
Selective dealkylation of alkyl polycyclic aromatic hydrocarbons towards innovative upgrading process of practical heavy oil†
Fumiya
Nakano
,
Tomohide
Goma
,
Satoshi
Suganuma
 *,
Etsushi
Tsuji
and
Naonobu
Katada
*,
Etsushi
Tsuji
and
Naonobu
Katada
Center for Research on Green Sustainable Chemistry, Tottori University, 4-101 Koyama-cho Minami. Japan. E-mail: Tottori 680-8552; suganuma@tottori-u.ac.jp; Fax: +81 857 31 5684; Tel: +81 857 31 5256
First published on 10th November 2020
Abstract
The dealkylation of alkyl polycyclic aromatic hydrocarbons (APAHs) in vacuum gas oil (VGO) was studied as a novel upgrading process alternative to conventional processes. A silica-monolayer loaded on alumina (SMA) with weak Brønsted acid sites and large pore size exhibited higher activity than amorphous silica-alumina and zeolites (USY and ZSM-5). The SMA almost completely converted the APAHs into aromatics and alkanes, but a small amount of APAHs with methyl and ethyl groups (short-chain) were unreacted. The larger pore size of the SMA was proposed to be enough for bulky APAHs to diffuse. The dealkylation by the SMA formed large amounts of long-chain alkanes, which can be utilized as light oil and kerosene for fuel, lubricating oil, etc. The cracking of long-chain alkanes did not proceed, and thus scarcely formed lighter alkanes. The SMA in the reaction adsorbed alkanes but did not form coke, and therefore exhibited continuous dealkylation activity. Additionally, it was revealed that the pore size of the SMA slightly affected the composition of the formed alkanes.
1. Introduction
Petroleum refining is generally developed for the production of transportation fuels, but is also used for minor but essential production of building blocks for petrochemicals, such as light alkenes (ethylene and propylene) and BTEX (benzene, toluene, ethylbenzene, and xylenes).1–4 The transportation fuels are produced by atmospheric distillation, while the atmospheric residue oil is processed by vacuum distillation. Vacuum gas oil (VGO) is converted into gasoline, light oil, and kerosene by fluid catalytic cracking (FCC), hydrocracking, or thermal cracking. However, the rate of demand for transportation fuels is declining due to improvement of fuel consumption of vehicles and control of carbon dioxide emission.5 The yield of petrochemicals is limited based on the operation conditions, which are designed to fit the demands of transportation fuels. Recently, an FCC process has been developed for increasing the production of light alkenes, but the yield of benzene derivatives cannot be increased by further reorganization of the process. On the other hand, the hydrocracking process consumes a large amount of hydrogen for hydrogenation of polycyclic aromatic hydrocarbons, which leads to a decrease of benzene derivative yields due to severe hydrogenation conditions. Benzene derivatives are applied as chemicals and their intermediates as synthetic fibers, resins, detergents, and various organic ingredients. Among the benzene derivatives, the manufacture volumes of benzene and para-xylene are notably increasing. However, the production of benzene derivatives is complicated for FCC and hydrocracking. Therefore, innovation of petroleum refining for increasing the flexibility in production is strongly required, in order to cope with the change of situation.We have studied an alternative process to FCC and hydrocracking,6–9 and an outline was given in a review.10 The components in VGO are categorized into 3 main types: aliphatics (alkanes as the main components), aromatics, and alkylaromatics. If alkyl polycyclic aromatic hydrocarbons (APAHs) are completely dealkylated to alkanes and PAHs with no alkyl groups, the components can be facilely separated into 2 types by methods such as solvent extraction.11–13 The dealkylation of an APAH via β-scission of a carbenium ion is catalyzed by Brønsted acid sites, like the cracking of cumene (2-phenylpropane),14,15 forming an aromatic hydrocarbon and a carbenium ion. Then, alkanes are formed from the carbenium ions through the presumed reaction pathways as follows: hydrogenation of the generated alkene from the carbenium ion or hydride transfer from the other alkane to the carbenium ion. If the alkane produced from APAHs is not cracked but keeps the long chain length, the yield of middle distillates such as light oil, kerosene, and >C20 alkanes for lubricating oil can be maximized. On the other hand, the produced polycyclic aromatic hydrocarbons such as naphthalene, anthracene, phenanthrene, pyrene, etc. can be converted by partial hydrogenation of the aromatic rings into tetralin derivatives, e.g., naphthalene into tetralin.16–18 Then, tetralin derivatives are converted into benzene derivatives through ring-opening.19,20 In fact, BTX production from PAHs has been recently reported.21–23 Thus, this alternative process to the conventional processes can maximize the amounts of long-chain alkanes and benzene derivatives with minimum consumption of hydrogen, compared to the hydrocracking process. We have previously reported dealkylation as a model reaction, in which only C16 and 18 alkylnaphthalenes were fed as reactants.6 The next report revealed the influence of coexisting substances in VGO as real heavy oil on the conversion of a C16 alkylnaphthalene added as an index reactant.7 However, the reactivity of the original hydrocarbons in VGO was not apparent. It is essential for a commercialized process to take into account the change of carbon balance in the reaction.
In this study, dealkylation of APAHs in practical VGO is made solely to proceed over various solid acids under a hydrogen atmosphere. However, the solid acids are poisoned by basic nitrogen-containing compounds in the original VGO. Before the dealkylation, the basic nitrogen-containing aromatic hydrocarbons were removed by an acidic ion-exchanged resin (containing a SO3H group).7 The industrial utilization of this resin may be difficult due to its high cost and laborious regeneration, but we have recently found that the alternative to the resin was inexpensive amorphous silica-alumina, which could be simply regenerated by heating.8 Benzene was added for increasing the fluidity. However, actual processes were operated in a flow of high-viscosity crude oil, and our dealkylation process should be also applied at large scales without addition of benzene. The dealkylation activity was compared among aluminosilicates: such as amorphous silica-alumina, zeolites, and a silica monolayer loaded on alumina (SMA) as a candidate catalyst. The SMA can be synthesized by chemical vapor deposition (CVD) of the Si precursor on the surface of alumina and has been found to have catalytic activity for Brønsted acid-catalyzed reactions.24,25 The SMA with 8–12 nm−2 Si concentration showed high activity and selectivity for dealkylation of C16 and -18 alkylnaphthalenes into naphthalene and C16 and -18 alkanes.6 Therefore, a SMA with 8 nm−2 Si concentration was prepared for the dealkylation of APAHs in the practical VGO. Additionally, it was presumed that the textural properties of the SMA affected the dealkylation activity in the practical VGO, which contained bulky hydrocarbons to prevent the diffusion of the reactants. In order to investigate the influence of pore size on dealkylation activity, SMAs with different pore sizes from 6.2 to 33 nm were synthesized. In the reaction, a small amount of hydrogen is fed for the hydrogenation of alkenes, which are produced by dealkylation and contained in the practical oil, because the formation of coke from the alkenes deactivates the solid acid. The previous study mainly reported the reactivity of C16 and -18 alkylnaphthalenes, as model reactants for indexing the dealkylation rate, in the coexistence of VGO components to quantitatively show the activity of the SMA catalyst,7 and here the reactivity and selectivity for dealkylation of practical VGO components are evaluated with the aid of the two-dimensional GC (2D-GC) technique, in order to clarify changes in compositions of existing hydrocarbons in VGO. The high dealkylation activity and selectivity to long-chain alkanes of the SMA, due to its weak Brønsted acidity and large pore size, will be focused on.
2. Experimental
2.1. Catalyst preparation
Thin silica layers were prepared by means of a CVD (chemical vapor deposition) method25 on the alumina materials JRC-ALO-6, -7, and -9, which had been supplied by Catalysis Society of Japan as reference catalysts, and a purchased sample from FUJIFILM Wako Pure Chemical Corp. The deposition was performed in a vacuum system using Si(OCH3)4 (tetramethoxysilane) as the precursor of silica. After the evacuation of the alumina support at 673 K for 2 h, the vapor of tetramethoxysilane was admitted to be in contact with alumina at 593 K, and the resultant increase of weight was monitored using a quartz spring balance. The supply of tetramethoxysilane vapor and evacuation were repeated to feed the precursor vapor and to remove the gaseous products continuously. The degree of vacuum was ca. 10−3 Torr (1 Torr = 133.3 Pa), and the vapor pressure of the alkoxide was kept at ca. 2.5 Torr by chilling the reservoir with an ice bath. After a weight increase corresponding to SiO2 with ca. 8 nm−2 Si concentration was observed, the sample was calcined in oxygen (200 Torr) at 673 K for 12 h. The amount of deposited Si was calculated from the weight increase while assuming that the formed material was SiO2, and the samples with ca. 8–9 Si atoms per nm2 were employed in the present study; hereafter, they are called SMA (silica monolayer loaded on alumina). As a comparison, an industrially available amorphous silica-alumina catalyst, N631-L (JGC Catalysts and Chemicals Ltd.), was tested for catalytic and analytical experiments, as well as the following zeolites. An H-USY zeolite CBV720 with FAU structure (Si/Al2 = 29) was supplied by Zeolyst. In addition, an NH4-ZSM-5 zeolite HSZ820NAA with MFI structure (Si/Al2 = 22) was supplied by Tosoh Corp; it was converted into H-ZSM-5 in the pretreatments for analytical and catalytic experiments.2.2. Structural analysis
The nitrogen adsorption isotherm was measured at 77 K using BELSorp-max equipment (Microtrac-BEL) after pretreatment at 573 K for 1 h. The surface area was calculated using the BET (Brunauer–Emmett–Teller) equation,26 and the pore diameter distribution was analyzed from the desorption branch using the BJH (Barrett–Joyner–Halenda) method.27The amount of chemisorbed benzoate anion was measured by the BAT (benzaldehyde-ammonia titration) method.28 About 40 mg of the sample was packed in a Pyrex tube (4 mm i.d.) and pretreated in an oxygen flow (27 μmol s−1, atmospheric pressure) at 673 K for 1 h. Benzaldehyde (9.8 μmol) was injected into the sample bed at 573 K in a helium flow (27 μmol s−1, atmospheric pressure), and the eluted aldehyde was monitored using a GC (GC-8A, Shimadzu) with a silicone SE-30 packed column and an FID (flame ionization detector). The injection of the aldehyde was repeated several times until saturation of adsorption was observed, and then ammonia (0.41 mmol) was repeatedly supplied at 673 K. The formed benzonitrile was quantified by the GC, and the coverage was calculated on the assumption that the benzoate anion was adsorbed only on the exposed alumina surface, not on the silica-covered surface.25
The ammonia IRMS (infrared/mass spectroscopy)-TPD (temperature-programmed desorption) method was applied to measure the number and strength distribution of each of the Brønsted and Lewis acid sites.29 A self-supporting disc (1 cm diameter), molded from the sample by compression, was held fixed by a set of metal rings into a cell of a Microtrac-BEL IRMS-TPD analyzer and pretreated at 823 K in oxygen. The IR (infrared) spectra were collected using an IR spectrometer (JASCO, FT/IR-4200) equipped with an MCT (mercury cadmium telluride detector) while heating the sample in a helium flow (68 μmol s−1, 6 kPa) at 2 K min−1. Then ammonia was adsorbed at 343 K, and the sample was again heated under the same conditions. The IR spectra and MS response (measured by a Pfeiffer Vacuum QMG220) were recorded. The ammonia TPD profile of each of the Brønsted and Lewis acid sites was analyzed according to our previous study.30 The number of acid sites was calculated from the peak intensity of the TPD, and the distribution of enthalpy of ammonia desorption (so-called adsorption heat) was analyzed by the curve fitting method.31
2.3. Dealkylation of APAHs
The reactant for dealkylation of APAHs was prepared as follows. An isomer mixture of hexadecylnaphthalene (HDN) with one alkyl branch, whose carbon number was 16, was purchased from Exxon Mobil Corp. as a commercial sample (Synesstic 5). VGO, obtained by vacuum distillation of a Middle East origin crude oil (final boiling point of 1008 K, specific gravity 0.915 g cm−3, C: 85.11 wt%, H: 12.35 wt%, S: 2.35 wt%, N: 0.06 wt%), was mixed with benzene and the above HDN. The composition of VGO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :benzene:HDN was 1:1:0.11 in weight ratio. Here benzene was added for increasing the fluidity, in order to mix the resulting VGO with the adsorbent for the removal of basic compounds and to feed it with a pump for the reaction tests. HDN was added as an index reactant for quantitative evaluation of the catalytic activity for the dealkylation of APAHs. Then, for the removal of basic compounds, a cation exchange resin (Amberlyst-15DRY, purchased from Organo) after pretreatment at 353 K overnight was added into the VGO; 10 wt% of Amberlyst-15DRY based on the VGO was used. The resulting mixture was stirred at room temperature for 1 h, and then Amberlyst-15DRY was removed by filtration. The above cation exchange resin treatment was repeatedly carried out (usually 2–3 times) until the pH trend indicated the neutralization point as observed using an automatic neutralization titrator (916Ti-Touch, Metrohm AG).7,8
:benzene:HDN was 1:1:0.11 in weight ratio. Here benzene was added for increasing the fluidity, in order to mix the resulting VGO with the adsorbent for the removal of basic compounds and to feed it with a pump for the reaction tests. HDN was added as an index reactant for quantitative evaluation of the catalytic activity for the dealkylation of APAHs. Then, for the removal of basic compounds, a cation exchange resin (Amberlyst-15DRY, purchased from Organo) after pretreatment at 353 K overnight was added into the VGO; 10 wt% of Amberlyst-15DRY based on the VGO was used. The resulting mixture was stirred at room temperature for 1 h, and then Amberlyst-15DRY was removed by filtration. The above cation exchange resin treatment was repeatedly carried out (usually 2–3 times) until the pH trend indicated the neutralization point as observed using an automatic neutralization titrator (916Ti-Touch, Metrohm AG).7,8
The reaction was carried out by a fixed-bed continuous-flow method. The catalyst was packed in a stainless steel tube (4 mm i.d.). The catalyst (210 mg) was pretreated in a flow of hydrogen (0.12 mol h−1) under atmospheric pressure at 773 K for 1 h. Then, the reactant was fed at 1.2 g h−1 with hydrogen flow (0.12 mol h−1) through the catalyst bed at 723 K. The total pressure in the tube was kept at 1 MPa using a back pressure valve connected to the outlet. The formed material was trapped in a glass tube chilled with an ice bath. The products were analyzed using a 2D-GC (Agilent 7890) with an FID and two capillary columns (0.25 μm thickness DB-5MS, length 30 m, i.d. 0.250 mm, and 0.15 μm thickness DB-17HT, length 5 m, i.d. 0.250 mm). An internal standard method was adopted with tetraethylene glycol dimethyl ether as the standard compound added after collecting the products. The composition of the components based on the number of carbon atoms was calculated from the relative intensities of GC peaks:

Light hydrocarbons were recovered by a gas pack and analyzed using GC equipment (GC-2014, Shimadzu) with an FID and a capillary column (InertCap1, 5.0 μm thickness, 30 m in length, and internal diameter of 0.53 mm). The composition averaged during 2–8 h of time on stream was calculated:
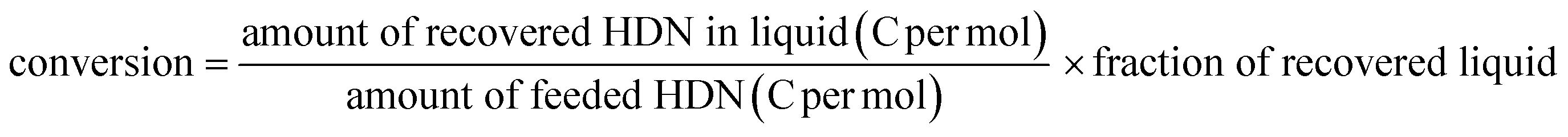
The structure and composition of the residues adsorbed on the catalysts after the reaction were investigated with elemental analysis and infrared (IR) spectroscopy. The catalysts were recovered after the reaction and dried at room temperature under vacuum. An elemental analysis of the catalysts was carried out with a Vario EL cube (Elementar Analytical). In the IR measurement, a self-supporting disc (1 cm diameter) was molded from a ground mixture of the catalyst and potassium bromide (1
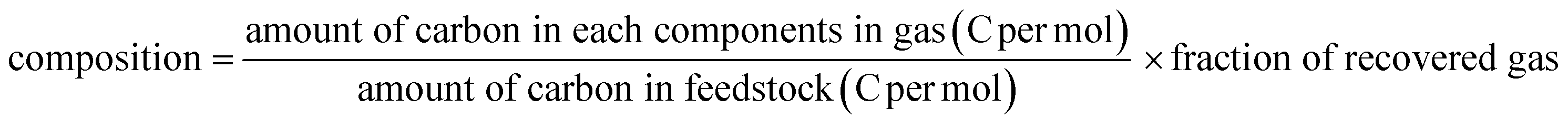 :10 in weight ratio). It was set in a Microtrac BEL, IRMS-TPD analyzer. After removal of water adsorbed on the catalyst under vacuum at 473 K for 0.5 h, the IR spectrum was recorded under vacuum at 293 K.
:10 in weight ratio). It was set in a Microtrac BEL, IRMS-TPD analyzer. After removal of water adsorbed on the catalyst under vacuum at 473 K for 0.5 h, the IR spectrum was recorded under vacuum at 293 K.
3. Results
3.1. Physical properties and acidity profiles
Table 1 shows the physical properties of the silica monolayer deposited on alumina (SMA). The amount of deposited material is here indicated by the surface concentration (Si atoms per nm2). The coverage of the surface by silica was calculated by the BAT method. At 8.3 ± 0.4 nm−2, the coverage of all the SMA samples exceeded 85%. It tells us that a monolayer of silica with 1:1 Al–O–Si bonding was formed to cover almost completely the Al2O3 surface.25,32 The BET surface areas of the SMAs before and after silica deposition are indicated in Tables S1† and 1, respectively. The BET surface area of the SMA (after CVD) divided by the weight of alumina was not largely different from the value before silica deposition, in all the cases, indicating that the surface area was unchanged by CVD. The mean pore diameter and mesopore volume of the SMAs before and after silica deposition are also shown in Tables S1† and 1, respectively, and the pore size distribution in the mesopore region is shown in Fig. S1.† The pore size and its distribution on the SMA were generally close to those of the original Al2O3. The order in mode value of pore size was: SMA-1 (6.2 nm) < SMA-2 (9.2 nm) < SMA-3 (24 nm) < SMA-4 (33 nm), according to the pore sizes of the parent Al2O3. The mesopore volume increased with increasing pore size. These textural profiles indicate that the morphology of particles was retained, consistent with the fact that a very thin layer of silica was formed on alumina. The physical properties of amorphous silica-alumina (N631-L) and zeolites (USY and ZSM-5) are indicated in Table 2. The BET surfaces of amorphous silica-alumina and zeolites were higher than those of the SMAs. The mesopore size of N631-L was smaller than those of the SMAs. On the other hand, most of the active sites on the zeolites are estimated to exist in the micropores, and the sizes of micropore channels in USY and ZSM-5 were ca. 0.7 nm and 0.6 nm, respectively, far smaller than the pores of the SMAs and N631-L. The mesopore volume of N631-L was similar to that of SMA-1, while the zeolites possessed small volumes of mesopores, mainly located between the crystal grains.
| Catalyst | Al2O3 | Amount of SiO2a/wt% | Deposited Si atoms per nm2 | Coverage/% | Surface areab/m2 g−1 | Pore sizec/nm | Mesopore volumed/cm3 g−1 |
|---|---|---|---|---|---|---|---|
| a Normalization by catalyst weight. b Calculated using the BET equation. c Mode value of pore size calculated by the BJH method. d Calculated by the BJH method. e Sample purchased from FUJIFILM Wako Pure Chemical Corp. f Normalized by catalyst weight. g Divided by the weight of alumina as a support. | |||||||
| SMA-1 | Purchased samplee | 14.1 | 8.5 | 86 | 184f (214g) | 6.2 | 0.36f (0.42g) |
| SMA-2 | ALO-9 | 14.2 | 8.1 | 90 | 182f (212g) | 9.2 | 0.58f (0.68g) |
| SMA-3 | ALO-6 | 13.3 | 7.9 | 91 | 169f (195g) | 24 | 0.83f (0.96g) |
| SMA-4 | ALO-7 | 13.1 | 8.7 | 95 | 147f (169g) | 33 | 0.95f (1.09g) |
| Catalyst | Surface areaa/m2 g−1 | Pore size/nm | Micropore volumeb/cm3 g−1 | Mesopore volumec/cm3 g−1 |
|---|---|---|---|---|
| a Calculated using the BET equation. b Calculated using the t-plot method. c Calculated by the BJH method. d Mode value of pore size calculated by the BJH method. e Estimated from the crystal structure of the zeolite. | ||||
| N631-L | 483 | 3.7d | <0.01 | 0.51 |
| USY | 756 | 0.74 × 0.74e | 0.30 | 0.24 |
| ZSM-5 | 308 | 0.53 × 0.56, 0.55 × 0.51e | 0.13 | 0.12 |
The ammonia IRMS-TPD determined the acidic properties.30 Fig. S2 and S3† show the IR spectra obtained on the catalysts during the TPD experiments. The sharp band at ca. 1450 cm−1 was assigned to the bending (ν4) vibration of NH4+ adsorbed on Brønsted acid sites (NH4+ (BAS)). The small peak at 1250–1330 cm−1 was assigned to the bending (δs) vibration of ammonia adsorbed on Lewis acid sites (ammonia (LAS)). Fig. S4† shows the TPD profiles of ammonia desorbed from the acid sites. MS-TPD indicates the profile of ammonia desorption evaluated with mass spectroscopy. The TPD profiles were calculated from the IR-TPD of the ca. 1450 cm−1-band (ν4, NH4+ (BAS)) and 1250–1330 cm−1-band (δs, ammonia (LAS)), respectively.30Table 3 shows the amount and strength of BAS. The mode value of enthalpy (ΔH) of ammonia desorption from the BAS on the catalysts is indicated as an index of Brønsted acid strength (the distribution of ΔH is shown in Fig. S5†).33 No difference was found in the strength of BAS on the SMAs as shown by ΔH. The amount of BAS on the SMA was distributed in the range of 0.06–0.11 mol kg−1 (0.25–0.36 nm−2 based on the surface area). The amorphous silica-alumina (N631-L) had more than 1.5 times the BAS of the SMAs, and the acid strength on N631-L was higher than those on the SMAs. The amount of BAS on the zeolites (USY and ZSM-5) was more than those on the SMAs and N631-L, and the BAS on the zeolites were much stronger as estimated from ΔH. In particular, ZSM-5 possessed stronger BAS in a high density.
| Catalyst | Brønsted acid amounta/mol kg−1 (sites per nm2) | ΔHb/kJ mol−1 |
|---|---|---|
| a Number of Brønsted acid sites normalized by the BET surface area. b Mode value of enthalpy of ammonia desorption from Brønsted acid sites. | ||
| SMA-1 | 0.11 (0.36) | 116 |
| SMA-2 | 0.08 (0.26) | 116 |
| SMA-3 | 0.10 (0.36) | 116 |
| SMA-4 | 0.06 (0.25) | 116 |
| N631-L | 0.17 (0.21) | 122 |
| USY | 0.34 (0.27) | 136 |
| ZSM-5 | 1.14 (2.23) | 148 |
3.2. Dealkylation of alkyl aromatic hydrocarbons
SMA-3 exhibited high activity and selectivity in the model reaction of HDN as previously reported,6 and was thus compared with various aluminosilicates as solid acids in this section. Hexadecylnaphthalene (HDN) was added to VGO as an index reactant, and the reactivity was assessed by the conversion of this compound. Fig. 1 shows the time courses of HDN conversion on various solid acids. At 2 h of time on stream, all the catalysts showed almost equivalent conversion values. The decline in the reaction rate was in the order of N631-L < SMA-3 < USY ≪ ZSM-5. Here it is noted that the previous studies have revealed that the SMA has the highest activity for the conversion of HDN at 673 K among the solid acids which were also tested in the present study.6,7 The present experiments were performed at 723 K for the evaluation of the catalytic performances under practical conditions, and therefore the apparent order of conversion was somewhat different.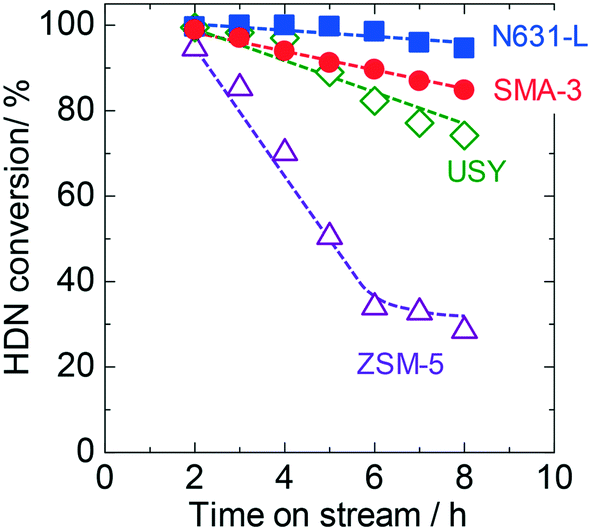 | ||
| Fig. 1 Time courses of HDN (hexadecylnaphthalene) conversion on SMA-3 (●), N631-L (■), USY (◊), and ZSM-5 (△) in the dealkylation of APAHs in the fed VGO at 723 K and LHSV = 5.7 gVGO gcat−1 h−1. | ||
This study quantified not only the HDN conversion but also the changes in hydrocarbon compositions by the reaction over aromatics and aliphatics. The compositions of the products in the reactions on the catalysts are shown in Tables S2–S5.† The time courses of changes in composition of APAHs in the outlet products are shown in Fig. 2. The original (fed) VGO contained 32 c-mol% of APAHs including HDN. SMA-3 reduced the APAH content down to 8 c-mol% constantly at 2–8 h of time on stream, indicating that the dealkylation of APAHs proceeded, and the catalytic activity was stably maintained. On N631-L and USY, the recovery of APAHs was high at the initial stage of the flow reaction, indicating low activity, and the recovery decreased with time on stream. On ZSM-5, more than half of the APAHs were eluted at the initial stage (2–4 h of time on stream), showing low activity, and the recovery gradually increased with time on stream, indicating the quick deactivation of the catalyst. These results indicate that the dealkylation activity of SMA-3 was higher than those of the amorphous silica-alumina and zeolites. However, 8 c-mol% of the APAHs was unreacted even on SMA-3. Fig. S6† shows the composition of alkyl groups in alkyl bicyclic and tricyclic aromatics in the products over SMA-3 during 2–8 h of time on stream. Most of the side chains before the reactions possessed >5 carbon atoms in an alkyl group. The dealkylation on SMA-3 reduced the carbon number down to <5. It means that the APAHs with long-chain alkyl groups were mainly dealkylated, while the methyl and ethyl groups attached to the polyaromatic rings showed lower reactivity on SMA-3.
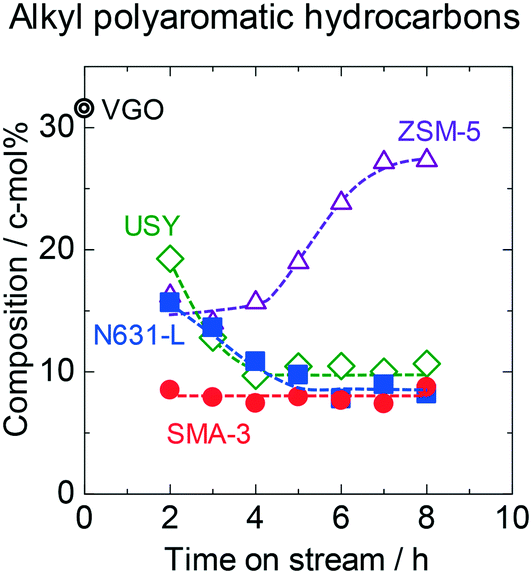 | ||
| Fig. 2 Time courses of changes in composition of APAHs in the products on SMA-3 (●), N631-L (■), USY (◊), and ZSM-5 (△) in the dealkylation of APAHs in the fed VGO at 723 K and LHSV = 5.7 gVGO gcat−1 h−1. Double circle indicates the amount of APAHs in the original (fed) VGO. | ||
Fig. 3 shows the time courses of changes in composition of alkylbenzenes (alkyl monocyclic aromatics) in the outlet products. The original VGO contained 9 c-mol% of alkylbenzenes. SMA-3 reduced the alkylbenzenes in a small amount down to 7 c-mol%, while the amorphous silica-alumina and zeolites obviously increased the alkylbenzenes. More than half of the outlet liquid and gas eluted in the reaction on ZSM-5 were alkylbenzenes in the initial period, but the yield decreased with time on stream. The composition of the side chains in alkylbenzenes in the products on SMA-3 is displayed in Fig. S7.† Most of the alkylbenzenes in the products possessed long-chain alkyl groups, meaning that the alkyl monoaromatics were inactive regardless of the long chain length on SMA-3. It was in contrast to the observations in the case of APAHs as stated above; the chain length of the alkyl group affected the reactivity in the case of APAHs. It is concluded that SMA-3 was active for the dealkylation of APAHs with long-chain alkyl groups but inactive for the reactions of alkyl monoaromatics and APAHs with short-chain alkyl groups.
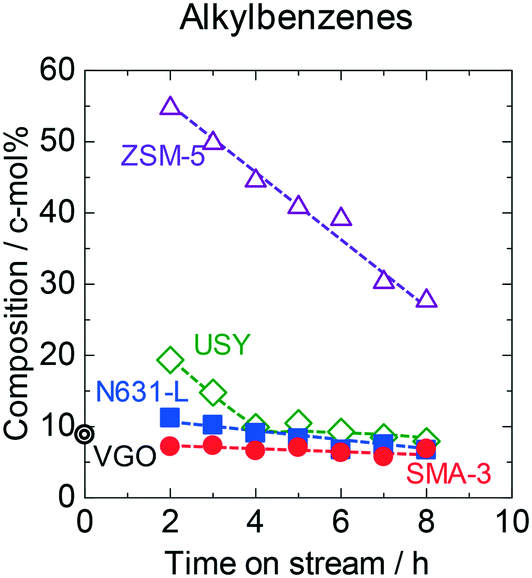 | ||
| Fig. 3 Time courses of changes in composition of alkylbenzenes in the products on SMA-3 (●), N631-L (■), USY (◊), and ZSM-5 (△) in the dealkylation of APAHs in the fed VGO at 723 K and LHSV = 5.7 gVGO gcat−1 h−1. Double circle indicates the amount of alkylbenzenes in the original (fed) VGO. | ||
Fig. 4 shows the time courses of changes in composition of the alkanes in the products. The original VGO contained 56 c-mol% of long-chain aliphatics (>C20); here it is noted that the original VGO contained alkanes and alkenes, whereas the outlet products contained mainly alkanes due to the reaction in pressurized hydrogen. ZSM-5 markedly decreased the >C20 alkanes, suggesting the progress of cracking of alkanes. USY and N631-L also showed decreases of >C20 alkane composition. SMA-3 also reduced the >C20 alkanes, but the extent was less than the amorphous silica-alumina and zeolites to keep the composition at 44 c-mol% during 5–8 h of time on stream (Fig. 4(A)). On the other hand, the original VGO contained only 4 c-mol% of C10–20 alkanes and no <C5 alkanes. The formation of <C20 alkanes, presumably due to the dealkylation of aromatics, was observed as shown in Fig. 4(B) and (C). SMA-3 and N631-L produced more C10–20 alkanes than the zeolites, while C5–9 alkanes were produced in the following order: N631-L > USY ≈ SMA-3 > ZSM-5. Fig. 5 shows the composition of light hydrocarbons during 2–8 h of time on stream. The yield of light hydrocarbons was in the following order: SMA-3 < N631-L < USY < ZSM-5. The composition of C4–6 in the light hydrocarbons over SMA-3, N631-L, and USY was >60%, while ZSM-5 produced excessive amounts of C1–3 alkanes. Here it is emphasized that ZSM-5 did not produce C5–20 alkanes but C3–4 alkanes in large quantities.
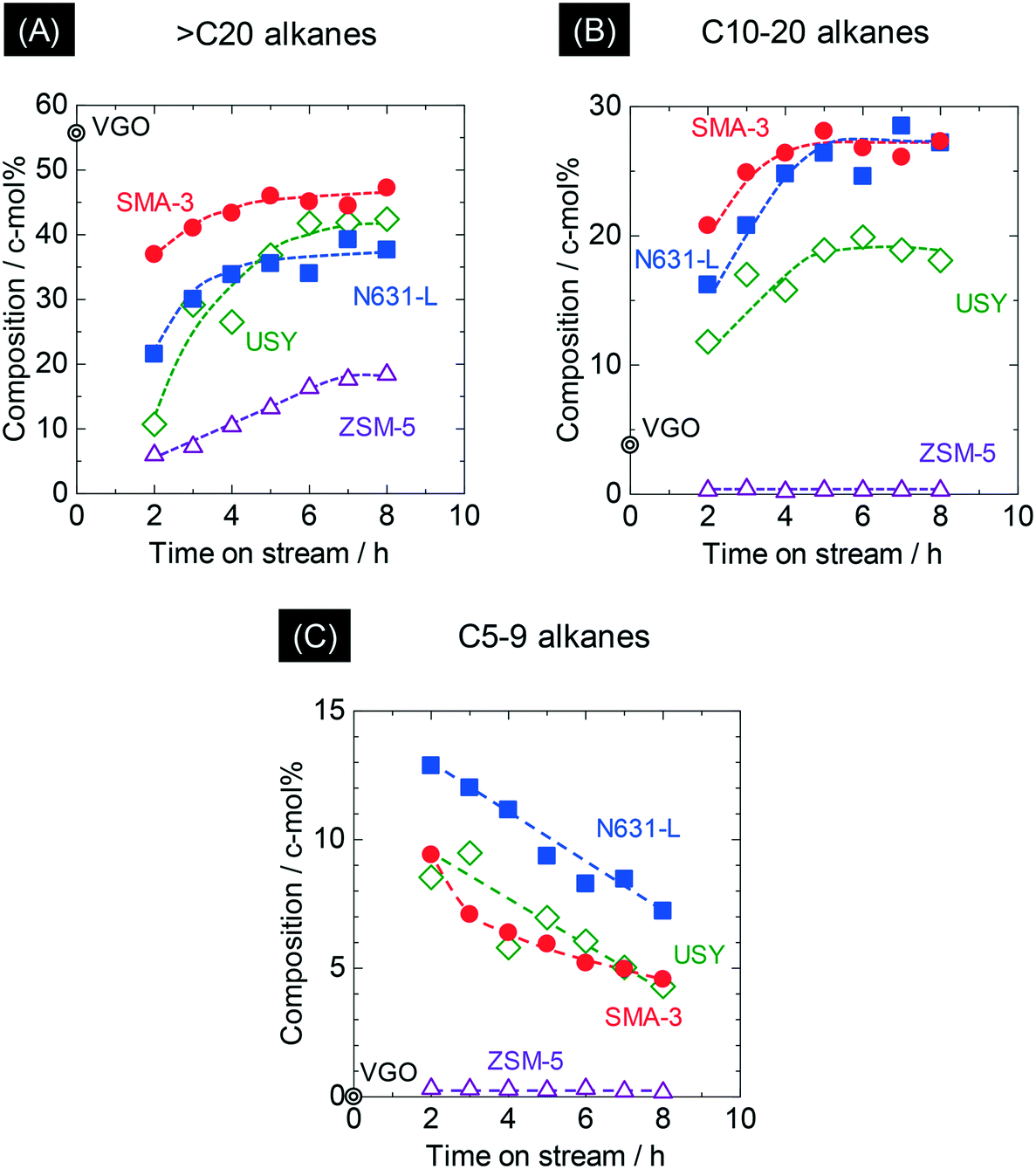 | ||
| Fig. 4 Time courses of changes in composition of alkanes ((A): >C20, (B): C10–20, and (C): C5–9) in the products on SMA-3 (●), N631-L (■), USY (◊), and ZSM-5 (△) in the dealkylation of APAHs in the fed VGO at 723 K and LHSV = 5.7 gVGO gcat−1 h−1. Double circle indicates the amounts of alkanes in the original (fed) VGO. | ||
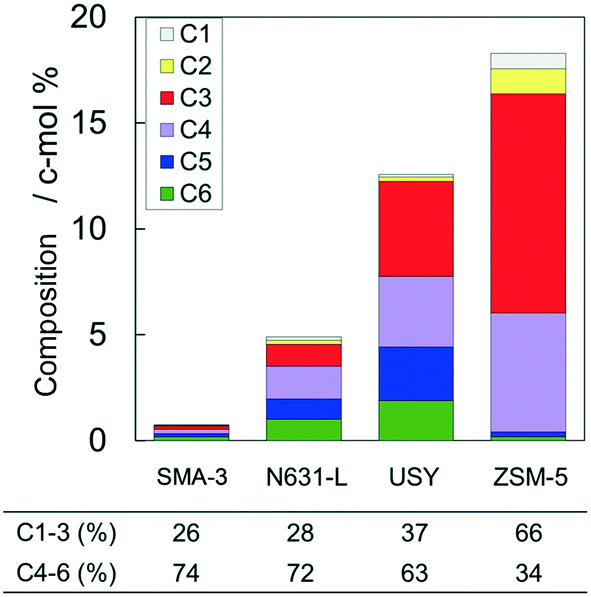 | ||
| Fig. 5 Composition of light hydrocarbons averaged during 2–8 h in the dealkylation of APAHs in the fed VGO at 723 K and LHSV = 5.7 gVGO gcat−1 h−1. | ||
A gradual decline in the dealkylation activity was observed as displayed in Fig. 1 and 2. The substances adsorbed on the catalysts during the reactions were analyzed by elemental analysis (Table 4) and IR measurements (Fig. 6). The amount of deposited carbon was in the following order: SMA-3 < ZSM-5 < N631-L < USY. Hydrogen was also detected, whereas trace amounts of nitrogen and sulfur were observed. The molar ratio of hydrogen to carbon (H/C) on USY was <1, indicating that the adsorbed hydrocarbons were polyaromatics (PAHs). The H/C ratio on SMA-3 and N631-L was close to 2, and therefore alkanes or long-chain alkylaromatics were presumed to be formed. ZSM-5 showed a higher H/C ratio than the other catalysts. The IR spectra of the catalysts after the reaction indicated the existence of aromatics and alkanes. The absorption band at 1600 cm−1 was assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration in aromatics, and the absorbance was in the following order: USY > ZSM-5 > N631-L > SMA-3. The pair of absorption bands at 2850 and 2900 cm−1 was assigned to the C–H stretching vibration in alkanes and/or alkyl groups (in alkylaromatics) and observed in all the spectra. The deposited substances on USY are thus revealed to be predominantly PAHs, while the substances on N631-L and SMA-3 were alkanes and small amounts of alkylaromatics. The adsorbed substances on ZSM-5 were mostly alkyl aromatics; too high a H/C ratio suggests the presence of water condensed in the micropores.
C stretching vibration in aromatics, and the absorbance was in the following order: USY > ZSM-5 > N631-L > SMA-3. The pair of absorption bands at 2850 and 2900 cm−1 was assigned to the C–H stretching vibration in alkanes and/or alkyl groups (in alkylaromatics) and observed in all the spectra. The deposited substances on USY are thus revealed to be predominantly PAHs, while the substances on N631-L and SMA-3 were alkanes and small amounts of alkylaromatics. The adsorbed substances on ZSM-5 were mostly alkyl aromatics; too high a H/C ratio suggests the presence of water condensed in the micropores.
| Wt% | Molar ratio | ||||
|---|---|---|---|---|---|
| C | H | N | S | H/C | |
| SMA-3 | 5.43 | 0.80 | Trace | Trace | 1.77 |
| N631-L | 13.85 | 5.10 | Trace | Trace | 1.82 |
| USY | 22.09 | 1.56 | Trace | Trace | 0.85 |
| ZSM-5 | 6.84 | 1.54 | Trace | Trace | 2.70 |
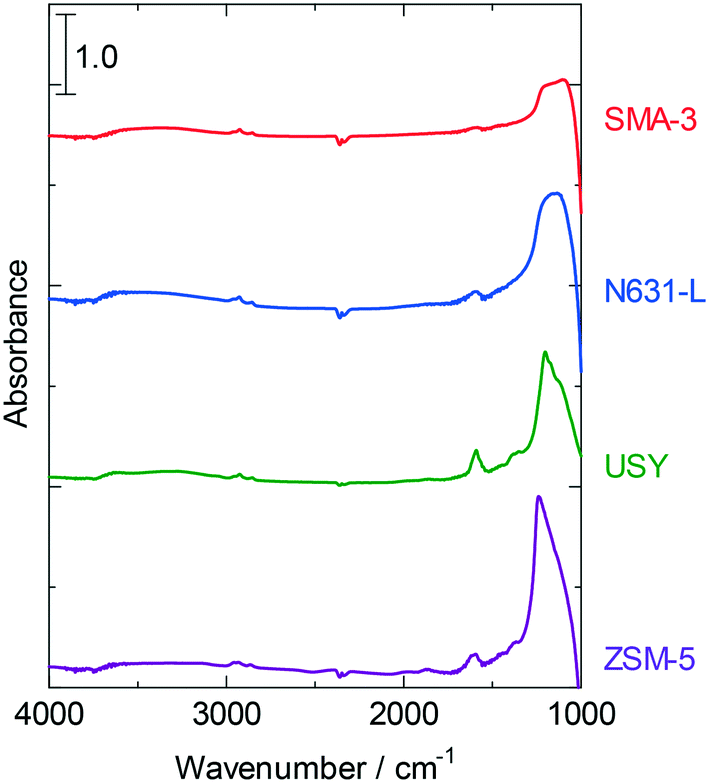 | ||
| Fig. 6 IR spectra of various solid acids after the dealkylation of APAHs in the fed VGO after 8 h time on stream at 723 K. Red line: SMA-3, blue line: N631-L, green line: USY, and purple line: ZSM-5. | ||
3.3. Influence of the textural properties of the SMA on activity and selectivity in the dealkylation
As stated in the last section, the SMA efficiently catalyzed the dealkylation of APAHs into PAHs and long-chain alkanes. As previously reported, the activity of the SMA for the dealkylation of a model substance was driven by the deposition of a silica monolayer with Brønsted acidity, and the activity was found to be at the maximum on the catalyst where the coverage by the monolayer was at the maximum, presumably due to the high density of Brønsted acid sites.6 Therefore, the importance of Brønsted acidity is undoubtable for the activity of the SMA in the dealkylation of APAHs. However, the low selectivity of amorphous silica-alumina (mesoporous) and zeolites (microporous) as shown in the last section suggests the influence of the porous nature on this reaction. It is presumed that the textural properties of the catalyst affected the activity for the dealkylation of APAHs in the practical VGO, which contained bulk hydrocarbon molecules, and their diffusion was prevented to some extent. Accordingly, the pore size of SMA was varied, and its influence was investigated. SMA-1 to -4 were prepared by the deposition of a silica monolayer on γ-alumina samples with different pore sizes from 6 to 33 nm; the resultant pore sizes of the SMAs were similar to the pore sizes of the support alumina samples (Tables 1 and S1†). The number of Brønsted acid sites normalized by the catalyst weight or surface area was not largely different (within 0.06–0.11 mol kg−1 or 0.25–0.36 nm−2); in addition, the Brønsted acid strength (ΔH) was similar as shown in Table 3. The similar acidic properties suggest principally the same chemical nature of the SMA surfaces due to the equivalent microstructures. It is concluded that SMA-1 to -4 had similar microstructures and Brønsted acidic properties, but the porous nature at 1–10 nm dimensions was different.The activities of the SMAs in the dealkylation are shown in Fig. S8–S11.† The composition of components in the reactions over all the catalysts displayed analogous changes over time on stream, and was steady during 5–8 h. Fig. 7 shows the average composition of components during 5–8 h in the dealkylation over the SMAs. Both the APAHs and PAHs were retrieved in the same amount over all the SMAs. This tells us that the dealkylation activities of all the SMA catalysts were comparable. The amount of alkylbenzenes after the reaction was almost the same as the original amount in VGO. Long-chain alkanes (>C20) relatively decreased over SMA-1 and -2, compared with SMA-3 and -4, while the composition of light hydrocarbons was in the following order: SMA-2 > -1 > -3 > -4. There were no differences in yields of C10–20 and C5–9 alkanes among all the catalysts. A part of long-chain alkanes was thus observed to be cracked into C5–9 alkanes, but deep cracking into C1–6 alkanes proceeded only on the SMA with a relatively small mesopore size.
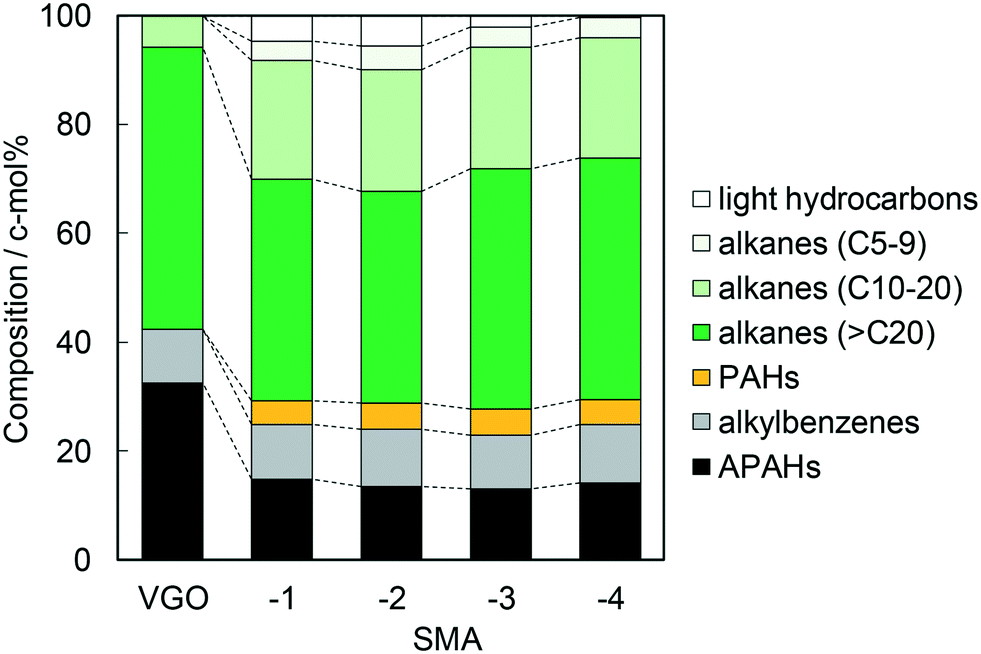 | ||
| Fig. 7 Compositions of outlet compounds averaged during 5–8 h on SMAs in the dealkylation of APAHs in the fed VGO at 723 K and LHSV = 5.7 gVGO gcat−1 h−1. The compositions in the original (fed) VGO are shown in the leftmost bar. | ||
4. Discussion
The assumed novel upgrading process consists of the dealkylation of APAHs in the feed mixture and the separation of aromatics (polar) and alkanes (non-polar) based on the large difference in polarity. The alkyl aromatic molecules possessing long alkyl chains with high compatibility should hinder the polar separation. Therefore, they need to be completely dealkylated. Alkanes are usually contained in the feed oil and produced by dealkylation. Among the alkanes, C5–10, C10–20, and larger alkanes are utilized as gasoline, diesel fuel, and lubricating oil, respectively, while short chain (<C4) alkanes, i.e., off-gas, are useless. The long-chain alkanes can be converted into alkanes suitable for gasoline, diesel fuel, and monocyclic aromatics by conventional FCC if necessary, while the off-gas components are difficult to convert into valued compounds. Therefore, in the dealkylation step, the chain length of alkanes and alkyl groups in the alkyl aromatics should be kept; in addition, cracking of alkanes, a side reaction, needs to be restricted, and a high yield of long-chain alkanes is desirable. For the above reactions, the research target is to find the catalyst which shows high conversion of APAHs and selectivity to long-chain alkanes in the dealkylation. A high HDN conversion and decrease of recovered APAHs show high catalytic activity for the desired reaction (dealkylation of APAHs), while the production of large amounts of recovered long-chain alkanes (C10–20 for diesel fuel and >C20 for lubricating oil) and small amounts of recovered C5–9 alkanes and light hydrocarbons shows high selectivity for the desired reaction.HDN conversion was in the order: ZSM-5 < USY < SMA-3 < N631-L, while the amounts of recovered APAHs were in the order: SMA-3 < N631-L ≈ USY < ZSM-5. HDN conversion on the SMAs gradually decreased as shown in Fig. 1 and S8.† The catalysts were deactivated by the adsorption of long-chain alkanes, but the deactivated catalysts can be regenerated by calcination at 773 K in oxygen flow.7 No difference among the SMAs was found in HDN conversion and amounts of recovered APAHs. In addition, the order of amounts of recovered alkylbenzenes was SMA-3 < N631-L < USY ≪ ZSM-5; the amount of alkylbenzenes increased from the original VGO on catalysts other than the SMAs, indicating the formation of alkylbenzenes. The amount of total recovered long-chain alkanes (C10–20 and >C20) was in the following order: ZSM-5 < USY < N631-L < SMA-3. In light alkanes, C5–9 alkanes were produced in the order: ZSM-5 < SMA-3 ≈ USY < N631-L, while the yield of light hydrocarbons was in the order: SMA-3 < N631-L < USY < ZSM-5. The total images of the reactions on the catalysts are shown in Fig. S12.† The bracketed values were calculated on the basis of Tables S2–S5† and averaged over 5–8 h in the reactions. The width of the arrows in the images indicates the amount of changes of hydrocarbons. SMA-3 reduced more APAHs and HDN (a total of 7.6 wt% from 30.6 c-mol%) and showed a higher yield of >C20 alkanes (45.7 c-mol%), indicating that SMA-3 exhibited high dealkylation activity and high selectivity to long-chain alkanes. N631-L and USY showed a higher yield of short-chain alkanes (C5–9 alkanes and light hydrocarbons) than SMA-3, due to the cracking of alkanes. ZSM-5 showed a higher yield of APAHs and HDN, but a lower yield of >C20 and C10–20 alkanes (a total of 16.8 c-mol%) than the other catalysts. On comparing the SMAs, the amounts of >C20 alkanes were lower over SMA-1 and -2 than those over SMA-3 and -4, while the yield of light hydrocarbons was in the following order: SMA-4 < -3 < -1 < -2. However, there were no differences in the yield of C10–20 and C5–9 alkanes among the SMAs.
The pore size was found to be ZSM-5 < USY < N631-L < SMAs, while the Brønsted acid strength was in the order: SMAs < N631-L < USY < ZSM-5. Among the SMAs, the mode value of pore size was in the order: SMA-1 < -2 < -3 < -4, whereas all the catalysts possessed a similar amount and strength of Brønsted acid sites.
The previous studies revealed the high activity of SMAs in the dealkylation of the model APAH reactant.6,7 This study confirms the higher conversion of APAHs in the practical oil and higher selectivity to long-chain alkanes on the SMAs than amorphous silica-alumina and zeolites. The high conversion on the SMAs is related to their large pore size. It is supposed that the bulky reactant molecules can penetrate into the large pore of the SMAs and then be dealkylated into alkanes and PAHs. On the other hand, the cracking of alkanes is more difficult, from the viewpoint of acid-catalysis chemistry, than the dealkylation of alkyl aromatics, because it needs a strong Brønsted acid site as follows. The dealkylation of APAHs proceeds through the attack of H+ on the aromatic ring and then formation of the carbenium ion (arenium ion) for relatively stable intermediates of the σ-complex, and finally produces PAHs and alkenes (subsequently hydrogenated into alkanes). The aromatic rings are subject to nucleophilic attack, and therefore the dealkylation proceeds even on weak Brønsted acid sites. On the other hand, the cracking of alkanes proceeds through the formation of an alkanium ion by the attack of H+ and then protolytic cracking into a carbenium ion and an alkane (or a dihydrogen). The alkanium ion is an unstable intermediate, and therefore the cracking proceeds only on strong Brønsted acid sites. The weak Brønsted acid sites on the SMAs cannot catalyze the cracking of both alkanes and alkyl groups in APAHs. According to these explanations, the high selectivity (low activity for alkane cracking) of the SMAs is considered to be due to the moderate Brønsted acid strength, and the alkyl groups in the reactants and the produced alkanes were difficult to crack into lighter alkanes. Thus, the large pore size and moderate Brønsted acid strength are concluded to generate the high activity and selectivity of the SMAs for the dealkylation of APAHs.
A difference between APAHs and alkylbenzenes was observed in the reaction over the SMAs; the SMA was inactive for the dealkylation of alkyl monocyclic aromatics but active for the dealkylation of APAHs. The arenium ion formed from APAHs holds a higher variety of resonance structures than that formed from alkylbenzenes. Due to the higher stability of the intermediate, the APAHs should be more reactive than the alkylbenzenes. As a result, the SMA catalyzed the conversion of APAHs but kept the alkylbenzene, which is directly useful for chemical resources, another advantage of the presently proposed process using the SMA.
A part of APAHs remained even on the SMAs, but most of them consisted of short (C1–4) alkyl groups. The binding energy between alkyl groups and aromatic rings is in the following order: propyl < ethyl ≪ methyl.34 In addition, the carbenium ion formed by the dealkylation is stable in the following order: tertiary carbon > secondary carbon > primary carbon > methylium. From the above principles, the removal of methyl and ethyl groups from the aromatic ring is estimated to be difficult compared to the >C3 alkyl groups. It is therefore reasonable to conclude that methyl and ethyl groups, in some cases multiple ones in a molecule, were unreacted on the SMAs and detected as the APAHs with C1–4 alkyl groups. The purpose of dealkylation is to separate the aromatic rings from alkanes which are useful as fuel and/or feeds of further treatment. The long-chain alkanes are useful as fuels and/or feeds, but C1–4 alkanes are not, and therefore the observed inactivity of C1–4 alkyl groups does not reduce the advantages of the SMAs for the dealkylation of APAHs.
N631-L and USY formed more APAHs in the initial period than during 6–8 h. The dealkylation of APAHs and the cracking of alkanes probably proceeded, and then polymerization to APAHs deactivated a part of the acid sites such as strong Brønsted acid sites. However, only the dealkylation proceeded with time. On the other hand, ZSM-5 substantially catalyzed the cracking of long-chain alkanes and produced lighter alkanes along with the formation of alkylbenzenes by swift cyclization of alkenes. N631-L exhibited higher activity in the dealkylation than the SMAs (Fig. 1 and 2) but formed APAHs by the cracking and polymerization of alkanes in the initial period (Fig. 2). Therefore, the order of conversion in Fig. 1 was different from the order of composition of APAHs in Fig. 2. However, carbon deposition on ZSM-5 also deactivated the acid sites, and the reaction rate of the dealkylation of APAHs decreased with time on stream. From the above results, more APAHs were recovered on N631-L, USY, and ZSM-5 than on the SMAs, and the cracking of long-chain alkanes proceeded.
The SMAs exhibited the same dealkylation activity, but a difference in alkane selectivity was observed. The selectivity to long-chain alkanes (>C20) on SMA-1 and -2 was less than that on SMA-3 and -4, while the composition of light hydrocarbons was in the following order: SMA-2 > -1 > -3 > -4. It was speculated that long-chain alkanes were prone to contact with acid sites due to the high surface area on SMA-1 and -2, otherwise due to capillary condensation in the smaller mesopores in SMA-1 and -2 than those in SMA-3 and -4. Therefore, SMA-3 and -4, which had weak Brønsted acid sites and large pores, exhibited higher dealkylation activity and selectivity to long-chain alkanes than the other solid acids including SMA-1 and -2. However, the pore size did not determinately influence the dealkylation activity. Therefore, the production of SMA catalysts for dealkylation should be commercially practicable.
5. Conclusions
A silica-monolayer loaded on alumina (SMA) with weak Brønsted acid sites and large pore size catalyzed the dealkylation of APAHs in a practical heavy oil such as VGO, and exhibited higher activity and selectivity to long-chain alkanes than amorphous silica-alumina (N631-L) and zeolites (USY and ZSM-5). The high activity on SMAs was proposed to be due to their larger pore size in which APAHs could sufficiently diffuse. Some APAHs were retrieved, but combined with multiple methyl or ethyl groups. The residual should not affect the separation of alkanes and PAHs by solvent extraction. On the one hand, the produced alkanes and alkyl groups in the reactants were not cracked on weak Brønsted acid sites in the SMAs. The retrieved >C20 and C10–20 alkanes can be utilized as lubricating oil and light oil or kerosene, respectively. On the other hand, C5–9 alkanes and light hydrocarbons were hardly formed. Analyses of the SMAs after the reaction found adsorbed alkanes, which did not lead to drastic deactivation. Additionally, the pore size of the SMAs increased with limiting cracking of long-chain alkanes, and the catalysts with larger pores exhibited higher selectivity to long-chain alkanes. The dealkylation produced long-chain alkanes and PAHs, which can be converted to benzene derivatives through further treatment. Catalytic reforming of long-chain alkanes may form benzene, toluene, and xylenes, whereas PAHs are converted into tetralin derivatives through partial hydrogenation, and further ring-opening of the naphthene in tetralin derivatives produces benzene and its derivatives. These processes for the production of aromatics will be studied as future work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this work was carried out as a part of projects for technological development entrusted by the Ministry of Economy, Trade and Industry, Japan to Japan Petroleum Energy Center. The other part was supported by JSPS KAKENHI Grant Number 16H04568 and JST CREST Grant Number JPMJCR17P1, Japan.References
- J. Chang, N. Tsubaki and K. Fujimoto, Fuel, 2001, 80, 1639–1643 CrossRef CAS.
- R. Yoshida, M. Miyazawa, H. Ishiguro, S. Itoh, K. Haraguchi, H. Nagaishi, H. Narita, T. Yoshida, Y. Maekawa and Y. Mitarai, Fuel Process. Technol., 1997, 51, 195–203 CrossRef CAS.
- R. Yoshida, M. Miyazawa, T. Yoshida, H. Narita and Y. Maekawa, Fuel, 1996, 75, 99–102 CrossRef CAS.
- R. Yoshida, T. Yoshida, H. Narita and Y. Maekawa, Fuel, 1986, 65, 425–428 CrossRef CAS.
- Organization of the Petroleum Exporting Countries, World Oil Outlook, 2019 Search PubMed.
- N. Katada, Y. Kawaguchi, K. Takeda, T. Matsuoka, N. Uozumi, K. Kanai, S. Fujiwara, K. Kinugasa, K. Nakamura, S. Suganuma and M. Nanjo, Appl. Catal., A, 2017, 530, 93–101 CrossRef CAS.
- K. Kinugasa, F. Nakano, S. Nagano, S. Suganuma, E. Tsuji and N. Katada, J. Jpn. Pet. Inst., 2018, 61, 294–301 CrossRef CAS.
- S. Suganuma, K. Arita, F. Nakano, E. Tsuji and N. Katada, Fuel, 2020, 266, 117055 CrossRef CAS.
- K. Nakajima, S. Suganuma, E. Tsuji and N. Katada, React. Chem. Eng., 2020, 5, 1272–1280 RSC.
- S. Suganuma and N. Katada, Fuel Process. Technol., 2020, 208, 106518 CrossRef CAS.
- S. W. Ferris, E. R. Birkhimer and L. M. Henderson, Ind. Eng. Chem., 1931, 23, 753–761 CrossRef CAS.
- A. De Lucas, L. Rodriguez, P. Sanchez and A. Carnicer, Sep. Sci. Technol., 1993, 28, 2465–2477 CrossRef CAS.
- B. Coto, R. van Grieken, J. L. Peña and J. J. Espada, Chem. Eng. Sci., 2006, 61, 8028–8039 CrossRef CAS.
- B. W. Wojciechowski, Catal. Rev.: Sci. Eng., 1982, 24, 1–65 Search PubMed.
- A. M. Youssef, A. I. Ahmed and S. E. Samra, Mater. Lett., 1990, 10, 175–180 CrossRef CAS.
- M. Du, Z. Qin, H. Ge, X. Li, Z. Lü and J. Wang, Fuel Process. Technol., 2010, 91, 1655–1661 CrossRef CAS.
- A. Stanislaus and H. C. Barry, Catal. Rev.: Sci. Eng., 1994, 36, 75–123 CrossRef CAS.
- Y. Choi, J. Lee, J. Shin, S. Lee, D. Kim and J. K. Lee, Appl. Catal., A, 2015, 492, 140–150 CrossRef CAS.
- A. Corma, V. González-Alfaro and A. V. Orchillés, J. Catal., 2001, 200, 34–44 CrossRef CAS.
- M. Santikunaporn, J. E. Herrera, S. Jongpatiwut, D. E. Resasco, W. E. Alvarez and E. L. Sughrue, J. Catal., 2004, 228, 100–113 CrossRef CAS.
- J. Shin, Y. Oh, Y. Choi, J. Lee and J. K. Lee, Appl. Catal., A, 2017, 547, 12–21 CrossRef CAS.
- Y. Oh, J. Shin, H. Noh, C. Kim, Y. S. Kim, Y. K. Lee and J. K. Lee, Appl. Catal., A, 2019, 577, 86–98 CrossRef CAS.
- Y. Oh, H. Noh, H. Park, H. Han, T. B. Nguyen and J. K. Lee, Catal. Today, 2020, 352, 329–336 CrossRef.
- M. Niwa, T. Hibino, H. Murata, N. Katada and Y. Murakami, J. Chem. Soc., Chem. Commun., 1989, 289–290 RSC.
- M. Niwa, N. Katada and Y. Murakami, J. Phys. Chem., 1990, 94, 6441–6445 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- M. Niwa, S. Inagaki and Y. Murakami, J. Phys. Chem., 1985, 89, 3869–3872 CrossRef CAS.
- M. Niwa, K. Suzuki, N. Katada, T. Kanougi and T. Atoguchi, J. Phys. Chem. B, 2005, 109, 18749–18757 CrossRef CAS.
- S. Suganuma, Y. Murakami, J. Ohyama, T. Torikai, K. Okumura and N. Katada, Catal. Lett., 2015, 145, 1904–1912 CrossRef CAS.
- N. Katada, T. Tsubaki and M. Niwa, Appl. Catal., A, 2008, 340, 76–86 CrossRef CAS.
- N. Katada, T. Fujii, K. Iwata, Y. Hibino and M. Niwa, J. Catal., 1999, 186, 478–480 CrossRef CAS.
- M. Niwa and N. Katada, Chem. Rec., 2013, 13, 432–455 CrossRef CAS.
- M. Szwarc, Chem. Rev., 1950, 47, 75–173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01590c |
| This journal is © The Royal Society of Chemistry 2021 |