DOI:
10.1039/D1DT00189B
(Paper)
Dalton Trans., 2021,
50, 4396-4407
Titanium complexes bearing oxa- and azacalix[4, 6]arenes: structural studies and use in the ring opening homo-/co-polymerization of cyclic esters†
Received
19th January 2021
, Accepted 4th March 2021
First published on 5th March 2021
Abstract
Reaction of excess [Ti(OiPr)4] with p-tert-butyltetrahomodioxacalix[6]areneH6 (L1H6) afforded, after work-up (MeCN), the complex [Ti2(OiPr)2(MeCN)L1]·3.5MeCN (1·3.5MeCN), whilst the oxo complex [Ti4(μ3-O)2(H2O)(L1)2]·MeCN (2·MeCN) was isolated via a fortuitous synthesis involving the use of two equivalents of [Ti(OiPr)4]. Reactions of p-methyl-dimethyldiazacalix[6]areneH6 (L2H6) with [TiF4] (four equivalents), [TiCl4(THF)2] (two equivalents) or [TiBr4] (>four equivalents) resulted in the titanium-based azacalix[n]arene complexes [Ti4F14L2H2(H)2]·2.5MeCN (3·2.5MeCN), [Ti2X4(H2O)2OL2H2(H)2] (X = Cl (4·5MeCN), Br (5·4.5MeCN) and [Ti4Br12L2(H)2(MeCN)6]·7MeCN (6·7MeCN), respectively. Reaction of four equivalents of [TiF4] with L3H4 (L3H4 = p-methyl-dimethyldiazacalix[4]areneH4) afforded the product [Ti2F2(μ-F)3L3(H)2(SiF5)]·2MeCN (7·2MeCN). These complexes have been screened for their potential to act as pre-catalysts in the ring opening polymerization (ROP) of ε-caprolactone (ε-CL), δ-valerolactone (δ-VL) and rac-lactide (r-LA). Generally, the titanium complexes bearing oxacalixarene exhibited better activities than the azacalixarene-based pre-catalysts. For ε-CL, δ-VL and r-LA, moderate activity at 130 °C over 24 h was observed for 1–6. In the case of the co-polymerization of ε-CL with r-LA, 1–6 afforded reasonable conversions and high molecular weight polymers; 7 exhibited lower catalytic performance due to low solubility. None of the complexes proved to be active in the polymerization of ω-pentadecalactone (ω-PDL) under the conditions employed herein.
Introduction
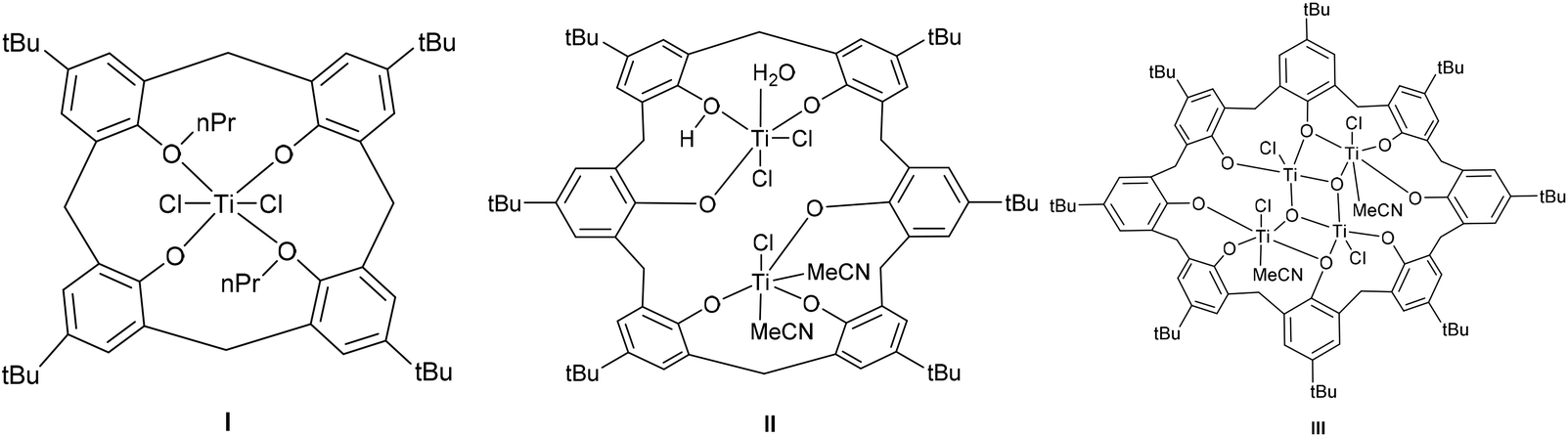
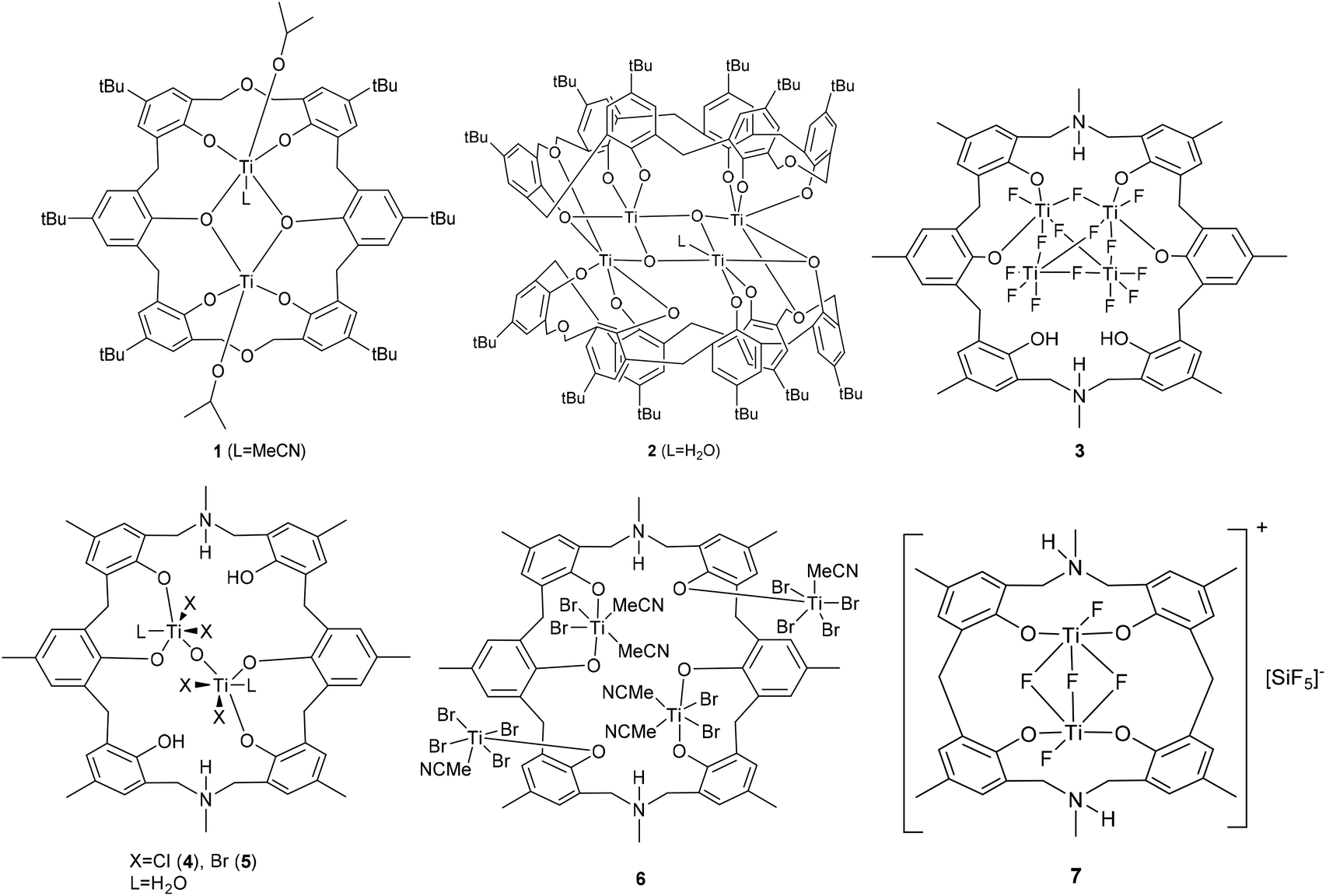
Calix[n]arenes are a family of macrocyclic molecules consisting of phenol units linked most commonly by methylene (–CH2–) bridges at their ortho positions, and have found widespread use in supramolecular chemistry.1 Investigations into their coordination chemistry have shown that their potential in areas such as catalysis can be greatly improved if the methylene bridges are replaced by heteroatom-containing bridges such as thia (–S–), sulfinyl (–SO–), sulfonyl (–SO2–) or dimethyleneoxa (–CH2OCH2–), which can potentially bind to the metal.2 Interestingly, there is a lack of such studies involving dimethyleneoxa (–CH2OCH2–) containing calix[n]arenes, where n ≥ 6.3 Moreover, there is even less data on azacalix[n]arenes, where the bridge (–NR–) has an addition group (R) bound to the nitrogen which can potentially be varied to control the sterics and/or electronics of the system.4 Given this, we have initiated a study of the coordination chemistry of both dimethyloxa- and azacalix[n]arenes with a view to investigating their potential as catalysts for the ring opening polymerization (ROP) of cyclic esters. Given recent successes using titanocalix[n]arenes for ROP (see Chart 1),5 we opted here to focus on titanium-containing dimethyloxa- and azacalixarenes, and have structurally characterized a number of interesting poly-metallic species (see Chart 2). The ability of these complexes to act as pre-catalysts for the ROP of ε-caprolactone (ε-CL), δ-valerolactone (δ-VL) and rac-lactide (r-LA) has been investigated; the copolymerization of ε-CL and r-LA was also investigated.
 |
| | Chart 1 Known calixarene pre-catalysts for the ROP of cyclic esters. | |
 |
| | Chart 2 Pre-catalysts prepared herein. | |
Results and discussion
Syntheses and solid-state structures
Dioxacalix[6]arene complexes.
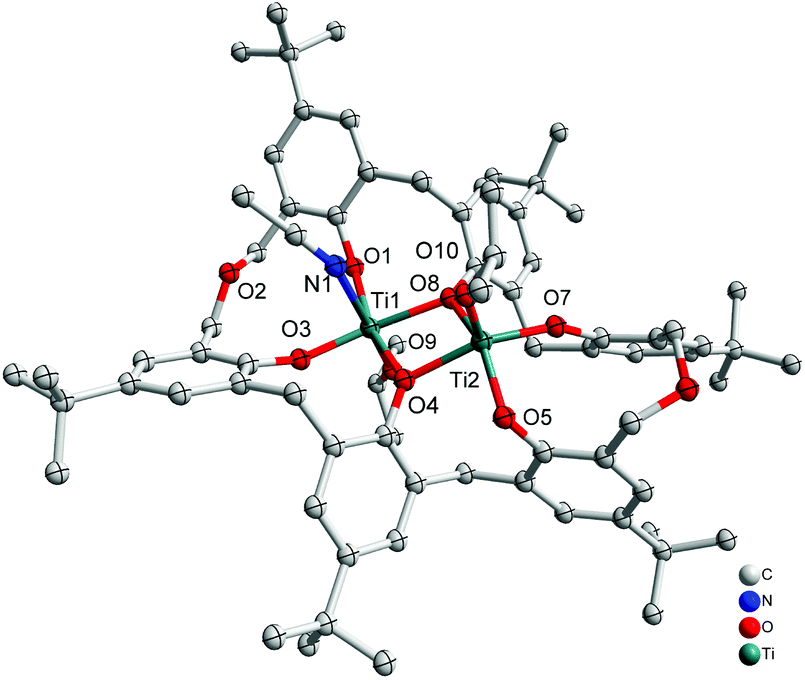
Interaction of an excess (3 equiv.) of [Ti(OiPr)4] with p-tert-butyltetrahomodioxacalix[6]areneH6 (L1H6) in refluxing toluene afforded, after work-up (MeCN), the orange complex [Ti2(OiPr)2(MeCN)L1]·3.5MeCN (1·3.5MeCN) in 52% yield. The molecular structure is shown in Fig. 1, with selected bond lengths and angles given in the ESI;† crystallographic data are given in Table 5. The complex contains two titanium centres, one of which, Ti(1), is distorted octahedral bound by an isopropoxide ligand, an acetonitrile ligand and four calixarene phenoxide oxygens in a square plane, two of which are shared with Ti(2). Ti(2) is five-coordinate and adopts a slightly distorted rectangular pyramidal (τ = 0.015).6
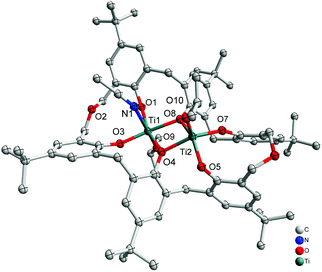 |
| | Fig. 1 Molecular structure of [Ti2(OiPr)2(MeCN)L1]·3.5MeCN (1·3.5MeCN). Solvent molecules and hydrogen atoms omitted for clarity. | |
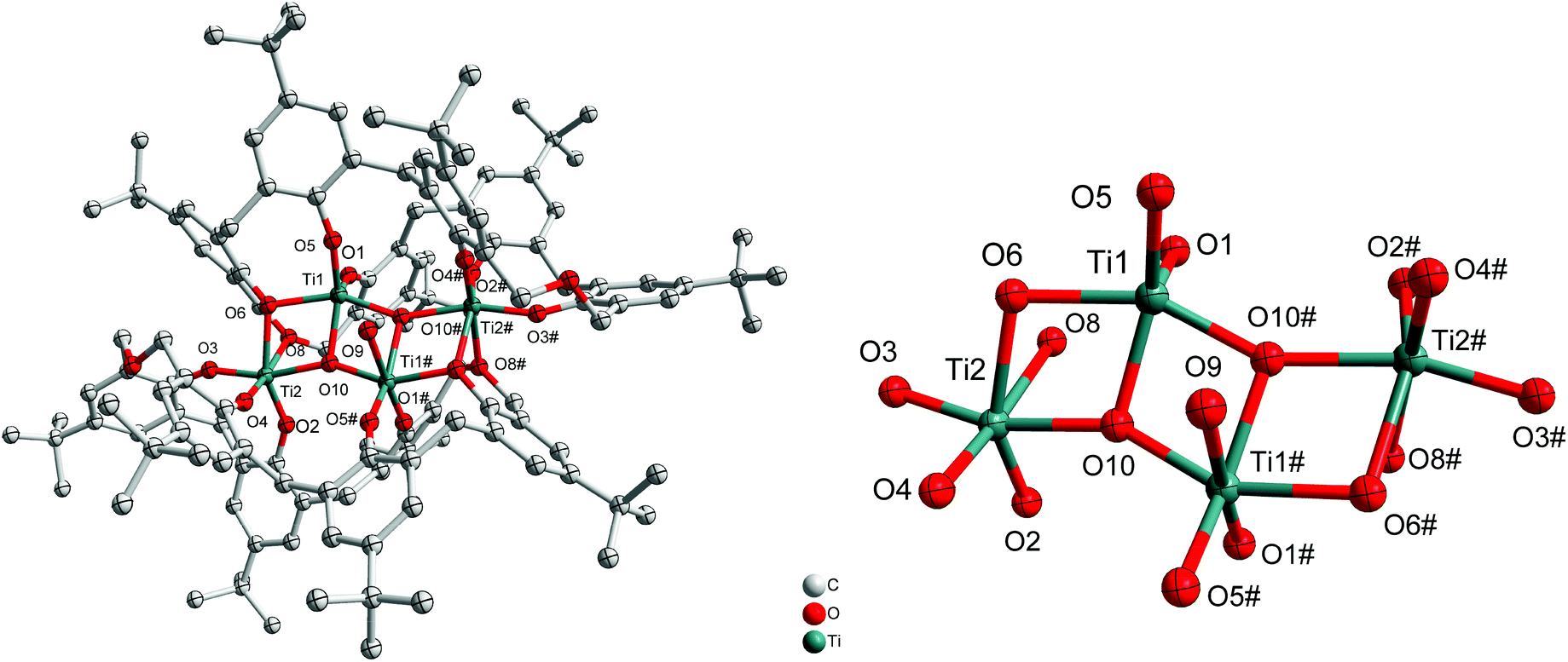
If the reaction is conducted in the presence of adventitious oxygen/water and using only two equivalents of [Ti(OiPr)4], then the isopropoxide groups are lost and a structure involving a titanium–oxygen Ti4O4 ladder sandwiched between two oxacalix[6]arenes is formed, namely [Ti4(μ3-O)2(H2O) (L1)2]·MeCN (2·MeCN). The molecular structure is shown in Fig. 2, with selected bond lengths and angles given in the ESI.† The water ligand partially occupies positions at Ti(1) and Ti(1#) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50). We note that calix[8]arene titanium ladder complexes have recently been isolated and utilized for ROP, CO2 photoreduction and photocatalytic H2 production.5b,7
:50). We note that calix[8]arene titanium ladder complexes have recently been isolated and utilized for ROP, CO2 photoreduction and photocatalytic H2 production.5b,7
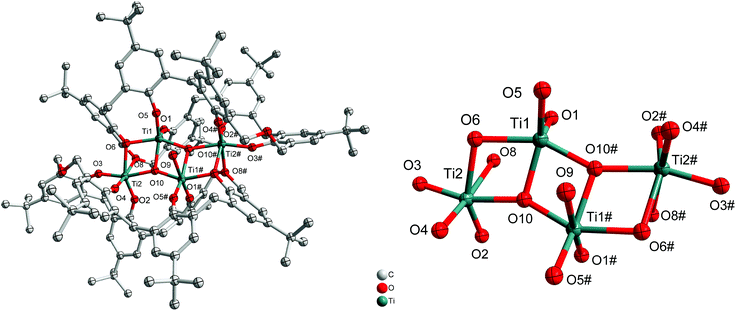 |
| | Fig. 2 Left: Molecular structure of [Ti4(μ3-O)2(H2O)(L1)2]·MeCN (2·MeCN); right: core of the structure. Solvent molecules and hydrogen atoms omitted for clarity. | |
Azacalixarene complexes.
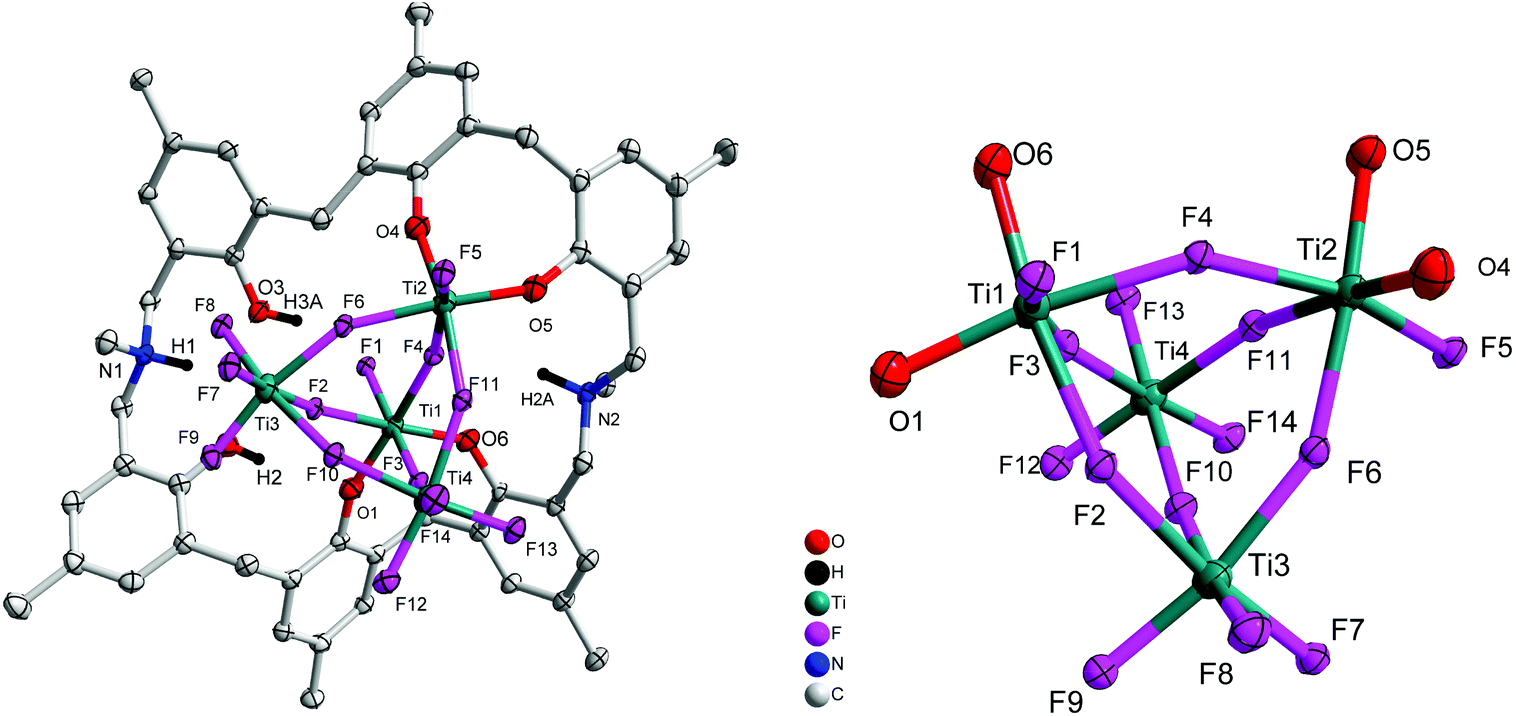
Reaction of p-methyl-dimethyldiazacalix[6]areneH6 (L2H6) with four equivalents of [TiF4] in refluxing toluene afforded, following work-up (MeCN), the orange complex [Ti4F14L2H2(H)2]·2.5MeCN (3·2.5MeCN). The molecular structure is shown in Fig. 3, with selected bond lengths and angles given in the ESI.† The complex contains four distorted octahedral titanium centres each linked via fluoride bridges to give a central Ti4F14 core. The azacalixarene acts as a bidentate O,O-chelate to Ti(1) and to Ti(2), leaving two uncoordinated phenolic groups on the macrocycle.
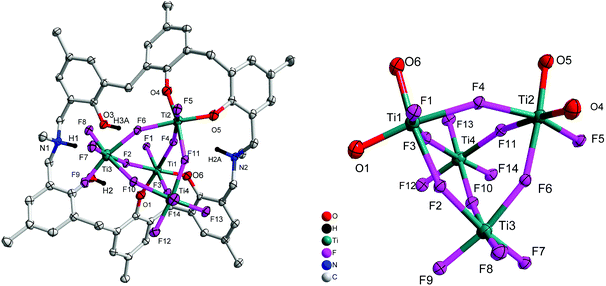 |
| | Fig. 3 Left: Molecular structure of [Ti4F14L2H2(H)2]·2.5MeCN (3·2.5MeCN); right: core of the structure. Solvent molecules and hydrogen atoms omitted for clarity. | |
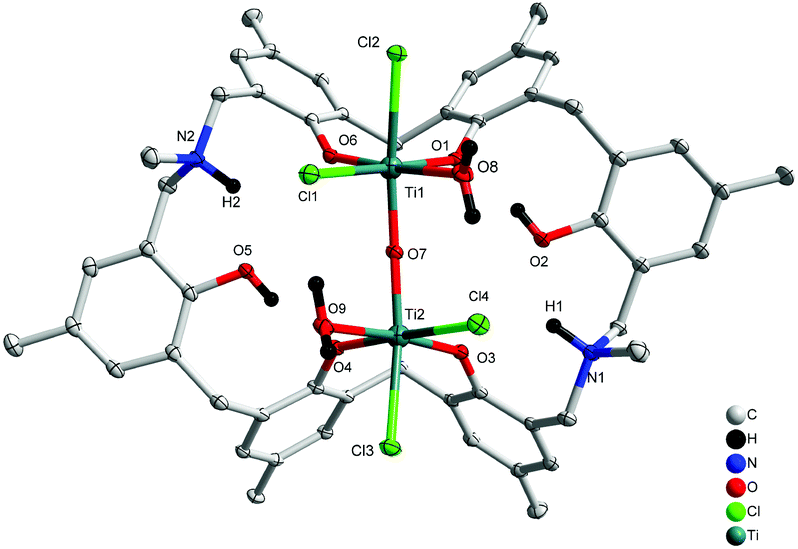
Treatment of L2H6 with two equivalents of [TiCl4(THF)2] in refluxing toluene afforded, after work-up (MeCN), dark red prisms of [Ti2Cl4(H2O)2OL2H2(H)2]·5MeCN (4·5MeCN) in 41% yield. The molecular structure is shown in Fig. 4, with selected bond lengths and angles given in the ESI.† The complex contains two distorted octahedral titanium centres linked via a near linear oxo bridge [Ti(1)–O(7)–Ti(2) 168.84(6)]. The coordination at each Ti centre is completed by two adjacent phenoxides of the calixarene, a water molecule and two chlorides, one of which is found trans to the oxo bridge. The titanium phenoxide bond lengths are typical [1.8324(11)–1.8975(11) Å], whilst those to the water ligands are, as expected, somewhat longer [2.1445(12) and 2.1570(11) Å].5b,8 The overall charge is balanced by the protonated aza bridges of the calixarene.
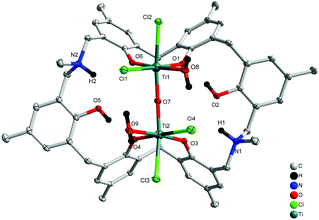 |
| | Fig. 4 Molecular structure of [Ti2Cl4(H2O)2OL2H2(H)2]·5MeCN (4·5MeCN). Solvent molecules and hydrogen atoms omitted for clarity. | |
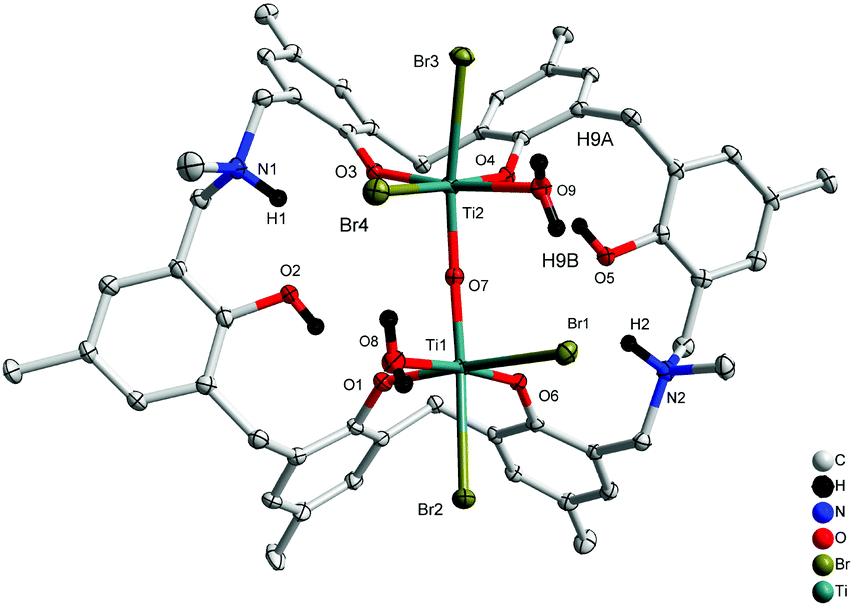
Similar treatment of L2H6 with two equivalents of [TiBr4] resulted in the isostructural complex [Ti2Br4(H2O)2OL2H2(H)2]·2MeCN 4.5MeCN (5·4.5MeCN) in 32% yield. The molecular structure is shown in Fig. 5, with selected bond lengths and angles given in the ESI.† As in 4, a linear oxo bridge [Ti(2)–O(7)–Ti(1) 172.65(17)] links the two distorted octahedral centres, and a bromide at each Ti centre can be found trans to the μ2-O. The Ti–O bond length range is similar to that in 4.
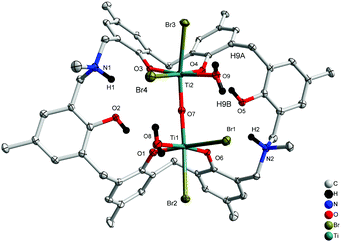 |
| | Fig. 5 Molecular structure of [Ti2Br4(H2O)2OL2H2(H)2]·4.5MeCN (5·4.5MeCN). Solvent molecules and hydrogen atoms omitted for clarity. | |
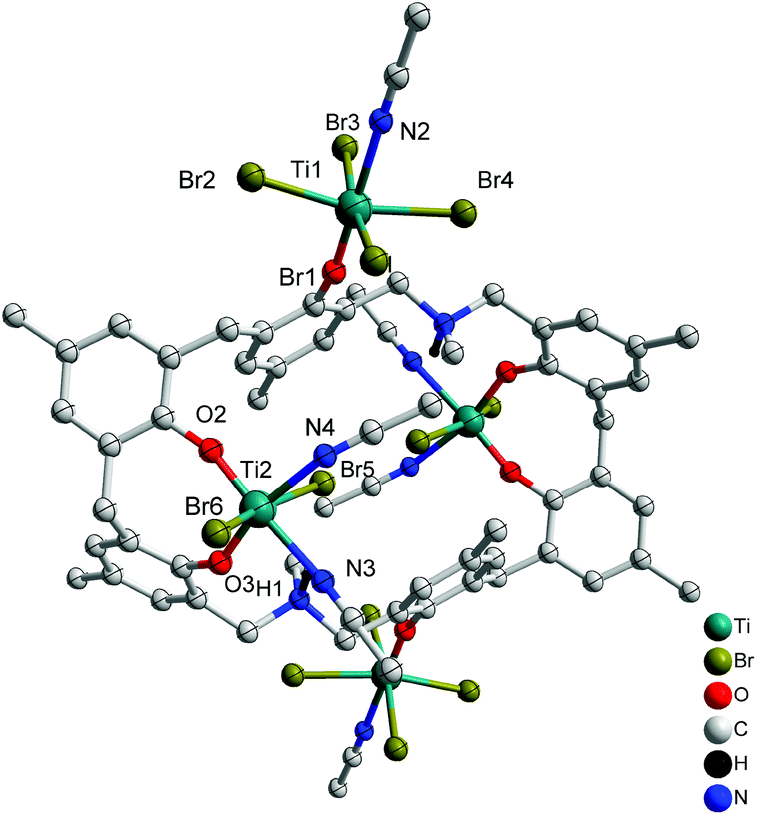
Reaction of L2H6 with excess [TiBr4] led to the isolation of the orange complex [Ti4Br12L2(H)2(MeCN)6]·7MeCN (6·7MeCN) in 26% isolated yield. A view of the molecular structure is shown in Fig. 6, with selected bond lengths and angles given in the ESI.† The asymmetric unit of 6.7MeCN contains two independent half molecules each comprising one half a p-methyldimethyldiazacalix[6]arene and two Ti ions (Ti1 and Ti2 in first half molecule; Ti3 and Ti4 in second half molecule). The complete molecule in each case is generated by the inversion centre. The two half molecules are similar but are not related by symmetry. In each, the two octahedral Ti ions have different environments. One (Ti1 or Ti3) is coordinated by four bromide ions in a square plane, with O from the calix and NCCH3 in trans arrangement. The calixarene is twisted so that the phenoxide points away from the centre of the molecule and places Ti1 and Ti1_i on opposite sides of the plane of the calixarene (symmop i = 1−x, −y, −z). A very similar arrangement is observed for Ti3 and Ti3_ii (symm. op. ii = 2−x, 1−y, 1−z). The second Ti ion (Ti2 & Ti4) is coordinated in an 8-membered chelate ring, by two geminal phenoxides from the calixarene, two cis NCCH3 in approximately the same plane, and two trans bromide ions. The twisted orientation of the calixarene allows for the formation of an N–H⋯Br hydrogen bond from the protonated aza linkage.
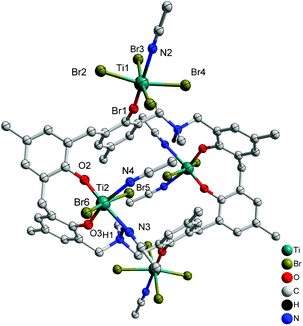 |
| | Fig. 6 Molecular structure of [Ti4Br12L2(H)2(MeCN)6]·7MeCN (6·7MeCN). Solvent molecules and hydrogen atoms omitted for clarity. | |
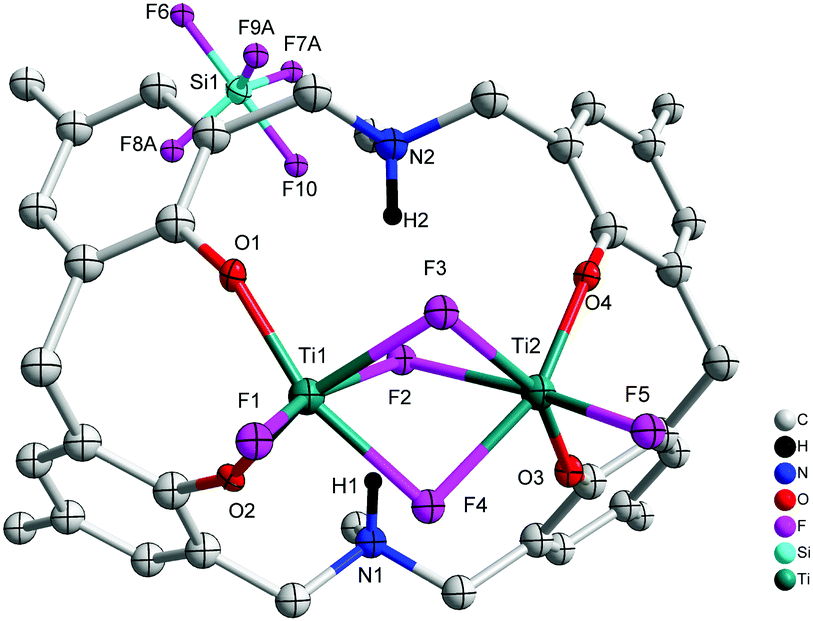
When L2H6 was treated with four equivalents of [TiF4], orange prisms were isolated on work-up, albeit in poor yield (<20%). A crystal structure determination revealed the complex to be [Ti2F2(μ-F)3L3(H)2(SiF5)]·2MeCN (7·2MeCN), see Fig. 7. In 7·2MeCN, triply bridging fluorides link two distorted octahedral Ti centres, with a terminal fluoride and two adjacent phenoxides of the macrocycle completing the coordination sphere at each metal centre. The SiF5− ion is thought to result from the scavenging of HF formed during the reaction. As noted previously, the scavenging of HF can occur via the reaction 5HF + SiO2 → 2H2O + H+ + SiF5−.9
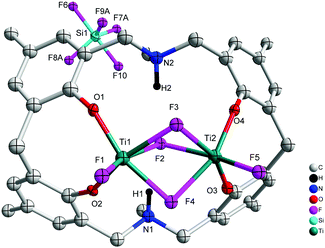 |
| | Fig. 7 Molecular structure of [Ti2F2(μ-F)3L3(H)2(SiF5)]·2MeCN (7·2MeCN). Solvent molecules and hydrogen atoms omitted for clarity. | |
Ring opening polymerization studies
General.
The performance of these complexes to act as pre-catalysts for the ring opening polymerization (ROP) of ε-caprolactone (ε-CL), δ-valerolactone (δ-VL) and rac-lactide (r-LA) with one equivalent of benzyl alcohol (BnOH) per titanium present, has been investigated. The co-polymerization of ε-caprolactone and rac-lactide (Table 4) has also been investigated.
ε-Caprolactone (ε-CL)
Complexes 1–7 were screened for their ability to polymerise ε-caprolactone and the results are collated in Table 1. The polymerization screening indicated that the best conditions were 250 equivalents of ε-caprolactone to titanium at 130 °C. In separate experiments, we re-recorded the 1H NMR spectra of 1–7 after prolonged heating at 130 °C to confirm their stability during the ROP conditions. The activity of complex 1 increased with temperature and peaked at 250 equivalents of monomer. Complex 1 was also active at low catalyst loading leading to 80.4% conversion after 8 h for 1000 equivalents of monomer. All polymers obtained were of relatively low polydispersity (PDI < 1.75), which suggested that these polymerizations occurred without significant side reactions. The Mn were found to be much lower than the calculated values. Interestingly, complex 1 proved to be active also under aerobic conditions achieving 84% conversion during 8 h (Table 1, run 7), which may suggest that the dioxacalix[6]arene based complexes 1 and 2 can tolerate air/water during the ROP catalysis.
Table 1 ROP of ε-CL using 1–7
| Run |
Cat. |
CL:Ti:BnOH |
T/°C |
t/h |
Conv.a (%) |
M
n,GPC × 10−3b |
M
w × 10−3b |
M
n,Cal × 10−3c |
PDId |
TONf |
|
Determined by 1H NMR spectroscopy.
M
n/w, GPC values corrected considering Mark–Houwink factor (0.56) from polystyrene standards in THF.
Calculated from ([monomer]0/Ti) × conv. (%) × monomer molecular weight (MCL = 114.14) + molecular weight of BnOH.
From GPC.
Reaction performed in air.
Turnover number (TON) = number of moles of ε-CL consumed/number of moles Ti.
|
| 1 |
1
|
1000:1:1 |
130 |
8 |
80.4 |
7.10 |
9.71 |
91.98 |
1.37 |
804 |
| 2 |
1
|
500:1:1 |
130 |
8 |
84.9 |
8.44 |
13.41 |
48.66 |
1.59 |
425 |
| 3 |
1
|
250:1:1 |
130 |
8 |
92.3 |
10.58 |
18.43 |
26.55 |
1.74 |
231 |
| 4 |
1
|
100:1:1 |
130 |
8 |
93.4 |
5.04 |
6.54 |
10.87 |
1.30 |
93 |
| 5 |
1
|
250:1:1 |
100 |
8 |
74.2 |
4.87 |
5.49 |
21.38 |
1.13 |
186 |
| 6 |
1
|
250:1:1 |
80 |
8 |
28.3 |
2.95 |
3.43 |
8.28 |
1.16 |
71 |
| 7 |
1
|
250:1:1 |
130 |
8 |
85.4 |
9.43 |
12.03 |
24.58 |
1.27 |
214 |
| 8 |
2
|
250:1:1 |
130 |
8 |
93.5 |
12.54 |
15.20 |
26.89 |
1.21 |
234 |
| 9 |
3
|
250:1:1 |
130 |
8 |
62.3 |
4.37 |
6.43 |
17.99 |
1.47 |
156 |
| 10 |
4
|
250:1:1 |
130 |
8 |
67.5 |
6.19 |
9.97 |
19.47 |
1.50 |
169 |
| 11 |
5
|
250:1:1 |
130 |
8 |
77.1 |
8.21 |
12.32 |
22.21 |
1.61 |
193 |
| 12 |
6
|
250:1:1 |
130 |
8 |
84.2 |
8.84 |
13.09 |
24.10 |
1.48 |
211 |
| 13 |
7
|
250:1:1 |
130 |
8 |
25.3 |
— |
— |
— |
— |
63 |
| |
| 14 |
1
|
250:1:1 |
130 |
24 |
>99 |
11.43 |
16.32 |
28.46 |
1.42 |
248 |
| 15 |
2
|
250:1:1 |
130 |
24 |
>99 |
13.34 |
23.14 |
28.46 |
1.73 |
248 |
| 16 |
3
|
250:1:1 |
130 |
24 |
76.4 |
6.78 |
7.93 |
22.01 |
1.17 |
191 |
| 17 |
4
|
250:1:1 |
130 |
24 |
82.5 |
8.40 |
11.11 |
23.75 |
1.32 |
206 |
| 18 |
5
|
250:1:1 |
130 |
24 |
96.4 |
8.28 |
11.64 |
27.72 |
1.41 |
241 |
| 19 |
6
|
250:1:1 |
130 |
24 |
>99 |
10.04 |
15.82 |
28.46 |
1.58 |
248 |
| 20 |
7
|
250:1:1 |
130 |
24 |
34.6 |
2.28 |
2.63 |
9.98 |
1.15 |
87 |
| |
| 21 |
1
|
250:1:0 |
130 |
24 |
71.6 |
4.30 |
5.20 |
20.49 |
1.20 |
179 |
| 22 |
2
|
250:1:0 |
130 |
24 |
74.7 |
6.13 |
7.01 |
21.37 |
1.14 |
187 |
| 23 |
3
|
250:1:0 |
130 |
24 |
59.1 |
2.38 |
3.62 |
16.92 |
1.52 |
148 |
| 24 |
4
|
250:1:0 |
130 |
24 |
63.8 |
3.42 |
5.51 |
18.26 |
1.61 |
160 |
| 25 |
5
|
250:1:0 |
130 |
24 |
69.2 |
4.18 |
5.79 |
19.80 |
1.39 |
173 |
| 26 |
6
|
250:1:0 |
130 |
24 |
62.8 |
4.85 |
5.78 |
17.97 |
1.19 |
157 |
| 27 |
7
|
250:1:0 |
130 |
24 |
— |
— |
— |
— |
— |
— |
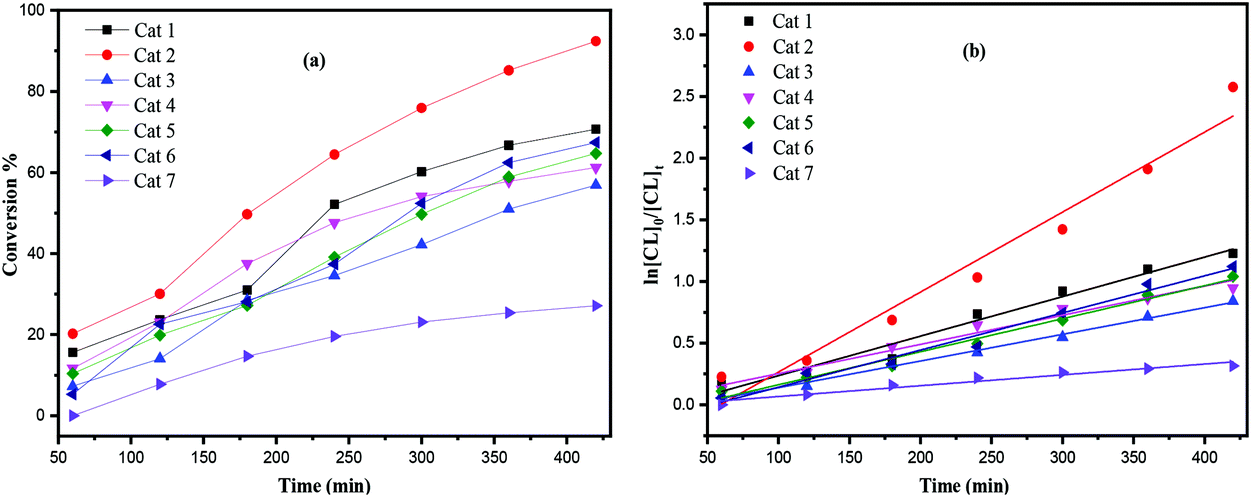
The screening of complexes 1–7 (Table 1) revealed that the titanium-based L1 complexes namely 1 and 2 herein, exhibited higher activities than other complexes under the conditions employed. After 24 h (Table 1), complexes 3, 4 and 7 afforded relatively lower conversions (<90%), whereas higher conversions (>90%) were reached using complexes 1, 2, 5 and 6, under similar conditions. From a kinetic study (Fig. 8), it was observed that the PCL polymerization rate followed the order: 2 > 1 > 5 ≈ 6 > 4 > 3 > 7. Compared with the larger titanocalix[6]arene complexes (complexes 1–6), complex 7 was found to be relatively inactive (Table 1, run 12 and 22), presumably due to the its low solubility in toluene. The observed activity of complex 2 surpassed that of the other complexes screened herein, and this may be attributed to the arrangement of and distance between the Ti centers.5b The higher activity of the chloro- (4) and bromo- (5, 6) azacalixarene titanium complexes compared with that of fluoro- (3) compound can be explained considering the lability of the ligands present. This is in line with our recent study on titanocalix[4]arenes, in which the presence of a labile ligand (i.e. MeCN and H2O) proved beneficial for the catalyst activity.51H NMR spectra of the PCL indicated the presence of an BnO end group (e.g. Fig. S4, ESI†), which agrees with the MALDI-ToF mass spectra (e.g. Fig. S1, ESI†) and indicates that the polymerization proceeded via a coordination insertion mechanism. Indeed, the MALDI-ToF spectrum of the sample displayed a major series of peaks separated by 114 m/z units accountable to two OH terminated PCL n-mers (M = 17 (OH) + 1(H) + n × 114.14 (CL) + 22.99 (Na+)) and there is a part of peaks consistent with the polymer terminated by OH and BnO end group (M = n × 114.12 (CL) + 108.05 (BnOH) + 22.99 (Na+)) and cyclic PCL (M = n × 114.12 (CL) + 22.99 (Na+)).
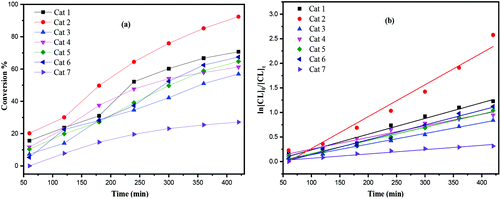 |
| | Fig. 8 (a) Relationship between conversion and time for the polymerization of ε-CL by using complex 1–7; (b) plot of ln[CL]0/[CL]tvs. time for the polymerization of ε-CL by using complexes 1–7; conditions: T = 130 °C, nMonomer:nTi:BnOH = 250:1:1. | |
δ-Valerolactone (δ-VL)
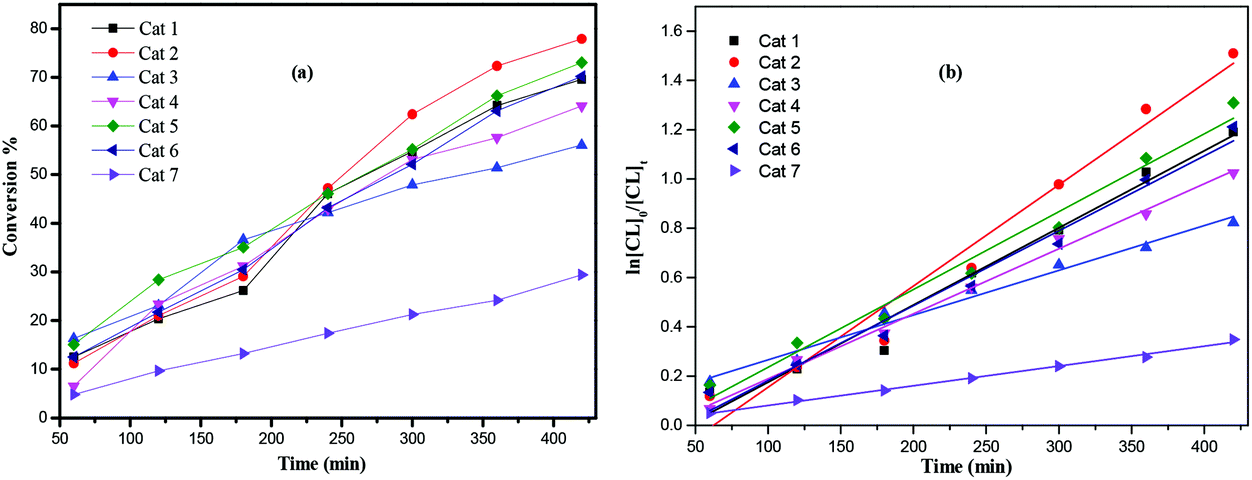
Furthermore, complexes 1–7 were also evaluated as pre-catalysts in the presence of one equivalent of BnOH for the ROP of δ-VL (Table 2). Using compound 1, the conditions of temperature and [Ti]:[δ-VL] were varied. On increasing the temperature to 130 °C and lowering the monomer to pre-catalyst ratio, best results were achieved at 130 °C using [Ti]:[δ-VL] at 1:250 over 8 h. As in the case of the ROP of ε-CL, kinetic studies (Fig. 9) revealed that the catalytic activities followed the order: 2 > 1 > 5 ≈ 6 > 4 > 3 > 7. As for the ROP of ε-Cl, nearly all observed Mn values were significantly lower than the calculated values. The MALDI-ToF mass spectra (Fig. S2, ESI†) exhibited a major family of peaks consistent with BnO end groups [M = 108.05 (BnOH) + n × 100.12 (VL) + 22.99 (Na+)], and a minor family assigned to cyclic PVL. 1H NMR spectra of the PVL also indicated the presence of an BnO end group (e.g. Fig. S5, ESI†).
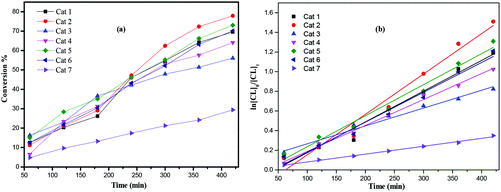 |
| | Fig. 9 (a) Relationship between conversion and time for the polymerization of δ-VL by using complex 1–7; (b) plot of ln[VL]0/[VL]tvs. time for the polymerization of δ-VL by using complexes 1–7; conditions: T = 130 °C, nMonomer:nTi:BnOH = 250:1:1. | |
Table 2 ROP of δ-VL using using 1–7
| Run |
Cat. |
VL:Ti:BnOH |
T/°C |
t/h |
Conv.a (%) |
M
n,GPC × 10−3b |
M
w × 10−3b |
M
n,Cal × 10−3c |
PDId |
TONf |
|
Determined by 1H NMR spectroscopy.
M
n/w, GPC values corrected considering Mark–Houwink factor (0.57) from polystyrene standards in THF.
Calculated from ([monomer]0/Ti) × conv. (%) × monomer molecular weight (MVL = 100.16) + molecular weight of BnOH.
From GPC.
Reaction performed in air.
Turnover number (TON) = number of moles of δ-VL consumed/number of moles Ti.
|
| 1 |
1
|
1000:1:1 |
130 |
8 |
81.2 |
11.56 |
18.23 |
81.30 |
1.58 |
812 |
| 2 |
1
|
500:1:1 |
130 |
8 |
80.1 |
12.27 |
17.54 |
40.10 |
1.43 |
401 |
| 3 |
1
|
250:1:1 |
130 |
8 |
89.4 |
13.49 |
27.46 |
22.38 |
2.03 |
224 |
| 4 |
1
|
100:1:1 |
130 |
8 |
86.7 |
5.84 |
8.12 |
8.68 |
1.38 |
87 |
| 5 |
1
|
250:1:1 |
100 |
8 |
68.1 |
4.06 |
4.67 |
17.05 |
1.15 |
170 |
| 6 |
1
|
250:1:1 |
80 |
8 |
— |
— |
— |
— |
— |
— |
| 7 |
1
|
250:1:1 |
130 |
8 |
74.6 |
10.23 |
14.56 |
18.67 |
1.42 |
187 |
| 8 |
2
|
250:1:1 |
130 |
8 |
90.8 |
16.44 |
32.07 |
22.73 |
1.95 |
227 |
| 9 |
3
|
250:1:1 |
130 |
8 |
64.6 |
6.36 |
8.02 |
16.17 |
1.26 |
162 |
| 10 |
4
|
250:1:1 |
130 |
8 |
70.3 |
7.10 |
9.71 |
17.60 |
1.36 |
176 |
| 11 |
5
|
250:1:1 |
130 |
8 |
83.3 |
9.58 |
14.59 |
20.85 |
1.52 |
208 |
| 12 |
6
|
250:1:1 |
130 |
8 |
80.6 |
10.76 |
15.96 |
20.28 |
1.48 |
202 |
| 13 |
7
|
250:1:1 |
130 |
8 |
38.1 |
2.64 |
3.24 |
9.54 |
1.22 |
95 |
ROP of r-lactide
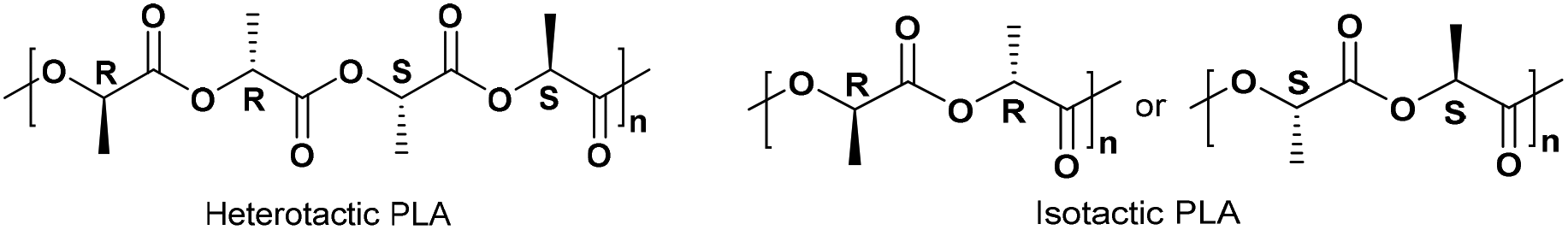
Selected complexes were also employed as pre-catalysts in the ROP of r-LA (Table 3). Best conversion was achieved in the presence of 2 (78.1%, run 2). The Mn of the polymer was lower than the calculated value albeit with narrow molecular weight distribution (7320 and 1.12, respectively). In the case of systems 1–7, all polymers obtained were of low polydispersity (PDI < 1.75), which suggested that there was reasonable control for polymerization. However, 7 only allowed for 25.6% monomer conversion affording low molecular weight species. 1H NMR spectra of the PLA indicated the presence of an BnO end group (e.g. Fig. S6, ESI†), which agrees with the MALDI-ToF mass spectra (e.g. Fig. S3, ESI†). The sample was analysed by MALDI-ToF mass spectra in positive-linear mode, the expected series corresponding to repeating unit mass of 72/144 for half/full LA was observed and the polymer chain was terminated by OH and BnO end group [M = 108.05 (BnOH) + n × 72.06 (C3H4O2) + 22.99 (Na+)]. The syndiotactic bias was determined by 2D J-resolved 1H NMR spectroscopy, investigating the methine area (5.13–5.20 ppm) of the spectra (e.g. Fig. S8, ESI†).10 The peaks were assigned to the corresponding tetrads according to the literature.10 For rac-lactide, when Pr = 0.5, the afforded PLA is an atactic polymer, and when Pr = 0, an isotactic polymer. The observed values herein (Pr = 0.39–0.52) suggested the catalysts afforded almost heterotactic polymers (Chart 3).
 |
| | Chart 3 Microstructure of heterotactic and isotactic poly-(rac-lactide).11 | |
Table 3 ROP of rac-lactide using complexes 1–7
| Run |
Cat. |
LA:Ti:BnOH |
T/°C |
t/h |
Conv.a (%) |
M
n,GPC × 10−3b |
M
w × 10−3b |
Prc |
M
n,Cal × 10−3d |
PDIe |
TONf |
|
Determined by 1H NMR spectroscopy on crude reaction mixture.
M
n/w, GPC values corrected considering Mark–Houwink factor (0.58) from polystyrene standards in THF.
From 2D J-resolved 1H NMR spectroscopy.
Calculated from ([monomer]0/Ti) × conv. (%) × monomer molecular weight (MLA = 144.13) + molecular weight of BnOH.
From GPC.
Turnover number (TON) = number of moles of r-LA consumed/number of moles Ti.
|
| 1 |
1
|
250:1:1 |
130 |
24 |
75.4 |
5.98 |
10.27 |
0.52 |
27.16 |
1.72 |
189 |
| 2 |
2
|
250:1:1 |
130 |
24 |
78.1 |
7.32 |
8.23 |
0.40 |
28.14 |
1.12 |
195 |
| 3 |
3
|
250:1:1 |
130 |
24 |
36.9 |
5.86 |
8.75 |
0.58 |
13.29 |
1.49 |
92 |
| 4 |
4
|
250:1:1 |
130 |
24 |
54.3 |
5.10 |
7.57 |
0.39 |
19.56 |
1.43 |
136 |
| 5 |
5
|
250:1:1 |
130 |
24 |
59.1 |
4.98 |
7.02 |
0.46 |
21.29 |
1.41 |
148 |
| 6 |
6
|
250:1:1 |
130 |
24 |
62.6 |
4.38 |
5.39 |
0.41 |
22.66 |
1.05 |
157 |
| 7 |
7
|
250:1:1 |
130 |
24 |
25.6 |
3.46 |
4.86 |
0.42 |
9.33 |
1.18 |
64 |
Co-polymerization of r-LA and ε-CL
The co-polymerization of r-LA and ε-CL was next investigated (Table 4). The complexes exhibited moderate conversions, with complex 2 performing best (85.1%), and with 1 and 3–6 also producing conversions >70%. In general, the systems appeared to be relatively well behaved with PDIs in the range 1.16–1.95; 1H NMR spectra were consistent with the presence of BnO and OH end groups (Fig. S7, ESI†). The composition of the copolymer was further investigated by 13C NMR spectroscopy. In fact, diagnostic resonances belonging to CL–CL–CL, LA–CL–CL, CL–CL–LA, LA–CL–LA, LA–LA–CL, CL–LA–LA and LA–LA–LA dyads can be observed in the region between δ 173.6 and 169.6 ppm (Fig. S9, ESI†). Based on the current results, the number-average sequence length was found to be 1.18 and 5.10 for CL and LA, respectively (Fig. S9, eqn (1)–(2), ESI†). Furthermore, no peaks corresponding to the CL–LA–CL triad at 171.1 ppm was observed. Such signals arise from the transesterification of the cleavage of the lactyl–lactyl bond in the lactidyl unit.12
Table 4 ROP of co-polymer (r-LA + ε-CL) using 1–7
| Runa |
Cat. |
LA:CL:Ti:BnOH |
t/h |
T/°C |
Conv.b (%) |
M
n,GPC × 10−3c,d |
M
w × 10−3c,d |
PDIc |
|
Testing method: rac-lactide was firstly added and heating for 24 h, then ε-caprolactone was added and heating for 24 h.
Determined by 1H NMR spectroscopy on crude reaction mixture based on ε-CL.
From GPC.
M
n values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor Mn/w GPC = [(Mn/w measured × 0.56 × (1 − %CL) + Mn/w measured × 0.58 × (1 − %LA)].
|
| 1 |
1
|
250:250:1:1 |
130 |
24 |
80.2 |
14.16 |
26.71 |
1.88 |
| 2 |
2
|
250:250:1:1 |
130 |
24 |
85.1 |
16.44 |
32.07 |
1.95 |
| 3 |
3
|
250:250:1::1 |
130 |
24 |
72.4 |
9.48 |
14.23 |
1.50 |
| 4 |
4
|
250:250:1:1 |
130 |
24 |
75.9 |
10.63 |
17.37 |
1.63 |
| 5 |
5
|
250:250:1:1 |
130 |
24 |
82.7 |
14.42 |
21.98 |
1.52 |
| 6 |
6
|
250:250:1:1 |
130 |
24 |
80.1 |
15.01 |
22.88 |
1.52 |
| 7 |
7
|
250:250:1:1 |
130 |
24 |
33.8 |
2.35 |
2.75 |
1.16 |
Table 5 Crystal structure data for 1·3.5MeCN, 2·MeCN, 3·2.5MeCN, 4·5MeCN, 5·4.5MeCN, 6·7MeCN, 7·2MeCN
| Compound |
1·3.5MeCN |
2·MeCN |
3·2.5MeCN |
4·5MeCN |
| Formula |
C83H109.5N4.5O10Ti2 |
C137H167NO18Ti4 |
C57H63.5F14N4.5O6Ti4 |
C62H75Cl4N7O9Ti2 |
| Formula weight |
1426.04 |
2307.30 |
1365.14 |
1299.86 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/c |
C2/c |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Unit cell dimensions |
|
a (Å) |
12.0347(2) |
28.1243(7) |
16.1213(2) |
12.6749(8) |
|
b (Å) |
28.3198(4) |
17.7721(4) |
22.8130(2) |
14.5410(8) |
|
c (Å) |
24.5367(3) |
31.2326(9) |
17.0185(2) |
18.7696(10) |
|
α (°) |
90 |
90 |
90 |
111.979 |
|
β (°) |
103.2370(10) |
113.009(3) |
102.381(10) |
95.829 |
|
γ (°) |
90 |
90 |
90 |
96.158 |
|
V (Å3) |
8140.4(2) |
14369.0(7) |
6113.42(12) |
3151.55(3) |
|
Z
|
4 |
4 |
4 |
2 |
| Temperature (K) |
100(2) |
100(2) |
100(2) |
100(2) |
| Wavelength (Å) |
1.54184 |
1.54178 |
1.54178 |
1.54178 |
| Calculated density (g cm−3) |
1.147 |
1.061 |
1.416 |
1.240 |
| Absorption coefficient (mm−1) |
2.112 |
2.275 |
5.091 |
4.142 |
| Transmission factors (min./max.) |
0.69725 and 1.0000 |
0.52429 and 1.000 |
0.6143 and 1.0000 |
0.679 and 1.0000 |
| Crystal size (mm3) |
0.200 × 0.120 × 0.040 |
0.160 × 0.030 × 0.010 |
0.150 × 0.080 × 0.050 |
0.090 × 0.060 × 0.020 |
|
θ(max) (°) |
66.6 |
66.5 |
70.4 |
68.2 |
| Reflections measured |
75678 |
46013 |
55482 |
134309 |
| Unique reflections |
14383 |
12487 |
11396 |
11490 |
|
R
int
|
0.0521 |
0.0985 |
0.0415 |
0.0270 |
| Reflections with F2 > 2σ(F2) |
12259 |
7981 |
9829 |
11030 |
| Number of parameters |
870 |
697 |
741 |
701 |
|
R
1 [F2 > 2σ(F2)] |
0.117 |
0.085 |
0.063 |
0.031 |
| wR2 (all data) |
0.368 |
0.234 |
0.178 |
0.091 |
| GOOF, S |
1.675 |
1.024 |
1.016 |
1.050 |
| Largest difference peak and hole (e Å−3) |
1.961 and −0.795 |
0.768 and −0.381 |
1.500 and −0.511 |
0.831 and −0.376 |
| Compound |
5·4.5MeCN |
6·7MeCN |
7·2MeCN |
| Formula |
C61H73.5Br4N6.5O9Ti2 |
C78H93Br12N15O6Ti4 |
C40H46F10N4O4SiTi2 |
| Formula weight |
1457.15 |
2487.02 |
960.70 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P |
P |
P |
| Unit cell dimensions |
|
a (Å) |
12.8059(9) |
17.7979(2) |
8.8857(4) |
|
b (Å) |
14.7294(9) |
18.1314(1) |
13.0733(5) |
|
c (Å) |
18.6847(13) |
18.4582(2) |
18.4231(11) |
|
α (°) |
111.546(5) |
71.110(1) |
84.872(4) |
|
β (°) |
94.299(6) |
74.116(1) |
82.208(4) |
|
γ (°) |
96.876(5) |
66.857(1) |
73.277(4) |
|
V (Å3) |
3227.4(4) |
5108.21(10) |
2027.83(18) |
|
Z
|
2 |
1 |
2 |
| Temperature (K) |
150(2) |
100(2) |
100(2) |
| Wavelength (Å) |
0.71073 |
1.54178 |
0.71075 |
| Calculated density (g cm−3) |
1.394 |
1.549 |
1.573 |
| Absorption coefficient (mm−1) |
2.775 |
8.449 |
0.517 |
| Transmission factors (min./max.) |
0.625 and 0.825 |
0.66859 and 1.0000 |
0.20966 and 1.0000 |
| Crystal size (mm3) |
0.360 × 0.260 × 0.200 |
0.120 × 0.080 × 0.050 |
0.100 × 0.060 × 0.020 |
|
θ(max) (°) |
26.373 |
68.236 |
27.529 |
| Reflections measured |
25790 |
237393 |
14106 |
| Unique reflections |
13079 |
18616 |
14106 |
|
R
int
|
0.0792 |
0.0693 |
0.1992 |
| Reflections with F2 > 2σ(F2) |
8355 |
18616 |
10493 |
| Number of parameters |
699 |
986 |
547 |
|
R
1 [F2 > 2σ(F2)] |
0.0458 |
0.0662 |
0.105 |
| wR2 (all data) |
0.1066 |
0.1895 |
0.2964 |
| GOOF, S |
0.859 |
1.021 |
1.026 |
| Largest difference peak and hole (e Å−3) |
0.894 and −0.841 |
1.542 and −1.156 |
2.991 and −0.829 |
Kinetics
From a kinetic study of the ROP of ε-CL using 1–7, it was observed that the polymerization rate exhibited first-order dependence on the ε-CL concentration (Fig. 8(a)), and the conversion of monomer achieved over 420 min was >25%. The activity trend in this case revealed that 2 was the most active and then 2 > 1 > 5 ≈ 6 > 4 > 3 > 7. An induction period of 2 hours observed for complexes 1–6 could be ascribed to the longer time required for the formation of the catalytically active species. A similar result was also observed in the polymerization of δ-VL (Fig. 9).
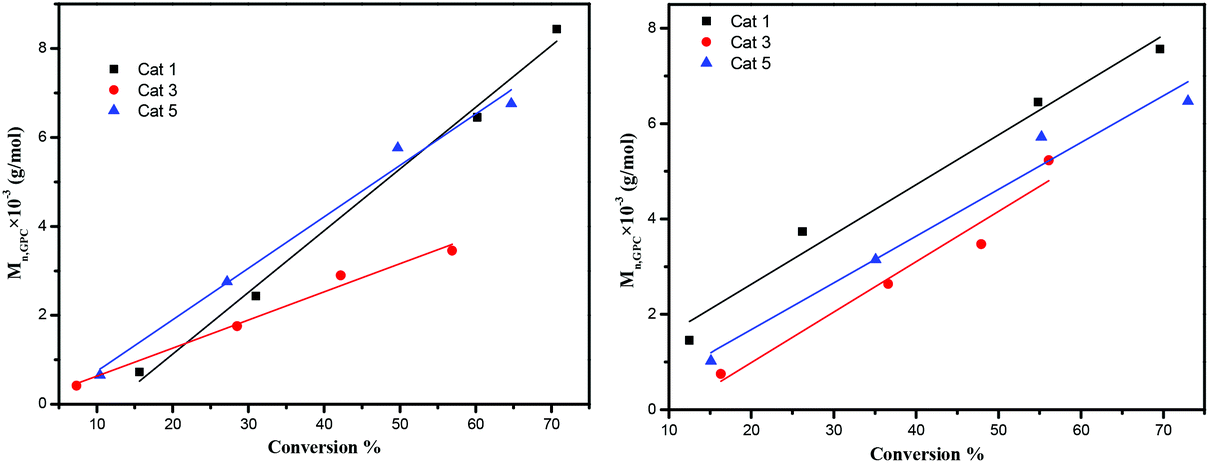
The dependence of the Mn and molecular weight distribution on the monomer conversion in the reactions catalyzed by 1, 3, 5 with BnOH was also investigated (Fig. 10). For the ROP of ε-CL, the polymer Mn was shown to increase linearly with the conversion, which suggested that the polymerization was well controlled (Fig. 10, left). A similar outcome was also observed in the reaction involving δ-VL (Fig. 10, right).
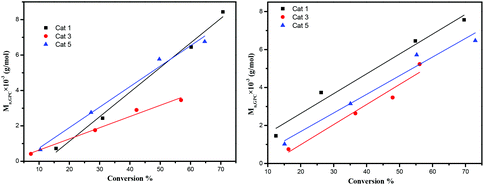 |
| | Fig. 10 Left: Mnvs. monomer conversion in the ROP of ε-CL by using 1, 3 and 5; right: Mnvs. monomer conversion in the ROP of δ-VL by using 1, 3 and 5; conditions: T = 130 °C, nMonomer:nTi:BnOH = 250:1:1. | |
ROP of ω-pentadecalactone
To enhance the thermal properties of the polymers obtained herein, we also investigated the ROP of the ω-pentadecalactone. Unfortunately, none of the systems herein proved to be effective as catalysts for the ROP of ω-pentadecalactone either in solution at high temperatures (130 °C) or as melts.
Conclusions
In this work, we report rare examples of metal (here titanium) complexes of larger dioxacalix[6]arenes and extend the work to include even rarer examples of titanium complexes bearing azacalixarenes. The molecular structures reveal how these macrocycles can support multiple metal centres which adopt some interesting structural motifs. The complexes are active for the ring opening polymerization of ε-caprolactone (ε-CL), δ-valerolactone (δ-VL) and rac-lactide (r-LA) but not ω-pentadecalactone. In all cases, the oxo complex [Ti4O4(L1)2]·MeCN (2·MeCN) proved to be the most active with first order kinetics.
Experimental
General
All reactions were conducted under an inert atmosphere using standard Schlenk techniques. Toluene was dried from sodium, acetonitrile was distilled from calcium hydride, diethylether was distilled from sodium benzophenone, and all solvents were degassed prior to use. The dioxacalix[6]arene and azacalixarenes were prepared according to the literature methods.13 All other chemicals were purchased from commercial sources. IR spectra (nujol mulls, KBr windows) were recorded on a Nicolet Avatar 360 FT IR spectrometer; 1H NMR spectra were recorded at room temperature on a Varian VXR 400 S spectrometer at 400 MHz or a Gemini 300 NMR spectrometer or a Bruker Advance DPX-300 spectrometer at 300 MHz. The 1H NMR spectra were calibrated against the residual protio impurity of the deuterated solvent. Elemental analyses were performed by the elemental analysis service at the University of Hull. Matrix Assisted Laser Desorption/Ionization Time of Flight (MALDI-TOF) mass spectrometry was performed in a Bruker autoflex III smart beam in linear mode, and the spectra were acquired by averaging at least 100 laser shots. 2,5-Dihydroxybenzoic acid was used as the matrix and THF as solvent. Sodium chloride was dissolved in methanol and used as the ionizing agent. Samples were prepared by mixing 20 μl of matrix solution in THF (2 mg mL−1) with 20 μL of matrix solution (10 mg mL−1) and 1 μL of a solution of ionizing agent (1 mg mL−1). Then 1 mL of these mixtures was deposited on a target plate and allowed to dry in air at ambient temperature.
Synthesis of [Ti2(OiPr)2(MeCN)L1]·3.5MeCN (1·3.5MeCN)
To [Ti(OiPr)4] (0.43 g, 1.50 mmol) and L1H6 (0.50 g, 0.49 mmol) was added toluene (30 mL) and then the system was refluxed for 12 h. On cooling, the volatiles were removed in vacuo, and the residue was extracted into warm MeCN (30 mL). On prolonged standing at 0 °C, an orange crystalline material formed (Fig. S10, ESI†), yield 0.32 g, 52%. Anal. calcd for C82H108N4O10Ti2 (sample dried in vacuo for 12 h, −3.5MeCN): C, 71.60; H, 7.06; found C, 71.92; H, 7.28%. IR (nujol mull, KBr): 3231w, 2923s, 2853s, 2349w, 1641.9w, 1461m, 1413w, 1377m, 1303w, 1260s, 1212w, 1092s, 1019s, 863w, 799s. 1H NMR (CDCl3) δ: 6.86–7.32 (m, 12H, arylH), 5.44 (m, 2H, –OCH(CH3)2), 5.04–5.21 (m, 4H, –OCH2–), 4.87 (d, J = 4.8 Hz, 4H, –OCH2–), 4.48 (m, 4H, –CH2–), 3.40 (d, J = 12.4 Hz, 4H, –CH2–), 2.03 (s, 3H, MeCN), 1.56 (m, 12H, –OCH(CH3)2), 2.03 (s, 3H, MeCN) 1.21–1.34 (m, 54H, –C(CH3)3). Mass Spec (EI): 1267.6 [M + Na+ − 3.5MeCN].
Synthesis of [Ti4(μ3-O)2(H2O)(L1)2]·MeCN (2·MeCN)
As for 1, but using [Ti(OiPr)4] (0.29 g, 1.00 mmol) and L1H6 (0.50 g, 0.49 mmol) affording 2 as orange prisms. Single orange prisms were grown from a saturated MeCN (30 mL) solution at 0 °C (yield 0.25 g, 44%). Anal. calcd for C135H164O18Ti4 (sample dried in vacuo for 12 h, –MeCN): C, 71.54; H, 7.29; found C, 71.91; H, 7.38%; IR (nujol mull, KBr): 2726w, 2359w, 2340w, 1651w, 1463s, 1377s, 1301m, 1260m, 1209m, 1080m, 1020m, 941w, 926w, 899w, 899m, 798s. 1H NMR (CDCl3) δ: 6.89–7.32 (m, 12H, arylH), 5.36 (d, J = 6.8 Hz, 4H, –OCH2–), 5.02–5.17 (m, 4H, –OCH2–), 4.88 (d, d, J = 6.8 Hz, 4H, –OCH2–), 4.64 (d, J = 6.8 Hz, 4H, –OCH2–), 4.34 (m, 4H, –CH2–), 3.49–3.87 (m, 8H, –CH2–), 2.01 (s, 3H, MeCN), 1.13–1.36 (m, 108H, –C(CH3)3). Mass Spec (EI): 2292 [M].
Synthesis of [Ti4F14L2H2(H)2]·2.5MeCN (3·2.5MeCN)
As for 1, but using [TiF4] (0.31 g, 2.48 mmol) and L2H6 (0.50 g, 0.62 mmol) affording 3 as red prisms. Single orange prisms were grown from a saturated MeCN (30 mL) solution at room temperature (yield 0.44 g, 56%). Anal. calcd for C52H56F14N2O6Ti4 (sample dried in vacuo for 12 h, −2.5MeCN): C, 49.47; H, 4.47; N, 2.22%. Found C, 50.03; H, 4.85; N, 2.06%. IR (nujol mull, KBr): 3849w, 3435s, 1737w, 1692w, 1552w, 1536s, 1461m, 1383m, 1260s, 1220w, 1093s, 1020s, 928w, 866m, 800m, 688m. 1H NMR (CDCl3) δ: 6.98–7.50 (m, 12H, arylH), 3.48–3.73 (m, 8H, –CH2–), 3.22–3.42 (m, 8H, –NCH2–), 2.34 (s, 6H, –NCH3), 2.05–2.18 (m, 18H, –CH3). 19F NMR (CDCl3) δ: −110.18 (bs, 6F), −98.32 (bs, 6F), −69.12 (bs, 2F). Mass Spec (EI): 1283 [M + Na+ − 2.5MeCN].
Synthesis of [Ti2Cl4(H2O)2OL2H2(H)2]·4.5MeCN (4·5MeCN)
As for 1, but using [TiCl4(THF)2] (0.41 g, 1.24 mmol) and L2H6 (0.50 g, 0.62 mmol) affording 4 as dark red prisms. Single orange prisms were grown from a saturated MeCN (30 mL) solution at room temperature (yield 0.28 g, 41%). Anal. calcd for C52H60Cl4N2O9Ti2 (sample dried in vacuo for 12 h, −5MeCN): C, 57.06; H, 5.53; N, 2.56%. Found C, 56.39; H, 5.25; N, 2.26%., IR (nujol mull, KBr): 3428m, 2745w, 2365w, 2278m, 1705m, 1628w, 1425s, 1357s, 1325m. 1232m, 1029s, 1015w, 799m, 754s. 1H NMR (CDCl3) δ: 6.88–7.49 (m, 12H, arylH), 3.71 (d, J = 4.8 Hz, 8H, –CH2–), 3.56 (d, J = 4.8 Hz, 8H, –NCH2–), 2.34 (s, 6H, –NCH3), 2.00–2.26 (m, 18H, –CH3), 1.53 (s, 2H, H2O). Mass Spec (EI): 1053 [M − 2H2O − 5MeCN].
Synthesis of [Ti2Br4(H2O)2OL2H2(H)2]·4.5MeCN (5·4.5MeCN)
As for 1, but using [TiBr4] (0.46 g, 1.24 mmol) and L2H6 (0.50 g, 0.62 mmol) affording 5 as dark orange prisms. Single orange prisms were grown from a saturated MeCN (30 mL) solution at room temperature (yield 0.25 g, 32%). Anal. calcd for C54H63Br4N3O9Ti2 (sample dried in vacuo for 12 h, −3.5MeCN): C, 49.38; H, 4.83; N, 3.20%. Found C, 49.62; H, 5.03; N, 2.86%. IR (nujol mull, KBr): 3427m, 2729w, 2348w, 2285m, 2248w, 1605m, 1461s, 1377s, 1302w. 1260s, 1156w, 1039m, 1021m, 863m, 800s. 1H NMR (CDCl3) δ: 1H NMR (CDCl3) δ: 6.80–7.38 (m, 12H, arylH), 3.79 (m, 8H, –CH2–), 3.52 (m, 8H, –NCH2–), 2.30 (s, 6H, –NCH3), 2.05–2.23 (m, 18H, –CH3), 1.51 (s, 2H, H2O). Mass Spec (EI): 1274 [M − 4.5MeCN].
Synthesis of [Ti4Br12L2(H)2(MeCN)6]·7MeCN (6·7MeCN)
As for 1, but using [TiBr4] (1.00 g, 2.69 mmol) and L2H6 (0.50 g, 0.62 mmol) affording 6 as orange/red prisms. Single orange prisms were grown from a saturated MeCN (30 mL) solution at 0 °C (yield 0.34 g, 26.0%). Anal. calcd for C60H66Br12N6O6Ti4 (sample dried in vacuo for 24 h, −3MeCN): anal. calcd for C, 34.03 H, 3.14; N, 3.97%. Found: C, 33.59 H, 3.10; N, 4.42%. IR (nujol mull, KBr): 2957s, 2852s, 2727w, 2350w, 2283w, 1693w, 1645m, 1567w, 1456s, 1377s, 1308w, 1259m, 1094m, 1019m, 927w, 856m, 800s. 1H NMR (CD2Cl2) δ: 7.24–6.81 (m, 12H, arylH), 3.91 (m, 8H, –CH2–), 3.41 (m, 8H, –NCH2–), 2.88 (s, 6H, –NCH3), 2.24 (m, 18H, –CH3), 1.95 (s, 12H, MeCN).
Synthesis of [Ti2F2(μ-F)3L3(H)2(SiF5)]·2MeCN (7·2MeCN)
As for 1, but using [TiF4] (0.44 g, 3.53 mmol) and L2H4 (0.50 g, 0.88 mmol) affording 7 as orange/red prisms. Single orange prisms were grown from a saturated MeCN (30 mL) solution at 0 °C (yield 0.15 g, 17%). Anal. calcd for C36H40F10N2SiO4Ti2 (sample dried in vacuo for 12 h, −2MeCN): anal. calcd for C, 49.22 H, 4.59; N, 3.19%. Found C, 49.82; H, 5.03; N, 3.59%. IR (nujol mull, KBr): 3353w, 2729w, 2360m, 2340w, 2251w, 1606w, 1463s, 1377s, 1304w, 1260m, 1231m, 1162w, 1093m, 1019m, 945w, 927w, 870m, 801m. 1H NMR (CDCl3) δ: 6.98–7.50 (m, 8H, arylH), 3.72 (m, 4H –CH2–), 3.46 (m, 8H, –NCH2–), 2.34(s, 6H, –CH3) 1.99 (s, 12H, –NCH3). 19F NMR (CDCl3) δ: −107.27 (bs, 5F), −82.16 (bs, 3F), −72.06 (bs, 2F). Mass Spec (EI): 879 [M − 2MeCN].
Procedure for ROP of ε-caprolactone, δ-valerolactone and rac-lactide
A toluene solution of pre-catalyst (0.010 mmol, 1.0 mL toluene) was added into a Schlenk tube in the glove-box at room temperature. The solution was stirred for 2 min, and then the appropriate equivalent of BnOH (from a pre-prepared stock solution of 1 mmol BnOH in 100 mL toluene) and the appropriate amount of ε-CL, δ-VL or r-LA along with 1.5 mL toluene was added to the solution. The reaction mixture was then placed into an oil/sand bath pre-heated at 130 °C, and the solution was stirred for the prescribed time (8 or 24 h). The polymerization mixture was quenched on addition of an excess of glacial acetic acid (0.2 mL) into the solution, and the resultant solution was then poured into methanol (200 mL). The resultant polymer was then collected on filter paper and was dried in vacuo.
Kinetic studies
The polymerizations were carried out at 130 °C in toluene (2 mL) using 0.010 mmol of complex. The molar ratio of monomer to initiator to co-catalyst was fixed at 250:1:1, and at appropriate time intervals, 0.5 μL aliquots were removed (under N2) and were quenched with wet CDCl3. The percent conversion of monomer to polymer was determined using 1H NMR spectroscopy.
X-ray crystallography
In all cases, crystals suitable for an X-ray diffraction study were grown from a saturated MeCN solution at either ambient temperature or 0 °C. Single crystal X-ray diffraction data (except 5) were collected at the UK National Crystallography service using Rigaku Oxford Diffraction ultra-high intensity instruments employing modern areas detectors. X-ray diffraction data for 5·4.5MeCN were collected using a Stoe IPDS2 image plate diffractometer operating with molybdenum radiation. In all cases standard procedures were employed for integration and processing of data.
Crystal structures were solved using dual space methods implemented within SHELXT.14 Completion of structures was achieved by performing least squares refinement against all unique F2 values using SHELXL-2018.15 All non-H atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed using a riding model. Where the location of hydrogen atoms was obvious from difference Fourier maps, C–H and O–H bond lengths were refined subject to chemically sensible restraints. Minor disorder was treated using standard methods.
SQUEEZE16 was used to model the disordered solvent in structures 1, 3, 4, 5 and 6.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the China Scholarship Council (CSC) for a PhD Scholarship to TX. The EPSRC Mass Spectrometry Service (Swansea, UK) and the EPSRC National X-ray Crystallography Service (Southampton) are thanked for data collection. CR also thanks the EPSRC (grants EP/S0225537/1 and EP/R023816/1) for financial support.
References
-
(a) D. H. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS PubMed;
(b)
Y. Li, K. Zhao, C. Redshaw, B. A. Ortega, A. Y. Nuñez and T. A. Hanna, in Coordination Chemistry and Applications of Phenolic Calixarene–metal Complexes, ed. Z. Rappoport, PATAI'S Chemistry of Functional Groups, 2014, pp. 1–164 Search PubMed.
-
(a) B. König and M. H. Fonseca, Eur. J. Inorg. Chem., 2000, 2303–2310 CrossRef;
(b) P. Lhoták, Eur. J. Org. Chem., 2004, 1675–1692 CrossRef;
(c) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291–5316 CrossRef CAS PubMed;
(d) H. Tsue, K. Ishibashi and R. Tamura, Top. Heterocycl. Chem., 2008, 17, 73–96 CAS;
(e) K. Cottet, P. M. Marcos and P. J. Cragg, Beilstein J. Org. Chem., 2012, 8, 201–226 CrossRef CAS PubMed;
(f)
Advances in Organic Crystal Chemistry, ed. R. Tamura and M. Miyata, 2015, ch. 13, pp. 241–261 Search PubMed;
(g) H. Takemura, T. Shinmyozu, H. Miura and I. U. Khan, J. Inclusion Phenom. Macrocyclic Chem., 1994, 19, 193–206 CrossRef CAS.
-
(a) C. Desroches, G. Pilet, S. A. Borshch, S. Parola and D. Luneau, Inorg. Chem., 2005, 44, 9112–9120 CrossRef CAS PubMed;
(b) R. Kuriki, T. Kuwabara and Y. i. Ishii, Dalton Trans., 2020, 49, 12234–12241 RSC;
(c) R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824–4870 RSC;
(d) P. Thuéry, M. Nierlich, J. Vicens and H. Takemura, Dalton Trans., 2000, 279–283 RSC;
(e) C. Redshaw, M. Rowan, L. Warford, D. M. Homden, A. Arbaoui, M. R. J. Elsegood, S. H. Dale, T. Yamato, C. P. Casas, S. Matsui and S. Matsuura, Chem. – Eur. J., 2007, 13, 1090–1107 CrossRef CAS PubMed;
(f) T. Xing, T. J. Prior, M. R. J. Elsegood, N. V. Semikolenova, I. E. Soshnikov, K. Bryliakov, K. Chen and C. Redshaw, Catal. Sci. Technol., 2021, 11, 624–636 RSC.
- M. Frediani, D. Sémeril, A. Marrioti, L. Rosi, P. Frediani, L. Rosi, D. Matt and L. Toupet, Macromol. Rapid Commun., 2008, 29, 1554–1560 CrossRef CAS.
-
(a) Z. Sun, Y. Zhao, O. Santoro, M. R. J. Elsegood, E. V. Bedwell, K. Zahra, A. Walton and C. Redshaw, Catal. Sci. Technol., 2020, 10, 1619–1639 RSC;
(b) O. Santoro, M. R. J. Elsegood, E. V. Bedwell, J. A. Pryce and C. Redshaw, Dalton Trans., 2020, 49, 11978–11996 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
-
(a) N. Li, J.-J. Liu, J.-W. Sun, B.-X. Dong, L.-Z. Dong, S.-J. Yao, Z. Xin, S.-L. Li and Y. Q. Lan, Green Chem., 2020, 22, 5325–5332 RSC;
(b) X.-X. Yang, W.-D. Yu, X.-Y. Yi and C. Liu, Inorg. Chem., 2020, 59, 7512–7519 CrossRef CAS PubMed;
(c) X. –X. Yang, W.-D. Yu, X.-Y. Yi, L. –J. Li and C. Liu, Chem. Commun., 2020, 56, 14035–14038 RSC.
- J. P. Fackler, Jr., F. J. Kristine, A. M. Mazany, T. J. Moyer and R. E. Shepherd, Inorg. Chem., 1985, 24, 1857–1860 CrossRef.
- A. C. Cooper, J. C. Bollinger, J. C. Huffman and K. G. Caulton, New J. Chem., 1998, 22, 473–480 RSC.
-
(a) C. Ludwig and M. R. Viant, Phytochem. Anal., 2010, 21, 22–32 CrossRef CAS PubMed;
(b) M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892–1898 CrossRef CAS.
-
(a) Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS PubMed;
(b) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed.
-
(a) F. D. Monica, E. Luciano, A. Buonerba, A. Grassi, S. Milione and C. Capacchione, RSC Adv., 2014, 4, 51262–51267 RSC;
(b) P. Vanhoorne, P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 1992, 25, 37–44 CrossRef CAS;
(c) J. Kasperczyk and M. Bero, Makromol. Chem., 1991, 192, 1777–1787 CrossRef CAS;
(d) J. Kasperczyk and M. Bero, Makromol. Chem., 1993, 194, 913–925 CrossRef CAS;
(e) N. Nomura, A. Akita, R. Ishii and M. Mizuno, J. Am. Chem. Soc., 2010, 132, 1750–1751 CrossRef CAS PubMed;
(f) G. Li, M. Lamberti, D. Pappalardo and C. Pellecchia, Macromolecules, 2012, 45, 8614–8620 CrossRef CAS.
-
(a) B. Dhawan and C. D. Gutsche, J. Org. Chem., 1983, 48(9), 1536–1539 CrossRef CAS;
(b) B. Masci, J. Org. Chem., 2001, 66, 1497–1499 CrossRef CAS PubMed;
(c) H. Takemura, A. Takahashi, H. Suga, M. Fukuda and T. Iwanaga, Eur. J. Org. Chem., 2011, 3171–3177 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2057320–2057326 (1·3.5MeCN, 2·MeCN, 3·2.5MeCN, 4·5MeCN, 5·4.5, 6·7MeCN, 7·2MeCN). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00189b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kai
Chen
a,
Kai
Chen