
Combining hydrogen bonding interactions with steric and electronic modifications for thermally robust α-diimine palladium catalysts toward ethylene (co)polymerization†
Handou
Zheng‡
,
Liu
Zhong‡
,
Cheng
Du
,
Wenbo
Du
,
Chi Shing
Cheung
,
Jingjing
Ruan
and
Haiyang
Gao
 *
*
School of Materials Science and Engineering, PCFM Lab, GD HPPC Lab, Sun Yat-sen University, Guangzhou 510275, China. E-mail: gaohy@mail.sysu.edu.cn; Fax: +86 20 84114033; Tel: +86 20 84113250
First published on 27th October 2020
Abstract
Development of thermally robust palladium-based catalysts for (co)polymerization of ethylene and polar monomers with high activities is a continuing challenge. Combining hydrogen bonding interactions with steric and electronic modifications, dibenzobarrelene-based α-diimine palladium complexes with different substituents (X = OMe, H, Cl, Br, and I) have been synthesized and characterized. The steric effect of the palladium complexes was elucidated by their buried volumes, and the electronic effect of the substituents was clarified by the Hammett constants (σ) of the substituents and 1H NMR analysis of the Pd-bound methyl. The hydrogen bonding interactions (H⋯Cl and H⋯OMe) were confirmed by single crystal structures of chloro- and methoxy-substituted neutral and cationic palladium complexes. Contributed by the steric and electronic effects as well as the hydrogen bonding, the chloro-substituted palladium catalyst was thermally robust at temperatures as high as 100 °C for ethylene polymerization, while the methoxy-substituted palladium catalyst showed excellent tolerance toward high temperature and polar groups and was able to copolymerize ethylene and methyl acrylate (MA) at 80 °C to produce the copolymer with high MA incorporation up to 9.5 mol%.
Introduction
Late transition metal catalysts, especially α-diimine nickel and palladium catalysts, have received intensive attention because of their unique catalytic properties and outstanding tolerance toward polar groups that allow for direct copolymerization of ethylene and polar monomers to prepare functional polyolefins.1–4 However, late transition metal catalysts have seen limited industrial application for the production of high-molecular-weight polyolefins. A significant source of this limited application is that late transition metal catalysts often show poor thermal stability and significant decay of catalytic activity at high temperatures, thus not being suitable for many industrial synthetic processes that usually operate at temperatures as high as 70–110 °C.5–7 In comparison with nickel-based catalysts, α-diimine palladium catalysts are inclined to show poorer thermal stability and lower activity.8 Conventional α-diimine palladium catalysts have been reported to undergo rapid decomposition above room temperature, which is attributed to the C–H activation of the metal center to the ligand originating from increasing N-aryl rotations from perpendicular to square-planar coordination planes at elevated temperatures.9–11 Therefore, the design and development of thermally robust α-diimine palladium catalysts for ethylene (co)polymerization remain a great challenge.Two main approaches including steric and electronic modifications are usually employed for enhancement of thermal stability and catalytic performance of α-diimine nickel and palladium catalysts.12–18 To restrict the N-aryl rotations, the utilization of N-aryl moieties containing sterically bulky or conformationally locked substituents has been successfully employed to enhance the stability of α-diimine nickel and palladium catalysts.19–42 Several representative anilines and their substituted derivatives include 2,6-diphenylanilines (Scheme 1A),19,20 2,6-dibenzhydrylanilines (Scheme 1B),21–31 8-phenyl-naphthylanilines (Scheme 1C),32–36 pentiptycenylanilines (Scheme 1D),37–40 and cyclophane-based anilines (Scheme 1E).41,42 In addition, the utilization of bulky backbone structures is also an effective approach to access thermally stable α-diimine derived catalysts. Our groups and other groups have developed camphyl (Scheme 1F),43–47 dibenzobarrelene (Scheme 1G),48–53 and dinaphthyobarrelene backbone-based α-diimine nickel and palladium catalysts (Scheme 1H)54 that can catalyze ethylene (co)polymerization at elevated temperatures, even in a living fashion.
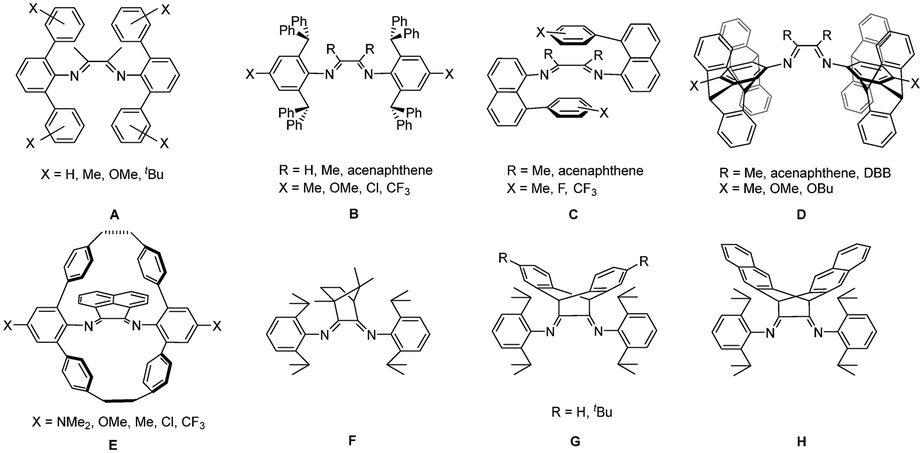 | ||
| Scheme 1 Selected examples of reported α-diimine ligands (A)–(H). | ||
The electronic modification has a perturbation on the electronic environment of the center metal, thereby impacting catalyst stability and catalytic activity. Two noteworthy examples are α-diimine palladium catalysts bearing electron-donating/withdrawing substituents at the para-position of the aniline reported by Guan and Chen groups (Scheme 1B and E).55–58 Generally, introduction of electron-withdrawing groups increases the positive charge of the metal center, thereby increasing catalytic activity. On the other hand, introduction of electron-withdrawing groups leads to poor catalyst stability, thereby reducing activity. In comparison with the electronic modification of aniline moieties, the electronic effect of the α-diimine ligand backbone has been rarely studied and reported.59,60 Hence, the single electronic modification has great difficulty in improving catalyst activity at high temperatures because of the competition between the two factors mentioned above.
Steric and electronic modifications have been combined together in the design of Ni/Pd based olefin polymerization catalysts for enhancing their thermal stability and catalytic performances,42,56,61 but over two approaches have been rarely employed together up to now. Besides steric and electronic modifications, weak noncovalent interactions are also employed in modulating catalytic olefin polymerization.62–64 As a kind of typical weak noncovalent interaction, the hydrogen bonding between hydrogen (H) and an electronegative atom such as F, Cl, and O is also employed in modulating catalytic olefin polymerization.65–69 A representative sample is phenoxy-imino titanium catalysts with ortho-fluoro aryls, which catalyze ethylene and propylene polymerization in a living fashion because the hydrogen bonding interaction between the ortho-F and the β-H of the growing polymer chain (C–H⋯F) inhibits chain transfer.65,66 In comparison with early transition metal catalysts (Ti and Zr), a few of the late transition metal (Ni and Pd) catalysts (e.g. phenoxy-iminato nickel catalysts) are observed to employ hydrogen bonding interaction in operating catalytic olefin polymerization.67–70 Hydrogen bonding has never been employed in modulating olefin polymerization with α-diimine type nickel and palladium catalysts prior to our work.71
In this contribution, we designed and synthesized a series of α-diimine palladium catalysts with electron-donating/withdrawing groups (OMe, Cl, Br, and I) on the dibenzobarrelene backbone by steric and electronic modifications in combination with hydrogen bonding interactions. The intra-ligand hydrogen bonding interactions (C–H⋯Cl and C–H⋯OMe) were fully confirmed by the single crystal analyses and variable-temperature 1H NMR spectra. In the designed palladium catalysts, hydrogen bonding interactions locked the conformation of the α-diimine palladium catalysts and restricted the N-aryl rotations, thereby enhancing the thermal stability of the palladium catalysts. The electronic modification improved the catalytic activity, and the steric modification inhibited chain transfer and enhanced the thermal stability of the palladium catalysts. The cooperative effect of the three factors endowed the designed palladium catalysts with thermally robust characteristics. The chloro-substituted palladium catalyst was able to catalyze ethylene living/controlled polymerization at 100 °C, while the methoxy-substituted palladium catalyst showed excellent copolymerization performance of ethylene and MA.
Results and discussion
Synthesis and characterization of palladium complexes
1,5-Disubstituted dibenzobarrelene α-diimine ligands with electron-donating/withdrawing groups (X = OMe, H, Cl, Br, and I) were prepared according to our reported synthetic routes.49,71 The complexation reactions of α-diimine ligands and Pd(COD)MeCl (COD: 1,5-cyclooctadiene) proceeded smoothly to produce neutral methyl chloride palladium complexes with electronic modification on the backbone. These methyl chloride palladium complexes were further treated with acetonitrile and sodium tetrakis (3,5-bis(trifluoromethyl)phenyl)borate (NaBArF) to yield cationic palladium complexes [(α-diimine)PdCH3(CH3CN)]+BArF− in high yields (Scheme 2). All of the new palladium complexes were fully characterized by NMR spectroscopy and elemental analysis (see the Experimental section).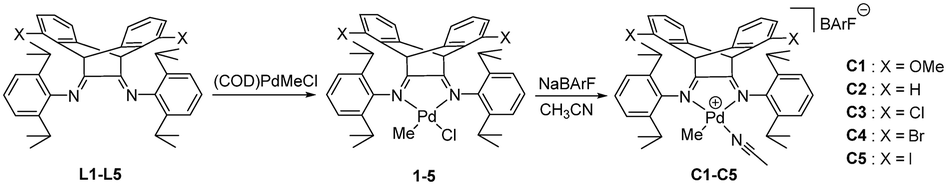 | ||
| Scheme 2 Synthesis of α-diimine palladium complexes. | ||
Single crystals of four neutral methyl chloride palladium complexes 1 and 3–5 suitable for X-ray analysis were grown from n-hexane/dichloromethane solutions. The cultivation of single crystals of cationic α-diimine palladium complexes was often difficult,45 and three high-quality single crystals of 1,5-disubstituted dibenzobarrelene based palladium complexes C1, C3, and C5 were herein obtained. All the palladium complexes adopted a distorted square-planar coordination on the basis of the distortions around the palladium metal center quantified by the τ4 parameter.72 As shown in Fig. 1 and 2, the dibenzobarrelene backbone completely shielded the back space of the palladium complexes, thereby providing more enclosed space around the palladium center.49,71
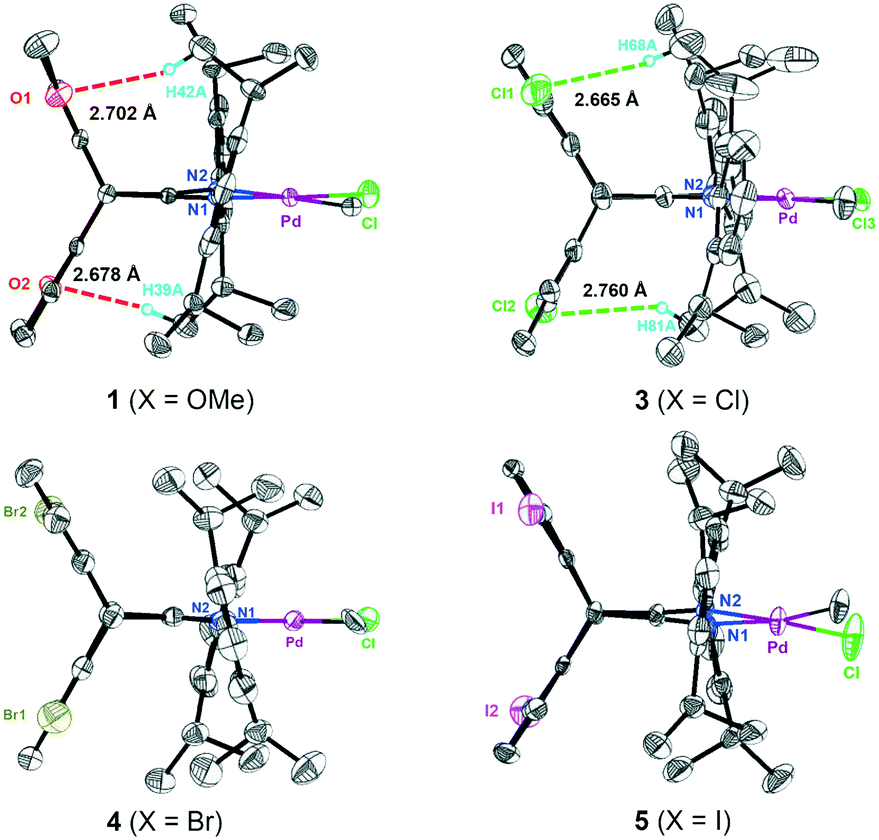 | ||
| Fig. 1 Crystal structures of neutral α-diimine palladium complexes 1 and 3–5 with thermal ellipsoids of 50% probability. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
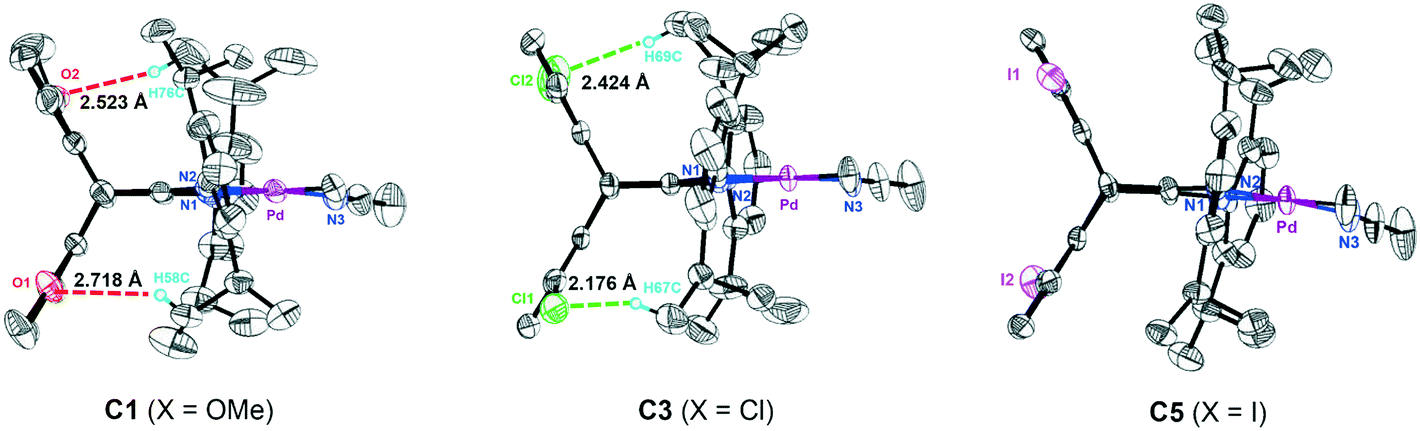 | ||
| Fig. 2 Crystal structures of cationic α-diimine palladium complexes C1, C3, and C5 with thermal ellipsoids of 50% probability. Hydrogen atoms, solvent molecules, and anion BArF− are omitted for clarity. | ||
Topographic steric maps calculated from single crystal structure data were also used to visualize catalytic pockets around the palladium center to address the effect of backbone substituents.73,74 The reported single crystal data of the classic α-diimine palladium complex 6 with a planar acenaphthene backbone were used as comparison.75 As shown in Fig. 3, the buried volumes of all the dibenzobarrelene-based palladium complexes are bigger than that of the acenaphthene-based α-diimine palladium complex 6, clearly supporting the steric effect of the dibenzobarrelene backbone (2: 49.4 and 6: 44.2 %VBur). In addition, the buried volumes of the five dibenzobarrelene-based palladium complexes are slightly changed (48.4–51.0 %VBur), indicating that the substituents on the dibenzobarrelene backbone have little effect on the steric hindrance around the palladium center. The buried volumes of cationic α-diimine palladium complexes C1, C2, and C3–C5 were also calculated from single crystal structure data and were bigger than those of the corresponding neutral palladium complexes (Table 1 and Fig. 4). Like the neutral palladium complexes, the substituents on the dibenzobarrelene backbone of the cationic palladium complexes nearly did not show a steric effect (see Table 1).
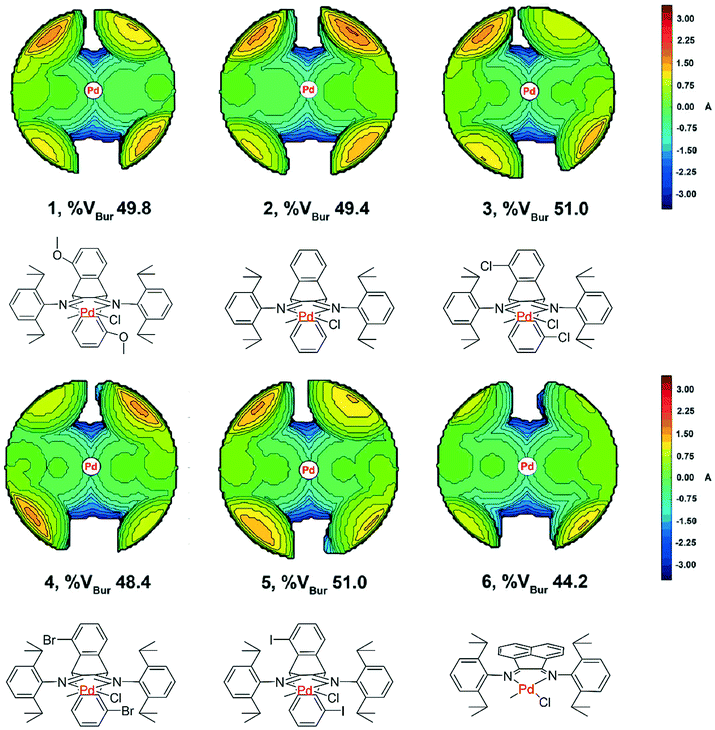 | ||
| Fig. 3 Topographic steric maps of neutral α-diimine palladium complexes with different backbones. | ||
| Pd complex | Substituent X | Hammett constant σpa | δ Pd–CH3b/ppm | X⋯CH(CH3)2 distance/Å | %VBur |
|---|---|---|---|---|---|
| a The electronic effect was quantified by the Hammett constant of the para-substituent. b Crystal data of complex C4 were unavailable. | |||||
| 1 | OMe | −0.268 | 0.49 | 2.678, 2.702 | 49.8 |
| 2 | H | 0 | 0.52 | — | 49.4 |
| 3 | Cl | 0.227 | 0.62 | 2.665/2.760 | 51.0 |
| 4 | Br | 0.232 | 0.64 | 3.117/3.338 | 48.4 |
| 5 | I | 0.276 | 0.66 | 3.325/3.457 | 51.0 |
| C1 | OMe | −0.268 | 0.50 | 2.523/2.718 | 51.7 |
| C2 | H | 0 | 0.53 | — | 50.4 |
| C3 | Cl | 0.227 | 0.64 | 2.176/2.424 | 52.0 |
| C4 | Br | 0.232 | 0.66 | —b | —b |
| C5 | I | 0.276 | 0.69 | 3.208/3.260 | 54.8 |
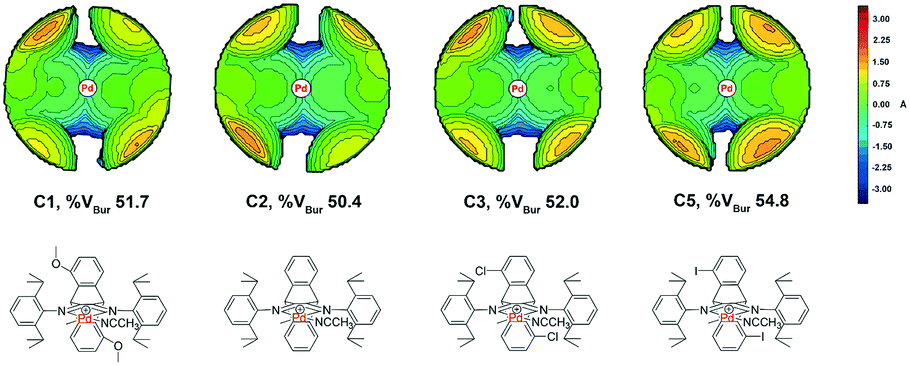 | ||
| Fig. 4 Topographic steric maps of cationic α-diimine palladium complexes with different backbones. | ||
Despite the negligible steric effect of the substituent on the dibenzobarrelene backbone, the substituent is able to impact the electronic properties of palladium complexes by a remote electronic effect.20,76 The Hammett constants (σ) of substituents of the phenyl group were usually used to quantify electronic effects.77,78 As shown in Table 1, the iodo substituent had the strongest electron-withdrawing effect among three halogens because of its very poor conjugation effect. The Pd-bound methyl signal was a good probe for assessing the electron density donated by the ligand to the palladium metal.581H NMR analysis of the Pd-bound methyl groups (Pd–CH3) in neutral palladium complexes 1–5 and cationic palladium complexes C1–C5 also clearly supported electronic effects. As expected, resonances of the Pd-bound methyl groups were overall shifted downfield for palladium complexes bearing electron-withdrawing ligands and upfield for those bearing electron-donating ligands.58 The chemical shift trends correlated well to the Hammett substituent constants (Fig. 5), proving the definite electronic effect of the substituents.
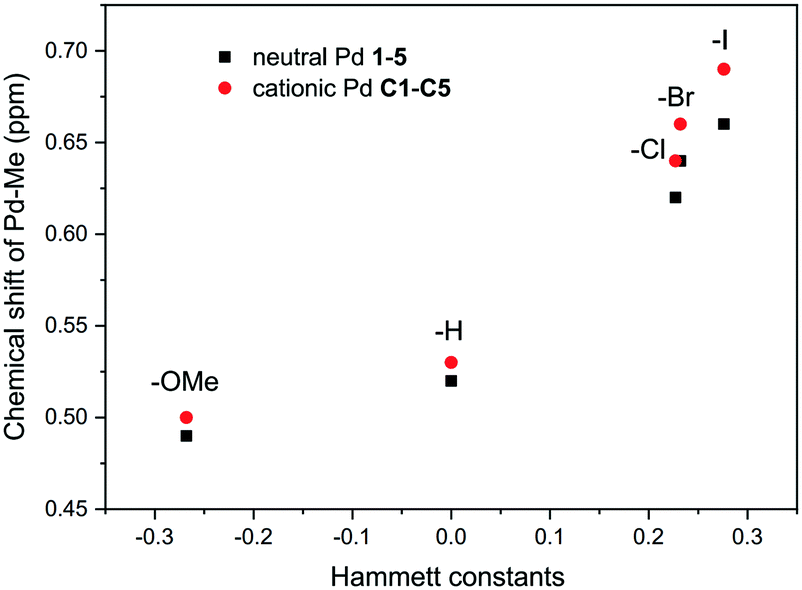 | ||
| Fig. 5 Chemical shifts for Pd–CH3 resonances of palladium complexes 1–5 and C1–C5 in 1H NMR spectra. | ||
More strikingly, the introduction of methoxy and chloro groups on the dibenzobarrelene backbone leads to weak noncovalent hydrogen bonding interactions. For neutral methyl chloride palladium complexes 1 and 3, the distances between the oxygen atoms of the methoxy as well as the chloro groups and the hydrogen atoms of the isopropyl CH(CH3)2 groups are within 3.0 Å, strongly indicating the existence of intramolecular interactions by O⋯H and Cl⋯H hydrogen bonds (Fig. 1). The hydrogen bond existence in solution was also proved by the 1H NMR results of the Pd complexes. The introduction of electron-withdrawing groups usually leads to the shift of the proton signals to downfield because of the decrease in electron cloud density. However, two methyl resonances of isopropyl groups of electron-deficient chloro-substituted palladium complex 3 appeared more upfield than expected (Fig. 6A), indicating increasing electron cloud density of the methyl groups. This result was attributed to the shielding effect of the bulky backbone on the methyl groups because of hydrogen bonding interactions.52,79 The intramolecular hydrogen bonding interactions were anticipated to restrict the N-aryl rotation and fix the perpendicular-coordination plane conformation of the α-diimine palladium complex at elevated temperatures, thereby enhancing the thermal stability of the palladium complexes.71
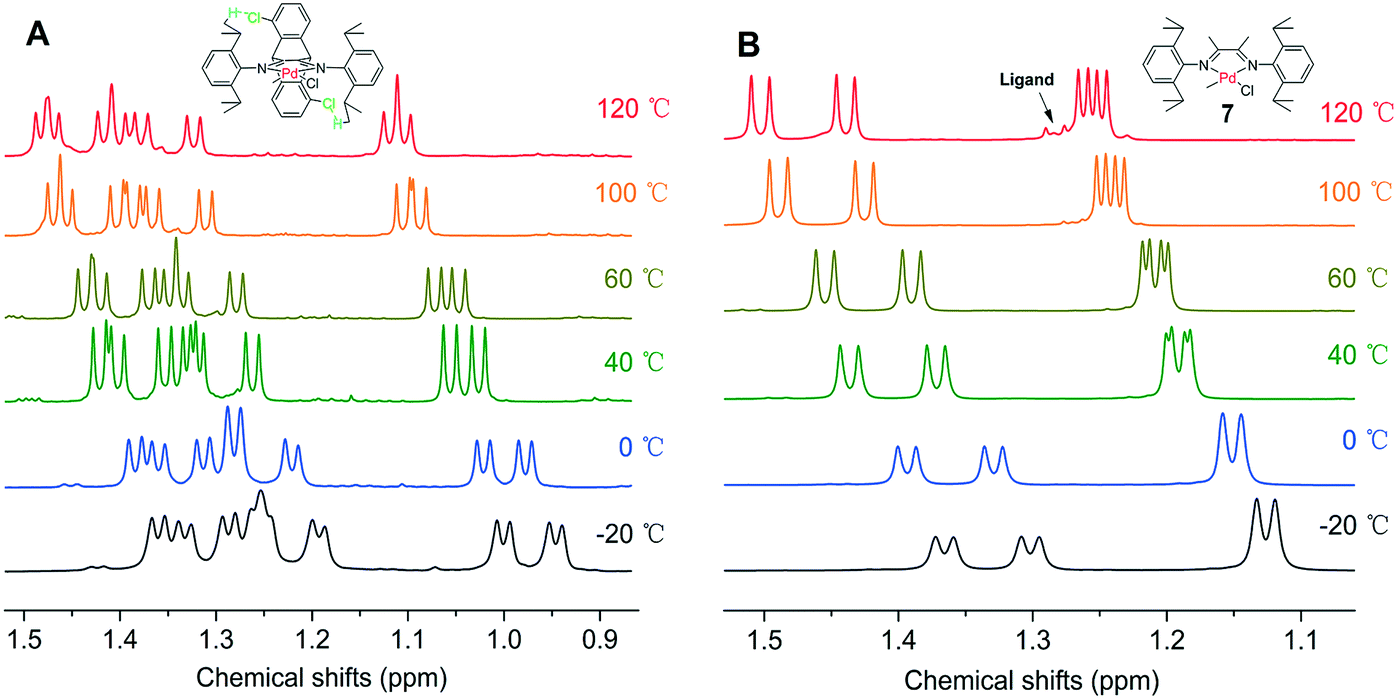 | ||
| Fig. 6 Variable-temperature 1H NMR spectra of the CH(CH3)2 of palladium complexes 3 (A) and 7 (B). | ||
To further verify the enhanced thermal stability of the palladium complex by hydrogen bonding interactions, we pursued a variable-temperature NMR spectroscopic experiment of complex 3 in C2D2Cl4 solution in a temperature range from −20 to 120 °C. As shown in Fig. 6A, two methyl resonances of isopropyl groups showed doublet peaks below 60 °C. When the set temperature exceeded 100 °C, two doublets began to merge to produce a multiple peak, which was attributed to the temperature effect. Differently, four methyl resonances of isopropyl groups of the dimethyl based α-diimine palladium complex 7 began to split doublet–doublet peaks above 40 °C because of the N-aryl rotation, which was consistent with previously reported decomposed temperature of an α-diimine palladium complex.9 Ligand signals were clearly observed when the temperature was over 100 °C, indicating the decomposition of dimethyl based α-diimine palladium complex 7. This comparison of variable-temperature NMR analysis confirmed that hydrogen bonding interactions significantly enhanced the thermal stability of the palladium complexes.
The noncovalent hydrogen bonding interactions between the substituted methoxy and chloro groups on the dibenzobarrelene backbone and hydrogen atoms of the isopropyl groups were also clearly observed for cationic palladium complexes C1 and C3 (Fig. 2). The variable-temperature 1H NMR spectra of cationic palladium complex C3 were obtained in a temperature range from −20 to 120 °C. Additionally, the variable-temperature 1H NMR spectra of cationic palladium complexes C2 and C7 were also collected as comparisons. As shown in Fig. 7, cationic palladium complex C3 was thermally stable at high temperature up to 120 °C, while ligand signals of C2 and C7 respectively appeared at 100 and 40 °C, indicating the decomposition of the palladium complexes. The comparison of variable-temperature NMR analysis between C2 and C3 clearly confirmed that hydrogen bonding interactions significantly enhanced the thermal stability of the palladium complexes. Besides, the high temperature 1H NMR spectra of cationic palladium complex C3 (100 °C and 120 °C) at different times were also obtained and studied (see Fig. S20†). Cationic palladium complex C3 was stable at 100 °C within 5 h but slightly decomposed at 120 °C for 5 h. Because cationic palladium complexes were able to directly catalyze ethylene polymerization, the hydrogen bonding interactions undoubtedly existed in olefin polymerization catalyzed by methoxy- and chloro-substituted palladium catalysts which were expected to be highly thermostable for ethylene polymerization.
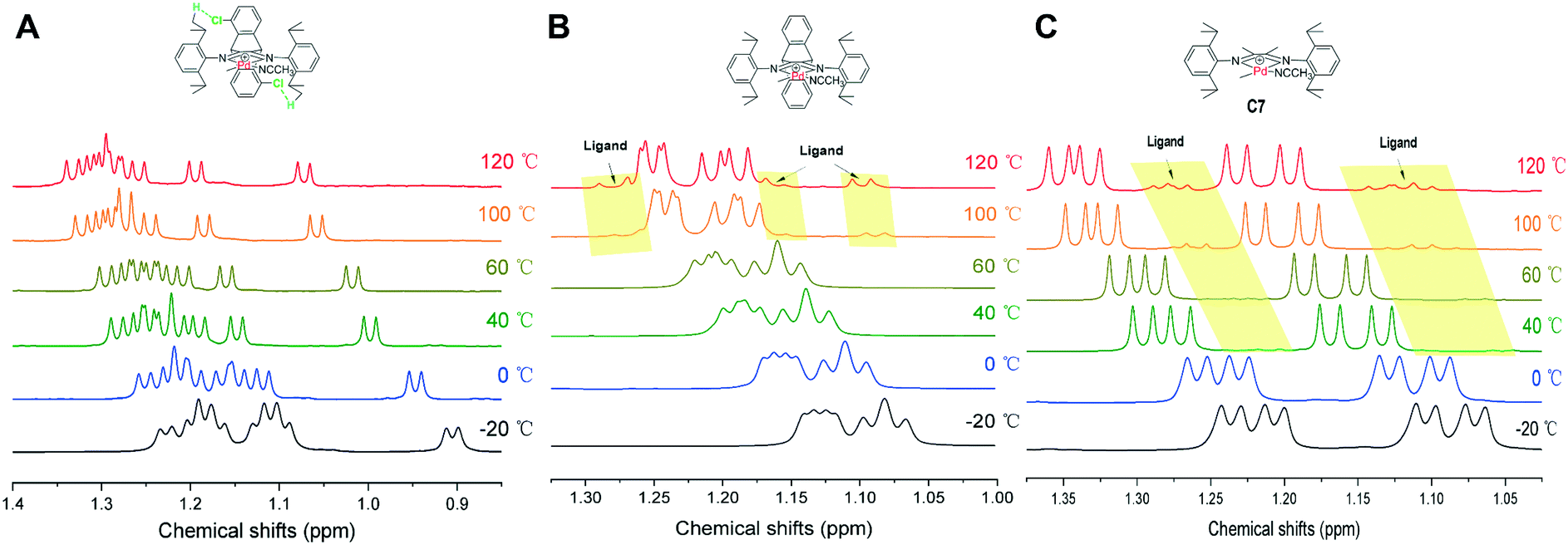 | ||
| Fig. 7 Variable-temperature 1H NMR spectra of the CH(CH3)2 of cationic palladium complexes C3 (A), C2 (B), and C7 (C). | ||
Ethylene polymerization with Pd catalysts
Neutral palladium complexes 1–5 after in situ activation by NaBArF were active for ethylene polymerization, while cationic palladium complexes C1–C5 were able to directly polymerize ethylene without any activators. In this work, ethylene polymerization was directly carried out using cationic palladium complexes C1–C5 to eliminate effects of activators.49,50 To evaluate the thermal stability of the palladium catalysts, ethylene polymerization was performed in a temperature range from 25 to 100 °C with small temperature intervals because α-diimine palladium-catalyzed ethylene polymerization was very sensitive to temperature.As shown in Table 2, palladium catalysts C1–C5 showed different temperature effects on ethylene polymerization. For palladium catalysts C1–C4, polymerization activities increased first and then began to decrease with increasing temperature. Methoxy-substituted palladium catalyst C1 with the strongest electron-donating groups showed the highest activity at 35 °C, while catalysts C2–C4 exhibited the highest activities at 55 °C. However, iodo-substituted palladium catalyst C5 with the strongest electron-withdrawing groups showed a gradually decreasing activity from 25 to 65 °C with increasing temperature although it was more active than C2 without substituents at 25 °C. Overall, the activity order of the palladium catalysts was C3 (X = Cl) > C4 (X = Br) > C2 (X = H) > C1 (X = OMe) > C5 (X = I) on the basis of their highest values toward ethylene polymerization. Considering the negligible steric effect of the substituents (see calculations of buried volumes), the substituent influence on polymerization activity was safely ascribed to the electronic effect. Commonly, introduction of electron-withdrawing groups increased the positive charge of the palladium center and was favorable for ethylene coordination and insertion, thereby improving catalytic activity.57,58,71 Therefore, the activity order C3 (X = Cl) > C4 (X = Br) > C2 (X = H) > C1 (X = OMe) was well explained by the electronic effect. Palladium catalyst C5 showed the lowest activity, this was because the introduction of strong electron-withdrawing iodo groups led to poor catalyst stability. This claim was clearly supported by the declined activity and the poorest thermal tolerance toward ethylene polymerization catalyzed by C5 (Table 2, entries 29–33).
| Entry | Pd | X | T (°C) | Yield (mg) | Act.b | M n (kg mol−1) | PDIc | BDd |
|---|---|---|---|---|---|---|---|---|
| a Conditions: 10 μmol Pd, 0.2 atm ethylene pressure, 2 h, 28 mL of toluene and 2 mL of CH2Cl2, results in entries 8–14 were from our reported results.49 b Act.: kg PE per mol Pd h. c Determined by GPC in 1,2,4-trichlorobenzene at 150 °C using a light scattering detector. d Branching density determined by 1H NMR in number of branches per 1000 carbons. | ||||||||
| 1 | C1 | OMe | 25 | 490 | 24.5 | 72.3 | 1.03 | 104 |
| 2 | C1 | OMe | 35 | 511 | 25.6 | 78.2 | 1.09 | 103 |
| 3 | C1 | OMe | 45 | 434 | 21.7 | 52.1 | 1.18 | 103 |
| 4 | C1 | OMe | 55 | 417 | 20.9 | 37.5 | 1.20 | 103 |
| 5 | C1 | OMe | 65 | 401 | 20.1 | 17.4 | 1.22 | 101 |
| 6 | C1 | OMe | 80 | 254 | 12.7 | 11.4 | 1.20 | 103 |
| 7 | C1 | OMe | 100 | 148 | 7.4 | 3.5 | 1.22 | 107 |
| 8 | C2 | H | 25 | 284 | 14.2 | 40.3 | 1.04 | 96 |
| 9 | C2 | H | 35 | 434 | 21.7 | 31.7 | 1.45 | 97 |
| 10 | C2 | H | 45 | 531 | 26.6 | 24.6 | 1.48 | 98 |
| 11 | C2 | H | 55 | 592 | 29.6 | 24.2 | 1.52 | 98 |
| 12 | C2 | H | 65 | 457 | 22.9 | 12.2 | 1.55 | 97 |
| 13 | C2 | H | 80 | 389 | 19.5 | 7.0 | 1.45 | 99 |
| 14 | C2 | H | 100 | 134 | 6.7 | 3.0 | 1.36 | 101 |
| 15 | C3 | Cl | 25 | 551 | 27.6 | 74.6 | 1.04 | 97 |
| 16 | C3 | Cl | 35 | 732 | 36.6 | 79.8 | 1.10 | 97 |
| 17 | C3 | Cl | 45 | 805 | 40.3 | 69.1 | 1.14 | 98 |
| 18 | C3 | Cl | 55 | 1052 | 52.6 | 49.7 | 1.15 | 96 |
| 19 | C3 | Cl | 65 | 916 | 45.8 | 30.9 | 1.17 | 99 |
| 20 | C3 | Cl | 80 | 686 | 34.3 | 14.0 | 1.18 | 100 |
| 21 | C3 | Cl | 100 | 287 | 14.4 | 3.7 | 1.20 | 103 |
| 22 | C4 | Br | 25 | 466 | 23.3 | 71.2 | 1.04 | 105 |
| 23 | C4 | Br | 35 | 478 | 23.9 | 66.9 | 1.11 | 96 |
| 24 | C4 | Br | 45 | 484 | 24.2 | 43.1 | 1.24 | 96 |
| 25 | C4 | Br | 55 | 561 | 28.1 | 34.2 | 1.24 | 97 |
| 26 | C4 | Br | 65 | 517 | 25.9 | 21.1 | 1.27 | 99 |
| 27 | C4 | Br | 80 | 291 | 14.6 | 7.7 | 1.29 | 101 |
| 28 | C4 | Br | 100 | 98 | 4.9 | 2.8 | 1.42 | 103 |
| 29 | C5 | I | 25 | 432 | 21.6 | 60.3 | 1.04 | 91 |
| 30 | C5 | I | 35 | 362 | 18.1 | 43.5 | 1.18 | 91 |
| 31 | C5 | I | 45 | 196 | 9.8 | 21.0 | 1.20 | 93 |
| 32 | C5 | I | 55 | 121 | 6.1 | 10.7 | 1.23 | 95 |
| 33 | C5 | I | 65 | 78 | 3.9 | 9.0 | 1.21 | 96 |
Additionally, it was noteworthy that palladium catalysts C1 and C3 showed good activity for ethylene polymerization among the five palladium catalysts at 100 °C while classic dimethyl based α-diimine palladium catalyst C7 was inactive above 45 °C under the same conditions. Although the electron-donating effect of the methoxy groups caused decreased activity, C1 still showed good activity at 100 °C. This was contributed by stabilization of the palladium center of the electron-donating methoxy groups and hydrogen bonding interactions (MeO⋯H). However, electron-deficient chloro-substituted palladium catalyst C3 showed a higher activity at 100 °C, which was ascribed to hydrogen bonding interactions (Cl⋯H). Although chloro and bromo groups had nearly the same Hammett constants (σp: 0.227 and 0.232, see Table 1), chloro-substituted palladium catalyst C3 exhibited a higher activity than bromo-substituted palladium catalyst C4 in a temperature range from 20 to 100 °C. This result clearly proved the effect of hydrogen bonding (Cl⋯H) on ethylene polymerization. A widely acceptable decomposition pathway of α-diimine palladium catalysts was the C–H activation of the palladium center to the ligand because of increasing N-aryl rotations from perpendicular to square-planar coordination planes at elevated temperatures.9 The intramolecular hydrogen bonding interaction restricted the rotation of the N-aryl bonds and locked the perpendicular-coordination plane conformation of the α-diimine palladium complexes, thereby enhancing the thermal tolerance of the α-diimine palladium catalyst.71 Higher molecular weight polymers with narrower polydispersities (PDIs) were produced by palladium catalysts C1 and C3, which further supported the hydrogen bonding effects on ethylene polymerization. To the best of our knowledge, this is the first report on hydrogen bond-modulated olefin polymerization with palladium-based catalysts.
Chloro-substituted palladium catalyst C3 was selected to further study its thermal stability and catalyst lifetime at 100 °C. Under atmospheric pressure and at 100 °C, ethylene polymerization with C3 was performed at different polymerization times ranging from 1 to 5 h. As shown in Fig. 8A, the turnover frequencies (TOFs) of catalyst C3 at different time periods remained relatively constant, while Mn increased linearly with the polymerization time and PDI (Mw/Mn) values were controlled to ∼1.20. These results strongly indicated that chloro-substituted palladium catalyst C3 was thermally stable and showed a living/controlled characteristic at 100 °C. Besides, increasing the ethylene pressure led to significantly improved catalytic activity and polymer molecular weight (Fig. 8B). At 20 atm and 100 °C, C3 produced highly branched polyethylene with a molecular weight up to 10 kg mol−1. Totally, palladium catalyst C3 was a thermally robust olefin polymerization catalyst by steric and electronic modifications in combination with hydrogen bonding interactions.
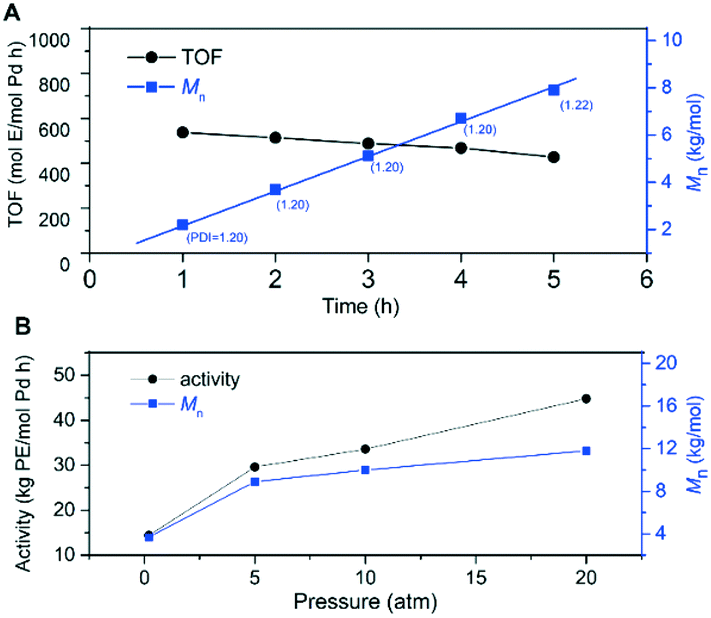 | ||
| Fig. 8 Ethylene polymerization by chloro-substituted palladium catalyst C3 at 100 °C, plots of TOF and Mn as a function of time (A) and activity and Mn as a function of pressure (B). | ||
Copolymerization of ethylene and methyl acrylate (MA)
The copolymerization of ethylene and methyl acrylate (MA) was also studied using cationic α-diimine palladium catalysts C1–C5 at various temperatures and monomer concentrations. A crucial question, the existence of hydrogen bonding in the presence of methyl acrylate, firstly needed to be answered. A control experiment was carried out by addition of methyl acrylate into the methoxy-substituted palladium catalyst C1 solution in CDCl3 (Fig. S17†). 1H NMR analysis showed that the presence of methyl acrylate had no influence on the chemical shifts of catalyst C1, which confirmed that the presence of methyl acrylate did not affect the hydrogen bonding, and the hydrogen bonding existed in copolymerization. The substituent effects on copolymerization were first screened under atmospheric pressure of ethylene and at 25 °C. As shown in Table 3, the copolymerization results had a significant dependence on the electronic effect of the substituents. An overall trend was that the introduction of more electronic-donating groups led to an increase in polymer molecular weight, activity, and incorporation of MA on the basis of the copolymerization activity order (C1 (X = OMe) > C2 (X = H) > C3 (X = Cl) > C4 (X = Br) > C5 (X = I)). The same electronic effect was observed in copolymerization of ethylene and MA using dimethyl based α-diimine palladium catalysts with para-substituted anilines.58 For catalysts C1–C3, raising the reaction temperature from 25 °C to 50 °C resulted in a decrease of activity and polymer molecular weight, but an increase of MA incorporation. Dimethoxy-substituted palladium catalyst C1 had the best thermal tolerance and still showed copolymerization activity to afford a narrowly dispersed copolymer (PDI < 1.2) at a high temperature of 80 °C. Besides, increasing the MA concentration also led to a decrease in activity of C1 and copolymer molecular weight, but an increase of MA incorporation. Catalyst C1 was still active at a high MA concentration of 4.4 M and afforded a copolymer with the highest MA incorporation of 9.52 mol%, indicating its good tolerance toward polar groups.| Entry | Pd | X | T (°C) | Conc. (M) | Yield (mg) | Act.b | I MA mol% | M n (kg mol−1) | PDId | BDe |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 20 μmol Pd, 0.2 atm ethylene pressure, 6 h, 30 mL CH2Cl2, results in entries 7–9 were from our reported results.49 b Act.: kg PE per mol Pd h. c Determined by 1H NMR spectroscopy. d Determined by gel permeation chromatography (GPC) in 1,2,4-trichlorobenzene at 150 °C using a light scattering detector. e Branching density determined by 1H NMR spectroscopy in number of branches per 1000 carbons. f 28 mL toluene and 2 mL CH2Cl2. | ||||||||||
| 1 | C1 | OMe | 25 | 1.1 | 444 | 3.7 | 2.07 | 38.4 | 1.06 | 103 |
| 2 | C1 | OMe | 35 | 1.1 | 287 | 2.4 | 4.03 | 19.5 | 1.09 | 96 |
| 3f | C1 | OMe | 50 | 1.1 | 176 | 1.5 | 5.00 | 13.1 | 1.18 | 88 |
| 4f | C1 | OMe | 80 | 1.1 | 83 | 0.7 | 7.64 | 5.9 | 1.19 | 84 |
| 5 | C1 | OMe | 25 | 2.2 | 197 | 1.6 | 4.34 | 18.7 | 1.08 | 95 |
| 6 | C1 | OMe | 25 | 4.4 | 68 | 0.6 | 9.52 | 9.6 | 1.30 | 103 |
| 7 | C2 | H | 25 | 1.1 | 430 | 3.6 | 1.84 | 36.3 | 1.08 | 99 |
| 8 | C2 | H | 35 | 1.1 | 239 | 2.0 | 3.14 | 17.9 | 1.19 | 93 |
| 9 | C2 | H | 50 | 1.1 | 145 | 1.2 | 4.64 | 12.6 | 1.30 | 91 |
| 10 | C3 | Cl | 25 | 1.1 | 420 | 3.5 | 1.24 | 35.3 | 1.12 | 100 |
| 11 | C3 | Cl | 35 | 1.1 | 142 | 1.2 | 2.71 | 17.3 | 1.16 | 97 |
| 12f | C3 | Cl | 50 | 1.1 | 92 | 0.8 | 3.22 | 9.9 | 1.20 | 92 |
| 13 | C4 | Br | 25 | 1.1 | 404 | 3.4 | 1.33 | 30.4 | 1.15 | 99 |
| 14 | C5 | I | 25 | 1.1 | 247 | 2.1 | 1.57 | 29.4 | 1.15 | 97 |
In general, methoxy-substituted palladium catalyst C1 showed the best catalytic properties toward copolymerization of ethylene and MA, which was attributed to the electron-donating effect of the methoxy and hydrogen bonding interactions together. In comparison with ethylene homopolymerization, the copolymerization by introduction of MA into ethylene polymerization systems led to an alternation of the optimized catalyst from C3 (X = Cl) to C1 (X = OMe) because binding of the electron-deficient MA monomer relative to ethylene on the electron-rich palladium center was favorable.15,80
Conclusions
In summary, we have developed thermally robust α-diimine palladium catalysts for (co)polymerization of ethylene and MA by steric and electronic modifications in combination with hydrogen bonding interactions. A series of neutral and cationic dibenzobarrelene-based α-diimine palladium complexes with different electron-donating/withdrawing substituents (X = OMe, H, Cl, Br, and I) were successfully synthesized and characterized. The intra-ligand hydrogen bonding interactions (H⋯Cl and H⋯OMe) were confirmed in the chloro- and methoxy-substituted neutral and cationic palladium complexes. The buried volumes of the palladium complexes proved the steric effect of the dibenzobarrelene backbone. The Hammett constants (σ) of the substituents (X = OMe, H, Cl, Br, and I) and 1H NMR analysis of the Pd-bound methyl groups clearly confirmed the electronic effect of the substituents. With the cooperative effect of the three factors, chloro-substituted palladium catalyst C3 was the most thermally robust for ethylene polymerization, while methoxy-substituted palladium catalyst C1 showed the best catalytic performance toward copolymerization of ethylene and MA. The tolerance of the designed palladium catalysts toward high temperatures and polar groups was superior to that of most of the palladium catalysts and comparable with the best performances reported in the literature. This study promoted advances in late transition metal catalysts and olefin polymerization, and these disciplines including the steric effect, electronic effect, and hydrogen bonding effect would enable the design and discovery of new thermally robust catalysts for olefin polymerization.Experimental section
General procedures
All manipulations involving air- and moisture sensitive compounds were carried out under an atmosphere of dried and purified nitrogen with standard vacuum-line, Schlenk, or glovebox techniques.Materials
Toluene and hexane were refluxed over a Na/K alloy before use, and dichloromethane was dried over P2O5 and was distilled under nitrogen. The α-diimine ligands L3–L5 and dimethyl-based α-diimine palladium complexes 7 and C7 were prepared according to literature procedures.49,71 Ethylene (99.99%) was purified by passing through Agilent moisture and oxygen traps. Methyl acrylate (MA) was dried over CaH2 and then freshly distilled under vacuum prior to use in copolymerization. Other commercially available reagents were purchased and used without purification.Characterization
Elemental analyses were performed on a Vario EL macro analyzer. The room temperature NMR data of organic compounds were obtained on a Bruker 400 MHz instrument in CDCl3, and the variable-temperature NMR data were recorded on a Bruker 600 MHz instrument in C2D2Cl4. The incorporation (mol%) of methyl acrylate (MA) in the E-MA copolymer was calculated by integrating the methyl proton signals (–OCH3) of inserted MA with respect to the signals of other protons in the 1H NMR spectrum (MA% = 4IOCH3/3(ICH3 + ICH2 + ICH) × 100%). The total branching density per 1000 carbon atoms of polymers was determined by integrating the methyl proton signals with respect to the signals of all protons in the 1H NMR spectrum. GPC analyses of the molecular weights and molecular weight distributions (PDI = Mw/Mn) of the polymer samples at 150 °C were performed on a PL-GPC 220 high-temperature chromatograph equipped with a triple-detection array, including a differential refractive-index detector, a two-angle light-scattering detector (15° and 90°), and a four-bridge capillary viscometer. 1,2,4-Trichlorobenzene (TCB) was used as the eluent at a flow rate of 1.0 mL min−1. The GPC was calibrated with narrow polystyrene standards (Polymer Laboratories).Crystal structure determination
The crystal was mounted on a glass fiber and transferred to a Bruker CCD platform diffractometer. The X-ray diffraction data were obtained in the ω–2θ scan mode on a Bruker SMART 1000 CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) or Cu Kα radiation (λ = 1.54178 Å) at 150 K. The structure was solved using direct methods, and further refinement with full-matrix least squares on F2 was obtained with the SHELXTL program package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were introduced in the calculated positions with the displacement factors of the host carbon atoms.Buried volume calculation
The %VBur (percentage of the sphere (r = 3.5 Å) around the metal occupied by a ligand) was quantified using the SambVca 2.1 tool, which is a web application to easily and effectively analyze and visualize the steric hindrance of organic ligands in metal coordination complexes, by making use of the buried volume values and of the steric maps. The steric maps report the value along the Z-axis at which the ligand starts to bury space in the coordination sphere around the metal center in the case of metal complexes.73,74Homo- and copolymerization of ethylene at atmospheric pressure
A round-bottom Schlenk flask with a stirring bar was heated for 2 h at 130 °C under vacuum and then cooled to room temperature. The flask was pressurized to 0.2 atm gauge pressure of ethylene and vented three times. Then the glass reactor was charged with the required amount of freshly distilled solvent (toluene/CH2Cl2) and methyl acrylate (MA) for copolymerization in sequence under 0.2 atm pressure of ethylene. The system was continuously stirred for 30 min at the desired temperature, and then 2 mL of palladium complex solution in CH2Cl2 was added using a syringe to the well-stirred solution. The ethylene pressure was kept constant at 0.2 atm by continuous feeding of ethylene throughout the reaction. After the polymerization process, the reaction was quenched by the addition of triethylsilane and acidic methanol. After the solvent was removed on a rotary evaporator, the produced polymer was dissolved in petroleum ether. The polymer solution was filtered through a plug of silica gel to remove palladium black before precipitating in methanol. The resulting precipitated polymers were collected and treated by filtration, washed with methanol several times, and dried in a vacuum oven to a constant weight.Ethylene polymerization at high pressure
A mechanically stirred Chemtron reactor was heated to 130 °C for 2 h under vacuum and then cooled to room temperature. The autoclave was pressurized with ethylene and vented three times. The autoclave was then charged with toluene solution under 0.2 atm of ethylene at initialization temperature. The system was maintained by continuously stirring for 30 min, and then 2 mL of palladium complex solution in CH2Cl2 was charged into the autoclave. The ethylene pressure was raised to the specified value, and stirred continuously for the desired time at a specific temperature. Polymerization was terminated by addition of triethylsilane and acidic methanol after releasing ethylene pressure. The resulting precipitated polymers were collected and treated by filtration, washed with methanol several times, and dried under vacuum at 45 °C to a constant weight.Synthesis of neutral α-diimine palladium complexes
The neutral α-diimine palladium complexes 1 and 3–5 were synthesized by the reaction of (COD)PdMeCl with the corresponding α-diimine ligands L1 and L3–L5 in dichloromethane. A typical synthetic procedure for methoxy-substituted palladium complex 1 was described as follows: 2.2 mmol of ligand L1 and 2.0 mmol of (COD)PdMeCl were added to a Schlenk tube together with 20 mL of dichloromethane, and the reaction mixture was then stirred for 12 h at room temperature. The solution was filtered with Celite, and then evaporated under vacuum to 5 mL and 50 mL hexane was added. Methoxy-substituted palladium complex 1 was obtained as an orange-red powder in 92% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.39–7.26 (m, 6H, Ar–H), 7.20–7.14 (m, 2H, Ar–H), 6.90–6.84 (m, 2H, Ar–H), 6.75–6.69 (m, 2H, Ar–H), 5.33 (s, 2H, CH), 3.68 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 2.96 (m, 2H, CH(CH3)2), 2.75 (m, 2H, CH(CH3)2), 1.42–1.28 (m, 12H, CH(CH3)2), 1.22 (d, 3H, CH(CH3)2), 1.17 (d, 3H, CH(CH3)2), 1.04–1.01 (m, 6H, CH(CH3)2), 0.49 (s, 3H, PdCH3). 13C NMR(100 MHz, CDCl3), δ (ppm): 172.14, 167.16 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 155.14, 154.95, 140.62, 140.58, 140.31, 140.00, 139.64, 139.46, 139.31, 138.85, 129.49, 129.33, 127.86, 127.13, 125.49, 123.82, 123.60, 123.17, 122.90, 118.40, 118.32, 110.32, 110.20 (Ar–C in backbone & aniline), 55.27 (NC–CH), 44.57, 43.80 (OCH3), 29.28, 28.90, 28.84, 28.45 (CH(CH3)2), 24.35, 23.98, 23.48, 23.17, 23.04, 23.01, 22.87, 22.62 (CH(CH3)2), 3.84 (PdCH3). Anal. calcd for C43H51ClN2O2Pd: C, 67.10; H, 6.68; N, 3.64; found: C, 67.38; H, 6.49; N, 3.51.
N), 155.14, 154.95, 140.62, 140.58, 140.31, 140.00, 139.64, 139.46, 139.31, 138.85, 129.49, 129.33, 127.86, 127.13, 125.49, 123.82, 123.60, 123.17, 122.90, 118.40, 118.32, 110.32, 110.20 (Ar–C in backbone & aniline), 55.27 (NC–CH), 44.57, 43.80 (OCH3), 29.28, 28.90, 28.84, 28.45 (CH(CH3)2), 24.35, 23.98, 23.48, 23.17, 23.04, 23.01, 22.87, 22.62 (CH(CH3)2), 3.84 (PdCH3). Anal. calcd for C43H51ClN2O2Pd: C, 67.10; H, 6.68; N, 3.64; found: C, 67.38; H, 6.49; N, 3.51.
Chloro-substituted palladium complex 3 was obtained as an orange-red powder in 85% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.43–7.18 (m, 12H, Ar–H), 5.38 (d, 2H, CH), 3.07 (m, 2H, CH(CH3)2), 2.71 (m, 2H, CH(CH3)2), 1.44–1.39 (m, 6H, CH(CH3)2), 1.35–1.29 (m, 9H, CH(CH3)2), 1.27–1.23 (d, 3H, CH(CH3)2), 1.05–0.97 (m, 6H, CH(CH3)2), 0.62 (s, 3H, PdCH3); 1.26 (m, hexane), 0.88 (t, hexane). 13C NMR(100 MHz, CDCl3), δ (ppm): 171.74, 166.99 (CN), 140.19, 140.14, 139.98, 139.53, 139.25, 138.79, 138.59, 138.09, 136.17, 135.43, 131.48, 131.31, 129.53, 129.39, 129.32, 129.16, 128.20, 127.44, 124.84, 124.73, 124.18, 123.82, 123.49, 122.78 (Ar–C in backbone & aniline), 47.95, 47.21 (NC–CH), 29.34, 28.96, 28.76, 28.38 (CH(CH3)2), 24.31, 23.75, 23.09, 23.05, 22.88, 22.72, 22.61 (CH(CH3)2), 4.49 (PdCH3). Anal. calcd for C41H45Cl3N2Pd: C, 63.25; H, 5.83; N, 3.60; found: C, 63.52; H, 5.59; N, 3.25.
Bromo-substituted palladium complex 4 was obtained as an orange-red powder in 80% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.44–7.27 (m, 10H, Ar–H), 7.16–7.10 (m, 2H, Ar–H), 5.38 (s, 1H, CH), 5.37 (s, 1H, CH), 3.19 (m, 2H, CH(CH3)2), 2.69 (m, 2H, CH(CH3)2), 1.45–1.39 (m, 6H, CH(CH3)2), 1.36–1.28 (m, 12H, CH(CH3)2), 1.01–0.95 (m, 6H, CH(CH3)2), 0.64 (s, 3H, PdCH3); 1.26 (m, hexane), 0.88 (t, hexane). 13C NMR(100 MHz, CDCl3), δ (ppm): 170.40, 165.68 (CN), 140.61, 140.36, 140.15, 139.97, 139.49, 138.86, 138.83, 138.49, 138.11, 137.72, 132.59, 132.41, 129.90, 129.77, 128.35, 127.60, 125.62, 125.50, 124.49, 124.00, 123.81, 123.58, 122.86, 121.24, 121.16, 101.02 (Ar–C in backbone & aniline), 50.83, 50.08 (NC–CH), 29.55, 29.14, 28.89, 28.53 (CH(CH3)2), 24.58, 23.84, 23.32, 23.25, 23.21, 23.12, 23.08, 22.61 (CH(CH3)2), 4.55 (PdCH3); 31.18, 22.77, 14.24 (hexane). Anal. calcd for C41H45Br2ClN2Pd: C, 56.77; H, 5.23; N, 3.23; found: C, 57.02; H, 5.07; N, 3.12.
Iodo-substituted palladium complex 5 was obtained as an orange-red powder in 92% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.66–7.57 (m, 2H, Ar–H), 7.43–7.27 (m, 8H, Ar–H), 7.00–6.93 (m, 2H, Ar–H), 5.29 (s, 1H, CH), 5.27 (s, 1H, CH), 3.35 (m, 2H, CH(CH3)2), 2.66 (m, 2H, CH(CH3)2), 1.41(d, 9H, CH(CH3)2), 1.37–1.28 (m, 9H, CH(CH3)2), 0.95–0.88 (m, 6H, CH(CH3)2), 0.66 (s, 3H, PdCH3). 13C NMR(100 MHz, CDCl3), δ (ppm): 170.86, 166.12 (CN), 142.39, 141.58, 140.95, 140.38, 1.09, 138.96, 138.79, 138.76, 138.59, 138.31, 137.94, 130.03, 129.90, 128.34, 127.60, 126.46, 126.31, 124.76, 124.10, 123.55, 122.79, 96.28, 96.20 (Ar–C in backbone & aniline), 55.81, 55.01 (NC–CH), 29.57, 29.10, 28.83, 28.50 (CH(CH3)2), 24.72, 23.82, 23.78, 23.69, 23.30, 23.12, 22.93, 22.36, 4.39 (CH(CH3)2), 4.39 (PdCH3); 31.71, 14.26 (hexane). Anal. calcd for C41H45ClI2N2Pd: C, 51.22; H, 4.72; N, 2.91; found: C, 51.49; H, 4.59; N, 2.73.
Synthesis of cationic α-diimine palladium complexes
Cationic α-diimine palladium catalysts C1 and C3–C5 were synthesized by the reaction of NaBArF and acetonitrile with palladium complexes 1 and 3–5 in dichloromethane. A typical synthetic procedure for methoxy-substituted palladium catalyst C1 can be described as follows: 1.0 mmol of palladium complex 1, 1.2 mmol of NaBArF, and 2 mL of acetonitrile were added to a Schlenk tube together with 20 mL of dichloromethane, and the reaction mixture was then stirred for 12 h at room temperature. The solution was filtered with Celite, and then evaporated under vacuum to 5 mL and 50 mL hexane was added. Methoxy-substituted palladium catalyst C1 was obtained as a yellow powder in 85% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.70 (s, 8H, Ar–H in BArF−), 7.52 (s, 4H, Ar–H in BArF−), 7.47–7.23 (m, 8H, Ar–H in backbone & aniline), 6.92–6.75 (m, 4H, Ar–H in backbone & aniline), 5.36 (s, 1H, CH), 5.34 (s, 1H, CH), 3.72 (s, 3H, OCH3), 3.68 (s, 3H, OCH3), 2.70 (m, 2H, CH(CH3)2), 2.59 (m, 2H, CH(CH3)2), 1.68 (s, 3H, CH3CN), 1.27–1.19 (m, 18H, CH(CH3)2), 1.14 (d, 3H, CH(CH3)2), 1.06 (d, 3H, CH(CH3)2), 0.50 (s, 3H, PdCH3); 5.30 (s, dichloromethane), 2.02 (s, free CH3CN). 13C NMR(100 MHz, CDCl3), δ (ppm): 176.55, 169.10 (CN), 162.60, 162.11, 161.61, 161.12 (Ar–C–B in BArF−), 155.35, 155.28, 155.14, 155.02, 139.89, 139.15, 139.04, 138.91, 138.58, 138.07, 134.96, 130.69, 130.56, 129.53, 129.27, 129.22 (Ar–C in backbone & aniline), 129.19, 129.16, 128.93, 128.91, 128.88, 128.85, 128.78, 128.32 (CF3 in BArF−), 126.07, 124.71, 124.40, 124.28, 124.21, 123.93 (Ar–C in backbone & aniline), 120.86, 120.65 (Ar–C in BArF−), 118.57, 118.38, 118.31 (Ar–C in backbone & aniline), 117.61 (CH3CN), 111.16, 111.11 (Ar–C in backbone), 55.47, 55.39 (NC–CH), 55.27 (OCH3), 29.31, 29.25, 28.88, 28.67 (CH(CH3)2), 23.92, 23.62, 23.28, 22.87, 22.85, 22.66, 22.64, 22.32 (CH(CH3)2), 8.81 (CH3CN), 1.78 (PdCH3). Anal. calcd for C77H66BF24N3O2Pd: C, 56.44; H, 4.06; N, 2.56; found: C, 56.64; H, 3.96; N, 2.42.
Chloro-substituted palladium catalyst C3 was obtained as a yellow powder in 72% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.70 (s, 8H, Ar–H in BArF−), 7.52 (s, 4H, Ar–H in BArF−), 7.49–7.22 (m, 12H, Ar–H in backbone & aniline), 5.41 (s, 1H, CH), 5.40 (s, 1H, CH), 2.82 (m, 1H, CH(CH3)2), 2.68 (m, 1H, CH(CH3)2), 2.57 (m, 2H, CH(CH3)2), 1.71 (s, 3H, CH3CN), 1.33 (d, 3H, CH(CH3)2), 1.30–1.21 (m, 15H, CH(CH3)2), 1.18 (d, 3H, CH(CH3)2), 1.04 (d, 3H, CH(CH3)2), 0.64(s, 3H, PdCH3). 13C NMR(100 MHz, CDCl3), δ (ppm): 174.67, 167.38 (CN), 162.44, 161.94, 161.45, 160.95 (Ar–C–B in BArF−), 139.79, 138.75, 138.56, 138.45, 137.94, 137.48, 134.83, 134.79, 134.37, 131.87, 131.78, 130.54, 130.48, 130.28, 130.17, 129.62 (Ar–C in backbone & aniline), 129.38, 129.07, 129.04, 128.76, 128.73, 128.69, 128.61, 128.44 (CF3 in BArF−), 125.90, 125.14, 124.95, 124.77, 124.20, 124.06, 123.91, 123.19 (Ar–C in backbone & aniline), 121.02, 120.48 (Ar–C in BArF−), 117.47 (CH3CN), 47.89, 47.14 (NC–CH), 29.43, 28.86, 28.62 (CH(CH3)2), 23.84, 23.50, 22.84, 22.75, 22.68, 22.54, 22.51, 22.42 (CH(CH3)2), 9.69 (CH3CN), 1.63 (PdCH3). Anal. calcd for C75H60BCl2F24N3Pd: C, 54.68; H, 3.67; N, 2.55; found: C, 54.87; H, 3.51; N, 2.51.
Bromo-substituted palladium catalyst C4 was obtained as a yellow powder in 70% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.70 (s, 8H, Ar–H in BArF−), 7.52 (s, 4H, Ar–H in BArF−), 7.48–7.45 (m, 2H, Ar–H in backbone & aniline), 7.41–7.26 (m, 8H, Ar–H in backbone & aniline), 7.24–7.17 (m, 2H, Ar–H in backbone & aniline), 5.41 (s, 1H, CH), 5.39 (s, 1H, CH), 2.92 (m, 1H, CH(CH3)2), 2.75 (m, 1H, CH(CH3)2), 2.57 (m, 2H, CH(CH3)2), 1.72 (s, 3H, CH3CN), 1.38 (d, 3H, CH(CH3)2), 1.32–1.21 (m, 15H, CH(CH3)2), 1.17 (d, 3H, CH(CH3)2), 1.00 (d, 3H, CH(CH3)2), 0.66 (s, 3H, PdCH3); 1.26 (m, hexane), 0.88 (t, hexane). 13C NMR(100 MHz, CDCl3), δ (ppm): 174.96, 167.66 (CN), 162.59, 162.10, 161.61, 161.11 (Ar–C–B in BArF−), 140.22, 138.98, 138.84, 138.62, 138.57, 138.15, 138.01, 137.73, 137.17, 136.68, 134.97, 133.60, 133.45 (Ar–C in backbone & aniline), 130.89, 130.85, 129.80, 129.55, 129.22, 128.89, 128.77, 128.59 (CF3 in BArF−), 126.06, 125.89, 125.74, 125.10, 124.31, 124.05, 123.3121.64, 121.41 (Ar–C in backbone & aniline), 121.10, 120.65 (Ar–C in BArF−), 117.61 (CH3CN), 50.79, 50.03 (NC–CH), 29.67, 29.66, 28.99, 28.75 (CH(CH3)2), 24.14, 23.65, 23.17, 23.00, 22.96, 22.91, 22.78, 22.58 (CH(CH3)2), 9.85 (CH3CN), 1.91 (PdCH3). Anal. calcd for C75H60BBr2F24N3Pd: C, 51.88; H, 3.48; N, 2.42; found: C, 51.67; H, 3.53; N, 2.47.
Iodo-substituted palladium catalyst C5 was obtained as a yellow powder in 80% yield. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.71 (s, 8H, Ar–H in BArF−), 7.52 (s, 4H, Ar–H in BArF−), 7.50–7.25 (m, 10H, Ar–H in backbone & aniline), 7.08–7.00 (m, 2H, Ar–H in backbone & aniline), 5.33 (s, 1H, CH), 5.28 (s, 1H, CH), 3.07 (m, 1H, CH(CH3)2), 2.86 (m, 1H, CH(CH3)2), 2.58 (m, 2H, CH(CH3)2), 1.73 (s, 3H, CH3CN), 1.44 (d, 3H, CH(CH3)2), 1.34–1.22 (m, 15H, CH(CH3)2), 1.14 (d, 3H, CH(CH3)2), 0.93 (d, 3H, CH(CH3)2), 0.69 (s, 3H, PdCH3). 13C NMR (100 MHz, CDCl3), δ (ppm): 175.41, 168.13 (CN), 162.59, 162.09, 161.60, 161.10 (Ar–C–B in BArF−), 141.11, 140.57, 139.83, 139.62, 139.09, 139.03, 138.57, 138.50, 137.93, 137.62, 137.26, 134.95 (Ar–C in backbone & aniline), 130.94, 129.76, 129.50, 129.22, 129.19, 128.87, 128.76, 128.59 (CF3 in BArF−), 126.69, 126.57, 126.05, 125.33, 1274.41, 124.28, 124.01, 123.35 (Ar–C in backbone & aniline), 121.15, 120.64 (Ar–C in BArF−), 117.62 (CH3CN), 96.62, 96.06 (Ar–C in backbone), 55.82, 55.03 (NC–CH), 29.74, 29.67, 28.90, 28.67 (CH(CH3)2), 24.25, 23.64, 23.56, 23.51, 23.04, 22.80, 22.67, 22.32 (CH(CH3)2), 9.64 (CH3CN), 1.92 (PdCH3). Anal. calcd for C75H60BF24I2N3Pd: C, 49.22; H, 3.30; N, 2.30; found: C, 49.49; H, 3.13; N, 2.15.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NSFC) (Project 51873234 and 21674130), the Basic and Applied Basic Foundation of Guangdong Province (2019B1515120063), and the Fundamental Research Funds for the Central Universities (19lgpy04).Notes and references
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS.
- A. Berkefeld and S. Mecking, Angew. Chem., Int. Ed., 2008, 47, 2538–2542 CrossRef CAS.
- Z. Chen and M. Brookhart, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS.
- L. Guo, S. Dai, X. Sui and C. Chen, ACS Catal., 2016, 6, 428–441 CrossRef CAS.
- T. Xie, K. B. McAuley, J. C. C. Hsu and D. W. Bacon, Ind. Eng. Chem. Res., 1994, 33, 449–479 CrossRef CAS.
- E. M. Ali, A. E. Abasaeed and S. M. Al-Zahrani, Ind. Eng. Chem. Res., 1998, 37, 3414–3423 CrossRef CAS.
- N. E. Mitchell and B. K. Long, Polym. Int., 2019, 68, 14–26 CrossRef CAS.
- D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS.
- D. J. Tempel, L. K. Johnson, R. L. Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686–6700 CrossRef CAS.
- L. H. Shultz, D. J. Tempel and M. Brookhart, J. Am. Chem. Soc., 2001, 123, 11539–11555 CrossRef CAS.
- D. H. Camacho and Z. Guan, Chem. Commun., 2010, 46, 7879–7893 RSC.
- Z. Wang, Q. Liu, G. A. Solan and W. Sun, Coord. Chem. Rev., 2017, 350, 68–83 CrossRef CAS.
- H. Hu, L. Zhang, H. Gao, F. Zhu and Q. Wu, Chem. – Eur. J., 2014, 20, 3225–3233 CrossRef CAS.
- H. Gao, H. Hu, F. Zhu and Q. Wu, Chem. Commun., 2012, 48, 3312–3314 RSC.
- H. Hu, D. Chen, H. Gao, L. Zhong and Q. Wu, Polym. Chem., 2016, 7, 529–537 RSC.
- S. Zai, H. Gao, Z. Huang, H. Hu, H. Wu and Q. Wu, ACS Catal., 2012, 2, 433–440 CrossRef CAS.
- H. Liao, L. Zhong, Z. Xiao, T. Zheng, H. Gao and Q. Wu, Chem. – Eur. J., 2016, 22, 14048–14055 CrossRef CAS.
- S. Zai, F. Liu, H. Gao, C. Li, G. Zhou, S. Cheng, L. Guo, L. Zhang, F. Zhu and Q. Wu, Chem. Commun., 2010, 46, 4321–4323 RSC.
- M. Schmid, R. Eberhardt, M. Klinga, M. Leskelä and B. Rieger, Organometallics, 2001, 20, 2321–2330 CrossRef CAS.
- D. Meinhard, M. Wegner, G. Kipiani, A. Hearley, P. Reuter, S. Fischer, O. Marti and B. Rieger, J. Am. Chem. Soc., 2007, 129, 9182–9191 CrossRef CAS.
- J. L. Rhinehart, L. A. Brown and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316–16319 CrossRef CAS.
- J. L. Rhinehart, N. E. Mitchell and B. K. Long, ACS Catal., 2014, 4, 2501–2504 CrossRef CAS.
- S. Dai, X. Sui and C. Chen, Angew. Chem., Int. Ed., 2015, 54, 9948–9953 CrossRef CAS.
- S. Du, S. Kong, Q. Shi, J. Mao, C. Guo, J. Yi, T. Liang and W. Sun, Organometallics, 2015, 34, 582–590 CrossRef CAS.
- S. Dai and C. Chen, Angew. Chem., Int. Ed., 2016, 55, 13281–13285 CrossRef CAS.
- L. A. Brown, W. C. Anderson, N. E. Mitchell, K. R. Gmernicki and B. K. Long, Polymer, 2018, 10, 41–49 Search PubMed.
- S. Dai and C. Chen, Macromolecules, 2018, 51, 6818–6824 CrossRef CAS.
- X. Chen, J. Gao, H. Liao, H. Gao and Q. Wu, Chin. J. Polym. Sci., 2018, 36, 176–184 CrossRef CAS.
- C. Tan, W. Pang and C. Chen, Chin. J. Polym. Sci., 2019, 37, 974–980 CrossRef CAS.
- S. Dai and C. Chen, Angew. Chem., Int. Ed., 2020, 59, 14884–14890 CrossRef CAS.
- S. Dai, S. Li, G. Xu and C. Chen, Macromolecules, 2020, 53, 2539–2546 CrossRef CAS.
- D. Zhang, E. T. Nadres, M. Brookhart and O. Daugulis, Organometallics, 2013, 32, 5136–5143 CrossRef CAS.
- T. Vaidya, K. Klimovica, A. M. LaPointe, I. Keresztes, E. B. Lobkovsky, O. Daugulis and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7213–7216 CrossRef CAS.
- K. E. Allen, J. Campos, O. Daugulis and M. Brookhart, ACS Catal., 2015, 5, 456–464 CrossRef CAS.
- K. S. O'Connor, A. Watts, T. Vaidya, A. M. LaPointe, M. A. Hillmyer and G. W. Coates, Macromolecules, 2016, 49, 6743–6751 CrossRef.
- O. Padilla-Velez, K. S. O'Connor, A. M. LaPointe, S. N. MacMillan and G. W. Coates, Chem. Commun., 2019, 55, 7607–7610 RSC.
- Y. Kanai, S. Foro and H. Plenio, Organometallics, 2019, 38, 544–551 CrossRef CAS.
- Y. Liao, Y. Zhang, L. Cui, H. Mu and Z. Jian, Organometallics, 2019, 38, 2075–2083 CrossRef CAS.
- X. Hu, Y. Zhang, Y. Zhang and Z. Jian, ChemCatChem, 2020, 12, 2497–2505 CrossRef CAS.
- Y. Zhang, C. Wang, S. Mecking and Z. Jian, Angew. Chem., Int. Ed., 2020, 59, 1–8 CrossRef.
- D. H. Camacho, E. V. Salo, J. W. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2004, 43, 1821–1825 CrossRef CAS.
- C. S. Popeney and Z. Guan, J. Am. Chem. Soc., 2009, 131, 12384–12393 CrossRef CAS.
- F. Liu, H. Hu, Y. Xu, L. Guo, S. Zai, K. Song, H. Gao, L. Zhang, F. Zhu and Q. Wu, Macromolecules, 2009, 42, 7789–7796 CrossRef CAS.
- J. Liu, D. Chen, H. Wu, Z. Xiao, H. Gao, F. Zhu and Q. Wu, Macromolecules, 2014, 47, 3325–3331 CrossRef CAS.
- L. Guo, H. Gao, Q. Guan, H. Hu, J. Deng, J. Liu, F. Liu and Q. Wu, Organometallics, 2012, 31, 6054–6062 CrossRef CAS.
- J. Gao, L. Zhang, L. Zhong, C. Du, H. Liao, H. Gao and Q. Wu, Polymer, 2019, 164, 26–32 CrossRef CAS.
- R. Gao, Z. Guo, J. Zhou, Y. Li, D. Liu and X. Zhang, J. Catal., 2020, 385, 103–106 CrossRef CAS.
- L. Zhong, G. Li, G. Liang, H. Gao and Q. Wu, Macromolecules, 2017, 50, 2675–2682 CrossRef CAS.
- S. Zhong, Y. Tan, L. Zhong, J. Gao, H. Liao, L. Jiang, H. Gao and Q. Wu, Macromolecules, 2017, 50, 5661–5669 CrossRef CAS.
- C. Du, L. Zhong, J. Gao, S. Zhong, H. Liao, H. Gao and Q. Wu, Polym. Chem., 2019, 10, 2029–2038 RSC.
- Z. Xiao, H. Zheng, C. Du, L. Zhong, H. Liao, J. Gao, H. Gao and Q. Wu, Macromolecules, 2018, 51, 9110–9121 CrossRef CAS.
- G. Liao, Z. Xiao, X. Chen, C. Du, L. Zhong, C. Cheung and H. Gao, Macromolecules, 2020, 53, 256–266 CrossRef CAS.
- B. K. Long, J. M. Eagan, M. Mulzer and G. W. Coates, Angew. Chem., Int. Ed., 2016, 55, 7106–7110 CrossRef CAS.
- L. Zhong, H. Zheng, C. Du, W. Du, G. Liao, C. Cheung and H. Gao, J. Catal., 2020, 384, 208–217 CrossRef CAS.
- F. Wang and C. Chen, Polym. Chem., 2019, 10, 2354–2369 RSC.
- L. Guo, S. Dai and C. Chen, Polymer, 2016, 8, 37–46 Search PubMed.
- C. S. Popeney, C. M. Levins and Z. Guan, Organometallics, 2011, 30, 2432–2452 CrossRef CAS.
- C. S. Popeney and Z. Guan, Organometallics, 2005, 24, 1145–1155 CrossRef CAS.
- W. Zou and C. Chen, Organometallics, 2016, 35, 1794–1801 CrossRef CAS.
- F. He, D. Wang, B. Jiang, Z. Zhang, Z. Cheng, Z. Fu, Q. Zhang and Z. Fan, Inorg. Chim. Acta, 2019, 486, 704–710 CrossRef CAS.
- Q. Muhammad, C. Tan and C. Chen, Sci. Bull., 2020, 65, 300–307 CrossRef CAS.
- C. Chen, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS.
- C. S. Popeney, A. L. Rheingold and Z. Guan, Organometallics, 2009, 28, 4452–4463 CrossRef CAS.
- D. H. Leung, J. W. Ziller and Z. Guan, J. Am. Chem. Soc., 2008, 130, 7538–7539 CrossRef CAS.
- M. Mitani, J. Mohri, Y. Yoshida, J. Saito, S. Ishii, K. Tsuru, S. Matsui, R. Furuyama, T. Nakano, H. Tanaka, S. Kojoh, T. Matsugi, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2002, 124, 3327–3336 CrossRef CAS.
- J. Tian and G. W. Coates, Angew. Chem., Int. Ed., 2000, 39, 3626–3629 CrossRef CAS.
- M. Delferro, J. P. McInnis and T. J. Marks, Organometallics, 2010, 29, 5040–5049 CrossRef CAS.
- M. P. Weberski, C. Chen, M. Delferro, C. Zuccaccia, A. Macchioni and T. J. Marks, Organometallics, 2012, 31, 3773–3789 CrossRef CAS.
- J. Wang, E. Yao, Z. Chen and Y. Ma, Macromolecules, 2015, 48, 5504–5510 CrossRef CAS.
- D. Zhang and C. Chen, Angew. Chem., Int. Ed., 2017, 56, 14672–14676 CrossRef CAS.
- L. Zhong, C. Du, G. Liao, H. Liao, H. Zheng, Q. Wu and H. Gao, J. Catal., 2019, 375, 113–123 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS.
- R. van Asselt, C. J. Elsevier, W. J. J. Smeets, A. L. Spek and R. Benedix, Recl. Trav. Chim. Pays-Bas, 1994, 113, 88–98 CrossRef CAS.
- L. Guo, K. Lian, W. Kong, S. Xu, G. Jiang and S. Dai, Organometallics, 2018, 37, 2442–2449 CrossRef CAS.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- M. Kang, A. Sen, L. Zakharov and A. Rheingold, J. Am. Chem. Soc., 2002, 124, 12080–12081 CrossRef CAS.
- M. Stȩpień, L. Latos-Grażyński, T. Lash and L. Szterenberg, Inorg. Chem., 2001, 40, 6892–6900 CrossRef.
- M. Szabo, R. Jordan, A. Michalak, W. Piers, T. Weiss, S. Yang and T. Ziegler, Organometallics, 2004, 23, 5565–5572 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of palladium complexes, crystallographic data of palladium complexes, and 1H and 13C NMR spectra of polymers. The crystal data of palladium complexes with deposition numbers 2021209–2021215. CCDC 2021209–2021215. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cy01617a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |