
Predicting the origin of selectivity in NHC-catalyzed ring opening of formylcyclopropane: a theoretical investigation†
Yang
Wang
 a,
Yan
Qiao
b,
Yu
Lan
*c and
Donghui
Wei
*c
a,
Yan
Qiao
b,
Yu
Lan
*c and
Donghui
Wei
*c
aDepartment of Material and Chemical Engineering, Zhengzhou University of Light Industry, 136 Science Avenue, Zhengzhou, Henan Province 450002, P.R. China. E-mail: wangyang@zzuli.edu.cn
bDepartment of Pathophysiology, School of Basic Medical Sciences, Zhengzhou University, 100 Science Avenue, Zhengzhou, Henan Province 450001, P.R. China
cCollege of Chemistry, Zhengzhou University, 100 Science Avenue, Zhengzhou, Henan Province 450001, P.R. China. E-mail: lanyu@cqu.edu.cn; donghuiwei@zzu.edu.cn
First published on 14th November 2020
Abstract
Using density functional theory, we investigated the origin of selectivity in the N-heterocyclic carbene (NHC)-catalyzed transformation of formylcyclopropane with an alkylidene oxindole. Our proposed mechanism includes the formation of a Breslow intermediate, ring opening of cyclopropane, formation of an enolate intermediate, formal [4 + 2] cycloaddition and disassociation of the catalyst. The transformation from an enolate intermediate into an alkylidene oxindole determines the chemoselectivity and stereoselectivity. Local reactivity analysis indicates that the chemoselectivity in the controlled transition state of the [4 + 2] cycloaddition is intrinsically favored by the higher reactivity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C moiety of the alkylidene oxindole. Electron localization function analysis indicates that the NHC alleviates the electronic density between Cα and Cγ, and thus facilitates opening of the cyclopropane ring. This knowledge of NHC organocatalysis helps predict the chemoselectivity of divergent reactions involving cyclopropane rings.
C moiety of the alkylidene oxindole. Electron localization function analysis indicates that the NHC alleviates the electronic density between Cα and Cγ, and thus facilitates opening of the cyclopropane ring. This knowledge of NHC organocatalysis helps predict the chemoselectivity of divergent reactions involving cyclopropane rings.
Introduction
Donor–acceptor cyclopropanes (DACs), substituted with electron-withdrawing and electron-donating groups, are a unique class of reactive molecules.1 DACs readily undergo ring opening because of this functional group synergy and thus generate an active intermediate that readily undergoes a wide range of reactions.2Researchers commonly use Lewis acids to promote the ring opening of DACs, and apply DACs to enantioselective reactions through incorporation of chiral ligands.3 In general, the DAC acts as a three-carbon synthon, which undergoes formal [3 + n] (n = 2, 3, and 4) annulation reactions with numerous reactive partners under Lewis acid catalysis.4 Related density functional theory (DFT) investigations on the Lewis acid-catalyzed ring opening of DACs indicated that GaCl3 promotes ring opening by changing the charge population (Scheme 1a).5 A Brønsted acid is used to promote the ring-opening of DACs by increasing the polarization of the C–C bond (Scheme 1b).6
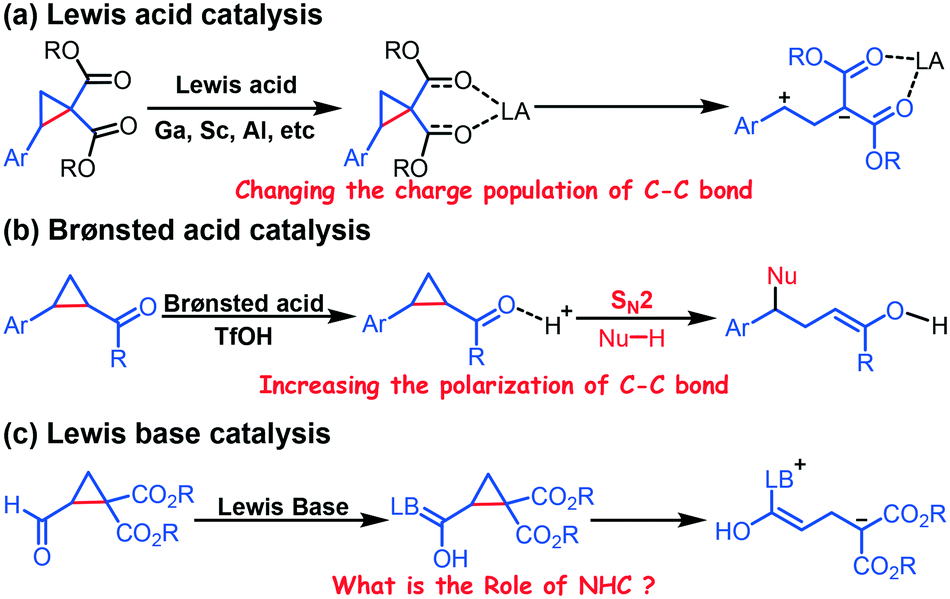 | ||
| Scheme 1 Representative strategies for the ring-opening of DACs. | ||
Recently, N-heterocyclic carbenes (NHCs), which have been frequently employed as an organocatalyst in many transformations,7 have also been successfully applied to promote the ring opening of formylcyclopropane (Scheme 1c).8 However, the specific role of NHCs is unknown. Therefore, elucidating the role of NHCs is foundational to optimizing such ring-opening/transformation reactions.
Vicario et al. reported an NHC-catalyzed chemoselective and stereoselective ring opening and transformation reaction of formylcyclopropane.8a This protocol provides a potential route for constructing diverse functional molecules (Scheme 2). We hypothesize that absorption of the NHC considerably affects the electronic density of the C–C bond in cyclopropane. Vicario et al. produced an ε-lactone product with excellent stereoselectivity (99% ee, Scheme 2); they did not observe the γ-lactone product obtained as the main species in other reports.9 Thus, we further hypothesize that the chemoselectivity depends on the reactivity of the interacting partners. To test our hypothesis, we used DFT, which has been commonly used to elucidate organocatalytic10 and organometallic11 reaction mechanisms, to simulate the ring-opening and transformation reaction of formylcyclopropane R1 and alkylidene oxindole R2 reported by Vicario and co-workers. Our goal was to predict the origin of the chemoselectivity and stereoselectivity, and elucidate the role of NHCs, in the ring-opening and transformation reactions of formylcyclopropane.
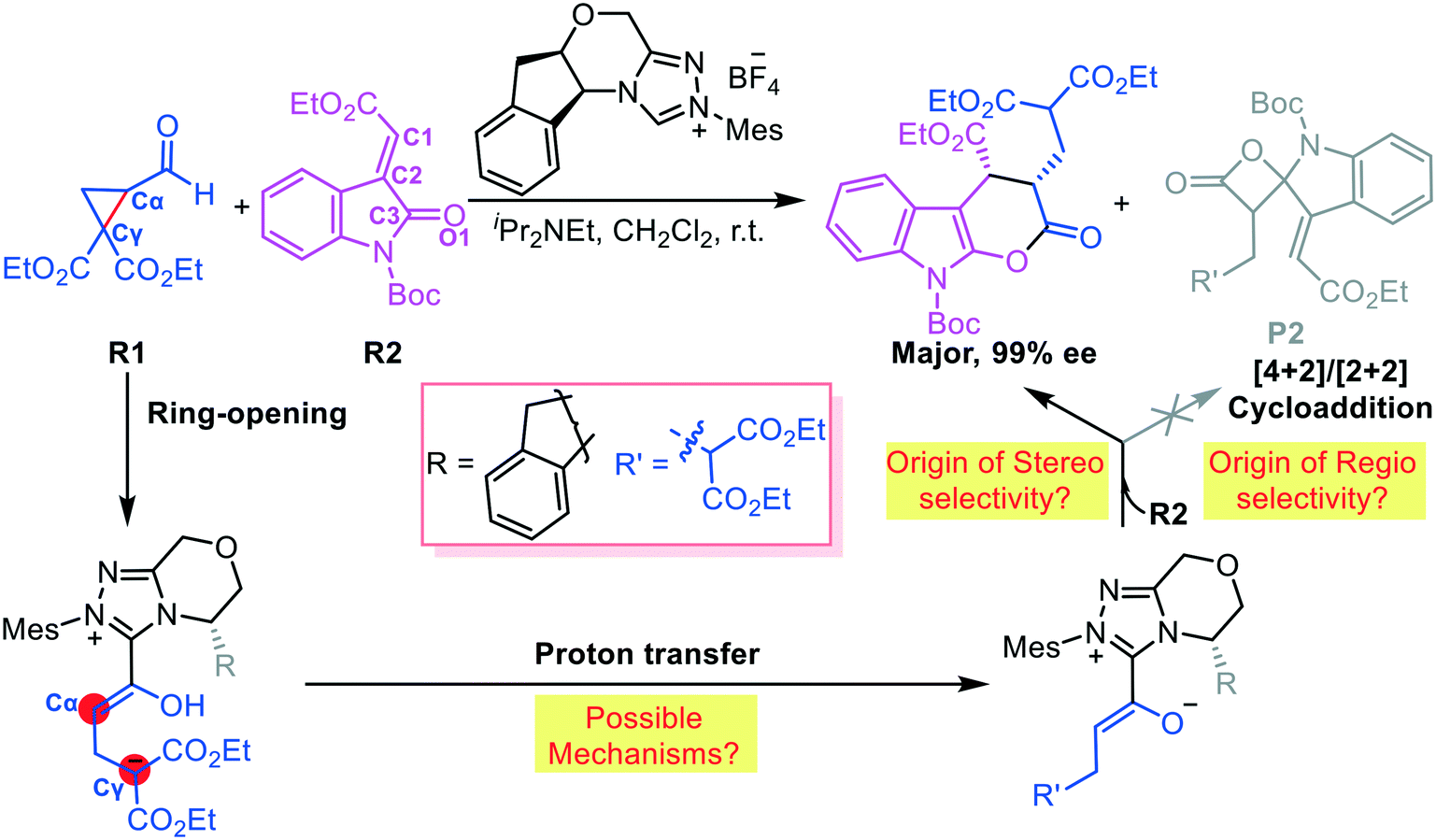 | ||
| Scheme 2 Possible activation and transformation pathways of cyclopropane under NHC organocatalysis. | ||
Results and discussion
Reaction mechanism
Fig. 1 shows the energy profile of the entire NHC-catalyzed ring-opening/transformation reaction of formylcyclopropane with an alkylidene oxindole. As shown in Fig. 1, nucleophilic addition of the NHC to the carbonyl atom of the formyl group inititates the reaction. This process costs an energy of 10.6 kcal mol−1 (TS1) to deliver intermediate M1. Then, a [1,2]-proton shift generates the Breslow-type intermediate M2. The energy barrier involved in the [1,2]-proton shift process is 20.7 kcal mol−1, associated with transition state TS2, which was considerably lowered by implicating the in situ generated Brønsted acid.12 Subsequently, the Breslow-type intermediate M2 quickly transforms into intermediate M3 via transition state TS3 through C–C bond cleavage. Then, γ-protonation gives enolate intermediate M4 via transition state TS4 through a [1,5]-proton shift, which costs an energy of 9.2 kcal mol−1. Subsequently, ε-lactonization ([4 + 2] cycloaddition) between the formed enolate intermediate M4 and alkylidene oxindole R2 takes place via transition state TS5. We considered and calculated four possible diastereoselective pathways, which led to four diastereoisomers (the stereochemistry is provided in Scheme S1 in the ESI†). The SR-configured products are the most energetically favored, with an energy barrier of 6.0 kcal mol−1 (the energy profiles of the other three pathways are provided in Fig. S1 in the ESI†). The last step of the catalytic cycle is desorption for catalyst turnover.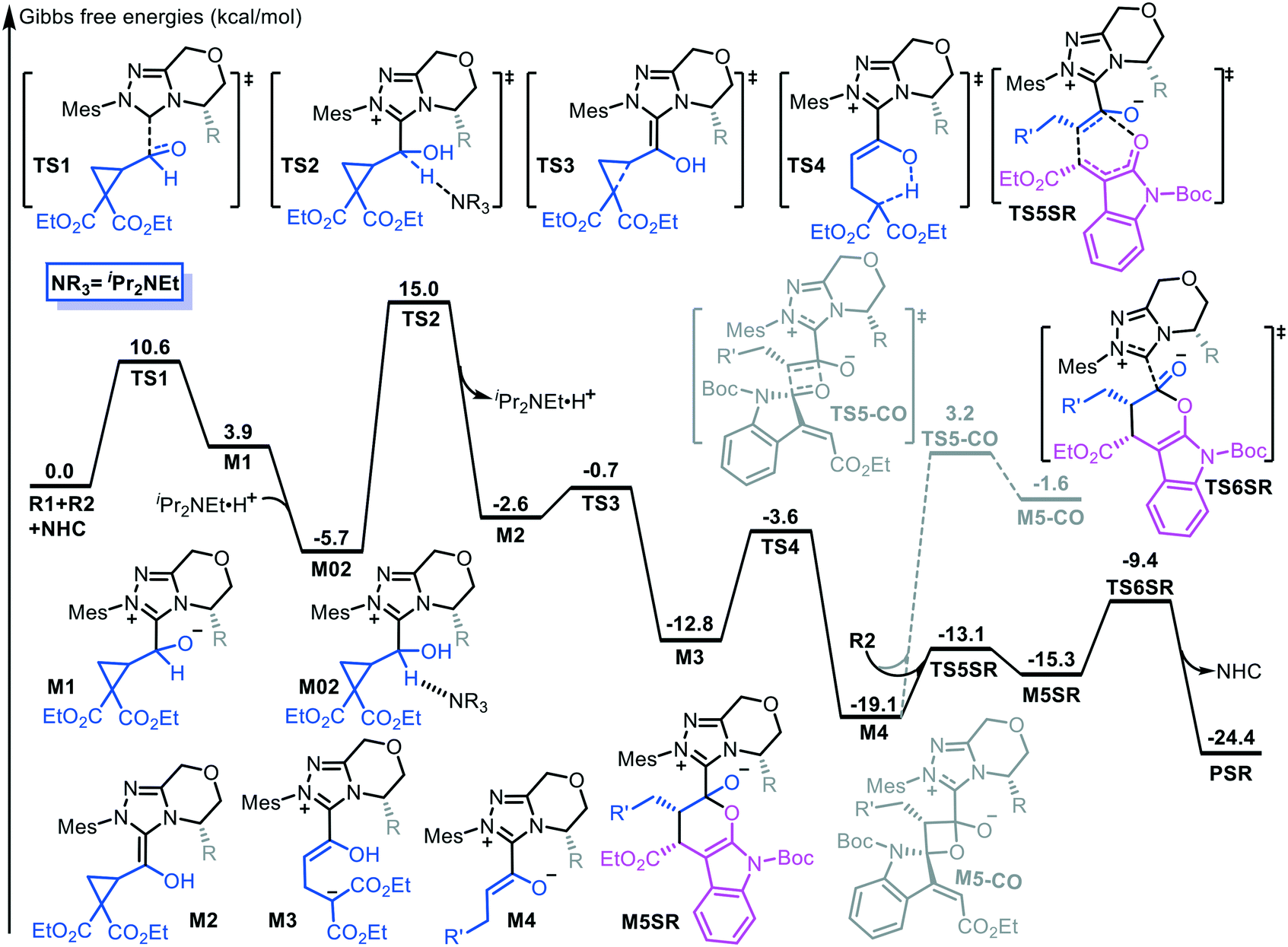 | ||
| Fig. 1 Energy profile of the NHC-catalyzed DAC ring-opening/[4 + 2] cycloaddition reaction. | ||
Alternatively, γ-lactonization, namely, the formal [2 + 2] cycloaddition, can occur between intermediate M4 and the CO moiety of R2 to form the γ-lactone product. The energy barrier of this process is 22.3 kcal mol−1 (TS5-CO, Fig. 1), which is much higher than that of ε-lactonization. Moreover, the Gibbs free energy of the [2 + 2] cycloproduct M5-CO is −1.6 kcal mol−1 and this is 13.7 kcal mol−1 higher than that of the [4 + 2] cycloproduct M5SR. These findings indicate that the [4 + 2] cycloaddition reaction is energetically favorable in terms of both kinetics and thermodynamics.
Origin of selectivities
We explored the mechanism of the NHC-catalyzed ring-opening/[4 + 2] cycloaddition reaction of formylcyclopropane with an alkylidene oxindole. Based on the computational results, we propose that the reaction between the enolate and alkylidene oxindole is the chemoselectivity- and stereoselectivity-determining step. The ε-lactonization reaction occurs prior to γ-lactonization and the SR-configured ε-lactone product formed predominately in ε-lactonization. As for the two reacting partners, the lower energy barrier of the corresponding transition state was mainly due to the higher nucleophilicity or electrophilicity.13 Therefore, we considered that the chemoselectivity could be modulated by the reactivity of the CC–CO and CO moieties; a higher electrophilicity or nucleophilicity of the reacting center would lead to a lower energy barrier. Thus, we predicted the chemoselectivity by comparing the Parr functions of the nucleophilicity (Pk−) and electrophilicity (Pk+) of the two interactive partners (M4 and R2).
As shown in Table 1, the nucleophilic and electrophilic values of Cα are 0.64 and 0.13, respectively; the corresponding values of Cγ are 0.02 and 0.01, respectively. Thus, Cα is nucleophilic and this atom has a higher nucleophilicity than Cγ. The computational results also indicate that C1 and C3 in R2 are electrophilic. The reactivity of C1 (Pk+ = 0.30) is higher than that of the C3 atom (Pk+ = 0.09), revealing that the [4 + 2] cycloaddition occurs prior to the [2 + 2] cycloaddition. Therefore, we conclude that the greater electrophilicity and nucleophilicity lead to a lower energy barrier through local electrophilicity and nucleophilicity analysis, which agrees with our previous studies.14
| R2 | M4 | |||||
|---|---|---|---|---|---|---|
| C1 | C2 | C3 | O1 | Cα | Cγ | |
| P k − | 0.14 | −0.07 | −0.04 | 0.09 | 0.64 | 0.02 |
| P k + | 0.30 | 0.15 | 0.09 | 0.12 | 0.13 | 0.01 |
In addition, some theoretical studies report that the electrostatic solvent–solute interaction is also responsible for the chemoselectivity.15 We tested whether the chemoselectivity is related to the intrinsic polarity of the corresponding transition state. Fig. 2 shows the electrostatic potential surfaces (EPSs) of the competing [4 + 2] and [2 + 2] cycloaddition transition states TS5SR and TS5-CO. In Fig. 2, blue represents the positively charged area, whereas red represents the negatively charged area. We found considerable charge separation in TS5-CO. Therefore, TS5-CO exhibits zwitterionic characteristics, which correspond to a larger dipole moment (μ = 13.99 D) and result in instability of the [2 + 2] cycloaddition transition state. In contrast, the [4 + 2] cycloaddition transition state TS5SR has a fairly neutral surface, which corresponds to a smaller dipole moment (μ = 2.38 D) and makes the [4 + 2] cycloaddition transition state stable. In order to clarify the effect of solvent polarity, we have additionally performed DFT calculations to re-compute the chemoselective [4 + 2] and [2 + 2] cyclization transition states in different solvents, including DMSO, cyclohexene, MeCN, toluene, and THF (the energy barriers in different solvents are summarized in Table S1 in the ESI†). The calculated results indicate that the main product always is the [4 + 2] cyclization product, so we can conclude that the solvent and its polarity should not be the main factor for controlling the chemoselectivity of the cyclization process.
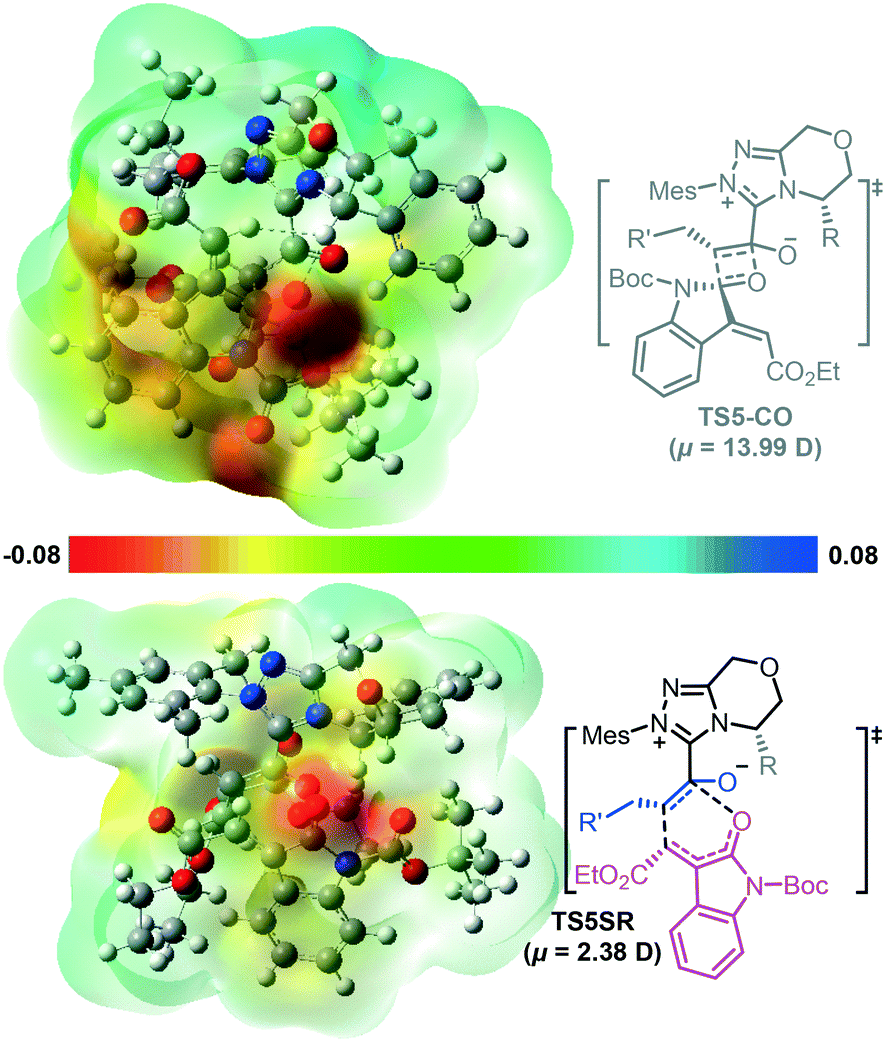 | ||
| Fig. 2 Electrostatic potential surfaces of the competing [4 + 2] and [2 + 2] cycloaddition transition states in dichloromethane with the NHC catalyst. | ||
Next, we turn to disclosing the origin of the stereoselectivity. Non-covalent interaction (NCI) analysis, which has been widely used to qualitatively analyse the types of weak interactions between two interactive fragments, was performed to address this point. As can be seen from Fig. 3 (the other two NCI pictures of transition states TS5RR and TS5SS are shown in Fig. S2 in the ESI†), C–H⋯π weak interactions existed in both transition structures, with that in TS5SR found to be stronger. The C–H⋯O interaction made an almost equal contribution in both transition states. Therefore, the C–H⋯π interaction was key to determining the stereoselectivity. Overall, NCI analysis implied that the weak interactions existing in TS5SR were more predominant, with the C–H⋯π interaction contributing significantly to stabilizing the structure.
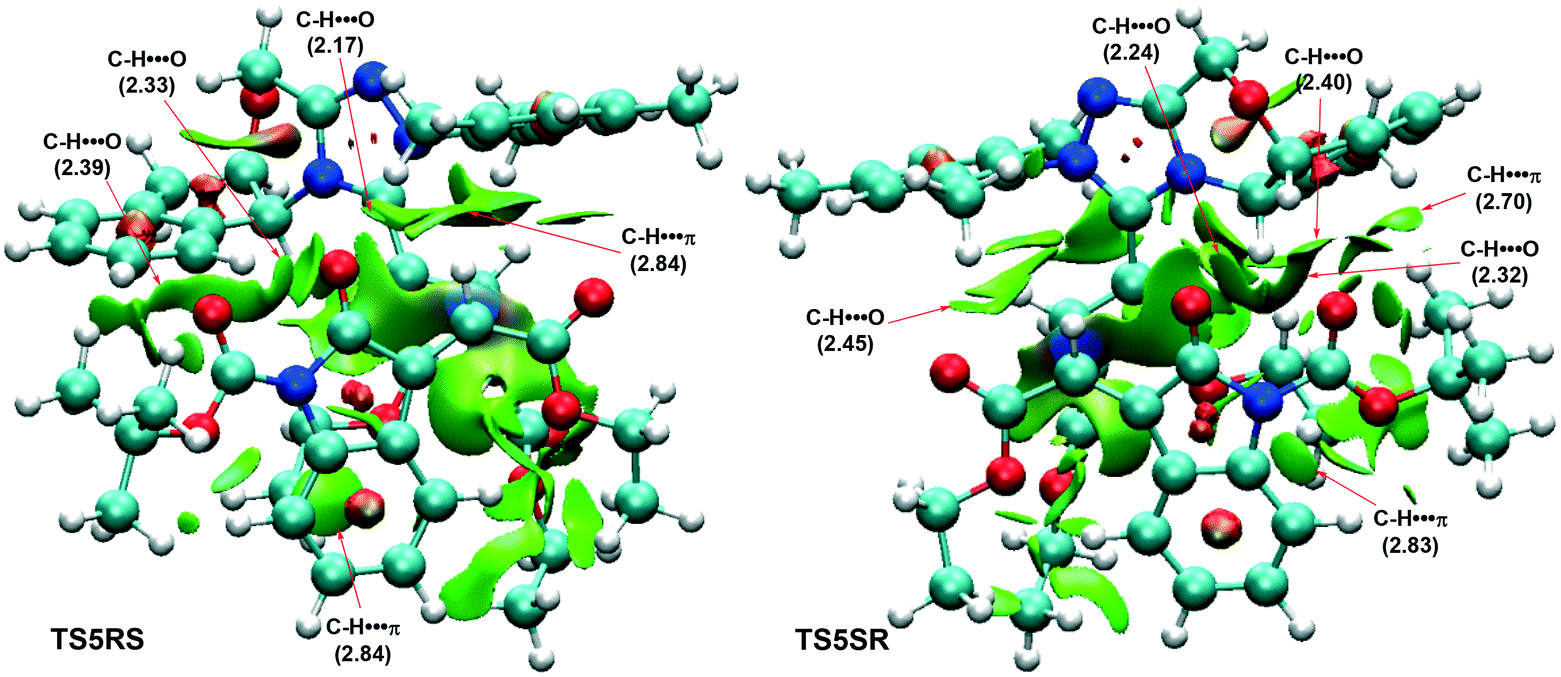 | ||
| Fig. 3 NCI analysis of stereoselective transition states TS5RS and TS5SR (distance in Å). | ||
Role of the catalyst
The literature indicates that NHC catalysts play a crucial role in opening three-membered rings. To understand the role of the NHC, we compared the electron density (∇2ρ), bond length (BL) and bond order (BD) between Cα and Cγ using electron localization function (ELF) analysis. The values of the electron density and bond order in formylcyclopropane R1 are 0.244 and 0.88, respectively; in M1, these are 0.228 and 0.89, respectively (Fig. 4). This is indicative of a strong σ-bond between Cα and Cγ. In Breslow intermediate M2, the values of the electron density and bond order are 0.193 and 0.77, respectively. This indicates that the Cα–Cγ σ-bond is weakened along with the Breslow intermediate formation and thus facilitates cleavage of the Cα–Cγ σ-bond. The electronic density in TS3 decreases to 0.009, indicating the C–C bond cleavage. Therefore, the NHC promotes the opening of the three membered-ring by alleviating the electron density between Cα and Cγ.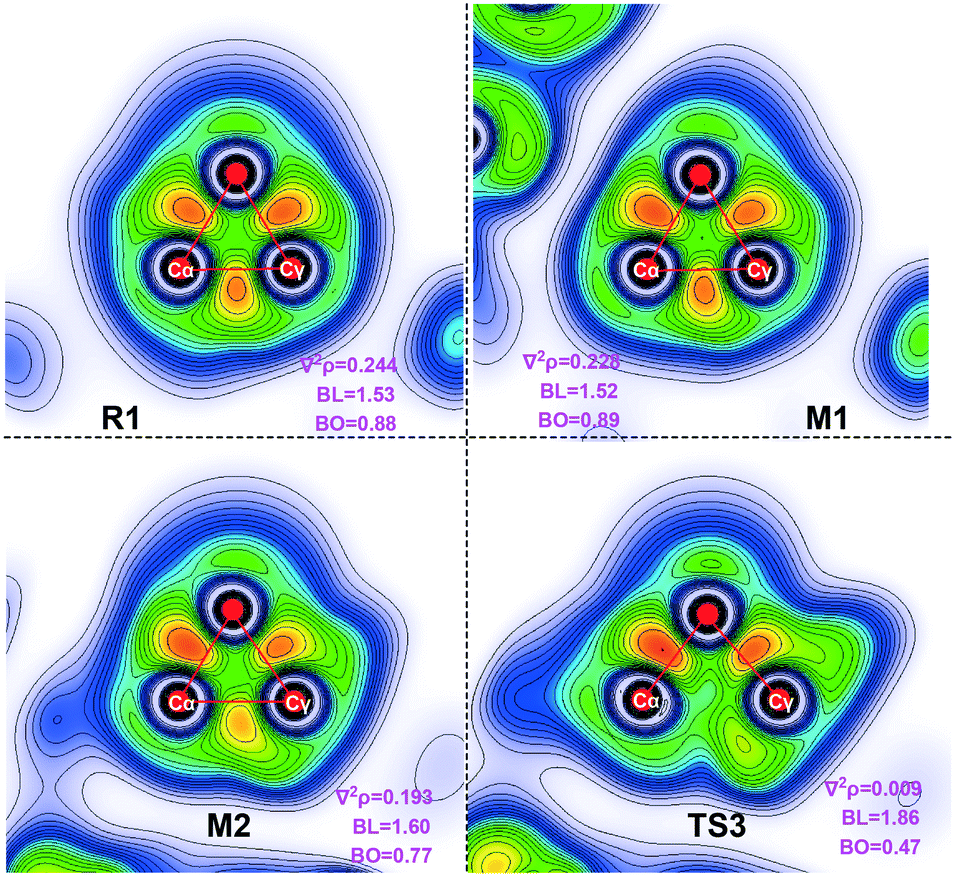 | ||
| Fig. 4 ELF analysis graphs derived from the Laplacian electron density function (∇2ρ) in a selected plane. | ||
Conclusions
We used DFT calculations to investigate the chemoselective and stereoselective mechanism of the NHC-catalyzed ring-opening/cycloaddition reaction of formylcyclopropane with an alkylidene oxindole. The reaction between the enolate intermediate and alkylidene oxindole is the chemoselectivity and stereoselectivity-determining step, and [4 + 2] cycloaddition occurs prior to [2 + 2] cycloaddition. The [2 + 2] cycloaddition pathway is unfavorable mainly because of the low reactivity of the CO moiety, as also indicated by the electrostatic solvent–solute interaction. Our NCI analysis indicates that the stereoselectivity is mainly controlled by C–H⋯π and C–H⋯O interactions. ELF analysis indicates that the NHC catalyst promotes opening of the three membered-ring by alleviating the electronic density between Cα and Cγ, which would provide a new method to predict the ring-opening position of cyclopropane rings.
Experimental
Computational details
The Gaussian 09 program16 was employed to perform the theoretical study. The geometry optimization and frequency calculations were carried out by using the M06-2X17 functional with the 6-31G(d,p) basis set18 and accounting for the CH2Cl2 (DCM, ε = 8.93) solvent effect by employing the IEF-PCM19 solvation model (M06-2X/6-31G(d,p)//IEF-PCMDCM, L1). Furthermore, single-point energy calculations were performed to refine the energies at the M06-2X-D3/6-311++G(2df,2pd)//IEF-PCMDCM level (L2). The free energies discussed in the text are obtained by the addition of Gibbs free energy correction calculated at L1 to the single-point energy calculated at L2. Moreover, the AIM and NCI analyses were plotted using Multiwfn.20Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank the financial support from the National Natural Science Foundation of China (No. 21773214, 21772020, 21822303, and 21903072), the Outstanding Young Talent Research Fund of Zhengzhou University (No. 1521316001), the Startup Fund of Zhengzhou University of Light Industry (No. 2017BSJJ036), and the Henan Province Supercomputing Center. We also thank Michael Scott Long, from Liwen Bianji, Edanz Editing China (http://www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.Notes and references
- For selected reviews: (a) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051–3060 RSC; (b) H. K. Grover, M. R. Emmett and M. A. Kerr, Org. Biomol. Chem., 2015, 13, 655–671 RSC; (c) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804–818 RSC; (d) P. Singh, R. K. Varshnaya, R. Dey and P. Banerjee, Adv. Synth. Catal., 2020, 362, 1447–1484 CrossRef CAS; (e) O. A. Ivanova and I. V. Trushkov, Chem. Rec., 2019, 19, 2189–2208 CrossRef CAS.
- (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS; (b) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS; (c) E. M. Budynina, K. L. Ivanov, I. D. Sorokin and M. Y. Melnikov, Synthesis, 2017, 49, 3035–3068 CrossRef CAS; (d) A. Kreft, P. G. Jones and D. B. Werz, Org. Lett., 2018, 20, 2059–2062 CrossRef CAS; (e) O. A. Ivanova, A. O. Chagarovskiy, A. N. Shumsky, V. D. Krasnobrov, I. I. Levina and I. V. Trushkov, J. Org. Chem., 2018, 83, 543–560 CrossRef CAS; (f) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00109.
- For selected examples: (a) Q.-K. Kang, L. Wang, Q.-J. Liu, J.-F. Li and Y. Tang, J. Am. Chem. Soc., 2015, 137, 14594–14597 CrossRef CAS; (b) Y. Xia, L. L. Lin, F. Z. Chang, X. Fu, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 13748–13752 CrossRef CAS; (c) S. M. Wales, M. M. Morgan and J. S. Johnson, Org. Lett., 2013, 15, 2558–2561 CrossRef CAS; (d) P.-J. Yang, L. Qi, Z. Liu, G. S. Yang and Z. Chai, J. Am. Chem. Soc., 2018, 140, 17211–17217 CrossRef CAS; (e) F. Z. Chang, L. L. Lin, Y. Xia, H. Zhang, S. X. Dong, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 2608–2612 Search PubMed.
- For selected examples: (a) A. U. Augustin, J. L. Merz, P. G. Jones, G. Mloston and D. B. Werz, Org. Lett., 2020, 22, 9405–9409 Search PubMed; (b) L. K. B. Garve, A. Kreft, P. G. Jones and D. B. Werz, J. Org. Chem., 2017, 82, 9235–9242 CrossRef CAS; (c) B. Q. Li, Z.-W. Qiu, A.-J. Ma, J.-B. Peng, N. Feng, J.-Y. Du, H.-P. Pan, X.-Z. Zhang and X.-T. Xu, Org. Lett., 2020, 22, 1903–1907 CrossRef CAS; (d) M. A. Zotova, R. A. Novikov, E. V. Shulishov and Y. V. Tomilov, J. Org. Chem., 2018, 83, 8193–8207 CrossRef CAS; (e) A. O. Chagarovskiy, V. S. Vasin, V. V. Kuznetsov, O. A. Ivanova, V. B. Rybakov, A. N. Shumsky, N. N. Makhova and I. V. Trushkov, Angew. Chem., Int. Ed., 2018, 57, 10338–10342 CrossRef CAS; (f) R. Dey and P. Banerjee, Org. Lett., 2017, 19, 304–307 CrossRef CAS; (g) A. U. Augustin, M. Sensse, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2017, 56, 14293–14296 CrossRef CAS.
- P.-C. Tu, L. Zhou, A. M. Kirillov, R. Fang and L. Z. Yang, Org. Chem. Front., 2018, 5, 1702–1712 RSC.
- (a) B. Cui, J. Ren and Z. W. Wang, J. Org. Chem., 2014, 79, 790–796 CrossRef CAS; (b) E. Richmond, V. D. Vukovic and J. Moran, Org. Lett., 2018, 20, 574–577 CrossRef CAS; (c) L. C. Irwin, C. R. Renwick and M. A. Kerr, J. Org. Chem., 2018, 83, 6235–6242 CrossRef CAS; (d) Y. Z. Zhou, R.-C. Xue, Y. Q. Feng and L. Zhang, Asian J. Org. Chem., 2020, 9, 311–316 CrossRef CAS.
- For selected examples: (a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS; (b) X.-Y. Chen, S. Li, F. Vetica, M. Kumar and D. Enders, iScience, 2018, 2, 1–26 CrossRef CAS; (c) Y. Wang, D. H. Wei and W. J. Zhang, ChemCatChem, 2018, 10, 338–360 CrossRef CAS; (d) X.-Y. Chen, Q. Liu, P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 3862–3873 CrossRef CAS; (e) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC.
- (a) L. Prieto, E. Sanchez-Diez, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, Adv. Synth. Catal., 2018, 359, 1678–1683 CrossRef; (b) S. S. Sohn and J. W. Bode, Angew. Chem., Int. Ed., 2006, 45, 6021–6024 CrossRef CAS; (c) L. Candish and D. W. Lupton, Chem. Sci., 2012, 3, 380–383 RSC; (d) D. Du and Z. W. Wang, Eur. J. Org. Chem., 2008, 2008, 4949–4954 CrossRef; (e) L. X. Li, D. Du, J. Ren and Z. W. Wang, Eur. J. Org. Chem., 2011, 2011, 614–618 CrossRef; (f) J. W. Bode and S. S. Sohn, J. Am. Chem. Soc., 2007, 129, 13798–13799 CrossRef CAS; (g) D. Du, L. X. Li and Z. W. Wang, J. Org. Chem., 2009, 74, 4379–4382 CrossRef CAS; (h) G.-Q. Li, L.-X. Dai and S.-L. You, Org. Lett., 2009, 11, 1623–1625 CrossRef CAS; (i) H. Lv, J. M. Mo, X. Q. Fang and Y. R. Chi, Org. Lett., 2011, 13, 5366–5369 CrossRef CAS.
- (a) A. Ametovski and D. W. Lupton, Org. Lett., 2019, 21, 1212–1215 CrossRef CAS; (b) S. Mondal, S. Mukherjee, T. K. Das, R. Gonnade and A. T. Biju, ACS Catal., 2017, 7, 3995–3999 CrossRef CAS.
- (a) P.-C. Tu, L. Zhou, A. M. Kirillov, R. Fang and L. Z. Yang, Org. Chem. Front., 2018, 5, 1356–1365 RSC; (b) T. Liu, S. M. Han, Y. P. Li and S. W. Bi, J. Org. Chem., 2016, 81, 9775–9784 CrossRef CAS; (c) P. Verma, P. A. Patni and R. B. Sunoj, J. Org. Chem., 2011, 76, 5606–5613 CrossRef CAS; (d) X. X. Wei, R. Fang and L. Z. Yang, Catal. Sci. Technol., 2015, 5, 3352–3362 RSC; (e) Y. Wang and D. H. Wei, Mol. Catal., 2020, 489, 110944 CrossRef CAS.
- (a) R. Fang, L. Yang, L. Zhou, A. M. Kirillov and L. Z. Yang, Org. Lett., 2020, 22, 4043–4048 CrossRef CAS; (b) Y. Liu, S. D. Feng and X. G. Bao, ChemCatChem, 2018, 10, 2817–2825 CrossRef CAS; (c) J. Zhang, C. H. Shan, K. Lv, L. Zhu, Y. Y. Li, T. Liu and Y. Lan, ChemCatChem, 2020, 11, 1228–1237 CrossRef; (d) T. Zhang, X. J. Wu and J. Li, ChemCatChem, 2020, 12, 1385–1393 CrossRef CAS; (e) B. Lian, L. Zhang and D.-C. Fang, Org. Chem. Front., 2019, 6, 2600–2606 RSC; (f) X. Y. Lv, X. T. Zhang, R. J. Sa, F. Huang and G. Lu, Org. Chem. Front., 2019, 6, 3629–3635 RSC.
- (a) X. H. Wang, Y. Wang, J. S. Song and D. H. Wei, Org. Chem. Front., 2020, 7, 1113–1121 RSC; (b) Q. L. Liu, Y. Y. Wang, X. Li, L.-B. Qu, Y. Lan and D. H. Wei, Mol. Catal., 2020, 484, 110723 CrossRef CAS; (c) Q. L. Liu, L. Sun, S.-J. Li, X. Li, L.-B. Qu, Y. Lan and D. H. Wei, Chem. – Asian J., 2019, 14, 2000–2007 CrossRef CAS; (d) Y. Wang, L.-B. Qu, D. H. Wei and Y. Lan, J. Org. Chem., 2020, 85, 8437–8446 CrossRef CAS; (e) X. Li, S.-J. Li, L.-B. Qu, Z. J. Li, Y. R. Chi, D. H. Wei and Y. Lan, Chem. Sci., 2020, 11, 7214–7225 RSC; (f) Y. Wang, L.-B. Qu and D. H. Wei, Chem. – Asian J., 2019, 14, 293–300 CAS; (g) J. X. Gao and Y. Wang, Org. Biomol. Chem., 2019, 17, 7442–7447 RSC; (h) Y. Wang and Y. Lan, Chin. Chem. Lett., 2020, 31, 736–738 CrossRef CAS; (i) Y. Wang, L.-B. Qu, Y. Lan and D. H. Wei, ChemCatChem, 2019, 11, 2919–2925 CrossRef CAS.
- (a) Y. Wang, Q.-Y. Wu, T.-H. Lai, K.-J. Zheng, L.-B. Qu and D. H. Wei, Catal. Sci. Technol., 2019, 9, 465–476 RSC; (b) Y. Wang, L.-B. Qu, Y. Lan and D. H. Wei, ChemCatChem, 2020, 12, 1068–1074 CrossRef CAS; (c) X. Li, R. H. Duan, Y. Y. Wang, L.-B. Qu, Z. J. Li and D. H. Wei, J. Org. Chem., 2019, 84, 6117–6125 CrossRef CAS.
- (a) L. J. Zheng, Y. Wang, D. H. Wei and Y. Qiao, Chem. – Asian J., 2016, 11, 3046–3054 CrossRef CAS; (b) X. Li, M. S. Tang, Y. Y. Wang, Y. Wang, Z. J. Li, L.-B. Qu and D. H. Wei, Chem. – Asian J., 2018, 13, 1710–1718 CrossRef CAS; (c) X. Li, Y. Y. Wang, Y. Wang, M. S. Tang, L.-B. Qu, Z. J. Li and D. H. Wei, J. Org. Chem., 2018, 83, 8543–8555 CrossRef CAS.
- (a) P.-P. Chen, H. Y. Zhang, B. Cheng, X. Chen, F. C. Cheng, S.-Q. Zhang, Z. Lu, F. K. Meng and X. Hong, ACS Catal., 2019, 9, 9589–9598 CrossRef CAS; (b) Y. Wang, C. Du, Y. Y. Wang, X.-K. Guo, L. Fang, M.-P. Song, J.-L. Niu and D. H. Wei, Adv. Synth. Catal., 2018, 360, 2668–2677 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr. , J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (b) W. Sang-Aroon and V. Ruangpornvisuti, Int. J. Quantum Chem., 2007, 108, 1181–1188 CrossRef.
- (a) B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS; (b) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01768j |
| This journal is © The Royal Society of Chemistry 2021 |