
Current status and perspectives in oxidative, non-oxidative and CO2-mediated dehydrogenation of propane and isobutane over metal oxide catalysts†
Tatiana
Otroshchenko
 a,
Guiyuan
Jiang
b,
Vita A.
Kondratenko
a,
Uwe
Rodemerck
a and
Evgenii V.
Kondratenko
*a
a,
Guiyuan
Jiang
b,
Vita A.
Kondratenko
a,
Uwe
Rodemerck
a and
Evgenii V.
Kondratenko
*a
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Strasse 29 a, D-18059 Rostock, Germany. E-mail: Evgenii.Kondratenko@catalysis.de
bState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing, Beijing, 102249, P. R. China
First published on 18th November 2020
Abstract
Conversion of propane or butanes from natural/shale gas into propene or butenes, which are indispensable for the synthesis of commodity chemicals, is an important environmentally friendly alternative to oil-based cracking processes. Herein, we critically analyse recent developments in the non-oxidative, oxidative, and CO2-mediated dehydrogenation of propane and isobutane to the corresponding olefins over metal oxide catalysts. Particular attention is paid to (i) comparing the developed catalysts in terms of their application potential, (ii) structure–activity–selectivity relationships for tailored catalyst design, and (iii) reaction-engineering aspects for improving product selectivity and overall process efficiency. On this basis, possible directions for further research aimed at the development of inexpensive and environmentally friendly catalysts with industrially relevant performance were identified. In addition, we provide general information regarding catalyst preparation and characterization as well as some recommendations for carrying out non-oxidative and CO2-mediated dehydrogenation reactions to ensure unambiguous comparison of catalysts developed in different studies.
 Tatiana Otroshchenko | Tatiana Otroshchenko graduated from the Moscow State University in 2012. In 2014 she started her PhD on the development of the catalysts for non-oxidative dehydrogenation of light alkanes in the Leibniz-Institute for Catalysis in Rostock under the supervision of associate Professor Evgenii V. Kondratenko. Her PhD was partially supported by DAAD scholarship and by the scholarship of the president of the Russian Federation for study abroad. In 2018 she defended her PhD and won a personal grant from DFG. Now she leads her own research project on metathesis of ethylene with butenes in the Leibniz-Institute for Catalysis. |
 Guiyuan Jiang | Guiyuan Jiang received his BS and MS degrees from China University of Petroleum, Beijing under the supervision of Prof. Chunming Xu, and PhD degree from the Institute of Chemistry, Chinese Academy of Sciences with Prof. Yanlin Song. He is currently a Professor in University of Petroleum, Beijing. He was a visiting postdoctor at the University of California, Riverside in 2010 with Professor Pingyun Feng, and visiting scholar in Prof. Yadong Li's group in Tsinghua University in 2013–2014. His research interests mainly focus on energy catalysis, including catalytic conversion of light hydrocarbons and artificial photosynthesis. |
 Vita A. Kondratenko | Vita A. Kondratenko graduated from the Novosibirsk State University in 1992. In 2000, she has started to work at the Institute for Applied Chemistry Berlin-Adlershof. She received her PhD from the Humboldt-Universität zu Berlin under supervision of Professor M. Baerns in 2005. In 2006 she won a personal grant from DFG. She is currently a senior scientist at the Leibniz-Institute for Catalysis in Rostock. Her research interests are mainly focused on the evaluation of mechanistic aspects of catalytic transformations of light hydrocarbons and determining kinetics parameters of single steps of these reactions. |
 Uwe Rodemerck | Uwe Rodemerck studied chemistry at the Technische Hochschule Merseburg and received his diploma in homogeneous catalysis under the supervision of Prof. R. Taube in 1985. After working on reaction engineering at the Academy of Sciences of the GDR he worked at the Institute for Applied Chemistry Berlin-Adlershof and received 1995 his PhD in heterogeneous catalysis from the University Bochum under the supervision of Prof. M. Baerns. Currently, he is leader of the research group “High-Throughput Technologies” at the Leibniz-Institute for Catalysis in Rostock. His research interests focus on high-throughput catalysis, reactions of small hydrocarbons (C1–C4) and CO2-based Fischer–Tropsch synthesis. |
 Evgenii V. Kondratenko | Evgenii V. Kondratenko graduated from the Novosibirsk State University in 1991. He earned his PhD in 1995 from the Institute of Chemistry of Natural Materials in Krasnoyarsk. In 1997, he was awarded Alexander von Humboldt Foundation fellowship at the Institute for Applied Chemistry Berlin-Adlershof (group of Prof. Baerns). In 2007, he obtained his habilitation degree from the Technical University Berlin. He is currently associate Professor at the University Rostock and leader of the group “Reaction mechanisms” at the Leibniz-Institute for Catalysis in Rostock. He is engaged in the development and understanding of catalysts for valorization of C1–C4 alkanes, CO2 and NH3. |
1 Introduction
The modern chemical industry is heavily relying on catalysis for the conversion of raw materials into various building blocks.1,2 Among them, propene and butenes are highly important due to their versatile applications (Fig. 1). Their demand is continuously increasing. At the present time, there are several commercial technologies for their production. They utilize natural gas, crude oil, or coal as raw materials (Fig. 1). Steam or fluid-catalytic-cracking of different oil fractions are widely used and provide the desired olefins but suffer from low and not easily adjustable selectivity to these products. Moreover, they cannot fulfil the steady increasing demand for propene. Another shortcoming of these processes is finite natural resources of crude oil. Natural gas and coal are expected to be longer available and can also be used for production of low olefins. Technologies on the basis of these raw materials can be divided into direct and indirect routes (Fig. 1). The latter option starts with the generation of syngas (a mixture of CO and H2) followed by Fischer–Tropsch (FT) or methanol synthesis. Methanol is further converted into lower olefins or propene through methanol to olefins3–5 or methanol to propene6 technologies. The FT process is not selective to the desired lower hydrocarbons, as long-chain hydrocarbons and unfortunately methane are also co-produced. The economy and environmental impact of the methanol and FT processes suffer from syngas production requiring high amounts of energy and resulting in strong CO2 emissions because of high endothermicity of steam reforming of methane or gasification of coal. The latter raw material is, in addition, not environmentally friendly. Some examples of damaging environmental influences of coal are emissions of particulate matter and methane upon mining, impurities such as heavy metals, sulphur- and nitrogen-containing compounds and radioactive as well as high CO2 emissions upon its gasification. Under this consideration, conversion of C2–C4 alkanes present in natural gas appears to be attractive. Steam cracking of ethane, second main component of natural/shale gas, is a mature technology for production of ethylene. This chemical can be dimerized to n-butenes followed by the metathesis of 2-butenes with ethylene to propene (Fig. 1). Although the metathesis reaction is highly selective and efficient,7,8 the above approach can be profitable only when an excess of ethylene is available.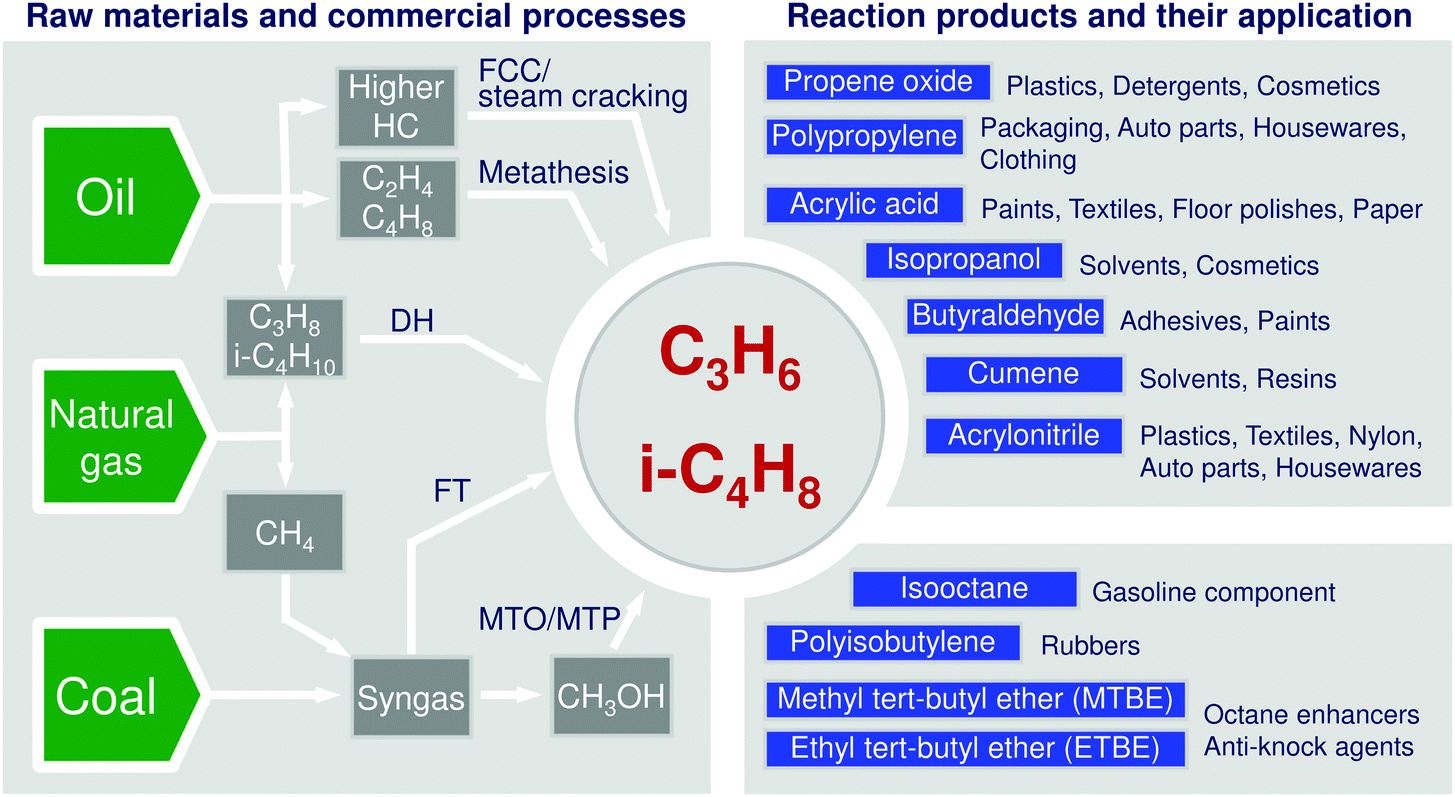 | ||
| Fig. 1 Raw materials, their main conversion ways into propene and isobutene as well as products formed from these olefins. | ||
In comparison with the above-mentioned commercial technologies, direct dehydrogenation of C3–C4 alkanes present in natural/shale gas or formed in oil-based cracking processes is attractive for on-purpose direct production of the corresponding olefins. There are three options for carrying out alkane dehydrogenation: (i) without any oxidant (non-oxidative dehydrogenation), (ii) with oxygen or air (oxidative dehydrogenation) and (iii) using CO2 as mild oxidant or for shifting the equilibrium in option (i) via reaction with H2. The oxidative approach is free of thermodynamic limitations, while thermodynamic constraints limit alkane conversion in the non-oxidative or CO2-mediated dehydrogenation. The latter reaction has the lowest equilibrium constant in the temperature range between 400 and 650 °C (Fig. 2).
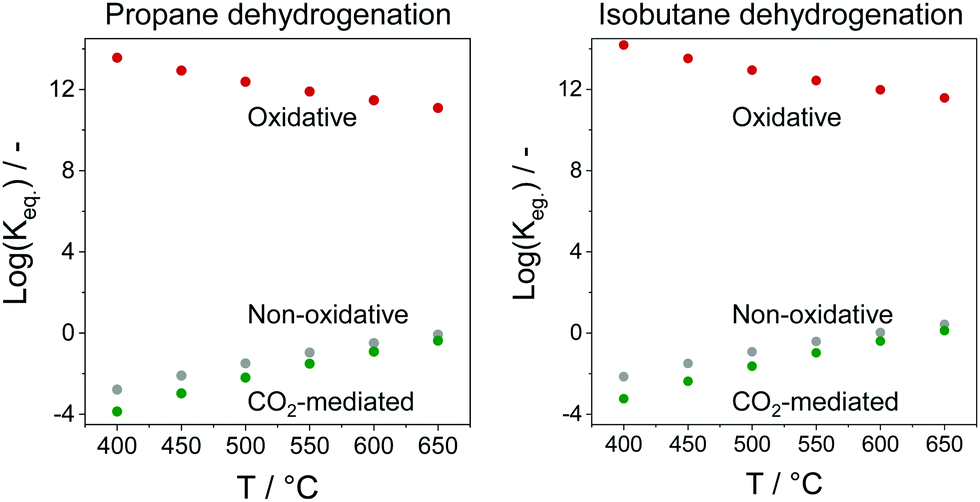 | ||
| Fig. 2 Equilibrium constants (Keq.) of non-oxidative, oxidative, or CO2-mediated dehydrogenation of propane or isobutane. | ||
The CO2-mediated dehydrogenation (CO2-DH) of C3–C4 alkanes has attracted attention of researchers due to possible application for control of global CO2 emissions. Such potential advantage of this approach can, however, be convincing if green (CO2-neutral) energy will be used for this strongly endothermic reaction. In addition, an efficient and CO2-neutral technology for separation of CO formed from CO2 in this reaction is required. These aspects, state of the art catalysts and mechanistic aspects of the CO2-DH of propane are discussed in recent reviews.9–11
Despite the energetic advantages, the oxidative alkane dehydrogenation is not commercialized. The main reason is too low selectivity to the desired olefins at industrially relevant degrees of alkane conversion and the necessity to use pure oxygen if the non-reacted alkane should be recycled. Large-scale air separation to obtain O2 is an expensive and energy-demanding technology. A most recent extensive review about the oxidative dehydrogenation of ethane and propane was published 13 years ago in 2007.10 Activity- and selectivity-governing properties as well as reaction engineering aspects have been reviewed. Seven years later, Carrero et al.12 have reviewed kinetic aspects of the oxidative dehydrogenation of propane over vanadium-based catalysts. In addition, approaches for catalyst preparation and characterization have also been discussed. Experimental requirements for carrying out and evaluating kinetic experiments have been defined. A brief analysis of the oxidative propane dehydrogenation to propene over V-based catalysts is given in a review published in 2018 and dealing with aerobic oxidations of C1–C4 alkanes.13 In general, due to a very large number of available data and sometimes contradictive statements in different studies, no statistically proven relationships between catalyst physicochemical property, reaction conditions and performance could be established up to now.
The non-oxidative dehydrogenation of propane or isobutane is applied on large scale and its share is expected to grow in the coming years.14–17 From a fundamental viewpoint, non-oxidative alkane dehydrogenation is also of great significance because it is a good model reaction for studying fundamentals of the activation of C–H bond. Commercially applied catalysts are composed of Al2O3 with supported Pt or CrOx species responsible for alkane dehydrogenation.15–18 There are, however, drawbacks related to the commercial catalysts. According to the U.S. Occupational Safety and Health Administration, workplace exposure to Cr(VI) may cause various health effects.19 High cost of platinum and the necessity to redisperse sintered Pt species in spent catalysts by ecologically harmful Cl2 or Cl-containing compounds are the main drawbacks of Pt-based catalysts.18 Nevertheless, a major part of the ongoing research is still dealing with Pt-based catalysts and is aimed at reducing Pt amount without losing high selectivity at improved on-stream stability and durability. To account for the weaknesses of the commercial catalysts, various groups around the globe try to develop alternative catalysts with supported VOx, GaOx, InOx, CoOx, ZnOx or SnOx species of different structure. Recently, bulk catalysts based on ZrO2, TiO2 or Al2O3 were also demonstrated to show promising performance. Challenges and developments in this area before 2019 are discussed in several representative reviews.15–18,20,21
Compared with published accounts on specific ways of production of propene and isobutene, this review discusses developments in the oxidative, non-oxidative, and CO2-mediated dehydrogenation of propane and isobutane over supported and bulk metal oxides. In addition, reaction engineering aspects are also analysed. The general aims are (i) to critically evaluate progress in catalyst development, (ii) to identify unifying guidelines for improving these processes, and (iii) to provide our personal view on future research directions concerning catalyst development and reactor operation. To avoid repetitions and to further distinguish our review from the relevant previous assays, we will analyse the developments in the non-oxidative and CO2-mediated dehydrogenation made in the last 5 years. As there are no recent reviews on the oxidative dehydrogenation, the relevant studies after 2007 will be reviewed. In particular, the present review is focused on (i) the application potential of catalysts developed, (ii) the structure–activity–selectivity relationships for tailored catalyst design, (iii) molecular level aspects of alkane dehydrogenation, (iv) reaction-engineering aspects, (v) factors relevant for preparation of certainly structured supported MOx (M stands for a metal) species or defective bulk MOx, and (vi) methods for catalyst characterisation to identify active sites and to analyse coke formation. As contradictory information is obtained from tests under different experimental conditions, we also provide some recommendations for carrying out alkane dehydrogenation tests and for their correct evaluation.
2 Non-oxidative propane dehydrogenation
2.1 General information about PDH process and applied catalysts
The non-oxidative propane dehydrogenation to propene (PDH) is a thermodynamically limited, highly endothermic reaction that requires temperatures above 550 °C to achieve industrially relevant degrees of propane conversion. According to Le Chatelier's principle, the equilibrium conversion increases with a decrease in partial pressure of propane, while co-feeding of H2 that is often used to suppress coke formation, has a negative effect on the conversion.18Fig. 3 shows the equilibrium conversion of propane as a function of partial pressure of this alkane in the absence of co-fed hydrogen at 550 °C or 600 °C. Commercial PDH processes typically operate at propane partial pressures above 0.3 bar.22 The Oleflex process utilizes a feed consisting of propane and hydrogen-rich recycle gas.22 The reaction feed used in the Snamprogetti/Yarsintez process can also be considered to contain hydrogen due to back mixing in a fluidized-bed reactor.23,24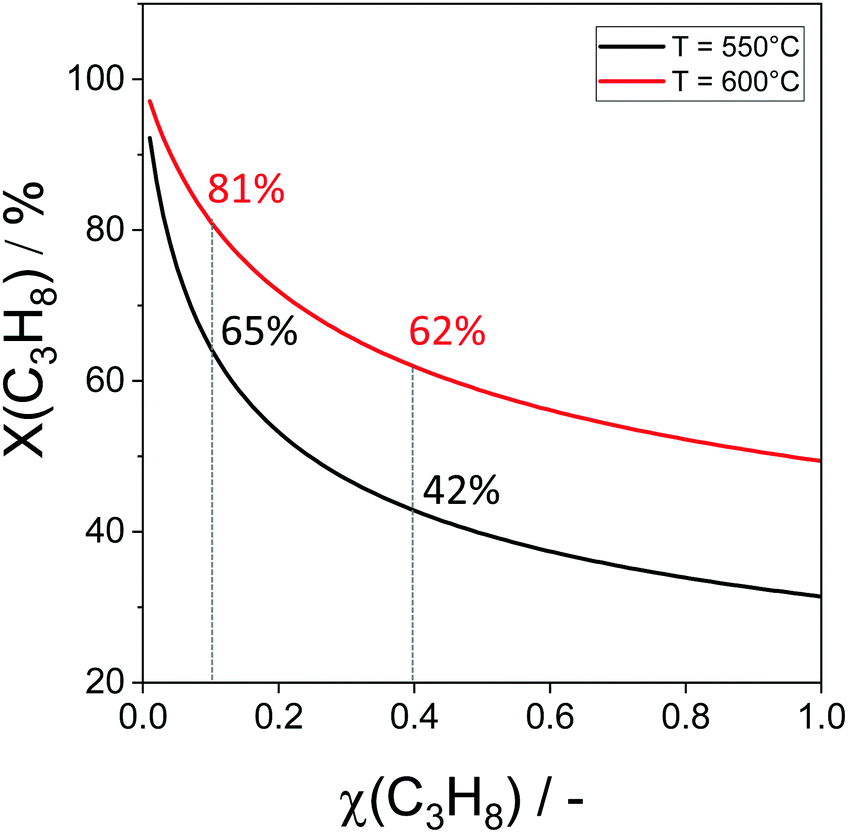 | ||
| Fig. 3 Equilibrium propane conversion as a function of propane content at total pressure of 1 bar and temperatures of 550 °C or 600 °C. No-co-fed H2 or C3H6, which negatively affect the conversion. The numbers stand for propane conversion degrees achieved using feeds with 10 or 40 vol% C3H8. | ||
Classically, catalysts composed of a supporting material and supported catalytically active metal or metal oxide species are used for the PDH reaction and dehydrogenation of other lower alkanes (Fig. 4). Their activity and selectivity are largely determined by the fine structure of supported active species and their interaction with the support (Fig. 4). One of the challenges upon catalyst development is to synthesize specific supported structures where the atoms are arranged in a predetermined way. Even when such structures are prepared, they may alter under severe reaction conditions thus resulting in a change of the catalyst performance. In addition, it is difficult to controllably place dopants/promoters in the vicinity of active PDH species to tune performance of the latter. Against this background, an alternative concept for design of alkane dehydrogenation catalysts has been introduced a few years ago.25 The dehydrogenation function is reserved to surface defects of bulk oxides of typically non-reducible metals and not to supported metal oxides or metals (Fig. 4). Representatives of such materials are oxides of zirconium, titanium, aluminium, europium, and gadolinium.26–29 The below discussion is aimed to demonstrate and to analyse recent developments including mechanistic aspects of propane dehydrogenation over conventional supported (Section 2.2) and alternative-type bulk (Section 2.3) catalysts. A major focus is put on the latter materials as they have not been thoroughly reviewed up to now.
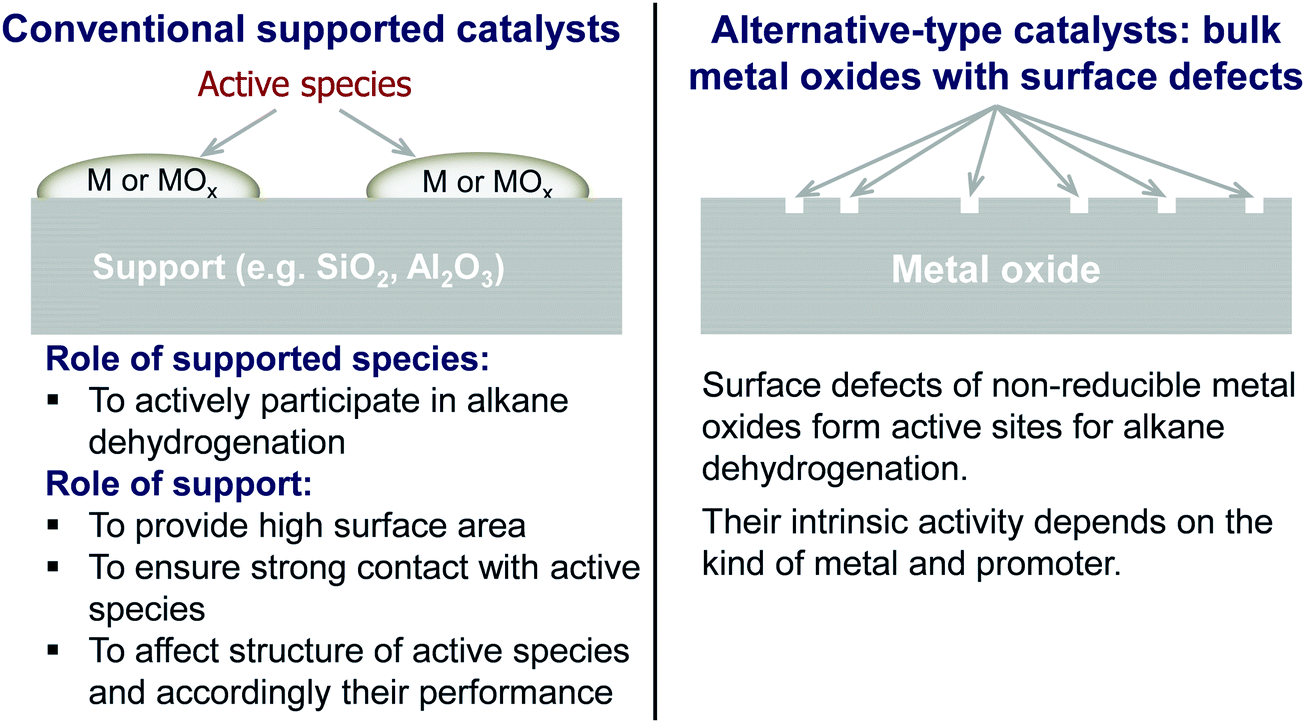 | ||
| Fig. 4 Conventional and alternative-type catalysts as well as principles of their functioning in non-oxidative alkane dehydrogenation. | ||
2.2 Conventional supported catalysts and their active species
Based on the available literature, we prepared Table 1 showing the nature of catalytically active species in the conventional catalysts with supported CrOx, VOx, ZnOx, GaOx, SnOx or CoOx species. As seen in this table, there are some contradictions in this regard. Thus, further studies are required to unambiguously identify the active sites and the ways for their purposeful creation as required for tailored catalyst design. Promoters applied in the last 5 years for the modification of such catalysts with respect to their physicochemical and catalytic properties are summarized in Table 2. After this general introduction, individual features of the above-mentioned catalysts are discussed in the below sections.| Catalyst | Suggested active species | Ref. |
|---|---|---|
| CrOx-Based | Four-coordinated Cr3+ | 30 and 31 |
| Redox and non-redox Cr3+ | 32 and 33 | |
| VOx-Based | V3+ is more active than V5+ and V4+ | 34 and 35 |
| V3+ and V4+ | 36 and 37 | |
| Isolated VOx species are more active than polymerized VOx species | 36 and 37 | |
| Polymerized VOx species are more active than isolated VOx species | 38 and 39 | |
| Monomeric and low-polymerized VOx species | 40 | |
| Al–O–V (for monomeric VOx), Al–O–V and coordinatively unsaturated V (for dimeric VOx), three-coordinated surface oxygen (for crystalline VOx) | 41 | |
| ZnOx-Based | Zn2+Ox | 42 and 43 |
| Zn(OH)+ | 44 | |
| GaOx-Based | Isolated four-coordinated Ga3+Ox | 45 |
| [GaH]2+ | 46–48 | |
| SnOx-Based | Metallic Sn | 49 |
| Isolated Sn2+Ox | 50 | |
| Polymeric Si–O–Sn2+ | 51 | |
| SnOx species with oxygen vacancies adjacent to the Lewis acidic sites | 52 | |
| Highly dispersed SnOx | 53 | |
| CoOx-Based | Metallic Co | 54 and 55 |
| Tetrahedral Co2+Ox species | 56–59 |
| Active species | Promoter | Promoter effect on physico-chemical properties | Promoter effect on catalytic properties | Ref. |
|---|---|---|---|---|
| CrOx | Zn | Enhancing dispersion of CrOx due to favouring the formation of spinel ZnCr2O4 phase | Enhancing activity | 60 |
| Ni | Favouring formation of oligomeric chromium species | Enhancing activity | 30 | |
| Ce | Decrease in the amount of inactive isolated Cr6+Ox, increase in the amount of oxygen vacancies, strong interaction between Ce and Cr | Enhancing activity, selectivity, stability | 61 | |
| VOx | P | Moderating surface acidity | Improving stability | 62 |
| Mg | Dispersing V2O5 nanoparticles into 2D VOx species | Improving stability | 63 | |
| SnOx | K | (1) Potassium interacts with Sn–O–Sn bonds | (1) Decreasing activity | 53 |
| (2) Decrease in surface acidity | (2) Enhancing selectivity | |||
| Ni | (1) Promoting complete reduction of SnOx to metallic Sn | (1) Decreasing activity | 51 | |
| (2) Formation of Ni3Sn2 alloy which promotes migration and recombination of hydrogen atoms | (2) Improving stability | |||
| Co, Cu | Promoting complete reduction of SnOx to metallic Sn | Decreasing activity | 51 | |
| Pd | Suppressing coke formation due to hydrogenation ability, Improving resistance of well-dispersed SnOx species against sintering | Improving on-stream stability | 49 | |
| ZnOx | Pt | Increasing dispersion of ZnOx, stabilizing Zn2+ | Enhancing activity and selectivity | 42 |
| CoOx | Fe | Formation of solid solution, generation of tetrahedral Co2+ species | Enhancing activity and selectivity | 59 |
| Mn, Cu | Generation of octahedral Co3+ species | Decreasing activity | 59 | |
| Zr | Co–O bond weakening | Enhancing activity and selectivity | 58 |
It has been demonstrated that surface acidity strongly influences the performance of CrOx-based catalysts. On the one hand, the catalysts with low acidity exhibit high on-stream stability and anti-coking ability.65 On the other hand, Brønsted acidic sites of support play an important role in propane activation thus enhancing catalyst activity.66 As for many supported catalysts, the nature of supported CrOx species and accordingly their PDH performance can be tuned through preparation method,31 support,32,65 loading of CrOx32,67 or promoter.30,60,61 Some examples about the influence of various promoters on physico-chemical and catalytic properties of CrOx-based catalysts are shown in Table 2. It should be mentioned that when CrOx is combined with ZrO2 to prepare supported CrOx/M(La or Y)ZrOx68 and Cr–Zr–Ox/MOx(SiO2)69,70 or bulk Cr–Zr–Ox68,71 materials, extremely high activity can be achieved as a result of synergetic effect between ZrO2 and CrOx. As surface defects of ZrO2 in these materials are crucial for the high activity of such catalysts, they were classified by us as alternative-type materials and will be discussed in Section 2.3.
It has been recently demonstrated that hydroxyl groups on supported VOx species strongly influence catalytic activity and coke formation.73 The catalysts containing hydroxyl groups on VOx species created after H2 treatment demonstrate lower PDH activity and lower coke deposition. It has been also shown that the performance of VOx-based catalysts is strongly influenced by the preparation method,74,75 the kind of vanadium precursor39 and support,76,77 vanadium loading,36,37,40 and the kind of promoter (Table 2). All these parameters affect the structure of supported VOx species and must be considered upon catalyst preparation (Section 8.1).
Zeolites were reported to stabilize small ZnO nanoclusters.78,79 The amount of highly dispersed ZnO clusters interacting with framework oxygen atoms increases with an increase in the SiO2/Al2O3 ratio in the zeolite.79 Accordingly, high activity of ZnO supported on high-silica HZSM-5 is related to the high dispersion of ZnOx species.
Very recently, Han et al.43 developed highly active ZrO2-supported catalysts with catalytically active ZnOx species. Binary MZrOx (M = Ce, La, Ti or Y) materials were used as supports. The most active Zn(4 wt%)/TiZrOx catalyst revealed higher activity than the state-of-the-art catalysts with supported CrOx, GaOx, ZnOx or VOx species as well as bulk ZrO2-based catalysts without ZnO. In contrast to ZrO2-based catalysts in Section 2.3, coordinatively unsaturated Zr cations were not concluded to be the main active sites in these novel catalysts. Isolated tricoordinated Zn2+Ox species are responsible for the high activity and on-stream stability. Their formation and intrinsic activity depend on the kind of metal oxide promoter for ZrO2 and the structure of the latter oxide. The stabilization of isolated Zn2+Ox species is favoured when ZrO2 has an amorphous structure. It should be also mentioned that the presence of TiO2 seems to increase the intrinsic activity of isolated Zn2+Ox species. Very recently, the same research group developed TiO2-supported catalysts with ZnOx and ZrO2.83 Small ZnOx clusters with 1 to 3 Zn atoms were suggested to be responsible for propane dehydrogenation, while ZrO2 enhances the intrinsic activity of ZnOx.
Although Ga-containing zeolites are known to be active in PDH, such catalysts show low propene selectivity at high degrees of propane conversion.87 Thereby, many recent works were focused on increasing the selectivity.87–89 The performance of Ga-containing catalysts could be mostly tuned by preparation method87–89 and/or support.84 It was suggested that strong metal–support interactions lead to higher dispersion of GaOx species and therefore play a crucial role for catalyst activity.84 Concerning propene selectivity, the presence of strong acidic sites has a negative effect since such sites provoke deep dehydrogenation, aromatization and coking reactions.84
The nature of the active sites and the mechanism of propane dehydrogenation over Ga-based catalysts are still under debate. Only four-coordinated Ga3+–Ox Lewis acidic sites were observed under reaction conditions on the surface of single-site GaOx/SiO2 catalyst. When the coordination of GaOx decreased, a significant decrease in catalytic activity was observed.45 Such phenomenon supports the assumption that four-coordinated Ga3+Ox sites are catalytically active. Recently, Schreiber et al.46 provided some evidence that Lewis–Brønsted acid pairs (Ga+ – Brønsted acidic site of support) are active sites for propane dehydrogenation over Ga/H-ZSM-5. Based on the results of DFT calculations, those authors proposed a bifunctional Lewis–Brønsted acid mechanism where the Brønsted acidic site protonates Ga+ forming [GaH]2+. A basic framework oxygen of Brønsted acidic site together with so-formed [GaH]2+ heterolytically breaks two C–H bonds in propane followed by elimination of H2 and propene in two sequential steps. Similarly, experimental data and DFT calculations carried out for Ga/H-MFI suggest that [GaH]2+ cations are the most active sites for dehydrogenation of light alkanes over Ga/H-MFI.47,48 It is proposed that the dehydrogenation reaction occurs via a heterolytic dissociation of C3H8 at [GaH]2+ site with a formation of [C3H7–GaH]+–H+ cation pairs.
The performance of Sn-containing catalysts can be affected by the presence of various additives. Some examples of their influence on physicochemical and catalytic properties of such catalysts are given in Table 2.
2.3 Alternative-type bulk catalysts
Alternative-type bulk catalysts composed of oxides of zirconium, titanium, aluminium, europium or gadolinium are a relatively new class of alkane dehydrogenation catalysts (Fig. 4). Unlike the conventional supported catalysts, such catalysts were only scarcely described in a recent review of Hu et al.21 Below, we discuss the kind of active sites in such materials, factors affecting their creation and intrinsic activity, molecular-level insights and catalytic performance. In a separate Section 2.4, such catalysts will be compared with conventional supported catalysts in terms of selectivity–conversion values and productivity under industrially relevant conditions. For benchmarking purposes, the most active catalysts from a previous review of Weckhuysen and co-workers18 will also be discussed.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11)) and oxygen-defective (d-ZrO2(11)) surfaces, it was established that Zrcus sites present in d-ZrO2(11) open an alternative pathway for PDH with a lower activation barrier in comparison with ZrO2(11).26 Two Zrcus4+ sites are required for the homolytic C–H bond activation on d-ZrO2(11), while Zr4+ and neighbouring lattice oxygen participate in this process on ZrO2(11) (Fig. 5). It is noticeable that the rate-determining step for PDH over d-ZrO2(11) is the formation of H2 (endothermic by 1.40 eV), while over ZrO2(11) it is methylene C–H bond activation (endothermic by 0.94 eV with an activation barrier of 1.25 eV).
11)) and oxygen-defective (d-ZrO2(11)) surfaces, it was established that Zrcus sites present in d-ZrO2(11) open an alternative pathway for PDH with a lower activation barrier in comparison with ZrO2(11).26 Two Zrcus4+ sites are required for the homolytic C–H bond activation on d-ZrO2(11), while Zr4+ and neighbouring lattice oxygen participate in this process on ZrO2(11) (Fig. 5). It is noticeable that the rate-determining step for PDH over d-ZrO2(11) is the formation of H2 (endothermic by 1.40 eV), while over ZrO2(11) it is methylene C–H bond activation (endothermic by 0.94 eV with an activation barrier of 1.25 eV).
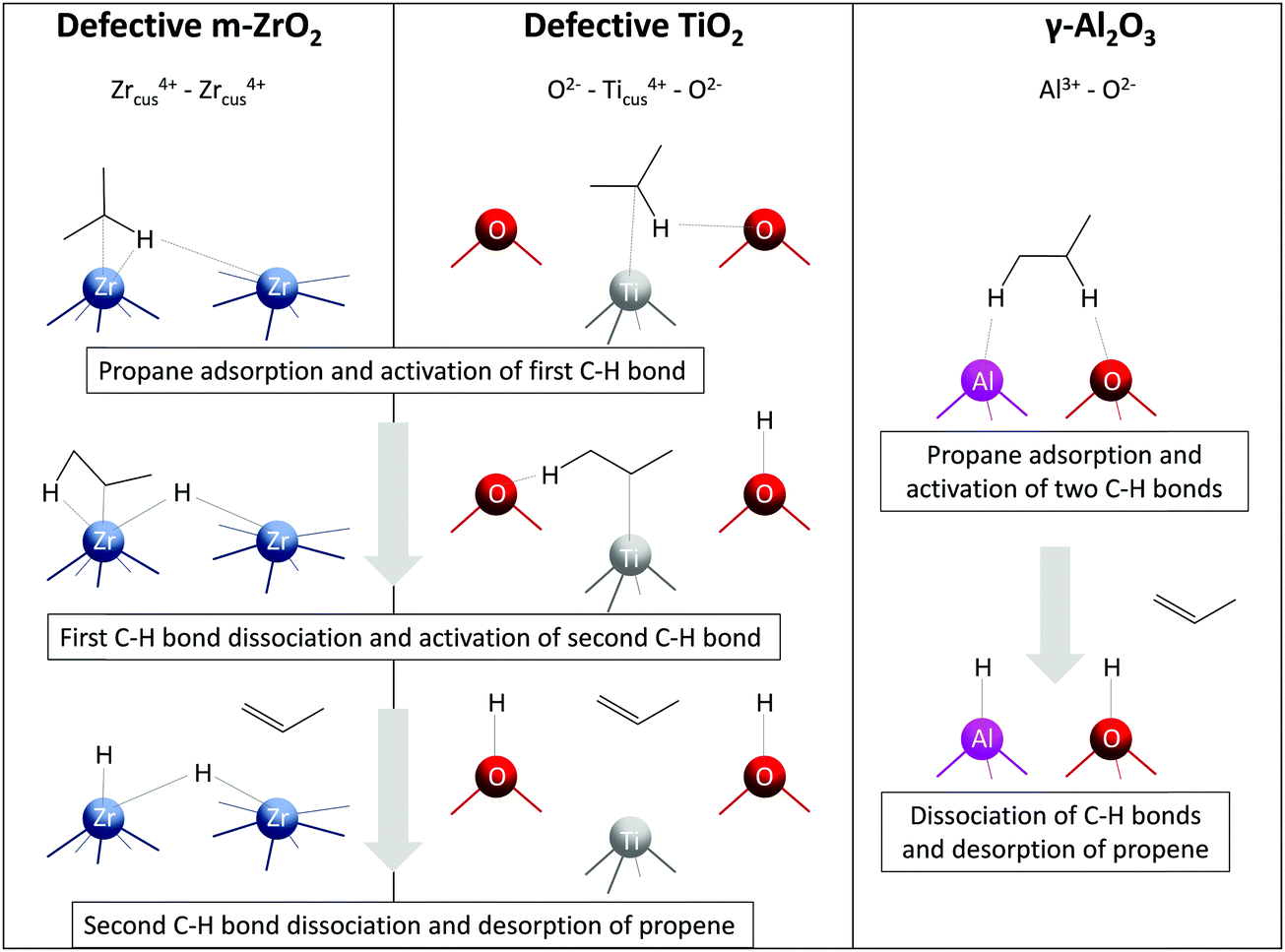 | ||
| Fig. 5 Schematic representation of C–H activation, dissociation, and propene formation steps of PDH catalysed by oxygen-defective m-ZrO2(11), oxygen-defective TiO2(101), and γ-Al2O3(110). | ||
The discovery of ZrO2-based catalysts prompted further search for other bulk catalysts. Bare TiO2, Al2O3, Eu2O3 and Gd2O3 were established to show reasonable activity in the non-oxidative dehydrogenation of propane and isobutane.27–29,94 Similarly to ZrO2, the activity of pristine TiO2 is strongly influenced by the presence of oxygen vacancies experimentally detected by the EPR technique.27 Based on the results of DFT calculations, the authors concluded that the presence of oxygen vacancies on the surface of TiO2 facilitates the adsorption of propane and its further dehydrogenation. Two fold-coordinated oxygen atoms were suggested to be responsible for the adsorption of the dissociated derivatives of propane on a perfect TiO2(101), while fourfold-coordinated titanium atoms surrounding the oxygen vacancy were the active sites for PDH over defective TiO2(101) (Fig. 5).
Similar to ZrO2- and TiO2-based catalysts, the active species for bare Al2O3 were also concluded to be coordinatively unsaturated Al3+ sites.28 Since such sites are Lewis acids, their presence was confirmed by Py-FTIR. Accordingly, the activity of bare Al2O3 was in a good correlation with the amount of Lewis acidic sites. DFT calculations performed by Dixit et al.95 on non-hydroxylated (100) and hydroxylated (110) facets of γ-Al2O3 for the PDH reaction considered two different mechanisms, namely concerted and stepwise. The former mechanism implies propene formation in a single step, while the latter considers a sequential abstraction of hydrogen atoms. Thus, the authors revealed that the PDH mechanism is site-dependent. The highest computed TOF of the reaction at 600 °C was obtained for AlIII–OIII (III means tricoordinated) site pair (Fig. 5) on the hydroxylated (110) facet implying a concerted mechanism.
Very recently, Perechodjuk et al.29 have demonstrated the application potential of Eu2O3 and Gd2O3 as catalysts for the PDH reaction. Similar to other previously discussed bulk catalysts, coordinatively unsaturated metal cations were suggested to participate in propane dehydrogenation.
Although coordinatively unsaturated cations in ZrO2, TiO2 and Al2O3 are essential for propane dehydrogenation, the nature of the active site on the surface of these oxides seems to be different (Fig. 5). Thus, two adjacent Zrcus4+ sites (five- and sixfold-coordinated) participate in the formation of propene over oxygen-defective m-ZrO2(11).26 Contrarily, one fourfold-coordinated Ticus4+ and twofold-coordinated O2− on the surface of oxygen-defective TiO2(101) are required for propane dehydrogenation to propene.27 For the most active AlIII–OIII site pair on γ-Al2O3(110), the formation of propene occurs in a single step with participation of threefold-coordinated Al3+ and threefold-coordinated O2− atoms.95 Noticeably, for the latter case, only perfect γ-Al2O3(110) surface facet was considered. The reaction mechanism and the kind of active site might change on an oxygen-defective surface.
Influence of phase composition and size of crystallites. Zhang et al.96 have recently shown that the activity of bare ZrO2 in PDH and the selectivity to propene can be tuned through the phase composition and/or the size of crystallites. Monoclinic ZrO2 was found to be more active and selective than ZrO2 stabilized in the tetragonal phase. To check if the phase composition influences the nature of active sites and individual reaction steps, elementary steps of propane activation and product formation over oxygen-defective monoclinic (m-ZrO2(
11)) and oxygen-defective tetragonal (t-ZrO2(101)) ZrO2 were elucidated by means of DFT calculations.96 Regardless of the phase composition, energy profiles during PDH reaction have the same shape and trend (Fig. 6) and two Zrcus4+ cations are required for the PDH reaction. However, the energy values are different for the individual pathways. The formation of propene over m-ZrO2(11) occurs easier than over t-ZrO2(101).
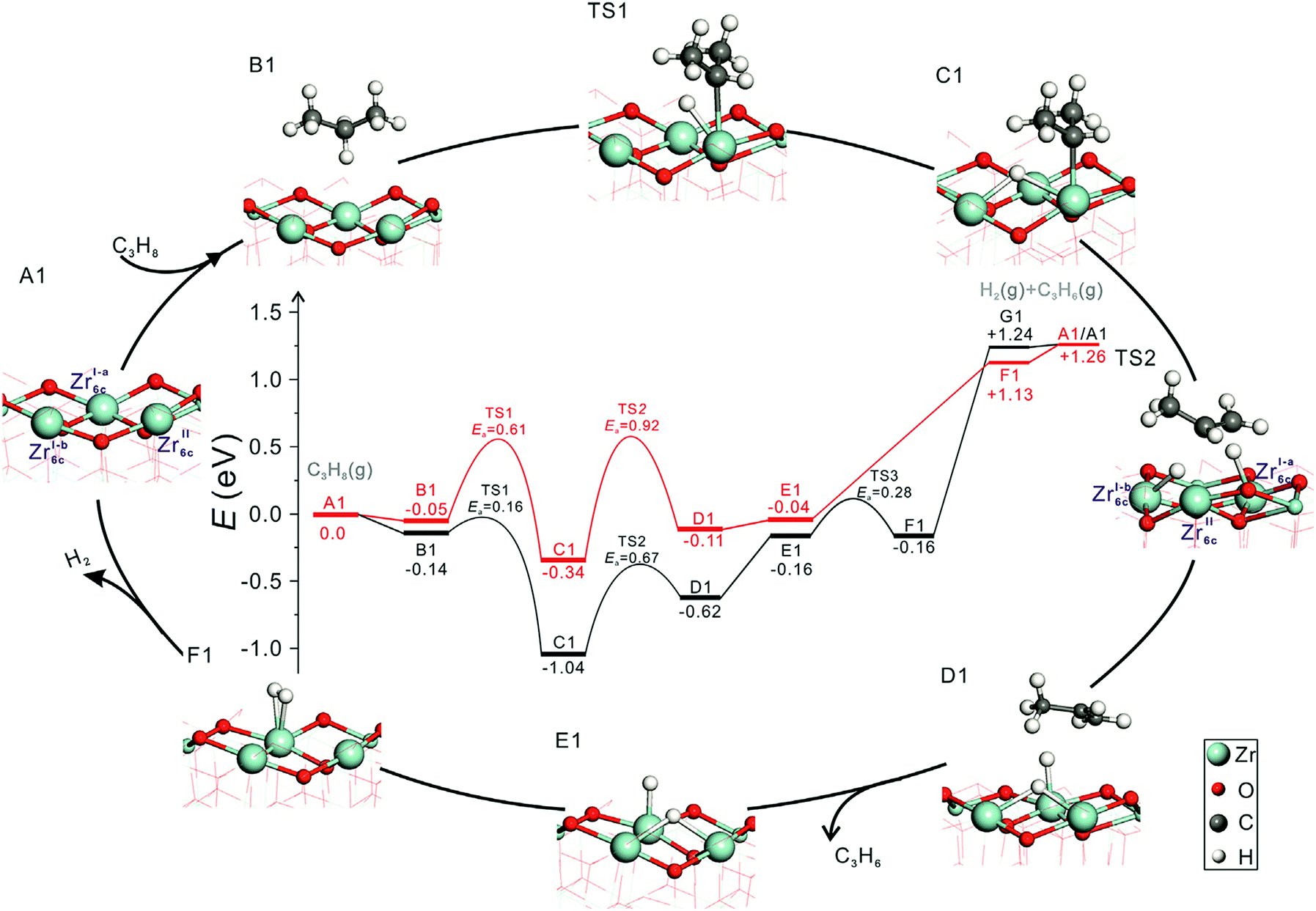 | ||
| Fig. 6 The calculated energy profiles and optimized structures of intermediates along the pathway of propane dehydrogenation to propene over defective t-ZrO2(101) (red) and m-ZrO2(11) (black) surfaces (Zr, light blue; C, grey; O, red; H, white). Reproduced from ref. 96 with permission from Elsevier, copyright 2020. | ||
The results of catalytic tests demonstrated that for both phases the activity and the selectivity to propene increased with a decrease in the size of crystallites (Fig. 7(a and b)). The reason for the different catalytic activity of ZrO2 samples was explained by their different ability to release lattice oxygen during reductive treatment. This catalyst property was determined by CO-TPR tests. The amount of consumed CO represents ZrO2 reducibility. The activity of the catalysts was in a good correlation with this parameter; the higher the reducibility, the more active the catalyst is.
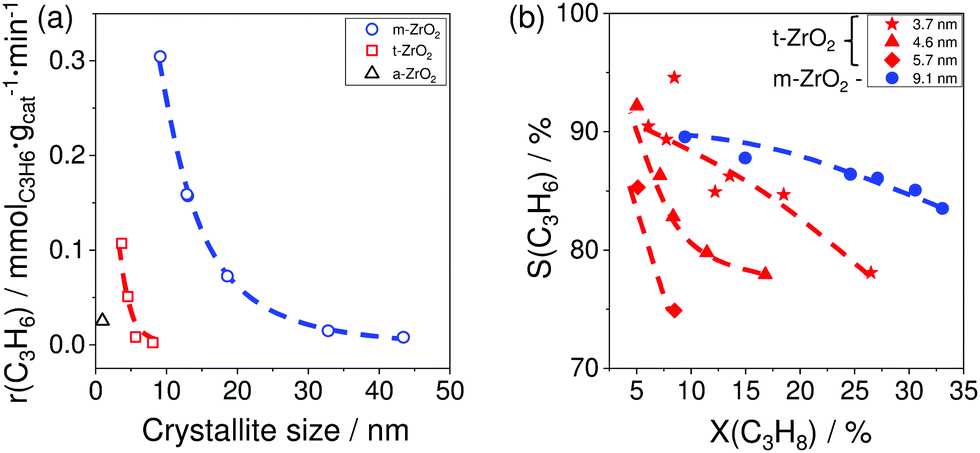 | ||
| Fig. 7 (a) The rate of propene formation (r(C3H6)) over monoclinic, tetragonal, and amorphous ZrO2 (m-ZrO2, t-ZrO2 and a-ZrO2, respectively) versus the size of crystallites; (b) the selectivity–conversion relationships for propene over m-ZrO2 with 9.1 nm crystallites and t-ZrO2 samples with 3.7, 4.6 or 5.7 nm crystallites. Reaction conditions: T = 550 °C, feed composition: 40 vol% C3H8 in N2. Adapted from ref. 96. | ||
The effect of ZrO2 reducibility on catalyst activity can be explained as follows. As above mentioned, the presence of oxygen vacancies is crucial for the activity of bulk catalysts because the active sites are coordinatively unsaturated metal cations located near such surface defects. Therefore, catalytic activity strongly depends on the concentration of oxygen vacancies. They can be produced during reductive catalyst treatment or in situ during the first minutes of PDH. It is obvious that the concentration of such defects would be higher for the sample with higher reducibility.
The increase in the selectivity to propene with a decrease in the size of crystallites was related to the lower ability of the samples with small crystallites to form coke. Propane conversion was about 15% for all samples with the exception for one t-ZrO2 which could achieve the conversion value of only 8.5%. Thus, it was shown that the ratio of the rate of propene formation to that of coke formation increases with decreasing crystallite size implying that the active sites for propene formation and coke formation are different. The desired reaction is catalysed by Zrcus sites, while regular surface Zr sites are responsible for coke formation.
Influence of treatment conditions. Since the active sites (coordinatively unsaturated metal cations) of bulk catalysts are located at oxygen vacancies, their creation should happen when lattice oxygen is removed. Such removal is possible during reductive treatment before catalytic reaction. The influence of different treatment conditions on the activity of bare and doped ZrO2 as well as of bare TiO2 was demonstrated in ref. 26, 27, 93, 97 and 98. In comparison with the air-treated samples, their H2-treated counterparts revealed significantly higher activity. For example, the rate of propene formation measured at 550 °C over bare ZrO2 pre-reduced in H2 at 600 °C was more than 4 times higher than that determined for the same catalyst but treated in air at 600 °C (0.109 versus 0.043 mmol(C3H6) gcat−1 min−1). For the bare TiO2, the rate of propene formation at 600 °C over the sample treated in H2 at 600 °C was about 3 times higher than over the sample treated in air at the same temperature (0.06 versus 0.02 mmol(C3H6) gcat−1 min−1). The positive effect of the reductive treatment is due to the removal of lattice oxygen and therefore formation of active coordinatively unsaturated Zrcus4+ or Ticus4+, which are present in much lower amounts in oxidized catalysts.
The amount of lattice oxygen removed during reductive treatment can be increased by (i) increasing reductive treatment temperature; (ii) increasing reductive treatment duration and/or (iii) using reducing agent(s) stronger than H2. The influence of reductive treatment temperature on the activity of bare ZrO2 or TiO2 is shown in Fig. 8. The rate of propene formation at 550 °C over bare ZrO2 gradually increases with increasing the temperature (Fig. 8(a)). It is however worth mentioning that for TiO2 the dependence is opposite (Fig. 8(b)). The negative effect of high-temperature treatment is due to the over-reduction of TiO2. Despite the removal of higher amount of lattice oxygen at higher reduction temperature, the creation of Ticus4+ does not happen since Ti4+ is reduced into inactive Ti3+. The negative effect of over-reduction was also observed for ZrO2-based catalysts with supported Rh or Ru species.93,99
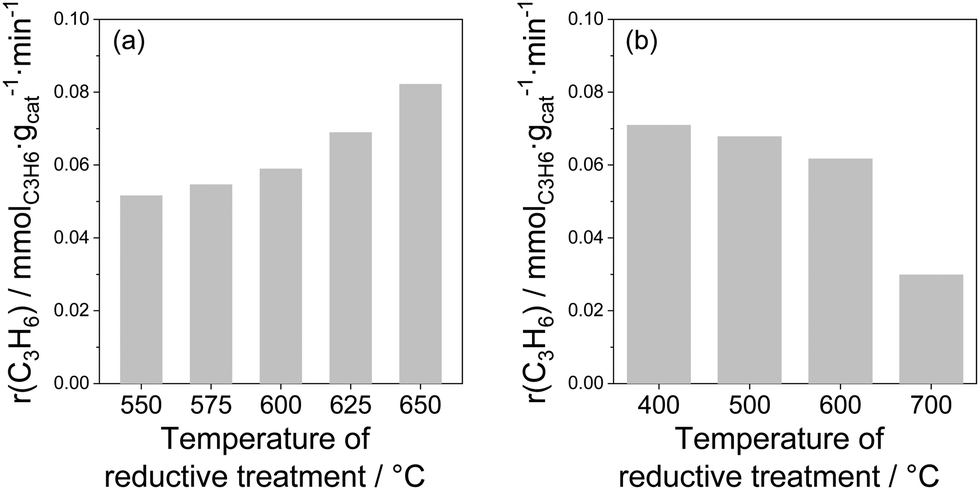 | ||
| Fig. 8 The rate of propene formation (a) at 550 °C over ZrO2 as a function of the temperature of H2 pre-treatment97 and (b) at 600 °C over TiO2 as a function of the temperature of H2 pre-treatment.27 | ||
The influence of reductive treatment time and the nature of the reducing agent on the rate of propene formation at 550 °C over bare ZrO2 was investigated in ref. 26. A gradual increase in the activity was observed when the duration of hydrogen treatment was extended to 7 hours. Such long treatment is not attractive from an applied viewpoint. When H2 was replaced by CO for the reduction purpose, the maximal activity was achieved after only 20 min treatment. Moreover, the CO-treated catalysts revealed about 3.5 times higher activity than their counterparts treated in H2 for 6 hours. In comparison with H2, CO can efficiently remove lattice oxygen and, more importantly, surface OH groups to generate Zrcus sites.
Influence of promoter for ZrO2. The kind of metal oxide dopant for ZrO2 is another important factor affecting the intrinsic activity of Zrcus sites. Thus, the presence of La3+, Y3+ or Sm3+ cations in the lattice of ZrO2 positively influences the activity of ZrO2, while the presence of Ca2+, Mg2+ or Li+ results in a decrease in the activity if compared with bare ZrO2 (Fig. 9(a)). The different effects of the dopants were explained by their dissimilar ability to generate/stabilize Zrcus active sites and their different effects on the intrinsic activity of Zrcus sites. Thus, it was shown that the activation energy (Ea) of propene formation over Y-doped ZrO2 is about 10 kJ mol−1 lower than that over bare ZrO2, La- or Sm-doped ZrO2, while the Ea values determined for Ca- or Mg-doped ZrO2 are 46 and 80 kJ mol−1 higher (Fig. 9(b)). Such results clearly demonstrate that the presence of Y3+ positively influences the intrinsic activity of Zrcus, while the presence of Ca2+ or Mg2+ has a negative effect. It is also worth mentioning that the dopant influences product selectivity. The selectivity to propene over doped ZrO2 is higher than over bare ZrO2 at a similar propane conversion. Such effect was related to a decrease in the concentration of strong acidic sites on the surface of ZrO2. Consequently, secondary transformations of propene into coke are partially hindered.
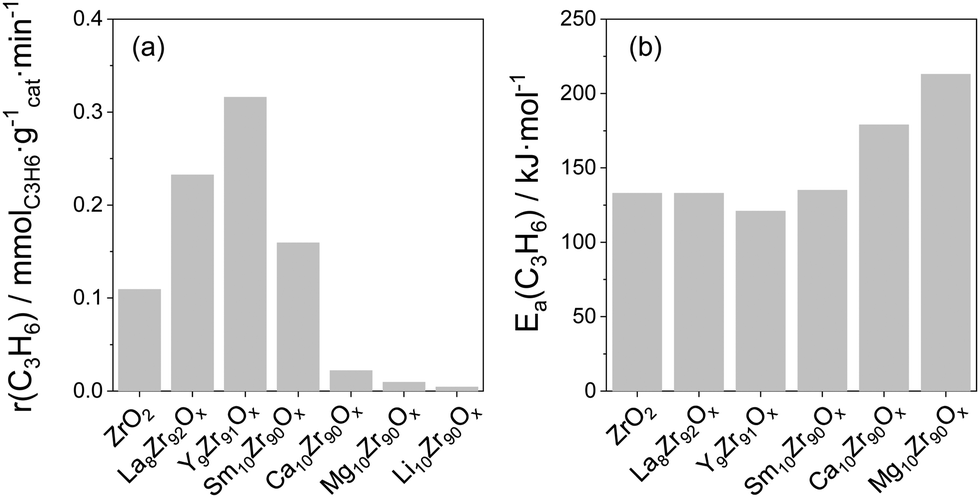 | ||
| Fig. 9 (a) The initial rate of propene formation at 550 °C over reduced ZrO2-based catalysts; (b) apparent activation energy of propene formation determined over reduced catalysts. Data are from ref. 98. | ||
Promoting ZrO2 with chromium oxide results in highly active catalysts, which significantly outperform a commercial analogue of CrOx–K/Al2O3.68 Moreover, in comparison with the latter catalyst, such catalysts have up to 40 times lower Cr content but show superior activity. Bulk binary CrZrOx or supported CrOx/LaZrOx catalysts which contain both Zrcus4+ and Cr3+ showed much higher rate of propene formation at similar Cr surface densities than CrOx/Al2O3 catalysts possessing only Cr3+ active sites (Fig. 10). Supported CrOx/LaZrOx catalysts demonstrated slightly higher propene selectivity (84–85%) than bulk binary CrZrOx (80–84%) but worse durability at a degree of propane conversion of about 30%. The on-stream stability of the latter materials can be improved through promoting with Cs, Ca and/or P.71
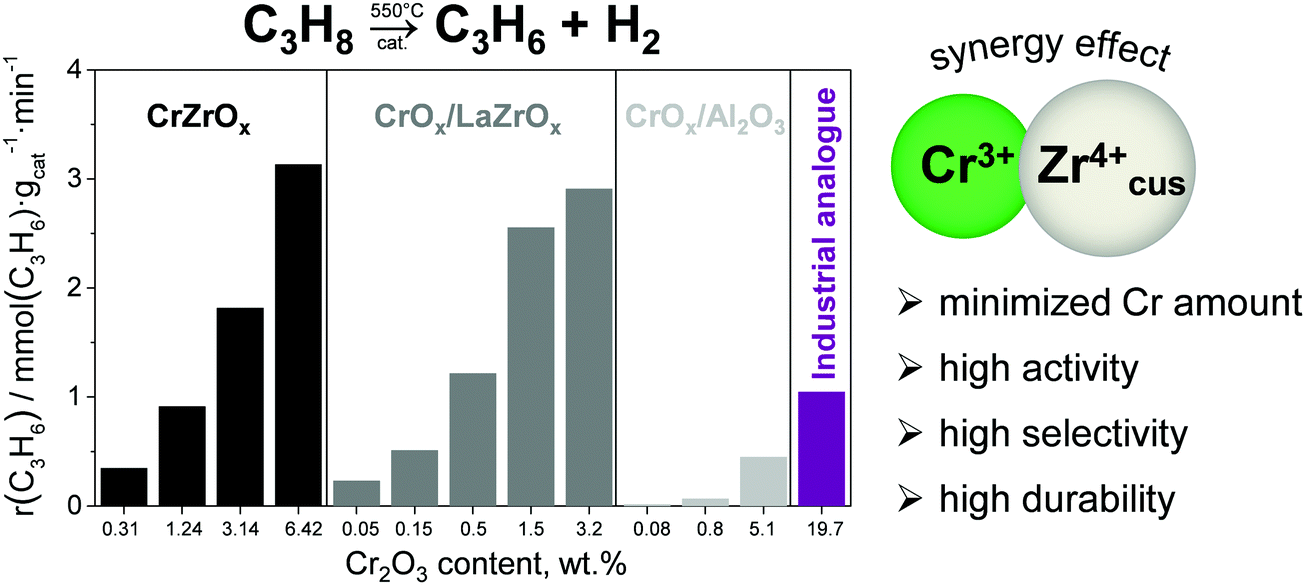 | ||
| Fig. 10 The rate of propene formation obtained at 550 °C over bulk CrZrOx, supported CrOx/LaZrOx and CrOx/Al2O3 and an analogue of industrial CrOx–K/Al2O3. Reproduced from ref. 68 with permission from Elsevier, copyright 2020. | ||
The origins of the high activity of CrOx-promoted ZrO2-based catalysts were thoroughly elucidated by Han et al.69 Those authors prepared catalysts with supported CrZrOx species. Mechanistic and kinetic tests were combined with characterization tests using the state-of-the art techniques, while molecular-level insights were derived from DFT calculations. Zrcus sites were concluded to be the main active sites. The role of CrOx promoter was explained as follows. Firstly, the promoter improves the ability of ZrO2 to release its lattice oxygen and accordingly to generate Zrcus sites. This positive effect also depends on the strength of interaction between CrOx, ZrO2 and the support. SiO2 weakly interacting with CrOx favours the stabilization of supported binary CrZrOx species that is also essential for the ability of ZrO2 to release its lattice oxygen. Secondly, CrOx enhances the intrinsic activity of Zrcus sites as concluded from DFT calculations. Fig. 11 shows the energy profile along the pathways in the course of propane dehydrogenation over CrZrOx and defective t-ZrO2(101). ZrO2 in CrZrOx is stabilized in the tetragonal phase. Regardless of the presence of promoter, two Zrcus cations form the active site. As seen in Fig. 11 the highest apparent barrier for PDH over 2Ov–CrZrOx(101) is lower than that over 1Ov–t-ZrO2(101) (0.95 versus 1.13 eV) implying that the presence of chromium provides more energetically favourable reaction pathway. It is noticeable that unlike PDH over t-ZrO2(101), where H2 formation is the rate-limiting step, PDH over 2Ov–CrZrOx(101) is limited by the breaking of the C–H bond in propane.
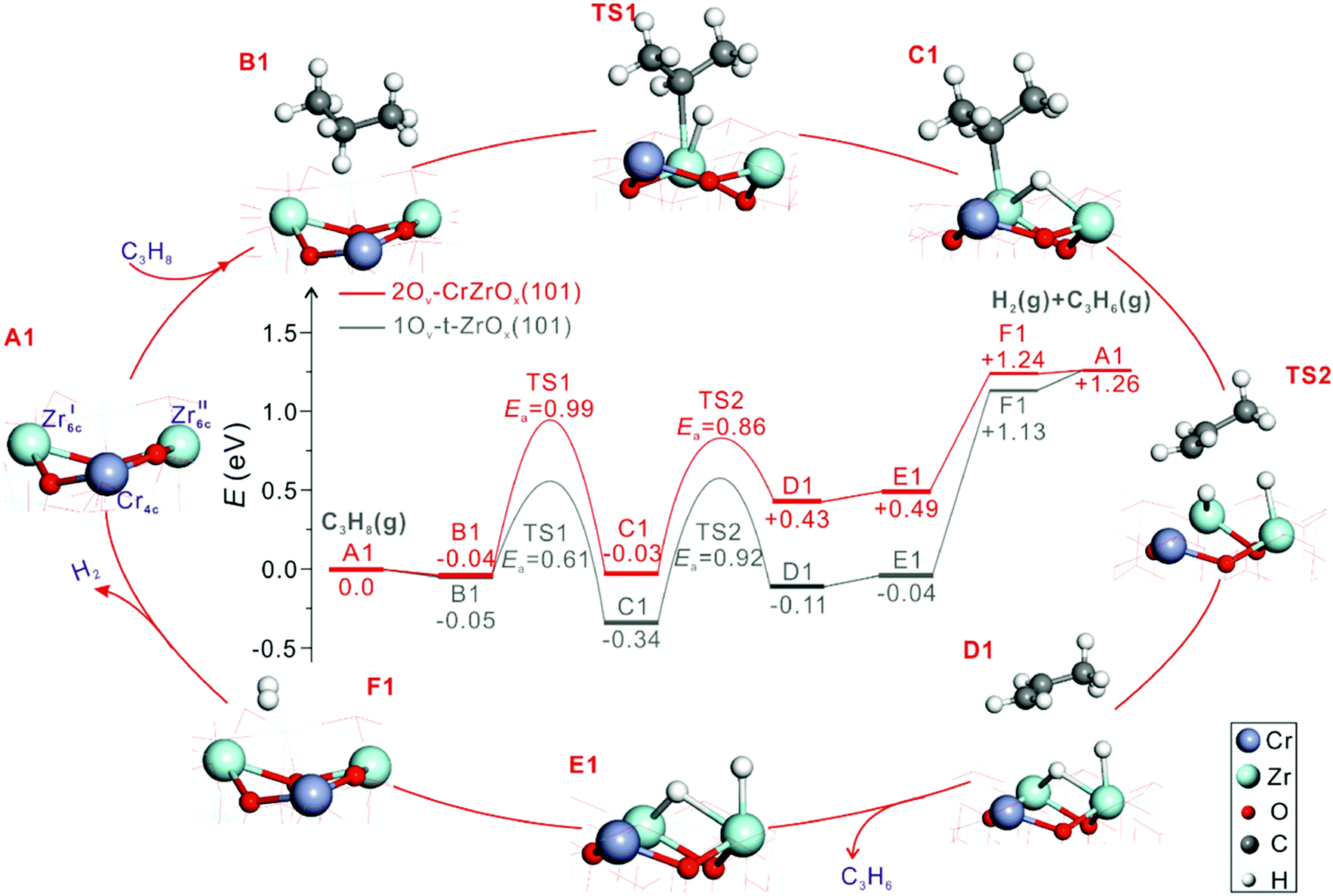 | ||
| Fig. 11 Calculated energy profiles and optimized structures of intermediates along the pathway of PDH to propene over 2Ov–CrZrOx(101) and 1Ov–t-ZrO2(101) surfaces (Cr, purple; Zr, light blue; C, grey; O, red; and H, white). Reproduced from ref. 69 with permission from American Chemical Society, copyright 2020. | ||
Influence of supported hydrogenation-active metal. It is well-known that the presence of hydrogenation-active metal on the surface of metal oxides enhances their reduction due to an easier generation of surface active hydrogen species from gas-phase H2.100–102 In case of bulk catalysts, where oxygen vacancies are crucial for the high activity, such property of the supported metals could be useful for promoting the generation of such defects. Indeed, recent studies on ZrO2-based catalysts proved that the presence of tiny (0.05 wt% and lower) amounts of Ru or Rh significantly increases the activity of ZrO2-based materials.93,99 In addition to this promoting effect, the supported metal can also indirectly participate in the PDH reaction. DFT calculations predict that the energy for H2 desorption (the rate-determining step) over Rh/d-ZrO2(
11) is lower than that over bare d-ZrO2(11).99 This result suggests that H2 desorption is accelerated in the presence of Rh. It should, however, be mentioned that the experimentally determined rate of propene formation over Rh/ZrO2 passes a maximum with Rh loading (Fig. 12(a)). According to DFT calculations, the positive effect of Rh on H2 formation disappears above a certain reduction degree of ZrO2 due to strong propene adsorption on Rh resulting in the blockage of sites required for hydrogen recombination. Very recently, this explanation was experimentally validated for bare ZrO2 and MZrOx (M = La or Y) with supported Ru or Rh nanoparticles.103
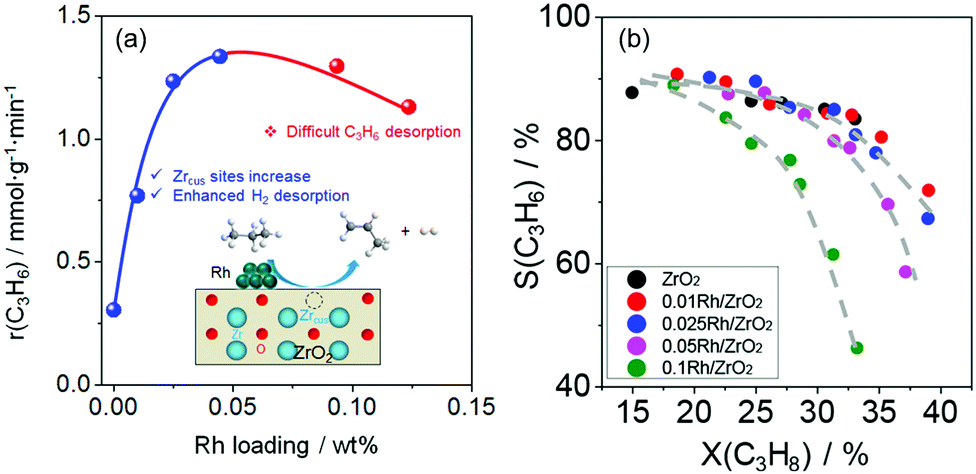 | ||
| Fig. 12 (a) The rate of propene formation (r(C3H6)) versus Rh loading of Rh/ZrO2; (b) dependence of selectivity to propene on propane conversion over bare ZrO2 and Rh/ZrO2 with different Rh loadings. Reproduced from ref. 99 with permission from American Chemical Society, copyright 2020. | ||
The presence of supported metal also influences product selectivity.99,103,104 As seen in Fig. 12(b), the selectivity to propene decreases with an increase in Rh loading in Rh/ZrO2 at a similar degree of propane conversion. The reason for such phenomenon is related to stronger propene adsorption on Rh than on bare ZrO2. Adsorbed propene can participate in undesired side reactions leading to coke. When using supported Pt or Ir species weakly interacting with propene, no negative effect of metal loading on propene selectivity was determined over bare Pt(Ir)/ZrO2 or Pt(Ir)/MZrOx (M = La or Y).103
2.4 Benchmarking and demonstrating catalyst development progress
As reviewed above, a large number of supported and bulk catalysts have been prepared and tested for their activity and selectivity during the last 5 years. Here, to demonstrate if there is any progress in catalyst development, we compare the most efficient catalysts with each other and with those described in a review of Weckhuysen and co-workers in 2014.18 The target characteristics were the selectivity to propene and the space time yield of propene formation STY(C3H6) at a propane conversion above 20%. Moreover, the catalysts tested with diluted reaction feeds, which are not attractive from an industrial viewpoint, were not used for such comparison. Only studies, which used reaction feeds with propane content of at least 20 vol%, were considered.STY(C3H6) values fulfilling the above criteria are summarized in Fig. 13(a). These criteria were also applied to the catalysts from ref. 18. The corresponding catalyst compositions and further relevant catalytic data are provided in Table 3. The highest STY(C3H6) values were achieved over recently developed binary Cr–Zr–Ox-based catalysts; 3.2CrOx/LaZrOx68 produced 2.6 kg(C3H6) kg(cat)−1 h−1 at 550 °C, while at 600 °C the value of about 3.5 kg(C3H6) kg(cat)−1 h−1 was obtained over Cr30Zr90/SiO2.69 ZrOx-, VOx-, CoOx- and ZnOx-based catalysts demonstrated lower productivity but still comparable with that of Pt-based catalysts described in ref. 18.
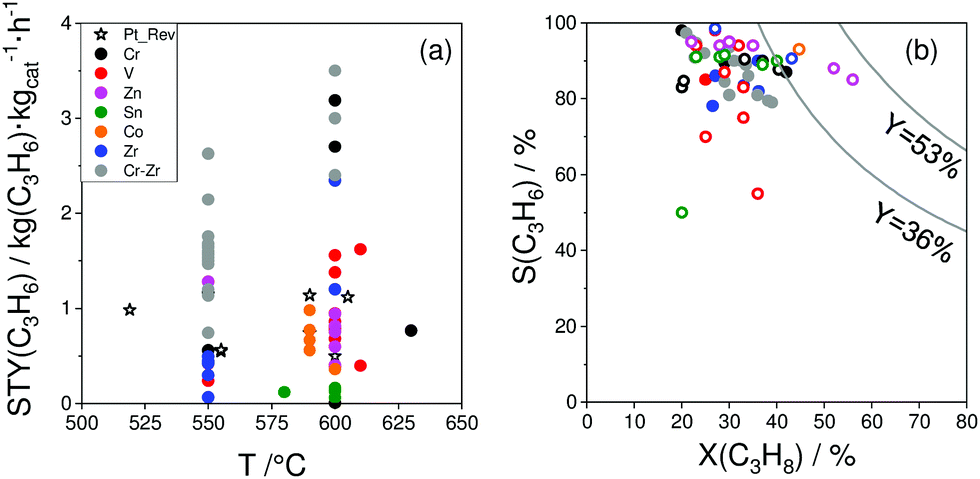 | ||
| Fig. 13 (a) Space time yield (STY(C3H6)) of propene obtained over the most perspective catalysts described for the last 5 years (circles) and over selected best-performing catalysts from ref. 18 (stars). All data were obtained at propane conversion of at least 20% using reaction feeds with at least 20 vol% C3H8; (b) dependence of selectivity to propene on propane conversion at 550 °C (closed symbols) and 600 °C (open symbols) for selected catalysts from (a). Catalyst composition and reaction conditions are given in Table 3. | ||
| Catalyst | T/°C | Feed composition | WHSV of C3H8/h−1 | X(C3H8)/% | S(C3H6)/% | Y(C3H6)/% | STY(C3H6)/kg(C3H6) kgcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| K–CrOx/Al2O3 | 550 | 40 vol% C3H8–60 vol% N2 | 4.71 | 29.0 | 89.5 | 26.0 | 1.17 | 68 |
| Cr-Al-800 | 600 | 100 vol% C3H8 | 9.43 | 33.2 | 90.4 | 30.0 | 2.70 | 65 |
| Cr-Al-Ref | 600 | 100 vol% C3H8 | 9.43 | 40.4 | 87.7 | 35.4 | 3.19 | 65 |
| 20CrOx/Al2O3 | 600 | 50 vol% C3H8–50 vol% N2 | 0.02 | 37.0 | 90.0 | 33.3 | 0.01 | 105 |
| K–CrOx/Al2O3 | 550 | 40 vol% C3H8–60 vol% N2 | 1.6 | 42.0 | 87.0 | 36.5 | 0.56 | 98 |
| Cr10ZrOx | 550 | 40 vol% C3H8–60 vol% N2 | 9.24 | 30.0 | 81.0 | 24.3 | 2.14 | 68 |
| 3.2CrOx/LaZrOx | 550 | 40 vol% C3H8–60 vol% N2 | 11.22 | 29.0 | 84.5 | 24.5 | 2.63 | 68 |
| Cr10Zr90/SiO2 | 550 | 40 vol% C3H8–60 vol% N2 | 4.32 | 35.9 | 81.0 | 29.1 | 1.20 | 69 |
| Cr10Zr90/SiO2 | 600 | 40 vol% C3H8–60 vol% N2 | 11.52 | 25.8 | 84.5 | 21.8 | 2.40 | 69 |
| Cr20Zr90/SiO2 | 550 | 40 vol% C3H8–60 vol% N2 | 5.18 | 38.2 | 79.5 | 30.4 | 1.50 | 69 |
| Cr20Zr90/SiO2 | 600 | 40 vol% C3H8–60 vol% N2 | 13.82 | 27.1 | 84.0 | 22.8 | 3.00 | 69 |
| Cr30Zr90/SiO2 | 550 | 40 vol% C3H8–60 vol% N2 | 5.44 | 39.0 | 79.0 | 30.8 | 1.60 | 69 |
| Cr30Zr90/SiO2 | 600 | 40 vol% C3H8–60 vol% N2 | 14.52 | 29.7 | 85.0 | 25.2 | 3.50 | 69 |
| Cr–Zr–Ox (Cr/Zr molar ratio = 1/9) | 550 | 40 vol% C3H8–60 vol% N2 | 5.89 | 34.0 | 86.0 | 29.2 | 1.64 | 71 |
| 1.25 wt% P/CZ | 550 | 40 vol% C3H8–60 vol% N2 | 5.89 | 31.0 | 90.0 | 27.9 | 1.57 | 71 |
| 1.5 wt% Cs/CZ | 550 | 40 vol% C3H8–60 vol% N2 | 5.89 | 29.0 | 94.0 | 27.3 | 1.53 | 71 |
| 1.5 wt% Cs–1.25 wt% P/CZ | 550 | 40 vol% C3H8–60 vol% N2 | 5.89 | 30.0 | 93.5 | 28.1 | 1.58 | 71 |
| 0.6 wt% Ca/CZ | 550 | 40 vol% C3H8–60 vol% N2 | 5.89 | 32.5 | 90.0 | 29.3 | 1.64 | 71 |
| 0.6 wt% Ca–1.25 wt% P/CZ | 550 | 40 vol% C3H8–60 vol% N2 | 5.89 | 33.5 | 89.0 | 29.8 | 1.68 | 71 |
| CrZrOx/SiO2_450 | 550 | 40 vol% C3H8–60 vol% N2 | 3.77 | 22.7 | 90.8 | 20.6 | 0.74 | 70 |
| CrZrOx/SiO2_500 | 550 | 40 vol% C3H8–60 vol% N2 | 5.66 | 22.0 | 95.3 | 21.0 | 1.13 | 70 |
| CrZrOx/SiO2_550 | 550 | 40 vol% C3H8–60 vol% N2 | 7.54 | 20.9 | 97.3 | 20.3 | 1.47 | 70 |
| CrZrOx/SiO2_600 | 550 | 40 vol% C3H8–60 vol% N2 | 8.08 | 23.0 | 94.7 | 21.8 | 1.68 | 70 |
| CrZrOx/SiO2_650 | 550 | 40 vol% C3H8–60 vol% N2 | 8.08 | 24.8 | 92.0 | 22.8 | 1.76 | 70 |
| 10 wt% V–K/meso-Al2O3-373 K | 610 | 80 vol% C3H8–20 vol% N2 | 2.83 | 69.0 | 87.0 | 60.0 | 1.62 | 38 |
| 6V/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3.3 | 23.0 | 94.0 | 21.6 | 0.68 | 34 |
| 12V/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3.3 | 32.0 | 94.0 | 30.1 | 0.95 | 34 |
| 20V/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3.3 | 29.0 | 87.0 | 25.2 | 0.79 | 34 |
| 6 wt% V/Al2O3 treated 1 h in H2 | 600 | 28 vol% C3H8–72 vol% N2 | 8.25 | 25.0 | 70.0 | 17.5 | 1.38 | 73 |
| 6 wt% V/Al2O3 treated 3 min in H2 | 600 | 28 vol% C3H8–72 vol% N2 | 8.25 | 36.0 | 55.0 | 19.8 | 1.56 | 73 |
| 12 wt% V/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3.3 | 33.0 | 75.0 | 24.8 | 0.78 | 63 |
| 12V1Mg/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3.3 | 33.0 | 83.0 | 27.4 | 0.86 | 63 |
| 10 wt% V/Al2O3 | 610 | 20 vol% C3H8–80 vol% N2 | 0.71 | 73.0 | 80.5 | 58.8 | 0.40 | 62 |
| 10 wt% Zn/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3 | 22.0 | 95.0 | 20.9 | 0.60 | 42 |
| 15 wt% Zn/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3 | 28.0 | 94.0 | 26.3 | 0.75 | 42 |
| 10 wt% Zn0.1 wt% Pt/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3 | 30.0 | 95.0 | 28.5 | 0.82 | 42 |
| 15 wt% Zn0.1 wt% Pt/Al2O3 | 600 | 28 vol% C3H8–28 vol% H2–44 vol% N2 | 3 | 35.0 | 94.0 | 32.9 | 0.94 | 42 |
| 4Zn/TiZrOx | 550 | 40 vol% C3H8–5 vol% H2–55 vol% N2 | 4.71 | 30.0 | 95.0 | 28.5 | 1.28 | 43 |
| 0.05Ru/YZrOx | 550 | 40 vol% C3H8–60 vol% N2 | 1.57 | 36.2 | 82.0 | 29.7 | 0.44 | 93 |
| 0.05Ru/YZrOx | 600 | 40 vol% C3H8–60 vol% N2 | 6.29 | 43.1 | 90.6 | 39.0 | 2.34 | 93 |
| Y9Zr91Ox | 550 | 40 vol% C3H8–60 vol% N2 | 1.6 | 36.0 | 90.0 | 32.4 | 0.49 | 98 |
| t-ZrO2-1 | 550 | 40 vol% C3H8–60 vol% N2 | 0.32 | 26.5 | 78.1 | 20.7 | 0.06 | 96 |
| m-ZrO2-1 | 550 | 40 vol% C3H8–60 vol% N2 | 1.89 | 27.1 | 86.1 | 23.3 | 0.42 | 96 |
| m-ZrO2-1 | 550 | 40 vol% C3H8–60 vol% N2 | 1.13 | 33.0 | 83.5 | 27.6 | 0.30 | 96 |
| 10.0SnO2/Si-2 | 580 | 99.9 vol% C3H8 | 0.65 | 20.0 | 96.0 | 19.2 | 0.12 | 51 |
| 1.5Ni10.0SnO2/Si-2 | 580 | 99.9 vol% C3H8 | 0.65 | 20.0 | 96.0 | 19.2 | 0.12 | 51 |
| 1.36-Sn/Si(2)_reduced | 600 | 99.9 vol% C3H8 | 0.65 | 23.0 | 91.0 | 20.9 | 0.13 | 50 |
| 1.78-Sn/Si(4)-(ws)_reduced | 600 | 99.9 vol% C3H8 | 0.65 | 28.0 | 91.0 | 25.5 | 0.16 | 50 |
| 2.57-Sn/Si(4)_oxidized | 600 | 99.9 vol% C3H8 | 0.65 | 20.0 | 50.0 | 10.0 | 0.06 | 50 |
| 3.82-Sn/Si(5)_reduced | 600 | 99.9 vol% C3H8 | 0.65 | 29.0 | 91.5 | 26.5 | 0.16 | 50 |
| Sn-HMS | 600 | 99.87 vol% C3H8–0.13 vol% C2H6 | 0.39 | 40.0 | 90 | 36.0 | 0.13 | 52 |
| 5Co–Al2O3–HAT | 590 | 20% vol C3H8–16 vol% H2–64 vol% N2 | 2.9 | 24.8 | 97.1 | 24.1 | 0.67 | 90 |
| Co/Al2O3-IMP | 590 | 20 vol% C3H8–16 vol% H2–64 vol% N2 | 2.9 | 21.2 | 95.5 | 20.2 | 0.56 | 90 |
| 7Co–Al2O3–HAT | 590 | 20 vol% C3H8–16 vol% H2–64 vol% N2 | 2.9 | 30.0 | 93.0 | 27.9 | 0.77 | 90 |
| Co/Al2O3 | 600 | 33.3 vol% C3H8–66.7 vol% N2 | 0.91 | 44.7 | 93 | 41.6 | 0.36 | 54 |
| 0.35 wt% Pt–1.26 wt% Sn/Al2O3 | 519 | 30 vol% C3H8–70 vol% N2 | 3.5 | 31.0 | 95.0 | 29.5 | 0.98 | 18 |
| 0.6 wt% Pt–5 wt% Ga/MgAl2O4 | 605 | 73 vol% C3H8–27 vol% H2 | 3.9 | 31.0 | 97.0 | 30.1 | 1.12 | 18 |
| 0.5 wt% Pt/Zn-Beta | 555 | 100 vol% C3H8 | 2.6 | 40.0 | 55 | 22.0 | 0.55 | 18 |
| 0.5 wt% Pt–Na/Sn-ZSM-5 | 590 | 75 vol% C3H8–25 vol% H2 | 3.0 | 41.7 | 95.3 | 39.7 | 1.14 | 18 |
| 0.5 wt% Pt–Zn/Na-Y | 555 | 100 vol% C3H8 | 2.6 | 24.8 | 91.6 | 22.7 | 0.56 | 18 |
| 0.5 wt% Pt–Sn–Na/Al-SBA-15 | 590 | 75% vol C3H8–25 vol% H2 | 3.0 | 27.5 | 94.0 | 25.9 | 0.74 | 18 |
| 0.7 wt% Pt/Mg(In)(Al)O | 600 | 20 vol% C3H8–25 vol% H2–55 vol% He | 2.6 | 20.4 | 98.0 | 20.0 | 0.50 | 18 |
For industrial application, not only productivity but also propene selectivity is an important parameter. To compare the catalysts from Fig. 13(a) in this performance, the reported selectivity–conversion values are shown in Fig. 13(b). It is obvious that all Zn-containing catalysts show propene selectivity above 80% at degrees of propane conversion up to about 40%. No data at higher conversion degrees are available. Some Zr-, Co-, Sn- or Cr-containing catalysts also show propene selectivity above 80% at degrees of propane conversion up to 50%.
In summary, an obvious progress in the development of metal oxide catalysts in terms of their productivity and selectivity at industrially relevant degrees of propane conversion was achieved during the last 5 years. However, to further check their application potential, tests with concentrated reaction feeds are required. It is also important to check catalysts durability, i.e. catalysts ability to restore their initial performance after oxidative regeneration in several dehydrogenation/regeneration cycles. Under this consideration, ZrO2-based catalysts should be especially mentioned since they demonstrate good durability under industrially relevant conditions.25,68,69,93
3 Non-oxidative isobutane dehydrogenation
As in the commercial PDH processes, catalysts with supported Pt or CrOx species are used for the non-oxidative dehydrogenation of isobutane to isobutene (BDH) on large scale. High cost of Pt and toxicity of Cr(VI) compounds motivated the researchers to develop low-cost and environmentally friendly alternatives. Among them, the catalysts based on oxides of vanadium,36,106–110 gallium,111–114 molybdenum,115 zirconium,98,116,117 iron118–120 and aluminium94 have been developed and investigated in the BDH reaction. None of them has so far found its commercial application. The most important tasks in the development of such catalysts include (i) maximizing their dehydrogenation performances (activity, selectivity and stability), (ii) identifying catalytically active sites, and (iii) establishing structure–activity–selectivity relationships. Herein, we describe and discuss the most recent achievements (2016–2020) related to catalysts based on metal oxides. Similar to the PDH reaction, the developed catalysts can be divided into two groups: conventional supported and alternative-type bulk catalysts (Fig. 4).3.1 Conventional supported catalysts
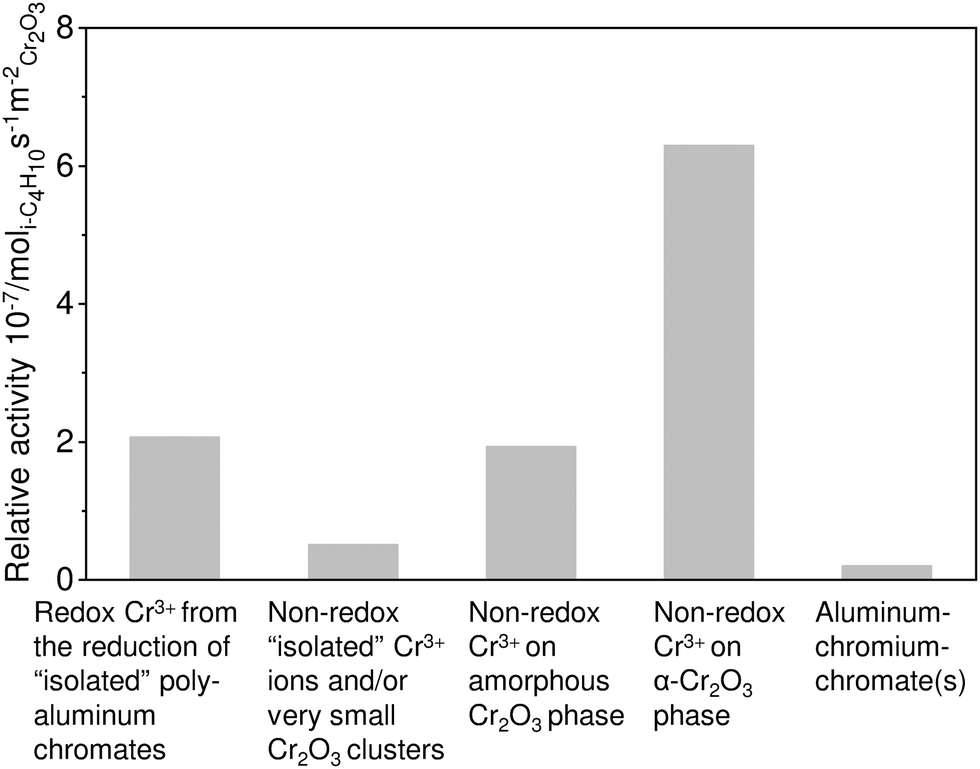 | ||
| Fig. 14 Relative activity of different Cr-containing species on the surface of the CrOx/Al2O3 catalysts. Adapted from ref. 122. | ||
Besides studies aimed at elucidating the nature of catalytically active sites, the on-going research also deals with analysing the influence of promoter(s) and support on catalytic performance. The promoters are selected to modify overall catalyst acidity for hindering coke formation and to stabilize specifically structured CrOx species and/or phases. Wang et al.123 reported that promoting CrOx/γ-Al2O3 with Ca improves both isobutane conversion and isobutene selectivity. These positive effects were related to regulation of acidic and redox properties. Lowering catalyst acidity inhibited side reactions such as cracking and isomerization. The presence of Ca is also decisive for suppressing over-reduction of CrOx species thus ensuring high activity.
K is another basic promoter widely used for preparation of CrOx/Al2O3 catalysts. The effect of K loading was investigated in ref. 124. When the loading is lower than 2%, both isobutane conversion and isobutene selectivity increase due to an improved dispersion of CrOx species. The promoters prefer to interact with CrOx species at higher loadings. This interaction decreases the intrinsic activity of such species without changing the selectivity.
The usage of non-basic promoters mainly affects redox properties of CrOx-species. Salaeva et al.125 established a synergistic effect between Cu and Zn on catalytic performance of CrOx/Al2O3. A decrease in the dehydrogenation activity of CrOx/γ-Al2O3 was found after promoting with Zn. When co-adding Cu, the activity was improved in comparison with CrOx/γ-Al2O3. Cu alone also has a positive effect. The introduction of Zn and/or Cu was concluded to contribute to the formation of defective spinels (CuAl2O4, ZnAl2O4). Such transformation promotes the formation of higher amounts of Cr(VI) species on the catalyst surface. It should also be noted that in contrast to Zn, which decreases the reducibility of supported chromium species, the presence of Cu or Cu–Zn facilitates the reduction of such species that is important for catalyst activity.
The origins of the positive effect of Cu promoter on catalytic properties of CrOx/Al2O3 were further investigated in a separate study.126 For Cu loading below 2.6 wt%, Cu is mainly incorporated into the structure of the alumina support. As a consequence, monomeric and dimeric Cr(VI) are stabilized on the catalyst surface, which are transformed into catalytically active mononuclear and dimeric Cr3+ sites under reaction conditions. A CuO phase is formed at higher Cu contents. This phase was suggested to decrease the amount of redox Cr(VI)Ox species and thus results in a decrease in the activity.
The kind of support is another decisive parameter affecting catalyst activity. The support typically affects the dispersion of CrOx species. For example, the usage of CeO2 as a support was established to worsen the activity due to favouring the formation of low-active α-Cr2O3 phase.127 In contrast, high dispersion can be achieved when using Al2O3, ZrO2 or CexZr1−xO2 as supports. Catalysts prepared on the basis of the two latter materials demonstrated the highest activity possibly due to the stabilization of Cr(VI)Ox strongly bound to the support surface. It cannot, however, be excluded that the high activity of ZrO2-based catalysts can be related to the synergy effect between Cr and Zrcus sites as recently was demonstrated for the PDH reaction over binary bulk or supported Cr–Zr–Ox.68,69
Promoting Al2O3 with SiO2 results in an increase in isobutane conversion and isobutene selectivity over K–CrOx/SiAlOx.128 In comparison with K–CrOx/Al2O3, these catalyst parameters were improved from 60.1 to 62.7% and from 87.3 to 90%, respectively. Moreover, the yield of coke decreased from 1.1 to 0.7%. The positive effect of SiO2 was related to the stabilization of χ-Al2O3 phase.
To elucidate the influence of the structure of VOx species on their intrinsic activity, isobutene selectivity and on-stream stability, a series of VOx/MCM-41 catalysts with different V loadings were prepared and investigated.36 MCM-41 was chosen because it does not have strong acidic or basic sites. V3+Ox and V4+Ox were concluded to be the main catalytically active sites. A correlation between the number of V–O–V bonds around a central V cation determined from UV-vis spectroscopic analysis and the apparent turnover frequency of isobutene formation (TOF(isobutene)) was established (Fig. 15). The higher the polymerisation degree (number of V–O–V bonds), the lower the TOF(isobutene) values are. This was explained by stronger Lewis acidity of isolated sites. This catalyst property has, however, a negative effect on isobutene selectivity (Fig. 15). Nevertheless, isolated VOx species deactivate slower than their polymerized VOx counterparts (Fig. 15). The increase in deactivation rate with an increase in the degree of VOx polymerization was related to an increase in the number of neighbouring V3+ or V4+ cations. For catalyst development, it was suggested that isolated VOx sites with balanced Lewis acidity are required for achieving high activity, selectivity and on-stream stability.
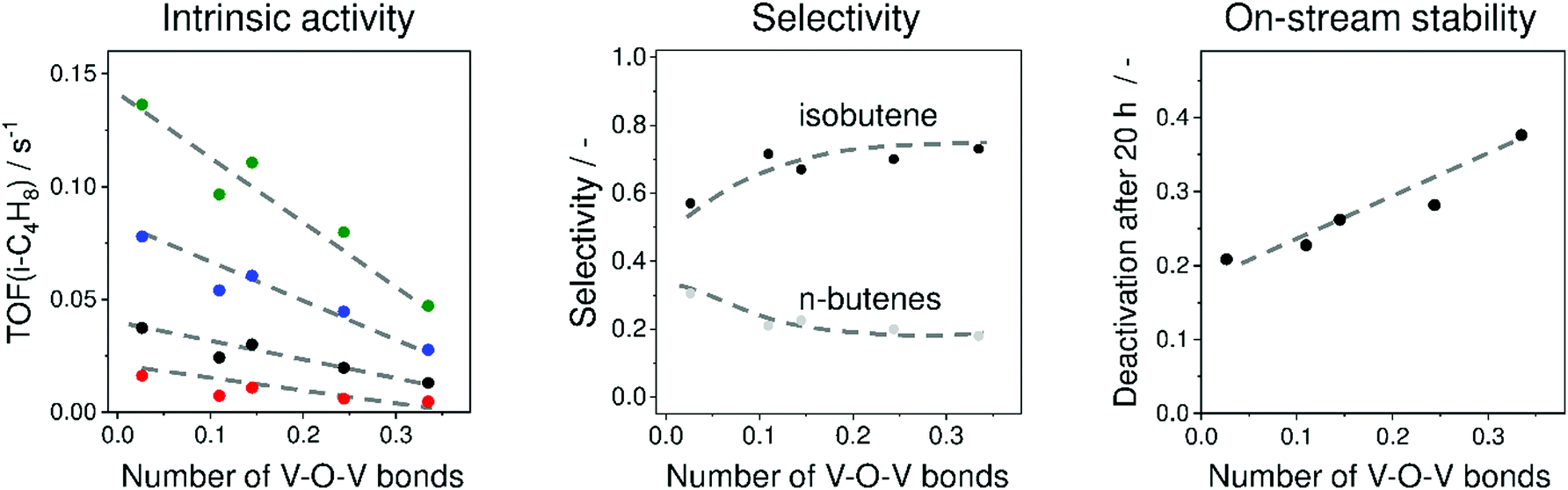 | ||
| Fig. 15 Influence of mean number of V–O–V bonds around a central V cation in VOx/MCM-41 on (left figure) turnover frequency of isobutene formation (TOF(i-C4H8)) at 525 °C (red circles), 550 °C (black circles), 575 °C (blue circles), and 600 °C (green circles), (middle figure) selectivity to isobutene and n-butenes at about 30% isobutane conversion at 550 °C and (right figure) on-stream stability at 550 °C. Adapted from ref. 36. | ||
Rodemerck et al.129 investigated the influence of the kind of support (MCM-41, Al2O3, mixed SiO2–Al2O3 oxides) on the nature of VOx species as well as physico-chemical and catalytic performance. The degree of polymerization of VOx species at similar V loading was concluded to depend on the support material. Thus, the lowest VOx polymerization degree was determined for VOx/SiO2 while it increased with aluminium content in the support. The support also influences overall catalyst acidity. All the prepared catalysts possess Lewis acidic sites. However, due to a similar electronegativity of V and Al, VOx/Al2O3 does not have Brønsted acidic sites, while such sites are present on the surface of VOx/AlSiOx because the electronegativity of Si is higher than that of V. Noticeably, the highest selectivity to isobutene of 85% at a degree of isobutane conversion of about 50% was achieved over VOx/Al2O3. A detailed study of the dependence of product selectivity on isobutane conversion using feeds with 1-butene, isobutene or isobutane showed that the low selectivity to isobutene over VOx/AlSiOx catalysts in BDH is related to their higher activity for isobutene isomerization to n-butenes that depends on the degree of polymerization of VOx and surface acidity. Catalyst isomerization ability affects the pathways of coke formation. This undesired reaction product is formed from isobutene over VOx/Al2O3 catalyst. Contrarily, butadiene is the main coke precursor in the course of BDH over VOx/AlSiOx.
Tian et al.106 reported that the distribution of VOx species such as isolated monomeric species, oligomeric species and polymeric vanadium oxide chains in K2O–VOx/γ-Al2O3 (vanadium loading of 5, 10, 15 or 20 wt%) depends on vanadium loading. Reduced VOx species containing V3+ and V4+ were proven to be the main catalytically active sites. Based on the results of FTIR, NMR and H2-TPR studies, it could be concluded that catalysts with vanadium loading up to 5 wt% possess only isolated monomeric and dimeric VOx species. Higher loaded materials contain polymerized VOx species. In contrast to ref. 36, oligomeric VOx species demonstrated an optimal dehydrogenation activity. This conclusion also disagrees with the results of Wang et al.,110 who investigated the effect of B modification of VOx/MCM-41 on catalytic performance. B was found to mainly exist in the form of [BO3]3− on the surface of the catalysts, with a small fraction residing in the framework of MCM-41. The introduction of B increased the dispersion of VOx sites as well as isobutane conversion and isobutene selectivity.
With respect to isobutene selectivity, this catalyst property decreases with an increase in the ratio of Lewis acidic sites to Brønsted acidic sites. Promoting K2O–VOx/γ-Al2O3 with sulphur was reported to also increase isobutene selectivity.109 XPS, Raman and 51VMAS NMR confirmed the existence of V–S bonds after sulfidation. As the bond energy of V–S is lower than that of V–O, vanadium species could be reduced more easily after sulfidation. Consequently, more catalytically active V3+/V4+ sites are produced. Noticeably, sulfidation led to an increase in the strength of acidic sites but to a decrease in their concentration. Such changes in the acidic properties resulted in increasing isobutane conversion and inhibiting cracking reactions.
Zn/ordered mesoporous alumina was studied in isobutane dehydrogenation by Cheng et al.131 It was found that the introduction of Zn with a content below 10% did not change the mesoporous structure and textural properties of the support. ZnOx species existed in highly dispersed forms and were incorporated into the support framework. With increasing the Zn content, the total number of acidic sites and that of weak sites increased, while those of strong acidic sites decreased. A correlation between the rate of isobutane conversion and the number of weak and medium acidic sites was established. The highest initial isobutane conversion and isobutene selectivity of 46.6% and 81.8%, respectively, were achieved over ZnO(10 wt%)/Al2O3 at 580 °C. This catalyst also showed good durability in 5 dehydrogenation–regeneration cycles.
Alumina-supported Ga2O3– or Ga2O3–Cr2O3-containing catalysts were investigated in a fluidized-bed reactor.112 It was found that the catalytic performances of Ga2O3/Al2O3 vary with gallium content. The conversion of isobutane at 580 °C increased from 42% to 55% with an increase in the content of gallium in Ga2O3/Al2O3 from 3 to 9 wt%. The conversion could be further improved after promoting Ga2O3(6 wt%)/Al2O3 with Cr (6 wt%) and ZrO2 (1 wt%). This catalyst showed same conversion but higher isobutene selectivity (90 vs. 87%) in comparison with an industrial CrOx-based catalyst. An outstanding stability during 60 cycles without any loss in catalyst performance was achieved.
Two recent studies dealt with kinetic aspects of the BDH reaction over Ga2O3/Al2O3113,114 in the temperature range from 520 to 580 °C. Based on the experimental results and proposed reaction network, six major components including isobutane, n-butane, isobutene, butene, propene and methane were included in the kinetic modelling. The rate equations were obtained, and the corresponding kinetic parameters were estimated. The much smaller value of apparent activation energy for isobutane dehydrogenation (195 kJ mol−1) than that of cracking (300 kJ mol−1) indicated the efficiency of Ga2O3/Al2O3 for this reaction.
Ordered mesoporous Al2O3-based catalysts containing both ZnOx and FeOx supported species were also tested in the BDH reaction.119 They contain FeOx species incorporated into the framework of Al2O3 and highly dispersed surface FeOx species. Zn species were present in the form of hexagonal ZnO. Fe3+ and Zn2+ are considered to dehydrogenate isobutane. The isobutane conversion of 50.7% and the isobutene yield of 37.8% were achieved at 580 °C over the catalyst with 1.9 and 7.2 at% of Fe and Zn respectively.
3.2 Alternative-type bulk catalysts
In order to understand the effect of dopant for ZrO2 on catalyst activity and product selectivity, a series of doped ZrO2 samples containing La3+, Y3+, Sm3+, Ca2+, Mg2+ or Li+ were prepared, characterized and investigated in the BDH reaction.98 The presence of dopant enhances the selectivity to isobutene in comparison with undoped ZrO2 due to a decrease in the concentration of strong acidic sites, which provoke coke formation. With respect to the activity, the introduction of La3+ or Y3+ results in a decrease in the activation energy of isobutene formation indicating that the presence of such dopants in the lattice of ZrO2 positively influences the intrinsic activity of Zrcus4+ sites.
Binary CrZrOx should be especially mentioned due to their unexpectedly high activity.116 The space time yield of isobutene formation at 550 °C over the most active Cr10Zr90Ox catalyst was two times higher than that over an analogue of commercial Cr–K/Al2O3. This catalyst also showed excellent stability in a series of 30 dehydrogenation–regeneration cycles at 550 °C and 600 °C without any changes in its initial performance from cycle to cycle.
Binary ZnZrOx materials are also active and selective for isobutene production.117 It was found that promoting ZrO2 with ZnO reduces surface acidity of ZrO2 and generates new Lewis acid–base pairs (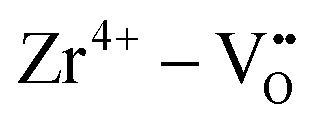 (
(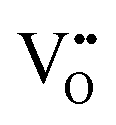 stands for oxygen vacancy), and Zn2+–O2−), which could efficiently hinder side reactions. In agreement with previous studies of BDH98,116 and PDH26,93 over ZrO2-based catalysts, coordinatively unsaturated Zr4+ sites were also suggested to be the active sites on the surface of ZnZrOx. Isobutene selectivity of about 95% at 32.5% isobutane conversion was obtained at 580 °C.
stands for oxygen vacancy), and Zn2+–O2−), which could efficiently hinder side reactions. In agreement with previous studies of BDH98,116 and PDH26,93 over ZrO2-based catalysts, coordinatively unsaturated Zr4+ sites were also suggested to be the active sites on the surface of ZnZrOx. Isobutene selectivity of about 95% at 32.5% isobutane conversion was obtained at 580 °C.
3.3 Benchmarking and demonstrating progress in the development of BDH catalysts
Similar to the PDH reaction (Section 2.4), we compare the most efficient catalysts developed for the BDH reaction in the last 5 years with each other and with those described in a review of Weckhuysen and co-workers in 2014.18 The catalysts are compared in terms of their productivity (space time yield of isobutene formation; STY(isobutene)) and the selectivity to isobutene at industrially relevant degrees of isobutane conversion. If the STY(isobutene) values were not provided in literature, we tried to calculate them when the necessary data (contact time, GHSV or WHSH, feed composition, isobutane conversion and isobutene selectivity) were given. Only studies using reaction feeds with isobutane content of at least 20 vol% were considered. The space time yield of isobutene formation STY(iso-C4H8) at isobutane conversion of at least 20% is shown in Fig. 16(a).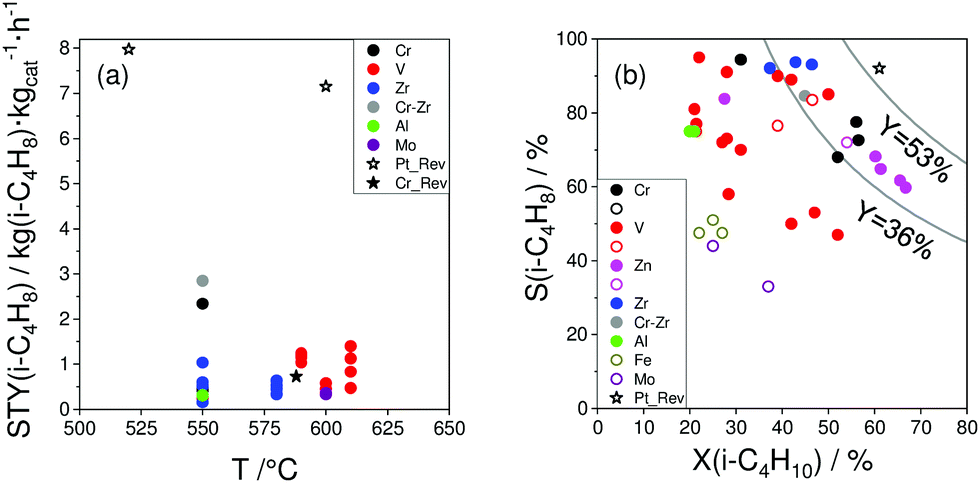 | ||
| Fig. 16 (a) Space time yield of isobutene formation (STY(i-C4H8)) obtained over selected catalysts reported within the last 5 years (circles) and over selected best-performing catalysts from ref. 18 (stars); (b) selectivity to isobutene (S(i-C4H8)) at certain degrees of isobutane conversion (X(C4H10)) at 550 °C (closed symbols) and 600 °C (open symbols) for selected catalysts. Requirements for the selected catalysts: reaction mixture with at least 20 vol% isobutane. Its conversion was at least 20%. Catalyst composition and reaction conditions are given in Table 4. | ||
Two Pt-containing and one Cr-containing catalysts from the previous review18 fulfilled the above-mentioned criteria, whereas 31 catalysts have been developed since 2016 (Fig. 16(a) and Table 4). Among them, there are four Cr-containing, eight ZrO2-based, one binary CrZrOx, fourteen V-containing, two bare Al2O3, and two Mo-containing catalysts. Noticeably, the Pt-based catalysts from ref. 18 outperform the developed metal oxide catalysts. The second most active catalyst is Cr10Zr90Ox. The corresponding STY(isobutene) value is (2.85 kg(isobutene) kgcat−1 h−1) at 550 °C. The productivity of ZrO2-based catalysts (not containing CrOx) at 550 °C only slightly exceeds 1 kg(isobutene) kgcat−1 h−1. VOx-based catalysts can achieve high STY(isobutene) values (up to 1.5 kg(i-C4H8) kgcat−1 h−1) at 610 °C. Other catalysts are less productive (lower than 1 kg(i-C4H8) kgcat−1 h−1).
| Catalyst | T/°C | Feed composition | WHSV of (i-C4H10)/h−1 | X(i-C4H10)/% | S(i-C4H8)/% | Y(i-C4H8)/% | STY (i-C4H8)/kg(i-C4H8) kgcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| Cr–K/Al2O3 | 550 | 40 vol% i-C4H10–60 vol% N2 | 6.3 | 31.0 | 94.4 | 29.3 | 2.34 | 98 |
| C–Cr/γ-Al2O3 | 550 | 100 vol% i-C4H10 | 52.0 | 68.0 | 35.4 | 0.42 | 123 | |
| Cr/γ-Al2O3 | 550 | 100 vol% i-C4H10 | 56.5 | 72.6 | 41.0 | 0.49 | 123 | |
| 2Ca–Cr/γ-Al2O3 | 550 | 100 vol% i-C4H10 | 56.0 | 77.5 | 43.4 | 0.52 | 123 | |
| V5–K2O/γ-Al2O3 | 590 | 50 vol% i-C4H10–50 vol% H2 | 54.0 | 68.2 | 36.8 | 1.04 | 106 | |
| V10–K2O/γ-Al2O3 | 590 | 50 vol% i-C4H10–50 vol% H2 | 62.5 | 68.0 | 42.5 | 1.20 | 106 | |
| V15–K2O/γ-Al2O3 | 590 | 50 vol% i-C4H10–50 vol% H2 | 62.9 | 68.9 | 43.3 | 1.22 | 106 | |
| V20–K2O/γ-Al2O3 | 590 | 50 vol% i-C4H10–50 vol% H2 | 59.4 | 74.3 | 44.1 | 1.24 | 106 | |
| VOx–K2O/γ-Al2O3 | 590 | 50 vol% i-C4H10–50 vol% H2 | 55.0 | 74.3 | 40.9 | 1.15 | 135 | |
| VOx/Al2O3 | 550 | 40 vol% i-C4H10–60 vol% N2 | 22.0 | 95.0 | 20.9 | 129 | ||
| VOx/Al2O3 | 550 | 40 vol% i-C4H10–60 vol% N2 | 28.0 | 91.0 | 25.5 | 0.31 | 129 | |
| VOx/Al2O3 | 550 | 40 vol% i-C4H10–60 vol% N2 | 39.0 | 90.0 | 35.1 | 129 | ||
| VOx/Al2O3 | 550 | 40 vol% i-C4H10–60 vol% N2 | 42.0 | 89.0 | 37.4 | 129 | ||
| VOx/Al2O3 | 550 | 40 vol% i-C4H10–60 vol% N2 | 50.0 | 85.0 | 42.5 | 129 | ||
| VOx/Si | 550 | 40 vol% i-C4H10–60 vol% N2 | 28.0 | 73.0 | 20.4 | 129 | ||
| VOx/Si | 550 | 40 vol% i-C4H10–60 vol% N2 | 47.0 | 53.0 | 24.9 | 129 | ||
| VOx/Si | 550 | 40 vol% i-C4H10–60 vol% N2 | 52.0 | 47.0 | 24.4 | 0.29 | 129 | |
| VOx/S10 | 550 | 40 vol% i-C4H10–60 vol% N2 | 27.0 | 72.0 | 19.4 | 129 | ||
| VOx/S10 | 550 | 40 vol% i-C4H10–60 vol% N2 | 31.0 | 70.0 | 21.7 | 129 | ||
| VOx/S10 | 550 | 40 vol% i-C4H10–60 vol% N2 | 42.0 | 50.0 | 21.0 | 0.25 | 129 | |
| 0.3V/MCM | 550 | 40 vol% i-C4H10–60 vol% N2 | 28.4 | 58.0 | 16.5 | 36 | ||
| 1.4V/MCM | 550 | 40 vol% i-C4H10–60 vol% N2 | 21.3 | 75.0 | 16.0 | 36 | ||
| 2.4V/MCM | 550 | 40 vol% i-C4H10–60 vol% N2 | 21.4 | 77.0 | 16.5 | 36 | ||
| 4.1V/MCM | 550 | 40 vol% i-C4H10–60 vol% N2 | 21.0 | 81.0 | 17.0 | 36 | ||
| V-1.5K/γ-Al2O3 | 610 | 50 vol% i-C4H10–50 vol% H2 | 40.1 | 73.8 | 29.6 | 0.83 | 109 | |
| V-1.5K-S/γ-Al2O3 | 610 | 50 vol% i-C4H10–50 vol% H2 | 56.8 | 87.7 | 49.8 | 1.4 | 109 | |
| V-3K/γ-Al2O3 | 610 | 50 vol% i-C4H10–50 vol% H2 | 27.0 | 62.0 | 16.7 | 0.47 | 109 | |
| V-3K-S/γ-Al2O3 | 610 | 50 vol% i-C4H10–50 vol% H2 | 50.9 | 78.5 | 40.0 | 1.12 | 109 | |
| V-0B-MCM-41 | 600 | 20 vol% i-C4H10–80 vol% N2 | 39.0 | 76.5 | 29.8 | 0.45 | 110 | |
| V-1.5B-MCM-41 | 600 | 20 vol% i-C4H10–80 vol% N2 | 46.5 | 83.5 | 38.8 | 0.58 | 110 | |
| ZnZrO-1 | 580 | 50 vol% i-C4H10–50 vol% H2 | 1.8 | 33.0 | 95.5 | 32.0 | 0.53 | 117 |
| ZnZrO-5 | 580 | 50 vol% i-C4H10–50 vol% H2 | 1.8 | 39.0 | 96.6 | 38.0 | 0.64 | 117 |
| ZnZrO-9 | 580 | 50 vol% i-C4H10–50 vol% H2 | 1.8 | 28.0 | 96.0 | 27.0 | 0.45 | 117 |
| Zn1/S-1 | 550 | 50 vol% i-C4H10–50 vol% N2 | 27.5 | 83.8 | 23.0 | 130 | ||
| Zn3/S-1 | 550 | 50 vol% i-C4H10–50 vol% N2 | 60.2 | 68.2 | 41.0 | 130 | ||
| Zn6/S-1 | 550 | 50 vol% i-C4H10–50 vol% N2 | 61.3 | 64.8 | 40.0 | 130 | ||
| Zn8/S-1 | 550 | 50 vol% i-C4H10–50 vol% N2 | 66.7 | 59.8 | 40.0 | 130 | ||
| Zn12/S-1 | 550 | 50 vol% i-C4H10–50 vol% N2 | 65.5 | 61.7 | 40.0 | 130 | ||
| 10Zn/Al2O3 | 600 | 100 vol% i-C4H10 | 54.0 | 72.0 | 38.0 | 131 | ||
| 5% Fe-SBA-15 | 600 | 100 vol% i-C4H10 | 22.0 | 47.5 | 10.0 | 120 | ||
| 10% Fe-SBA-15 | 600 | 100 vol% i-C4H10 | 27.0 | 47.5 | 13.0 | 120 | ||
| 15% Fe-SBA-15 | 600 | 100 vol% i-C4H10 | 25.0 | 51.0 | 13.0 | 120 | ||
| MoAl(C) | 600 | 100 vol% i-C4H10 | 25.0 | 44.0 | 11.0 | 0.33 | 115 | |
| MoAl | 600 | 100 vol% i-C4H10 | 37.0 | 33.0 | 12.2 | 0.37 | 115 | |
| ZrO2 | 550 | 40 vol% i-C4H10–60 vol% N2 | 0.8 | 21.0 | 75.0 | 15.8 | 0.16 | 98 |
| La8Zr92Ox | 550 | 40 vol% i-C4H10–60 vol% N2 | 1.2 | 42.9 | 93.7 | 40.2 | 0.6 | 98 |
| Y9Zr91Ox | 550 | 40 vol% i-C4H10–60 vol% N2 | 1.9 | 46.4 | 93.1 | 43.2 | 1.04 | 98 |
| Sm10Zr90Ox | 550 | 40 vol% i-C4H10–60 vol% N2 | 1.2 | 37.3 | 92.1 | 34.4 | 0.52 | 98 |
| ZrO2 | 580 | 50 vol% i-C4H10–50 vol% H2 | 1.8 | 21 | 95.0 | 20.0 | 0.34 | 117 |
| Cr10Zr90Ox | 550 | 40 vol% i-C4H10–60 vol% N2 | 5.9 | 44.9 | 84.6 | 38.0 | 2.85 | 116 |
| Al2O3, 10 h_600 °C H2/N2 | 550 | 40 vol% i-C4H10–60 vol% N2 | 20.0 | 75.0 | 15.0 | 0.3 | 134 | |
| Al2O3, 5 h_700 °C N2 | 550 | 40 vol% i-C4H10–60 vol% N2 | 21.0 | 75.0 | 16.0 | 0.32 | 134 | |
| 2 wt% Pt–1 wt% Sn/CeO2/C | 520 | 33 vol% i-C4H10–7 vol%H2–60 vol% He | 24.8 | 37.0 | 90.0 | 33.3 | 7.97 | 18 |
| 0.58 wt% Pt–Sn/K–L | 600 | 33 vol% i-C4H10–67 vol%H2 | 13.2 | 61.0 | 92.0 | 56.1 | 7.15 | 18 |
| 40% Cr2O3/60% Al2O3 | 588 | 100 vol% i-C4H10 | 3.3 | 23.0 | 99.4 | 22.9 | 0.73 | 18 |
The selectivity to isobutene over selected catalysts tested at 550 °C or 600 °C is compared in Fig. 16(b). The black lines in this figure stand for the yield of isobutene of 36 and 53%. Noticeably, apart from the catalysts based on MoOx, FeOx or Al2O3, their counterparts with catalytically active VOx, ZnOx species or bulk ZrO2-based catalysts can ensure the selectivity to isobutene above 80% at degrees of isobutane conversion higher than 20%. Moreover, industrially relevant isobutene yields were achieved over these catalysts.
4 Coke formation in PDH and BDH and characterization
In addition to activity and product selectivity, catalyst on-stream stability is another important property relevant for industrial applications. There are several origins of deactivation of dehydrogenation catalysts (Fig. 17).92,105,136 Some of them are more specific and observed only for a certain kind of catalysts. For example, the reduction of ZnO into metallic Zn followed by its volatilization is a typical reason for the deactivation of ZnO-based catalysts.80 The others are more general and can be observed for the most of the catalysts. One of such phenomena typical for all dehydrogenation catalysts including those applied on large scale is coke formation. Against this background, the purposes of the below section are to discuss mechanistic aspects of coke formation and methods for monitoring coke formation and characterising this undesired product. | ||
| Fig. 17 The most common reasons for deactivation of alkane dehydrogenation catalysts. | ||
4.1 Mechanistic concepts on coke formation
Regardless of the kind of alkane dehydrogenation catalysts, coke deposits can be formed directly from the feed alkane and through consecutive transformations of the desired olefins. The latter reactions seem to dominate. Apart some general considerations/assumptions concerning coke formation, detailed mechanistic aspects of coke formation are scarcely studied on metal-oxide catalysts unlike their Pt-based counterparts. It is supposed that coke species sequentially transform from aliphatic into aromatic, and finally into graphite-like deposits with proceeding DH reaction.135,137 It is also assumed that surface acidic sites contribute to coke formation.18 Only a few detailed studies dealing with elucidation of the mechanism of coke formation over metal-oxide catalysts have been reported up to now. They analysed supported catalysts with active VOx species129,135,138 or bulk ZrO2-based catalysts.43,69,97,98In contrast to the generally accepted concept relating coke formation to catalyst acidity, no correlation between the concentration of acidic sites determined through NH3-TPD and the amount of coke formed (coke selectivity) could be established in the PDH reaction over VOx/SiO2–Al2O3,138 ZnOx/MZrOx43 and CrZrOx/SiO2.69 For the VOx/SiO2–Al2O3 catalysts, VOx species were found to be more active for the formation of coke than acidic sites of support. The rate of coke formation determined from in situ TGA tests and the rate of catalyst deactivation in PDH correlate with the edge energies of supported VOx species determined from UV-vis spectra.138 On this basis, it was concluded that the degree of polymerisation of VOx species affects coke formation. The effect of the VOx distribution was explained as follows. To form aromatic structures followed by their further oligomerization and condensation to large graphitic structures, adsorbed propene molecules should be near to each other. Such situation is difficult to realize for isolated VOx sites. Upon increasing the size of VOx species, a probability of interaction between several adsorbed propene molecules increases resulting in a higher rate of carbon deposition.
A similar concept was very recently suggested for ZnOx/MZrOx43 and CrZrOx/SiO2,69 where coke formation takes place over supported ZnOx and CrOx species respectively. Those authors used time-resolved operando UV-vis spectroscopy for analysing coke formation. The degree of polymerisation of ZnOx species at same Zn loading can be tuned through the kind of metal oxide dopant for ZrO2 in MZrOx. Cr content in CrZrOx/SiO2 determines CrOx distribution.
It should be especially mentioned that in the case of isobutane dehydrogenation, coke species are formed via multiple mechanisms, e.g. directly from isobutene on the same sites as for the BDH reaction, or through isomerization of isobutene into n-butenes followed by dehydrogenation of the latter to butadiene, which undergoes oligomerization and cyclization reactions.129,135 The contribution of different pathways to coke formation is mostly influenced by the nature of support material. Since skeletal isomerization of isobutene is catalysed by Brønsted acidic sites, the supports possessing such sites provoke coke formation through opening the route through n-butenes/butadiene.
Concerning PDH over bulk ZrO2-based catalysts, several factors were identified to influence coke formation: (i) surface acidity;98 (ii) phase composition and crystallite size of ZrO2;26,96 (iii) reducibility of ZrO2.96,97 In general, coke can originate directly from propane or via reactions with participation of propene. The contribution of each pathway depends on phase composition and crystallite size of ZrO2.96 DFT calculations predict that desorption of propene formed from propane is easier from t-ZrO2 than from m-ZrO2. Longer residence time of propene on catalyst surface can lead to its transformation into coke. This might be a reason that propane-aided pathway predominates over m-ZrO2. It should be noted that there are at least two types of active sites on the surface of ZrO2 which are able to adsorb and convert propane: (i) Zrcus located at steps, kinks or corners of the lattice of ZrO2 and (ii) regular surface zirconium cations. The former sites are responsible for the transformation of propane into propene and in a lesser degree into coke, while the latter sites produce coke. Accordingly, increasing the ratio between Zrcus and regular sites is beneficial for decreasing coke selectivity. The concentration of regular sites and therefore the probability of interaction of adsorbed propene molecules with each other leading to coke formation can be decreased by decreasing the size of ZrO2 crystallites. Another possible way for decreasing the concentration of regular sites is the removal of lattice oxygen from ZrO2 during reductive treatment. Such idea is supported by the fact that coke formation is suppressed with increasing reducibility of ZrO2.97
4.2 Characterization of coke species
Since deposition of coke is the main reason for catalyst deactivation in PDH and BDH, it is highly important to understand factors affecting coke formation with the purpose to develop catalysts with suppressed ability to form coke. To help researchers aiming to elucidate coke formation, now we discuss methods suitable for characterising coke formation. Their specific characteristics are briefly introduced. There are a lot of methods which can be applied for characterization of coke (Fig. 18). They can be divided into two groups: those used directly under experimental conditions (in situ/operando) and those applied to characterize spent catalysts after the reaction (ex situ).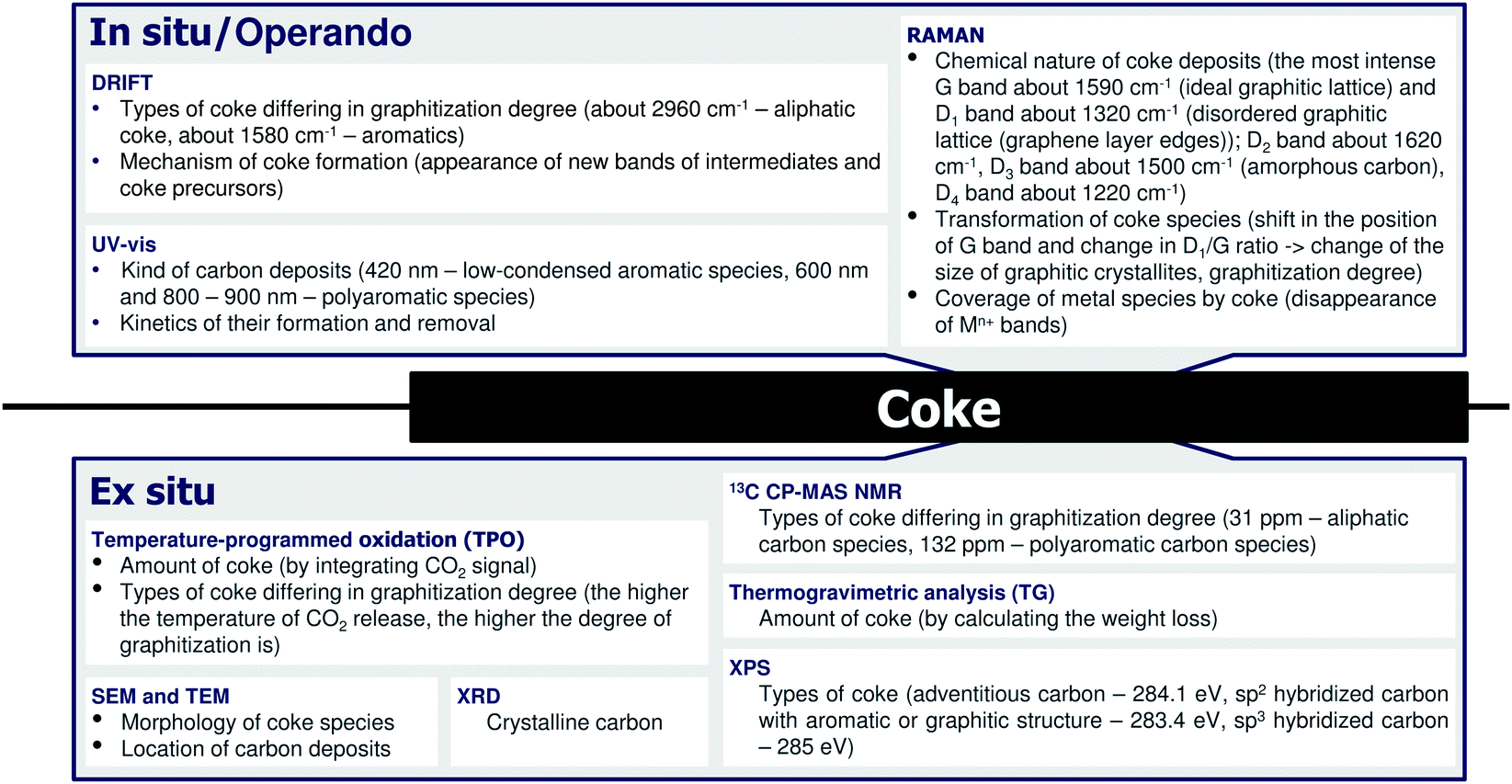 | ||
| Fig. 18 In situ/operando and ex situ methods for coke characterization. | ||
Raman spectroscopy has been widely used to study the chemical nature of coke species formed on catalyst surface.139,140 Raman bands related to coke species at ∼1325 cm−1 and ∼1590 cm−1 are assigned to D (disorder-allowed vibration modes) and G (E2g optical mode of graphitic carbon) bands respectively. The D band is usually deconvoluted into 4 peaks: D1 band (∼1320 cm−1) related to ring vibrations of defects and edges of graphitic lattice and attributed to the in-plane imperfection such as defects and heteroatoms. The D2 band (∼1620 cm−1) is assigned to disordered graphitic lattice vibration mode with E2g symmetry. The D3 band (∼1500 cm−1) corresponds to amorphous carbon, while the D4 band (∼1220 cm−1) is characteristic for the disordered graphitic lattice of an A1g symmetry.135,139 The ratio of ID1/IG intensities is assumed to indicate the degree of coke graphitization. An increase in the ID1/IG ratio implies either increasing the number of defects in the graphitic lattice or decreasing the size of graphitic crystallites. It has also been reported that the size of graphitic crystallites influences the position of G band. Thus, with decreasing the size of crystallites, G band shifts to higher wavenumber.139,141
DRIFT spectroscopic analysis of adsorbed propene at different temperatures allows to investigate the mechanism of coke formation from propene since different coke precursors will be observed at different temperatures. It is generally assumed that during PDH reaction aliphatic structures of coke (band at ∼2960 cm−1) are firstly formed via deep dehydrogenation reactions and then gradually transform into aromatic structures.141
UV-vis spectroscopy can be applied for analysing the kinds of coke species and the kinetics of their formation and removal during PDH reaction and regeneration stages, respectively. It is supposed that UV-vis bands with maxima at 600 nm and 800–900 nm are related to polyaromatic species, while the band with the maximum at 420 nm can be assigned to low-condensed aromatic species.70 The kinetics of coke formation/removal can be evaluated by analysing temporal changes of Kubelka–Munk function at certain wavelengths.
In situ/operando methods provide qualitative information about coke formation: composition of coke precursors (DRIFT spectroscopy), kinetics of coke formation (Raman and UV-vis spectroscopy) and graphitization degree (Raman and UV-vis spectroscopy). Such methods alone however cannot give quantitative information. To this end, ex situ analysis of spent catalysts is required.
Solid-state 13C NMR allows to distinguish different carbon species. A resonance centred at around 31 ppm is generally assigned to aliphatic carbon species related to –CH2 groups, while a peak at around 132 ppm is related to polyaromatic carbonaceous species (Fig. 19).135
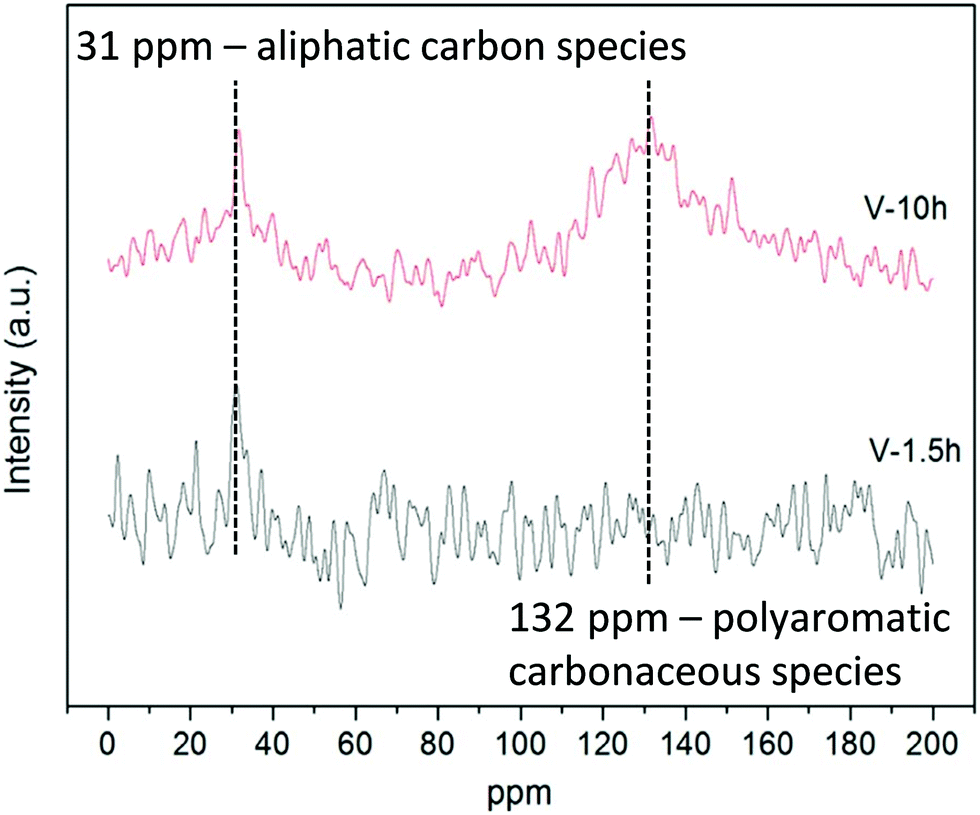 | ||
| Fig. 19 Solid-state 13C NMR spectra of VOx–K2O/Al2O3 catalyst tested in isobutane dehydrogenation after different times on stream. Reproduced from ref. 135 with permission from Elsevier, copyright 2020. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) can be used to investigate the morphology of coke species and their location.
Temperature-programmed oxidation (TPO) is a common method for quantitative analysis of coke deposits. The amount of carbon oxides (COx) released during TPO experiment represents the amount of carbon deposits. The TPO profile can also give information about the kind of carbon species. The temperature of the maximum of COx release gives information about graphitization degree of coke deposits. Thus, the higher the temperature, the higher the graphitization degree is. When H2O is also measured upon TPO, the ratio of H/C can be determined from the concentration of released COx and H2O. The ratio is an indicator for the kind of carbon deposits, i.e. olefinic, aromatic or graphitic. However, precautions must be considered when measuring COx and particularly H2O. When spent catalysts are characterized after their exposure to air and storage before starting TPO, adsorbed water and carbon dioxide from the air might worsen the correct quantification of carbon and hydrogen in surface carbon-containing deposits. Thus, the best way is to perform TPO tests in the same set-up used for alkane dehydrogenation without catalyst exposure to air.
The amount of coke can also be determined by thermogravimetric analysis (TG). Such method implies the registration of changes in catalyst weight during combustion of carbon deposits.
X-ray photoelectron spectroscopy (XPS) is typically used for characterizing and quantifying surface chemical composition of spent catalysts. Thus, carbon deposits can also be analysed. Typically, the C 1s signal at 283.4 eV is related to sp2 hybridized carbon, while such signal at around 285 eV is attributed to sp3 hybridized carbon.135 The signal at around 284 eV is assigned to adventitious carbon.
X-ray diffraction (XRD) method can give information about the existence of crystalline graphitic carbon.
5 Oxidative propane dehydrogenation
Since the beginning of nineties of the last century, the oxidative dehydrogenation of propane to propene (ODP) has been intensively investigated to establish a cost-efficient and environmentally friendly alternative to the commercially applied cracking processes and the non-oxidative dehydrogenation of propane. Despite the enormous efforts made by various research groups, the ODP reaction is still not commercialized due to too low selectivity to propene at industrially relevant degrees of propane conversion. The reason is higher reactivity of the desired olefin in comparison with the feed alkane. Thus, this reaction is also intriguing from a fundamental viewpoint, i.e. selective activation of stronger C–H bond in C3H8 with suppressed ability for the undesired cleavage of weaker C–C bond. Below, we briefly analyse the progress in catalyst development with respect to fulfilling industrially relevant requirements (Section 5.1) and summarize fundamentals upon which the scientific community has agreed (Section 5.2).5.1 Is there any progress in catalyst development since 2008?
Based on the results of our search for the relevant literature from 2008 on, about 1300 different catalysts have been synthesized and tested for their activity and selectivity in the ODP reaction up to now (Fig. 20(a)). The catalysts are divided into various groups with respect to the nature of active component. A major part (in total about 700) of these catalysts contain vanadium as the active component (Fig. 20(b)). Other relatively often investigated catalysts comprise B, Co, Cr, Fe or Ni. To check if any progress in catalyst development has been achieved, we proceeded as follows. The selectivity–conversion data for different V-containing or V-free catalysts from ref. 10 were used as the state-of-the-art performance reported before 2007. These data highlighted by black circles are given in Fig. 20(c and d). This figure also shows the performance of commercial PDH technologies, which is the target for the ODP reaction. The grey lines stand for the yield of propene of 35 and 53% typical for these technologies.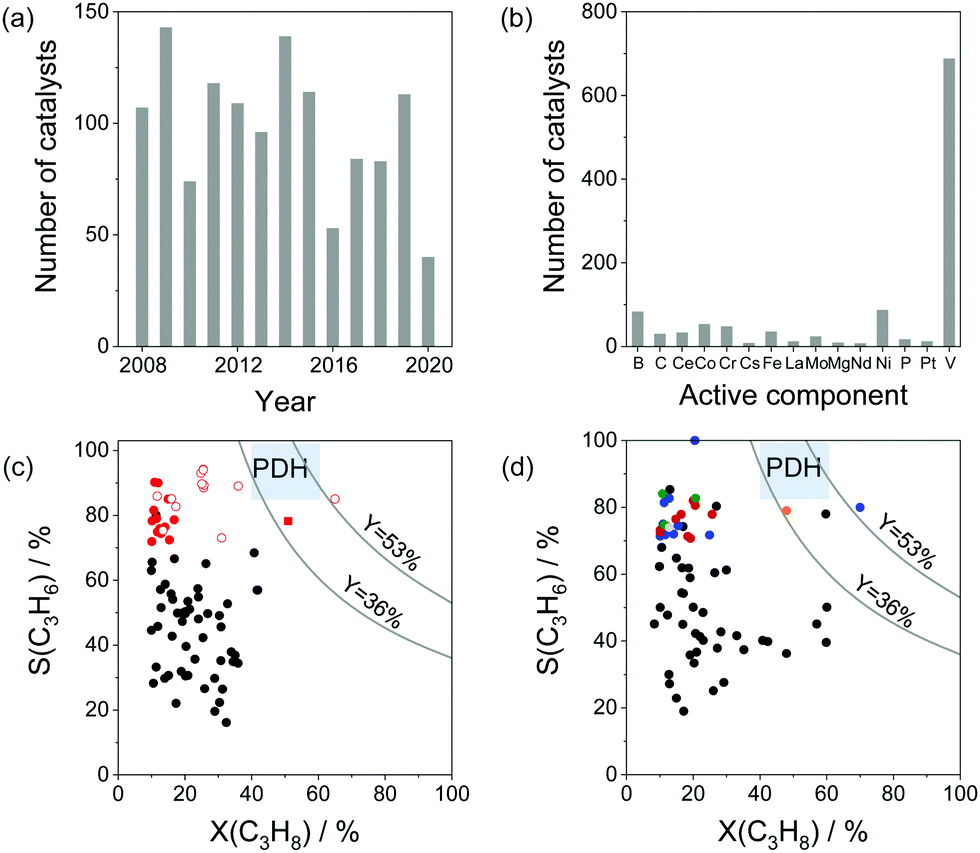 | ||
Fig. 20 (a) Annual number of catalysts tested and reported in literature from 2008 up to August 2020, (b) overall number of catalysts developed since 2008 with a certain active component. Selectivity–conversion plots for propene over (c) V-based or (d) V-free catalysts tested under different reaction conditions. Black circles in (c) and (d) represent the catalysts from ref. 10. Closed and open red symbols in (c) stand for the catalysts developed after 2007 with propene selectivity above 70% at propane conversion above 10% using C3H8/O2 = 2–0.5 ( ) or C3H8/O2 > 10 ( ) or C3H8/O2 > 10 ( ) feeds or upon alternating feeding of C3H8 and air ( ) feeds or upon alternating feeding of C3H8 and air ( ). Coloured circles in (d) distinguish catalysts with different active components: NiOx ( ). Coloured circles in (d) distinguish catalysts with different active components: NiOx ( ), B ( ), B ( ), CrOx ( ), CrOx ( ), Pt ( ), Pt ( ) or C ( ) or C ( ). Catalyst composition and reaction condition for the datapoints after 2007 are given in Tables S1 and S2 (ESI†) with experimental details related to tests with V-containing and V-free catalysts in (c) and (d) respectively. Experimental details of tests with alternating feeds of propane and air in (c) are given in Table S3 (ESI†). ). Catalyst composition and reaction condition for the datapoints after 2007 are given in Tables S1 and S2 (ESI†) with experimental details related to tests with V-containing and V-free catalysts in (c) and (d) respectively. Experimental details of tests with alternating feeds of propane and air in (c) are given in Table S3 (ESI†). | ||
To demonstrate the developments after 2007, we decided to apply a certain criterion for selecting promising catalysts. If we showed all the selectivity–conversion datapoints reported in this period of time, Fig. 20(c and d) would be reader unfriendly. Thus, the catalysts with propene selectivity above 70% at propane conversion larger than 10% have been selected and are shown as filled not-black circles in Fig. 20(c and d). Only a few catalysts among about 700 V-containing catalysts reported in literature75,142–149 fulfilled this criterion. Moreover, none of them revealed the target propene selectivity at propane conversion above 20% (Fig. 20(c)). Thus, since 2008 no significant progress in the development of V-containing catalysts with industrially relevant performance has been achieved when performing the ODP reaction with stoichiometric O2–C3H8 feeds. Reaction engineering aspects seem to be promising for improving catalyst performance. As seen in Fig. 20(c), propene selectivity of around 80% at propane conversion of above 50% can be achieved when operating under O2-lean (strong excess of C3H8 over O2)150 or O2-free (chemical looping or redox operation)151–156 conditions. The developments in these fields are discussed in Sections 7.3 or 7.4.
In comparison with V-containing catalysts, some improvements in the performance of V-free catalysts have been achieved (Fig. 20(d)). For example, Xie et al.157 reported propene selectivity of 80% at propane conversion of about 70% over a NiO-modified CeO2 catalyst at 500 °C. The latter oxide has nanorod morphology. A stable operation over about 100 hours on stream was reported. It should, however, be mentioned that such high performance was achieved when using a reaction feed containing HCl. No propene was formed in the absence of HCl. This additive affected not only the selectivity to propene but also increased propane conversion. Based on the results of catalyst characterization and DFT calculations, Cl˙-like species were suggested to be responsible for the selective conversion of propane to propene. They may be formed upon reaction of HCl with surface O22− species generated upon activation of gas-phase oxygen on an anion vacancy. From an application viewpoint, special requirements with respect to reactor material, safety operation and environmental aspects should be fulfilled when operating with HCl.
Another catalytic system to be mentioned is PtSn/SiO2.158 The support is a dealuminated beta zeolite. The highest propene selectivity of 79% at propane conversion of 48% was achieved at 550 °C. The catalyst slowly deactivated with time on propane stream due to conversion of the bimetallic PtSn species into the individual components. Boron-containing catalysts introduced a few years ago also show high propene selectivity above 70% but at degrees of propane conversion not higher than 25%.159–166 Other catalysts in (Fig. 20(d)), which fulfil the above defined criterion for propene selectivity, contain Ni,157,167–172 Cr,173,174 Mn,175 or C176 as active components.
We also compared catalysts in terms of their productivity, i.e. space time yield of propene STY(C3H6) formation (kgC3H6 kgcatalyst h−1). The STY(C3H6) values of the recently developed catalysts from Fig. 20(c and d) are shown in Fig. 21 together with such values obtained over the catalysts developed before 2007 from ref. 10. For brevity, the latter materials with STY values larger than 0.5 kgC3H6 kgcatalyst h−1 were used. Like the selectivity–conversion relationship, no improvements in the productivity of V-containing catalysts have been achieved after 2007. Contrarily, some progress could be identified for V-free catalysts. Boron-based catalysts should be especially mentioned. The very high activity reported for a NiO-modified CeO2157 is probably related to the presence of HCl.
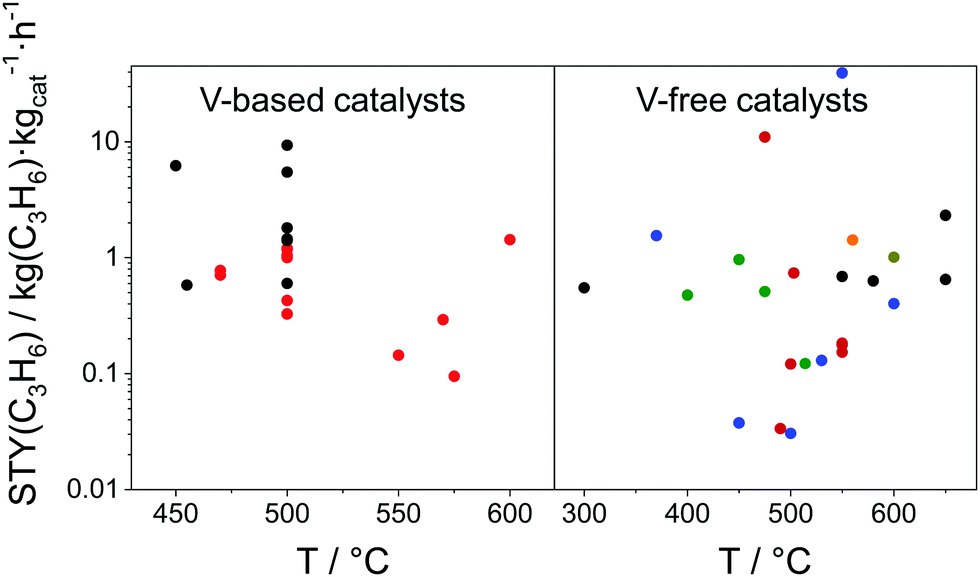 | ||
| Fig. 21 Space time yield of propene formation (STY(C3H6)) determined over different catalysts developed before 2007 (black) and after 2007 (coloured symbols). The black datapoints are from ref. 10. The coloured symbols stand for the catalysts in Fig. 20(c and d). Catalyst composition and reaction conditions for the datapoints after 2007 are given in Tables S1 and S2 (ESI†) for V-containing and V-free catalysts, respectively. | ||
The above discussion can be summarized as follows. The recently introduced boron-based catalysts demonstrate the potential of developing materials with uncommon active components for improving propene selectivity. Despite a great number of studies dealing with V-based catalysts, no evident improvements in propene selectivity at industrially relevant degrees of propane conversion could be achieved through catalyst design. This statement also arises a question, whether the desired progress can be realized. Probably, we need deeper understanding of the relationships between catalyst physicochemical properties including the structure of VOx species and the kinetics of selective and unselective reaction pathways. The state-of-the-art knowledge about the fundamentals of the ODP reaction over catalysts based on metal oxides is, therefore, described and discussed in the next section.
5.2 Fundamental aspects of ODP
In general, it is well accepted that the ODP reaction over V-based catalysts occurs through a Mars–van Krevelen redox mechanism involving lattice oxygen of VOx species for activating two C–H bonds in propane to yield propene. In a second step, the reduced VOx species is reoxidized by gas-phase oxygen or another oxidant. Lattice oxygen is also involved into consecutive conversion of the desired olefin to carbon oxides. These mechanistic considerations should also be valid for other catalysts based on reducible metal oxides. In addition to lattice oxygen, adsorbed oxygen species are also assumed to contribute to the undesired side reactions.177,178 The extent of reduction of vanadium under ODP conditions is another selectivity-governing factor.179–182 VOx dispersion and the kind of support are also considered to be decisive descriptors for catalyst activity and selectivity as supported by the below analysis.As the highest number of differently supported and loaded catalysts with VOx,183–190 ZnOx,191 B,192 CoOx193 or FeOx194 were tested at 450 °C, we plotted the corresponding TOF values in Fig. 22 as a function of apparent surface density of the active component. Due to the high activity of CrOx-174 or NiOx-195 containing catalysts, it was not possible to calculate the TOF values at 450 °C. The lowest possible temperature is 300 °C. Thus, the latter data are given in Fig. 22. Cr loading above 5 atoms nm−2 has a negative effect on the TOF values due to the presence of Cr2O3. For NiOx/CeO2, a continuous decrease in the intrinsic activity of NiOx species with rising Ni loading was established. The strength of the decrease is significantly stronger for these materials in comparison with CrOx/nanodiamonds.
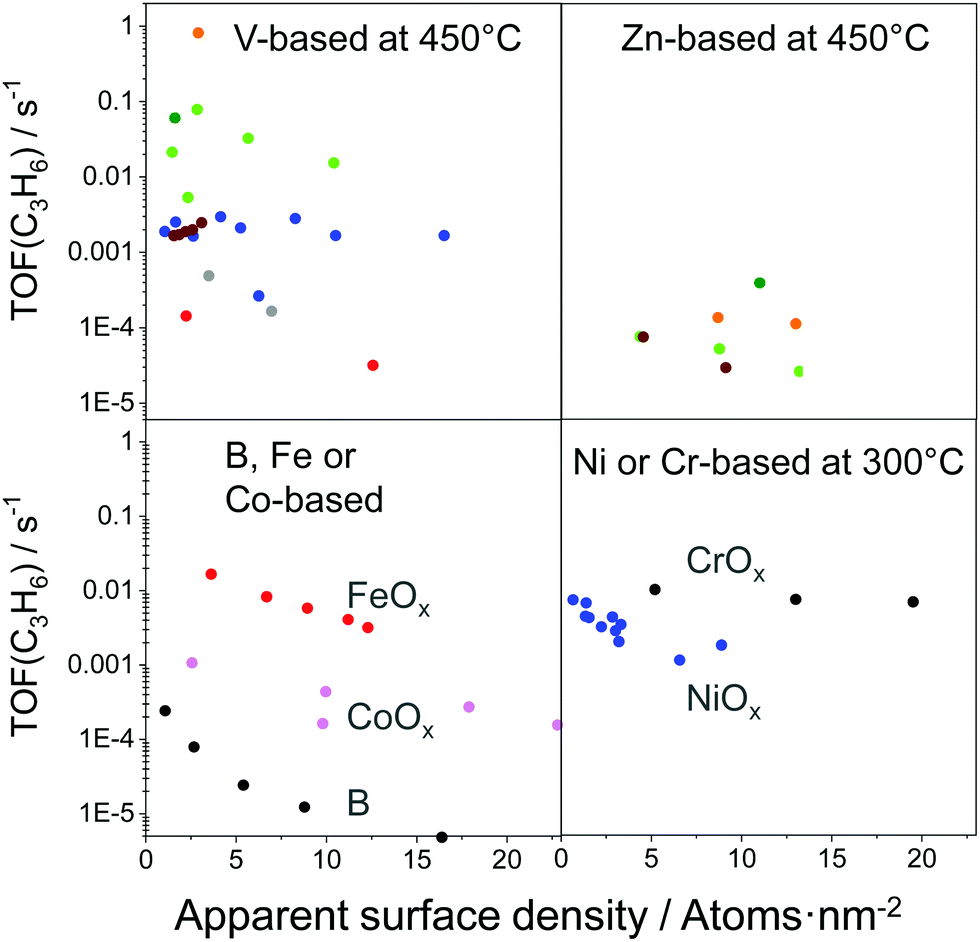 | ||
| Fig. 22 Turnover frequency of propene formation (TOF(C3H6)) versus apparent density of V,183–188,190,196 Zn,191 Fe,194 Co,193 B,192 Ni195 or Cr174 in different catalysts. The colours distinguish different supports, which are Al2O3 (light green circles), carbon (black circles), Ca5[OH|(PO4)3] (red circles), CeO2 (blue circles), MgO (grey circles), MgAlOx (pink circles), SiO2 (brown circles), TiO2 (dark green circles) or ZrO2 (orange circles). The kind of active metal oxide and reaction temperature are given in each plot. Further details are provided in Table S4 (ESI†). | ||
The intrinsic activity of B, FeOx and CoOx also decreases with rising content of the active component (Fig. 22). Although a direct comparison is not fully justified due to different kinds of supports, FeOx species should show higher activity than CoOx and B. ZnOx species are intrinsically less active than FeOx and CoOx and their activity also decreases with rising Zn loading regardless the kind of supporting material. For achieving high activity, TiO2 and ZrO2 supports should be applied for catalyst preparation.
The V-related TOF values for catalysts based on Al2O3,183,184 calcium hydroxyapatites (Ca5[OH|(PO4)3]),185 CeO2,186,187 MgO,188 SiO2196 and TiO2190 supports are given in Fig. 22 as a function of V loading. The lowest TOF values were obtained over Ca5[OH|(PO4)3]-185 or MgO-supported188 catalysts. In agreement with a previous review on the kinetics of the ODP reaction,12 Al2O3, TiO2, and ZrO2 appear to be the most efficient supports in terms of intrinsic activity of supported VOx species.
No effect of V loading up to 10 Atom nm−2 on the TOF values of CeO2-supported catalysts could be identified. The datapoint for 6.5 V nm−2 was calculated from ref. 186, while other points are from ref. 187. As all the values follow same trend, the absence of any effect of V loading on the TOF values of VOx/CeO2 materials might be justified. This may be related to high dispersion of VOx on the surface of CeO2. Isolated VOx and polyvanadate species but no V2O5 were detected in VOx/CeO2 with 6.5 V nm−2.186 It can be assumed from Fig. 22 that the activity of MgO- or Ca5[OH|(PO4)3]-supported VOx species decreases with rising V loading, although there are only two loading datapoints for each catalyst have been reported. A negative effect of the loading may exist for VOx/Al2O3. The intrinsic activity of VOx species on the surface of SiO2 does not seem to depend on V loading below 2 V nm−2.
To further analyse the effect of the kind of support and V loading on TOF, we prepared Table 5 containing the values reported for VOx/SiO2 and VOx/SiTiOx catalysts at 500 °C. The catalysts contain less than 3.1 V nm−2 and should not possess three-dimensional VOx particles. Interestingly, although the VOx/SiO2 catalysts were tested at different partial pressures of propane (0.05–0.63 bar) and oxygen (0.05–0.33 bar), the TOF values did not significantly depend on the pressures. No obvious dependence on V loading could also be deduced. However, when comparing the TOF values additionally related to the partial pressures of propane and oxygen, a negative effect of the loading will be obtained. Such dependence may be explained as follows: (i) any direct comparison of TOF values obtained over differently prepared but similarly loaded VOx/SiO2 is not possible as they were prepared from different chemicals by different researchers as well as tested in different laboratories under different reaction conditions or (ii) isolated VOx species have higher intrinsic activity than their polymerized counterparts.
| Support | ω V/nm−2 | p(C3H8)/bar | p(O2)/bar | TOF(C3H6)/s−1 | S(C3H6)/% | Ref. |
|---|---|---|---|---|---|---|
| Si | 1.64 × 10−1 | 0.05 | 0.05 | 2.91 × 10−3 | 80.0 | 198 |
| Si | 2.89 × 10−1 | 0.05 | 0.05 | 1.92 × 10−3 | 89.0 | 198 |
| Si | 1.74 × 100 | 0.48 | 0.06 | 1.72 × 10−3 | 63.0 | 184 |
| Si | 2.35 × 100 | 0.10 | 0.10 | 2.74 × 10−3 | 88.2 | 147 |
| Si | 2.19 × 100 | 0.10 | 0.10 | 3.31 × 10−3 | 91.0 | 147 |
| Si | 1.90 × 100 | 0.10 | 0.10 | 4.33 × 10−3 | 89.3 | 147 |
| Si | 1.23 × 100 | 0.10 | 0.10 | 6.21 × 10−3 | 86.5 | 147 |
| Si | 9.90 × 10−1 | 0.10 | 0.10 | 5.85 × 10−3 | 91.9 | 147 |
| Si | 6.64 × 10−1 | 0.10 | 0.10 | 5.34 × 10−3 | 90.3 | 147 |
| Si | 1.55 × 100 | 0.67 | 0.33 | 8.57 × 10−3 | 80.0 | 196 |
| Si | 1.83 × 100 | 0.67 | 0.33 | 9.85 × 10−3 | 74.0 | 196 |
| Si | 2.19 × 100 | 0.67 | 0.33 | 1.04 × 10−2 | 72.0 | 196 |
| Si | 2.58 × 100 | 0.67 | 0.33 | 1.13 × 10−2 | 76.0 | 196 |
| Si | 3.08 × 100 | 0.67 | 0.33 | 9.14 × 10−3 | 58.0 | 196 |
| Si | 5.86 × 10−1 | 0.29 | 0.15 | 1.10 × 10−2 | 75.1 | 197 |
| SiTi(168) | 6.95 × 10−1 | 0.29 | 0.15 | 1.16 × 10−2 | 78.6 | 197 |
| SiTi(55) | 7.59 × 10−1 | 0.29 | 0.15 | 6.66 × 10−3 | 64.8 | 197 |
| SiTi(22) | 1.49 × 100 | 0.29 | 0.15 | 1.01 × 10−2 | 69.7 | 197 |
| SiTi(10) | 1.11 × 100 | 0.29 | 0.15 | 4.55 × 10−2 | 50.9 | 197 |
| SiTi(8) | 1.37 × 100 | 0.29 | 0.15 | 4.05 × 10−2 | 38.4 | 197 |
| SiTi(5.7) | 1.75 × 100 | 0.29 | 0.15 | 3.02 × 10−2 | 40.2 | 197 |
| SiTi(8.3) | 0.59 × 100 | 0.40 | 0.20 | 1.50 × 10−2 | 68.0 | 179 |
| SiTi(1.6) | 0.66 × 100 | 0.40 | 0.20 | 1.40 × 10−1 | 75.0 | 179 |
| SiTi(0.7) | 1.10 × 100 | 0.40 | 0.20 | 2.60 × 10−1 | 90.0 | 179 |
The effect of Si/Ti ratio in SiaTibO2 supports on the activity of VOx species was thoroughly investigated in two independent studies.179,197 As a general trend, the reported TOF values increased with a decrease in the ratio at a close V loading (Table 5). The positive effect is due to improved reducibility of VOx species through the presence of Ti.
We also tried to validate if reported positive effects of some dopants (supported metal oxides, additives or mixed phase) on the intrinsic activity of VOx species are of general character or were simply determined in single studies. To this end, we calculated average V-related TOF values for catalysts based on a certain support (Al2O3,144,183,184,199–203 SiO2,144,147,150,184,196,198,203–208 TiO2,184,199,209 ZrO2,209 SiZrOx210) with V density below 7 mn−2 and free of any promoter. The error bars were also determined as a standard deviation. So obtained TOF values are compared in Fig. 23 with the values reported for catalysts based on same support but containing a promoter for VOx. The following promoted catalytic systems have been previously investigated: Mg–VOx/Al2O3,201,202 P–VOx/Al2O3211 (vanadyl phosphate), F–VOx/SiO2,205 K–VOx/SiO2,212 Mo–VOx/SiO2,208 Nb–VOx/SiO2,213 Sb–VOx/SiO2,214 P–VOx/TiO2211 (vanadyl phosphate), P–VOx/ZrO2211 (vanadyl phosphate), Sb–VOx/SiZrO2(3.5 wt% SiO2).210 Although the uncertainty of such comparison is high, it can be concluded that all promoters probably with exception of Nb have rather negative effect on the intrinsic activity of VOx species. According to ref. 208 the negative effect of Mo on the V-related TOF values of Mo–VOx/SiO2 is related to the structure of VOx species. Addition of MoOx prevents the formation of V–O–V bond in favour of isolated VOx species. The latter species were concluded to be less active than their oligomerized counterparts. This statement agrees with the results in Fig. 22. An increase in the TOF values of VOx/SiO2 with rising V loading is seen in this figure. Interesting, such behaviour could not be identified for VOx on other supports and for other supported metal oxides and boron (Fig. 22). Further studies are, however, required for understanding if and how the intrinsic activity of supported metal oxide species depends on their structure and the kind of support.
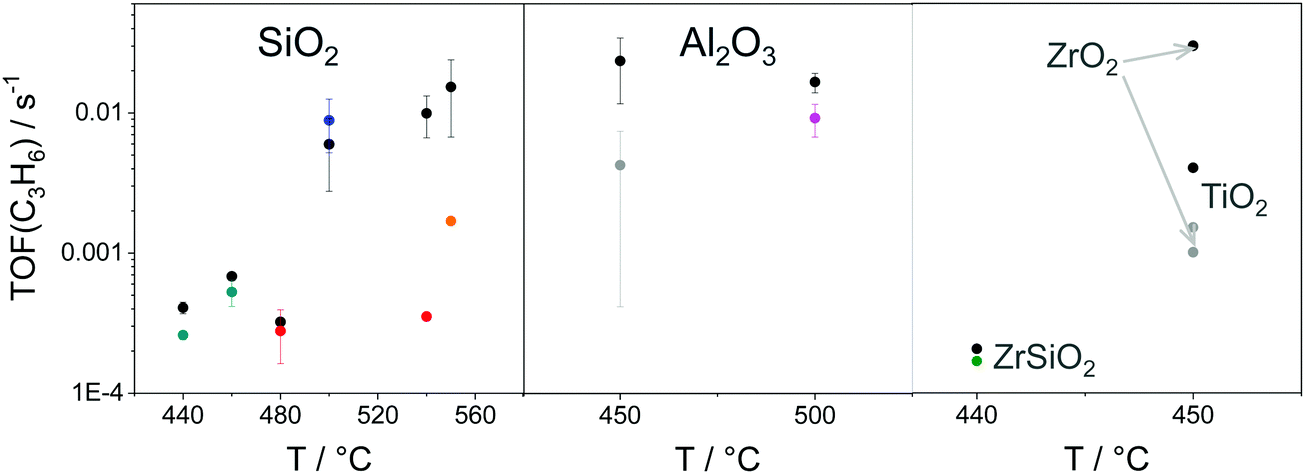 | ||
Fig. 23 Averaged V-related turnover frequencies of propene formation over differently supported V-containing catalysts in the absence (●) or the presence of different promoters (F –  , K – , K –  , Mo – , Mo –  , Nb – , Nb –  , Mg – , Mg –  , P – , P –  , Sb – , Sb –  ). The kind of support is written in each figure. Table S5 (ESI†) contains full catalyst compositions and reaction conditions. ). The kind of support is written in each figure. Table S5 (ESI†) contains full catalyst compositions and reaction conditions. | ||
Although the addition of K to VOx/SiO2 has a negative effect on the V-related TOF value (Fig. 23), a positive effect of Na presence was very recently reported in ref. 215. Those authors investigated the ODP reaction over Na6V10O28/SiO2, α-NaVO3/SiO2 and VOx/SiO2. The two latter catalysts showed lower activity, too. If Na6V10O28/SiO2 transforms to α′-NaV2O5/SiO2, the activity decreases. However, no control test with Na-promoted VOx/SiO2 has been carried out.
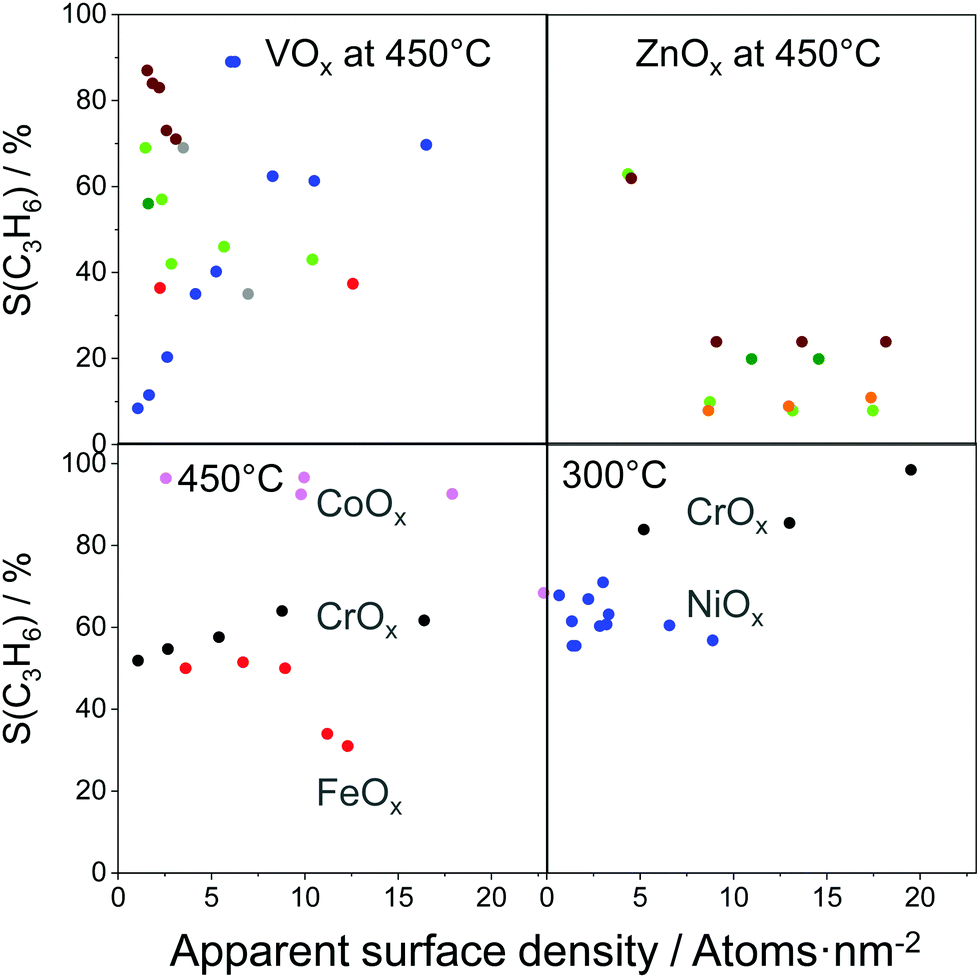 | ||
| Fig. 24 Primary (X(C3H8) < 10%) propene selectivity (S(C3H6)) versus apparent density of metal in active metal oxide species. The colours distinguish different supports (Al2O3 (light green circles), carbon (black circles), Ca5[OH|(PO4)3] (red circles), CeO2 (blue circles), MgO (grey circles), MgAlOx (pink circles), SiO2 (brown circles), TiO2 (dark green circles) or ZrO2 (orange circles)), while the kind of active metal oxide and reaction temperature are given in each figure. Further details are provided in Table S4 (ESI†). | ||
In comparison with V-containing catalysts, the selectivity reported for Zn-based catalysts is significantly lower. Co/MgAlOx catalysts reveal the highest (above 90%) primary propene selectivity, which does not depend on Co loading up to about 17 Co nm−2. B/C and FeOx/CaPOx show significantly lower selectivity. CrOx/C catalysts seem to be efficient for the dehydrogenation reaction and the desired selectivity does not decrease with Cr loading. This selectivity is also not affected by Ni content in NiOx/CeO2, although NiOx species are intrinsically less selective.
In order to check if the conclusions made in one specific study are of general character, various VOx/SiO2 catalysts in Table 5 are compared in terms of the effect of V loading on the primary propene selectivity at 500 °C. Although the reported datapoints scatter, no general negative effect could be identified for the catalysts with up to 5 V nm−2 as one could assume from Fig. 24, where the results from only one study are shown. In this view, the effect of Ti/Si in VOx/SiTiOx with a similar (between 0.7 and 1.7 V nm−2) V loading on the primary propene selectivity is also contradictive. One research group197 established a negative effect, while a positive effect was reported in another study.179 Such obvious discrepancies may derive from different reaction conditions including catalyst treatment procedures as well as different methods of catalyst synthesis.
Further, available literature is analysed to check whether and how the support and the kind of promoter for VOx as well as the type of active component affect the primary selectivity to propene. Another question to be clarified is the effect of temperature on this catalyst performance. To this end, an average selectivity at a certain reaction temperature was calculated for (i) VOx-containing (without any promoter) catalysts based on Al2O3,144,162,183,184,200–203,216–220 CeO2,186,187,216,219 SiO2,75,147,150,184,196,198,204,207,216,217,219,221–223 TiO2144,184,190,205,206,213,216,217,219,224–227 or MgO188,219 supports or (ii) all catalysts containing V,75,142,144,147,150,162,183,184,186–188,190,196,198,200–207,213,214,216–229 B,160,162–164,166,192,218,220,230,231 Co,175,193,219,227,232–237 Cr,173,174,238–240 Mo208,241–244 or Ni157,167,168,171,172,189,195,219,233,235,245–252 without or with one or several promoters. For the calculations, the selectivity values obtained at a propane conversion below 10% were used. Fig. 25 was constructed on this basis. The corresponding values also containing error bars for each averaged datapoint are shown in Fig. S1 (ESI†).
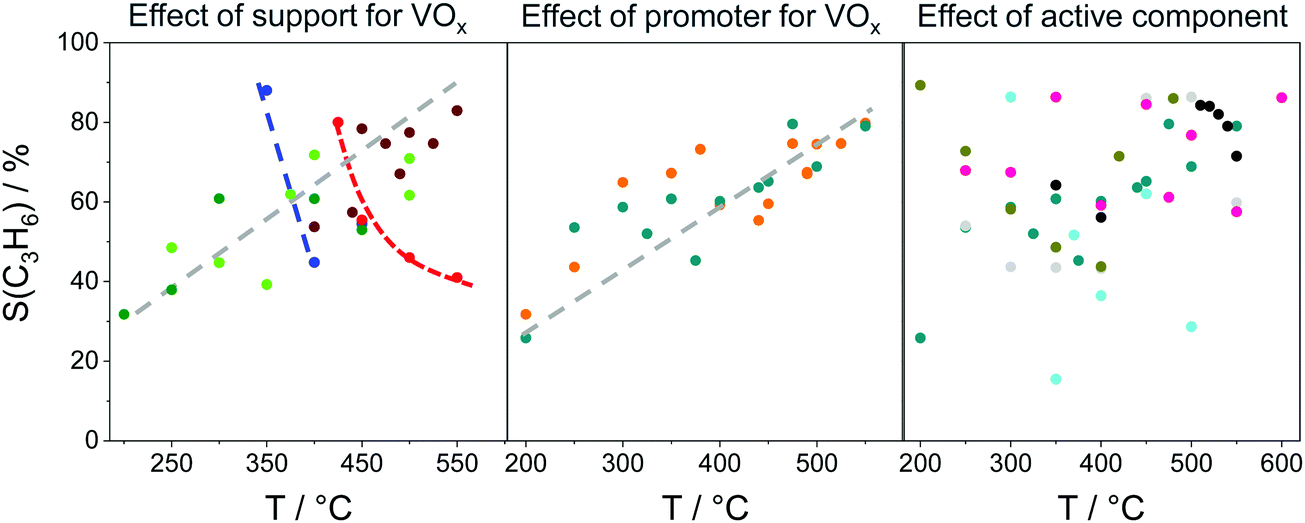 | ||
Fig. 25 Averaged propene selectivity calculated from literature data obtained at a propane conversion below 10% at different temperatures. Effect of support for VOx: the colours distinguish different supports (Al2O3 (light green circles), CeO2 (blue circles), SiO2 (brown circles), TiO2 (dark green circles) or MgO (grey circles).) for VOx species. Effect of promoter for VOx: supported catalysts with VOx exclusively ( ) or all catalysts containing V ( ) or all catalysts containing V ( ). Effect of active component: B (●), Co ( ). Effect of active component: B (●), Co ( ), Cr ( ), Cr ( ), Mo ( ), Mo ( ) or Ni ( ) or Ni ( ). ). | ||
We start our discussion with the effects of temperature and support on the selectivity to propene over VOx species. For the catalysts based on the SiO2, TiO2 or Al2O3 supports, the desired selectivity increases with rising reaction temperature. Such effect can be due to different activation energies for propene formation and for propane/propene conversion into carbon oxides. The latter reaction should have lower activation energy. The positive effect of temperature on the selectivity to propene is also valid when considering both supported and bulk V-containing catalysts with or without any promoter. It is also obvious that any effect of promoter on the selectivity to propene could be hardly seen. This means that the improvements reported in various papers are not significant or the reference V-containing (without any promoter) catalysts used in those papers show lower performance in comparison with similarly composed catalysts tested in other studies. Concerning the effect of the kind of support on product selectivity, it is difficult to distinguish between the SiO2, TiO2 or Al2O3 supports when bearing in mind the error bars calculated for each catalyst (Fig. S1, ESI†). Opposite to these supports, the propene selectivity over the catalysts based on the CeO2 or MgO supports decreases with an increase in temperature. A possible reason may be the formation of mixed phases at high temperature. It is, however, worth mentioning that only a few studies fulfilled the above-mentioned criterion for selecting them for our analysis. Thus, further studies are required.
Regarding the effect of active component on product selectivity, the datapoints in Fig. 25 are too strongly scattered to draw a definitive conclusion. Nevertheless, when analysing the datapoints at propene selectivity above 80%, some hints can be obtained. Regardless of the reaction temperature, an averaged propene selectivity over V-based catalysts is below this value. The selectivity above 80% achieved over Mo-containing catalysts at 200 and 480 °C were reported in single studies only, i.e.ref. 244 and 243, respectively. Reaction temperatures of 350 and 400 °C are more representative, as they were applied in several studies dealing with the ODP reaction.241,242,244 On this basis, one can conclude that up to now tested Mo-based catalysts do not show high primary propene selectivity. B-, Co- or Ni-containing catalysts seem to be suitable for achieving the desired performance. Cr-containing catalysts are also promising when they operate at about 300 °C.
In summary, the support material, the kind of active component and its loading are decisive for the primary propene selectivity over different supported catalysts. Such conclusion is actually not novel. When considering the discussion of catalyst development in Section 5.1, one may assume that this knowledge is not enough to ensure the design and the preparation of catalysts with high propene selectivity. Some progress can be achieved when deeper fundamental correlations between the real structure of supported active species, the kinetics of their reduction/reoxidation, the nature of oxygen species formed upon activation of gas-phase oxygen and the kinetics of selective and unselective pathways of propane conversion can be established. In this regards, density functional theory calculations (DFT) have the potential for providing molecular-level details relevant for catalyst design as discussed in the next section.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) is responsible for the first activation step, with the vanadyl-O mediated mechanism being dominant. With respect to product selectivity, both the desired pathway and side reactions leading to iso-propanol or acetone are equivalently possible over V2O5.255 This was concluded to be the main reason for low propene selectivity over catalysts containing even nanosized V2O5. In contrast to the latter material, the dehydrogenation reaction seems to dominate over supported catalysts as calculated for one monolayer VOx/TiO2.255 Supported catalysts also reveal higher reactivity due to reducing the activation barrier for the rate-limiting step that is breaking C–H bond in propane.
O) is responsible for the first activation step, with the vanadyl-O mediated mechanism being dominant. With respect to product selectivity, both the desired pathway and side reactions leading to iso-propanol or acetone are equivalently possible over V2O5.255 This was concluded to be the main reason for low propene selectivity over catalysts containing even nanosized V2O5. In contrast to the latter material, the dehydrogenation reaction seems to dominate over supported catalysts as calculated for one monolayer VOx/TiO2.255 Supported catalysts also reveal higher reactivity due to reducing the activation barrier for the rate-limiting step that is breaking C–H bond in propane.
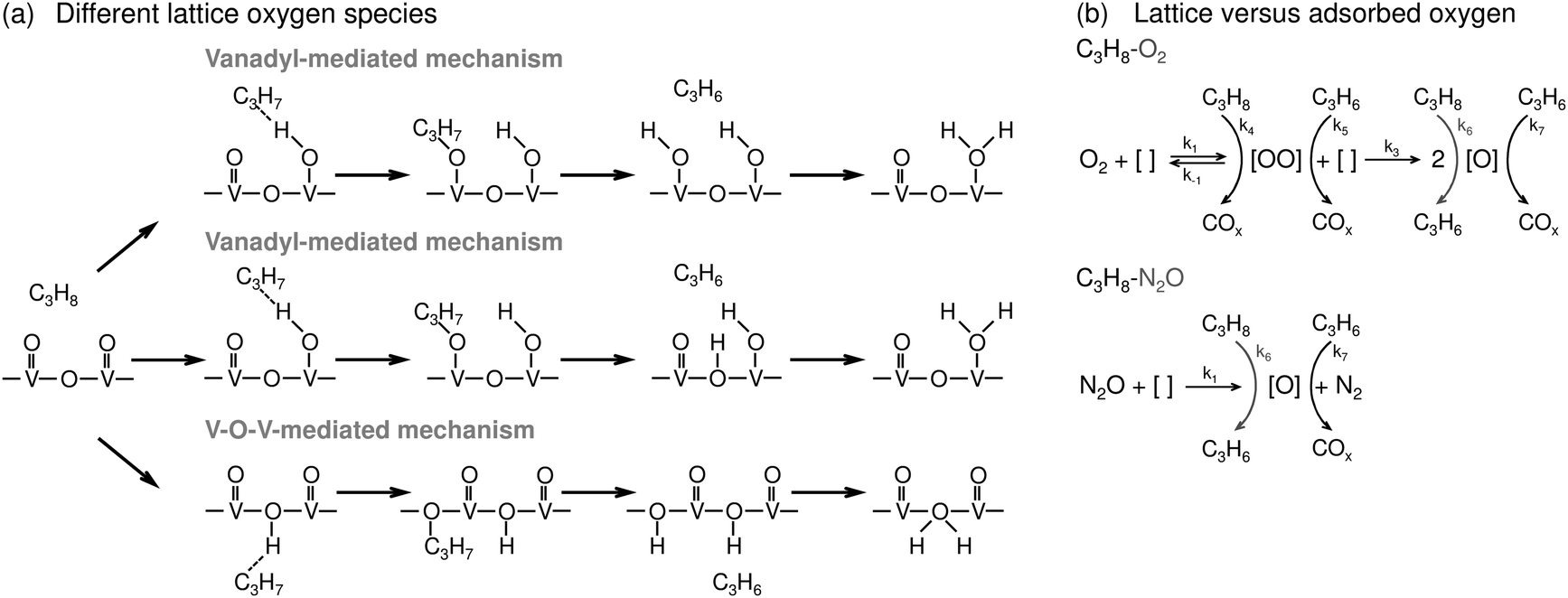 | ||
| Fig. 26 (a) Possible mechanisms for the oxidative dehydrogenation of propane to propene over V2O5 with participation of lattice oxygen. (b) Oxygen species and their participation in propane/propene oxidation. Adapted from ref. 174 and 256 respectively. | ||
The important role of the vanadyl group for the first propane activation step was also confirmed for the ODP reaction over supported catalysts. In addition to different lattice oxygen species (vanadyl or bridged), peroxo-oxygen species can be formed upon activation of gas-phase O2 over reduced VOx species.178,259 Such species reveal high activity for undesired propene oxidation to carbon oxides. Thus, to control this pathway, the catalyst should be able to quickly convert this unselective oxygen species into lattice oxygen. This conclusion is indirectly supported by the fact that the selectivity to propene increases when O2 oxidant is replaced by N2O. The latter can generate lattice oxygen exclusively.
The kind of support or supported VOx species (isolated or dimeric) is decisive for the second C–H cleavage. It is predicted that a HOV(IV)Ox site formed upon propane activation over VOx/SiO2 with isolated sites does not subtract another hydrogen from the generated iso-propyl radical. The latter should desorb and react with VO of another VOx species yielding propene and a second HOV(IV)Ox site. The situation changes when analysing the reaction pathways of propane dehydrogenation over dimeric VOx species on SiO2.178 Bridged oxygen (V–O–V) or neighbouring vanadyl oxygen can participate in the second hydrogen abstraction. It was also concluded that a V(V)/V(IV) cycle should prevail over a V(V)/V(III) cycle. These calculations also predict independence of the V-related TOF on V loading upon propane dehydrogenation over highly dispersed VOx sites (from isolated VOx to those with a fewer number of V atoms). This conclusion agrees with our analysis of experimental literature data (Fig. 22 and Table 5).
The effect of anatase TiO2 crystalline face on the reactivity of isolated and dimeric VOx species in the ODP reaction was investigated in ref. 261. VOx/TiO2(100) and VOx/TiO2(001) models were applied. Bridged (V–O–Ti) of isolated VOx on the surface of TiO2(100) is predicted to show higher activity than the vanadyl oxygen, while these both species in isolated VOx on the surface of TiO2(001) should have similar reactivity. In comparison with the isolated species, propane dehydrogenation over dimeric VOx species on the surface of TiO2(100) or TiO2(001) is initiated through breaking the methylene C–H bond by the vanadyl oxygen. In terms of intrinsic activity, VOx/TiO2(100) should perform superior to VOx/TiO2(001). This difference was related to higher ability of the former system to bond H atoms and to lower stability of supported VOx species on the surface of TiO2(100).
All the above discussed studies with supported catalysts considered one structure of VOx species. Such species is terminated by vanadyl oxygen and possesses connections with the support through three bridged oxygen. Cheng et al.262 constructed possible monomeric and dimeric structures after H3VO4 grafting on the surface of anatase TiO2(001). The hydroxylated surface was used for calculations. Six monomeric and two dimeric VOx species were constructed (Fig. 27). They were used for calculating reaction pathways and energies for propanol formation from propane. No difference in the overall reaction mechanism between these differently structured VOx species could be established. The first step is the activation of propane C–H bond by the vanadyl group in agreement with several previous studies discussed above. In a second step, an O–C bond is formed. The reactivity was not found to depend on the size of the active species. However, its molecular structure (coordination environment) seems to play an important role for catalyst activity. VOx species with square pyramidal coordination environment (M-molecular or M-tridentate structures in Fig. 27) should possess higher reactivity in comparison with their tetrahedrally coordinated counterparts (M-bidentate and M-bidentate_2 in Fig. 27). Such difference was explained by more efficient stabilization of reduced square pyramidal coordinated VOx species in the reaction intermediate structures. These theoretical results demonstrate the potential in catalyst development when controlling the structure of supported VOx species and may also explain different experimental TOF values determined for V-containing catalysts prepared with a certain support in different studies.
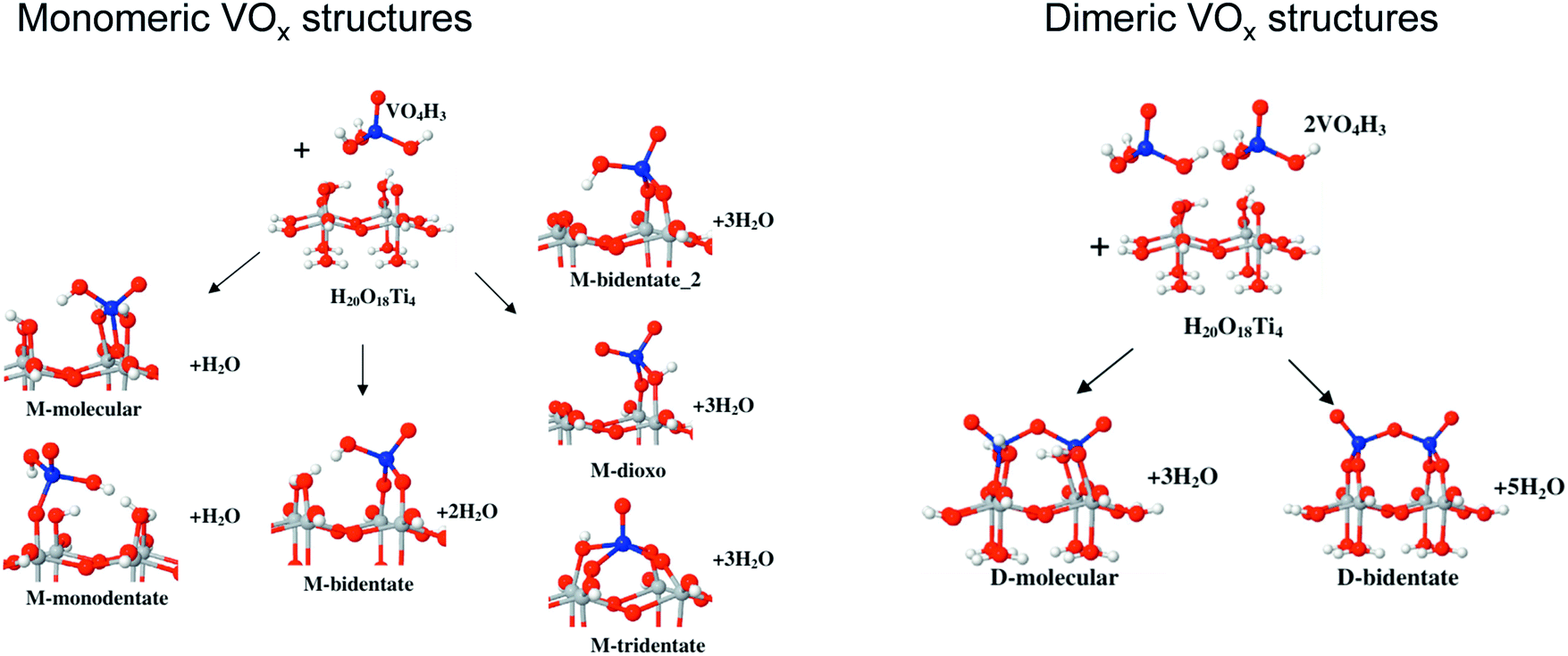 | ||
| Fig. 27 Possible structures of monomeric and dimeric VOx species on the TiO2 anatase (001) surface. Reproduced from ref. 262 with permission from Elsevier, copyright 2020. | ||
Du et al.261 have carried out a systematic study of possible structures of supported VOx species formed upon reaction of H3VO4 with the TiO2 anatase (001), (100) and (101) surfaces. Surprisingly, five-coordinated VOx species were identified to be the most stable structures on the (001) surface. Surface stress upon adsorption of H3VO4 determines the final structures. The stress does not seem to play an important role for the (100) and (101) surfaces. Consequently, tetrahedrally coordinated VOx species on the latter faces are the dominant structures. It was also concluded that VOx species on the (001) surface are more stable than those on the (100) and (101) surfaces. Thus, the latter surfaces are less suitable for stabilizing isolated VOx species and will favour their polymerization.
The surface structures of isolated, diatomic and polymerized VOx species on the (101) and (001) tetragonal ZrO2 surfaces were examined.263 The effect of water was also analysed. Differently structured species were formed upon adsorption of V2O5 and H2O on these single surfaces. For the lowest number of V atoms (0.25 per eight Zr cations), a dimer is the most stable structure on the (101) surface. Each V cation has a terminal oxygen (vanadyl) and is connected via a bridge oxygen with another V cation. Contrarily, monoatomic species is the dominant structure on the (001) tetragonal ZrO2 surface at same V loading. A dimeric species is formed at higher V loading. No stable structures, where V5+ replaces Zr4+ could be identified. However, a structure with one vanadyl oxygen can be stabilized. In addition, a monolayer structure is more preferable than V2O5 crystallites. For all analysed structures, vanadium atoms are connected with zirconium atoms through lattice oxygen. Water was concluded to be a decisive parameter for stabilizing a certain VOx structure and its reducibility. The diatomic structures on the (101) surface are transformed into the isolated species in the presence of about 0.01 bar water. The latter species show lower reducibility than the dimers.
Different isolated VOx·Ce12O24 (x = 0–4) structures were modelled as a function of temperature (130–630 °C) at very low oxygen partial pressure (about 10−9 bar).264 The (111) CeO2 surface was used for calculations. The most stable structure was concluded to be VO2·Ce12O24. Ce4+ is reduced to Ce3+, while the oxidation state of vanadium is +5. The most stable structure is composed of one vanadyl group and three V–O–Ce bonds. It was also concluded that the reducibility of ceria increases in the presence of supported VOx species. Any consequences for the ODP reaction were not considered.
Summarising this part, the real structure of supported VOx species on the surface of a certain support, their stability and reactivity strongly depend on the kind of support face, temperature, and water presence. As different faces can be exposed for adsorption of VOx species upon catalyst preparation, the resulting catalysts will probably contain differently structured VOx species. Thus, it is practically impossible to determine the activity and selectivity of a certain structure in the ODP reaction. Such information may become available if supports with a preferential orientation of exposed faces will be prepared and applied for adsorption of VOx species.
6 Oxidative isobutane dehydrogenation
In comparison with the ODP reaction, the oxidative dehydrogenation of isobutane to isobutene (ODB) is less investigated. Our search of literature on the latter topic resulted in only 52 articles published since 2000. About 190 different catalysts have been developed and tested. In contrast to the ODP reaction, the largest number of the prepared catalysts contain chromium as active component.265–278 In total, about 49 such catalysts were synthesized in comparison with 45 catalysts based on vanadium.269,272,279–288 Surprisingly, carbon-based catalysts (in total 21 materials) build the third group of mostly investigated catalysts.289–296 Another important class of catalysts is based on molybdenum typically in form of molybdates.297–301 Phosphates and pyrophosphates of different metals were also tested for the ODB reaction.302–305 Against this background, we initially discuss developments within each group of the catalysts and then compare their application potential.6.1 Active components and mechanistic aspects of ODB
With respect to the effect of support, Jibril et al.270 concluded that γ-Al2O3 is the best choice among MgO, TiO2, SiO2 and γ-Al2O3. Calcination and pre-treatment temperatures have a minor effect on the performance of VOx/Al2O3 catalysts.267 However, the surface area of the support seems to have a positive effect on the selectivity to isobutene. The highest selectivity of ca. 68% at isobutane conversion of about 10% was obtained at 250 °C over the catalysts with Puralox 150/170 and high-surface-area aluminium oxide support.267 Therefore, not only the surface area but also the type of support material should be considered for preparation of active and selective catalysts. A Cr-containing catalyst prepared using high surface area (787 m2 g−1) SiO2 support (SBA-15) converted 14% isobutane with 79% selectivity at 540 °C.277 Since formation of highly dispersed mono and polychromate domains was mentioned to be an important requirement for effective catalyst,277 high surface area of the support could be a factor facilitating formation of such domains upon catalyst synthesis.
Different dopants were investigated for their effect on catalytic performance of Al2O3-supported catalysts. Using promoters on the basis of oxides of Ni, Co, Mo, Na, W, V, Li, La or Bi for synthesis of Cr–M–O/Al2O3 catalysts with a total content of metal of 10 wt% did not result in any significant improvement of catalytic performance.276 Contrarily, promoting CrOx/γ-Al2O3 with CaO was established to positively affect the selectivity to isobutene when the content of the promoter was below 2 wt%.274,275 The promoter facilitated the formation of highly dispersed Cr6+ species, which are the active sites. In addition, catalyst acidity decreased, thus facilitating isobutene desorption and hindering its readsorption. A bulk Ca-chromate phase was formed with an increase in Ca content that was detrimental for catalyst performance. Although promoting CrOx/Al2O3 with K2O was also established to reduce the overall acidity, the selectivity to isobutene decreased.278 The same study demonstrated that this promoter improved the selectivity over CrOx/TiO2 from 34.8 to 55.5%.278 An opposite effect was observed for CrOx/Al2O3. Such a contradictory effect was related to the changes in the rate of oxygen chemisorption and coverage by oxygen species. These parameters increased for CrOx/TiO2 and decreased for CrOx/Al2O3 when these catalysts were doped with K, while acid–base properties changed in the same way upon potassium addition. Thus, the effect of the acidity on isobutene desorption/readsorption and accordingly on the selectivity to isobutene should be further elucidated.
In comparison with standard metal oxides, mixed metal oxide supports seems to be more suitable for achieving high isobutene selectivity. A catalyst containing chromium oxide (8 wt%) on the surface of the fluorite-type Ce0.60Zr0.35Y0.02O2 support resulted in 93% selectivity at isobutane conversion of 11% at 540 °C.266 Very auspicious results (isobutane conversion of 52.4% and isobutene selectivity of 84%) were obtained over CrOx(18 wt%)/ZnAlLaOx at even higher temperature of 580 °C.265 It is still to be proven if the reaction temperature or/and the support are important for the high catalyst performance.
To incorporate vanadium into the structure of SiO2 (MCM-41), two different methods were used: (i) direct hydrothermal synthesis using VOSO4282 or VOC2O4279 as vanadium source or (ii) template-ion exchange using MCM-41 containing ca. 50 wt% template and VOC2O4. The catalysts prepared according to the former method selectively converted isobutane to isobutene when the vanadium content exceeded 1 wt%. Contrarily, the template-ion exchange method resulted in non-selective catalysts. The effect of the preparation method was related to the location/structure of vanadium species. Isolated tetrahedrally coordinated VOx species sites dispersed on the wall of MCM-41 were produced by the template-ion exchange method, while the selective sites formed through the direct hydrothermal synthesis are incorporated inside the walls of MCM-41. The conclusion about the low selectivity of supported isolated VOx species seems to be contradictive. Actually, selective catalysts could be synthesized by simple impregnation of MCM-41 with NH4VO3.281 The selectivity was further increased when using V(t-BuO)3O as a source of vanadium. The highest selectivity value of about 84% at isobutane conversion of 48% was achieved at 560 °C. Such high performance was explained by high dispersion of surface VOx species.
Another interesting approach for enhancing dispersion of VOx species and their resistance against sintering is a grafting technique and the usage of promoters. Using this method, two series of VOx–TiO2–SiO2 materials were prepared: (i) titanium and vanadyl alkoxides were simultaneously grafted on SiO2 and (ii) vanadyl tri-isopropoxide was grafted on a TiO2–SiO2 support.280 The former materials showed superior selectivity. The improvement was related to the kind of catalytically active species. Isolated bridged oxygen species (V–O–Ti) formed on the silica surface via method i are less active but more selective than polyvanadylic V–O–V formed when vanadyl tri-isopropoxide was grafted on the TiO2–SiO2 support.
Depending on the kind of support, its shape can be decisive for the performance of supported VOx species. For example, the same species were obtained through deposition of vanadium on nano-shaped TiO2 with different surface facets.285 Accordingly, these catalysts revealed similar activity and selectivity. Contrarily, when CeO2 rods or octahedra were used as a support for VOx,287 the octahedra-based catalyst showed higher selectivity and lower activation energy for isobutane conversion. The observed effects were explained by the difference in surface oxygen vacancy formation energy, number of defect sites and surface O–O distance of different surface CeO2 planes.
Another group of V-based catalysts intensively tested in the ODB reaction are vanadium-containing mixed oxides. For example, the performance of Cr–V–Nb mixed oxides depends strongly on the content of components.272 Isobutane conversion increases from 20% to 50% upon increasing the ratio of Cr/V from 2.3 to 4.7. The selectivity to isobutene also increases. The highest selectivity of 90% was achieved at isobutane conversion of 45% at 573 °C using a feed mixture with O2/i-C4H10 ratio of 1.06. When Cr content is further increased the selectivity decreased. Such effect of Cr loading was assumed to be due to the changes of active surface areas and redox properties of the catalysts upon variation of the catalyst composition.
Bulk V–Sb mixed oxides show rather low selectivity to isobutene, i.e. about 11% at isobutane conversion of ca. 13%. Their performance could be slightly increased upon promoting with niobium oxide (22% selectivity to isobutene at isobutane conversion of 15%).283 Further improvements were achieved when depositing VSbOx on microspheric γ-Al2O3.283 The selectivity of 68% at isobutane conversion of 36.5% was reported. The usage of other supports like α-Al2O3, Si–Al–O, SiO2 or MgO for preparation of VSbOx-containing catalysts worsened the performance.284,306 Promoting VSbOx/γ-Al2O3 with Ni resulted in an increase in the conversion to 42–44% and keeping the selectivity on the same level of ca. 70%.283 The positive effect of Ni was explained by the formation of the nickel vanadate NiV2O6 phase. The catalyst containing this phase is easier reducible and possesses higher amount of mobile oxygen species. Thus, more facile redox cycle of active vanadium species was considered to be responsible for the enhancement of the catalyst performance.
For supported VMoOx, Al2O3 was also found to be a more suitable support than CeO2 or TiO2.288 Since Al2O3 possesses significantly higher surface area, better performance of Al2O3-supported catalysts was related to the higher dispersion of the active sites on the catalyst surface. VMoOx/Al2O3 with V2O5–MoO3 content of 5 wt% converted 22.2% of isobutane with isobutene selectivity of 72.3% at 500 °C. The selectivity decreased upon increasing the content of the active components or reaction temperature. As isobutane conversion also increased, it is not clear if the decrease in the selectivity was really related to the content of the active component or temperature or simply to higher conversion. It is well known that the selectivity decreases with rising isobutane conversion over same catalyst under isothermal conditions due to higher formation of consecutive reaction products (COx).
Bulk Mo–V–Sb mixed oxides also produce methacrolein with a selectivity of 9.3% at 440 °C. At isobutane conversion of 7.6% the corresponding isobutene selectivity amounts to 25.9%. The isobutene selectivity decreases upon an increase in Mo content, while selectivity towards methacrolein increases.286
Carbon catalysts possess many different surface functional groups like quinonic, ether, carbonylic, phenolic, carboxylic, lactone, etc. (Fig. 28). Some of these groups negatively influence catalyst performance. So carboxylic anhydride groups formed on the surface of carbon xerogel during the ODB reaction lowered the TOF values.296 Acidic groups on the surface of activated carbon fibre reduced catalytic activity.292 Therefore, modifying carbon surface is an attractive way to control the nature of surface functional groups and respectively catalyst performance. Such modifications could, for example, be achieved through incorporation of nitrogen into the structure of carbon xerogel.296 Treatment of synthesized materials under different conditions is an useful tool for controlling the type and the amount of surface functional groups or improving their accessibility.289,292–296 So, closed fullerene-like cavities of graphitized mesoporous carbon are uniformly opened through air oxidation at 500 °C without affecting the graphitic structure but enhancing catalyst activity. These cavities can be closed during oxidative treatment at 600 °C.
 | ||
| Fig. 28 Oxygen-containing functional groups on the surface of carbon catalysts. | ||
Regardless of the type of carbon material, the ODB reaction is catalysed by carbonyl-quinone groups.291–296 DFT calculations295 predict that dicarbonyls at zigzag edges and quinones at arm chair edges provide the main contribution to the isobutene formation. Although some experimental results indicate that total oxidation occurs on other type of surface sites,289 these sites were not still reliably identified. There are some debates about mechanistic aspects of regeneration of selective sites. The regeneration of active sites occurs by reaction with gas-phase O2. Similar to the mechanistic concept suggested for the metal oxides this reaction yields water.293 However, DFT calculations predicted a very high activation barrier (>150 kJ mol−1) for regeneration step via water removal.295 So, it was suggested that hydrogenated functionality reacted with O2 forming H2O2.
Not only the temperature but also the duration of thermal treatment and the kind of treating atmosphere can affect catalyst activity.289,291–293 Carbon catalysts exposed to N2O, O2 or H2 at the same temperature for the same time performed differently.293 The O2-treated catalyst was more active, but less selective than its counterparts treated in N2O or H2. An increase in treatment time in air at 500 °C from 24 to 48 h led to ca. two fold increase in the rate of isobutane conversion over graphitic carbon.289 This enhancement was related to the generation of active quinone-type functional groups. Additional active carbonyl-quinone groups can be also incorporated upon exposure of carbon materials to supercritical CO2 or water.292 Contrarily, treatment of activated carbon catalysts in peroxyacetic or nitric acid or in ammonium peroxydisulfate decreased isobutene yield.291 The negative effect of nitric acid treatment was explained by destroying of micropores and generation of oxygen-containing functionalities, especially carboxylic acid groups.292 An increase in isobutene selectivity can also be achieved through blocking active sites responsible for the formation of COx. Treatment of graphitic mesoporous carbon by phosphoric acid caused such changes.294 However, the positive effect was observed just until a certain coverage by phosphorous surface groups was achieved. This coverage corresponds to P content of 0.19 at%.
An ODB study over magnesium molybdates possessing different crystalline phases has shown that MgMoO4 is the most selective due to decreased ability for cracking of isobutane/isobutene to propene and methane.298 So, at 530 °C selectivity to isobutene over 1.0MgO/1.0MoO3 was amounted to 84% at isobutene conversion of 6% using reaction mixture containing 2.7 vol% O2 and 8.2% of isobutane balanced by He.
Similar to the ODB over V- or Cr-based catalysts, the formation of isobutene occurred via a Mars–van Krevelen mechanism.307 Chemisorbed oxygen species are responsible for COx formation. This mechanistic scheme provided the best fit of experimental kinetic data obtained for CoMoO4 and NiMoO4307 as well as for Co0.95MoO4, NiMoO4 and MnMoO4.300 The highest rates of isobutene and propene formation were achieved over Co0.95MoO4. It was explained by an optimal strength of olefin-catalyst bond and surface concentration of strong acidic sites.
6.2 Catalyst comparison in terms of their industrial relevance
To demonstrate an industrial relevance of different catalysts, the catalysts with isobutene selectivity above 70% at isobutane conversion higher than 10% have been selected. The corresponding selectivity–conversion data are shown in Fig. 29.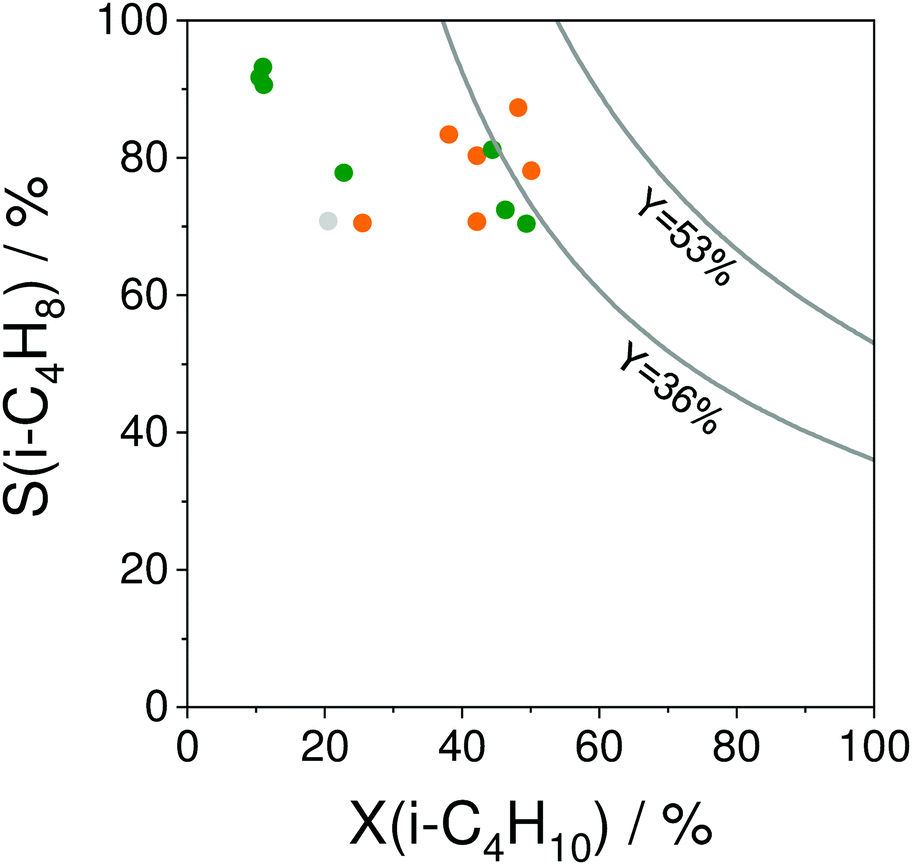 | ||
Fig. 29 Selectivity–conversion plots for V- ( ),272,281,283,288 Cr- ( ),272,281,283,288 Cr- ( )265,266,272 or Ce-based ( )265,266,272 or Ce-based ( )302 catalysts with isobutene selectivity above 70% at isobutane conversion above 10%. Catalyst composition and reaction conditions are given in Table 6. )302 catalysts with isobutene selectivity above 70% at isobutane conversion above 10%. Catalyst composition and reaction conditions are given in Table 6. | ||
Different symbols correspond to catalysts with different active components. The grey lines stand for the yield of propene of 35 and 53%, which are typical for commercial BDH process. In total, 14 catalysts fulfilled the above requirement. Six catalysts contain vanadium as active component. Seven catalysts are based on chromium. Cerium phosphate also showed relatively high performance. Their exact composition and reaction conditions are given in Table 6. The catalysts Cr0.67V0.24Nb0.09, Cr0.74V0.19Nb0.07 and Cr0.78V0.16Nb0.06 from ref. 272 are not shown in this table because the reaction feed applied for ODHB tests was not clearly defined. The reported conversion/selectivity values are 22.8/77.8, 44.4/81.2 and 49.4/70.4%, respectively. They were tested at 300 °C. In comparison with the ODP reaction, several catalysts show the selectivity–conversion data, which are close to those of commercial BDH catalysts (isobutane conversion of 64–65%, isobutene selectivity of 88–89%, 580 °C112). Nevertheless, the ODB is still not commercialized.
| Catalysts | T/°C | p(i-C4H10)/bar | p(O2)/bar | X(i-C4H10)/% | S(i-C4H8)/% | STY(i-C4H8)/kg kg−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|
| a Prepared using V(t-BuO)3O. b Prepared using NH4VO3. c V/Sb/Ni = 8.8/1/3.6. d V/Sb/Ni = 8.8/1/7.0. | |||||||
| 18 wt% Cr/ZnAlLaOx | 580 | 0.79 | 0.23 | 52.4 | 84.1 | 1.71 × 10−1 | 265 |
| 6 wt% CrOx/Ce0.60Zr0.35Y0.05O2 | 540 | 0.04 | 0.08 | 10.5 | 91.7 | 2.89 × 10−4 | 266 |
| 8 wt% CrOx/Ce0.60Zr0.35Y0.05O2 | 540 | 0.04 | 0.08 | 11.0 | 93.2 | 3.08 × 10−4 | 266 |
| 10 wt% CrOx/Ce0.60Zr0.35Y0.05O2 | 540 | 0.04 | 0.08 | 11.1 | 90.6 | 3.02 × 10−4 | 266 |
| CeP2O7 | 500 | 0.27 | 0.07 | 20.5 | 70.8 | 7.21 × 10−3 | 302 |
| 10.7 wt% V2O5/MCM-41a | 560 | 0.12 | 0.06 | 48.2 | 87.3 | 1.51 × 100 | 281 |
| 10.7 wt% V2O5/MCM-41b | 560 | 0.12 | 0.06 | 50.1 | 78.1 | 1.40 × 100 | 281 |
| V–Sb–Ni–O/Al2O3c | 550 | 0.2 | 0.12 | 43.6 | 69.6 | 1.37 × 10−1 | 283 |
| V–Sb–Ni–O/Al2O3d | 550 | 0.2 | 0.12 | 46.6 | 66.3 | 1.39 × 10−1 | 283 |
| 5 wt% V2O5–MoO3/Al2O3 | 500 | 0.18 | 0.09 | 22.2 | 72.3 | 1.02 × 10−1 | 288 |
| 10 wt% V2O5–MoO3/Al2O3 | 500 | 0.18 | 0.09 | 25.5 | 70.5 | 1.14 × 10−1 | 288 |
7 Alternative oxidising agents and reaction engineering aspects for improving product selectivity
The main drawback of the oxidative dehydrogenation of C3–C4 alkanes to the corresponding olefins is low selectivity to the desired products due to their conversion into carbon oxides. In addition to catalyst, the selectivity can be controlled through reaction engineering aspects or the usage of alternative oxidants. Among the latter, the soft oxidants CO2 and N2O were applied. The corresponding results are presented and discussed in Sections 7.1 and 7.2. Another opportunity to improve overall process economy and the selectivity to the olefins is to carry out the dehydrogenation reaction under O2-lean conditions (alkane/O2 > 10) or using alternating feeds of alkane and air. The developments in this area are discussed in Sections 7.3 and 7.4.7.1 General knowledge about alkane dehydrogenation with CO2
CO2-DH of propane (eqn (1)) or isobutane can be considered as a combination of non-oxidative alkane dehydrogenation (eqn (2) for propane) and reversed water gas shift reaction (RWGS, eqn (3)). Besides these two reactions, dry reforming of alkane/olefin can occur as a side reaction (eqn (4) for propane). This reaction is thermodynamically favoured at 600 °C309 but seems to be kinetically hindered over most of the catalysts studied.Different positive effects of CO2 on alkane dehydrogenation are possible and discussed in literature.
i. Enhancing equilibrium conversion in the non-oxidative dehydrogenation due to H2 removal through RWGS.
ii. Improving the selectivity to olefins through site isolation caused by partial blockage of the active sites by adsorbed CO2 (competition with propane adsorption and consequently lowering catalyst activity).
iii. Increasing on-stream stability due to removal of coke through Boudouard reaction.
For the PDH reaction, an increase in thermodynamic limit of propane conversion by about 10% can be reached at 600 °C or the reaction temperature can be lowered by about 50 °C to reach the same conversion when performing the dehydrogenation reaction in CO2 presence at same partial pressure of propane.310 Mechanistically, the CO2-DH reaction can proceed either through oxidative dehydrogenation with participation of lattice oxygen of metal oxide species followed by reoxidation of the reduced sites by CO2, or by simple combination of non-oxidative hydrogenation and RWGS. Literature data are controversial in this respect.
| C3H8 + CO2 ⇆ C3H6 + CO + H2O ΔH298K = +165 kJ mol−1 CO2-PDH | (1) |
| C3H8 ⇆ C3H6 + H2 ΔH298K = +124 kJ mol−1 PDH | (2) |
| CO2 + H2 ⇆ CO + H2O ΔH298K = +41 kJ mol−1 RWGS | (3) |
| C3H8 + 3CO2 ⇆ 6CO + 4H2 ΔH298K = +620 kJ mol−1 dry reforming | (4) |
 | ||
| Fig. 30 Comparison of propene yields obtained in traditional PDH vs. CO2-PDH over metal-oxide based catalysts. For clarity reasons in the catalyst names only the element symbols of active metals and supports are given, i.e. Ga/Zr means GaOx species supported on ZrO2 and Ga80–Al20 means mixed-metal oxide containing 80 wt% Ga2O3 and 20 wt% Al2O3. Data are from literature, for references see Table 7. | ||
| Active material | Support | T/°C | PDH | CO2-PDH | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p(C3H8)/bar | X(C3H8)/% | S(C3H6)/% | Y(C3H6)/% | p(C3H8)/bar | p(CO2)/bar | X(C3H8)/% | S(C3H6)/% | Y(C3H6)/% | ||||
| CrOx | SiO2 | 550 | 0.2 | 12.7 | 92.1 | 11.7 | 0.2 | 0.8 | 17.2 | 94.4 | 16.3 | 334 |
| 5CrOx | SBA-1 | 550 | 0.067 | 33.0 | 86.0 | 28.4 | 0.067 | 0.333 | 37.2 | 85.4 | 31.8 | 314 |
| CrOx | Al2O3 | 600 | 0.2 | 22.1 | 93.1 | 20.5 | 0.2 | 0.8 | 12.7 | 84.4 | 10.7 | 310 |
| CrOx | Al2O3 | 550 | 0.2 | 14.0 | 95.8 | 13.4 | 0.2 | 0.8 | 12.9 | 88.3 | 11.4 | 334 |
| 4Cr | Al2O3 | 550 | 0.125 | 9.6 | 93.6 | 9.0 | 0.125 | 0.375 | 5.2 | 89.8 | 4.7 | 309 |
| CrOx | ZrO2 | 550 | 0.025 | 69.0 | 74.0 | 51.1 | 0.025 | 0.065 | 41.0 | 62. | 25.4 | 335 |
| CrOx | ZrO2 | 550 | 0.025 | 69.0 | 67.0 | 46.2 | 0.025 | 0.065 | 58.0 | 52.0 | 30.2 | 335 |
| CrOx | ZrO2 | 550 | 0.025 | 74.0 | 76.0 | 56.2 | 0.025 | 0.065 | 59.0 | 55.0 | 32.5 | 335 |
| CrOx | ZrO2 | 550 | 0.025 | 69.0 | 65.0 | 44.9 | 0.025 | 0.065 | 58.0 | 57.0 | 33.1 | 335 |
| 6.8V | MCM-41 | 600 | 0.11 | 23.9 | 76.4 | 18.2 | 0.11 | 0.55 | 53.5 | 84.8 | 45.4 | 322 |
| 6.8V | MCM-41 | 600 | 0.11 | 23.9 | 76.4 | 18.2 | 0.11 | 0.33 | 43.2 | 89.2 | 38.5 | 322 |
| 6.8V | MCM-41 | 600 | 0.11 | 23.9 | 76.4 | 18.2 | 0.11 | 0.11 | 40.6 | 88.6 | 36.0 | 322 |
| VOx | SiO2 | 600 | n.r. | 47.0 | 74.8 | 35.1 | 0.111 | 0.444 | 59.1 | 81.9 | 48.4 | 323 |
| 7V | Al2O3 | 550 | 0.125 | 7.2 | 96.3 | 6.9 | 0.125 | 0.375 | 6.7 | 96.7 | 6.5 | 309 |
| 7V4Cr | Al2O3 | 550 | 0.125 | 9.2 | 95.4 | 8.8 | 0.125 | 0.375 | 10.0 | 99.6 | 9.9 | 309 |
| 7V5Mo | Al2O3 | 550 | 0.125 | 10.3 | 96.7 | 9.9 | 0.125 | 0.375 | 10.1 | 96.6 | 9.7 | 309 |
| 7V7W | Al2O3 | 550 | 0.125 | 6.9 | 97.3 | 6.7 | 0.125 | 0.375 | 9.9 | 97.1 | 9.6 | 309 |
| 5Mo | Al2O3 | 550 | 0.125 | 2.6 | 96.9 | 2.5 | 0.125 | 0.375 | 2.2 | 96.3 | 2.1 | 309 |
| 7W | Al2O3 | 550 | 0.125 | 0.6 | 90.6 | 0.6 | 0.125 | 0.375 | 0.6 | 90.9 | 0.5 | 309 |
| Ga2O3 | 500 | 0.025 | 41.3 | 93.3 | 38.5 | 0.025 | 0.05 | 35.9 | 97.2 | 34.9 | 325 | |
| Ga2O3 | TiO2 | 600 | 0.025 | 23.0 | 85.0 | 19.6 | 0.025 | 0.05 | 32.0 | 73.0 | 23.4 | 326 |
| Ga2O3 | Al2O3 | 600 | 0.2 | 9.6 | 89.3 | 8.6 | 0.2 | 0.8 | 5.4 | 90.8 | 5.3 | 310 |
| Ga2O3 | Al2O3 | 600 | 0.025 | 33.0 | 92.0 | 30.4 | 0.025 | 0.05 | 26.0 | 94.0 | 24.4 | 326 |
| Ga2O3 | ZrO2 | 600 | 0.025 | 39.0 | 74.0 | 28.9 | 0.025 | 0.05 | 30.0 | 65.0 | 19.5 | 326 |
| Ga2O3 | SiO2 | 600 | 0.025 | 7.2 | 92.0 | 6.6 | 0.025 | 0.05 | 6.4 | 92.0 | 5.9 | 326 |
| Ga2O3 | MgO | 600 | 0.025 | 5.3 | 34.0 | 1.8 | 0.025 | 0.05 | 4.3 | 29.0 | 1.2 | 326 |
| Ga8Al2O15 | 500 | 0.025 | 51.7 | 91.6 | 47.4 | 0.025 | 0.05 | 49.7 | 91.7 | 45.6 | 325 | |
| Ga5Al5O15 | 500 | 0.025 | 38.4 | 92.3 | 35.4 | 0.025 | 0.05 | 33.7 | 92.9 | 31.3 | 325 | |
| Ga2Al8O15 | 500 | 0.025 | 22.8 | 94.9 | 21.6 | 0.025 | 0.05 | 19.3 | 92.9 | 17.9 | 325 | |
| Ga–Al–O ht | 550 | 0.032 | 40.5 | 92.1 | 37.3 | 0.032 | 0.097 | 35.2 | 95.1 | 33.4 | 327 | |
| Ga–Al–O prec. | 550 | 0.032 | 33.6 | 91.2 | 30.6 | 0.032 | 0.097 | 26.2 | 95.2 | 24.9 | 327 | |
| Ga–Al–O grind. | 550 | 0.032 | 11.4 | 90.8 | 10.4 | 0.032 | 0.097 | 8.7 | 95.8 | 8.3 | 327 | |
| In2O3 | 600 | 0.025 | 3.0 | 69.0 | 2.1 | 0.025 | 0.1 | 1.0 | 55.0 | 0.6 | 329 | |
| In-Al-40 | 600 | 0.025 | 12.0 | 80.0 | 9.6 | 0.025 | 0.1 | 22.5 | 72.0 | 16.2 | 329 | |
| In-Al-20 | 600 | 0.025 | 16.0 | 84.0 | 13.4 | 0.025 | 0.1 | 27.5 | 78.0 | 21.4 | 329 | |
| In-Al-10 | 600 | 0.025 | 14.0 | 86.0 | 12.0 | 0.025 | 0.1 | 26.0 | 78.0 | 20.3 | 329 | |
Cr-Containing catalysts. CrOx-Based catalysts have been most frequently applied for the CO2-PDH. In the first review published in 2004 only such catalysts have been analysed.9 This may be because Cr2O3 supported on alumina is one of the two types of commercially applied catalysts for the PDH15–18 and water gas shift312 reactions. Meanwhile, catalysts containing CrOx on different supports (mesoporous silica, alumina, zirconia) have been studied.
Owing to the neutral (neither strong acidic nor strong basic sites) character of SiO2, CrOx/SiO2 catalysts have been investigated in several studies focused on various mechanistic aspects.313–318 Michorczyk et al.319 applied operando UV-vis spectroscopy for determining the oxidation state of chromium in CrOx/SiO2(SBA-1) under reaction conditions. It was shown that the initial Cr6+ species are reduced to Cr3+/Cr2+ ones already within the first 10 minutes of PDH or CO2-PDH. Such changes indicate that the oxidative DH over Cr6+ plays only a minor role. Similar results were obtained for CrOx/SiO2 (TUD-1 mesoporous silica matrix) prepared by microwave-assisted method.315 Propene selectivity was above 90% (propane conversion between 5 and 24%) up to 550 °C but decreased to 75% (propane conversion of 45%) at 650 °C. The catalysts initially deactivate rapidly due to coke formation but became more stable after about 2 hours for additional 6 hours. The deactivation rate is lower for the CO2-assisted reaction what is explained by the lower rate of coke formation.
Wang et al.316 studied the influence of preparation method (Cr addition at different stages of synthesis of mesoporous silica from TEOS) on the catalytic performance in CO2-PDH. The method affects dispersion of CrOx species as well as surface physicochemical properties. The sample, where Cr was added after TEOS, showed the highest activity, and possesses the lowest surface acidity and the highest Cr6+/Cr3+ ratio at the surface. For this sample, propene selectivity of about 90% at propane conversion of 40% was obtained. The catalysts deactivate but reactivation with O2 restored the initial activity over 5 cycles. Agafonov et al.317 compared the catalytic performance of silica-supported CrOx catalyst with CrOx/Al2O3 and GaOx supported on both silica and alumina. The highest propene yield of about 40% was obtained over CrOx/SiO2, however the propene selectivity was only 50%. This is due to a high activity of the fresh catalyst towards methane and coke formation. CrOx/Al2O3 was even less active and selective. CO2 was concluded to actively participate in the RWGS reaction and in coke removal through the Boudouart reaction. Very recently, Jin et al.318 studied Ru-promoted CrOx/SiO2 catalysts for CO2-PDH. The activity showed a volcano-type curve with respect to Ru loading. Small amounts of Ru promote the activation of both propane and CO2 as well as help to shift the equilibrium to higher conversions through removal of adsorbed H2 by RWGS reaction. At higher Ru loading (3 wt% Ru), however, CO2 adsorption increases and hinders propane adsorption. Moreover, the high Ru loaded catalyst shows high activity for dry reforming of propene as a side reaction that decreases propene selectivity.
Very recently, Michorczyk et al.320 investigated Cr-containing aluminium-free (Si/Al = 1000) beta zeolite prepared by dealumination followed by its incipient wetness impregnation. Low acidity of the dealuminated zeolite is responsible for high propene selectivity (>80%) at propane conversion of up to 33%. When switching between CO2-free and CO2-containing feeds it could be shown that higher propene formation rate was obtained over the dealuminated catalysts with CO2 whereas the rate over the Al-containing catalysts decreased in CO2 presence. Actually, as seen in Fig. 30, other SiO2-supported Cr-containing catalysts revealed higher propene yield in CO2-PDH than in PDH, while an opposite effect is valid for their Al2O3-supported counterparts. Thus, the kind of support seems to be decisive for the effect of CO2 on propene production.
De Oliveira et al.321 studied PDH and CO2-PDH over CrOx/ZrO2 prepared by conventional and microwave-assisted hydrothermal preparation methods. The yield of propene is lower for all catalysts when adding CO2 to the feed (Fig. 30). This is due to a decrease in both propane conversion and propene selectivity (see Table 7). The lower activity is caused by blocking active sites due to strong CO2 adsorption whereas the decrease in the selectivity is due to blocking basic sites by CO2 that favours acidity and leads to increased methane formation. Additional experiments showed that reduced CrOx can partly be re-oxidized by CO2. Some of the catalysts deactivate slower when CO2 is present in the feed. Regeneration of deactivated catalysts with CO2 is not as effective as with O2 because only part of coke can be removed.
CO2-Assisted PDH over CrOx/Al2O3 was very recently studied by Sandupatla et al.309 The propene yield is lower for the CO2-PDH in comparison with the conventional PDH (Fig. 30) what is mainly due to decreased activity (Table 7). Reduced CrOx sites present under reaction conditions catalyse the PDH reaction but can also strongly adsorb CO2 what explains the reduced activity in CO2 presence.
V-Containing catalysts. Supported V-containing materials are the most intensively studied catalysts both in the PDH (Section 2) and ODP (Section 5) reactions. Therefore, it is not surprising that this type of catalysts has also been applied for CO2-PDH. Different reaction mechanisms are proposed for the latter reaction; either oxidative propane activation followed by re-oxidation of reduced VOx species by CO2 or the PDH reaction assisted by RWGS that can shift the equilibrium to higher conversion.
The direct comparison of PDH and CO2-PDH under identical reaction conditions (Fig. 30 and Table 7) shows that the yield of propene over VOx supported on mesoporous silica322,323 is strongly increased when adding CO2 to the feed. However, no increase in the yield was found over VOx/Al2O3.309 To analyse the positive effect of CO2 on propene production in detail we plotted propene selectivity versus propane conversion over VOx/SiO2 in conventional PDH and CO2-PDH322,323 (Fig. 31). Both the selectivity and the conversion are increased when adding CO2 to the feed. The propane conversion could be further improved upon increasing CO2/propane ratio at a constant partial pressure of propane in the feed. Thus, CO2 enhances both the turnover frequency and the selectivity. Unfortunately, the corresponding selectivity–conversion relationships were not provided to understand which undesired pathways are suppressed in CO2 presence.
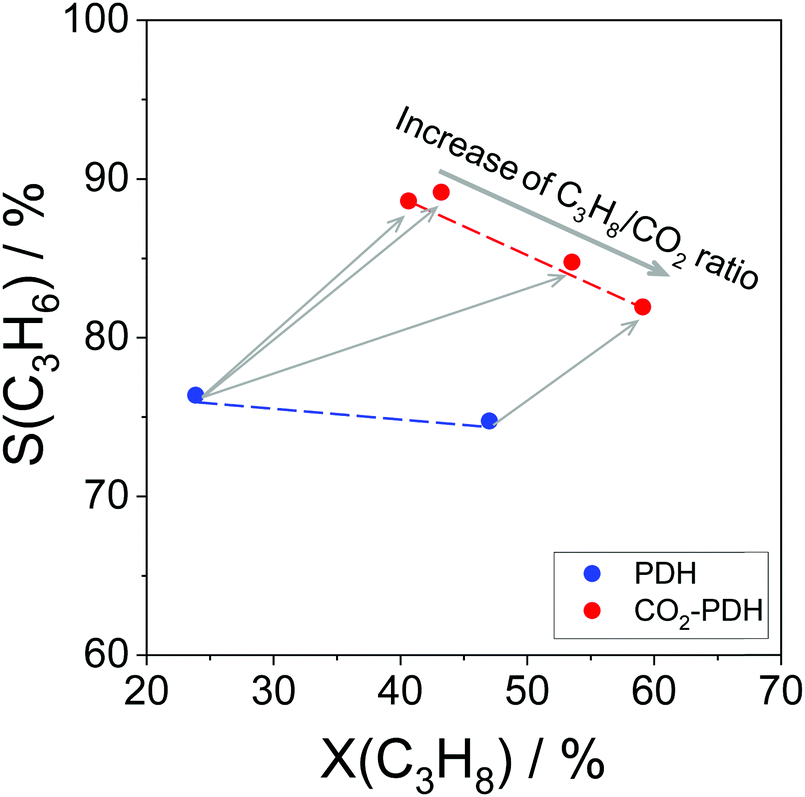 | ||
| Fig. 31 Selectivity–conversion plot for propene in PDH and CO2-PDH over VOx/SiO2. Data are from ref. 322 and 323. | ||
The influence of V loading on activity was very similar for VOx either incorporated in MCM-41322 or in three-dimensional dendritic mesoporous silica nanospheres.323 The propane conversion passes a maximum with increasing V loading. This was explained by the higher intrinsic activity of lower aggregated 2D VOx species in comparison with 3D V2O5 aggregates. The catalysts deactivated partly within 2 hours on stream due to coke formation. They could restore their activity after regeneration in O2 at the reaction temperature. In these both studies, the oxidative mechanism of CO2-PDH was suggested, although RWGS was not excluded. However, no solid experimental proof was presented in ref. 322 and 323. Such information was provided in a separate study, where WOx–VOx/SiO2 catalysts (0–8.8 wt% W and 3–4 wt% V) prepared by incipient wetness impregnation were tested for CO2-PDH over the samples treated with O2, reduced by H2 or reoxidized by CO2 after the dehydrogenation reaction.324 Moreover, pure RWGS reaction and CO2-PDH with a feed mixture D2:C3H8:CO2 were analysed over one catalyst sample. It was concluded that the CO2-PDH reaction proceeds through a Mars–van Krevelen mechanism. Besides, RWGS happens in combination with the PDH reaction, too. The experimental findings were supported by DFT calculations which show that the C–H activation over V5+Ox is the rate limiting step, while reoxidation of the reduced VOx by CO2 is fast.
Sandupatla et al.309 studied the CO2-PDH over VOx(7 wt%)/Al2O3 prepared by incipient wetness impregnation. The propene yield could not be increased in comparison with the PDH. However, when doping VOx/Al2O3 by CrOx or WOx, higher yield was obtained in the CO2-PDH (Fig. 30 and Table 7). This result is surprising because VOx/Al2O3 CrOx/Al2O3 and WOx/Al2O3 becomes less active in CO2 presence (Table 7).
Ga-Containing catalysts. Ga-Containing catalysts have been frequently studied for CO2-PDH.11 Bulk Ga2O3 deactivates fast,325 while its supported counterparts or Ga-containing mixed-metal oxides reveal higher on-stream stability.11 Tetrahedrally coordinated Ga3+ Lewis sites are assumed to be responsible for propane activation. As mentioned above, CO2 does not improve the yield of propene over SiO2-, Al2O3- or ZrO2-supported catalysts (Fig. 30 and Table 7). A slight increase in the yield was reported only for GaOx/TiO2.326 This was explained by the high resistance of this catalysts against blocking sites by CO2 and its high activity in RWGS. Nevertheless, CO2 can have a positive influence on the selectivity to propene and on-stream stability as seen from the below discussion.
The influence of different supports (SiO2, Al2O3, TiO2, ZrO2, MgO) on catalytic performance of supported GaOx species was studied in detail by Xu et al.326 The usage of SiO2 and MgO as supports resulted in low activity. Agafonov et al.317 confirmed low activity of GaOx/SiO2. The catalysts based on other supports were active what was related to their acidic properties.
Despite the fact that propene yield over Ga–Al-based catalysts (supported GaOx and mixed-metal oxide) is lower in CO2 presence (Fig. 30 and Table 7), the selectivity to propene is increased for several catalysts and the lower yield is only due to lower activity (Fig. 32). It is explained by blockage of active sites by CO2 (see below). Ga2O3–Al2O3 solid solutions were studied by Chen et al.325 With increasing Ga2O3 content (from 20 over 50 to 80 wt%) higher propene yields were obtained (see Fig. 30 and Table 7). The lowest deactivation rate over 50 hours at high conversion was found for 50 wt% Ga2O3–50 wt% Al2O3. In comparison with the PDH reaction, higher propene selectivity was achieved in CO2 presence (Fig. 32 and Table 7). This conclusion, however, needs validation through, for example, analysis of selectivity–conversion profiles obtained for individual catalysts at same temperature and using a feed mixture with same propane partial pressure.
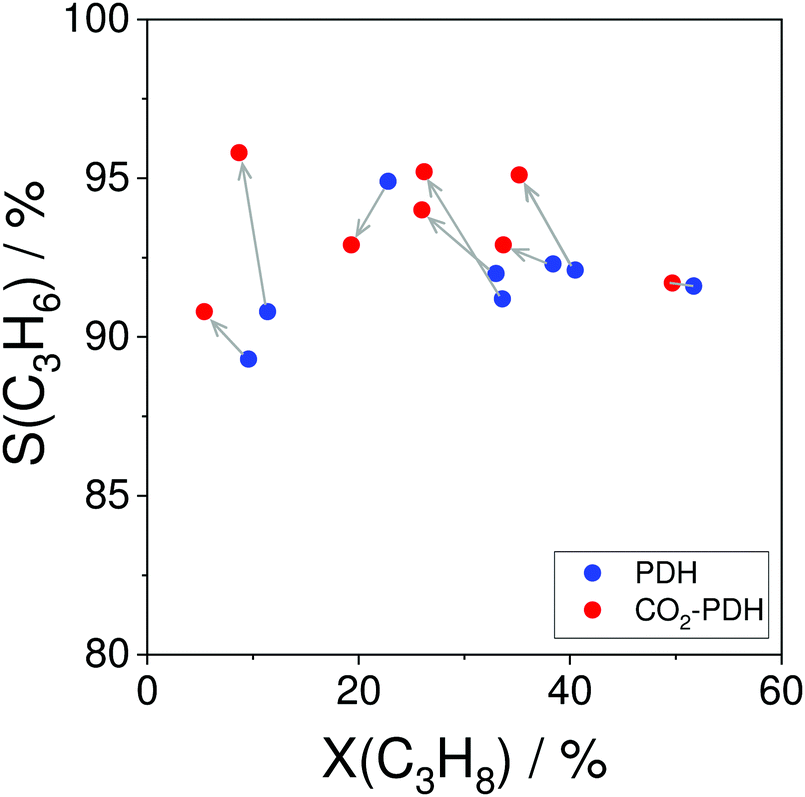 | ||
| Fig. 32 Selectivity–conversion plot for propene in PDH and CO2-PDH over GaOx/Al2O3 and GaAlOx. Data are from ref. 310 and 325–327. | ||
The effect of preparation method (hydrothermal, co-precipitation and grind-mixing) on catalytic performance of Ga–Al mixed oxides was investigated by Xiao et al.327 The hydrothermally prepared sample showed higher and more stable propane conversion whereas the propene selectivity was initially 95% for all samples. Importantly, the selectivity did not decrease with time on stream over this catalyst only. Its superior performance was attributed to higher concentration of medium strong Lewis-acidic tetrahedral Ga3+ on the surface. In addition, a small fraction of reduced Gaδ+ (δ < 2) sites was identified by XPS on the surface of this catalyst. It is, however, not clear how and if these species besides Ga3+ contribute to the high catalyst activity and selectivity. XPS spectra of gallium after both PDH and CO2-PDH are similar indicating the presence of Ga3+ on the surface what let the authors conclude that no redox mechanism occurs over their catalysts.
In-Containing catalysts. Bulk In2O3 shows only low activity and selectivity to propene.328,329 However, the catalytic performance is enhanced when InOx species are highly dispersed either on the surface of an oxidic support material328,330,331 or within a mixed-metal oxide.328,329,331 This is due to easier reducibility of dispersed InOx species in comparison with bulk In2O3.328,329 For binary mixed metal oxides the following activity order was determined: InAlOx > InZnOx > InZrOx and InTiOx ≫ InMeOx (Me = Fe, Mg, Si, or Ce). The latter materials show as low propene yield as bulk In2O3.328 Metallic In0 surface species are identified as catalytically active species. This was also concluded in a study published quite recently dealing with CO2-PDH over In/HZSM-5 catalysts prepared by incipient wetness impregnation.332 In general, the following four experimental findings support the important role of In0 for propene formation:
(i) In3+ is reduced to In0 under reaction conditions and In0 cannot be re-oxidized by CO2.328,329
(ii) In0 was identified by XPS on the surface of catalysts after CO2-PDH or reduction by H2 and treated afterwards by CO2.329,330
(iii) The catalytic activity and product selectivity depend on the concentration of surface In0 species.328–330
(iv) Fresh calcined mixed In2O3–Al2O3 catalysts show an induction period of about three hours where activity increases slightly, and selectivity is enhanced drastically (from 25 to more than 80%). High activity and selectivity are achieved over reduced catalysts from the beginning of the reaction.328
The catalytic performance of In-based catalysts is strongly influenced by acidic and basic properties of the catalyst that can be adjusted by the ratio of In2O3 to the second metal oxide and the nature of the second metal oxide itself. In contrast to bulk In2O3, the number of acidic sites in In–Al–O catalysts increases due to acidic sites of alumina. The total amount of basic sites (mmol gcat−1) increases also but the specific basicity (μmol gcat−1) is lower what is consistent with the higher specific basicity of the low-surface area In2O3 compared to alumina.325 The activity of In–Al–O catalysts increases with temperature but at temperatures above 600 °C the selectivity to propene decreases due to higher cracking activity. On-stream stability also decreased at high temperatures.328
Very recently, In/HZSM-5 catalysts prepared by incipient wetness impregnation were studied for CO2-PDH.332 Small In2O3 crystallites (20–30 nm) are supported on the outer surface of HZSM-5. As already described for supported and mixed-metal oxide catalysts, a part of In3+ is reduced at low temperature (150–300 °C) to In0 and the reduction temperature increases with In2O3 loading. Thus, In0 species are present under reaction conditions as confirmed by XPS of spent catalysts. Catalytic results are similar compared to the catalysts described above. Studying different propane/CO2 ratios showed that activity and selectivity were not much influenced.
Other catalysts. Besides catalysts based on Cr, V, Ga or In, other active elements have been described in literature, too. MoOx or WOx supported on alumina show much lower propane conversion compared to VOx or CrOx deposited on the same support. Propene yield was lower for CO2-PDH compared to conventional PDH (ref. 309, Fig. 30 and Table 7).
ZnO supported on HZSM-5 with the Si/Al ratio of 160 was studied for CO2-PDH.333 Initial propane conversion of 68% decreases to 41% after 30 hours. However, propene selectivity was low (47–62%) due to formation of aromatics and cracking products. A promoting effect of CO2 on propene yield was found and attributed to RWGS reaction. Moreover, catalyst stability was enhanced by CO2.
In summary, several catalysts based on oxides of Cr, V, Ga, In, Mo, W or Zn have been studied for CO2-PDH. Except for In, that is metallic, all other active sites are present as metal-oxide species on the catalyst surface under reaction conditions. Depending on the kind of support and active component, CO2 may have either positive or negative effects on propene yield. This is due to different catalyst resistance against blockage of catalytically active sites by CO2. For some catalysts, CO2 positively affects the selectivity and on-stream stability. With this respect, supported or mixed metal oxides on Ga basis perform superior to other metal oxides.
Several studies published catalytic data obtained under the same reaction conditions (temperature and partial pressure of isobutane) both in the presence and the absence of CO2. Fig. 33 shows the corresponding yields to isobutene to illustrate if the activity or the selectivity is changed by the presence of CO2 in the feed. Additional experimental data are given in Table 8. As seen in Fig. 33, the isobutene yield over several catalysts is higher in the presence of CO2. This is in most cases due to higher activity (Table 8). However, the presented initial data are measured at the beginning of the reaction and very often the catalysts deactivate faster when CO2 is in the feed. Isobutene selectivity in CO2-BDH is in most studies equal or lower (due to the higher conversion) than in BDH. This selectivity is improved by CO2 only in CO2-BDH over VOx supported on silica (SBA-15 or silicalite-1) (Table 8).
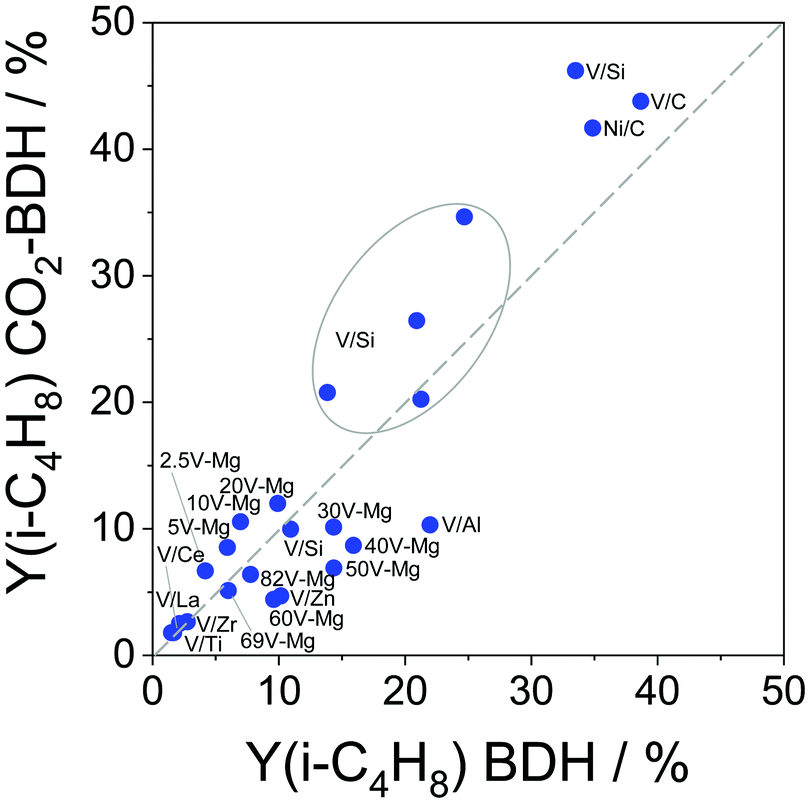 | ||
| Fig. 33 Comparison of isobutene yields for BDH vs. CO2-BDH over metal-oxide based catalysts. For clarity reasons only the element symbols of active metals and supports in the catalyst names are given, i.e. V/Si means VOx species supported on SiO2 and 10V–Mg means V2O5–MgO mixed-metal oxide with 10 wt% V2O5. Data are from literature, for references see Table 8. | ||
| Active material | Support | T/°C | BDH | CO2-BDH | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p(i-C4H10)/bar | X(i-C4H10)/% | S(i-C4H8)/% | Y(i-C4H8)/% | p(i-C4H10)/bar | p(CO2)/bar | X(i-C4H10) 7% | S(i-C4H8)/% | Y(i-C4H8)/% | ||||
| a Measured after 4 h on stream. b Measured after 6 h on stream. All other values were measured at initial time on stream after 0.1–0.2 hours. | ||||||||||||
| 20% VOx | CeO2 | 600 | 0.14 | 2.9 | 73.9 | 2.1 | 0.14 | 0.86 | 3.9 | 64.4 | 2.5 | 337 |
| 20% VOx | La2O3 | 600 | 0.14 | 4.2 | 65.4 | 2.7 | 0.14 | 0.86 | 4.2 | 63.2 | 2.7 | 337 |
| 20% VOx | SiO2 | 600 | 0.14 | 12.1 | 90.4 | 10.9 | 0.14 | 0.86 | 11.6 | 86.0 | 10.0 | 337 |
| 20% VOx | TiO2 | 600 | 0.14 | 2.2 | 68.5 | 1.5 | 0.14 | 0.86 | 2.8 | 63.6 | 1.8 | 337 |
| 20% VOx | Al2O3 | 600 | 0.14 | 26.2 | 83.9 | 22.0 | 0.14 | 0.86 | 13.2 | 78.1 | 10.3 | 337 |
| 20% VOx | ZrO2 | 600 | 0.14 | 2.4 | 69.7 | 1.7 | 0.14 | 0.86 | 2.7 | 65.7 | 1.8 | 337 |
| 20% VOx | ZnO | 600 | 0.14 | 11.5 | 88.3 | 10.2 | 0.14 | 0.86 | 7.9 | 59.3 | 4.7 | 337 |
| 20% VOx | Activated C | 600 | 0.14 | 45.5 | 85.0 | 38.7 | 0.14 | 0.86 | 54.8 | 79.9 | 43.8 | 337 |
| 12VOx | MSU-1 | 600 | 0.25 | 40.5 | 82.8 | 33.5 | 0.25 | 0.75 | 58.8 | 78.5 | 46.2 | 336 |
| 7VOx | SBA-15 | 570 | n.r. | 32.5 | 76.0 | 24.7 | 0.17 | 0.83 | 41.0 | 84.5 | 34.6 | 338 |
| 7VOx | SBA-15 | 570 | n.r. | 31.5a | 67.5a | 21.3a | 0.17 | 0.83 | 23.0a | 88a | 20.2a | 338 |
| 2.5V–Mg–Ox | 600 | 0.17 | 5.8 | 72.0 | 4.2 | 0.17 | 0.83 | 9.8 | 68.0 | 6.7 | 339 | |
| 5V–Mg–Ox | 600 | 0.17 | 7.8 | 76.0 | 5.9 | 0.17 | 0.83 | 12.0 | 71.0 | 8.5 | 339 | |
| 10V–Mg–Ox | 600 | 0.17 | 8.5 | 82.0 | 7.0 | 0.17 | 0.83 | 13.7 | 77.0 | 10.5 | 339 | |
| 20V–Mg–Ox | 600 | 0.17 | 12.4 | 80.0 | 9.9 | 0.17 | 0.83 | 16.0 | 75.0 | 12.0 | 339 | |
| 30V–Mg–Ox | 600 | 0.17 | 17.5 | 82.0 | 14.4 | 0.17 | 0.83 | 13.5 | 75.0 | 10.1 | 339 | |
| 40V–Mg–Ox | 600 | 0.17 | 18.5 | 86.0 | 15.9 | 0.17 | 0.83 | 11.9 | 73.0 | 8.7 | 339 | |
| 50V–Mg–Ox | 600 | 0.17 | 17.1 | 84.0 | 14.4 | 0.17 | 0.83 | 10.0 | 69.0 | 6.9 | 339 | |
| 60V–Mg–Ox | 600 | 0.17 | 12.0 | 80.0 | 9.6 | 0.17 | 0.83 | 6.3 | 70.0 | 4.4 | 339 | |
| 69V–Mg–Ox | 600 | 0.17 | 8.0 | 75.0 | 6.0 | 0.17 | 0.83 | 6.4 | 80.0 | 5.1 | 339 | |
| 82V–Mg–Ox | 600 | 0.17 | 9.7 | 80.0 | 7.8 | 0.17 | 0.83 | 7.7 | 83.0 | 6.4 | 339 | |
| 3Cr | Silicalite-1 | 570 | 0.5 | 31.0 | 67.5 | 20.9 | 0.5 | 0.5 | 37.0 | 71.5 | 26.5 | 343 |
| 3Cr | Silicalite-1 | 570 | 0.5 | 19.5b | 71.0b | 13.8b | 0.5 | 0.5 | 27.5b | 75.5b | 20.8b | 343 |
| NiO | Activated C | 550 | 0.14 | 40.5 | 86.1 | 34.9 | 0.14 | 0.86 | 48.0 | 86.8 | 41.7 | 345 |
The results recently obtained in CO2-BDH over VOx/SiO2 are not consistent. Whereas the isobutane conversion over VOx/MSU-1336 is much higher with CO2, it is not changed over VOx/SiO2.337 Over VOx/SBA-15338 it is only higher for the beginning of the reaction and then decreases rapidly and becomes much lower than without CO2. The same behaviour was determined over VMgOx mixed-metal oxides with V loading up to 20 wt% but not for their higher loaded counterparts where the conversion is always lower for CO2-BDH in comparison with BDH.339 The isobutene selectivity is increased in the presence of CO2 in ref. 336 but decreased in ref. 337 and 338. The catalyst stability is improved in ref. 336 but worsen in ref. 338.
The influence of support on the activity and the selectivity to isobutene in the CO2-BDH was studied by Ogonowski and Skrzynska.337 The activity decreased in the order: VOx/activated carbon ≫ VOx/Al2O3 > VOx/SiO2 > VOx/ZnO. VOx supported on CeO2, TiO2 or ZrO2 showed very low activity. Among these catalysts only VOx/activated carbon revealed an increase in isobutene selectivity in CO2-BDH in comparison with BDH. An increase in isobutane conversion was found over VOx supported on or incorporated into CeO2–ZrO2 as was shown by lower reaction temperature needed in TPRS experiment.340 The selectivity to C4 olefins was above 85% at isobutane conversion up to 20%.
CO2-BDH over VMgOx mixed metal oxides with V2O5 content between 2.5 and 82 wt% was studied in ref. 339 and 341. High isobutene selectivity of 75–85% over these catalysts was obtained at isobutane conversion below 20% for both CO2-BDH and BDH. The selectivity was slightly higher without CO2 (Table 8). For lower V2O5 loading (<30 wt%) the isobutane conversion is only initially higher with CO2 but then decreases with a higher rate than in BDH. The higher deactivation rate is due to higher rate of coke formation in CO2-BDH caused by the higher surface acidity of the catalyst in presence of CO2. The acidity was determined by means of isopropanol decomposition measurements. Although some coke is removed by CO2 through the Boudouard reaction, the rate of its removal is lower than the formation rate. Mechanistic study over the same catalyst (VMgOx)341 showed that two-step dehydrogenation (BDH and RWGS) occurs whereas the redox mechanism was excluded because oxidizing properties of CO2 are insufficient.
For CrOx supported on SBA-15 (partly doped by Ce)342 or silicalite-1,343 it was shown that the activity in CO2-BDH is lower over the catalysts pre-reduced with H2 in comparison with the oxidized ones. The conversion is higher in the presence than in the absence of CO2.342 This indicates that Cr6+ is more active than reduced Cr species and the latter can be partly reoxidized in situ by CO2 as shown by XPS.343 A direct correlation was established between isobutane conversion and content of Cr6+.343 From these results, it was concluded that CO2-BDH follows a redox mechanism. However, because H2 was also detected among the reaction products, the two-step reaction pathway (non-oxidative BDH and RWGS) also occurs. The isobutene selectivity is between 90 and 92% at isobutane conversion up to 35% over Cr–Ce/SBA-15 catalyst342 but between 70 and 80% at 21–36% conversion over Cr/silicalite-1 catalysts.343
An increase in both activity and selectivity over NiO/Al2O3 in CO2-BDH (CO2/isobutane ratio of 15) in comparison with the BDH reaction was reported in ref. 344. However, it was not indicated if the isobutane was diluted to the same degree in both reaction feeds. NiO supported on activated carbon was studied for the CO2-BDH by the same group.345 NiO is partly reduced to Ni by the carbon support during calcination in Ar atmosphere at higher temperature and, thus, by variation of calcination temperature the different catalytic performance of NiO and Ni could be studied. Whereas the former catalyses BDH and RWGS, the latter is only active for BDH.
MoO3 catalysts either undoped or doped with Fe, Ce or Sn were prepared by decomposition of heteropoly molybdates and tested for CO2-BDH.346 With increasing CO2/isobutane ratio the conversion increased. The selectivity to isobutene over these catalysts was very low (below 25%) because of high activity for cracking what results in selectivity to cracking products up to more than 90%. Experiments without CO2 were not reported.
Summarising the attempts of CO2-mediated dehydrogenation of propane and isobutane, one can state that promising results were obtained over some catalysts. For different mesoporous silica with supported VOx species, olefin yield in the presence of CO2 is higher than in the CO2-free dehydrogenation and reaches the industrially relevant level of 36–53% (see Fig. 13 and 16). However, co-feeding of CO2 does not help to reduce catalyst deactivation. Although it was shown that regeneration in air is possible for some cycles, long-time studies are missing up to now. Further studies in this respect are necessary before drawing a definitive conclusion on industrial applicability of such approach. Moreover, economic evaluation of a CO2-assisted process is necessary that compares the benefit from higher olefin yield with the additional effort for removing co-formed CO from the product mixture. Since to date only a few studies on CO2-assisted dehydrogenation of propane or isobutane over the promising VOx/SiO2 catalysts have been published, even better-performing catalysts and deeper understanding of the function of CO2 in the reaction mechanism including coke formation can be achieved in the nearer future.
7.2 Dehydrogenation with N2O
A few studies have used N2O as an oxidising agent in the ODP reaction using V-based catalysts,179,211,347 Cr-based239 catalysts or Fe-containing zeolites.348–352 The main purpose of the studies with V-based catalysts was to elucidate factors affecting selective and non-selective reaction pathways. When using N2O instead of O2 for the ODP reaction, the selectivity to propene increases due to an increase in the reduction degree of bulk and surface VOx.179,181,182 In addition, non-selective biatomic (peroxo) adsorbed species cannot be formed from N2O that also has a positive effect on the desired selectivity.177,178Fe-Containing zeolites of the MFI type have been often applied for the ODP reaction in the past. Such studies have been stimulated by the fact that a certain kind (so-called alpha oxygen) of oxygen species is formed from N2O over FeOx sites and can selectively oxidize benzene to phenol.353 We now discuss the results reported after 2010. Similar to the ODP reaction with O2, we plot available selectivity–conversion data obtained at 450 and 500 °C over differently prepared catalysts. The results are shown in Fig. 34.
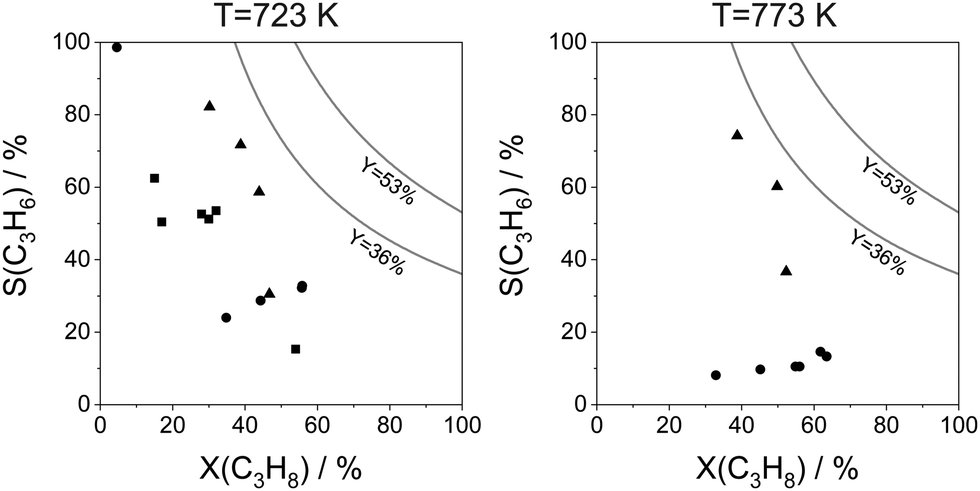 | ||
| Fig. 34 Selectivity–conversion data obtained in the ODP reaction with N2O over FeOx/ZSM-5 catalysts prepared through grafting352 (▲), solid348 (■) or liquid ion350 (●) exchange methods. The lines show the yield of propene. | ||
Propene selectivity above 70% was achieved at degrees of propane conversion between 30 and 40%. This performance is superior to that achieved with O2 (Fig. 20). The results in Fig. 34 also provide some hints about the importance of method of deposition of FeOx species, iron loading and zeolite structure. The highest yield was obtained over the catalysts prepared through a grafting method,352 while those synthesised through solid348 or liquid350 ion exchange methods were less selective. In the grafting method, ferrocene (iron precursor) sublimated at high temperatures and then reacted with the Brønsted acidic sites of zeolite supports, which were H-ZSM-5, H-mordenite, H-USY, H-ZSM-35 and H-beta. In contrast to all other catalysts, FeOx/ZSM-35 showed very low activity. The highest activity and propene selectivity were achieved over FeOx/ZSM-5. For the latter catalyst, the effect of calcination temperature on the ODP performance was investigated. As a general trend, the conversion of propane decreased with increasing calcination temperature from 600 to 900 °C, while the selectivity to propene increased. Importantly, when these catalysts were used in the ODP reaction with O2, carbon oxides were the main reaction products. It was concluded that N2O provides an atomic oxygen species that facilitate the desired dehydrogenation reaction. Extra-framework Fe–O–Al species were suggested to be responsible for generation of such species from N2O. FeOx/ZSM-5 catalysts prepared through a solid-state exchange technique using FeCl2·4H2O as the iron precursor possess differently structured FeOx species ranging from isolated to large Fe2O3 particles.348 The presence of the latter is probably a reason for low propene selectivity obtained over such catalysts.
A simple impregnation method with FeCl2 dissolved in acetylacetone was used to prepare two FeOx/ZSM-5 catalysts. They were tested in the ODP reaction at 400 °C using a diluted feed (1.5 vol% N2O and 1.5 vol% C3H8). Propene selectivity between 40–50% was achieved at about 30% propane conversion. Under such reaction conditions, coke formation, which is a major undesired reaction, is strongly suppressed. Nevertheless, the catalysts lose their activity with rising time on stream. Protonic zeolite sites were concluded to contribute to coke formation and accordingly affect catalyst deactivation.
Although the usage of N2O has a positive effect on propene selectivity, this oxidant is not attractive for large-scale applications. First of all, it is very expensive in comparison with O2 and not available in large amounts as required for propene production. Moreover, N2O decomposition is an exothermic process and thus precautions should be taken into account for heat management. In case of propane recycling (typically propane conversion is below 50%), non-reacted propane must be separated from N2 formed from N2O.
7.3 O2-Lean dehydrogenation
The oxidative dehydrogenation of propane and isobutane is typically carried out using feeds with the ratio of C3H8(C4H10)/O2 between 2 and 0.5 to achieve high propane conversion. However, the reported conversion degrees are significantly lower than the expected stoichiometric values due to formation of carbon oxides requiring between 7 and 13 oxygen atoms in comparisons to 0.5 for the desired selective reaction. The oxidative dehydrogenation of propane and isobutane was also investigated under O2-lean conditions (C3H8(C4H10)/O2 > 10).150,354 For the oxidative dehydrogenation of isobutane, an overall C3–C4 olefin selectivity of 97% (85% to isobutene, 9% to n-butenes and 2% to propene) at isobutane conversion of 54% was achieved at 560 °C over VOx(5 wt%)/MCM-41.354 When performing the ODP reaction under O2-lean conditions at same temperature and over same catalyst, the selectivity to propene of 80% at 50% propane conversion was reported.150 From a mechanistic viewpoint, it was suggested that the oxidative alkane dehydrogenation takes place in the up-stream catalyst layer when gas-phase O2 is still present. After the oxidant was completely consumed, olefin is formed through the non-oxidative dehydrogenation in the bottom part of catalyst layer. In comparison with the pure PDH reaction, heat required for this reaction can be provided by the ODP reaction in the upper catalyst layer. Another advantage of O2-lean operation is the low concentration of gas-phase oxygen. Thus, safer and cheaper operation can be ensured than in the case of oxidative dehydrogenation with O2-rich feeds.Similar to ref. 354, Fan et al.281 also investigated the ODB reaction over VOx/MCM-41 with different V loading under O2-lean conditions. The catalyst with V content of 6 wt% showed the highest yield of isobutene of 42.1% at the corresponding selectivity of 87.3%. Isolated VOx species were concluded to reveal the highest activity, selectivity and on-stream stability.
7.4 Propane oxidative dehydrogenation upon alternating propane and air feeds
Another opportunity to improve overall economy of the ODP reaction and the selectivity to propene is the usage of air instead of pure oxygen for eliminating expensive air separation. Such operation can be achieved through usage of oxygen-conducting membranes or upon alternating feeding of air and propane. The latter operation mode is also called chemical looping. Propane and air are separately fed to the reactor, where the alkane is oxidized by lattice oxygen of reducible metal oxide followed by catalyst reoxidation with air. Using a membrane (BaBi0.05Co0.8Nb0.15O3−δ perovskite) reactor with CoOx/SiO2 catalyst, propane yield of about 50% at propene selectivity of 74% was obtained over 50 h on stream at 650 °C.355 Unfortunately, such high temperatures are required to ensure reasonable flux of lattice oxygen through the membrane. No data about catalyst productivity was provided.In comparison with membrane reactors, chemical looping approach can be applied at low temperatures as typically surface or near surface lattice oxygen is consumed for propane oxidation. As lattice oxygen diffusion is suppressed under such conditions, catalysts treated in propane must be reoxidized after a few minutes on stream. Several studies elucidated the application potential of such approach with V-based,151–156,356–359 Pb-based,360 Ce-based,361 Co-based234 or Ni-based catalysts.227,362–364 As seen in Fig. 20, an attractive performance, i.e. propene selectivity above 80% at propane conversion above 60%, can be achieved when using a V-based catalyst.
It is, however, worth mentioning that the published studies on this topic do not always exactly report how product selectivity was calculated. For example, no reports about formation of carbon oxides upon catalyst reoxidation are provided. It is well known that reduced V-based catalysts can form coke. As this product was formed from fed propane it must also be taken into consideration. Thus, it is important to determine overall amounts of propane consumed and products formed both in propane and air cycles through integration of the corresponding temporal profiles. On this basis, the selectivity to different products should be calculated. In some papers, it is not reported if such approach was used. If the selectivity values are determined after a certain reaction time or without considering coke formation, they may be overestimated. For example, based on propane converted into gaseous carbon-containing products only, Rostom and de Lasa153 obtained 93% propene selectivity at 25% propane conversion. This high selectivity value drops to 85% when considering coke formation.
A recent study156 should be especially mentioned as it provides some mechanistic insights into the factors governing product selectivity. Those authors investigated MoVOx binary materials. In agreement with various previous studies, lattice oxygen is responsible for the oxidative propane dehydrogenation to propene. Importantly, the binding energy of V–O bonds increased upon addition of Mo promoter. Consequently, non-selective propane conversion into carbon oxides could be slightly hindered. The most selective MoVOx (V/Mo = 6) catalysts resulted in 89% propene selectivity at 36% propane conversion at 500 °C. This catalyst showed stable operation over 100 dehydrogenation/reoxidation cycles. Unfortunately, it is not clear if coke formation was considered for calculating the selectivity.
Despite the above approaches are attractive they suffer from low productivity, which is limited by the ability of the materials to provide their lattice oxygen. Thus, novel materials with high oxygen-storage capacity and oxygen mobility are needed to ensure high productivity.
7.5 Alkane dehydrogenation with selective hydrogen combustion
Selective hydrogen combustion (SHC) was proposed to be used in combination with non-oxidative alkane dehydrogenation in order to increase olefin yield.365,366 Since the non-oxidative dehydrogenation reaction is endothermic and requires a lot of energy, combusting a part of hydrogen obtained during dehydrogenation process produces the required energy making the overall process heat-balanced or even exothermic. Moreover, consumption of hydrogen in SHC reaction shifts the reaction equilibrium towards the desired olefin. As a result, higher olefin yields can be obtained. The SHC catalysts typically described in literature consist of oxides of metals of group IIIA, IVA and VA (Bi, In, Pb, Sb, etc.), mixed oxides based on Mo, W, Mn as well as Pt-containing materials.365,367–369There are two operation modes for the performing DH reaction coupled with SHC: (i) sequential co-fed DH→SHC→DH mode and (ii) redox DH + SHC mode.365,366 The first configuration is continuous and implies spatial separation of DH and SHC catalysts. The DH and SHC catalysts are arranged in series, high oxidation state of SHC catalyst is achieved by continuous co-feeding of oxygen or air into the SHC reactor. Hazard arising from mixing oxygen with hydrogen and hydrocarbons at high temperature as well as low selectivity and stability of hydrogen combustion catalysts are the main disadvantages of such mode. In the second mode, the DH and SHC catalysts are physically mixed. Alkane dehydrogenation and hydrogen combustion proceeds in the same reactor in the absence of gaseous oxygen. Hydrogen combustion occurs with participation of lattice oxygen of the SHC catalyst. Regeneration of the catalysts is performed in oxygen or air flow in the oxidation step. Dehydrogenation and oxidation steps are separated by purge cycles with inert gas to avoid contact of hydrocarbons and hydrogen with oxygen.367
8 Synthesis and characterization of supported and bulk catalysts
As demonstrated in Sections 2–7, the performance (activity, selectivity and on-stream stability) of differently composed catalysts in both non-oxidative, oxidative and CO2-mediated dehydrogenation reactions is strongly influenced by their various physicochemical properties and the structure of supported metal oxide species or surface defects (Fig. 35). All these catalyst characteristics are affected by preparation method, loading of active species, the kind of support, the presence and the kind of promoter as well as catalyst treatment conditions. All these factors could have different influence on the properties of different catalysts. Nevertheless, some general features could be identified within one preparation method. Below we will discuss the most common methods for the preparation of supported and bulk catalysts with the purpose to highlight their applicability for controlling specific solid material properties.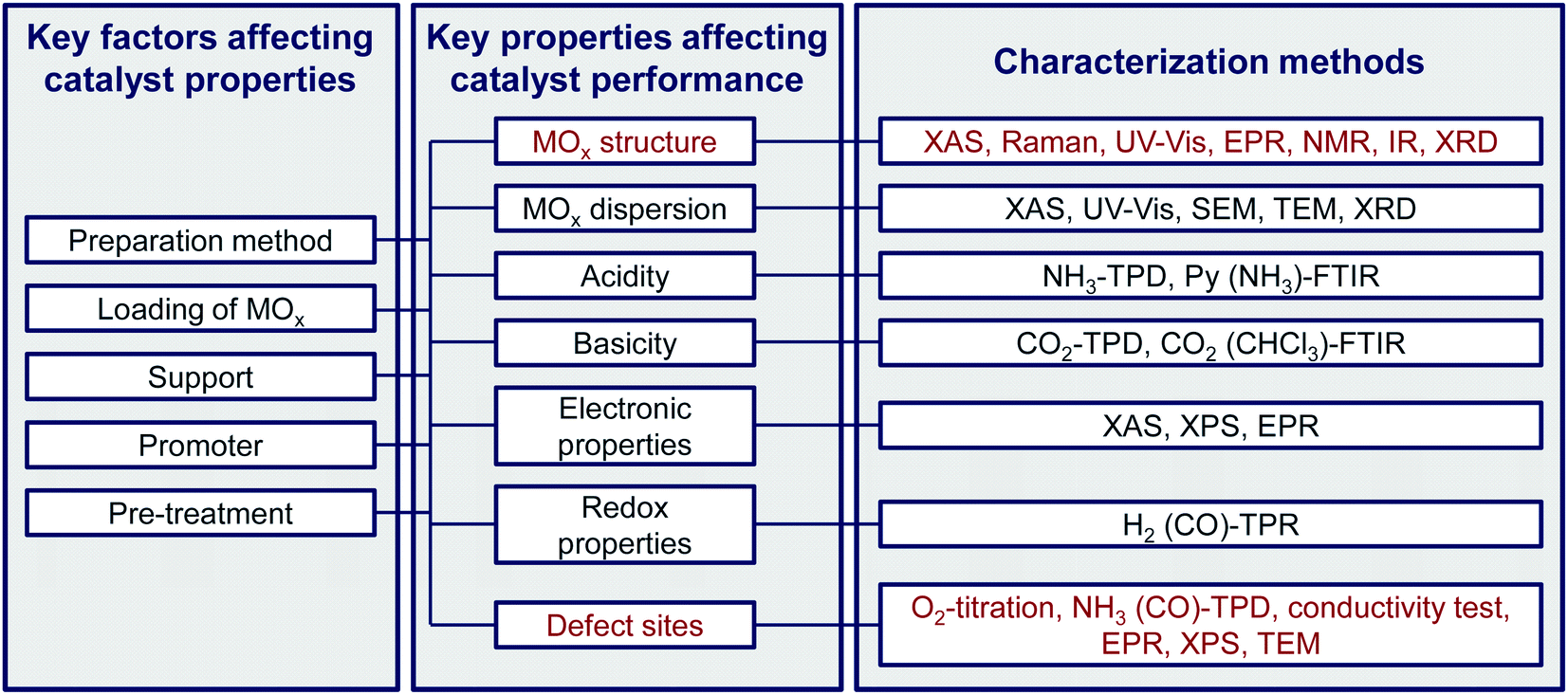 | ||
| Fig. 35 Key factors and properties affecting catalyst performance as well as characterization methods for analysing such properties. The most common methods applied for analysing the structure of supported species and for detection of defect sites are written in red and will be discussed in detail in Sections 8.3 and 8.4. | ||
8.1 Methods for preparation of supported catalysts
Impregnation is one of the easiest and frequently used preparation methods.370 It implies adding the solution (typically aqueous solution) of metal precursor(s) to a support material followed by drying (and calcination if necessary). Two different impregnation strategies are applied, i.e. (i) wet impregnation performed with an excess of a solvent and (ii) incipient wetness impregnation (or dry impregnation), which implies the usage of a certain amount of solvent required only to fill the pore volume of a support material. Although each impregnation method is simple and produces low waste amounts, it offers only little control over the structure of supported metal oxide species and their dispersion. The structure of the final catalyst can be tuned upon adjusting the pH and ionic strength.371Deposition–precipitation (DP) method was initially developed for the synthesis of catalysts with high loading of active components which is not possible to achieve via the impregnation method due to the limited solubility of precursor components.370,372 The DP method ensures the formation of metal oxide compound with low solubility (often metal hydroxides) on the surface of a support material from a solution of precursor usually by a change of pH. The surface of the support acts as a seed for the nucleation. Typically, this method cannot guarantee uniform size distribution of deposited metal oxide species. However, catalysts with highly dispersed metal oxide species can be prepared through varying conditions of the synthesis. It is also worth mentioning that the interaction between the precursor and the support plays a crucial role in the production of the final catalyst. Typically, when the interaction is too weak, large particles are formed, while too strong interacting can result in the formation of metal–support phases.370
Chemical vapor deposition (CVD) method is carried out by reaction/decomposition of a gaseous metal precursor stream onto the support.371 An obvious advantage of this method in comparison with the impregnation and DP methods is the ability to control coverage, nucleation rate and particle size. Usually, the catalysts prepared by CVD method are characterized by higher dispersion and better catalytic activity, however, particle size is not always uniform.
Organometallic grafting is applied to produce catalysts possessing well-defined supported species with a certain structure and specific, tailored surface coordination.371,373,374 It is a molecular-level technique which implies the reaction of organometallic precursors with the surface of a support material followed by removal of the organic ligands. So prepared catalysts are often used for investigating the correlation between the supported structures and spectroscopic characteristics as well as for studying reaction mechanisms and understanding structure–performance relationships. This method does not seem to be suitable for large-scale applications.
Atomic layer deposition method (ALD) is a method, where two self-limiting reactions occur sequentially between gas precursors and a substrate.371,375,376 Typically, two types of gaseous precursors are pulsed alternately into the preparation chamber. In each new pulse, precursor molecules react with the surface functional groups generated in a previous pulse. Unreacted precursor and gaseous by-products are removed by purging with an inert gas. At the end of each cycle, functional groups are generated on the surface of support material. They can again react with the gas-phase precursor. Thus, the desired precursor is deposited in a layer-by-layer fashion. By varying the number and type of ALD cycles, desired loading and surface structure can be obtained. The method allows the production of highly dispersed catalysts with uniform structure of supported species with near atomic precision. The usage of ALD method is, however, limited by the availability of suitable gaseous precursors, which can react with support functional groups but are thermally stable at growth temperature. Moreover, the limited size of conventional ALD chamber does not allow the production of high catalyst amount. For addressing this question, spatial ALD with continuous operation and/or ALD based on fluidized bed reactor have been proposed.377,378
In summary, Table 9 was prepared and provides a brief description of each of the above-mentioned methods including their advantages and drawbacks.
| Impregnation | Deposition–precipitation | Chemical vapor deposition | Organometallic grafting | Atomic layer deposition (ALD) | |
|---|---|---|---|---|---|
| Description | Impregnation of a support material with a solution of active component precursor | Catalyst precursor is deposited on the surface of support material by precipitation | Gaseous precursor is decomposed on the surface of support material | Molecular organometallic precursor reacts with the surface of support material followed by removal of the organic ligands | Deposition of gaseous precursor on the surface of support material in a layer-by-layer fashion |
| Advantages | Simple execution, low waste streams | Simple execution, allows to produce highly loaded catalysts | Ability to control coverage, nucleation rate and particle size; usually high dispersion of supported species | Well-defined isolated surface species with uniform structure | Ability to control loading and surface structures resistant to aggregation. High dispersion |
| Disadvantages | Little control of the kind of surface species and their dispersion | Usually nonuniform distribution of particle size and structures | Special equipment is needed; particle size is not always uniform | Expensive, difficult to execute | Expensive; limited by availability of suitable precursors; high amount of catalyst is difficult to be produced via conventional ALD |
8.2 Methods for preparation of bulk catalysts
Precipitation is one of the most frequently applied methods to prepare bulk catalysts and supports.379,380 It can be used for the preparation of either single metal oxide or mixed metal oxide catalysts. Usually hydroxides or carbonates are the most preferable substances to be precipitated due to their low solubility, simplicity of their decomposition into oxides and minimal toxicity.372 Depending on precipitation conditions, it is possible to vary crystallinity and crystallite size of the resulting precipitant. The most significant factors that affect the properties of the precipitant include pH, residence time, temperature, agitation, nature and concentration of precursors as well as the usage of various additives.380 Usually, high concentration of precursors, low temperature and short ageing time lead to the formation of finely crystalline or amorphous substances, which are difficult to filtrate. Low concentration of precursors, high temperature, and extended ageing result in coarse crystalline materials, which are easier to filtrate. The formation of large amounts of salt-containing waste solutions is the main drawback of this preparation method.Sol–gel method implies the formation of sol (liquid suspension of solid particles smaller than 1 μm) followed by its transformation into gel.380–382 The sol is formed as a result of hydrolysis of an inorganic salt or a metal alkoxide. Further polymerization through condensation of hydroxyl and/or alkoxy groups results in the formation of gel. It should be mentioned that such method allows control of the texture, composition, homogeneity and structural properties of the final materials. It offers better control over surface area, pore volume and pore size distribution in comparison with the precipitation method.372 Generally, if the initial gel contains polymeric branched and cross-linked chains, it has large void regions and is structurally rigid.381 Macroporous and mesoporous oxides are usually formed after calcination of such gel. The gel with a low amount of branched and cross-linked chains has smaller void regions and is structurally weak. Such gel collapses readily during calcination and results in the formation of microporous oxide with low surface area. pH value during gel formation and ageing of gel strongly influence structural properties of the final metal oxide. The rate of condensation process at low pH values is slow in comparison with the hydrolysis, that results in the formation of weakly cross-linked gel and final metal oxide with low surface area. The rate increases with rising pH. This results in the formation of high amount of cross-linkages in the gel and in an increase in pore volume and surface area of resulting metal oxides. In most cases, ageing also enhances cross-linking of the gel.
Hydrothermal method is a technology for crystallizing materials from an aqueous solution in an autoclave above 100 °C.383,384 The procedure includes three basic steps: achievement of supersaturation, nucleation and crystal growth.380 The properties of the resulting material are affected by pH, temperature, pressure, time and concentration of precursors.372
Templating method implies the usage of a porous polymeric matrix as a template.385,386 The preparation includes impregnation of a template with an aqueous solution of metal-oxide precursor, followed by its combustion. The pore structure of the resulting oxide is strongly influenced by template, concentration of precursor, heating rate, and combustion temperature. Typically, so prepared oxides are characterized by high surface area, presence of mesopores and good thermal stability.
8.3 Characterization of supported species
Characterization of supported species participating in catalytic reaction is of a great importance for identifying the kind of active sites, elucidating the fundamentals for their formation and transformation as well as for understanding reaction mechanism and structure–activity–selectivity relationships. The most common methods applied for analysing the structure of supported species are X-ray absorption spectroscopy (XAS), electronic paramagnetic resonance (EPR), UV-vis spectroscopy, IR spectroscopy, Raman spectroscopy and NMR. A brief description of the methods is given below.XAS is a powerful tool for determining the local geometric and electronic structure of a matter on an atomic scale.387,388 The method is element specific and does not require a long range order. An XAS spectrum can be divided into two regions: X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). XANES data are related to the electronic transitions from core electronic states of the metal to the higher energy excited electronic states (lowest unoccupied molecular orbital). Thus, the information about oxidation state and geometry of the metal site can be obtained. As EXAFS signal is observed because of an interaction of the outgoing photoelectrons and backscattered electrons from the near neighbour atoms, the numbers and types of neighbouring atoms as well as interatomic distances can be determined. Importantly, XAS spectra can be collected under in situ or even operando conditions.
EPR is a technique for detection of paramagnetic species with unpaired electrons and analysing their coordination.389 The concentration of such species can be determined by comparing their EPR signal with that of a standard with known spin concentration. In situ or operando EPR allows investigation of the changes occurring with the species upon their treatment in various atmospheres at different temperatures as well as kinetics of such transformations.390
UV-vis spectroscopy is widely applied for analysing oxidation state of metals and molecular structure of MOx (M is a metal) species.391,392 However, some UV-vis bands can overlap with each other and other can be too weak, therefore, distinguishing between multiple species can be complicated.393 The UV-vis edge energy (Eg) can be calculated from the spectra to estimate oligomerization degree of MOx species: the higher the Eg value is, the higher is the dispersion of the species. The UV-vis technique can be applied under in situ and operando conditions.
IR spectroscopy provides molecular vibrational information and is more sensitive to asymmetric vibrations. It allows the detection of anchoring OH sites for MOx species on the surface of support and provides some information about molecular structure of MOx species. The method can be used in situ or operando for deriving insights into surface intermediates and reaction products formed on the surface of catalysts tested.393 The usage of probe molecules in IR spectroscopy is a powerful tool to investigate the surface properties of the catalysts (acidity, basicity, dispersion of supported component, structure of supported species, etc.) as well as the nature of adsorbates under reaction conditions.394,395 Some probe molecules frequently applied in IR studies include pyridine, NH3, CO2, CO, H2, NO, C2H4. It should be mentioned that probe molecules must satisfy several requirements: they should be detectable by IR, have relatively small size (with some exceptions) and high extinction coefficients, the probe–surface interaction should be ideally weak. Moreover, the selection of a probe molecule is based on assigned task and the features of the sample under the study. Some specific examples of the usage of probe molecules in IR spectroscopy can be found in ref. 394 and 395.
Similar to IR, Raman spectroscopy also provides molecular vibrational information, but is more sensitive to symmetric vibrations. It is typically applied for determining molecular structure of supported MOx species and their transformations under different conditions.393,396,397
Solid-state NMR is used to study the substances possessing nuclei with a non-zero nuclear magnetic moment.398 The method is often combined with DFT calculations and gives information about the coordination and chemical environment of specific species.399
In summary, the described methods have been often used for the characterization of supported MOx species. However, no single technique is fully enough for obtaining a complete picture about the nature of supported MOx species. Each method has its own requirements and limitations. Moreover, the interpretation of the results obtained with most of these methods is based on the comparison of the obtained data with those of reference materials of known structure. The divergence of the measured parameter from that of a standard can be interpreted in a different way leading to different conclusions. Accordingly, only the usage of multiple characterization techniques can give true insights into the structure of MOx species.
8.4 Methods for detection of surface defects of bulk catalysts
In general, various defects on the surface of metal oxide catalysts have higher reactivity in comparison with non-defective sites and thus play an important role in heterogeneous catalysts.400–402 For the PDH (Section 2.3) and BDH (Section 3.2) reactions, coordinatively unsaturated metal cations were concluded to be the active sites on the surface of alternative-type bulk catalysts (Fig. 4). They are located near to oxygen vacancy. Thus, for further development of such catalysts, it is important to identify and quantify such defects. Several characterization methods can be applied for such purposes. They are pulse titration tests with probe molecules, e.g. O2 or N2O, ionic conductivity measurements, NH3-TPD, CO-TPD, UV-vis spectroscopy, HAADF-STEM, XPS and EPR. Some of them can provide quantitative information, while others can only identify the presence of such defect sites. Below we provide a brief description of each method.Titration pulse experiments with an oxidant are based on the phenomenon that anion vacancies can react with the pulsed probe molecule. O2 or N2O are typically used for such purposes. The experiments can be carried out with any set-up enabling to pulse a known amount of such molecules and their quantification at the reactor outlet. When the number of anion vacancies is very low, that is typically the case, the temporal analysis of products (TAP) reactor is an ideal equipment for such purposes.403–405 The lowest pulse size of this technique is about 0.2 nmol. Regardless of the used technique, an on-line mass spectrometer is typically applied for detection of pulsed gases. A certain number of pulses is sequentially introduced until all oxygen vacancies are filled due to their reaction with the pulsed oxidant. This situation is realized when the number of pulsed molecules is equal to the number of these molecules at the reactor outlet. Some precautions should be considered when performing such tests. Reaction temperature should be identified, at which surface vacancies can react with pulsed probe molecules, while diffusion of surface lattice oxygen into metal oxide bulk is hindered. When N2O is used as a probe molecule, it is important to check if oxygen species formed from N2O can also decompose N2O. The latter process must be avoided. The O2-pulse titration can be considered as semiquantitative method for the detection of active sites in ZrO2-based catalysts as it is difficult to suppress lattice oxygen diffusion.93
Ionic (lattice oxygen) conductivity measurements of metal oxides can also provide information about the presence of oxygen vacancies in these materials. Such type of conductivity correlates with the number of oxygen vacancies.26,96 The higher the conductivity, the more oxygen vacancies are present in the sample. This method is, however, not quantitative. Typically, the measurements of overall electrical conductivity of oxides are performed with two inert electrodes upon variation of partial pressure of oxygen. The conductivity of a pure ionic conductor does not depend on the pressure, while the conductivity of n- or p-conductors decreases or increases. A more detailed description of such experiments and the main concepts related to the electronic properties of metal oxides are provided in ref. 406 and 407.
The number of coordinatively unsaturated metal cations can be determined from temperature-programmed desorption tests using probe molecules selectively adsorbing on such sites. It was demonstrated that the formation of Lewis sites in bare alumina is mainly due to the defective Alcus3+.28,134 Such sites can bind basic probe molecules such as pyridine and NH3.28,94 Accordingly, the amount of Alcus3+ can be determined by calculating the amount of desorbed NH3 from the surface of pre-treated alumina during NH3-TPD experiment. It should be however mentioned that the presence of Brønsted acidic sites in the sample results in overestimating the amount of Alcus3+ determined by NH3-TPD. To ensure the absence of Brønsted acidic sites a Pyridine-adsorbed Fourier Transform Infrared Spectroscopy (Py-FTIR) can be applied. Such technique distinguishes Brønsted (the band at around 1540 cm−1) and Lewis (the band at around 1450 cm−1) acidic sites. Similarly, CO-TPD technique was successfully applied for the titration of Zrcus4+ sites in ZrO2-based catalysts.26,99 Being a Lewis acidic site, Zrcus4+ adsorbs CO molecule. The amount of desorbed CO should be equal to the amount of Zrcus4+ sites.
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) can provide limited information about the amount and distribution of oxygen vacancies.26 The X-ray photoelectron spectroscopy (XPS) and electron paramagnetic resonance (EPR) provide information about the presence of oxygen vacancies.27 The XPS spectra of samples possessing oxygen vacancies are usually characterized by a shift in the binding energy of O 1s peak and/or of metal cation, while the EPR spectra of such samples possess the corresponding signal related to the oxygen vacancies (g = 2.001–2.005).
8.5 General experimental requirements
After analysing the literature in the preceding sections, we have noticed that sometimes not all required information is provided in the published articles for a proper comparison of different catalysts. For example, degree of alkane conversion, contact time, WHSV or GHSV are not provided. As a consequence, it is impossible to estimate catalyst activity when such values are not reported. In some studies, WHSV and GHSV are mixed up.In addition to some missing information, conditions applied for catalytic tests are not industrially relevant and so-obtained data may result in wrong conclusions. Particularly for the non-oxidative or CO2-mediated dehydrogenation of propane and isobutane, a lot of studies have been carried out under conditions with very diluted (10 vol% of alkane and lower) reaction feeds. As the equilibrium conversion strongly depends on alkane partial pressures (Fig. 3), this catalyst characteristics cannot be used for catalyst comparison. Such conditions are also industrially irrelevant and not favourable for coke formation. Thus, catalysts tested with diluted feeds may artificially show higher selectivity in comparison to those tested with alkane-rich feeds. Moreover, many researchers calculate product selectivity on the basis of gas-phase components without considering coke formation. Such calculation way overestimates the selectivity values. Against this background, we would like to highlight some general recommendations for investigating the non-oxidative or CO2-mediated dehydrogenation of propane and isobutane as well as for evaluation of experimental data. Some experimental requirements for carrying out the oxidative propane dehydrogenation to propene are published in a review of Carrero et al.12 Our recommendations are as follows:
i. Ensuring that the tests are carried out under conditions free of any mass-transport limitations and strong contributions of gas-phase reactions. The latter play a role at high temperatures and long contact times.
ii. When determining reaction rates and other parameters related to the rate (e.g. turn over frequency or activation energy), the degree of alkane conversion should be below 10% to ensure a pseudo differential reactor operation regime.
iii. For demonstrating application potential of the developed catalysts, it is highly recommended to use industrially relevant feeds (alkane partial pressure higher than 0.3 bar). Comparison with the state-of-the-art catalysts in terms of their selectivity and productivity (space time yield) should be made under similar experimental conditions and degrees of alkane conversion, which should be at least larger than 20%.
iv. Coke cannot be ignored as a reaction product when alkane conversion is higher than 10%. Otherwise, the selectivity to gas-phase products is overestimated. Thus, carbon balance is highly important for ensuring precision in the obtained data. Eqn (3) can be used for calculating coke selectivity.
v. For analysing coke formation, diluted feeds (alkane partial pressure below 0.3 bar) should be avoided. As olefin is the main precursor of coke, which is actually a minor product, high olefin partial pressures are required for precise determining the selectivity to this side product.
vi. Reaction induced changes in the number of moles (two molecules, i.e. H2 and C3H6, are formed from one C3H8) must be taken into account when particularly operating with alkane-rich feeds. The changes can be easily considered when using reaction feeds with an inert standard, e.g. N2. Alkane conversion and selectivity to gas-phase products and coke can be calculated as following:
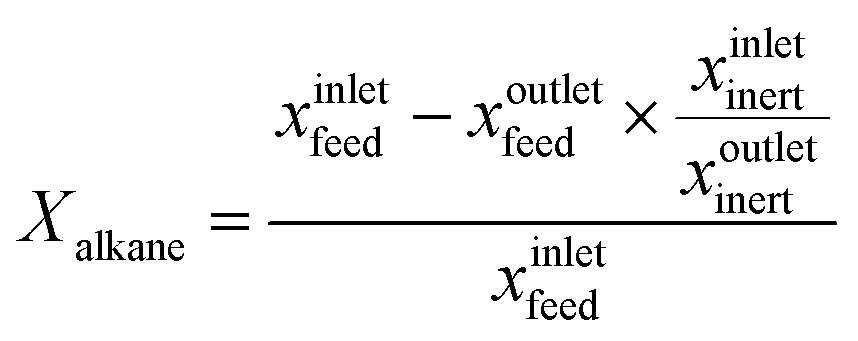 | (1) |
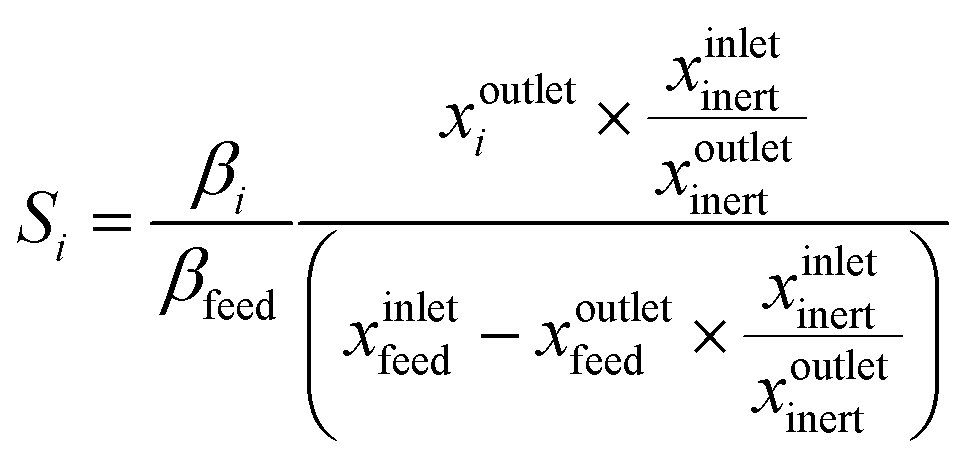 | (2) |
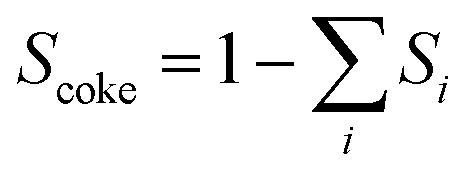 | (3) |
9 Conclusions
The present review has provided a critical analysis of the current status on the conversion of propane and isobutane into the corresponding olefins through non-oxidative, oxidative and CO2-mediated routes. A particular focus was put on comparing the developed catalysts in terms of their application potential. Catalyst design concepts and mechanistic aspects of product formation including the kind of active sites and ways for their purposeful creation have also been thoroughly reviewed with the purpose to identify fundamentals required for tailored catalyst design. Some reaction engineering concepts have been briefly discussed.From a mechanistic viewpoint, some general similarities, and differences between non-oxidative, oxidative, and CO2-mediated dehydrogenation of propane/isobutane have been identified. For catalysts with supported metal oxide species, it is undisputable that all these reactions are initiated through breaking strong C–H bonds in these alkanes with participation of M-O2− pairs (H2 is formed and the metal does not change its oxidation state) or lattice/adsorbed oxygen (H2O is formed, the metal changes its oxidation state). The structure of supported MOx species and the kind of support material are decisive for catalyst activity and olefin selectivity. Although there are still some controversies, isolated metal oxide species seem to reveal higher selectivity, activity, and on-stream stability in all these dehydrogenation reactions in comparison with polymerized species. According to DFT calculations, the structure and consequently the reactivity of MOx species depend on the face of support material, where they are located. The knowledge about the exact structure of supported species and their catalytic performance provides some hints for catalyst design.
In comparison with supported catalysts, recently introduced bulk catalysts on the basis of non-reducible metal oxides, such as ZrO2, TiO2, Al2O3, Gd2O3 and Eu2O3 have other active sites. Alkane activation occurs with participation of coordinatively unsaturated metal cations at anion vacancies. For TiO2 and Al2O3, the corresponding cation and one neighbouring lattice oxygen participate in breaking C–H bonds in propane. Owing to combining experimental studies with DFT calculations, an alternative kind of active sites was discovered for ZrO2-based catalysts; two coordinatively unsaturated Zr cations form the active site. Fundamentals affecting intrinsic activity of coordinatively unsaturated metal cations and ways for their creation have been elucidated and build the basis for purposeful catalyst preparation.
On the basis of literature analysis in Fig. 13 and 16, we can safely assert that there is a significant progress in the development of catalysts for the PDH and BDH reactions. Both industrially relevant olefin selectivity and productivity were achieved using different catalysts. For some catalyst systems, long-term stability performance has been validated over several dehydrogenation/regeneration cycles lasting in total for a few days. Among the developed catalysts, those based on bulk ZrO2 should be especially mentioned. In comparison with V- or Cr-containing catalysts often used in these dehydrogenation reactions, these novel catalysts are environmentally friendly. Moreover, they have been introduced for the first time just five years ago. Thus, further improvements in the performance of such catalysts are expected.
Despite continuous studies on the ODP reaction using stoichiometric mixtures of propane and oxygen, no visible progress in the development of catalysts with propene selectivity above 80% at propane conversion higher than 30% has been achieved over the last 20 years. The low selectivity to propene is related to the interplay between the kinetics of selective (formation of propene) and non-selective consecutive reactions of the target olefin to carbon oxides. In comparison with the ODP reaction, some progress has been achieved in the ODB reaction. The isobutene yield values reported are comparable with those typical for BDH. Nevertheless, the necessity to separate air for obtaining pure oxygen is a negative cost factor limiting the commercialization of ODP and ODB. This drawback can be overcome and the undesired oxidation of propene/isobutene to carbon oxides can be partially hindered, when alkane dehydrogenation is carried out over reducible metal oxides using alternating feeds containing alkane or air. Such process operation, however, suffers from the limited ability of the catalysts to provide their lattice oxygen for the dehydrogenation reaction and from the necessity to interrupt the desired reaction after a relatively short period of time for catalyst reoxidation. Carrying out the ODP/ODB reactions under O2-lean (strong excess of alkane over oxygen) seems to be an attractive approach from cost (lower amounts of expensive oxygen are required), productivity (no often process interruption for catalyst regeneration), on-stream stability and selectivity viewpoints. Under such conditions, the oxidative dehydrogenation occurs in the upstream part of catalyst bed till complete consumption of oxygen and provides heat for the non-oxidative reaction taking place in the downstream catalyst bed.
Generally, materials with supported metal oxides used for the non-oxidative and oxidative dehydrogenation reactions can also catalyse the CO2-assisted alkane dehydrogenation. Studies on the latter subject are also focused on understanding the role of the kind of supported metal oxide species and support material for product selectivity, catalyst activity and on-stream stability. The derived conclusions are, however, controversial. In terms of application potential, some promising results have been obtained in terms of olefin yield.
10 Outlook
Our personal view on possible developments related to improving catalysts performance in the dehydrogenation of propane/isobutane to the corresponding olefins is provided below. To overcome the main drawback (low selectivity at industrially relevant degrees of alkane conversion) of the ODP and ODB reactions through catalyst design, new preparation methods enabling creation of certain structures of supported metal oxides on specific faces of support materials are required. So-designed catalysts should be tested for product selectivity in a broad range of alkane conversion to understand their ability/activity for consecutive oxidation of the target olefins. On this basis, common structure–selectivity–catalyst property relationships can be established. Such experimental approach should be supported by DFT calculations. The latter should be focused on understanding factors affecting the undesired oxidation reactions. We expect, however, that the most potential improvements can be achieved through reaction engineering aspects (Sections 7.3 and 7.4). O2-Lean or O2-free (chemical looping, redox process) dehydrogenation seems to be the most promising options. For the latter mode, catalysts with high capacity to release high amounts of lattice oxygen and to quickly refill over several thousand dehydrogenation/regeneration cycles have to be developed. Oxygen-conducting membranes can also be applied for carrying out ODP/ODB with air without or with an additional catalyst. They should, however, ensure high enough flux of lattice oxygen to meet industrial productivity requirements.Although there are various catalytic materials showing industrially relevant performance with respect to activity and olefin selectivity in the PDH and BDH reactions, their industrial potential has not been validated in long-term tests. In addition, tests with reaction feeds representative for the commercial products are scarcely reported. Concerning development of novel materials, bulk catalysts based on oxides of non-reducible metal oxides seem to be attractive, as only a few such materials have been prepared and tested up to now. Their performance can be tuned when controlling the coordination number of surface cations, their location on certain facets and/or the shape of crystallites. Regardless of catalyst type, further deeper understanding of catalyst property–selectivity–activity relationships is still required. With this respect, our knowledge about the mechanism of coke formation on a molecular level and factors affecting this process is really superficial, as a major part of papers dealing with catalyst development is focused on mechanistic and kinetic aspects of alkane activation and olefin formation. DFT calculations related to coke formation, which are missing in open literature up to now, could support catalyst development. The above recommendations are also valid for CO2-mediated alkane dehydrogenation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Deutsche Forschungsgemeinschaft (KO 2261/8-1), the National Natural Science Foundation of China (Grant No. 21961132026, 21878331, 91645108), Science Foundation of China University of Petroleum, Beijing (C201604), Ministry of Science and Technology of PRC (National Key Research and Development Program Nanotechnology Specific Project (No. 2020YFA0210900)) and the State of Mecklenburg-Vorpommern is gratefully acknowledged. The authors are also thankful to A. Skrypnik, Z. Aydin, N. Ortner, A. Perechodjuk, Q. Yang, K. Wu, Y. M. Li and D. Zhao for assistance with analysing literature data.References
- J. Hagen, in Industrial Catalysis, ed. J. Hagen, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 261–298 DOI:10.1002/3527607684.ch8.
- J. Hagen, Industrial Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 47–80 DOI:10.1002/3527607684.ch3.
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- P. Tian, Y. X. Wei, M. Ye and Z. M. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- https://www.engineering-airliquide.com/de/lurgi-mtp-methanol-zu-propylen, Lurgi MTP™ – Methanol-zu-Propylen.
- J. C. Mol and P. W. N. M. van Leeuwen, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, vol. 6, pp. 3420–3456 Search PubMed.
- S. Lwin and I. E. Wachs, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- S. Wang and Z. H. Zhu, Energy Fuels, 2004, 18, 1126–1139 CrossRef.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- M. A. Atanga, F. Rezaei, A. Jawad, M. Fitch and A. A. Rownaghi, Appl. Catal., B, 2018, 220, 429–445 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- J. T. Grant, J. M. Venegas, W. P. McDermott and I. Hermans, Chem. Rev., 2018, 118, 2769–2815 CrossRef CAS.
- C. Boswell, On-purpose Technologies ready to fill Propylene gap, http://www.icis.com/Articles/2012/04/16/9549968/on-purpose-technologies-ready-to-fill-propylene-gap.html CAS.
- B. V. Vora, Top. Catal., 2012, 55, 1297–1308 CrossRef CAS.
- J. C. Bricker, Top. Catal., 2012, 55, 1309–1314 CrossRef CAS.
- K. J. Caspary, H. Gehrke, M. Heinritz-Adrian and M. Schwefer, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, ch. 14.6, vol. 7, pp. 3206–3228 Search PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS.
- https://www.osha.gov/SLTC/hexavalentchromium/index.html, accessed October 20, 2020.
- G. Wang, X. Zhu and C. Li, Chem. Rec., 2020, 20, 604–616 CrossRef CAS.
- Z.-P. Hu, D. Yang, Z. Wang and Z.-Y. Yuan, Chin. J. Catal., 2019, 40, 1233–1254 CrossRef CAS.
- C. Sim-Yee, A. Hisyam and H. Prasetiawan, Int. J. Chem. React. Eng., 2015, 14 Search PubMed.
- K. J. Caspary, H. Gehrke, M. Heinritz-Adrian and M. Schwefer, Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 DOI:10.1002/9783527610044.hetcat0162.
- Z. Nawaz, Rev. Chem. Eng., 2015, 31, 413–436 CAS.
- T. Otroshchenko, S. Sokolov, M. Stoyanova, V. A. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, Angew. Chem., Int. Ed., 2015, 54, 15880–15883 CrossRef CAS.
- Y. Zhang, Y. Zhao, T. Otroshchenko, H. Lund, M.-M. Pohl, U. Rodemerck, D. Linke, H. Jiao, G. Jiang and E. V. Kondratenko, Nat. Commun., 2018, 9 Search PubMed.
- C.-F. Li, X. Guo, Q.-H. Shang, X. Yan, C. Ren, W.-Z. Lang and Y.-J. Guo, Ind. Eng. Chem. Res., 2020, 59, 4377–4387 CrossRef CAS.
- H. Zhu, P. Wang, Z. Xu, T. Wang, Y. Yue and X. Bao, Catal. Sci. Technol., 2020, 10, 3537–3541 RSC.
- A. Perechodjuk, V. A. Kondratenko, H. Lund, N. Rockstroh and E. V. Kondratenko, Chem. Commun., 2020, 56, 13021–13024 RSC.
- P.-P. Li, W.-Z. Lang, K. Xia, L. Luan, X. Yan and Y.-J. Guo, Appl. Catal., A, 2016, 522, 172–179 CrossRef CAS.
- M. F. Delley, M.-C. Silaghi, F. Nunez-Zarur, K. V. Kovtunov, O. G. Salnikov, D. P. Estes, I. V. Koptyug, A. Comas-Vives and C. Coperet, Organometallics, 2017, 36, 234–244 CrossRef CAS.
- A. Wegrzyniak, S. Jarczewski, A. Wegrzynowicz, B. Michorczyk, P. Kustrowski and P. Michorczyk, Nanomaterials, 2017, 7 Search PubMed.
- A. Węgrzyniak, A. Rokicińska, E. Hędrzak, B. Michorczyk, K. Zeńczak-Tomera, P. Kuśtrowski and P. Michorczyk, Catal. Sci. Technol., 2017, 7, 6059–6068 RSC.
- G. Liu, Z.-J. Zhao, T. Wu, L. Zeng and J. Gong, ACS Catal., 2016, 6, 5207–5214 CrossRef CAS.
- V. V. Kaichev, Y. A. Chesalov, A. A. Saraev and A. M. Tsapina, J. Phys. Chem. C, 2019, 123, 19668–19680 CrossRef CAS.
- U. Rodemerck, M. Stoyanova, E. V. Kondratenko and D. Linke, J. Catal., 2017, 352, 256–263 CrossRef CAS.
- C. Chen, M. Sun, Z. Hu, Y. Liu, S. Zhang and Z.-Y. Yuan, Chin. J. Catal., 2020, 41, 276–285 CrossRef CAS.
- P. Bai, Z. Ma, T. Li, Y. Tian, Z. Zhang, Z. Zhong, W. Xing, P. Wu, X. Liu and Z. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 25979–25990 CrossRef CAS.
- P. Hu, Y. Chen, X. Yan, W.-Z. Lang and Y.-J. Guo, Ind. Eng. Chem. Res., 2019, 58, 4065–4073 CrossRef CAS.
- P. Hu, W.-Z. Lang, X. Yan, X.-F. Chen and Y.-J. Guo, Appl. Catal., A, 2018, 553, 65–73 CrossRef CAS.
- C. Xiong, S. Chen, P. Yang, S. Zha, Z.-J. Zhao and J. Gong, ACS Catal., 2019, 9, 5816–5827 CrossRef CAS.
- G. Liu, L. Zeng, Z.-J. Zhao, H. Tian, T. Wu and J. Gong, ACS Catal., 2016, 6, 2158–2162 CrossRef CAS.
- S. Han, D. Zhao, T. Otroshchenko, H. Lund, U. Bentrup, V. A. Kondratenko, N. Rockstroh, S. Bartling, D. E. Doronkin, J.-D. Grunwaldt, U. Rodemerck, D. Linke, M. Gao, G. Jiang and E. V. Kondratenko, ACS Catal., 2020, 10, 8933–8949 CrossRef CAS.
- T. Gong, L. Qin, J. Lu and H. Feng, Phys. Chem. Chem. Phys., 2016, 18, 601–614 RSC.
- V. J. Cybulskis, S. U. Pradhan, J. J. Lovon-Quintana, A. S. Hock, B. Hu, G. Zhang, W. N. Delgass, F. H. Ribeiro and J. T. Miller, Catal. Lett., 2017, 147, 1252–1262 CrossRef CAS.
- M. W. Schreiber, C. P. Plaisance, M. Baumgärtl, K. Reuter, A. Jentys, R. Bermejo-Deval and J. A. Lercher, J. Am. Chem. Soc., 2018, 140, 4849–4859 CrossRef CAS.
- N. M. Phadke, E. Mansoor, M. Bondil, M. Head-Gordon and A. T. Bell, J. Am. Chem. Soc., 2019, 141, 1614–1627 CrossRef CAS.
- E. Mansoor, M. Head-Gordon and A. T. Bell, ACS Catal., 2018, 8, 6146–6162 CrossRef CAS.
- G. Wang, H. Zhang, H. Wang, Q. Zhu, C. Li and H. Shan, J. Catal., 2016, 344, 606–608 CrossRef CAS.
- H. Wang, H. Huang, K. Bashir and C. Li, Appl. Catal., A, 2020, 590, 117291 CrossRef CAS.
- H. Wang, H. Wang, X. Li and C. Li, Appl. Surf. Sci., 2017, 407, 456–462 CrossRef CAS.
- G. Wang, H. Zhang, Q. Zhu, X. Zhu, X. Li, H. Wang, C. Li and H. Shan, J. Catal., 2017, 351, 90–94 CrossRef CAS.
- Q. Liu, M. Luo, Z. Zhao and Q. Zhao, Chem. Eng. J., 2020, 380, 122423 CrossRef CAS.
- X. Li, P. Wang, H. Wang and C. Li, Appl. Surf. Sci., 2018, 441, 688–693 CrossRef CAS.
- C. Chen, S. Zhang, Z. Wang and Z.-Y. Yuan, J. Catal., 2020, 383, 77–87 CrossRef CAS.
- D. P. Estes, G. Siddiqi, F. Allouche, K. V. Kovtunov, O. V. Safonova, A. L. Trigub, I. V. Koptyug and C. Coperet, J. Am. Chem. Soc., 2016, 138, 14987–14997 CrossRef CAS.
- B. Hu, W.-G. Kim, T. P. Sulmonetti, M. L. Sarazen, S. Tan, J. So, Y. Liu, R. S. Dixit, S. Nair and C. W. Jones, ChemCatChem, 2017, 9, 3330–3337 CrossRef CAS.
- Y. Zhao, H. Sohn, B. Hu, J. Niklas, O. G. Poluektov, J. Tian, M. Delferro and A. S. Hock, ACS Omega, 2018, 3, 11117–11127 CrossRef CAS.
- C.-W. Zhang, J. Wen, L. Wang, X.-G. Wang and L. Shi, New J. Chem., 2020, 44, 7450–7459 RSC.
- J. Liu, Y. Liu, Y. Ni, H. Liu, W. Zhu and Z. Liu, Catal. Sci. Technol., 2020, 10, 1739–1746 RSC.
- Y. Zhang, S. Yang, J. Lu, Y. Mei, D. He and Y. Luo, Ind. Eng. Chem. Res., 2019, 58, 19818–19824 CrossRef CAS.
- Y. Gu, H. Liu, M. Yang, Z. Ma, L. Zhao, W. Xing, P. Wu, X. Liu, S. Mintova, P. Bai and Z. Yan, Appl. Catal., B, 2020, 274, 119089 CrossRef CAS.
- T. Wu, G. Liu, L. Zeng, G. Sun, S. Chen, R. Mu, S. Agbotse Gbonfoun, Z.-J. Zhao and J. Gong, AIChE J., 2017, 63, 4911–4919 CrossRef CAS.
- O. O. James, S. Mandal, N. Alele, B. Chowdhury and S. Maity, Fuel Process. Technol., 2016, 149, 239–255 CrossRef CAS.
- X.-Q. Gao, W.-D. Lu, S.-Z. Hu, W.-C. Li and A.-H. Lu, Chin. J. Catal., 2019, 40, 184–191 CrossRef CAS.
- Z.-P. Hu, Y. Wang, D. Yang and Z.-Y. Yuan, J. Energy Chem., 2020, 47, 225–233 CrossRef.
- D. He, Y. Zhang, S. Yang, Y. Mei and Y. Luo, ChemCatChem, 2018, 10, 5434–5440 CrossRef CAS.
- T. P. Otroshchenko, U. Rodemerck, D. Linke and E. V. Kondratenko, J. Catal., 2017, 356, 197–205 CrossRef CAS.
- S. Han, Y. Zhao, T. Otroshchenko, Y. Zhang, D. Zhao, H. Lund, T. H. Vuong, J. Rabeah, U. Bentrup, V. A. Kondratenko, U. Rodemerck, D. Linke, M. Gao, H. Jiao, G. Jiang and E. V. Kondratenko, ACS Catal., 2020, 10, 1575–1590 CrossRef CAS.
- S. Han, T. Otroshchenko, D. Zhao, H. Lund, N. Rockstroh, T. H. Vuong, J. Rabeah, U. Rodemerck, D. Linke, M. Gao, G. Jiang and E. V. Kondratenko, Appl. Catal., A, 2020, 590, 117350 CrossRef CAS.
- S. Han, T. Otroshchenko, D. Zhao, H. Lund, U. Rodemerck, D. Linke, M. Gao, G. Jiang and E. V. Kondratenko, Catal. Commun., 2020, 138, 105956 CrossRef CAS.
- S. Sokolov, M. Stoyanova, U. Rodemerck, D. Linke and E. V. Kondratenko, J. Catal., 2012, 293, 67–75 CrossRef CAS.
- Z.-J. Zhao, T. Wu, C. Xiong, G. Sun, R. Mu, L. Zeng and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 6791–6795 CrossRef CAS.
- P. Hu, W.-Z. Lang, X. Yan, L.-F. Chu and Y.-J. Guo, J. Catal., 2018, 358, 108–117 CrossRef CAS.
- Q. Liu, Z. Yang, M. Luo, Z. Zhao, J. Wang, Z. Xie and L. Guo, Microporous Mesoporous Mater., 2019, 282, 133–145 CrossRef CAS.
- Y. Xie, R. Luo, G. Sun, S. Chen, Z.-J. Zhao, R. Mu and J. Gong, Chem. Sci., 2020, 11, 3845–3851 RSC.
- S. Sokolov, M. Stoyanova, U. Rodemerck, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2014, 4, 1323–1332 RSC.
- C. Chen, Z. Hu, J. Ren, S. Zhang, Z. Wang and Z.-Y. Yuan, ChemCatChem, 2019, 11, 868–877 CrossRef CAS.
- C. Chen, Z.-P. Hu, J.-T. Ren, S. Zhang, Z. Wang and Z.-Y. Yuan, Mol. Catal., 2019, 476, 110508 CrossRef CAS.
- D. Zhao, Y. Li, S. Han, Y. Zhang, G. Jiang, Y. Wang, K. Guo, Z. Zhao, C. Xu, R. Li, C. Yu, J. Zhang, B. Ge and E. V. Kondratenko, iScience, 2019, 13, 269–276 CrossRef CAS.
- C. Chen, M. Sun, Z. Hu, J. Ren, S. Zhang and Z.-Y. Yuan, Catal. Sci. Technol., 2019, 9, 1979–1988 RSC.
- J. Camacho-Bunquin, P. Aich, M. Ferrandon, A. “Bean” Getsoian, U. Das, F. Dogan, L. A. Curtiss, J. T. Miller, C. L. Marshall, A. S. Hock and P. C. Stair, J. Catal., 2017, 345, 170–182 CrossRef CAS.
- S. Han, D. Zhao, H. Lund, N. Rockstroh, S. Bartling, D. E. Doronkin, J.-D. Grunwaldt, M. Gao, G. Jiang and E. V. Kondratenko, Catal. Sci. Technol., 2020, 10, 7046–7055 RSC.
- C.-T. Shao, W.-Z. Lang, X. Yan and Y.-J. Guo, RSC Adv., 2017, 7, 4710–4723 RSC.
- Y. Xiang, H. Wang, J. Cheng and J. Matsubu, Catal. Sci. Technol., 2018, 8, 1500–1516 RSC.
- A. Bhan and W. Nicholas Delgass, Catal. Rev., 2008, 50, 19–151 CrossRef CAS.
- S.-W. Choi, W.-G. Kim, J.-S. So, J. S. Moore, Y. Liu, R. S. Dixit, J. G. Pendergast, C. Sievers, D. S. Sholl, S. Nair and C. W. Jones, J. Catal., 2017, 345, 113–123 CrossRef CAS.
- W.-g. Kim, J. So, S.-W. Choi, Y. Liu, R. S. Dixit, C. Sievers, D. S. Sholl, S. Nair and C. W. Jones, Chem. Mater., 2017, 29, 7213–7222 CrossRef CAS.
- M. Nakai, K. Miyake, R. Inoue, K. Ono, H. Al Jabri, Y. Hirota, Y. Uchida, S. Tanaka, M. Miyamoto, Y. Oumi, C. Y. Kong and N. Nishiyama, Catal. Sci. Technol., 2019, 9, 6234–6239 RSC.
- Y. Dai, J. Gu, S. Tian, Y. Wu, J. Chen, F. Li, Y. Du, L. Peng, W. Ding and Y. Yang, J. Catal., 2020, 381, 482–492 CrossRef CAS.
- D. P. Estes, Chimia, 2017, 71, 177–180 CrossRef CAS.
- N. Dewangan, J. Ashok, M. Sethia, S. Das, S. Pati, H. Kus and S. Kawi, ChemCatChem, 2019, 11, 4923–4934 CrossRef CAS.
- T. Otroshchenko, V. A. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, J. Catal., 2017, 348, 282–290 CrossRef CAS.
- U. Rodemerck, E. V. Kondratenko, T. Otroshchenko and D. Linke, Chem. Commun., 2016, 52, 12222–12225 RSC.
- M. Dixit, P. Kostetskyy and G. Mpourmpakis, ACS Catal., 2018, 8, 11570–11578 CrossRef CAS.
- Y. Zhang, Y. Zhao, T. Otroshchenko, S. Han, H. Lund, U. Rodemerck, D. Linke, H. Jiao, G. Jiang and E. V. Kondratenko, J. Catal., 2019, 371, 313–324 CrossRef CAS.
- T. Otroshchenko, O. Bulavchenko, H. V. Thanh, J. Rabeah, U. Bentrup, A. Matvienko, U. Rodemerck, B. Paul, R. Kraehnert, D. Linke and E. V. Kondratenko, Appl. Catal., A, 2019, 585 Search PubMed.
- T. P. Otroshchenko, V. A. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2017, 7, 4499–4510 RSC.
- Y. Zhang, Y. Zhao, T. Otroshchenko, A. Perechodjuk, V. A. Kondratenko, S. Bartling, U. Rodemerck, D. Linke, H. Jiao, G. Jiang and E. V. Kondratenko, ACS Catal., 2020, 10, 6377–6388 CrossRef CAS.
- D. Hoang and H. Lieske, Catal. Lett., 1994, 27, 33–42 CrossRef CAS.
- J. A. Wang, T. López, X. Bokhimi and O. Novaro, J. Mol. Catal. A: Chem., 2005, 239, 249–256 CrossRef CAS.
- C. Berger-Karin, J. Radnik and E. V. Kondratenko, J. Catal., 2011, 280, 116–124 CrossRef CAS.
- A. Perechodjuk, Y. Zhang, V. A. Kondratenko, U. Rodemerck, D. Linke, S. Bartling, C. R. Kreyenschulte, G. Jiang and E. V. Kondratenko, Appl. Catal., A, 2020, 602, 117731 CrossRef CAS.
- T. Otroshchenko and E. V. Kondratenko, Catal. Commun., 2020, 144, 106068 CrossRef CAS.
- S. Sim, S. Gong, J. Bae, Y.-K. Park, J. Kim, W. C. Choi, U. G. Hong, D. S. Park, I. K. Song, H. Seo, N. Y. Kang and S. Park, Mol. Catal., 2017, 436, 164–173 CrossRef CAS.
- Y.-P. Tian, P. Bai, S.-M. Liu, X.-M. Liu and Z.-F. Yan, Fuel Process. Technol., 2016, 151, 31–39 CrossRef CAS.
- A. Rodriguez-Gomez, A. D. Chowdhury, M. Caglayan, J. A. Bau, E. Abou-Hamad and J. Gascon, Catal. Sci. Technol., 2020, 10, 6138–6150 RSC.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS.
- Y.-P. Tian, Y.-A. Liu, X.-M. Liu and Z.-F. Yan, Catal. Sci. Technol., 2018, 8, 5473–5481 RSC.
- X.-s. Wang, G.-l. Zhou, Z.-w. Chen, Q. Li, H.-j. Zhou and C.-m. Xu, Appl. Catal., A, 2018, 555, 171–177 CrossRef CAS.
- G. Wang, C. Li and H. Shan, Catal. Sci. Technol., 2016, 6, 3128–3136 RSC.
- A. N. Matveyeva, N. A. Zaitseva, P. Mäki-Arvela, A. Aho, A. K. Bachina, S. P. Fedorov, D. Y. Murzin and N. A. Pakhomov, Ind. Eng. Chem. Res., 2018, 57, 927–938 CrossRef CAS.
- A. N. Matveyeva, S. O. Omarov, D. A. Sladkovskiy and D. Y. Murzin, Chem. Eng. J., 2019, 372, 1194–1204 CrossRef CAS.
- A. N. Matveyeva, J. Wärnå, N. A. Pakhomov and D. Yu. Murzin, Chem. Eng. J., 2020, 381 Search PubMed.
- H. Zhao, H. Song, L. Chou, J. Zhao, J. Yang and L. Yan, Catal. Sci. Technol., 2017, 7, 3258–3267 RSC.
- T. Otroshchenko, J. Radnik, M. Schneider, U. Rodemerck, D. Linke and E. V. Kondratenko, Chem. Commun., 2016, 52, 8164–8167 RSC.
- Y. Liu, C. Xia, Q. Wang, L. Zhang, A. Huang, M. Ke and Z. Song, Catal. Sci. Technol., 2018, 8, 4916–4924 RSC.
- B. Liu, H. Zhao, J. Yang, J. Zhao, L. Yan, H. Song and L. Chou, Microporous Mesoporous Mater., 2020, 293 Search PubMed.
- M. Cheng, H. Zhao, J. Yang, J. Zhao, L. Yan, H. Song and L. Chou, Catal. Lett., 2019, 149, 1326–1336 CrossRef CAS.
- M. Cheng, H. Zhao, J. Yang, J. Zhao, L. Yan, H. Song and L. Chou, Microporous Mesoporous Mater., 2018, 266, 117–125 CrossRef CAS.
- V. Z. Fridman, R. Xing and M. Severance, Appl. Catal., A, 2016, 523, 39–53 CrossRef CAS.
- V. Z. Fridman and R. Xing, Appl. Catal., A, 2017, 530, 154–165 CrossRef CAS.
- G. Wang, N. Song, K. Lu, W. Wang, L. Bing, Q. Zhang, H. Fu, F. Wang and D. Han, Catalysts, 2019, 9 Search PubMed.
- D. A. Nazimov, O. V. Klimov, A. V. Saiko, S. N. Trukhan, T. S. Glazneva, I. P. Prosvirin, S. V. Cherepanova and A. S. Noskov, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.03.005.
- A. A. Salaeva, M. A. Salaev, O. V. Vodyankina and G. V. Mamontov, Appl. Catal., A, 2019, 581, 82–90 CrossRef CAS.
- A. A. Salaeva, M. A. Salaev and G. V. Mamontov, Chem. Eng. Sci., 2020, 215 Search PubMed.
- T. A. Bugrova and G. V. Mamontov, Kinet. Catal., 2018, 59, 143–149 CrossRef CAS.
- A. A. Shutilov and G. A. Zenkovets, Mater. Today: Proc., 2020, 25, 483–486 CAS.
- U. Rodemerck, S. Sokolov, M. Stoyanova, U. Bentrup, D. Linke and E. V. Kondratenko, J. Catal., 2016, 338, 174–183 CrossRef CAS.
- G. Liu, J. Liu, N. He, C. Miao, J. Wang, Q. Xin and H. Guo, RSC Adv., 2018, 8, 18663–18671 RSC.
- M. Cheng, H. Zhao, J. Yang, J. Zhao, L. Yan, H. Song and L. Chou, RSC Adv., 2019, 9, 9828–9837 RSC.
- J. Mu, J. Shi, L. J. France, Y. Wu, Q. Zeng, B. Liu, L. Jiang, J. Long and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 23112–23121 CrossRef CAS.
- E. Cheng, L. McCullough, H. Noh, O. Farha, J. Hupp and J. Notestein, Ind. Eng. Chem. Res., 2020, 59, 1113–1122 CrossRef CAS.
- U. Rodemerck, E. V. Kondratenko, T. Otroshchenko and D. Linke, Chem. Commun., 2016, 52, 12222–12225 RSC.
- Y.-P. Tian, X.-M. Liu, M. J. Rood and Z.-F. Yan, Appl. Catal., A, 2017, 545, 1–9 CrossRef CAS.
- R. L. Puurunen, B. G. Beheydt and B. M. Weckhuysen, J. Catal., 2001, 204, 253–257 CrossRef CAS.
- S. M. K. Airaksinen, M. A. Bañares and A. O. I. Krause, J. Catal., 2005, 230, 507–513 CrossRef CAS.
- S. Sokolov, V. Y. Bychkov, M. Stoyanova, U. Rodemerck, U. Bentrup, D. Linke, Y. P. Tyulenin, V. N. Korchak and E. V. Kondratenko, ChemCatChem, 2015, 7, 1691–1700 CrossRef CAS.
- J. J. H. B. Sattler, A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2013, 15, 12095–12103 RSC.
- J. J. H. B. Sattler, A. M. Mens and B. M. Weckhuysen, ChemCatChem, 2014, 6, 3139–3145 CrossRef CAS.
- H.-Z. Wang, L.-L. Sun, Z.-J. Sui, Y.-A. Zhu, G.-H. Ye, D. Chen, X.-G. Zhou and W.-K. Yuan, Ind. Eng. Chem. Res., 2018, 57, 8647–8654 CrossRef CAS.
- B. R. Kumar and R. Kumar, J. Nat. Gas Chem., 2008, 17, 256–263 CrossRef CAS.
- K. Scheurell, G. Scholz, A. Pawlik and E. Kemnitz, Solid State Sci., 2008, 10, 873–883 CrossRef CAS.
- C. Oliva, S. Cappelli, I. Rossetti, N. Ballarini, F. Cavani and L. Forni, Chem. Eng. J., 2009, 154, 131–136 CrossRef CAS.
- J. Xu, M. Chen, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Microporous Mesoporous Mater., 2009, 118, 354–360 CrossRef CAS.
- M. D. Putra, S. M. Al-Zahrani and A. E. Abasaeed, Catal. Commun., 2011, 14, 107–110 CrossRef CAS.
- H. Zhu, S. Ould-Chikh, H. Dong, I. Llorens, Y. Saih, D. H. Anjum, J. L. Hazemann and J. M. Basset, ChemCatChem, 2015, 7, 3332–3339 CrossRef CAS.
- Q. X. Luo, X. K. Zhang, B. L. Hou, J. G. Chen, C. Zhu, Z. W. Liu, Z. T. Liu and J. Lu, Catal.: Sci. Technol., 2018, 8, 4864–4876 RSC.
- J. Guo, X. Li, Y. Tang and J. Zhang, ChemistrySelect, 2019, 4, 13576–13581 CrossRef CAS.
- O. Ovsitser, R. Schomaecker, E. V. Kondratenko, T. Wolfram and A. Trunschke, Catal. Today, 2012, 192, 16–19 CrossRef CAS.
- S. A. Al-Ghamdi and H. I. De, Lasa, Fuel, 2014, 128, 120–140 CrossRef CAS.
- A. A. Ayandiran, I. A. Bakare, H. Binous, S. Al-Ghamdi, S. A. Razzak and M. M. Hossain, Catal.: Sci. Technol., 2016, 6, 5154–5167 RSC.
- S. Rostom and H. I. De Lasa, Ind. Eng. Chem. Res., 2017, 56, 13109–13124 CrossRef CAS.
- M. M. Hossain, Ind. Eng. Chem. Res., 2017, 56, 4309–4318 CrossRef CAS.
- S. Rostom and H. De Lasa, Ind. Eng. Chem. Res., 2018, 57, 10251–10260 CrossRef CAS.
- S. Chen, L. Zeng, R. Mu, C. Xiong, Z. J. Zhao, C. Zhao, C. Pei, L. Peng, J. Luo, L. S. Fan and J. Gong, J. Am. Chem. Soc., 2019, 141, 18653–18657 CrossRef CAS.
- Q. Xie, H. Zhang, J. Kang, J. Cheng, Q. Zhang and Y. Wang, ACS Catal., 2018, 8, 4902–4916 CrossRef CAS.
- L. Sun, Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Catal.: Sci. Technol., 2018, 8, 3044–3051 RSC.
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Science, 2016, 354, 1570–1573 CrossRef CAS.
- J. T. Grant, W. P. McDermott, J. M. Venegas, S. P. Burt, J. Micka, S. P. Phivilay, C. A. Carrero and I. Hermans, ChemCatChem, 2017, 9, 3623–3626 CrossRef CAS.
- L. Shi, D. Wang, W. Song, D. Shao, W. P. Zhang and A. H. Lu, ChemCatChem, 2017, 9, 1788–1793 CrossRef CAS.
- J. Tian, J. Tan, M. Xu, Z. Zhang, S. Wan, S. Wang, J. Lin and Y. Wang, Sci. Adv., 2019, 5 Search PubMed.
- A. M. Love, M. C. Cendejas, B. Thomas, W. P. McDermott, P. Uchupalanun, C. Kruszynski, S. P. Burt, T. Agbi, A. J. Rossini and I. Hermans, J. Phys. Chem. C, 2019, 123, 27000–27011 CrossRef CAS.
- Q. Liu, Y. Wu, F. Xing, Q. Liu, X. Guo and C. Huang, J. Catal., 2020, 381, 599–607 CrossRef CAS.
- B. Qiu, F. Jiang, W. D. Lu, B. Yan, W. C. Li, Z. C. Zhao and A. H. Lu, J. Catal., 2020, 385, 176–182 CrossRef CAS.
- Y. Wang, W. C. Li, Y. X. Zhou, R. Lu and A. H. Lu, Catal. Today, 2020, 339, 62–66 CrossRef CAS.
- Q. Zhang, C. Cao, T. Xu, M. Sun, J. Zhang, Y. Wang and H. Wan, Chem. Commun., 2009, 2376–2378, 10.1039/b823369a.
- M. Chen, J. L. Wu, Y. M. Liu, Y. Cao, L. Guo, H. Y. He and K. N. Fan, J. Solid State Chem., 2011, 184, 3357–3363 CrossRef CAS.
- L. Wang, W. Chu, C. Jiang, Y. Liu, J. Wen and Z. Xie, J. Nat. Gas Chem., 2012, 21, 43–48 CrossRef CAS.
- B. Farin, P. Eloy, C. Poleunis, M. Devillers and E. M. Gaigneaux, Catal.: Sci. Technol., 2016, 6, 6046–6056 RSC.
- S. Baoyi, X. Aiju and W. Jiang, Integr. Ferroelectr., 2016, 171, 16–22 CrossRef.
- B. Farin, M. Devillers and E. M. Gaigneaux, Microporous Mesoporous Mater., 2017, 242, 200–207 CrossRef CAS.
- F. Ma, S. Chen, Y. Li, H. Zhou, A. Xu and W. Lu, Appl. Surf. Sci., 2014, 313, 654–659 CrossRef CAS.
- F. Ma, S. Chen, H. Zhou, Y. Li and W. Lu, RSC Adv., 2014, 4, 40776–40781 RSC.
- S. Tanasoi, G. Mitran, N. Tanchoux, T. Cacciaguerra, F. Fajula, I. Sndulescu, D. Tichit and I. C. Marcu, Appl. Catal., A, 2011, 395, 78–86 CrossRef CAS.
- L. Cao, P. Dai, L. Zhu, L. Yan, R. Chen, D. Liu, X. Gu, L. Li, Q. Xue and X. Zhao, Appl. Catal., B, 2020, 262 Search PubMed.
- E. V. Kondratenko and A. Brückner, J. Catal., 2010, 274, 111–116 CrossRef CAS.
- X. Rozanska, E. V. Kondratenko and J. Sauer, J. Catal., 2008, 256, 84–94 CrossRef CAS.
- O. Ovsitser, M. Cherian, A. Brückner and E. V. Kondratenko, J. Catal., 2009, 265, 8–18 CrossRef CAS.
- O. Ovsitser, M. Cherian and E. V. Kondratenko, J. Phys. Chem. C, 2007, 111, 8594–8602 CrossRef CAS.
- E. V. Kondratenko, O. Ovsitser, J. Radnik, M. Schneider, R. Kraehnert and U. U. Dingerdissen, Appl. Catal., A, 2007, 319, 98–110 CrossRef CAS.
- E. V. Kondratenko, M. Cherian, M. Baerns, D. S. Su, R. Schlogl, X. Wang and I. E. Wachs, J. Catal., 2005, 234, 131–142 CrossRef CAS.
- O. Schwarz, B. Frank, C. Hess and R. Schomäcker, Catal. Commun., 2008, 9, 229–233 CrossRef CAS.
- A. Dinse, C. Carrero, A. Ozarowski, R. Schomäcker, R. Schlögl and K. P. Dinse, ChemCatChem, 2012, 4, 641–652 CrossRef CAS.
- S. Sugiyama, T. Osaka, Y. Ueno and K. I. Sotowa, J. Jpn. Pet. Inst., 2008, 51, 50–57 CrossRef CAS.
- M. N. Taylor, A. F. Carley, T. E. Davies and S. H. Taylor, Top. Catal., 2009, 52, 1660–1668 CrossRef CAS.
- X. Gao, C. Chen, S. Ren, J. Zhang and D. Su, Cuihua Xuebao, 2012, 33, 1069–1074 CAS.
- I. V. Mishakov, A. A. Vedyagin, A. F. Bedilo, V. I. Zaikovskii and K. J. Klabunde, Catal. Today, 2009, 144, 278–284 CrossRef CAS.
- G. Mitran, A. Urda, N. Tanchoux, F. Fajula and I. C. Marcu, Catal. Lett., 2009, 131, 250–257 CrossRef CAS.
- A. H. S. Kootenaei, J. Towfighi, A. Khodadadi and Y. Mortazavi, Appl. Surf. Sci., 2014, 298, 26–35 CrossRef CAS.
- S. Arndt, B. Uysal, A. Berthold, T. Otrebma, Y. Aksu, M. Driess and R. Schomäcker, J. Nat. Gas Chem., 2012, 21, 581–594 CrossRef CAS.
- X. Sun, Y. Ding, B. Zhang, R. Huang and D. S. Su, Chem. Commun., 2015, 51, 9145–9148 RSC.
- G. Mitran, T. Cacciaguerra, S. Loridant, D. Tichit and I. C. Marcu, Appl. Catal., A, 2012, 417-418, 153–162 CrossRef CAS.
- M. Khachani, M. Kacimi, A. Ensuque, J. Y. Piquemal, C. Connan, F. Bozon-Verduraz and M. Ziyad, Appl. Catal., A, 2010, 388, 113–123 CrossRef CAS.
- X. Zhang, R. You, D. Li, T. Cao and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 35897–35907 CrossRef CAS.
- G. Mitran, R. Ahmed, E. Iro, S. Hajimirzaee, S. Hodgson, A. Urdă, M. Olea and I. C. Marcu, Catal. Today, 2018, 306, 260–267 CrossRef CAS.
- N. Hamilton, T. Wolfram, G. Tzolova Müller, M. Hävecker, J. Kröhnert, C. Carrero, R. Schomäcker, A. Trunschke and R. Schlögl, Catal.: Sci. Technol., 2012, 2, 1346–1359 RSC.
- S. A. Karakoulia, K. S. Triantafyllidis, G. Tsilomelekis, S. Boghosian and A. A. Lemonidou, Catal. Today, 2009, 141, 245–253 CrossRef CAS.
- C. A. Carrero, C. J. Keturakis, A. Orrego, R. Schomäcker and I. E. Wachs, Dalton Trans., 2013, 42, 12644–12653 RSC.
- O. Schwarz, D. Habel, O. Ovsitser, E. V. Kondratenko, C. Hess, R. Schomäcker and H. Schubert, J. Mol. Catal. A: Chem., 2008, 293, 45–52 CrossRef CAS.
- T. Kharlamova, E. Sushchenko, T. Izaak and O. Vodyankina, Catal. Today, 2016, 278, 174–184 CrossRef CAS.
- E. D. Sushchenko, T. S. Kharlamova, T. I. Izaak and O. V. Vodyankina, Kinet. Catal., 2017, 58, 630–641 CrossRef CAS.
- I. Rossetti, E. Bahadori, A. Tripodi and G. Ramis, Materials, 2019, 16 Search PubMed.
- S. A. Karakoulia, K. S. Triantafyllidis and A. A. Lemonidou, Microporous Mesoporous Mater., 2008, 110, 157–166 CrossRef CAS.
- Y. Liu, C. Jiang, W. Chu, W. Sun and Z. Xie, React. Kinet., Mech. Catal., 2010, 101, 141–151 CrossRef CAS.
- B. Schimmoeller, Y. Jiang, S. E. Pratsinis and A. Baiker, J. Catal., 2010, 274, 64–75 CrossRef CAS.
- K. Chalupka, C. Thomas, Y. Millot, F. Averseng and S. Dzwigaj, J. Catal., 2013, 305, 46–55 CrossRef CAS.
- T. Fu, Y. Wang, A. Wernbacher, R. Schlögl and A. Trunschke, ACS Catal., 2019, 9, 4875–4886 CrossRef CAS.
- P. R. Shah, I. Baldychev, J. M. Vohs and R. J. Gorte, Appl. Catal., A, 2009, 361, 13–17 CrossRef CAS.
- S. A. D'Ippolito, M. A. Bañares, J. L. G. Fierro and C. L. Pieck, Catal. Lett., 2008, 122, 252–258 CrossRef.
- M. P. Casaletto, G. Landi, L. Lisi, P. Patrono and F. Pinzari, J. Mol. Catal. A: Chem., 2010, 329, 50–56 CrossRef CAS.
- Q. Liu, M. Luo, Z. Zhao and L. Guo, Catal. Lett., 2019, 149, 1345–1358 CrossRef CAS.
- W. Ruettinger, A. Benderly, S. Han, X. Shen, Y. Ding and S. L. Suib, Catal. Lett., 2011, 141, 15–21 CrossRef CAS.
- H. Zhang, S. Cao, Y. Zou, Y. M. Wang, X. Zhou, Y. Shen and X. Zheng, Catal. Commun., 2014, 45, 158–161 CrossRef CAS.
- M. Nadjafi, P. M. Abdala, R. Verel, D. Hosseini, O. V. Safonova, A. Fedorov and C. R. Müller, ACS Catal., 2020, 10, 2314–2321 CrossRef CAS.
- A. Dinse, B. Frank, C. Hess, D. Habel and R. Schomäcker, J. Mol. Catal. A: Chem., 2008, 289, 28–37 CrossRef CAS.
- A. Dinse, A. Ozarowski, C. Hess, R. Schomäcker and K. P. Dinse, J. Phys. Chem. C, 2008, 112, 17664–17671 CrossRef CAS.
- B. Frank, J. Zhang, R. Blume, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2009, 48, 6913–6917 CrossRef CAS.
- K. Fukudome and T. Suzuki, Catal. Surv. Asia, 2015, 19, 172–187 CrossRef CAS.
- J. Tian, J. Lin, M. Xu, S. Wan, J. Lin and Y. Wang, Chem. Eng. Sci., 2018, 186, 142–151 CrossRef CAS.
- S. Barman, N. Maity, K. Bhatte, S. Ould-Chikh, O. Dachwald, C. Haeßner, Y. Saih, E. Abou-Hamad, I. Llorens, J. L. Hazemann, K. Köhler, V. D'Elia and J. M. Basset, ACS Catal., 2016, 6, 5908–5921 CrossRef CAS.
- J. T. Grant, A. M. Love, C. A. Carrero, F. Huang, J. Panger, R. Verel and I. Hermans, Top. Catal., 2016, 59, 1545–1553 CrossRef CAS.
- S. Zhang and H. Liu, Appl. Catal., A, 2019, 573, 41–48 CrossRef CAS.
- A. Löfberg, T. Giornelli, S. Paul and E. Bordes-Richard, Appl. Catal., A, 2011, 391, 43–51 CrossRef.
- A. Essakhi, A. Löfberg, S. Paul, P. Supiot, B. Mutel, V. Le Courtois and E. Bordes-Richard, Top. Catal., 2011, 54, 698–707 CrossRef CAS.
- H. Kazerooni, J. Towfighi Darian, Y. Mortazavi, A. A. Khadadadi and R. Asadi, Catal. Lett., 2020, 150, 2807–2822 CrossRef CAS.
- K. Fukudome, N. O. Ikenaga, T. Miyake and T. Suzuki, Catal.: Sci. Technol., 2011, 1, 987–998 RSC.
- K. C. Szeto, B. Loges, N. Merle, N. Popoff, A. Quadrelli, H. Jia, E. Berrier, A. De Mallmann, L. Delevoye, R. M. Gauvin and M. Taoufik, Organometallics, 2013, 32, 6452–6460 CrossRef CAS.
- C. Carrero, M. Kauer, A. Dinse, T. Wolfram, N. Hamilton, A. Trunschke, R. Schlögl and R. Schomäcker, Catal.: Sci. Technol., 2014, 4, 786–794 RSC.
- Y. Marco, L. Roldán, E. Muñoz and E. García-Bordejé, ChemSusChem, 2014, 7, 2496–2504 CrossRef CAS.
- W. D. Lu, D. Wang, Z. Zhao, W. Song, W. C. Li and A. H. Lu, ACS Catal., 2019, 9, 8263–8270 CrossRef CAS.
- S. Pei, B. Zhang, K. Jiao, R. Bao, B. Yue and H. He, Acta Phys.-Chim. Sin., 2008, 24, 561–564 CrossRef CAS.
- M. Salamanca, Y. E. Licea, A. Echavarría, A. C. Faro Jr and L. A. Palacio, Phys. Chem. Chem. Phys., 2009, 11, 9583–9591 RSC.
- S. Crapanzano, I. V. Babich and L. Lefferts, Appl. Catal., A, 2011, 391, 70–77 CrossRef CAS.
- J. Velasquez, A. Echavarria, A. Faro and L. A. Palacio, Ind. Eng. Chem. Res., 2013, 52, 5582–5586 CrossRef CAS.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. Destefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS.
- M. Salamanca-Guzmán, Y. E. Licea-Fonseca, A. Echavarría-Isaza, A. Faro and L. A. Palacio-Santos, Rev. Fac. Ing., 2017, 2017, 97–104 Search PubMed.
- C. Boucetta, M. Kacimi, A. Ensuque, J. Y. Piquemal, F. Bozon-Verduraz and M. Ziyad, Appl. Catal., A, 2009, 356, 201–210 CrossRef CAS.
- J. Janas, J. Gurgul, R. P. Socha, J. Kowalska, K. Nowinska, T. Shishido, M. Che and S. Dzwigaj, J. Phys. Chem. C, 2009, 113, 13273–13281 CrossRef CAS.
- E. Rombi, D. Gazzoli, M. G. Cutrufello, S. De Rossi and I. Ferino, Appl. Surf. Sci., 2010, 256, 5576–5580 CrossRef CAS.
- Z. Bai, P. Li, L. Liu and G. Xiong, ChemCatChem, 2012, 4, 260–264 CrossRef CAS.
- G. Xiong and J. Sang, J. Mol. Catal. A: Chem., 2014, 392, 315–320 CrossRef CAS.
- Z. Zhai, X. Wang, R. Licht and A. T. Bell, J. Catal., 2015, 325, 87–100 CrossRef CAS.
- E. Goudarzi, R. Asadi, J. T. Darian and A. Shahbazi Kootenaei, RSC Adv., 2019, 9, 11797–11809 RSC.
- D. Shee and G. Deo, J. Mol. Catal. A: Chem., 2009, 308, 46–55 CrossRef CAS.
- J. Li, C. Wang, C. Huang, W. Weng and H. Wan, Catal. Lett., 2010, 137, 81–87 CrossRef CAS.
- J. H. Li, C. C. Wang, C. J. Huang, Y. F. Sun, W. Z. Weng and H. L. Wan, Appl. Catal., A, 2010, 382, 99–105 CrossRef CAS.
- K. Fukudome, A. Kanno, N. O. Ikenaga, T. Miyake and T. Suzuki, Catal. Lett., 2011, 141, 68–77 CrossRef CAS.
- W. Fang, Q. J. Ge, J. F. Yu and H. Y. Xu, Ind. Eng. Chem. Res., 2011, 50, 1962–1967 CrossRef CAS.
- T. Li, J. Wang, B. Zhaorigetu and A. Xu, React. Kinet., Mech. Catal., 2013, 110, 421–435 CrossRef CAS.
- X. Fan, J. Li, Z. Zhao, Y. Wei, J. Liu, A. Duan and G. Jiang, J. Energy Chem., 2014, 23, 171–178 CrossRef CAS.
- B. Farin, C. Swalus, M. Devillers and E. M. Gaigneaux, Catal. Today, 2013, 203, 24–31 CrossRef CAS.
- S. Wannakao, B. Boekfa, P. Khongpracha, M. Probst and J. Limtrakul, ChemPhysChem, 2010, 11, 3432–3438 CrossRef CAS.
- N. H. Nguyen, T. H. Tran, M. T. Nguyen and M. C. Le, Int. J. Quantum Chem., 2010, 110, 2653–2670 CrossRef CAS.
- M.-F. R. Konstantinos Alexopoulos and G. B. Marin, J. Catal., 2012, 289, 127–139 CrossRef.
- J. M. H. Lo, Z. A. Premji, T. Ziegler and P. D. Clark, J. Phys. Chem. C, 2013, 117, 11258–11274 CrossRef CAS.
- Y. J. Du, Z. H. Li and K. N. Fan, J. Mol. Catal. A: Chem., 2013, 379, 122–138 CrossRef CAS.
- X. Rozanska, R. Fortrie and J. Sauer, J. Am. Chem. Soc., 2014, 136, 7751–7761 CrossRef CAS.
- J. Liu, F. Mohamed and J. Sauer, J. Catal., 2014, 317, 75–82 CrossRef CAS.
- V. I. Avdeev and A. F. Bedilo, Res. Chem. Intermed., 2016, 42, 5237–5252 CrossRef CAS.
- Y.-J. Du, Z. H. Li and K.-N. Fan, Surf. Sci., 2012, 606, 956–964 CrossRef CAS.
- L. Cheng, G. A. Ferguson, S. A. Zygmunt and L. A. Curtiss, J. Catal., 2013, 302, 31–36 CrossRef CAS.
- A. Hofmann, M. V. Ganduglia-Pirováno and J. Sauer, J. Phys. Chem. C, 2009, 113, 18191–18203 CrossRef CAS.
- C. Popa, M. V. Ganduglia-Pirovano and J. Sauer, J. Phys. Chem. C, 2011, 115, 7399–7410 CrossRef CAS.
- L. He, L. Fu and Y. Tang, Catal.: Sci. Technol., 2015, 5, 1115–1125 RSC.
- G. Wang, H. Dai, L. Zhang, J. Deng, C. Liu, H. He and C. T. Au, Appl. Catal., A, 2010, 375, 272–278 CrossRef CAS.
- S. M. Al-Zahrani, N. O. Elbashir, A. E. Abasaeed and M. Abdulwahed, Ind. Eng. Chem. Res., 2001, 40, 781–784 CrossRef CAS.
- M. Hoang, J. F. Mathews, K. C. Pratt and Z. Xie, Kinet. Catal., 2010, 51, 398–403 CrossRef CAS.
- S. M. Al-Zahrani, N. O. Elbashir, A. E. Abasaeed and M. Abdulwahed, J. Mol. Catal. A: Chem., 2004, 218, 179–186 CrossRef CAS.
- B. Y. Jibril, N. O. Elbashir, S. M. Al-Zahrani and A. E. Abasaeed, Chem. Eng. Process., 2005, 44, 835–840 CrossRef CAS.
- K. Samson, B. Grzybowska, R. Grabowski, M. A. Banares and E. Lozano Diz, Pol. J. Chem., 2009, 83, 1977–1992 CAS.
- G. Karamullaoglu, S. Onen and T. Dogu, Chem. Eng. Process., 2002, 41, 337–347 CrossRef CAS.
- Y. Kato, S. Nitta, S. Shimazu, M. Kurashina, M. Katoh, W. Ninomiya and S. Sugiyama, J. Chem. Eng. Jpn., 2019, 52, 99–105 CrossRef CAS.
- G. Neri, A. Pistone, S. De Rossi, E. Rombi, C. Milone and S. Galvagno, Appl. Catal., A, 2004, 260, 75–86 CrossRef CAS.
- P. Moriceau, B. Grzybowska, L. Gengembre and Y. Barbaux, Appl. Catal., A, 2000, 199, 73–82 CrossRef CAS.
- N. O. Elbashir, S. M. Al-Zahrani, A. E. Abasaeed and M. Abdulwahed, Chem. Eng. Process., 2003, 42, 817–823 CrossRef CAS.
- G. Wang, L. Zhang, J. Deng, H. Dai, H. He and C. T. Au, Appl. Catal., A, 2009, 355, 192–201 CrossRef CAS.
- J. Słoczyński, B. Grzybowska, A. Kozłowska, K. Samson, R. Grabowski, A. Kotarba and M. Hermanowska, Catal. Today, 2011, 169, 29–35 CrossRef.
- Q. Zhang, Y. Wang, Y. Ohishi, T. Shishido and K. Takehira, J. Catal., 2001, 202, 308–318 CrossRef CAS.
- V. Iannazzo, G. Neri, S. Galvagno, M. Di Serio, R. Tesser and E. Santacesaria, Appl. Catal., A, 2003, 246, 49–68 CrossRef CAS.
- Z. Fan, T. Zeng, W. Wu, D. Jiang and C. Miao, Ind. Eng. Chem. Res., 2019, 58, 10249–10254 CrossRef CAS.
- B. Sulikowski, Z. Olejniczak, E. Wloch, J. Rakoczy, R. X. Valenzuela and V. Cortés Corberán, Appl. Catal., A, 2002, 232, 189–202 CrossRef CAS.
- V. P. Vislovskiy, N. T. Shamilov, A. M. Sardarly, R. M. Talyshinskii, V. Y. Bychkov, P. Ruiz, V. Cortés Corberán, Z. Schay and Z. Koppany, Appl. Catal., A, 2003, 250, 143–150 CrossRef CAS.
- N. T. Shamilov and V. P. Vislovskiy, J. Korean Chem. Soc., 2011, 55, 812–818 CrossRef CAS.
- S. Kraemer, A. J. Rondinone, Y. T. Tsai, V. Schwartz, S. H. Overbury, J. C. Idrobo and Z. Wu, Catal. Today, 2016, 263, 84–90 CrossRef CAS.
- T. Shishido, A. Inoue, T. Konishi, I. Matsuura and K. Takehira, Catal. Lett., 2000, 68, 215–221 CrossRef CAS.
- Z. Wu, V. Schwartz, M. Li, A. J. Rondinone and S. H. Overbury, J. Phys. Chem. Lett., 2012, 3, 1517–1522 CrossRef CAS.
- G. Mitran, I. C. Marcu, A. Urdă and I. Săndulescu, J. Serb. Chem. Soc., 2010, 75, 1115–1124 CrossRef CAS.
- H. Xie, Z. Wu, S. H. Overbury, C. Liang and V. Schwartz, J. Catal., 2009, 267, 158–166 CrossRef CAS.
- C. Liang, H. Xie, V. Schwartz, J. Howe, S. Dai and S. H. Overbury, J. Am. Chem. Soc., 2009, 131, 7735–7741 CrossRef CAS.
- I. Gniot, P. Kirszensztejn and M. Kozłowski, Appl. Catal., A, 2009, 362, 67–74 CrossRef CAS.
- N. Martin-Sanchez, O. S. G. P. Soares, M. F. R. Pereira, M. J. Sanchez-Montero, J. L. Figueiredo and F. Salvador, Appl. Catal., A, 2015, 502, 71–77 CrossRef CAS.
- J. de Jesús Díaz Velásquez, L. M. C. Suárez and J. L. Figueiredo, Appl. Catal., A, 2006, 311, 51–57 CrossRef.
- V. Schwartz, H. Xie, H. M. Meyer Iii, S. H. Overbury and C. Liang, Carbon, 2011, 49, 659–668 CrossRef CAS.
- G. K. P. Dathar, Y. T. Tsai, K. Gierszal, Y. Xu, C. Liang, A. J. Rondinone, S. H. Overbury and V. Schwartz, ChemSusChem, 2014, 7, 483–491 CrossRef CAS.
- I. Pelech, O. S. G. P. Soares, M. F. R. Pereira and J. L. Figueiredo, Catal. Today, 2015, 249, 176–183 CrossRef CAS.
- R. Zãvoianu, C. R. Dias and M. F. Portela, Catal. Commun., 2001, 2, 37–42 CrossRef.
- Y. J. Zhang, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Catal. Today, 2000, 61, 377–382 CrossRef CAS.
- Y. A. Agafonov, N. V. Nekrasov, N. A. Gaidai, M. A. Botavina, P. E. Davydov and A. L. Lapidus, Kinet. Catal., 2009, 50, 577–582 CrossRef CAS.
- Y. A. Agafonov, N. V. Nekrasov, N. A. Gaidai and A. L. Lapidus, Kinet. Catal., 2007, 48, 255–264 CrossRef CAS.
- C. R. Dias, R. Zavoianu and M. F. Portela, Catal. Commun., 2002, 3, 85–90 CrossRef CAS.
- S. M. Al-Zahrani, N. O. Elbashir, A. E. Abasaeed and M. Abdulwahed, Catal. Lett., 2000, 69, 65–70 CrossRef CAS.
- I. C. Marcu, J. M. M. Millet and I. Săndulescu, J. Serb. Chem. Soc., 2005, 70, 791–798 CrossRef CAS.
- Y. Takita, X. Qing, A. Takami, H. Nishiguchi and K. Nagaoka, Appl. Catal., A, 2005, 296, 63–69 CrossRef CAS.
- I. C. Marcu, M. N. Urlan, A. Rédey and I. Săndulescu, C. R. Chim., 2010, 13, 365–371 CrossRef CAS.
- N. T. Shamilov and V. P. Vislovskiy, J. Korean Chem. Soc., 2011, 55, 81–85 CrossRef CAS.
- Y. A. Agafonov, N. V. Nekrasov and N. A. Gaidai, Kinet. Catal., 2001, 42, 821–827 CrossRef CAS.
- K. Lohbeck, H. Haferkorn, W. Fuhrmann and N. Fedtke, Ullmann's Encyclopedia of Industrial Chemistry, 2000 DOI:10.1002/14356007.a16_053.
- A. S. Sandupatla, K. Ray, P. Thaosen, C. Sivananda and G. Deo, Catal. Today, 2020, 354, 176–182 CrossRef CAS.
- P. Michorczyk and J. Ogonowski, React. Kinet. Catal. Lett., 2003, 78, 41–47 CrossRef CAS.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- K.-O. Hinrichsen, K. Kochloefl and M. Muhler, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, 2008, pp. 2905–2920 DOI:10.1002/9783527610044.hetcat0147.
- Y. A. Agafonov, N. A. Gaidai and A. L. Lapidus, Russ. Chem. Bull., 2014, 63, 381–388 CrossRef CAS.
- P. Michorczyk, P. Pietrzyk and J. Ogonowski, Microporous Mesoporous Mater., 2012, 161, 56–66 CrossRef CAS.
- A. Burri, M. A. Hasib, Y. H. Mo, B. M. Reddy and S. E. Park, Catal. Lett., 2018, 148, 576–585 CrossRef CAS.
- H. M. Wang, Y. Chen, X. Yan, W. Z. Lang and Y. J. Guo, Microporous Mesoporous Mater., 2019, 284, 69–77 CrossRef CAS.
- Y. A. Agafonov, N. A. Gaidai and A. L. Lapidus, Kinet. Catal., 2018, 59, 744–753 CrossRef CAS.
- R. Jin, J. Easa, D. T. Tran and C. P. O'Brien, Catal.: Sci. Technol., 2020, 10, 1769–1777 RSC.
- P. Michorczyk, P. Pietrzyk and J. Ogonowski, Microporous Mesoporous Mater., 2012, 161, 56–66 CrossRef CAS.
- P. Michorczyk, K. Zenczak-Tomera, B. Michorczyk, A. Wegrzyniak, M. Basta, Y. Millot, L. Valentin and S. Dzwigaj, J. CO2 Util., 2020, 36, 54–63 CrossRef CAS.
- J. F. S. de Oliveira, D. P. Volanti, J. M. C. Bueno and A. P. Ferreira, Appl. Catal., A, 2018, 558, 55–66 CrossRef CAS.
- Z. F. Han, X. L. Xue, J. M. Wu, W. Z. Lang and Y. J. Guo, Chin. J. Catal., 2018, 39, 1099–1109 CrossRef CAS.
- X. L. Xue, W. Z. Lang, X. Yan and Y. J. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 15408–15423 CrossRef CAS.
- I. Ascoop, V. V. Galvita, K. Alexopoulos, M. F. Reyniers, P. Van Der Voort, V. Bliznuk and G. B. Marin, J. Catal., 2016, 335, 1–10 CrossRef CAS.
- M. Chen, J. Xu, F.-Z. Su, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Catal., 2008, 256, 293–300 CrossRef CAS.
- B. Xu, B. Zheng, W. Hua, Y. Yue and Z. Gao, J. Catal., 2006, 239, 470–477 CrossRef CAS.
- H. Xiao, J. F. Zhang, P. Wang, X. X. Wang, F. Pang, Z. Z. Zhang and Y. S. Tan, Catal. Sci. Technol., 2016, 6, 5183–5195 RSC.
- M. Chen, J. Xu, Y. M. Liu, Y. Cao, H. Y. He and J. H. Zhuang, Appl. Catal., A, 2010, 377, 35–41 CrossRef CAS.
- M. Chen, J. Xu, Y. Cao, H.-Y. He, K.-N. Fan and J.-H. Zhuang, J. Catal., 2010, 272, 101–108 CrossRef CAS.
- M. Chen, J. L. Wu, Y. M. Liu, Y. Cao, L. Guo, H. Y. He and K. N. Fan, Appl. Catal., A, 2011, 407, 20–28 CrossRef CAS.
- M. R. Kantserova, N. V. Vlasenko, S. M. Orlyk, K. Veltruska and I. Matolinova, Theor. Exp. Chem., 2019, 55, 207–214 CrossRef CAS.
- H. F. Tian, J. K. Liao, F. Zha, X. J. Guo, X. H. Tang, Y. Chang and X. X. Ma, ChemistrySelect, 2020, 5, 3626–3637 CrossRef CAS.
- Y. Ren, J. Wang, W. Hua, Y. Yue and Z. Gao, J. Ind. Eng. Chem., 2012, 18, 731–736 CrossRef CAS.
- P. Michorczyk, K. Zeńczak, R. Niekurzak and J. Ogonowski, Pol. J. Chem. Technol., 2012, 14, 77 Search PubMed.
- J. F. S. de Oliveira, D. P. Volanti, J. M. C. Bueno and A. P. Ferreira, Appl. Catal., A, 2018, 558, 55–66 CrossRef CAS.
- G. Sun, Q. Huang, S. Huang, Q. Wang, H. Li, H. Liu, S. Wan, X. Zhang and J. Wang, Catalysts, 2016, 6 Search PubMed.
- J. Ogonowski and E. Skrzyńska, React. Kinet. Catal. Lett., 2006, 88, 293–300 CrossRef CAS.
- C. L. Wei, F. Q. Xue, C. X. Miao, Y. H. Yue, W. M. Yang, W. M. Hua and Z. Gao, Catalysts, 2016, 6 Search PubMed.
- J. Ogonowski and E. Skrzyńska, Catal. Lett., 2008, 121, 234–240 CrossRef CAS.
- R. Yuan, Y. Li, H. Yan, H. Wang, J. Song, Z. Zhang, W. Fan, J. Chen, Z. Liu, Z. Liu and Z. Hao, Cuihua Xuebao, 2014, 35, 1329–1336 CAS.
- J. Ogonowski and E. Skrzynska, Catal. Commun., 2009, 11, 132–136 CrossRef CAS.
- C. Wei, F. Xue, C. Miao, Y. Yue, W. Yang, W. Hua and Z. Gao, Chin. J. Chem., 2017, 35, 1619–1626 CrossRef CAS.
- Y. J. Luo, C. X. Miao, Y. H. Yue, W. M. Yang, W. M. Hua and Z. Gao, Catalysts, 2019, 9 Search PubMed.
- J. F. Ding, R. Shao, J. Wu, Z. F. Qin and J. G. Wang, React. Kinet., Mech. Catal., 2010, 101, 173–181 CrossRef CAS.
- J. F. Ding, Z. F. Qin, X. K. Li, G. F. Wang and J. G. Wang, J. Mol. Catal. A: Chem., 2010, 315, 221–225 CrossRef CAS.
- A. Aouissi, D. Aldhayan and S. Alkahtani, Chin. J. Catal., 2012, 33, 1474–1479 CrossRef CAS.
- I. V. Mishakov, E. V. Ilyina, A. F. Bedilo and A. A. Vedyagin, React. Kinet. Catal. Lett., 2009, 97, 355–361 CrossRef CAS.
- A. Ates, C. Hardacre and A. Goguet, Appl. Catal., A, 2012, 441–442, 30–41 CrossRef CAS.
- P. Sazama, N. K. Sathu, E. Tabor, B. Wichterlová, Š. Sklenák and Z. Sobalík, J. Catal., 2013, 299, 188–203 CrossRef CAS.
- G. Wu, F. Hei, N. Guan and L. Li, Catal.: Sci. Technol., 2013, 3, 1333–1342 RSC.
- G. Wu, Y. Hao, N. Zhang, N. Guan, L. Li and W. Grünert, Microporous Mesoporous Mater., 2014, 198, 82–91 CrossRef CAS.
- G. Wu, F. Hei, N. Zhang, N. Guan, L. Li and W. Grünert, Appl. Catal., A, 2013, 468, 230–239 CrossRef CAS.
- G. I. Panov, CATTECH, 2000, 4, 18–32 CrossRef CAS.
- O. Ovsitser and E. V. Kondratenko, Chem. Commun., 2010, 46, 4974–4976 RSC.
- Z. Wang, Z. Bian, N. Dewangan, J. Xu and S. Kawi, J. Membr. Sci., 2019, 578, 36–42 CrossRef CAS.
- I. Rossetti, L. Fabbrini, N. Ballarini, C. Oliva, F. Cavani, A. Cericola, B. Bonelli, M. Piumetti, E. Garrone, H. Dyrbeck, E. A. Blekkan and L. Forni, J. Catal., 2008, 256, 45–61 CrossRef CAS.
- V. Balcaen, I. Sack, M. Olea and G. B. Marin, Appl. Catal., A, 2009, 371, 31–42 CrossRef CAS.
- S. Al-Ghamdi, J. Moreira and H. De Lasa, Ind. Eng. Chem. Res., 2014, 53, 15317–15332 CrossRef CAS.
- S. Rostom and H. de Lasa, Chem. Eng. Process., 2019, 137, 87–99 CrossRef CAS.
- J. Beckers and G. Rothenberg, Green Chem., 2009, 11, 1550–1554 RSC.
- J. Beckers and G. Rothenberg, Dalton Trans., 2009, 5673–5682, 10.1039/b904681j.
- S. Crapanzano, I. V. Babich and L. Lefferts, Catal. Today, 2013, 203, 17–23 CrossRef CAS.
- S. Crapanzano, I. V. Babich and L. Lefferts, Appl. Catal., A, 2010, 385, 14–21 CrossRef CAS.
- S. Crapanzano, I. V. Babich and L. Lefferts, Appl. Catal., A, 2010, 378, 144–150 CrossRef CAS.
- R. K. Grasselli, D. L. Stern and J. G. Tsikoyiannis, Appl. Catal., A, 1999, 189, 1–8 CrossRef CAS.
- R. K. Grasselli, D. L. Stern and J. G. Tsikoyiannis, Appl. Catal., A, 1999, 189, 9–14 CrossRef CAS.
- L. M. van der Zande, E. A. de Graaf and G. Rothenberg, Adv. Synth. Catal., 2002, 344, 884–889 CrossRef CAS.
- M.-L. Yang, C. Fan, Y.-A. Zhu, Z.-J. Sui, X.-G. Zhou and D. Chen, J. Phys. Chem. C, 2015, 119, 21386–21394 CrossRef CAS.
- R. B. Dudek, Y. Gao, J. Zhang and F. Li, AIChE J., 2018, 64, 3141–3150 CrossRef CAS.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS.
- S. L. Wegener, T. J. Marks and P. C. Stair, Acc. Chem. Res., 2012, 45, 206–214 CrossRef CAS.
- C. Perego and P. Villa, Catal. Today, 1997, 34, 281–305 CrossRef CAS.
- S. Barman, N. Maity, K. Bhatte, S. Ould-Chikh, O. Dachwald, C. Haeßner, Y. Saih, E. Abou-Hamad, I. Llorens, J.-L. Hazemann, K. Köhler, V. D’Elia and J.-M. Basset, ACS Catal., 2016, 6, 5908–5921 CrossRef CAS.
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef.
- J. Lu, J. W. Elam and P. C. Stair, Surf. Sci. Rep., 2016, 71, 410–472 CrossRef CAS.
- A. M. Johnson, B. R. Quezada, L. D. Marks and P. C. Stair, Top. Catal., 2014, 57, 177–187 CrossRef CAS.
- J. R. v. Ommen, D. Kooijman, M. d. Niet, M. Talebi and A. Goulas, J. Vac. Sci. Technol., A, 2015, 33, 021513 CrossRef.
- W. Hengwei and L. Junling, Acta Phys.-Chim. Sin., 2018, 34, 1334–1357 Search PubMed.
- M. Campanati, G. Fornasari and A. Vaccari, Catal. Today, 2003, 77, 299–314 CrossRef CAS.
- H. K. Olaf Deutschmann, K. Kochloefl and T. Turek, Ullmann's Encyclopedia of Industrial Chemistry, 2009 DOI:10.1002/14356007.a05_313.pub2.
- H. H. Kung and E. I. Ko, Chem. Eng. J. Biochem., 1996, 64, 203–214 CrossRef CAS.
- J. A. Schwarz, C. Contescu and A. Contescu, Chem. Rev., 1995, 95, 477–510 CrossRef CAS.
- M. Shandilya, R. Rai and J. Singh, Adv. Appl. Ceram., 2016, 115, 354–376 CrossRef CAS.
- A. Rabenau, Angew. Chem., Int. Ed. Engl., 1985, 24, 1026–1040 CrossRef.
- A. N. Shigapov, G. W. Graham, R. W. McCabe and H. K. Plummer, Appl. Catal., A, 2001, 210, 287–300 CrossRef CAS.
- R. Liu and C.-a. Wang, Microporous Mesoporous Mater., 2014, 186, 1–6 CrossRef CAS.
- J. Yano and V. K. Yachandra, Photosynth. Res., 2009, 102, 241 CrossRef CAS.
- E. E. Alp, S. M. Mini and M. Ramanathan, X-ray absorption spectroscopy: EXAFS and XANES - A versatile tool to study the atomic and electronic structure of materials, United States, 1990 Search PubMed.
- M. Che and E. Giamello, in Stud. Surf. Sci. Catal., ed. J. L. G. Fierro, Elsevier, 1990, vol. 57, pp. B265–B332 Search PubMed.
- A. Brückner, Chem. Soc. Rev., 2010, 39, 4673–4684 RSC.
- X. Gao, S. R. Bare, B. M. Weckhuysen and I. E. Wachs, J. Phys. Chem. B, 1998, 102, 10842–10852 CrossRef CAS.
- E. L. Lee and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 14410–14425 CrossRef CAS.
- A. Chakrabarti and I. E. Wachs, Catal. Lett., 2015, 145, 985–994 CrossRef CAS.
- J. Ryczkowski, Catal. Today, 2001, 68, 263–381 CrossRef CAS.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- I. E. Wachs and C. A. Roberts, Chem. Soc. Rev., 2010, 39, 5002–5017 RSC.
- M. A. Bañares and I. E. Wachs, J. Raman Spectrosc., 2002, 33, 359–380 CrossRef.
- J. J. Fitzgerald and S. M. DePaul, in Solid-State NMR Spectroscopy of Inorganic Materials, American Chemical Society, 1999, ch. 1, vol. 717, pp. 2–133 Search PubMed.
- M. de Oliveira, D. Seeburg, J. Weiß, S. Wohlrab, G. Buntkowsky, U. Bentrup and T. Gutmann, Catal. Sci. Technol., 2019, 9, 6180–6190 RSC.
- J. A. Dumesic, G. W. Huber and M. Boudart, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, 2008, vol. 1, pp. 1–15 Search PubMed.
- J. C. Védrine, Catalysts, 2017, 7, 341 CrossRef.
- Y. Bai, H. Huang, C. Wang, R. Long and Y. Xiong, Mater. Chem. Front., 2017, 1, 1951–1964 RSC.
- J. T. Gleaves, G. S. Yablonskii, P. Phanawadee and Y. Schuurman, Appl. Catal., A, 1997, 160, 55–88 CrossRef CAS.
- J. Pérez-Ramírez and E. V. Kondratenko, Catal. Today, 2007, 121, 160–169 CrossRef.
- K. Morgan, N. Maguire, R. Fushimi, J. T. Gleaves, A. Goguet, M. P. Harold, E. V. Kondratenko, U. Menon, Y. Schuurman and G. S. Yablonsky, Catal. Sci. Technol., 2017, 7, 2416–2439 RSC.
- Z. Zhang, X. E. Verykios and M. Baerns, Catal. Rev., 1994, 36, 507–556 CrossRef CAS.
- C. Ahamer, A. K. Opitz, G. M. Rupp and J. Fleig, J. Electrochem. Soc., 2017, 164, F790–F803 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs01140a |
| This journal is © The Royal Society of Chemistry 2021 |