
In situ probing of Pt/TiO2 activity in low-temperature ammonia oxidation†
Lidiya S.
Kibis
 a,
Dmitry A.
Svintsitskiy
a,
Andrey I.
Stadnichenko
a,
Elena M.
Slavinskaya
a,
Anatoly V.
Romanenko
a,
Elizaveta A.
Fedorova
a,
Olga A.
Stonkus
a,
Valery A.
Svetlichnyi
b,
Elena D.
Fakhrutdinova
b,
Mykhailo
Vorokhta
c,
Břetislav
Šmíd
c,
Dmitry E.
Doronkin
de,
Vasyl
Marchuk
d,
Jan-Dierk
Grunwaldt
*de and
Andrei I.
Boronin
*a
a,
Dmitry A.
Svintsitskiy
a,
Andrey I.
Stadnichenko
a,
Elena M.
Slavinskaya
a,
Anatoly V.
Romanenko
a,
Elizaveta A.
Fedorova
a,
Olga A.
Stonkus
a,
Valery A.
Svetlichnyi
b,
Elena D.
Fakhrutdinova
b,
Mykhailo
Vorokhta
c,
Břetislav
Šmíd
c,
Dmitry E.
Doronkin
de,
Vasyl
Marchuk
d,
Jan-Dierk
Grunwaldt
*de and
Andrei I.
Boronin
*a
aBoreskov Institute of Catalysis, pr. Lavrentieva 5, 630090, Novosibirsk, Russia. E-mail: boronin@catalysis.ru
bTomsk State University, pr. Lenina 36, 634050, Tomsk, Russia
cDepartment of Surface and Plasma Science, Faculty of Mathematics and Physics, Charles University, V Holešovičkách 2, 180 00, Prague 8, Czech Republic
dInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology, Engesserstr. 20, 76131, Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
eInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344, Eggenstein-Leopoldshafen, Germany
First published on 10th November 2020
Abstract
The improvement of the low-temperature activity of the supported platinum catalysts in selective ammonia oxidation to nitrogen is still a challenging task. The recent developments in in situ/operando characterization techniques allows to bring new insight into the properties of the systems in correlation with their catalytic activity. In this work, near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) and operando X-ray absorption spectroscopy (XAS) techniques were applied to study Pt/TiO2 catalysts in ammonia oxidation (NH3 + O2 reaction). Several synthesis methods were used to obtain samples with different size of Pt particles, oxidation state of Pt, and morphology of the support. Metal platinum particles on titania prepared by pulsed laser ablation in liquids exhibited the highest activity at lower temperatures with the temperature of 50% conversion of NH3 being 150 °C. The low-temperature activity of the catalysts synthesized by impregnation can be improved by the reductive pretreatment. NAP-XPS and operando XANES data do not show formation of PtOx surface layers or PtO/PtO2 oxides during NH3 + O2 reaction. Despite the differences in the oxidation state of platinum in the as-prepared catalysts, their treatment in the reaction mixture results in the formation of metallic platinum particles, which can serve as centers for stabilization of the adsorbed oxygen species. Stabilization of the bulk platinum oxide structures in the Pt/TiO2 catalysts seems to be less favorable due to the metal–support interaction.
Introduction
Pt-Containing catalysts are important components of the complex systems used for the exhausts gas aftertreatment.1–4 Supported platinum systems are utilized in diesel vehicles, among others, as a part of ammonia slip catalysts responsible for NH3 oxidation to molecular nitrogen.5,6Both model platinum systems – single-crystals, foils, and gauzes,7–12 as well as supported platinum catalysts,13–20 have been studied in detail as catalysts for the ammonia oxidation reaction, mostly within the Ostwald process for nitric acid production. DFT calculations21–25 and kinetic modeling26–28 were performed to gain more insight into the mechanism. However, ways to improve the low-temperature activity (T < 250 °C) of the catalysts and the selectivity of the reaction towards N2 formation (required for exhaust gas aftertreatment) are still under discussion. For supported Pt systems, activity and selectivity of the catalysts are influenced by many factors: the size of the platinum particles, Pt oxidation state, the structure and nature of the support, metal–support interactions, etc. Also, the state of the active component can change during the reaction process, which requires special experimental approaches to obtain relevant results. This makes it difficult to draw reliable conclusion on “structure” – “activity/selectivity” correlations.
The recent development of in situ and operando techniques opens new opportunities to get information about the systems under reaction conditions. In situ methods play a key role in studying reaction mechanisms and identifying the main factors affecting the activity and selectivity of the catalysts. Thus, in situ X-ray photoelectron spectroscopy (XPS) gives information about processes taking place on the surface of the catalysts during the reaction: the reaction intermediates formed on the surface can be identified; the change of the charge state of the active component can be analyzed. In situ XPS data for ammonia oxidation reaction on the surface of bulk platinum systems have already been reported.10 The authors used stepped platinum surface which might to some extent simulate the structural heterogeneity of nanoparticles present in real powder catalysts. However, to obtain more relevant results, a further transition to the study of supported systems is necessary.
In this paper, we present a study of Pt/TiO2 catalysts by in situ/operando methods in the ammonia oxidation reaction. The Pt/TiO2 systems are less investigated for this reaction than the traditionally used Pt/Al2O3.29,30 However, unlike the Pt/Al2O3 catalysts, for the Pt/TiO2 system, there is a possibility of the metal–support interaction, which can influence the catalytic properties.31–36 It was shown that the metal–support interaction can provide stabilization of the metal platinum particles.31,33 Oxidation of the metal platinum particles with the formation of oxide structures on the surface leads to deactivation of the catalysts of the ammonia oxidation reaction.13,19,37 Therefore, stabilization of metallic Pt species might be beneficial for the NH3 + O2 catalytic process. As potential catalysts for the selective NH3 oxidation at low temperature, the Pt/TiO2 systems should be investigated in detail using the modern physicochemical methods.
Two techniques for the synthesis of Pt/TiO2 catalysts were applied in this work. Apart from the traditional impregnation technique, pulsed laser ablation in liquids (LAL) was used for the synthesis of the catalysts. The LAL method leads to the formation of dispersed particles with defective, distorted structure.38–41 As the defects of the TiO2 support might stimulate the metal–support interaction,42–44 the LAL can be a promising method for the preparation of the Pt/TiO2 catalysts active in the ammonia oxidation reaction. Previously, it was shown that the composites prepared by LAL are active in CO oxidation reaction,38,39 low-temperature ammonia oxidation,45 phenol photodegradation process under visible light.41
The supported Pt/TiO2 catalysts were studied using NAP-XPS and operando XAS – powerful complementary tools for the investigation of the catalysts under working conditions. The application of these methods provided insights into the evolution of the surface and bulk Pt particle structure under NH3 + O2 conditions showing that the metal platinum particles do not undergo significant oxidation under the reaction conditions, regardless of the preparation method.
Experimental
Samples preparation
Two approaches were used for the synthesis of Pt/TiO2 samples. The first was the incipient wetness impregnation of the commercial TiO2 support (AEROXIDE P25, Evonik) with a solution of platinum nitrate Pt(NO3)x (17.87 wt%, KZCM). The resulted sample was dried at room temperature (for 16 h) with further increase of the temperature to 60 °C (heating for 1 h) and 120 °C (heating for 2 h). The dried catalyst was calcined in air at 400 °C for 4 h. The platinum content in the sample was 2 wt%. This sample is denoted by Pt/TiO2-IMP in the text.The second method for the synthesis of the catalysts was pulsed laser ablation in liquids. The detailed description of the preparation of the composite catalysts by the LAL is given in ref. 38 and 39. Briefly, the Pt and TiOx dispersions were obtained separately by ablation of platinum and titanium metal plates in liquids. The ablation of the targets was carried out by focused radiation of a Nd:YAG laser (1064 nm, 7 ns, 150 mJ imp., 20 Hz). Based on the earlier study,39 an ethanol solution (96.3 vol%) and distilled water were taken as solvents to obtain platinum and TiOx dispersions, respectively. The concentration of the components in the resulting dispersions was determined by the loss of the mass of the corresponding target. The resulting solutions were mixed in such a proportion so as to obtain 2 wt% of platinum in the catalyst. The Pt and TiOx dispersions were mixed using a magnetic stirrer at room temperature and dried in air at 60 °C. The air-dried powder was calcined in air at 400 °C for 4 h. The LAL-prepared sample is denoted in the text by Pt/TiO2-LAL.
Physicochemical characterization
To characterize the structure of the catalysts X-ray diffraction (XRD) and transmission electron microscopy (TEM) were used. The XRD data for the catalysts were obtained on a Bruker D8 instrument (Germany) using Cu Kα radiation in a 2θ range 15° < 2θ < 90° (step size 0.05°, 5 s per step). Diffraction intensities were measured with the LynxEye position sensitive detector. For crystalline phase analysis, the ICDD PDF-2 powder database was used. The refinement of the structure and the profile analysis were carried out with the TOPAS software package (Bruker, Germany).46TEM experiments were performed on a JEM-2200FS electron microscope (JEOL Ltd., Japan) at the “VTAN” resource center of Novosibirsk State University, Russia. To collect the high-resolution (HRTEM) and HAADF-STEM images with a spatial resolution of 1 Å an accelerating voltage of 200 kV was applied. TEM data were analyzed using “ImageJ” software47 to determine the particle size distribution. For TEM experiments samples were deposited on 3 mm copper grids coated with a carbon film by dispersing in ethanol and sputtering the dispersion on the grids.
The specific surface area of the samples (SBET) was determined by the standard Brunauer–Emmett–Teller method using the low-temperature adsorption isotherms of N2 collected at an ASAP-2400 setup (Micromeritics Instrument. Corp., Norcross, GA, USA).
Analysis of the samples by X-ray photoelectron spectroscopy (XPS) under UHV (p ∼ 10−8 mbar) conditions (ex situ XPS experiments) was performed using an ES-300 spectrometer (KRATOS Analytical). To record the spectra a non-monochromatic Mg Kα source (hν = 1253.6 eV) was employed.
In situ XPS data were collected at a near ambient pressure XPS setup (SPECS Surface Nano Analysis, GmbH Germany) at the Charles University (Prague, Czech Republic). A monochromatic Al Kα source (hν = 1486.6 eV) was used to collect XPS spectra. The powder catalysts were pressed to pellets and fixed on the sample holder using spot welding of Ta foil stripes. The sample holder contained a K-type thermocouple welded near the sample pellet for temperature control. Heating was performed by the electron bombardment at the back of the sample stage, which was in thermal contact with the sample holder while mounted on the NAP manipulator. XPS analysis of the samples before their exposure to the reaction mixture was performed at room temperature (RT) in Ar flow (p(Ar) ∼ 1 mbar) to prevent the surface charging. The in situ experiments were performed by stepwise heating the samples in the NH3 + O2 reaction mixture (NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 ratio of 1:40 and total pressure p∑ ∼ 1.4 mbar) from 50 °C to 400 °C with simultaneous acquisition of XPS data. XPS spectra were collected after heating the samples to a certain temperature and stabilization in the reaction mixture for 20 min. XPS experiments were complemented with simultaneous analysis of the gas mixture by mass spectrometry (Prisma Pro, Pfeiffer Vacuum, Germany) to follow the change of concentrations of the reagents (NH3, O2) and products (H2O, N2O, N2, NOx) with temperature increase.
:O2 ratio of 1:40 and total pressure p∑ ∼ 1.4 mbar) from 50 °C to 400 °C with simultaneous acquisition of XPS data. XPS spectra were collected after heating the samples to a certain temperature and stabilization in the reaction mixture for 20 min. XPS experiments were complemented with simultaneous analysis of the gas mixture by mass spectrometry (Prisma Pro, Pfeiffer Vacuum, Germany) to follow the change of concentrations of the reagents (NH3, O2) and products (H2O, N2O, N2, NOx) with temperature increase.
The spectra energy calibration was done using a Ti2p3/2 line as an internal standard with a binding energy value Eb(Ti2p3/2) = 458.6 eV for TiO2. Such a calibration procedure gave Eb(C1s) = 284.7 eV for the residual amorphous carbon present on the surface. The Pt4f/Ti2p ratio was calculated based on the area of the corresponding spectral lines. The Shirley background was subtracted before processing the spectra. Ti2p, O1s, C1s, and N1s spectra were fitted with a combination of the Gaussian and Lorentzian functions. The Pt4f spectra fitting was performed using a combination of the Gaussian and Lorentzian functions for oxidized components, while Doniach–Šunjić function was used to describe the asymmetry of metallic Pt0 doublet. Also, Pt4f spectra fitting was performed with consideration of the FWHM (full width at half maximum) values of the corresponding Ti2p lines. The XPS-Calc program38,39 was used for processing the XPS data.
Ex situ and operando X-ray absorption near edge structure (XANES) spectra at Pt L3 absorption edge were recorded at the P65 beamline of the PETRA III synchrotron radiation source (DESY, Hamburg, Germany) in transmission mode. Energy of the X-rays was selected using a Si(111) double crystal monochromator and higher harmonics were rejected by a pair of Si mirrors. Beam size was selected by slits as 0.2 (vert.) × 1.0 (hor.) mm2. Catalyst samples were measured as 100–200 μm sieved powders packed in quartz capillaries (o.d. = 1.5 mm, 0.02 mm wall thickness) heated by a hot air blower. The catalyst bed length was approx. 5 mm and the X-ray beam was probing part near inlet of the catalyst bed. The gas mixture contained 880 ppm NH3, 10 vol% O2, and 10 vol% N2 in He with a flow of 70 ml min−1. Outlet gas composition was analyzed by an MKS Multigas 2030 FTIR analyzer. The spectra were corrected for the energy shift using a spectrum of Pt foil measured simultaneously and then normalized using the Athena program from the IFFEFIT software package.48 The average oxidation state of Pt was determined by linear combination analysis (LCA) of the XANES data in Athena48 with Pt foil and PtO2 reference spectra in the energy range between 11550 and 11600 eV. For the analysis of extended X-ray absorption fine structure (EXAFS), the spectra were background-subtracted, k2-weighted, multiplied by a Hanning window with sill size of 1 Å−1, and Fourier-transformed in the k-range 2.5–11.0 Å−1. The amplitude reduction factor S02 = 0.74 was obtained by fitting the Pt foil and the PtO2 (Alfa Aesar, 99.95%) reference spectra to structural models as reported in the Inorganic Crystal Structure Database (ICSD, CCs = 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 923 and 4415). The fits were performed on k1, k2, and k3-weighted data using Artemis48 by a least square method in R-space between 1.0 and 3.0 Å. Coordination numbers, interatomic distances, energy shift (δE0) and mean square deviation of interatomic distances (σ2) were refined during the fitting. The absolute misfit between theory and experiment was expressed by ρ.
923 and 4415). The fits were performed on k1, k2, and k3-weighted data using Artemis48 by a least square method in R-space between 1.0 and 3.0 Å. Coordination numbers, interatomic distances, energy shift (δE0) and mean square deviation of interatomic distances (σ2) were refined during the fitting. The absolute misfit between theory and experiment was expressed by ρ.
Catalytic experiments
The catalytic properties of the Pt/TiO2 samples were studied in the temperature-programmed reaction of ammonia oxidation (TPR-NH3 + O2). The experiments were performed in a plug flow quartz reactor. 0.145 g of the catalyst was analyzed in the catalytic experiments. Heating of the samples in the NH3 + O2 mixture was performed from room temperature to 400 °C at a rate of 10 °C min−1. The reaction mixture containing 0.1 vol% NH3, 4.0 vol% O2 (balance He) was fed at a flow of 500 ml min−1. Before TPR-NH3 + O2 experiments, Pt/TiO2-LAL and Pt/TiO2-IMP samples were heated in 20%O2/He flow at 400 °C for 2 h to remove the surface admixtures. A gas-phase FTIR spectrometer (I1801, MIDAC Corp., USA) was used to measure concentrations of NH3, N2O, NO, NO2, while O2 and N2 concentrations were analyzed using a gas chromatograph (Crystal 2000M, CHROMATEC, Russia). Conversion (in %) of ammonia was calculated as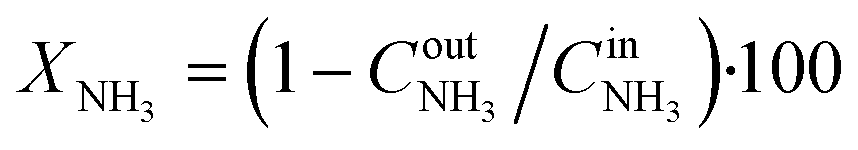 , where
, where 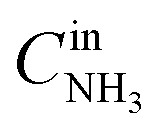 and
and 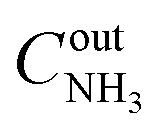 are the inlet and outlet NH3 concentrations, respectively. Selectivity (in %) to the reaction products (N2, N2O, NO, or NO2) was calculated as Si = (ni/∑ni)·100, where ni – the concentration of the corresponding product.
are the inlet and outlet NH3 concentrations, respectively. Selectivity (in %) to the reaction products (N2, N2O, NO, or NO2) was calculated as Si = (ni/∑ni)·100, where ni – the concentration of the corresponding product.
Results and discussion
Structural characterization of Pt/TiO2 samples
Fig. 1 shows the X-ray diffraction patterns for Pt/TiO2-LAL (purple line (1)) and Pt/TiO2-IMP (green line (2)) samples. For obtained XRD patterns, a full profile Rietveld refinement was performed. The XRD profile Rietveld modeling for the samples are given in ESI† file (Fig. S1). The Pt/TiO2-LAL sample contains an X-ray amorphous phase as well as the crystalline phases attributed to metallic Pt (ICDD PDF-2 # 04-0802) and anatase structure of TiO2 (ICDD PDF-2 # 21-1272). The calculated diffraction curve poorly describes the experimental pattern in the region of the anatase reflections (see Fig. S1†), which suggests that the anatase phase might be defective. The structural characteristics of the TiO2 phases (lattice parameters (a, c), the average crystallite size (D)) calculated from XRD data are given in Table 1. There is a difference between the values of the TiO2-anatase lattice parameters for the Pt/TiO2-LAL sample and the data reported for the single crystal anatase TiO2 (a = 3.784 Å, c = 9.515 Å).49 Ballirano et al.50 observed the modified lattice parameters (a = 3.784 Å, c = 9.500 Å) for the nanosized TiO2-anatase particles (D ∼ 21 nm). For the Pt/TiO2-LAL sample, the calculated average crystallite size is ∼5 nm. Thus, the change of the lattice parameters of the anatase phase might be related to the small size and defective structure of the TiO2 crystallites. Contribution from the metallic Pt in the Pt/TiO2-LAL sample was refined with the introduction of two phases corresponding to large (D ∼ 25 nm) and small particles (D ∼ 2 nm).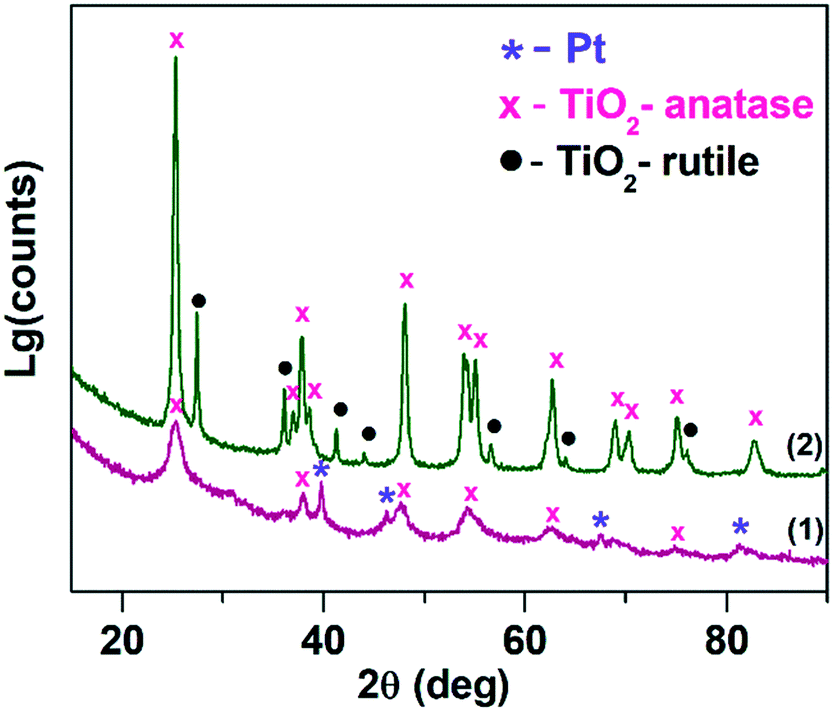 | ||
| Fig. 1 X-ray diffraction patterns for (1) Pt/TiO2-LAL, (2) Pt/TiO2-IMP. | ||
| S BET, m2 g−1 | TiO2-Anatase | TiO2-Rutile | |||||||
|---|---|---|---|---|---|---|---|---|---|
| a, Å | c, Å | D, nm | W, wt% | a, Å | c, Å | D, nm | W, wt% | ||
| Pt/TiO2-LAL | 152 | 3.804(3) | 9.47(1) | 4.7(3) | 100 | — | — | — | — |
| Pt/TiO2-IMP | 63 | 3.786(1) | 9.508(1) | 18.8(3) | 84.1(6) | 4.595(1) | 2.960(1) | 29(2) | 15.9(6) |
The X-ray diffraction pattern of the Pt/TiO2-IMP sample can be described with a mixture of anatase (ICDD PDF-2 # 21-1272) and rutile (ICDD PDF-2 # 21-1276) phases. The amount of the anatase phase is greater than the rutile phase (see Table 1). The average crystallite sizes of anatase and rutile phases are ∼19 nm and ∼30 nm, respectively. The lattice parameters of the TiO2 phases are similar to those reported for the nanosized TiO2.50 Platinum-containing phases were not detected by the XRD method allowing us to propose the presence of the highly dispersed Pt species in the Pt/TiO2-IMP sample. The smaller size of the TiO2 crystallites in the Pt/TiO2-LAL sample results in a higher value of the surface area (SBET) in comparison with the Pt/TiO2-IMP sample (see Table 1).
Fig. 2 shows TEM data for the Pt/TiO2-LAL sample. Most of the TiO2 particles are 5–10 nm in size, but larger particles from 10 to 200 nm in diameter are also observed. Both the crystalline phase of anatase (Fig. 2d) and amorphous TiOx particles (Fig. 2e) can be identified using HRTEM. Platinum nanoparticles in the Pt/TiO2-LAL sample have a spherical-like shape. Their distribution over the support is uniform. Most of the platinum particles have a size of 1–3 nm (Fig. 2b). However, there are some larger particles with a size of 10–20 nm. The results are in a good agreement with the XRD data.
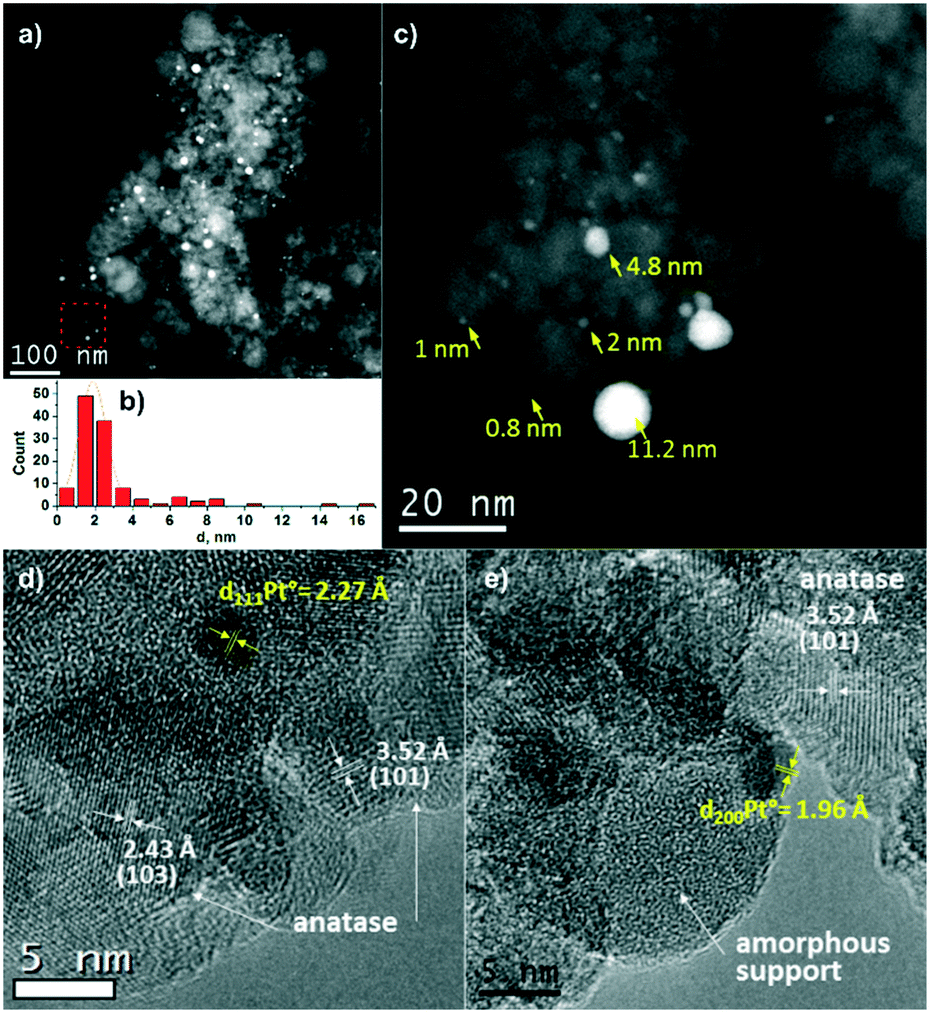 | ||
| Fig. 2 STEM and HRTEM study of the Pt/TiO2-LAL sample: (a) HAADF-STEM data, (b) Pt particle size distribution, (c) HAADF-STEM data of the area marked in (a) with red square, (d and e) HRTEM data of selected regions. | ||
TEM data for the Pt/TiO2-IMP sample are given in Fig. 3. The size of the TiO2 particles varies from 10 to 40 nm. The Pt nanoparticles have a spherical-like shape (Fig. 3a). The average size of platinum particles is 1.2 nm (Fig. 3c). Also, highly dispersed platinum particles less than 1 nm in size (marked with yellow arrows in Fig. 3b) can be well detected due to the high contrast of platinum on the TiO2 surface in the dark-field images (Z-contrast).
 | ||
| Fig. 3 TEM study of Pt/TiO2-IMP sample: (a) TEM image, (b) HAADF-STEM image (yellow arrows mark the presence of highly dispersed Pt species), (c) Pt particle size distribution. | ||
XPS analysis of the Pt/TiO2 samples in UHV
Fig. 4 shows the Pt4f X-ray photoelectron (XP) spectra of the Pt/TiO2 samples fitted to the individual components. The Ti2p and O1s spectra of the samples are given in ESI,† Fig. S2. For Pt/TiO2-LAL sample the main Pt4f doublet has a Eb(Pt4f7/2) value 70.6 eV (Fig. 4a). The Eb(Pt4f7/2) value typical for bulk metal Pt0 surfaces (foils, single-crystals etc.) is usually considered to be 71.0–71.2 eV.51–53 It cannot be excluded that the interaction of the Pt0 nanoparticles with the TiO2 support results in a slight decrease of the observed Eb(Pt4f7/2) value for the Pt/TiO2-LAL sample. Metal–support interaction is known for the supported catalytic systems.42,44,54–57 It can cause a change in the electronic state of the active component and the support, structure, and morphology of the catalyst. For the Pt–TiO2 systems, metal–support interaction is most frequently discussed in terms of the charge transfer from TiO2 to the small Pt particles32,55,58 with accumulation of the charge at the Pt–TiO2 interface55 or encapsulation of the platinum particles by the support.32,34,54,59 Metal–support interaction is usually observed when samples are heated in a reductive atmosphere. Under these conditions, the reduced, defective TiO2−δ structures might be formed facilitating the interaction with Pt particles.42–44 In our case, the LAL method leads to the formation of defective, amorphous structures of titanium oxide, hence, a metal–support interaction might occur. The accumulation of the charge at the Pt0–TiO2 interface can cause the inhomogeneous charging of the sample surface, thus resulting in the observed negative shift of Pt0 peaks.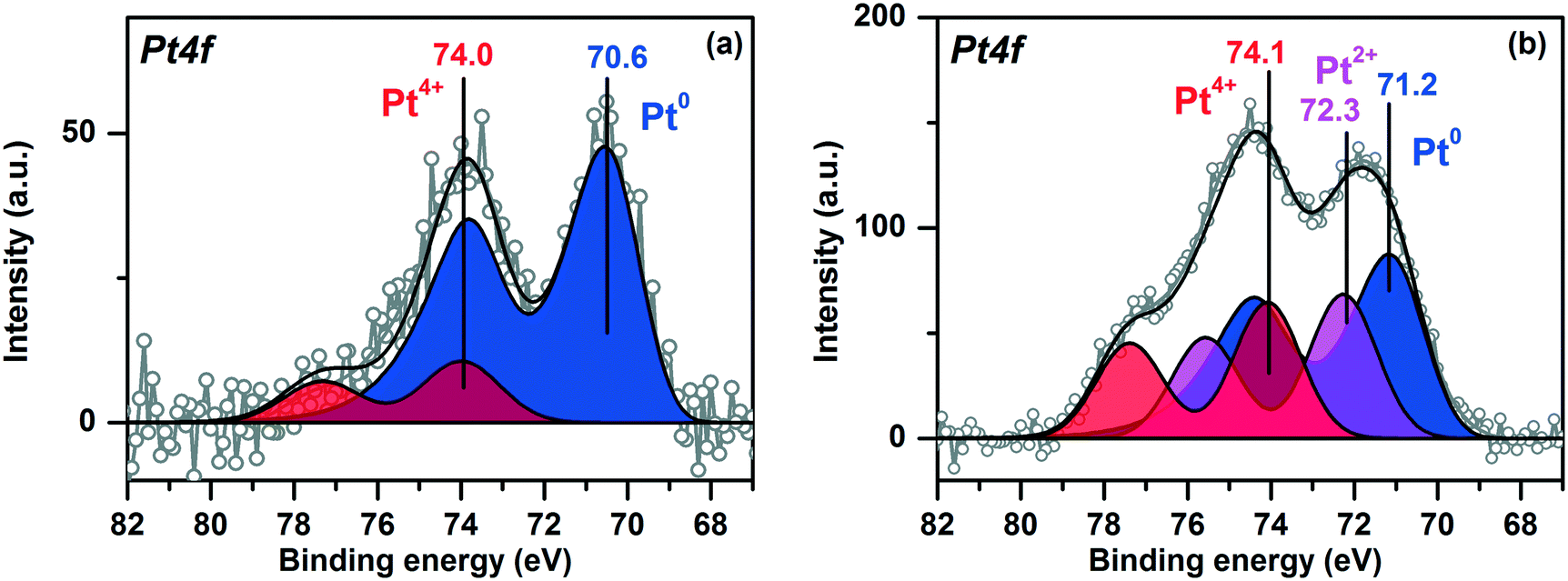 | ||
| Fig. 4 Pt4f XP spectra for (a) Pt/TiO2-LAL, (b) Pt/TiO2-IMP samples. | ||
A Pt4f7/2 peak with a lower intensity at ∼74.0 eV (Fig. 4a) can be related to oxidized Pt4+ species.51,60 It is known that small platinum particles can be easily oxidized with the formation of PtOx structures.61–65 TEM data for the Pt/TiO2-LAL sample show the presence of a certain amount of the highly dispersed platinum species. We can propose that the highly dispersed Pt species are in Pt4+ oxidation state.
The Pt4f region for the Pt/TiO2-IMP catalyst reveals three platinum states with the Eb(Pt4f7/2) values of 71.2, 72.3 and 74.1 eV (Fig. 4b). The first state might be attributed to the metallic platinum species.51,53,60 The second and third states of platinum can be related to Pt2+ and Pt4+ species, respectively.51,53,60 The stoichiometry of the PtOx species formed by interaction of small platinum particles with oxygen depends on the particle size. According to Wang et al.,66 the x value changes from 1 to 2 when particle size decreases below 2 nm. Since we have a distribution of the size of the Pt particles, it can be proposed that the smallest platinum particles oxidize to PtO2, while the larger particles give PtO species. The formation of a PtO oxide layer covering the surface of the metal platinum particles is also possible. Thus, the observed Pt2+ oxidized species can be present as a PtO layer on the surface of Pt0 particles and/or as oxidized PtO particles. Zhang et al. also observed peaks at 71.2 eV and 72.3 eV for a Pt–TiO2 film calcined at 400 °C.67 The authors proposed oxidation of Pt0 sites to Pt2+ species which substitute Ti4+ ions in TiO2 lattice. Hence, the stabilization of the Pt2+ species due to the interaction with the TiO2 lattice cannot be ruled out.
The contribution of the oxidized platinum species to the Pt4f spectrum for Pt/TiO2-IMP sample is higher than in the case of the Pt/TiO2-LAL sample. Such a result is in a good agreement with the TEM data which show higher amount of the highly dispersed platinum species in the Pt/TiO2-IMP sample.
The ratio of the Pt4f and Ti2p line intensities (Pt4f/Ti2p) in the Pt/TiO2-LAL is significantly lower (about 3.5 times) than for the Pt/TiO2-IMP. This can be explained by the presence in the Pt/TiO2-LAL sample a certain amount of the large platinum particles which results in low intensity of the platinum signal. A partial encapsulation of the platinum particles by a TiOx layer is also possible. Furthermore, the SBET of the Pt/TiO2-LAL sample is almost 2.5 times higher than the one of the Pt/TiO2-IMP sample (see Table 1). This can lead to a higher intensity of the titanium signal due to larger surface.
To sum up, the ex situ characterization shows that the sample prepared using the LAL technique comprises mostly metallic platinum nanoparticles in contact with TiO2. The Pt/TiO2-IMP sample shows more dispersed and oxidized platinum particles.
Catalytic activity of the Pt/TiO2 samples in NH3 + O2 reaction
The activity of the samples in ammonia oxidation was studied using the temperature-programmed reaction NH3 + O2 (TPR-NH3 + O2). Data on the ammonia conversion and the selectivity to the reaction products as function of the temperature are given in Fig. 5.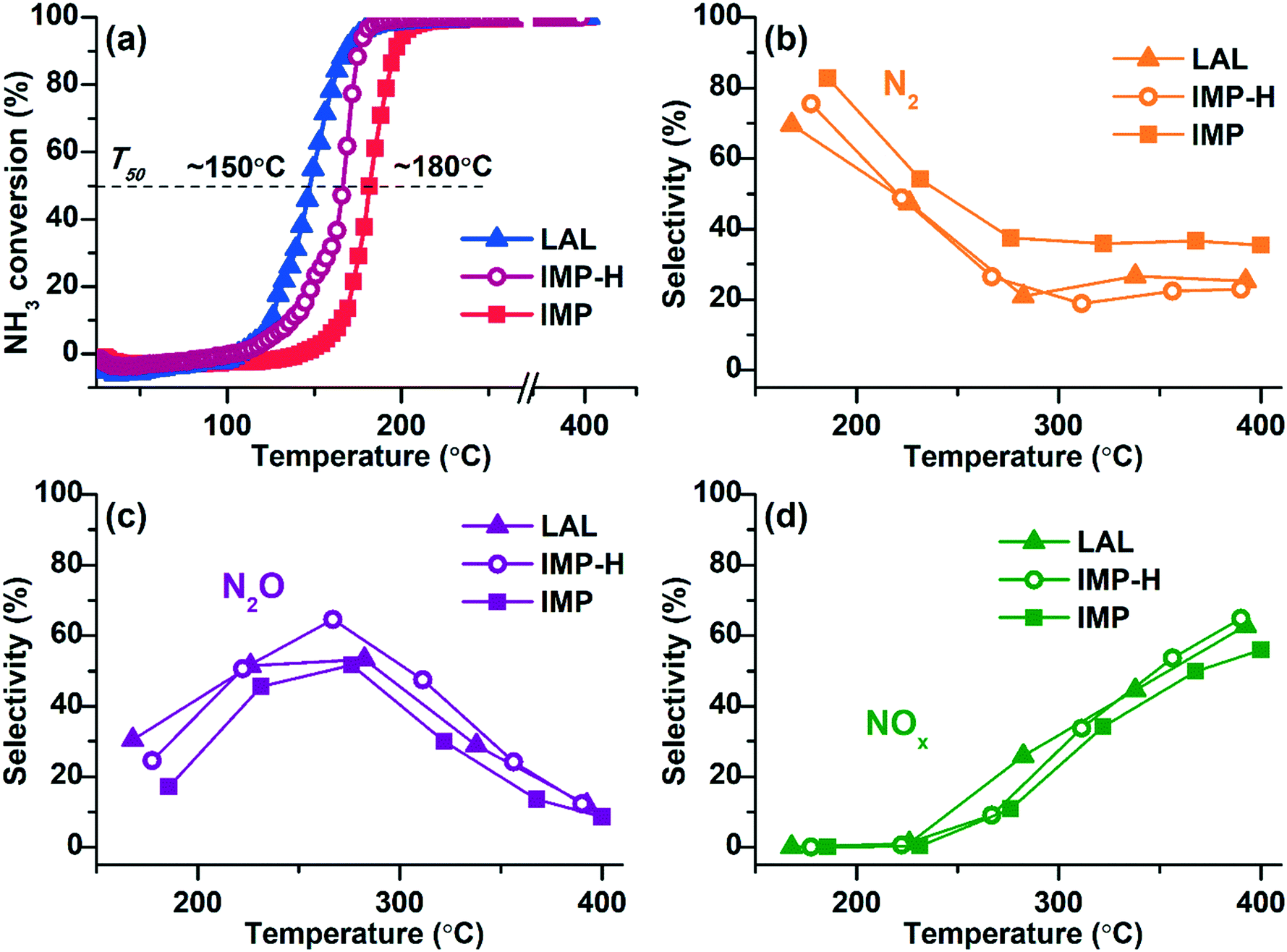 | ||
| Fig. 5 TPR-NH3 + O2 data for Pt/TiO2 catalysts: (a) NH3 conversion; products selectivity: (b) N2, (c) N2O, (d) NOx. Sample Pt/TiO2-LAL marked as triangles, Pt/TiO2-IMP as squares, and Pt/TiO2-IMP-H as circles. The reaction mixture: 0.1 vol% NH3 and 4.0 vol% O2 (balance He). | ||
All samples show low-temperature activity reaching 100% conversion of ammonia at T < 200 °C. Comparison of the NH3 conversion curves shows that the Pt/TiO2-LAL sample is more active at low temperatures. The temperature of the 50% ammonia conversion (T50) is ∼150 °C, while for the Pt/TiO2-IMP sample the T50 value is ∼180 °C. Metallic platinum is proposed to be more active in the oxidation of ammonia in O2 excess.68,69 Therefore, higher low-temperature activity observed for the Pt/TiO2-LAL sample can be attributed to the presence of higher amount of the metallic platinum particles.
To confirm this assumption, the Pt/TiO2-IMP sample was reduced in 20%H2/He flow at 250 °C for 2 h in a catalytic reactor directly before the catalytic experiments (the reduced sample is denoted by Pt/TiO2-IMP-H). This procedure should lead to an increase of the amount of the reduced platinum forms, and, accordingly, contribute to the improvement of the low-temperature activity. Indeed, one can see a decrease of T50 value (Fig. 5a, the IMP-H curve) for the Pt/TiO2-IMP-H sample in comparison with the initial Pt/TiO2-IMP catalyst.
The selectivity to the reaction products is similar for all samples. In the temperature range up to 250 °C, the main products are molecular nitrogen and N2O. The maximum selectivity to N2 is observed in the region of NH3 conversion below 100% (T < 200 °C). At higher temperature the contribution of nitrogen oxides becomes more significant: N2O at lower temperatures, and NO and NO2 oxides at temperatures above 320–350 °C. Such temperature-dependent product distribution is typical for bulk Pt structures8,10 as well as platinum particles stabilized on oxide supports.15,69,70
Thus, although all samples show activity in the NH3 + O2 reaction at T < 200 °C, NH3 conversion for the Pt/TiO2-LAL and Pt/TiO2-IMP-H samples is observed at lower temperatures. To follow the evolution of the oxidation/charge state of Pt during the catalytic reaction the samples were analyzed by NAP-XPS.
NAP-XPS study of the Pt/TiO2 samples
Fig. 6 presents the Pt4f spectra fitted into the individual components for the Pt/TiO2-IMP and the Pt/TiO2-IMP-H samples collected at a NAP-XPS setup before exposure to the reaction mixture. The Pt/TiO2-IMP-H sample was in this case prepared directly in the chamber of the NAP-XPS setup by reduction of the Pt/TiO2-IMP in H2 flow at 350 °C for 2 h (p(H2)∼1 mbar). Ti2p and O1s spectra for Pt/TiO2-IMP and Pt/TiO2-IMP-H samples are given in ESI,† Fig. S3.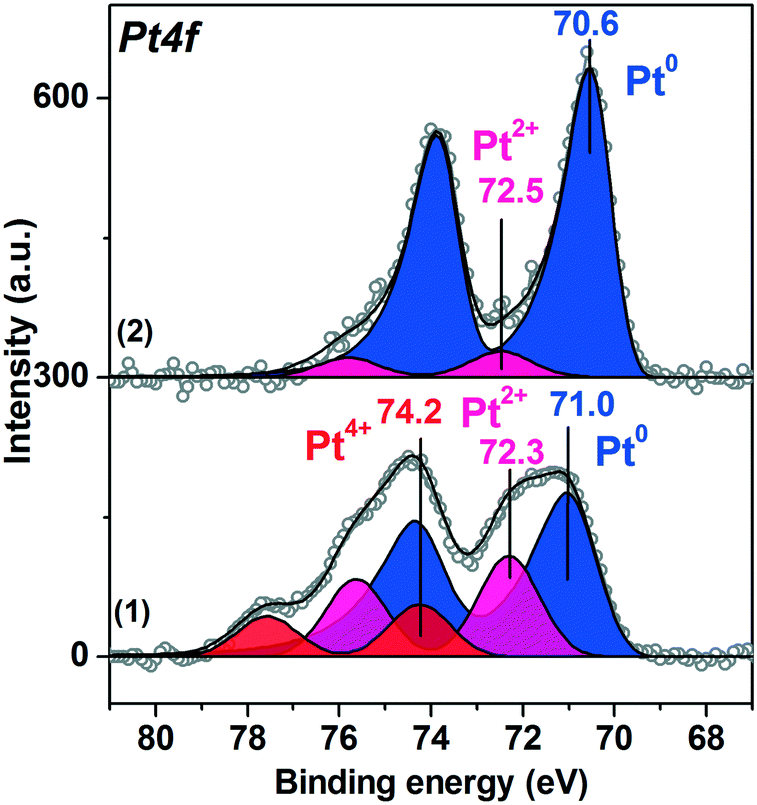 | ||
| Fig. 6 Curve-fitted in situ Pt4f XP spectra of (1) Pt/TiO2-IMP and (2) Pt/TiO2-IMP-H samples acquired in Ar (p(Ar)∼1 mbar, RT). | ||
Similar to the ex situ XPS data, the in situ Pt4f spectrum of the Pt/TiO2-IMP sample shows three doublets with maxima of the Pt4f7/2 peaks at binding energies (Eb(Pt4f7/2)) = 71.0 eV, 72.3 eV, and 74.2 eV. These doublets can be related to the Pt0, Pt2+ and Pt4+ platinum species, respectively.51,53,60
Treatment of the Pt/TiO2-IMP sample in H2 flow results in reduction of Pt2+ and Pt4+ species. The Pt4f spectrum of the Pt/TiO2-IMP-H sample (Fig. 6 curve (2)) shows the main peak of the reduced Pt species at 70.6 eV. Heating of Pt/TiO2 in a reductive atmosphere is known to promote the metal–support interaction.42–44 We do not observe a noticeable change in the Pt4f/Ti2p intensity ratio upon sample heating in H2, which suggests that the encapsulation of the platinum particles by the titania does not take place. Nevertheless, the charge transfer between the support and the platinum particles might take place influencing the Eb(Pt4f7/2) value. It can be assumed that the reductive treatment results in the formation of the defects on the surface of titania, which promotes the Pt–TiO2 interaction. Unfortunately, due to the morphology of the supported powder catalysts and the rather low concentration of the active component, we could not detect any noticeable changes in the Ti2p spectra which might give information on the possible appearance of Ti3+ species due to metal–support interaction (see ESI,† Fig. S3).
The state of platinum in the Pt/TiO2-IMP-H sample is similar to the Pt/TiO2-LAL catalyst (see Fig. 4a). Pt4f spectra for both catalysts show the main Pt state with the Eb(Pt4f7/2) ∼ 70.6 eV, indicating a similar nature of active sites in these catalysts, namely: Pt0 species in contact with TiO2 surface. This is also confirmed by similar catalytic properties of these samples.
The Pt/TiO2-IMP and Pt/TiO2-IMP-H samples were heated in the NH3 + O2 mixture with simultaneous collection of the NAP-XPS data. The change of the concentrations of the reagents and products with the increase of the reaction temperature was monitored with a mass-spectrometer. MS-data are given in ESI† (Fig. S4). Both samples under given reaction conditions showed similar MS results. Conversion of NH3 was observed at T > 175 °C. At temperature below 225 °C, the main products were N2 and N2O, while at T ∼ 225 °C partial pressure of NO increased and became predominant at 400 °C. These results are in a good agreement with data of the catalytic experiments performed in a plug flow reactor (see Fig. 5).
Fig. 7 shows the evolution of the Pt4f XP spectra collected in situ during heating the Pt/TiO2-IMP (Fig. 7a) and Pt/TiO2-IMP-H (Fig. 7b) samples in the NH3 + O2 mixture. Treatment of the Pt/TiO2-IMP sample in the reaction mixture results in a slight increase of the Eb(Pt4f7/2) value of the peak related to Pt0 species. The conductivity of the sample surface might slightly change during the reaction due to the removal or accumulation of O-containing surface groups. Thus, the shift of the Pt0 peaks can be caused by the variation of the charge over the sample surface.
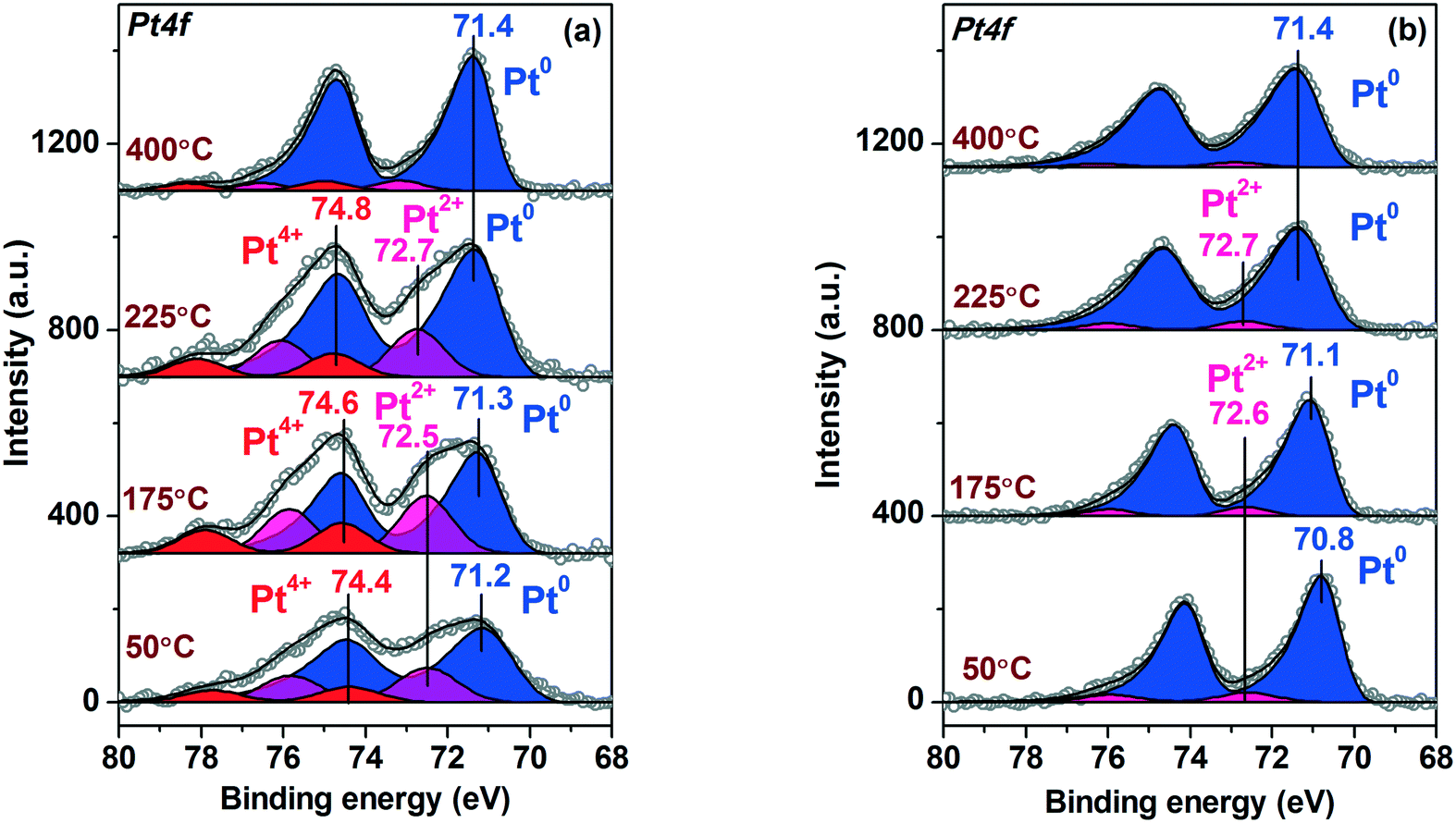 | ||
| Fig. 7 Curve-fitted in situ Pt4f XP spectra of (a) Pt/TiO2-IMP and (b) Pt/TiO2-IMP-H samples acquired during heating of the samples in the NH3 + O2 mixture (NH3:O2 ratio of 1:40, total pressure p∑ ∼ 1.4 mbar). | ||
Also, interaction of the platinum particles with oxygen can be the reason for the slight increase of the Eb(Pt4f7/2) value. Different oxygen species can be formed on the Pt surface: adsorbed oxygen,8,10,66,71 subsurface oxygen72 or platinum oxide-like structures.73–76 The conditions under which various surface oxide structures occur were studied in detail for Pt single-crystals,73–75,77–79 foils, and nanoparticles.60,72,80–83
For surface and bulk platinum oxides, the binding energy of the Pt4f7/2 peak is usually considered to be more than 72.0 eV.73–76 Since we do not observe a strong shift of the Pt4f line (the binding energy does not exceed 71.4 eV), the formation of fully oxidized PtOx layers or bulk oxides seems unlikely. Smirnov et al.72,80 showed that dissolution of oxygen in platinum leads to an increase in the Eb value of the platinum line, while its metal nature is preserved. However, they also observed quite significant shifts of the Pt4f line (∼1 eV) for small Pt particles (d ∼ 1–2.5 nm).80
Gland et al.7 observed adsorption of oxygen atoms on edge sites of the stepped Pt single-crystal surface under ammonia oxidation reaction. Günther et al.10 detected in their study by in situ XPS of ammonia oxidation over a Pt(533) surface the formation of a 1D platinum oxide phase. Imbihl et al.11 also showed that OHad as well as Oad species should be considered during ammonia oxidation. Based on this, we can propose that the Eb(Pt4f7/2) shift observed in our work might be caused by the formation of adsorbed oxygen species (Oad and/or OHad) on the Pt0 surface or at the Pt0–TiO2 boundary. Unfortunately, analysis of the O1s spectra of the samples (see ESI,† Fig. S5) does not allow to reliably detect signals related to the oxygen species adsorbed on the platinum surface due to low concentration of Pt in the samples, high intensity of the titania oxygen, and variety of the oxygen-containing species adsorbed on the sample surface.
The increase of the Eb(Pt4f7/2) value of the peaks related to the oxidized Pt2+ and Pt4+ species can be explained by the formation of the Pt(OH)x structures51,84 due to the interaction with the reaction products and intermediates (H2O, OHads). Also, a slight variation of the Pt4f line width during sample treatment in NH3 + O2 mixture can be observed. The broadening of the Pt4f spectra was caused by the non-uniform charging of the particles surface during the in situ NAP-XPS experiments. See ESI† for the details.
In case of the Pt/TiO2-IMP-H sample the shift of the Pt4f7/2 peak corresponding to the reduced Pt species is more noticeable. Some authors have shown that the encapsulation of the platinum particles by a support with the formation of TiOx suboxide layer on the surface of the platinum particles can lead to a positive shift of Eb(Pt4f7/2).85,86 The encapsulation of the platinum particles by TiO2−δ when heated in oxygen containing atmosphere is usually observed at rather high temperatures (T > 400 °C).67,81 In our work lower temperatures were used, and the Pt4f/Ti2p intensity ratio changed insignificantly during heating of the sample in the reaction medium. Based on this, we believe that the encapsulation of the platinum particles by TiOx suboxide should be excluded. Moreover, encapsulated Pt0 particles are usually considered catalytically inactive,44 while we did not observe decrease of the NH3 conversion during heating. Similar to the Pt/TiO2-IMP sample, we can assume that the shift of the Pt4f line can be either associated with the slight change of the surface conductivity during the reaction or with the formation of adsorbed oxygen species (Oad and/or OHad) on the Pt0 surface or within the Pt0–TiO2 boundary.
Thus, the NAP-XPS data do not show the formation of strongly oxidized platinum species (PtOx layers or PtO/PtO2 oxides) during NH3 oxidation. The obtained data are in a good agreement with the results of the in situ study of the interaction of the bulk platinum surface with NH3 + O2 reaction mixture,10 pointing to similarity of the active sites.
Fig. 8 presents in situ N1s spectra fitted to the individual components collected for the Pt/TiO2-IMP (Fig. 8a) and the Pt/TiO2-IMP-H (Fig. 8b) samples heated in the NH3 + O2 mixture at different temperatures. The N1s spectra of the Pt/TiO2-IMP sample treated in the NH3 + O2 mixture at 50 °C show the main peak with Eb(N1s) ∼ 400.0 eV. Such Eb value was observed after treatment of the TiO2 support in NH3 flow at 600 °C.87 The authors assign this N1s peak to Ti–O–N bond. Since we use much lower temperatures, doping of the TiO2 surface under such conditions is unlikely. NH3 species adsorbed on the surface of metallic Pt are also characterized by Eb(N1s) ∼ 400.0 eV.8,10,88 Adsorption of NH3 on the TiO2 support surface should also take place. The increase of the temperature results in the appearance of the peak at 398.6–398.9 eV corresponding to adsorbed NHx (x = 1, 2) forms.8,88 The appearance of such forms indicates dehydrogenation of the NH3 species, which is a first step of the NH3 oxidation reaction.11,89
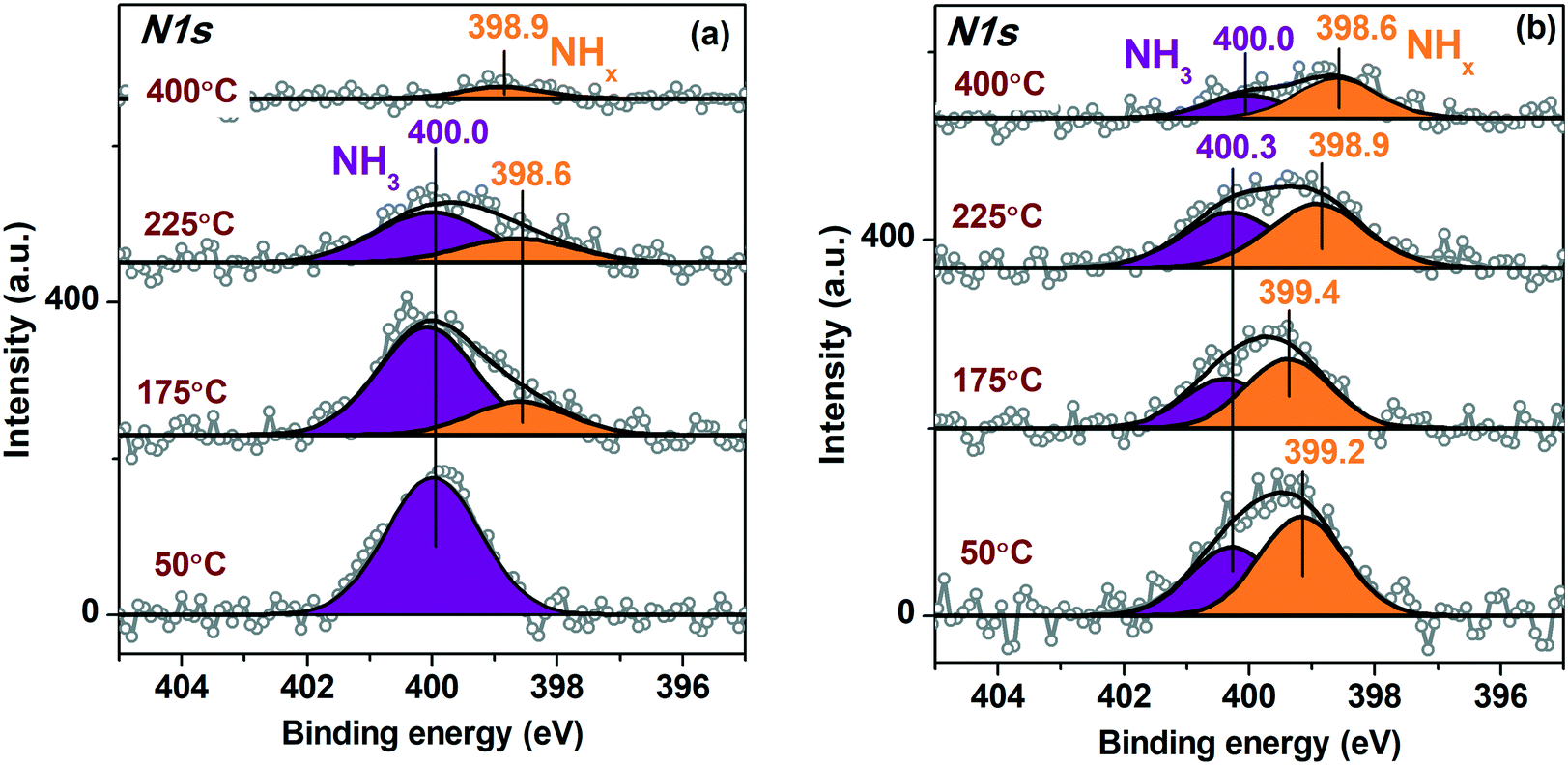 | ||
| Fig. 8 Curve-fitted in situ N1s XP spectra of (a) Pt/TiO2-IMP and (b) Pt/TiO2-IMP-H samples acquired during heating of the samples in the NH3 + O2 mixture (NH3:O2 ratio of 1:40, total pressure p∑ ∼ 1.4 mbar). | ||
It should be noted that for the sample heated in the NH3 + Ar mixture the N1s spectra also show the peak of the adsorbed NH3 species (Eb(N1s) ∼ 399.9–400.2 eV), as well as a peak with Eb(N1s) ∼ 398.2–398.4 eV, corresponding to the NHxad (x = 1, 2) species (see ESI,† Fig. S6). However, the impact of the adsorbed NHx on the overall N1s spectrum at all temperatures is low. This result is in a good agreement with the literature data. It is known that the presence of oxygen species adsorbed on the surface of Pt0 enhances dissociation of NH3.8,89
At temperatures above 225 °C the amount of the adsorbed NH3 and NHx decreases (Fig. 8). In the same temperature range, the selectivity of the reaction changes towards the formation of NOx (Fig. S4†). Desorption of NHxad provides free sites for the oxygen adsorption thus causing a shift of the selectivity towards NOx formation.8 For the reduced Pt/TiO2-IMP-H sample, the formation of the adsorbed NHx is observed already at 50 °C, and the contribution of these species to the overall N1s spectra is higher compared to the Pt/TiO2-IMP sample in the entire temperature range. This may be related to the large number of platinum metal species that provide sites for adsorption of ammonia and oxygen. The predominant presence of the dehydrogenated NHxad species rather than NH3ad on the surface was reported for bulk metal Pt treated in NH3 + O2 mixture.10,90 Note that no NOad species were detected on the samples surface in a good agreement with literature data.10 The NO desorption is considered to be much faster process than NO formation. It cannot be excluded that at the partial pressure used in NAP-XPS experiments the amount of the NOx species adsorbed on the surface is not sufficient for the detection.
Fig. 9 shows variation of the fraction of Pt0 species for Pt/TiO2-IMP and Pt/TiO2-IMP-H samples during heating in the NH3 + O2 mixture. The fraction of Pt0 species was calculated as a ratio of the area of the Pt4f peak with Eb(Pt4f7/2) = 70.8–71.4 eV to the overall area of Pt4f spectrum. For the Pt/TiO2-IMP sample, the fraction of Pt0 species decreases slightly with an increase of the reaction temperature from 50 °C to 175 °C, but increases at higher temperatures. The study of the thermal stability of the Pt/TiO2-IMP sample (see ESI,† Fig. S7) shows that PtO structures are not stable at temperature higher than 175 °C, in a good agreement with literature data.60,81,82 However, the contribution of the Pt4+ signal to the Pt4f spectrum remains practically unchanged up to 200 °C (Fig. S7†). We can propose that in O2 flow the highly dispersed Pt sites remain in the Pt4+ state. The higher stability of PtOx is known for smaller Pt particles.62 Also, heating of the Pt/TiO2-IMP sample in the reaction mixture from 50 to 225 °C results in a slight increase of the Pt4f/Ti2p intensity ratio (from 0.07 to 0.1). The variation of the fraction of Pt0 species together with the change in the Pt4f/Ti2p intensity ratio points to the transition of the oxidized PtO to the Pt0 sites at a temperature range up to 225 °C. Such transition might be accompanied by the change of the sample morphology which results in the increase of the Pt4f/Ti2p intensity ratio. Temperature increase to 400 °C leads to a significant increase of the fraction of Pt0 species as a result of the reduction of PtO and PtO2 species and increase of the number of Pt0 sites. A slight decrease of the Pt4f/Ti2p ratio at this temperature range (from 0.1 to 0.07) might indicate sintering of the platinum particles under treatment in the reaction mixture at higher temperatures. TEM data for the Pt/TiO2-IMP sample after the reaction confirms a slight increase of the average size of the platinum particles (see ESI,† Fig. S8).
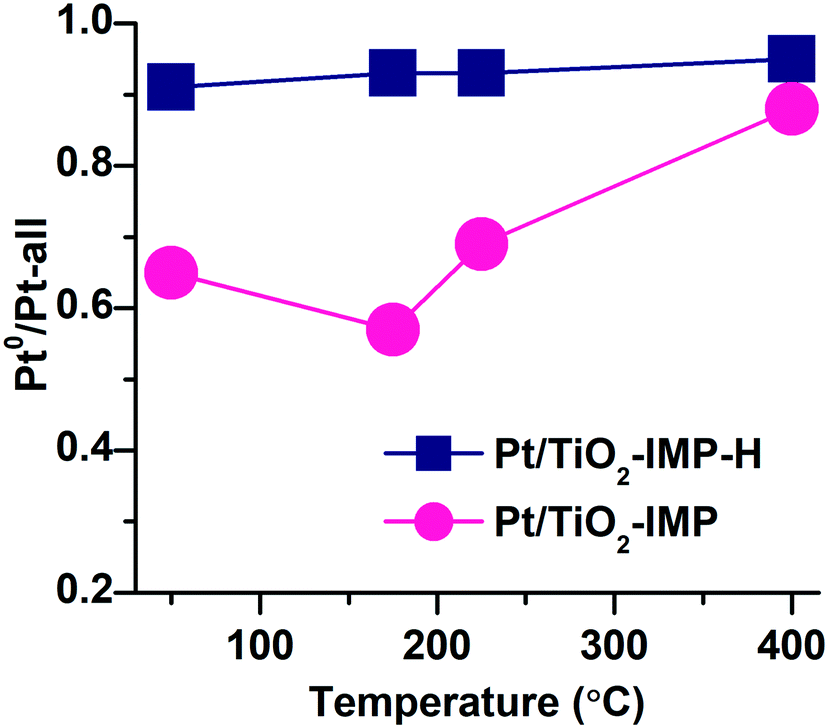 | ||
| Fig. 9 Variation of the fraction of Pt0 species (Pt4f peak with Eb(Pt4f7/2) = 70.8–71.4 eV) to the overall Pt4f signal intensity for Pt–TiO2-IMP and Pt–TiO2-IMP-H samples during NAP-XPS study in the NH3 + O2 mixture. | ||
For the reduced Pt/TiO2-IMP-H sample the fraction of Pt0 species remains practically unchanged with the temperature increase. No significant oxidation of the metal Pt0 is taking place.
Thus, the NAP-XPS data show that the states of Pt in the analyzed samples upon their treatment in the NH3 + O2 mixture at T > 225 °C are very similar and best described by Pt0 surface. One can conclude that, despite the difference in the charge states of Pt in the as-prepared samples, the “active” states of the surface observed during the treatment of the samples in the NH3 + O2 mixture are similar. The oxidized Pt species initially present in the Pt/TiO2-IMP sample decompose with the temperature increase providing the metallic platinum sites available for oxygen adsorption.
XANES/EXAFS study of the Pt/TiO2 samples
To follow the change of the platinum charge state during the reaction in the bulk, XANES and EXAFS spectroscopy were employed. The experiments were performed for the most active of the studied samples – Pt/TiO2-LAL.XANES and Fourier transformed (FT) extended X-ray absorption fine structure (EXAFS) spectra collected ex situ for the Pt/TiO2-LAL sample are given in Fig. 10. In a good agreement with the data of other methods, the sample contains mostly platinum in the metallic state. The average oxidation state determined by linear combination analysis (LCA) of the XANES data in the energy range between 11550 and 11600 eV is 0.6 ± 0.1. These results are in a good agreement with the XPS data from which the average oxidation state can be estimated as 0.8. Somewhat lower oxidation state observed by XANES may be due to unaccounted number of large Pt NPs (see Fig. 2b). FT k2-weighted EXAFS spectra (Fig. 10b) and the corresponding analysis (Table 2) reveal features very similar to metallic Pt with slightly lower intensity implying rather large (>2 nm) Pt nanoparticles.91 No Ti neighbor in the second shell was required to fit the experimental data (k2-weighted EXAFS functions and EXAFS fits for all analyzed samples are given in ESI,† Fig. S9, and S10). So, EXAFS data do not clearly show the interaction of Pt particles with TiO2 support (due to very low fraction of Pt atoms at the Pt–TiO2 interface which would be invisible in the averaged signal). However, the Pt–TiO2 interaction at the contact of the platinum particles with a TiO2 surface cannot be ruled out.
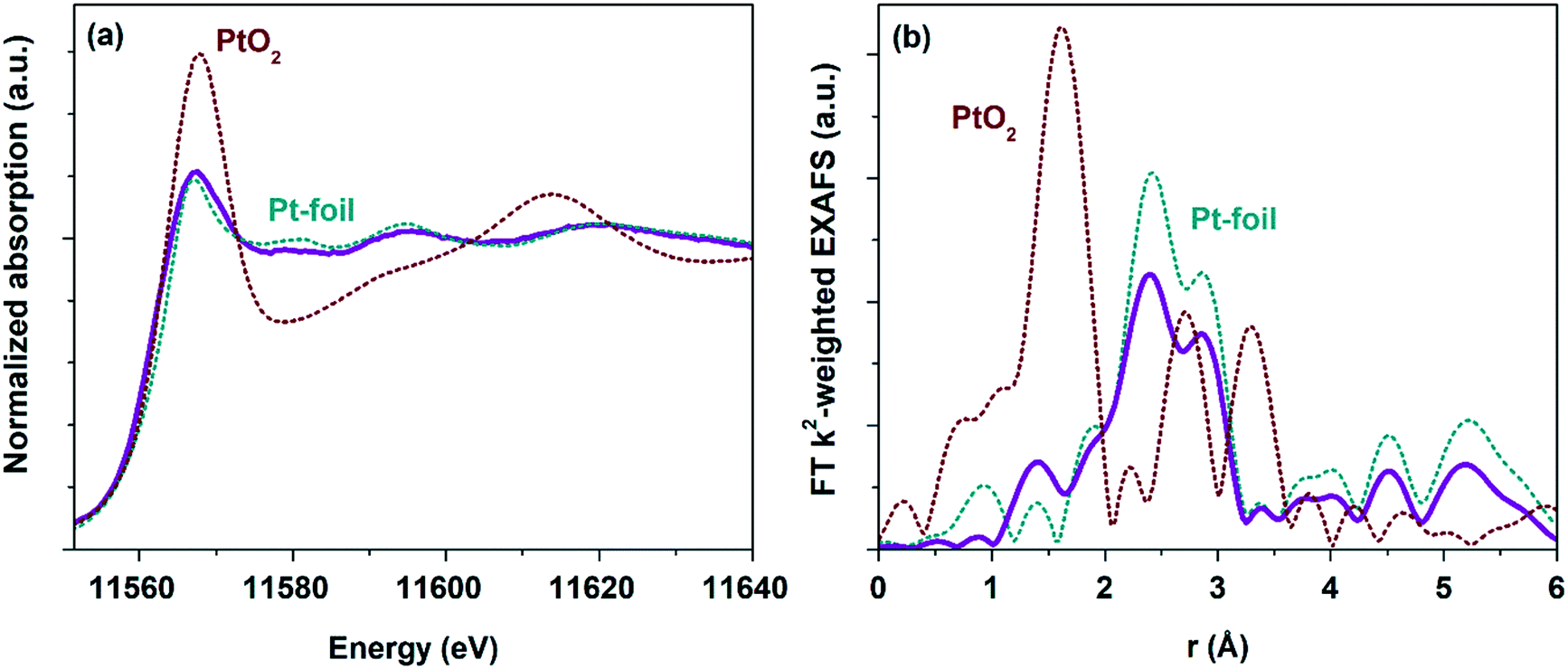 | ||
| Fig. 10 (a) Ex situ Pt L3-XANES and (b) FT k2-weighted EXAFS spectra of the Pt/TiO2-LAL sample (alongside the data for PtO2 and metallic Pt reference samples). The EXAFS is Fourier-transformed in the k-range of: 2.5–11.0 Å−1 and is not corrected for the phase shift. | ||
| Experiment | Pt–O | Pt–Pt | DW factor (10−3 Å2) | δE 0 (eV) | ρ (%) | ||
|---|---|---|---|---|---|---|---|
| R (Å) | CN | R (Å) | CN | ||||
| Ex situ | 1.96 ± 0.03 | 0.9 ± 0.2 | 2.76 ± 0.01 | 9.9 ± 1.4 | 4.7 ± 1.0 | 6.2 ± 1.2 | 0.7 |
| In situ (50 °C) | 2.03 ± 0.05 | 0.9 ± 0.5 | 2.76 ± 0.02 | 11.1 ± 3.3 | 6.2 ± 2.2 | 7.8 ± 2.2 | 1.6 |
| In situ (400 °C) | 1.98 ± 0.04 | 1.5 ± 0.5 | 2.75 ± 0.01 | 12.3 ± 3.1 | 12.1 ± 2.7 | 6.1 ± 1.7 | 2.5 |
| In situ (50 °C), after heating–cooling cycle | 2.04 ± 0.07 | 1.1 ± 0.8 | 2.76 ± 0.02 | 10.2 ± 4.6 | 5.5 ± 3.2 | 8.5 ± 3.7 | 2.9 |
Fig. 11a presents a trend of the average oxidation state of platinum in the Pt/TiO2-LAL sample during the heating in the NH3 + O2 mixture (derived from LCA of operando XANES (see Fig. S11†)), as well as the change of the NH3 conversion and the yield of reaction products. According to the operando XANES analysis, the oxidation state of platinum does not change significantly during the catalytic reaction.
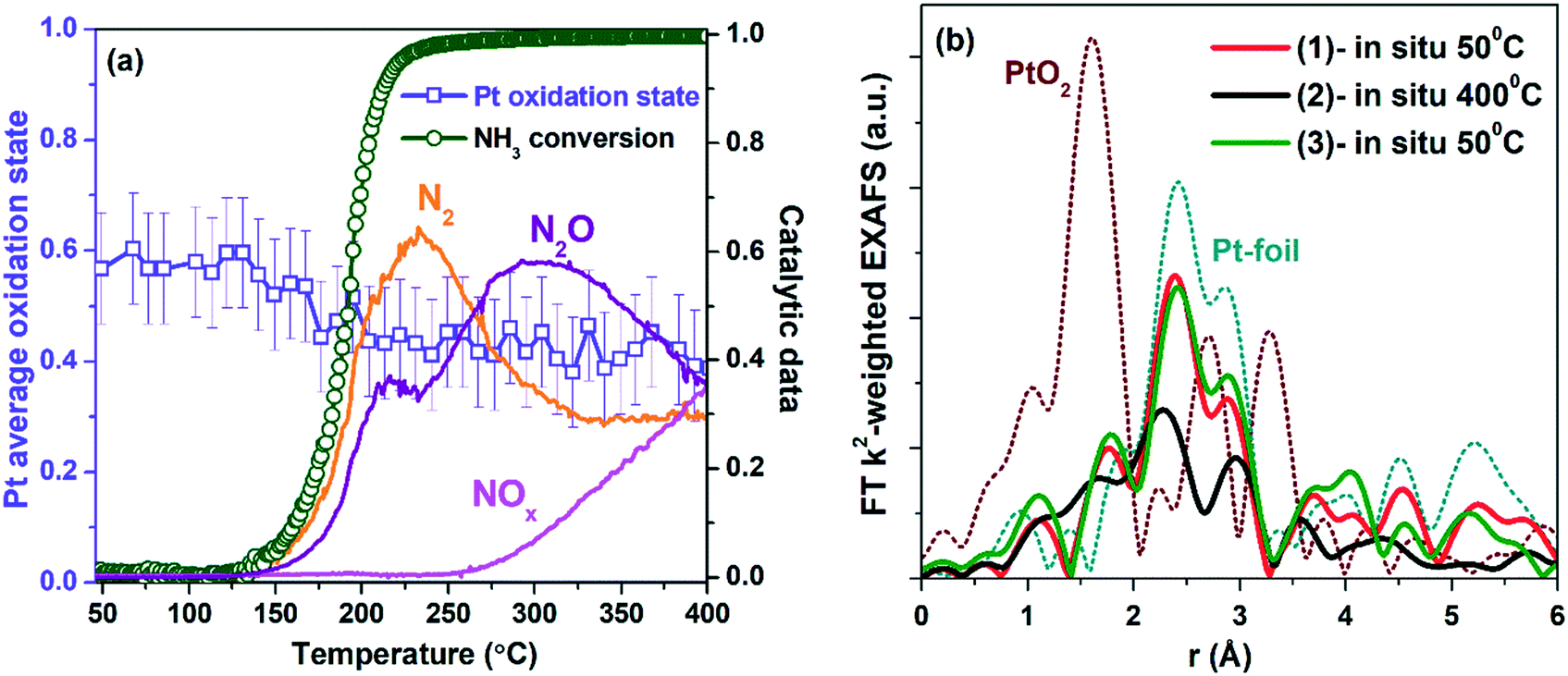 | ||
| Fig. 11 (a) NH3 conversion, N2, N2O, and NOx yields, and Pt average oxidation state profiles obtained from operando XANES experiments during heating of Pt/TiO2-LAL in the NH3 + O2 mixture (the reaction mixture: 880 ppm NH3, 10 vol% O2, and 10 vol% N2 in He). (b) FT k2-weighted EXAFS spectra of the Pt/TiO2-LAL sample in NH3 oxidation feed at (1) 50 °C and (2) 400 °C, and (3) after cooling down to 50 °C. The EXAFS is Fourier-transformed in the k-range of: 2.5–11.0 Å−1 and is not corrected for the phase shift. | ||
Fig. 11b shows Fourier transformed extended X-ray absorption fine structure (EXAFS) spectra for the Pt/TiO2-LAL sample at temperature plateaus during NH3 oxidation (during heating – at 50 °C and 400 °C, after cooling down in reaction mixture to 50 °C). Platinum nanoparticles are rather stable during the experiment. The observed decrease in the backscattering intensity in the spectrum measured at 400 °C can be attributed only to the increased disorder due to thermal motion (Debye–Waller factor). Qualitatively, one may notice an increased backscattering on the second Pt shell (3.5–4.5 Å) which may indicate sintering of Pt nanoparticles due to thermal cycling. Quantification of this effect by EXAFS is not possible due to limited data quality resulting in no statistically significant changes.
Hence, in situ/operando methods applied in our study allowed us to follow the evolution of Pt oxidation states under catalytic conditions. The use of NAP-XPS technique revealed the preservation of metallic Pt species on the surface of TiO2 under NH3 + O2 conditions even at high reaction temperatures (Fig. 7). To bridge the pressure gap between NAP-XPS experiments and realistic catalytic conditions operando XANES/EXAFS method was applied. Similar to the results of surface sensitive NAP-XPS, analysis of the average oxidation state of Pt within the bulk of the sample by operando XANES/EXAFS does not reveal significant oxidation of the metallic platinum species during the reaction. However, this does not exclude the presence of adsorbed oxygen species on the platinum surface.
Previously, we performed operando XANES study of the Pt/Al2O3 samples in the NH3 + O2 reaction.69 In contrast to the Pt/TiO2 system, it was shown that during the reaction at least 40% of the platinum surface remains oxidized in the form of a PtOx layer regardless of the charge state of platinum in the initial Pt/Al2O3 catalysts.69 We can propose that metal–support interaction in Pt/TiO2 samples that is practically absent in case of Al2O3 support might influence the charge state of platinum and its reducibility.
Comparison of the results collected for Pt/TiO2 samples prepared by different preparation techniques allowed us to conclude that for the stabilization of the catalytically active metallic Pt species through the metal–support interaction several approaches can be applied: a) the use of defective TiO2 as a support that can be provided by application of the pulsed laser ablation in liquids; or b) the reductive pretreatment of highly dispersed Pt nanoparticles prepared by impregnation. In situ/operando methods revealed that, in contrast to Pt/Al2O3 samples,69 thus stabilized metallic Pt remains preserved during the reaction. The obtained results show that Pt/TiO2 has a high potential for the catalytic application in ASC systems as a catalyst for efficient ammonia oxidation at low temperatures.
Conclusions
The Pt/TiO2 samples with different sizes of Pt particles and oxidation state of platinum were prepared by LAL and impregnation methods and tested as catalysts for the ammonia oxidation reaction. The catalysts containing metallic platinum particles showed activity at a lower temperature (T50 ∼ 150 °C) than the catalysts initially containing oxidized platinum species (T50 ∼ 180 °C). A combination of NAP-XPS and operando XAS methods was applied to follow the changes on the surface of the samples and within their bulk during the NH3 + O2 reaction. NAP-XPS experiments allowed to differentiate the oxidation states of Pt on the surfaces and follow their evolution under the reaction conditions, as well as detect the surface intermediates. The results showed that decomposition of the PtOx species with the formation of the metallic sites is required to initiate the NH3 + O2 reaction. Only metallic Pt species were observed during the reaction without the formation of a stable PtOx surface oxide layer. Based on the analysis of N1s spectra the NH3ad and NHxad species were detected on the samples surface during the NH3 + O2 exposure. Operando XAS experiments allowed to analyze the sample under more realistic conditions at a higher partial pressure of the reagents. They unraveled that, similar to NAP-XPS results, the average oxidation state of Pt did not change significantly during the reaction with the metallic platinum being the main Pt state. Combination of operando XAS and NAP-XPS methods allowed to address the “pressure” gap problem. Similarity of the NAP-XPS and operando XAS results confirms the applicability of the NAP-XPS method for the analysis of the processes taking place on the samples surface during the catalytic reaction.The NAP-XPS experiments were performed for the first time for the supported Pt catalysts in NH3 + O2 reaction. The obtained results demonstrate a good correlation to the data obtained earlier during in situ XPS studies of ammonia oxidation over the model Pt systems (single crystals and foils). Thus, we address the “material” gap problem known for the model catalytic studies and confirm the relevance of the results collected on the model catalytic systems for the analysis of the powder catalysts.
A comparison with the results obtained previously for the Pt/Al2O3 catalysts suggests that the higher activity of the Pt/TiO2 systems at lower temperatures might be associated with the metal–support interaction providing the stabilization of Pt in a metallic state. Thus, Pt/TiO2 system should be further studied in details as a potential catalyst for low-temperature NH3 oxidation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by Helmholtz – Russian Science Foundation Joint Research Groups grant #18-43-06201 from 03.09.2018 (RSF)/HRSF-0046 from 01.09.2018 (HGF). We acknowledge the CERIC-ERIC Consortium for the access to near ambient pressure XPS facilities at Charles University; DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provided beamtime at PETRA III for operando XANES/EXAFS experiments; the resource center “VTAN” (Novosibirsk State University, Russia) for the access to the TEM equipment. We would like to thank Dr. Edmund Welter for assistance in using beamline P65 at DESY. We further thank Dr. Paolo Dolcet (KIT) for his support during the XAS measurements.References
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS
.
-
P. L. Silveston and W. S. Epling, in Periodic Operation of Chemical Reactors, Elsevier Inc., 2013, pp. 141–170 Search PubMed
- T. Johnson, SAE Int. J. Engines, 2013, 6, 699–715 CrossRef
- O. Deutschmann and J.-D. Grunwaldt, Chem. Ing. Tech., 2013, 85, 595–617 CrossRef CAS
- M. Colombo, I. Nova, E. Tronconi, V. Schmeißer, B. Bandl-Konrad and L. Zimmermann, Appl. Catal., B, 2013, 142–143, 861–876 CrossRef CAS
- S. Shrestha, M. P. Harold, K. Kamasamudram and A. Yezerets, Catal. Today, 2014, 231, 105–115 CrossRef CAS
- J. L. Gland and V. N. Korchak, J. Catal., 1978, 53, 9–23 CrossRef CAS
- C. J. Weststrate, J. W. Bakker, E. D. L. Rienks, C. P. Vinod, A. V. Matveev, V. V. Gorodetskii and B. E. Nieuwenhuys, J. Catal., 2006, 242, 184–194 CrossRef CAS
- J. Pérez-Ramírez, E. V. Kondratenko, V. A. Kondratenko and M. Baerns, J. Catal., 2004, 227, 90–100 CrossRef
- S. Günther, A. Scheibe, H. Bluhm, M. Haevecker, E. Kleimenov, A. Knop-Gericke, R. Schlögl and R. Imbihl, J. Phys. Chem. C, 2008, 112, 15382–15393 CrossRef
- R. Imbihl, A. Scheibe, Y. F. Zeng, S. Günther, R. Kraehnert, V. A. Kondratenko, M. Baerns, W. K. Offermans, A. P. J. Jansen and R. A. Van Santen, Phys. Chem. Chem. Phys., 2007, 9, 3522–3540 RSC
- Y. Yao and K. P. Giapis, Phys. Chem. Chem. Phys., 2016, 18, 29858–29863 RSC
- J. J. Ostermaier, J. R. Katzer and W. H. Manogue, J. Catal., 1974, 33, 457–473 CrossRef CAS
- M. Sun, S. Wang, Y. Li, H. Xu and Y. Chen, Appl. Surf. Sci., 2017, 402, 323–329 CrossRef CAS
- M. Sun, J. Liu, C. Song, Y. Ogata, H. Rao, X. Zhao, H. Xu and Y. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 23102–23111 CrossRef CAS PubMed
- J. Schäffer, V. A. Kondratenko, N. Steinfeldt, M. Sebek and E. V. Kondratenko, J. Catal., 2013, 301, 210–216 CrossRef
- G. Olofsson, L. R. Wallenberg and A. Andersson, J. Catal., 2005, 230, 1–13 CrossRef CAS
- Y. Li and J. N. Armor, Appl. Catal., B, 1997, 13, 131–139 CrossRef CAS
- J. J. Ostermaier, J. R. Katzer and W. H. Manogue, J. Catal., 1976, 41, 277–292 CrossRef CAS
- M. Jabłońska, Catal. Commun., 2015, 70, 66–71 CrossRef
- D. A. Daramola and G. G. Botte, Comput. Theor. Chem., 2012, 989, 7–17 CrossRef CAS
- M. Rafti, J. Luis, A. Albesa, A. Scheibe and R. Imbihl, Surf. Sci., 2012, 606, 12–20 CrossRef CAS
- G. Novell-Leruth, A. Valcárcel, A. Clotet, J. M. Ricart and J. Pérez-Ramírez, J. Phys. Chem. B, 2005, 109, 18061–18069 CrossRef CAS PubMed
- H. Ma and W. F. Schneider, J. Catal., 2020, 383, 322–330 CrossRef CAS
- J. D. Gonzalez, K. Shojaee, B. S. Haynes and A. Montoya, Phys. Chem. Chem. Phys., 2018, 20, 25314–25323 RSC
- E. V. Rebrov, M. H. J. M. De Croon and J. C. Schouten, Chem. Eng. J., 2002, 90, 61–76 CrossRef CAS
- R. Kraehnert and M. Baerns, Chem. Eng. J., 2008, 137, 361–375 CrossRef CAS
- A. Scheuer, M. Votsmeier, A. Schuler, J. Gieshoff, A. Drochner and H. Vogel, Top. Catal., 2009, 52, 1847–1851 CrossRef CAS
-
BASF Corporation, US Pat., US8524185B2, Florham Park, NJ (US), 2013 Search PubMed
- T. Lan, Y. Zhao, J. Deng, J. Zhang, L. Shi and D. Zhang, Catal. Sci. Technol., 2020, 5792–5810 RSC
- H. Huang, D. Y. C. Leung and D. Ye, J. Mater. Chem., 2011, 21, 9647–9652 RSC
- A. Lewera, L. Timperman, A. Roguska and N. Alonso-Vante, J. Phys. Chem. C, 2011, 115, 20153–20159 CrossRef CAS
- L. Y. Lin, S. Kavadiya, X. He, W. N. Wang, B. B. Karakocak, Y. C. Lin, M. Y. Berezin and P. Biswas, Chem. Eng. J., 2020, 389, 123450 CrossRef CAS
- Z. Rui, L. Chen, H. Chen and H. Ji, Ind. Eng. Chem. Res., 2014, 53, 15879–15888 CrossRef CAS
- Y. Zhou, D. E. Doronkin, M. Chen, S. Wei and J.-D. Grunwaldt, ACS Catal., 2016, 6, 7799–7809 CrossRef CAS
- In Studies in Surface Science and Catalysis, ed. B. Imelik, C. Naccache, G. Coudurier, H. Praliaud, P. Meriaudeau, P. Gallezot, G. A. Martin and J. C. Vedrine, 1982, pp. 1–384 Search PubMed
- D. P. Sobczyk, E. J. M. Hensen, A. M. De Jong and R. A. Van Santen, Top. Catal., 2003, 23, 109–117 CrossRef CAS
- E. M. Slavinskaya, T. Y. Kardash, O. A. Stonkus, R. V. Gulyaev, I. N. Lapin, V. A. Svetlichnyi and A. I. Boronin, Catal. Sci. Technol., 2016, 6, 6650–6666 RSC
- E. M. Slavinskaya, A. I. Stadnichenko, V. V. Muravyov, T. Y. Kardash, E. A. Derevyannikova, V. I. Zaikovskii, O. A. Stonkus, I. N. Lapin, V. A. Svetlichnyi and A. I. Boronin, ChemCatChem, 2018, 10, 2232–2247 CrossRef CAS
- E. Ogel, M. Casapu, D. E. Doronkin, R. Popescu, H. Störmer, C. Mechler, G. Marzun, S. Barcikowski, M. Türk and J.-D. Grunwaldt, J. Phys. Chem. C, 2019, 123, 5433–5446 CrossRef CAS
- E. D. Fakhrutdinova, A. V. Shabalina, M. A. Gerasimova, A. L. Nemoykina, O. V. Vodyankina and V. A. Svetlichnyi, Materials, 2020, 13, 4–7 CrossRef PubMed
- G. L. Haller and D. E. Resasco, Adv. Catal., 1989, 36, 173–235 CAS
- K. Fujiwara, K. Okuyama and S. E. Pratsinis, Environ. Sci.: Nano, 2017, 4, 2076–2092 RSC
- X. Y. Shi, W. Zhang, C. Zhang, W. T. Zheng, H. Chen and J. G. Qi, J. Microsc., 2016, 262, 203–215 CrossRef CAS PubMed
- E. A. Fedorova, A. I. Stadnichenko, E. M. Slavinskaya, L. S. Kibis, O. A. Stonkus, D. A. Svintsitskiy, I. N. Lapin, A. V. Romanenko, V. A. Svetlichnyi and A. I. Boronin, J. Struct. Chem., 2020, 61, 316–329 CrossRef CAS
-
Bruker, TOPAS, Version 4.2, Bruker AXS Inc., Madison, WI, USA, 2009
- M. D. Abramoff, P. J. Magalhaes and S. J. Ram, Biophotonics Int., 2004, 11, 36–42 Search PubMed
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- M. Horn, C. F. Schwebdtfeger and E. P. Meagher, Z. Kristallogr., 1972, 136, 273–281 CrossRef CAS
- P. Ballirano and R. Caminiti, J. Appl. Crystallogr., 2001, 34, 757–762 CrossRef CAS
- M. Peuckert and H. P. Bonzel, Surf. Sci., 1984, 145, 239–259 CrossRef CAS
- K. Vijay Kumar, Z. Phys. Chem., 1991, 173, 105–113 CrossRef
- A. V. Kalinkin, M. Y. Smirnov, A. I. Nizovskii and V. I. Bukhtiyarov, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 15–18 CrossRef CAS
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS
- T. Ioannides and X. E. Verykios, J. Catal., 1996, 161, 560–569 CrossRef CAS
- Q. Fu and T. Wagner, Surf. Sci. Rep., 2007, 62, 431–498 CrossRef CAS
- C. J. Pan, M. C. Tsai, W. N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S. Y. Cheng and B. J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154–186 CrossRef CAS
- J. Ohyama, A. Yamamoto, K. Teramura, T. Shishido and T. Tanaka, ACS Catal., 2011, 1, 187–192 CrossRef CAS
- L. Nie, P. Zhou, J. Yu and M. Jaroniec, J. Mol. Catal. A: Chem., 2014, 390, 7–13 CrossRef CAS
- D. A. Svintsitskiy, L. S. Kibis, A. I. Stadnichenko, S. V. Koscheev, V. I. Zaikovskii and A. I. Boronin, ChemPhysChem, 2015, 16, 3318–3324 CrossRef CAS PubMed
- D. A. Svintsitskiy, E. M. Slavinskaya, O. A. Stonkus, A. V. Romanenko, A. I. Stadnichenko, L. S. Kibis, E. A. Derevyannikova, A. A. Evtushkova and A. I. Boronin, J. Struct. Chem., 2019, 60, 919–931 CrossRef CAS
-
A. Y. Stakheev, D. A. Bokarev, I. P. Prosvirin and V. I. Bukhtiyarov, in Advanced Nanomaterials for Catalysis and Energy, Elsevier, 2019, pp. 295–320 Search PubMed
- A. Naitabdi, R. Fagiewicz, A. Boucly, G. Olivieri, F. Bournel, H. Tissot, Y. Xu, R. Benbalagh, M. G. Silly, F. Sirotti, J. J. Gallet and F. Rochet, Top. Catal., 2016, 59, 550–563 CrossRef CAS
- A. Boubnov, S. Dahl, E. Johnson, A. P. Molina, S. B. Simonsen, F. M. Cano, S. Helveg, L. J. Lemus-Yegres and J.-D. Grunwaldt, Appl. Catal., B, 2012, 126, 315–325 CrossRef CAS
- A. M. Gänzler, M. Casapu, P. Vernoux, S. Loridant, F. J. Cadete Santos Aires, T. Epicier, B. Betz, R. Hoyer and J.-D. Grunwaldt, Angew. Chem., Int. Ed., 2017, 56, 13078–13082 CrossRef PubMed
- C. Wang and C. Yeh, J. Catal., 1998, 178, 450–456 CrossRef CAS
- M. Zhang, Z. Jin, Z. Zhang and H. Dang, Appl. Surf. Sci., 2005, 250, 29–34 CrossRef CAS
- B. Bahrami, V. G. Komvokis, M. S. Ziebarth, O. S. Alexeev and M. D. Amiridis, Appl. Catal., B, 2013, 130–131, 25–35 CrossRef CAS
- D. A. Svintsitskiy, L. S. Kibis, A. I. Stadnichenko, E. M. Slavinskaya, A. V. Romanenko, E. A. Fedorova, O. A. Stonkus, D. E. Doronkin, V. Marchuk, A. Zimina, M. Casapu, J.-D. Grunwaldt and A. I. Boronin, ChemCatChem, 2020, 12, 867–880 CrossRef CAS
- S. Hinokuma, H. Shimanoe, S. Matsuki, M. Kawano, Y. Kawabata and M. Machida, Chem. Lett., 2015, 45, 179–181 CrossRef
- J. G. Wang, W. X. Li, M. Borg, J. Gustafson, A. Mikkelsen, T. M. Pedersen, E. Lundgren, J. Weissenrieder, J. Klikovits, M. Schmid, B. Hammer and J. N. Andersen, Phys. Rev. Lett., 2005, 95, 256102 CrossRef CAS PubMed
- E. I. Vovk, A. V. Kalinkin, M. Y. Smirnov, I. O. Klembovskii and V. I. Bukhtiyarov, J. Phys. Chem. C, 2017, 121, 17297–17304 CrossRef CAS
- D. J. Miller, H. Öberg, S. Kaya, H. Sanchez Casalongue, D. Friebel, T. Anniyev, H. Ogasawara, H. Bluhm, L. G. M. Pettersson and A. Nilsson, Phys. Rev. Lett., 2011, 107, 195502–195507 CrossRef CAS PubMed
- C. R. Parkinson, M. Walker and C. F. McConville, Surf. Sci., 2003, 545, 19–33 CrossRef CAS
- D. R. Butcher, M. E. Grass, Z. Zeng, F. Aksoy, H. Bluhm, W.-X. Li, B. S. Mun, G. A. Somorjai and Z. Liu, J. Am. Chem. Soc., 2011, 133, 20319–20325 CrossRef CAS PubMed
- D. Miller, H. Sanchez Casalongue, H. Bluhm, H. Ogasawara, A. Nilsson and S. Kaya, J. Am. Chem. Soc., 2014, 136, 6340–6347 CrossRef CAS PubMed
- C. Puglia, A. Nilsson, B. Hernnäs, O. Karis, P. Bennich and N. Mårtensson, Surf. Sci., 1995, 342, 119–133 CrossRef CAS
- S. P. Devarajan, J. A. Hinojosa and J. F. Weaver, Surf. Sci., 2008, 602, 3116–3124 CrossRef CAS
- Z. Zhu, F. Tao, F. Zheng, R. Chang, Y. Li, L. Heinke, Z. Liu, M. Salmeron and G. A. Somorjai, Nano Lett., 2012, 12, 1491–1497 CrossRef CAS PubMed
- M. Y. Smirnov, A. V. Kalinkin and V. I. Bukhtiyarov, J. Struct. Chem., 2007, 48, 1053–1060 CrossRef CAS
- L. K. Ono, B. Yuan, H. Heinrich and B. R. Cuenya, J. Phys. Chem. C, 2010, 114, 22119–22133 CrossRef CAS
- L. K. Ono, J. R. Croy, H. Heinrich and B. R. Cuenya, J. Phys. Chem. C, 2011, 115, 16856–16866 CrossRef CAS
- Y. Xu, W. A. Shelton and W. F. Schneider, J. Phys. Chem. A, 2006, 110, 5839–5846 CrossRef CAS PubMed
- J. E. Drawdy, G. B. Hoflmd, S. D. Gardner and E. Yngvadottir, Surf. Interface Anal., 1990, 16, 369–374 CrossRef CAS
- F. Pesty, H.-P. Steinrück and T. E. Madey, Surf. Sci., 1995, 339, 83–95 CrossRef CAS
- Y. M. Sun, D. N. Belton and J. M. White, J. Phys. Chem., 1986, 90, 5178–5182 CrossRef CAS
- Y. Wang, C. Feng, M. Zhang, J. Yang and Z. Zhang, Appl. Catal., B, 2011, 104, 268–274 CrossRef CAS
- C. J. Weststrate, J. W. Bakker, E. D. L. Rienks, C. P. Vinod, S. Lizzit, L. Petaccia, A. Baraldi and B. E. Nieuwenhuys, Surf. Sci., 2006, 600, 1991–2001 CrossRef CAS
- D. P. Sobczyk, A. M. De Jong, E. J. M. Hensen and R. A. Van Santen, J. Catal., 2003, 219, 156–166 CrossRef CAS
- D. P. Sobczyk, J. Van Grondelle, P. C. Thüne, I. E. Kieft, A. M. De Jong and R. A. Van Santen, J. Catal., 2004, 225, 466–478 CrossRef CAS
- A. Jentys, Phys. Chem. Chem. Phys., 1999, 1, 4059–4063 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S11. See DOI: 10.1039/d0cy01533d |
| This journal is © The Royal Society of Chemistry 2021 |