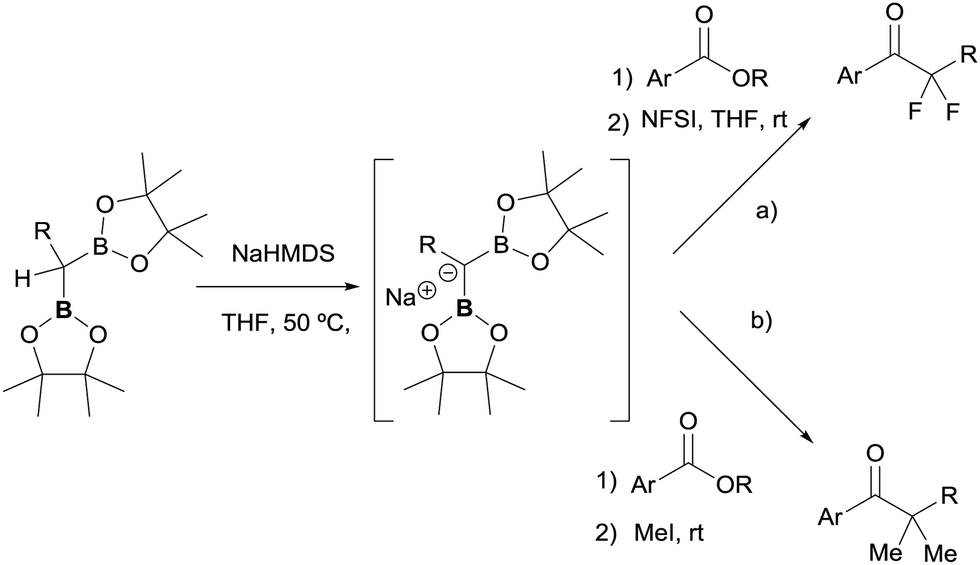
The great advantage of this conceptual methodology came from the group of Pattison and co-workers who made a significant contribution and extension in 2018,39 promoting the condensation of esters with gem-bis(pinacolboryl)alkanes, to generate an α,α-bis(enolate) equivalent which can be trapped with electrophiles including alkyl halides and fluorinating agents. This works represents an efficient, convergent synthetic strategy for the synthesis of unsymmetrical blocked ketones. The authors discovered that benzyl-substituted geminal bis(pinacolboryl) compound reacted with NaHMDS as base to be deprotonated. The in situ formed carbanion reacted with esters via addition/elimination/enolization sequence to generate an α,α-bis(enolate) that can be further doubly trapped by addition of an electrophilic fluorinating reagent such N-fluorobenzenesulfonimide (NFSI) (Scheme 34a).39,40 For enolizable esters, the used of LiTMP was required. A similar strategy developed by the same authors allowed the dimethylative coupling using as base either NaHMDS for nonenolizable esters or LiTMP for enolizable esters (Scheme 34b). For the most reliable dimethylation of the resultant α,α-bis(enolate), up to 5 equiv. of iodomethane were added for trapping.39
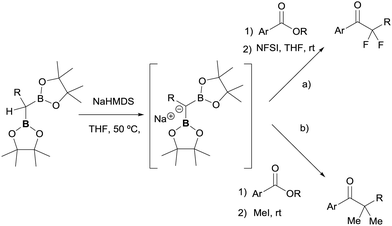 |
| | Scheme 34 Nucleophilic attack of diborylcarbanions to esters and subsequent double trapping of α,α-bis(enolate) equivalents with electrophilic fluorinating agents (a) and methyl iodide (b). | |
Chirik and co-workers have extended the conversion of esters to ketones using benzyldiboronates including substrates with acidic α-protons adjacent to carbonyl groups complementing the concept towards tertiary amides and carbonates.41 The authors also support the formation of an α-boryl carbanion through an alkoxide-mediated deborylation of gem-bis(pinacolato)diboron reagents. The α-boryl carbanion serves as a reactive intermediate for C–C bond formation towards ketone synthesis (Scheme 35).
 |
| | Scheme 35 Nucleophilic attack of benzyldiboryl carbanions to esters, tertiary amides and carbonates. | |
Xia and Liu have reported a dual functionalization of gem-bis(pinacolboryl)alkanes through deoxygenative enolization with carboxylic acids (Scheme 36).42 This reaction is considerably challenging in this context because of the plausible quenching effect of the carboxylic acidic proton. Still, the authors realised on the bases of a FTIR study that MeLi was able both, to deprotonate the acid and to deborylate the gem- bis(pinacolboryl)alkane generating an α-monoboryl carbanion that react with the lithium carboxylate salts to afford a boron enolate. Electrophilic trapping of enolate species with various electrophiles provides dual functionalization to afford a variety of α-mono, di-, and tri-substituted ketones.
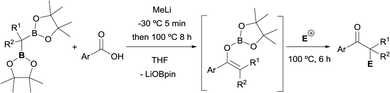 |
| | Scheme 36 Dual functionalization of 1,1-diborylalkanes via a deoxygenative enolization with carboxylic acids. | |
6. Applications of boron-Wittig olefination to C–C cross-coupling reactions
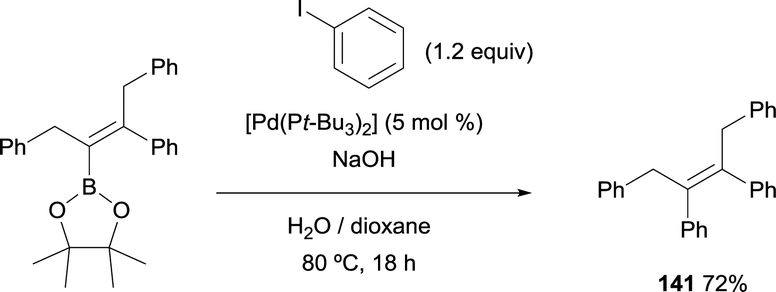
Matteson and co-workers were pioneer testing the Suzuki–Miyaura cross-coupling following the boron-Wittig reaction.8 Within the last decade, Endo and Shibata demonstrated that the tetrasubstituted alkenylboronate, containing the stable Bpin moiety, could proceed through C–C coupling using catalytic amounts of [Pd(tBu3)2] and NaOH. (E)-2-(1,3-Diphenylpent-2-en-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane could be transformed into (Z)-but-2-ene-1,2,3,4-tetrayltetrabenzene (141) in 72% yield, heating the reaction at 80 °C for 18 h (Scheme 37).15
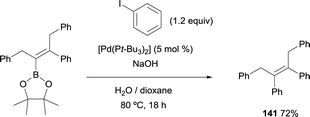 |
| | Scheme 37 Suzuki–Miyaura cross-coupling with [Pd(tBu3)2] and NaOH. | |
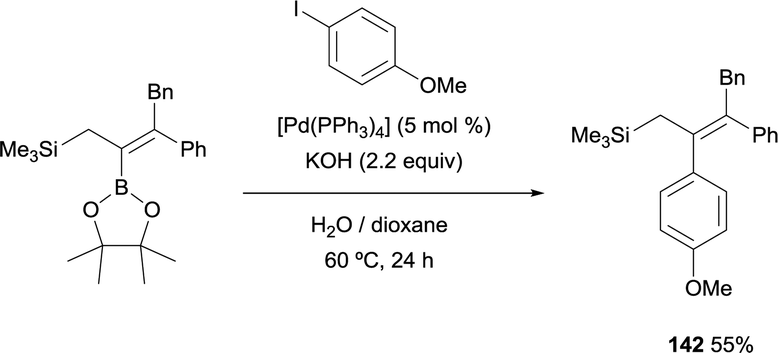
Alternatively, Endo and Shibata also found that 5 mol% [Pd(PPh3)4] and KOH (2.2 equiv.) catalyzed the cross-coupling of (E)-(3,4-diphenyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)but-2-en-1-yl)trimethylsilane with 4-iodoanisole at 60 °C. The trimethyl silyl moiety was unaltered under these reaction conditions (Scheme 38).18
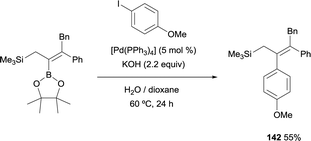 |
| | Scheme 38 Suzuki–Miyaura cross-coupling with [Pd(PPh3)4] and KOH. | |
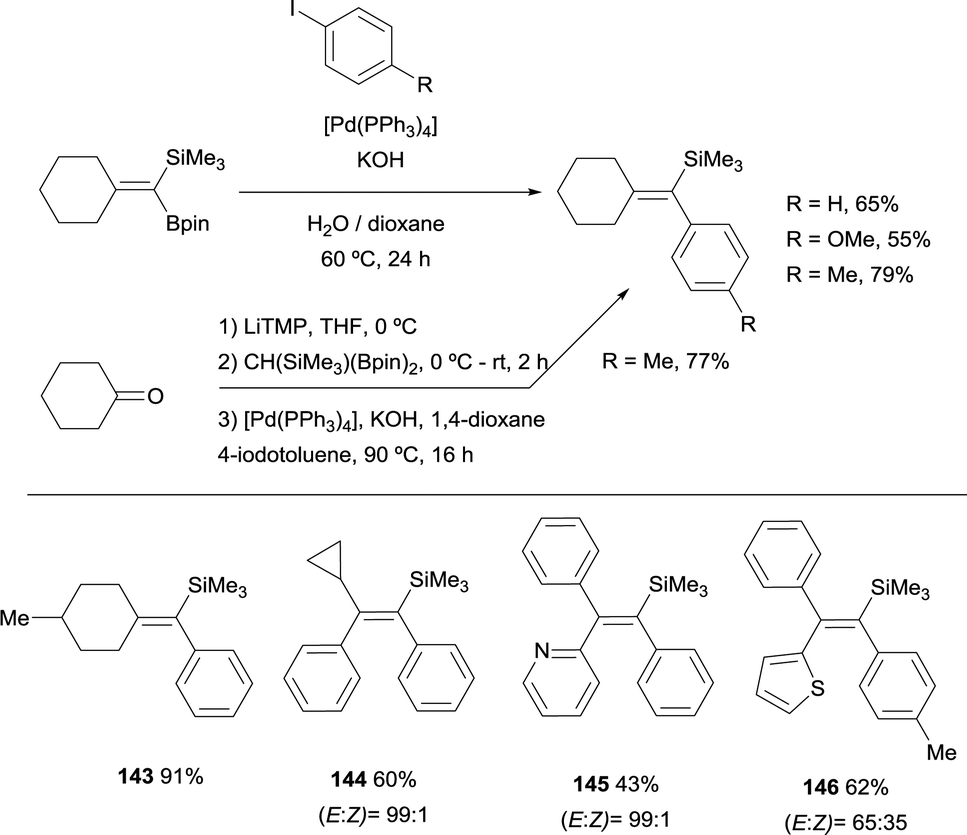
As an extension of these valuable precedents, Fernández, Cuenca and co-workers designed a route to transform 1,1-(boryl)(silyl)-alkenes, prepared via boron-Wittig reaction, into fully substituted vinyl silanes 143–146. The protocol could be applied via stepwise or one pot protocol, using [Pd(PPh3)4] and KOH as the catalytic system, with 1,4-dioxane as solvent working at 90 °C (Scheme 39).20 Since the silyl group resulted unaltered under these reaction conditions, the gem-silylboronates were efficiently transformed into 1-aryl,1-trimethylsilylalkenes following the sequence gem-silylborylation/cross-coupling for a representative types of cyclic and acyclic ketones. This methodology complements previous reported challenging protocols to synthesize all-substituted vinyl silanes.43
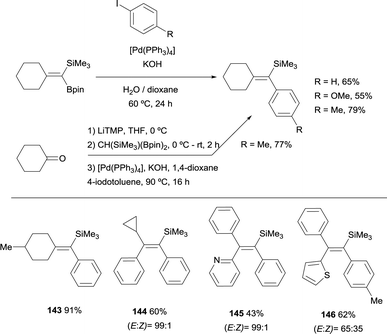 |
| | Scheme 39 Boron-Wittig/Suzuki–Miyaura cross-coupling through 1,1-(boryl)(silyl)-alkenes intermediates. | |
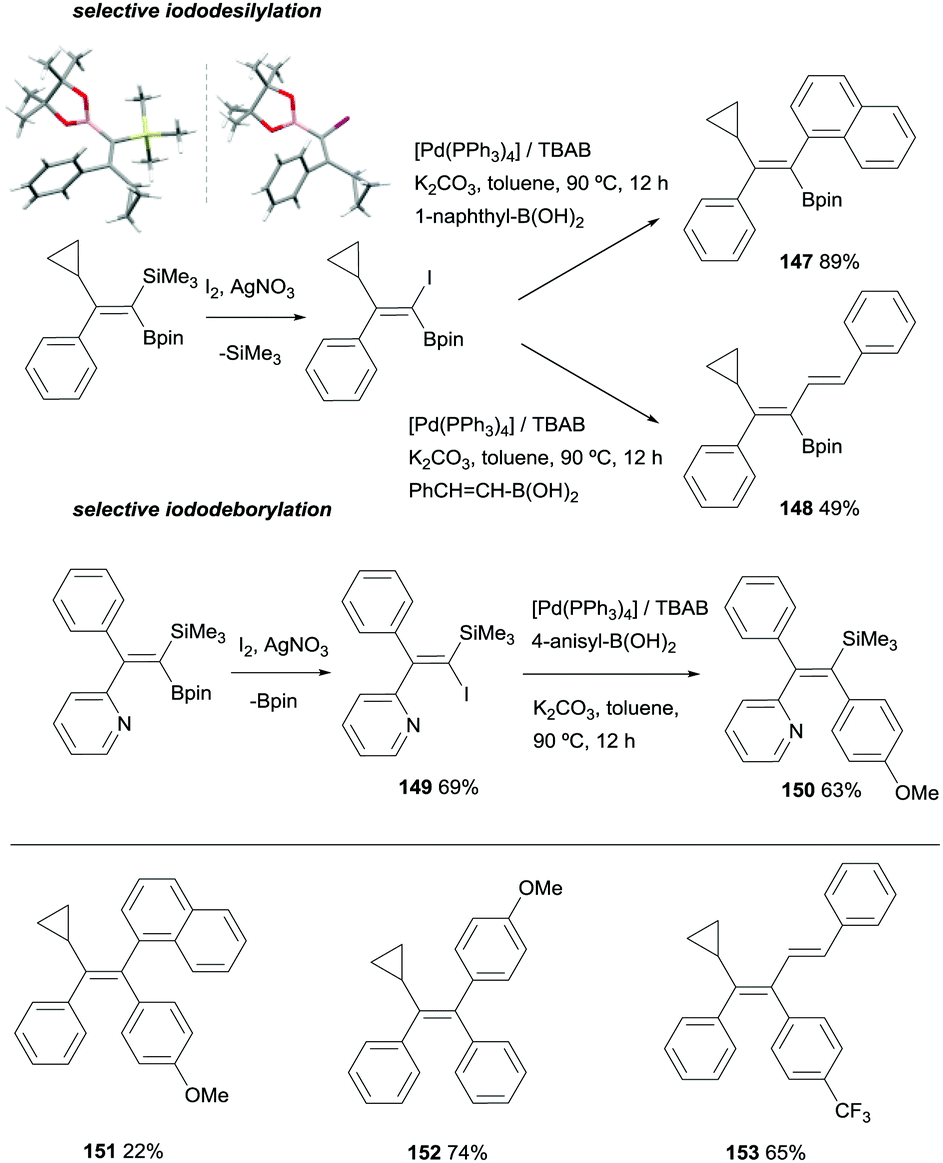
Fernández, Cuenca and co-workers also explored an alternative cross-coupling based on the most challenging silicon-based arylation strategy, keeping the Bpin moiety untouched. To this end, a iododesilylative protocol44 was designed using I2/AgNO3 demonstrating the viability of the procedure towards the transformation of the C-SiMe3 into C-I, that was further reacted with 1-naphthylboronic acid or trans-styrylboronic acid in the presence of [Pd(PPh3)4] catalyst and tetrabutylammonium bromide (TBAB)/K2CO3 to obtain 147 and 148 respectively (Scheme 40). These reactions represented the first attempt towards iododesilylation/cross coupling in silylboronated products with the extra value of the stereoselective control on the isolation of trisubstituted 1-borylalkenes. To make the process even more versatile, iododeborylation could be performed efficiently in gem-silylborylated alkenes containing a pyridyl moiety, suggesting that the intramolecular interaction of N with B assists the Bpin release keeping the SiMe3 group unaltered.20 Subsequent cross-coupling of 149 with 4-anisylboronic acid in the presence of [Pd(PPh3)4] catalyst and TBAB/K2CO3 allowed the isolation of trisubstituted vinyl silane 150 with total stereocontrol (Scheme 40). Based on this new stepwise protocol, the stereoselective synthesis of tetrasubstituted olefins 151–152 could be achieved from the cross-coupling of trisubstituted 1-borylalkenes with arylhalide reagents and [Pd(tBu3)2] as catalytic system (Scheme 40 bottom).20
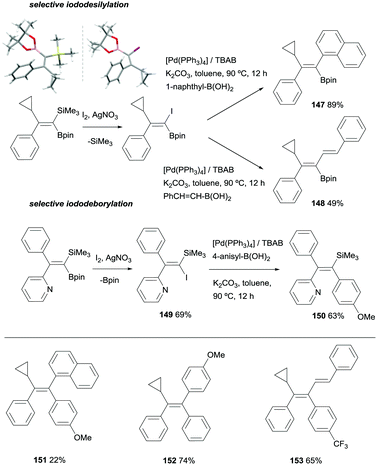 |
| | Scheme 40 Divergent iododesilylation or iododeborylation/cross-coupling reactions. | |
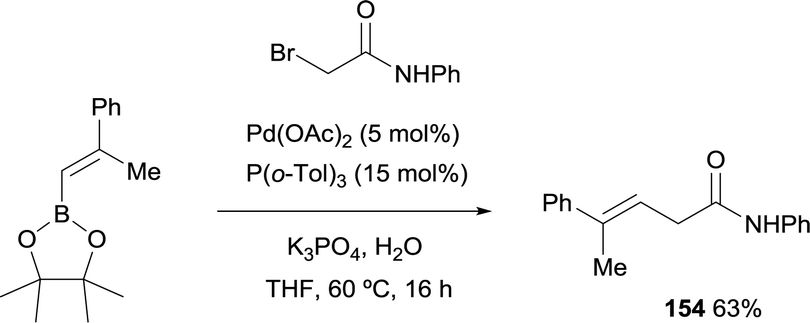
Morken and co-workers studied the cross-coupling of trisubstituted alkenyl boronate with α-bromoamide generating stereodefined β,γ-unsaturated amides of type 154 with preservation of the alkene geometry (Scheme 41).23 The catalyst used for the Suzuki–Miyaura coupling was Pd(OAc)2 modified with P(o-Tol)3.
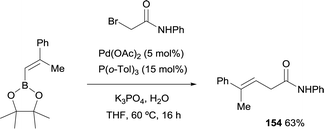 |
| | Scheme 41 Suzuki–Miyaura cross-coupling of trisubstituted alkenyl boronate with α-bromoamide. | |
The ability to transform the vinyl boronate through Suzuki–Miyaura coupling, allowed to address the application of the boron-Wittig/cross-coupling sequence to prepare derivatives of tamoxifen, an antagonist of the estrogen receptor in breast tissue. The first attempt was made by Endo and Shibata, obtaining the desired tetrasubstituted derivative in good yield, as a sole stereoisomer (Scheme 42) [Pd(tBu3)2] as catalytic system. Interestingly, the biological activity of this derivative was even higher than that of tamoxifen, but its synthesis could not be achieved previously.45
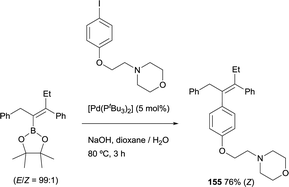 |
| | Scheme 42 Stereoselective synthesis of tamoxifen derivative all-substituted vinyl boronates prepared by boron-Wittig reaction. | |
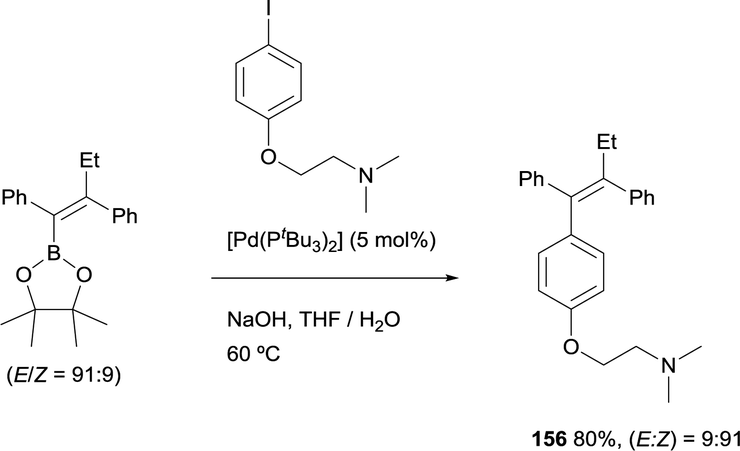
Similarly, Hartwig and co-workers developed a straightforward synthesis of tamoxifen, that was prepared in good yield from the tetrasubstituted alkenylboronate ester (E)-2-(1,2-diphenylbut-1-en-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane by Suzuki–Miyaura cross-coupling with the corresponding aryl iodide.26 This reaction occurred in the presence of [Pd(tBu3)2] as catalytic system and with retention of the ratio of E to Z configuration of the double bond (Scheme 43). This method complements previous approaches towards tamoxifen that required multistep synthesis.46
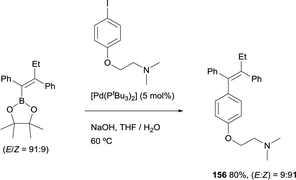 |
| | Scheme 43 Highly stereoselective synthesis of tamoxifen. | |
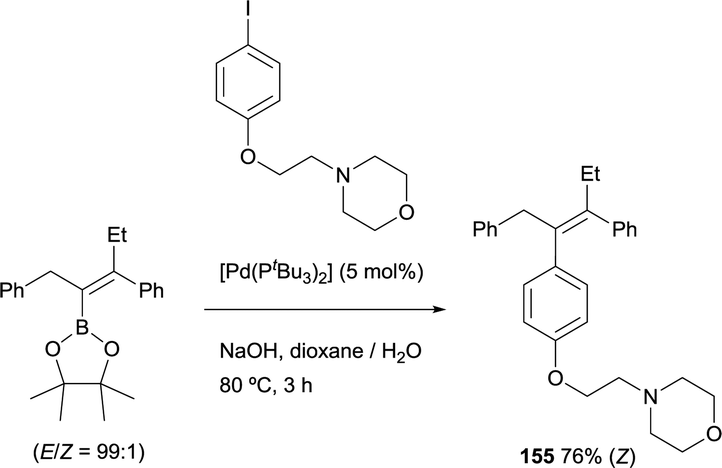
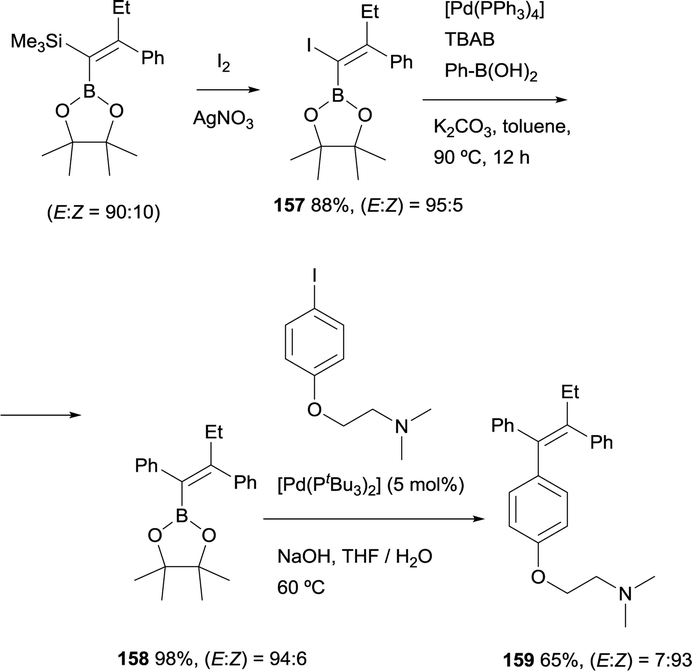
Alternatively, Cuenca and Fernández found a convenient pathway to transform gem-silylborylated alkenes into (Z)-tamoxifen with more than 93% stereoselection.20 As Scheme 44 shows, iododesilylation of (E)-trimethyl(2-phenyl-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)but-1-en-1-yl)silane (obtained with a E/Z ratio = 90:10), provides the intermediate 1-iodo-1-borylalkene 157 in 88% yield and high stereoselectivity. The required trans Ph group was introduced by Suzuki–Miyaura cross-coupling between the previous intermediate 157 and Ph-B(OH)2 using [Pd(PPh3)4] as catalytic system. The synthesis was completed by a second cross-coupling from 158 that leads to tamoxifen 159 (>93% Z) in 65% yield (Scheme 44). This modular stereoselective synthesis constitutes one of the most step and cost-economic routes to this antagonistic prodrug.
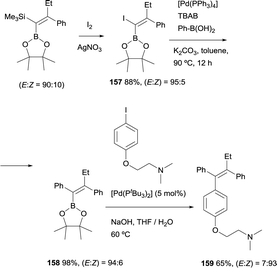 |
| | Scheme 44 Stereoselective synthesis of tamoxifen from gem-silylborylated alkenes obtained in the boron-Wittig reaction. | |
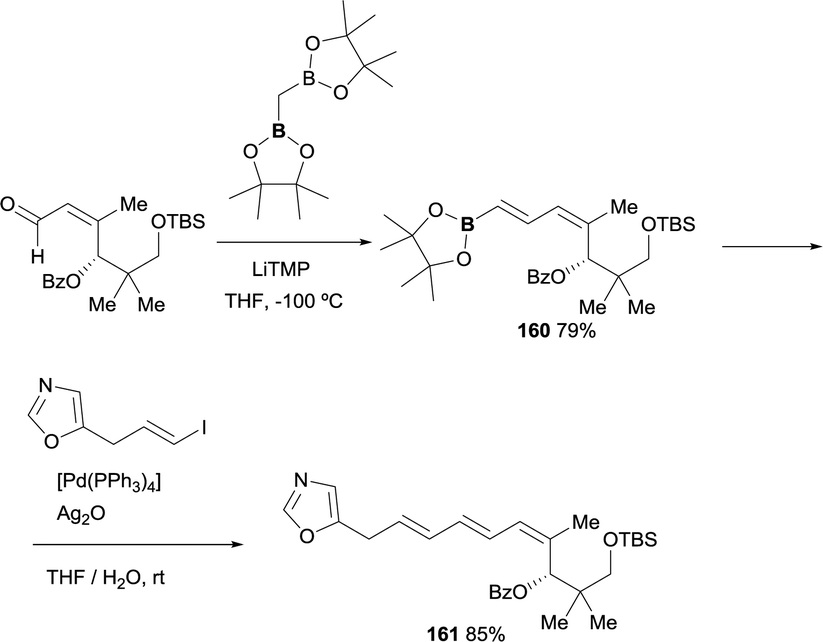
The use of boron-Wittig reaction has also served as a convenient approach for key intermediates involved in the synthesis of antibiotic inthomycin.47 The reactivity of bis(pinacolboryl)methane with the α,β-unsaturated motif provided a direct access to the corresponding E-vinyl boronate 160 in 79% yield in perfect control of stereoselectivity. Further reactivity with the E-vinyl iodide under Suzuki–Miyaura reaction conditions, in presence of [Pd(PPh3)4], afforded geometrically pure 4Z,6E,8E-triene 161 intermediate for the inthomycin synthesis, diminishing the number of steps and guarantying the stereochemical control of the reaction (Scheme 45).
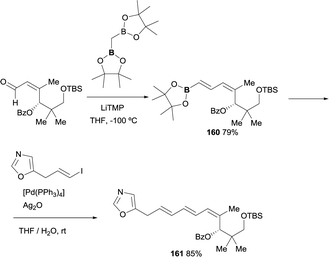 |
| | Scheme 45 Stereoselective synthesis of inthomycin intermediate via boron-Wittig/cross coupling strategy. | |
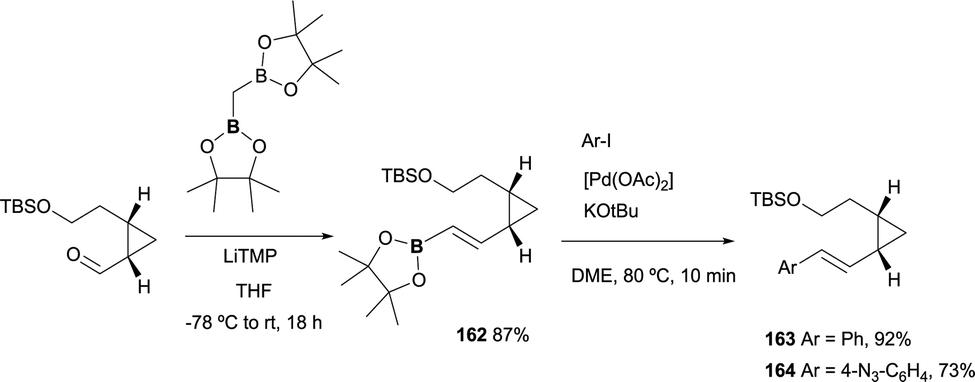
Recently, a new lysine-reactive cyclopropropyl aldehyde has been developed for the covalent modification of proteins. Towards this end, a divinylcyclopropane–cycloheptadiene rearrangement has been studied to render the condensation irreversible.48 The boron-Wittig reactions/followed by cross-coupling approach has served as the practical methodology to achieve the starting material through the condensation of the aldehyde functionality and CH2(Bpin)2 ion the presence of LiTMP. The corresponding (E)-vinyl boronate 162 was isolated in 87%. Subsequently, styryl derivatives were obtained in good to excellent yields under Suzuki–Miyaura conditions, completing the synthesis with the deprotection/oxidation and amination ring closing (Scheme 46).
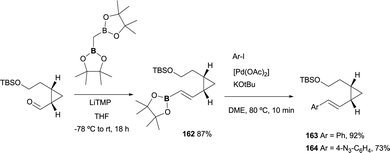 |
| | Scheme 46 Stereoselective boron-Wittig reaction for the synthesis of the substrates to prepare new lysine-reactive cyclopropropyl compounds. | |
7. Conclusions
In summary, the boron-Wittig olefination reaction that takes place between gem-bis(boryl)alkanes and carbonyl compounds has recently become a standard methodology to prepare vinyl boronates in a stereoselective manner. The transformation builds upon the ready generation of α-boron-stabilized lithium carbanions which are then capable of undergoing nucleophilic addition to carbonyl derivatives, eventually giving rise to the corresponding tri and tetrasubstituted alkenyl boronates, often in a stereoselective manner. In the case of ketones, the generally observed formation of the (E)-vinyl boronates has been rationalised on the bases of lithium coordinative effects, either due to the boronate oxygen atom or to the heteroatoms present at the substrate. The presence of hindered tertiary amines as additives showed also to be determinant for the stereochemical outcome of some of these reactions. This reactivity has also been extended to the gem-bis(boryl)silyl methane, providing a straightforward entry to synthetically appealing gem-silylborylalkenes, an interesting family of building blocks which may enable a direct and stereoselective access to all-carbon tetrasubstituted alkenes. This olefination has also been applied to aldehydes, although to a lesser extent and with overall lower levels of stereoselectivity. Through the addition of lithium α-boryl carbanions to primary amides the corresponding enamides can be easily obtained. In contrast, the boron-Wittig reaction applied to carbonyl derivatives able to undergo acylic substitution reactivity, such as N,N-dimethylformamide, esters or carboxylic acids, leads to the generation of the corresponding α-boryl ketones, which are typically in equilibrium with O-bound boron enolates. These versatile nucleophilic species, in turn, can be subsequently trapped with electrophiles to afford a variety of α-mono, di-, and tri-substituted ketones. In the last section, we present a selected group of applications in which the boron-Wittig olefination, followed by ensuing cross-coupling reactivity, provides an efficient route to a series of target compounds. Some straightforward synthesis of the well-known estrogen receptor modulator tamoxifen and the stereoselective synthesis of an inthomycin intermediate are included for a shake of illustration. This Tutorial summarises the extent of the applicability of the boron-Wittig process so as to the reaction conditions and the factors that control the stereochemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by MICINN through project PID2019-109674GB-I00 and CTQ2017-86936-P.
Notes and references
- M. Vaultier and G. Alcaraz, Sci. Synth., 2005, 6, 721 Search PubMed.
- G. Cainelli, G. Dal Bello and G. Zubiani, Tetrahedron Lett., 1966, 36, 4315 CrossRef.
- D. S. Matteson and P. B. Tripathy, J. Organomet. Chem., 1970, 21, 6 CrossRef.
- R. B. Castle and D. S. Matteson, J. Organomet. Chem., 1969, 20, 19 CrossRef CAS.
- D. S. Matteson and P. B. Tripathy, J. Organomet. Chem., 1974, 69, 53 CrossRef CAS.
- J. Royes, A. B. Cuenca and E. Fernández, Eur. J. Org. Chem., 2018, 2728 CrossRef CAS.
- D. S. Matteson, R. J. Moody and P. K. Jesthi, J. Am. Chem. Soc., 1975, 97, 5608 CrossRef CAS.
- D. S. Matteson, Synthesis, 1975, 147 CrossRef CAS.
- D. S. Matteson and P. K. Jesthi, J. Organomet. Chem., 1976, 110, 25 CrossRef CAS.
- D. S. Matteson, M. S. Biernbaum, R. A. Bechtold, J. D. Campbell and R. J. Wilcsek, J. Org. Chem., 1978, 43, 950 CrossRef CAS.
- D. S. Matteson and R. J. Moody, J. Org. Chem., 1980, 45, 1091 CrossRef CAS.
- M. W. Rathke and R. Kow, J. Am. Chem. Soc., 1972, 94, 6854 CrossRef CAS.
- D. S. Matteson and R. J. Moody, J. Am. Chem. Soc., 1977, 99, 3196 CrossRef CAS.
- D. S. Matteson and R. J. Moody, Organometallics, 1982, 1, 20 CrossRef CAS.
- K. Endo, M. Hirokami and T. Shibata, J. Org. Chem., 2010, 75, 3469 CrossRef CAS PubMed.
- K. Itami, T. Nokami and J. Yoshida, Org. Lett., 2000, 2, 1299 CrossRef CAS PubMed.
- A. Pelter, D. Buss, E. Colclough and B. Singaram, Tetrahedron, 1993, 49, 7077 CrossRef CAS.
- K. Endo, A. Sakamoto, T. Ohkubo and T. Shibata, Chem. Lett., 2011, 40, 1440 CrossRef CAS.
- J. Carreras, A. Caballero and P. J. Pérez, Chem. – Asian J., 2019, 14, 329 CrossRef CAS PubMed.
- E. La Cascia, A. B. Cuenca and E. Fernández, Chem. – Eur. J., 2016, 22, 18737 CrossRef CAS PubMed.
- D. J. Peterson, J. Org. Chem., 1968, 33, 780 CrossRef CAS.
- M. Kovalenko, D. V. Yarmoliuk, D. Serhiichuk, D. Chernenko, V. Smyrnov, A. Breslavskyi, O. V. Hryshchuk, I. Kleban, Y. Rassukana, A. V. Tymtsunik, A. A. Tolmachev, Y. O. Kuchkovska and O. O. Grygorenko, Eur. J. Org. Chem., 2019, 5624 CrossRef CAS.
- S. Namirembe, C. Gao, R. P. Wexler and J. P. Morken, Org. Lett., 2019, 21, 4392 CrossRef CAS PubMed.
- A. R. Bassindale, R. J. Ellis, J. C.-Y. Lau and P. G. Taylor, J. Chem. Soc., Chem. Commun., 1986, 98 RSC.
- K. J. Kolonko, M. M. Biddle, I. A. Guzei and H. J. Reich, J. Am. Chem. Soc., 2009, 131, 11525 CrossRef CAS PubMed.
- K. J. Kolonko, I. A. Guzei and H. J. Reich, J. Org. Chem., 2010, 75, 6163 CrossRef CAS PubMed.
- S. H. Cho and J. F. Hartwig, Chem. Sci., 2014, 5, 694 RSC.
- H. Chen, T. Zhang, Ch Shan, S. Liu, Q. Song, R. Bai and Y. Lan, Org. Lett., 2019, 21, 4924 CrossRef CAS PubMed.
- A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415 RSC.
- T. C. Stephens and G. Pattison, Org. Lett., 2017, 19, 3498 CrossRef CAS PubMed.
- J. R. Coombs, L. Zhang and J. P. Morken, Org. Lett., 2015, 17, 1708 CrossRef CAS PubMed.
- T. Kawashima and R. Okazaki, Synlett, 1996, 600 CrossRef CAS.
- T. Kawashima, N. Yamashita and R. Okazaki, J. Am. Chem. Soc., 1995, 117, 6142 CrossRef CAS.
- T. Wang, S. Kohrt, C. G. Daniliuc, G. Kehr and G. Erker, Org. Biomol. Chem., 2017, 15, 6223 RSC.
- W. Sun, L. Wang, Y. Hu, X. Wu, Ch Xia and Ch Liu, Nat. Commun., 2020, 11, 3113 CrossRef CAS PubMed.
- P. Zheng, Y. Zhai, X. Zhao and T. Xu, Chem. Commun., 2018, 54, 13375 RSC.
- P. Zheng, Y. Zhai, X. Zhao and T. Xu, Org. Lett., 2019, 21, 393 CrossRef CAS PubMed.
- T. Mukaiyama, M. Murakami, T. Oriyama and M. Yamaguchi, Chem. Lett., 1981, 1193 CrossRef CAS.
- C. E. Iacono, T. C. Stephens, T. S. Rajan and G. A. Pattison, J. Am. Chem. Soc., 2018, 140, 2036 CrossRef CAS PubMed.
- G. Pattison, Eur. J. Org. Chem., 2018, 3520 CrossRef CAS.
- B. Lee and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 2429 CrossRef CAS PubMed.
- W. Sun, L. Wang, C. Xia and C. Liu, Angew. Chem., Int. Ed., 2018, 57, 5501 CrossRef CAS PubMed.
- T. Hata, H. Kitagawa, H. Masai, T. Kurahashi, M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2001, 40, 790 CrossRef CAS PubMed.
- R. Nakajima, C. Delas, Y. Takayama and F. Sato, Angew. Chem., Int. Ed., 2002, 41, 3023 CrossRef CAS PubMed.
- M. J. Meegan, R. B. Hughes, D. G. Lloyd, D. C. Williams and D. M. Zisterer, J. Med. Chem., 2001, 44, 1072 CrossRef CAS PubMed.
- T. Kames, K. Itami and J. Yoshida, Adv. Synth. Catal., 2004, 346, 1824 CrossRef.
- J. H. Kim, Y. Song, M. J. Kim and S. Kim, J. Org. Chem., 2020, 85, 4795 CrossRef CAS PubMed.
- C. Apel, M.-A. Kasper, C. E. Stieger, C. P. R. Hackenberger and M. Christmann, Org. Lett., 2019, 21, 10043 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Elena
Fernández
a and
Elena
Fernández