
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold(I) bridged dimeric and trimeric heterometallic {Cr7Ni}-based qubit systems and their characterization†
Deepak
Asthana
 ,
Selena J.
Lockyer
,
Selina
Nawaz
,
Robert J.
Woolfson
,
Grigore A.
Timco
,
Christopher A.
Muryn
,
Iñigo J.
Vitorica-Yrezabal
,
David
Collison
,
Neil A.
Burton
and
Richard E. P.
Winpenny
*
,
Selena J.
Lockyer
,
Selina
Nawaz
,
Robert J.
Woolfson
,
Grigore A.
Timco
,
Christopher A.
Muryn
,
Iñigo J.
Vitorica-Yrezabal
,
David
Collison
,
Neil A.
Burton
and
Richard E. P.
Winpenny
*
The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: richard.winpenny@manchester.ac.uk
First published on 2nd March 2021
Abstract
Gold(I) bridged dimeric and trimeric structures of a ground state spin S = 1/2 heterometallic {Cr7Ni} wheel have been prepared and studied by continuous wave (CW) and pulsed wave EPR spectrometry. The {Cr7Ni} relaxation time constants (T1 and Tm) show rates matching well with previous observations. Four pulse Double Electron Resonance (DEER) studies suggest presence of more than one conformations. Small Angle X-ray Scattering (SAXS) in conjunction with Molecular Dynamic (MD) Simulations were performed to look at the possible conformations in solution. In line with DEER results, simulation data further indicated more flexible molecular geometry in solution than the one in solid state.
Introduction
The family of {Cr7M} rings has been investigated since the first report in 2003;1 one major aim of the research has been as possible qubits for quantum information processing.2–4 {Cr7Ni} has attracted special interests due to its overall S = 1/2 ground state and tunable coherence times of the spin states.5 In the recent past, we have linked {Cr7Ni} rings into large assemblies6,7 and proposed possible logic operations.8,9The high stability of these {Cr7Ni} rings makes it possible to functionalize them. Previously we have focused on inclusion of a pyridine ligand by displacement of a pivalate ligand using iso-nicotinic acid.7 We have also examined using iso-phthalic acid to produce a ring functionalised with a carboxylate,10 and done a substitution reaction followed by organic functionalisations to give an extended bi-pyridyl ligand to produce an 8 nm molecular assembly.11
Here, we have extend the range of groups we can introduce to the {Cr7Ni} ring to include a sterically hindered phosphine ligand. We then show this very bulky phosphine ligand can act as a ligand for gold(I). The compounds are characterised by single crystal diffraction in the solid state, but small angle X-ray scattering studies in solution indicate the compounds dissociate in solution.
Results
The starting compound is [NH2nPr2][Cr7NiF8(O2CtBu)16] (1), and this was reacted with diphenylphosphino-benzoic acid to produce [NH2nPr2][Cr7NiF8(O2CtBu)15(O2C-C6H4-PPh2)] (2) (Scheme 1). The structure of 2 was confirmed by single crystal diffraction and we predict that the incoming ligand replaces a carboxylate bound to a Cr⋯·Ni edge in the octanuclear ring (Fig. 1a) due to the much higher reactivity of Ni(II) compared to Cr(III). Metric parameters within the ring are unchanged from 1 and the X-ray crystallography, combined with mass spectroscopy and elemental analysis, confirm the presence of pure phosphine rather than phosphine oxide.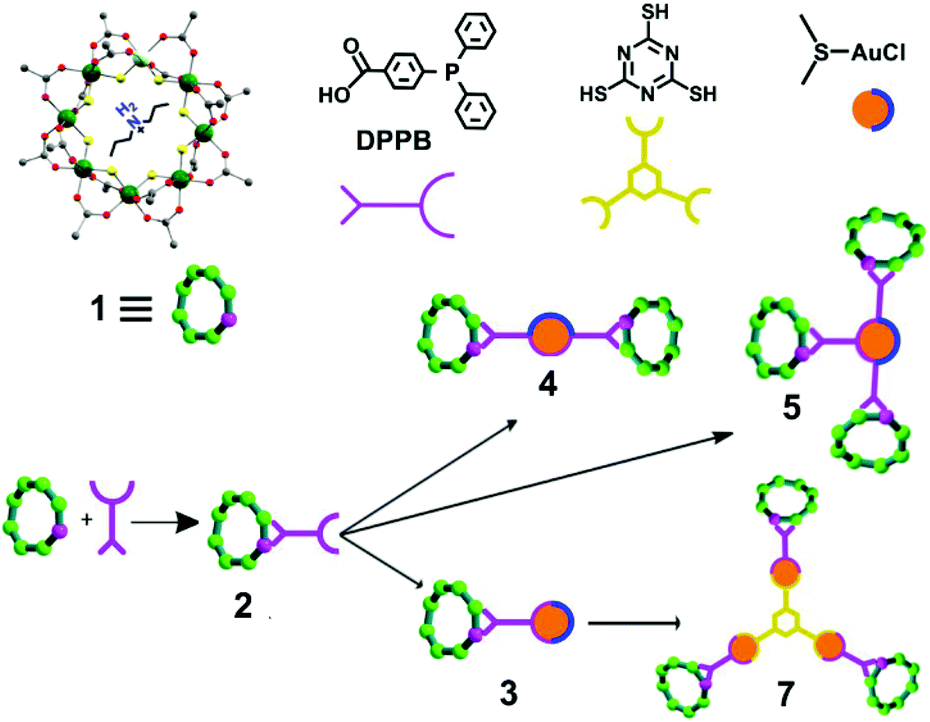 | ||
| Scheme 1 Schematic representation of the functionalization of 1 with DPPB to form 2, followed by reactions with gold chloride and trithiocyanuric acid to form 3–5, and 7. | ||
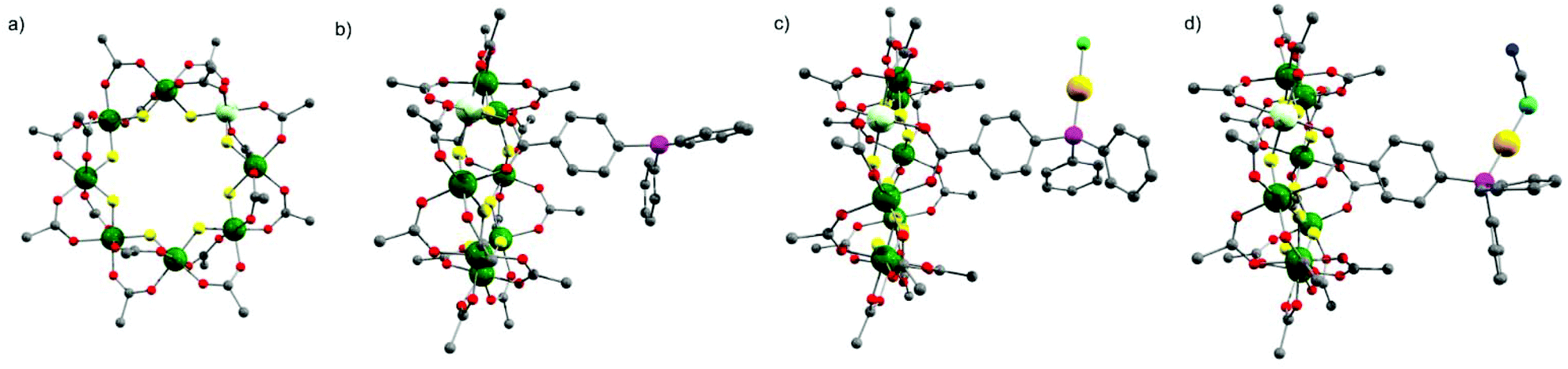 | ||
| Fig. 1 Single crystal X-ray structure of (a) 1 (b) 2 (c); 3 (d) 6. Colours: Cr (green), Ni (light green), Au (gold), P (purple), F (yellow), O (red), C (grey), Cl (bright green), N (blue), S (aqua). Tert-butyl methyl groups of pivalic acid, all the hydrogen atoms and dipropylammonium cation at the ring centre have been omitted for clarity. | ||
Stirring 2 with one equivalent of (Me2S)AuCl resulted in the crystallisation of [NH2nPr2][Cr7NiF8(O2CtBu)15(O2C-C6H4-PPh2)(AuCl)] (3) from CH2Cl2/MeCN. The phosphine from 2 displaces the Me2S from gold giving a linear two-coordinate Au(I) complex (Fig. 1b). The reaction is sensitive to the 2:(Me2S)AuCl ratio; if this is changed to 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 a gold bridged compound {[NH2nPr2][Cr7NiF8(O2CtBu)15(O2C-C6H4-PPh2)]}2AuCl (4) crystallises (Fig. 2a). A lower yield of 4 is found at a 2:1 ratio. Higher ratios than 2.5:1 would bring the possibility of forming other compounds such as 5 described below.
:1 a gold bridged compound {[NH2nPr2][Cr7NiF8(O2CtBu)15(O2C-C6H4-PPh2)]}2AuCl (4) crystallises (Fig. 2a). A lower yield of 4 is found at a 2:1 ratio. Higher ratios than 2.5:1 would bring the possibility of forming other compounds such as 5 described below.
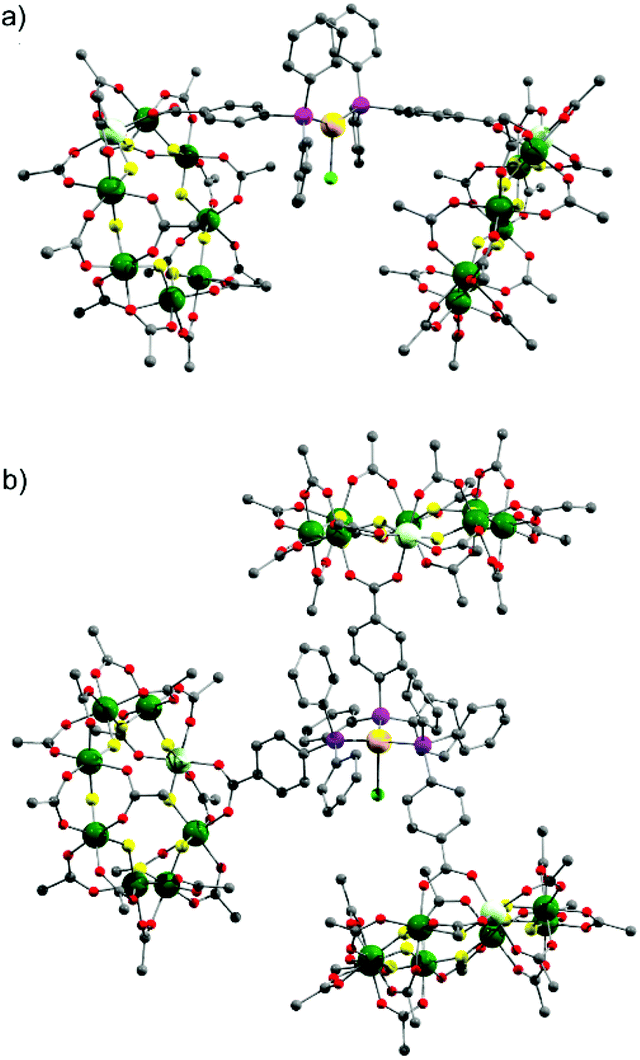 | ||
| Fig. 2 X-ray crystal structure of: (a) 4 and (b) 5. Colours as Fig. 1. Disordered tert.-butyl methyl groups, hydrogens and dipropylammonium cation are omitted for clarity. | ||
X-ray quality crystals of 4 could be grown from warm acetone or CH2Cl2/MeCN solutions. Complex 4 is an unusual example of a three-coordinate gold chloride structure with a bulky monodentate phosphine ligand.12 On one occasion while attempting to remake 4, we crystallised the {[NH2nPr2][Cr7NiF8(O2CtBu)15(O2C-C6H4-PPh2)]}3AuCl (5) (Fig. 2b) where three {Cr7Ni} rings are bound to the Au(I) centre. This forms in low yield and we have not attempted to optimise the synthesis as 5 does not appear to be present in solution (see below).
This chemistry produces three Au(I) compounds each featuring the same ligands but with different coordination numbers. This is known for simple phosphines such as triphenylphosphine, but is not common. Comparing metric parameters between 3, 4 and 5 (Table 1) the Au–P bond lengths increase from 2.241 Å to 2.311 Å to 2.369–2.381 Å and there is a concomitant increase in the Au–Cl bond lengths.
| Compound | Bond lengths/Å | Bond angles/° | ||
|---|---|---|---|---|
| Au–Cl | Au–P | Cl–Au–P | P–Au–P | |
| 3 | 2.273 | 2.241 | 176.32 | n.a. |
| 4 | 2.500 | 2.311 | 106.91 | 146.17 |
| 5 | 2.685 | 2.371 | 103.44 | 121.05 |
| 2.381 | 93.24 | 117.72 | ||
| 2.369 | 99.98 | 114.27 | ||
These angles show that while 2 would initially appear to be a very large phosphine, in fact as the {Cr7Ni} ring is attached to the 4-position of one phenyl it is quite distant from the Au centre. We calculate the Tolman cone angle for 2 as 171° (see experimental part).
We will return to the solution equilibria between these gold(I) complexes below.
The presence of a chloride on the Au(I) centre in 3 suggested the possibility of further reaction chemistry. Reaction of 3 with a stoichiometric amount of KSCN resulted in the formation of the product, [NH2nPr2][Cr7NiF8(O2CtBu)15(O2C-C6H4-PPh2)AuSCN] 6 that grew as thin green plates from CH2Cl2/MeCN (Fig. 1d). This suggested we could use trithiocyanuric acid as a bridging ligand (Scheme 1). The tri-sodium salt of trithiocyanuric acid was reacted with three equivalents of 3 giving [{[NH2nPr2][Cr7NiF8(O2CtBu)15(O2C-C6H4-PPh2Au)}3(S3C3N3)] (7) (Fig. 3).
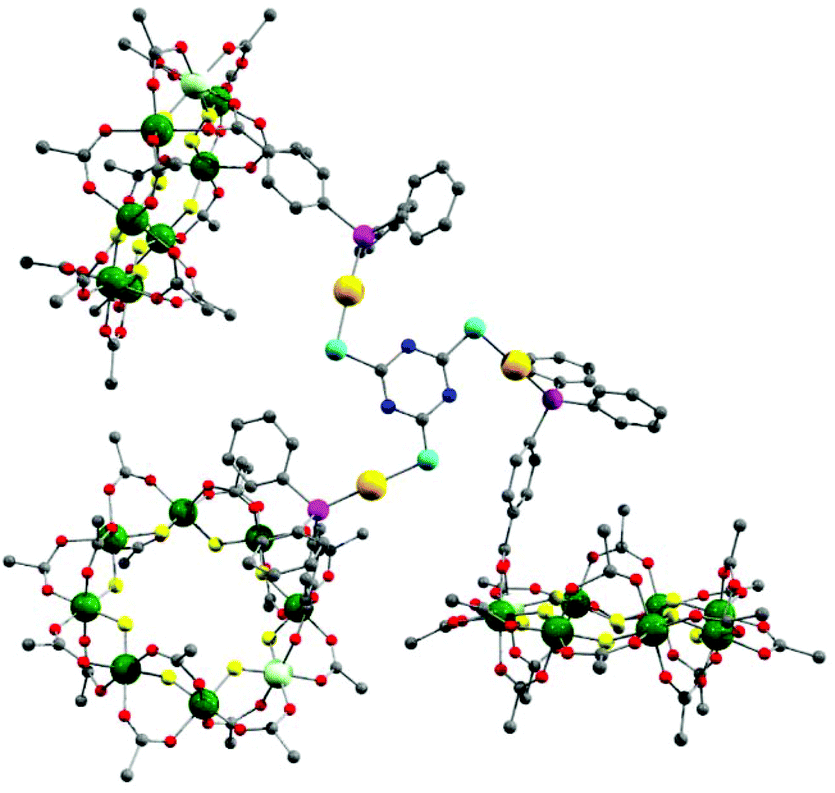 | ||
| Fig. 3 X-ray crystal structure of 7. Colours as Fig. 1. Disordered tert.-butyl methyl groups, hydrogens and dipropylammonium cation are omitted for clarity. | ||
The three 2 moieties in 7 are found to be non-equivalent, with each showing different bond lengths and angles. Two of the {Cr7Ni} rings lie almost perpendicular to the plane of the central heterocycle with torsion angles of 74.76° and 84.17°, whilst the third one lies closer to co-parallel with a torsion angle of 31.94° (ESI Fig. S3†).
Solution state behaviour by SAXS and molecular dynamics
We have previously used small angle X-ray scattering (SAXS) to demonstrate that supramolecular assemblies of heterometallic rings maintain their integrity in solution. Therefore, we investigated the SAXS of toluene solutions of 3, 4 and 5 at 290 K. The result is disappointing as a single major peak is seen at around 8 Å (Fig. 4) for all three compounds. This is consistent with the presence of a single ring, as in 2 or 3 but does not appear consistent with more than one ring as seen in the crystal structures of 4 or 5.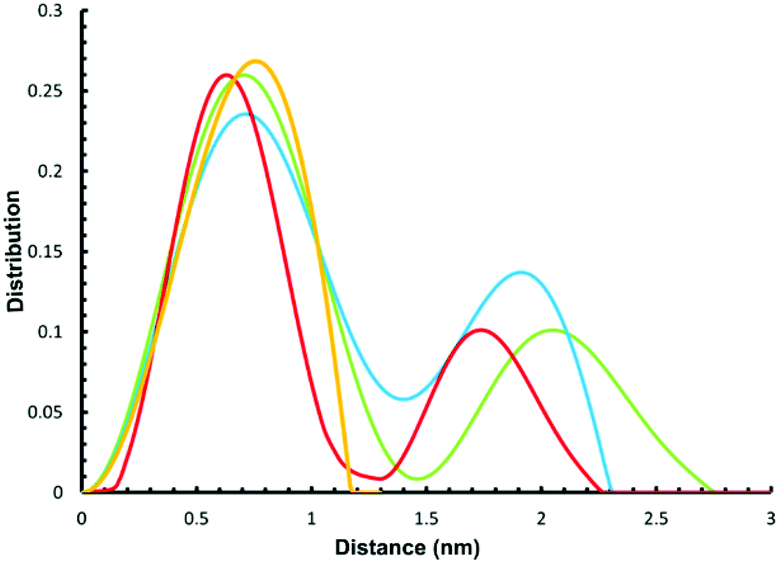 | ||
| Fig. 4 Experimental (yellow) and simulated SAXS plots for three conformations 4-A (red), 4-B (blue) and 4-C (green). | ||
To understand this result molecular dynamics simulations were performed using GROMACS 5.1.4.13,14 We began with 4 and paid attention to how the two metal rings might move relative to each other. Three simulations were set up using different starting structures as shown in Scheme 2. We began with the molecular structure seen in the crystal (Scheme 2a) but also rotated the {Cr7Ni} with respect to one another (Scheme 2b). A third conformation was produced by tilting the rings to obtain a structure where the two rings lie in the same plane (Scheme 2c). All-atom simulations of the three conformations of 4 in toluene solvent were set up. An energy minimized system of single 4 molecule in a solution of 10000 toluene molecules at a density of 865 kg m−3 in a cubic box size of ∼10.00 nm3 was used to obtain SAXS data and pair distribution functions. SAXS data were calculated for all three conformers (Fig. 4). For each conformation (4-A, 4-B and 4-C), two MD simulations were run. One where the four central atoms of the bridging ligand were frozen in place (i.e. Au+, Cl− and both P atoms) and a second one where those atoms were unfrozen.
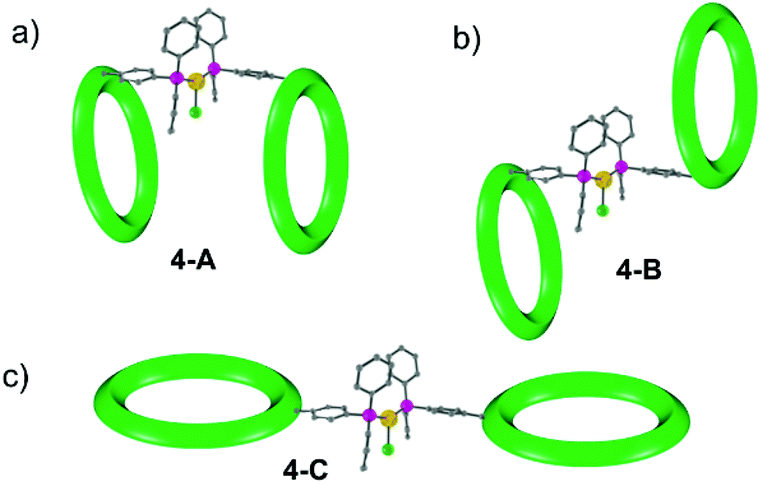 | ||
| Scheme 2 Schematic representation of the three conformations of 4 used for MD simulation: (a) regular structure found in solid state, (b) & (c) hypothetical structures obtained by re-orienting {Cr7Ni} rings. | ||
The calculations all predict two maxima for the 4 structure (Fig. 4). The maximum close to 8 Å is a result of contacts within the rings and corresponds to the maxima observed in the SAXS data. The second maximum is due to contacts between the two rings in 4 and predicts distances around 20 Å, just like the average distance distribution function (ESI Fig. S9†). This peak is absent in the experimental data, indicating that the majority of 4 is dissociated in solution, presumably into 2 and 3.
EPR measurements
The EPR spectra of 3 and 4 were recorded at Q- (34 GHz, Fig. 5a) and X-Band (9.5 GHz, ESI Fig. S5†) at 5 K as powders and as 3.0 mM solution in dichloromethane:toluene (1:1). The spectra are very similar with a signal centred at around 13600 Gauss arising from the {Cr7Ni} rings. This feature is known and is due to S = 1/2 ground state of the heterometallic ring which centres on a g ≈ 1.78.15 A well resolved gz feature of the {Cr7Ni} ring is seen in the powder spectra of both compounds, but is absent in solution spectra (Fig. 5a). The full width of the S = 1/2 signal is the same in both powder and solution spectra.
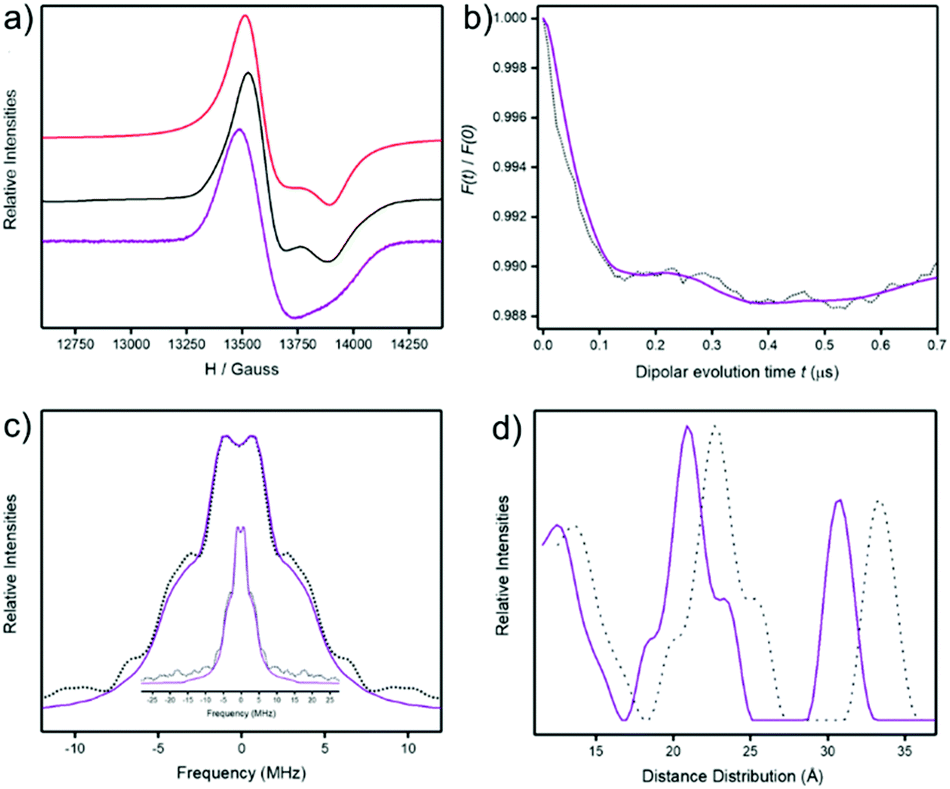 | ||
| Fig. 5 EPR spectroscopy at Q-Band (ca. ∼34 GHz) for 4. (a) CW EPR spectra at 5 K; 3 mM solution (purple line), powder (black) and simulation of powder (red). (b) DEER trace for a 0.2 mM solution of 4 at 3 K; after background subtraction (black dots) and fitted data (purple line). (c) Pake pattern from Fourier transformation of dipolar evolution (dotted black line) and fitted data (solid purple line). (d) Distance distribution; with default g values (dotted black line) and corrected g values (solid purple line). See SI for full details. | ||
The powder spectra were simulated16 using a simple spin-Hamiltonian incorporating only the individual g-matrices of the {Cr7Ni} ring to give g-values = 1.795, 1.786, 1.745 (Fig. 5a). The simulation provides a good fit for both 3 and 4. The solution spectra are less well-resolved and lines are very slightly shifted down field, however the spectra are all typical of {Cr7Ni} rings when isolated3 or when in weakly coupled oligomers.15 Therefore the c.w. EPR spectra do not show whether the structures are maintained in solution.
Pulsed EPR measurements at 13701 Gauss were used to measure relaxation times and at 3 K T1 = 124(1) μS, Tm = 713(1) ns for 4; these numbers are typical for {Cr7Ni} rings. In order to investigate any weak dipolar interactions between the two {Cr7Ni} rings a 4-pulse DEER experiment was performed with the pump pulse at 13701 Gauss and the observer pulse set −100 MHz from the pump pulse. We observed oscillations in the DEER trace, though distorted with multiple oscillation frequencies (Fig. 5b). Fourier transformation gives a Pake pattern consisting of a peak at ±1 MHz and shoulders at ±3.2 MHz and ±6.9 MHz, with a broad wing centred at 10 MHz (Fig. 5c). Tikhonov computations17 provide distance distributions with three main components; the middle one of the main components matches the distances predicted for the ring⋯ring contacts by molecular dynamics simulations.
Discussion
The initial intention of this work was to link together {Cr7Ni} rings via a single Au(I) centre and perhaps exploit the luminescence properties that have been observed in complexes such as [Au(PPh3)2Cl]18 to switch on/off the interactions between the rings. The result has been the crystallisation of large assemblies but it appears these are not stable in solution, at least not in mobile solutions. The experimental SAXS data measured at 290 K on 4 dissolved in toluene are only consistent with presence of single rings such as 2 or 3. This strongly suggests in the equilibrium shown in Scheme 3 the equilibrium lies on the side of 2 and 3:We would expect 2 and 3 to give similar SAXS results as the additional AuCl would make only a minor difference to the X-ray scattering in solution. All three species would give the peak centred at 8 Å and only 4 would give the peak at higher distances. Even if 4 were intact we would expect the shorter distance peak to be more intense based on precedent.15 Here where we have significant dissociation we are unable to measure any ring⋯ring distances. A further complication may be the possibility of different conformers of 4 (see Scheme 2). This would broaden the SAXS data for the higher distance peak. We attempted to make the solution more concentrated and as a result crystallised 5. Even from that solution the SAXS remained that of a single ring.
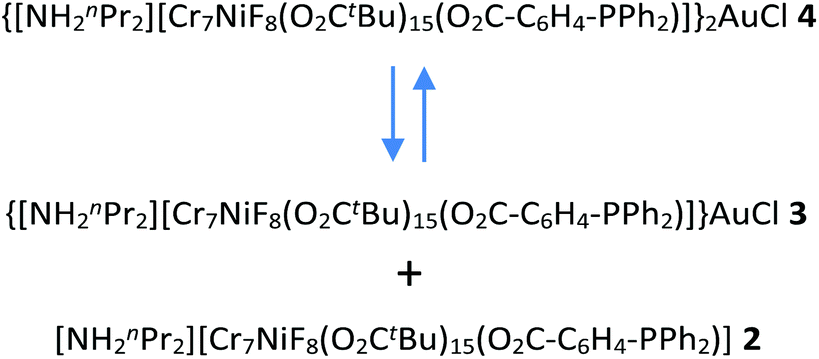 | ||
| Scheme 3 | ||
By contrast we see some evidence of ring⋯ring interactions by DEER spectroscopy (Fig. 5). Given the weak modulation observed we must be very careful not to overinterpret the data. The presence of any modulation suggests the presence of some ring⋯ring interactions in the frozen solution at 5 K.
Our explanation is that in mobile solution at 290 K the di-ring species 4 is in very rapid equilibrium with the monomers 2 and 3. At 5 K we have frozen out the exchange and sufficient molecules of 4 remain in the sample at the freezing point that we are able to measure ring⋯ring distances. While the explanation accounts for the data it also means the system is useless for our original intention of making switchable dimers of {Cr7Ni} rings.
The results also indicate that DEER must be used with some caution in supramolecular systems. Previously15 we have published examples where SAXS demonstrates that a complicated assembly exists in solution, and the DEER is then consistent with the SAXS. Here we demonstrate by SAXS that the oligomers do not exist in mobile solution but that we can freeze them out and see the dipolar interaction by DEER even in these equilibrium mixtures. DEER measures an interaction energy but it needs to be complimented by another solution technique to understand nucleation.
Experimental
General information
All reagents were purchased from commercial sources and used as received. Column chromatography was performed using a Grace Reverelis® Autocolumn with Reverelis® Flash Cartridges. NMR spectroscopy was performed using a Bruker AVIII HD 500 spectrometer with a CryoProbe™ Prodigy BBO probe at room temperature (unless otherwise specified). Mass spectrometry was performed with a Thermo Fisher Scientific “Exactive Plus EMR orbitrap” in electrospray mode. Elemental analysis for carbon, nitrogen, phosphorus and hydrogen analysis was performed using a Flash 200 elemental analyser. Metal elemental analysis was performed by Thermo iCap 6300 Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES).Single crystal X-ray structures were collected either on a Bruker Apex II Prospector with a Cu source at 100 K (2, 3, 4, 5 and 6) or on beamline i19 at the Diamond Light Source19 (7). Data sets were reduced with CrysAlisPro then solved and refined by full matrix least squares techniques on F2 using the Shelxl20 suite in Olex2.21 Hydrogen atoms were included in the refinement process as per the riding model. CCDC 1499587–1499591 and 2052037–2052038† contain the supplementary crystal data for all compounds.
:1) at 5 K using liquid helium cooling. Pulse Q-Band (∼34 GHz) EPR data were collected on a Bruker ELEXSYS E580Q FT spectrometer. The measurements were collected from a dry and degassed solutions of toluene at 3 K (unless otherwise stated) using a Cryogenic cryogen free variable temperature cryostat incorporating a closed helium circuit. Spectral simulations were performed using the EasySpin 5.2.28 simulation software.16
Molecular dynamic simulations
All-atom simulations of the systems were set up using Gromacs 5.1.4 molecular dynamics package.13 The initial crystal structure Cartesian coordinates for structure 4-A was obtained experimentally from the crystal structure and parameterized according to the General Amber Forcefield.22 minimized with the conjugate gradient algorithm. The minimized structures were run for 10 ns using an NPT ensemble (T = 298 K and p = 1.01325 bar) using the Nosé–Hoover thermostat23,24 and the Parrinello–Rahman barostat.25 A time step for integration was fixed at 1 fs with the neighbour list updated every 10 fs and periodic boundary conditions were employed. The electrostatics were evaluated using the particle-mesh Ewald method.26Cone angle determination
Tolman cone angle27 for 2 was estimated from the percentage buried volume (%Vbur) using the freely available online software package SambVca.28,29 Values were calculated using the crystal structures discussed above but the metal centre was set at 2.28 Å from the phosphorus site. The calculated %Vbur for PPh3 showed a good fit to that previously reported (29.9% vs. 29.6%). Calculated %Vbur for 2 rises significantly to 35.3%. The cone angle of 2 was then estimated using linear correlation between Tolman cone angle and %Vbur.Synthetic procedures
1 was synthesised as previously reported.12 was synthesised by refluxing [NH2nPr2][Cr7NiF8(O2CtBu)16] (1Ni) (1.0 g, 0.43 mmol) with DPPB (0.4 g, 1.29 mmol) in dry xylenes (15 mL) at 160 °C for 18 h under a nitrogen atmosphere. After cooling, the mixture was dried by rotary evaporation, dissolved in hexane, filtered and purified by column chromatography. Using a 40 g silica cartridge and 98% hexane: 2% diethyl ether as the eluent, a mixture of unreacted 1Ni and 2Ni eluted followed closely by 0.054 g (5.02%) of pure 2Ni ESMS+: 2498.618 (M + H); 2521.611 (M + Na). Elemental Anal. (calc.): C, 48.04 (48.06); H, 6.79 (6.65); N, 0.54 (0.56); P, 1.44 (1.25); Cr, 13.88 (14.56); Ni, 2.17 (2.34). Crystals suitable for X-ray diffraction were grown from slow evaporation of a CH2Cl2/MeCN solution.
3 was synthesised by reacting 2 (0.1 g, 0.04 mmol) with AuCl(SMe2) (12.9 mg, 0.044 mmol) in dry CH2Cl2 (10 mL). The reaction was filtered over Celite and the solvent removed by rotary evaporation yielding a green powder (0.108 g, 0.0395 mmol, 99% yield). Crystals suitable for X-ray diffraction were grown from slow evaporation of a CH2Cl2/MeCN solution. ESMS+: 2732.560 (M + H). Elemental Anal. (calc.): C, 44.15 (43.72); H 6.28 (6.12); N, 0.51 (0.52); Cl, 1.62 (1.30); P, 1.04 (1.14); Cr, 12.90 (13.38); Ni, 2.12 (2.16); Au, 7.41 (7.26). Crystals suitable for X-ray diffraction were grown from slow evaporation of a CH2Cl2/MeCN solution.
4 was synthesised by dissolving 2 (0.1 g, 0.04 mmol) in CH2Cl2 and adding AuCl(SMe2) (4.7 mg, 0.016 mmol). The resulting solution was stirred for 1 h and filtered over Celite. Crystals suitable for single crystal X-ray diffraction were grown from slow evaporation of a CH2Cl2/MeCN solution. ESMS+: 5193.260 (M − Cl). Elemental Anal. (calc.): C; 47.92 (46.93); H, 6.79 (6.73); N, 0.56 (0.54); P, 0.93 (1.18); Cr, 13.49 (13.98); Ni, 2.23 (2.24); Au, 3.03 (3.77).
5 was obtained by reacting 2 with AuCl(SMe2) under similar conditions as for synthesis of 4. Reaction mixture was filtered over Celite and filtrate was dried using rotary evaporator. Obtained solid was dissolved in CH2Cl2 and layered with MeCN. Crystals grown in overnight time were immediately studied under single crystal X-ray spectrometer.
6 was synthesised by adding KSCN (10.7 mg, 0.110 mmol) to a suspension of 3 (0.3 g, 0.110 mmol) in acetone and stirring for 1 h, filtered over Celite and dried by rotary evaporation. Crystals suitable for X-ray crystallography were grown from slow evaporation of a CH2Cl2/MeCN solution. ESMS+: 2776.531 (M + H). Elemental Anal. (calc.): C, 43.82 (44.05); H, 5.86 (6.04); N, 0.89 (1.02); Cl, 0 (0); Cr, 13.66 (13.22); Ni, 2.14 (2.13); Au, 7.42 (7.15); S, 1.10 (1.16); P, 1.16 (1.12).
7 was synthesised in a two-step procedure. Trithiocyanuric acid (0.5 g, 2.82 mmol) was dissolved in methanol (25 mL) and sodium methoxide (0.457 g 8.46 mmol) added. The solution was stirred for 1 h, filtered and dried by rotary evaporation yielding the tri-sodium salt. 3 (0.2 g, 0.07 mmol) was dissolved in CH2Cl2 (10 mL) and trisodium-trithiocyanuric acid (6 mg, 0.024 mmol) in CH2Cl2 (2 ml) was added. The solution was stirred at room temperature for 4 h and dried by rotary evaporation yielding the product in quantitative yields. ESMS+: 8262.439 (M + H), 8285.492 (M + Na) Elemental Anal. (calc.): C, 44.53 (44.05); H, 6.44 (6.05); N, 1.03 (1.02); Cl, 0 (0); Cr, 13.70 (13.22); Ni, 2.18 (2.13); Au, 7.64 (7.15); S, 1.22 (1.16); P, 1.09 (1.12).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC (grant EP/P000444/1). We also acknowledge the EPSRC National EPR Facility and Service (EP/NS/A000055/1). We also thank EPSRC (UK) for funding an X-ray diffractometer (EP/K039547/1). This work was supported by an EPSRC Established Career Fellowship (EP/R011079/1) to REPW, We also thank the European Research Council for an Advanced Grant (ERC-2017-ADG-786734). We thank Diamond Light Source for access to synchrotron X-ray facilities. RJW acknowledges a studentship from Centre for Doctoral Training North-West Nanoscience Doctoral Training Centre, NOWNANO (EPSRC grant EP/G03737X/1).Notes and references
- F. K. Larsen, E. J. L. McInnes, H. Mkami, J. Overgaard, S. Piligkos, G. Rajaraman, E. Rentschler, A. A. Smith, G. M. Smith, V. Boote, M. Jennings, G. A. Timco and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 101–105 CrossRef CAS PubMed.
- G. A. Timco, S. Carretta, F. Troiani, F. Tuna, R. J. Pritchard, C. A. Muryn, E. J. L. McInnes, A. Ghirri, A. Candini, P. Santini, G. Amoretti, M. Affronte and R. E. P. Winpenny, Nat. Nanotechnol., 2009, 4, 173–178 CrossRef CAS PubMed.
- G. A. Timco, T. B. Faust, F. Tuna and R. E. P. Winpenny, Chem. Soc. Rev., 2011, 40, 3067–3075 RSC.
- R. E. P. Winpenny, Angew. Chem., Int. Ed., 2008, 47, 7992–7994 CrossRef CAS PubMed.
- C. J. Wedge, G. A. Timco, E. T. Spielberg, R. E. George, F. Tuna, S. Rigby, E. J. L. McInnes, R. E. P. Winpenny, S. J. Blundell and A. Ardavan, Phys. Rev. Lett., 2012, 108, 107204 CrossRef CAS PubMed.
- J. Ferrando-Soria, A. Fernandez, D. Asthana, S. Nawaz, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, C. A. Muryn, F. Tuna, G. A. Timco, N. D. Burton and R. E. P. Winpenny, Nat. Commun., 2019, 10, 3720 CrossRef PubMed.
- G. F. S. Whitehead, F. Moro, G. A. Timco, W. Wernsdorfer, S. J. Teat and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2013, 52, 9932–9935 CrossRef CAS PubMed.
- A. Fernandez, J. Ferrando-Soria, E. M. Pineda, F. Tuna, I. J. Vitorica-Yrezabal, C. Knappke, J. Ujma, C. A. Muryn, G. A. Timco, P. E. Barran, A. Ardavan and R. E. P. Winpenny, Nat. Commun., 2016, 7, 10240 CrossRef CAS PubMed.
- J. Ferrando-Soria, E. Moreno Pineda, A. Chiesa, A. Fernandez, S. A. Magee, S. Carretta, P. Santini, I. J. Vitorica-Yrezabal, F. Tuna, G. A. Timco, E. J. L. McInnes and R. E. P. Winpenny, Nat. Commun., 2016, 7, 11377 CrossRef CAS PubMed.
- G. F. S. Whitehead, J. Ferrando-Soria, L. G. Christie, N. F. Chilton, G. A. Timco, F. Moro and R. E. P. Winpenny, Chem. Sci., 2014, 5, 235–239 RSC.
- J. Ferrando-Soria, A. Fernandez, E. M. Pineda, S. A. Varey, R. W. Adams, I. J. Vitorica-Yrezabal, F. Tuna, G. A. Timco, C. A. Muryn and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137, 7644–7647 CrossRef CAS PubMed.
- M. C. Gimeno and A. Laguna, Chem. Rev., 1997, 97, 511–522 CrossRef CAS PubMed.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CrossRef CAS.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- S. J. Lockyer, S. Nawaz, A. Brookfield, A. J. Fielding, I. J. Vitorica-Yrezabal, G. A. Timco, N. A. Burton, A. M. Bowen, R. E. P. Winpenny and E. J. L. McInnes, J. Am. Chem. Soc., 2020, 142, 15941–15949 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- J. Weese, Comput. Phys. Commun., 1992, 69, 99–111 CrossRef.
- C. King, M. N. I. Khan, R. J. Staples and J. P. Fackler Jr., Inorg. Chem., 1992, 31, 3236–3238 CrossRef CAS.
- H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435–441 CrossRef CAS PubMed.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- S. A. Nose, Mol. Phys., 1984, 52, 255–268 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- U. Essmann, L. Perera and M. L. Berkowitz, J. Chem. Phys., 1995, 103, 8577–8592 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2052037 and 2052038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00150g |
| This journal is © The Royal Society of Chemistry 2021 |