
Aqueous solubility of Pb at equilibrium with hydroxypyromorphite over a range of phosphate concentrations
Yuting
Zhou
b,
Xinxin
Li
c and
Murray B.
McBride
 *a
*a
aSection of Soil and Crop Sciences, Cornell University, Ithaca, NY 14850, USA. E-mail: mbm7@cornell.edu; Fax: +1-607-255-2644; Tel: +1-607-255-1728
bCollege of Environmental and Resource Science, Zhejiang University, Hangzhou, China
cSchool of Environmental Science and Engineering, Shandong University, Qingdao, China
First published on 23rd December 2020
Abstract
Hydroxypyromorphite (HPM) is a low-solubility Pb phosphate mineral that has the potential to limit solubility and bioavailability of Pb in soils and water. Because of reported uncertainty regarding the solubility product of this important mineral, we re-evaluated the solubility of Pb and activity of the free Pb2+ ion in aqueous suspensions of microcrystalline HPM equilibrated up to 30 days over a wide range of added soluble phosphate. A small addition of phosphate (0.1 mM) reduced Pb solubility as measured by ICP-OES, but greater phosphate additions (up to 50 mM) had no further effect in lowering HPM solubility. However, free Pb2+ ion activity measured by ion-selective electrode progressively decreased from about 10−6.5 with no added phosphate to 10−9 as soluble phosphate was increased. The effect of soluble phosphate in lowering Pb2+ activity is attributed to inhibited dissolution of HPM as well as increased Pb2+-phosphate ion pair formation in solution at higher solution concentrations of phosphate. Measurement of the ion activity products (IAP) of the solutions at equilibrium with HPM gave highly variable IAP values that were sensitive to pH and were generally not consistent with the reported solubility product of this mineral. The high variability of the IAPs for solutions with variable pH and phosphate concentrations indicates that dissolution–precipitation reactions of HPM are not described by a constant solubility product at equilibrium, possibly because of the incongruent dissolution behavior of this mineral at near-neutral pH.
Environmental significanceThe solubility, lability and bioavailability of Pb in soils and water can potentially be reduced by treatment with phosphate to produce the highly stable hydroxypyromorphite. Because past research has shown this Pb apatite to display incongruent dissolution behavior in water, we tested the classical thermodynamic model of constant solubility product for hydroxypyromorphite over a range of dissolved phosphate concentrations. Small additions of soluble phosphate reduced dissolved Pb as well as free Pb2+ activity. However, the measured ion activity products (IAPs) of the solution phase in equilibrium with crystalline pyromorphite varied widely as a function of pH and soluble phosphate, indicating that Pb solubility and free Pb2+ ion activity cannot be predicted accurately using the standard assumption of a constant solubility product. |
Introduction
Pyromorphites are low-solubility Pb phosphate minerals of the apatite-group that may limit Pb solubility in contaminated soils and water when significant concentrations of soluble phosphate are present.1,2 For example, hydroxypyromorphite (HPM), Pb5(PO4)3OH, is commonly formed under natural conditions in soils,3 and is also reported to be involved in limiting Pb solubility in phosphate-treated drinking water.4,5 Although chloropyromorphite, Pb5(PO4)3Cl, is considered to be more stable than HPM in natural waters containing chloride based on its lower calculated solubility, the former mineral is not generally detected in phosphate-dosed “hard” water in lead pipes. In fact, mixed Ca–Pb apatite minerals tend to be formed rather than pure Pb mineral phases in these natural waters.2,6 Solid solutions of Pb with Ca could theoretically limit Pb solubility to lower levels than pure Pb5(PO4)3OH.7HPM has a dissolution reaction which can be described as follows:
Pb5(PO4)3OH (s) + 7H+ ⇌ 5Pb2+ + 3H2PO4− + H2O, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KSP = −4.14 KSP = −4.14 | (1) |
Therefore, the equation for the solubility product, KSP, of HPM is expressed as:
| logKSP = 5log(Pb2+) + 3.0log(H2PO4−) + 7pH = −4.14 | (2) |
The solubility and dissolution kinetics of this mineral could be of great importance in limiting human exposure to Pb. The equilibrium constant given in eqn (1) originates from the early research of Nriagu,8,9 and has persisted in the thermodynamic database of a number of computer programs used to calculate speciation of metal ions in water such as Visual MINTEQ10 despite the fact that it was measured at high ionic strength and chloride concentration and extremely low pH (<3), and therefore might not be expected to be valid under natural conditions of soils or water. The above reaction is written with H2PO4− for convenience in calculations because it is the predominant form of phosphate in the environmentally relevant pH range of 3 to 7.
Despite their potential importance to human health and the environment, the solubilities and dissolution characteristics of Pb phosphates, and more specifically the pyromorphite minerals, remain uncertain. Solubilities of pyromorphites and reports of congruent dissolution behavior have been based on behavior in solution at very low pH (2–3), conditions of little relevance in most soil and water environments.8,11,12 Furthermore, chloropyromorphite, Pb5(PO4)3Cl, has been frequently reported to have a solubility product several orders of magnitude greater than the value published in various standard compilations of mineral stabilities and employed in chemical speciation calculations.12–14 These experimental discrepancies in measured solubility products have led some researchers to conclude that the widely accepted assumption of invariant solubility products for minerals of the apatite group, including pyromorphite, is invalid, and that a surface coating compositionally different from the ideal mineral phase forms and controls solubility.15,16 A recent study of HPM precipitation and solubility over a range of pH at high dissolved phosphate is consistent with this hypothesis,17 as is an earlier study of Pb phosphates revealing discrepancies between ion activity products (IAPs) in solution and the actual minerals precipitated from solution.18
Measuring the true ion activity products (IAPs) of solutions in equilibrium with pyromorphites (and other relatively insoluble Pb minerals) under environmentally realistic conditions (e.g., pH 5–8) where Pb solubility and Pb2+ ion activities are expected to be very low is difficult for a number of reasons. First, because few methods are available to directly measure or estimate the activity of the free Pb2+ cation in solution, IAPs are typically estimated by calculating the Pb2+ activity from total dissolved Pb after estimating the fraction of dissolved Pb in soluble complexes with phosphate, bicarbonate, hydroxide and other ligands that might complex or pair with the metal cation. Incorrect estimates of the formation constants of one or more of these ion pairs or complexes would lead to erroneous values for ion activity products of solutions. In addition, very fine colloidal forms of Pb that bypass filtration could be erroneously included in the measure of dissolved Pb by methods such as ICP-MS and ICP-OES, leading to overestimation of the true dissolved concentration. The filtration procedure itself further introduces uncertainty about true Pb solubility because Pb2+ ions are particularly prone to retention on filter membranes, a process that seems to relate to cation exchange properties of the membrane materials.19,20 An additional difficulty with the definition of solubility products for mineral structures of the apatite group is the tendency of these structures to dissolve incongruently as a result of the exchange of surface metal cations (Ca2+ and Pb2+) into solution by H+ ions.21 As a consequence, the measured IAPs of solutions in equilibrium with Pb phosphates are commonly not in accordance with the reported solubility products of the mineral phases actually present,18 and the assumption of invariant solubility products for the apatite mineral structural group including pyromorphites may be invalid.15
A final likely source of apparent solubility product variability of pyromorphites is the inability to accurately estimate concentrations of the various ion pairs formed with Pb2+ in solution, a difficulty caused in large part by the very low solubility of Pb phosphate in water over the pH range of environmental relevance. Consequently, there are no published direct measurements of the stability constants of dissolved PbHPO04 and PbH2PO4+ ion pairs, species that could be very important in controlling Pb solubility in aqueous solutions with large phosphate/Pb mole ratios. Instead, these constants have been estimated in databases of chemical equilibrium programs by analogy with the measured stabilities of other presumeably analogous metal-phosphate ion pairs.22 Although Nriagu8 reported log formation constants of 1.50 and 3.10 for PbH2PO4+ and PbHPO04, respectively, these estimates were calculated from solutions equilibrated in very acidic solutions (pH 2–4) that would be unfavorable to ion pair formation. Nriagu8 considered the calculated constants to be reasonable given their similarity to earlier estimates of phosphate ion pair stabilities with divalent metals such as Ca2+ (log formation constants for CaH2PO4+ and CaHPO40 are reported to be 1.40 and 2.74, respectively). However, this analogy must be questioned given later evidence that Pb2+ bonding with organophosphates is stronger than that of alkaline earth metals.23 Saha et al.24 determined the stability constants of metal-HPO4 ion pairs (KMHPO4) by potentiometric titration, finding metals such as Cu2+ and Cd2+ to have KMHPO4 constants at least one log unit greater than Ca2+. Although they did not report a stability constant for PbHPO04, it seems likely that KPbHPO4 would be at least as large as the stability constant for CdHPO04.
Pb2+-nitrate ion pair formation constants in databases of speciation programs, obtained from quite old studies, indicate that Pb2+ complexes more strongly than most other divalent metal ions with nitrate.25,26 More recent spectroscopic studies have confirmed that Pb2+ has a stronger tendency to form inner-sphere ion pairs with NO3− than Ca2+ or Mg2+.27,28 The fact that the formation constants for PbNO3+ and Pb(NO3)02 are reported to be nearly 6 log units greater than those for the analogous Ca-nitrate ion pairs25 means that other more strongly basic oxyanions such as phosphate are also likely to form ion pairs with Pb2+ much more strongly than with Ca2+, and that the commonly assumed stability constants for Pb-phosphate ion pairs have underestimated Pb2+ complexation with this anion.
The solubilities of Pb measured in high-phosphate solutions at pH 6–8 are greater than predicted from the reported solubility product of HPM,17 suggesting that the contribution of PbH2PO4+ and particularly PbHPO04 ion pairs to the solubility of Pb has been underestimated in chemical speciation programs. Miller found that the addition of phosphate to water at pH 9.0 beyond 1.0 mg L−1 increased Pb solubility, reversing the trend for reduced Pb solubility by dosing drinking water with phosphate.29 In addition, as greater phosphate concentrations (3 and 5 mg L−1) were introduced into the water, the dissolved Pb increasingly exceeded that expected from the solubility product of HPM. Zhao et al.5 concluded that dosing drinking water with excess phosphate lowers dissolved Pb concentrations but may increase concentrations of particulate Pb by increasing Pb phosphate particle charge and inhibiting aggregation. These variable effects of phosphate addition on measured soluble Pb levels are of concern to human health because phosphate addition to municipal water is the primary treatment method used to reduce human exposure to Pb in drinking water.
The uncertainties that exist about factors involved in limiting the aqueous solubility of Pb by HPM precipitation need to be better understood. Therefore, the present investigation of HPM solubility was designed to test whether the widely employed model of constant ion activity product (IAP) is valid for this mineral over a range of phosphate concentrations in the solution phase in the pH range of 6–7.
Materials and methods
Hydroxypyromorphite (HPM) was prepared according to a method modified from Zhu et al.21 Specifically, 4.14 g of Pb(NO3)2 and 0.9905 g of (NH4)2HPO4 salt (5/3 mole ratio of Pb to P) were dissolved in 100 mL of 0.62 M NH4 acetate (pH 4.8). The white suspension formed immediately was adjusted to pH to 7.5 with concentrated NH4OH and stirred for 10 minutes. The suspension was placed in a pressure bomb at 100 °C for 72 hours, then washed 4 times in deionized water to remove salts, and finally dried to a powder at 70 °C. The mineral product consisted of micron-sized columnar crystallites as shown by optical microscopy (Fig. 1A), and confirmed by XRD analysis using a Brucker D8 Advance diffractometer with Cu Kα radiation (λ = 1.54060 Å) to be crystalline HPM (Fig. 1B). This mineral product was used for all subsequent studies of Pb phosphate dissolution in aqueous solutions containing a range of phosphate concentrations.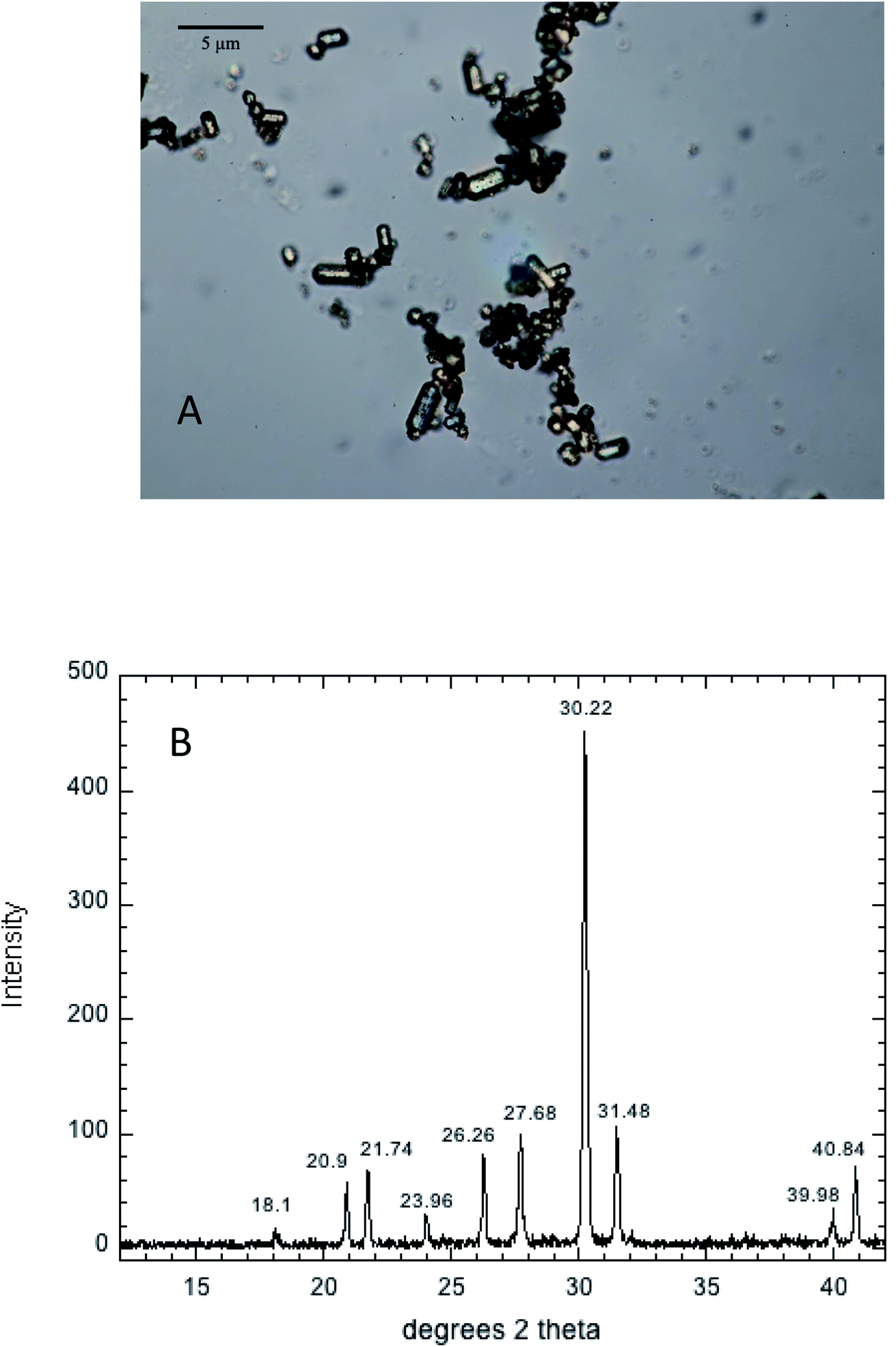 | ||
| Fig. 1 Optical microscopic image (A) and XRD pattern (B) of synthetic hydroxypyromorphite. | ||
Stock aqueous solutions of 0.50 M Na2HPO4 and 0.50 M NaNO3 were prepared from reagent grade salts. The 0.50 M Na2HPO4 stock was adjusted to pH 7.00 using concentrated NaOH, and a 0.10 M Na2HPO4 solution was prepared from this stock. Quantities of the 0.50 M NaNO3 and 0.10 M Na2HPO4 were pipetted into 125 mL. Erlenmeyer flasks containing 0.100 g of the synthetic OH-pyromorphite and diluted to 100 mL total volume with deionized water to create 3 replicates each at phosphate concentrations of 0, 0.1, 1.0, 10 and 50 mM, all in a electrolyte medium of 0.05 M NaNO3.
The flasks containing these suspensions were shaken continuously on an orbital shaker at 22 °C, with pH measured with a Thermo Scientific combination glass electrode and Pb activity measured using a Cole Parmer solid-state ion-selective combination electrode at 8, 15 and 30 days reaction time. The pH electrode was calibrated using commercial pH 4.00 and 7.00 buffers, while the Pb electrode was calibrated using six Pb2+ activity buffers (with calculated free Pb2+ activities varying from 10−4.04 to 10−9.43) prepared from oxalate solutions of different concentrations with Pb in 0.01 M Na acetate buffer at pH 5.0. The free Pb2+ ion activities were calculated for these solutions using Visual MINTEQ and corrected stability constants for Pb-oxalate 1:1 and 1:2 complexes taken from Xiong et al.30 The relationship between millivolt (mV) response of the Pb electrode and calculated −logPb2+ ion activity (pPb) varied somewhat from day to day, with a representative electrode response averaged over a week being:
| pPb = −0.207 (±0.134) − 0.0356 (±0.00028) mV, R = 0.994 | (3) |
As shown by eqn (3), the slope of the millivolt response is quite stable over several days, but the absolute mV reading as reflected by the variability in the intercept of the equation can differ from day to day. Therefore, calibration of the electrode was done immediately before and after sample measurements.
At each of the sampling times up to 30 days equilibration, replicate (n = 3) 10 mL samples of the supernatants were taken, passed through Whatman #42 paper filters (pore size 2.5 μm), stabilized with 0.1 mL concentrated HNO3 and analyzed for dissolved Pb by ICP-OES (Spectro Arcos) at 220.35 nm. Although smaller pore size membrane filters were also tested for their ability to remove all Pb phosphate particles from the solution phases, preliminary experiments using 0.2 μm nitrocellulose membrane filters showed high variability in replicated measurements of dissolved Pb in filtrates at all dissolved phosphate levels tested. Because this result suggested a contamination issue with the nitrocellulose filters or filter holders, a systematic test of Pb mineral filtration by nylon 0.2 μm membrane filters was undertaken. It was found during the filtration of Pb sulfate suspensions that 90% or more of dissolved Pb in the first 10 mL passed through the filter was retained by the membrane filter at pH 4–5, and this removal efficiency increased to nearly 100% at higher pH (6–8). Retention of Pb decreased markedly as additional 10 mL volumes of the same solutions were passed through the filters, indicating saturation of the adsorptive capacity of the filter membranes, with little or no additional Pb retention once 30 mL total volume passed through the filters. Thus, the 0.2 μm membrane filters were determined to be unsatisfactory for Pb filtration in the present experiment, producing non-reproducible results in measuring soluble Pb of filtered Pb mineral suspensions. Conversely, filtrates collected from Whatman #42 filters showed very good reproducibility in replicate samples at all phosphate concentrations except the lowest (no added phosphate). In fact, measured Pb approached the detection limit of the ICP-OES instrument (4 μg L−1) when phosphate concentrations were adjusted to 0.1 mM and greater. Nevertheless, a statistically significant correlation (R = 0.891) between measured Pb and P in the filtrates collected from the Pb phosphate suspensions with zero added phosphate suggests that some of the smaller pyromorphite particles may have passed through the Whatman filters at the lowest phosphate level tested. However, with 0.1 mM phosphate added, filtration using Whatman #42 paper appeared to be effective as indicated by reproducibly low filtrate Pb concentrations and the fact that shaking time (8, 15, 30 days) had no measurable effect on dissolved Pb or P. Also, increasing or decreasing the quantity of HPM in suspensions had no discernible effect in changing the measured Pb or P in filtrates at the 0.1 mM phosphate level.
The logarithm of the ion activity product (IAP) for OH-pyromorphite from the mineral dissolution reaction (eqn (1)) is given by the expression:
| logIAP = 5log(Pb2+) + 3log(H2PO4−) + 7pH | (4) |
IAP for each solution equilibrated with HPM, we first measured the solution pH directly using a glass electrode. The H2PO4− activity was calculated from the ICP-measured total dissolved phosphate using the chemical speciation simulation program Visual MINTEQ10 to correct for pH-dependent speciation and ionic strength effects. Finally, the free Pb2+ ion activity in equilibrated aqueous suspensions of HPM was calculated by 2 methods:
(1) Using the Pb ion-selective electrode to measure free Pb2+.
(2) Using Visual MINTEQ to speciate the total dissolved Pb in solution measured by ICP.
The Visual MINTEQ program was additionally used to predict dissolved Pb as a function of soluble phosphate based on the solubility product of HPM in the database and the inputs of measured pH and dissolved phosphate. It is worth noting that the stability constants for Pb phosphate minerals and ion pairs of Pb with anions such as phosphate and nitrate imbedded in the thermodynamic databases of Visual MINTEQ, MINEQL+, and other commonly used chemical speciation programs are derived in most cases from quite old experimental studies,8,25 many of which have not been verified by more recent research.
Visual MINTEQ predicts that the presence of dissolved carbonate species as a result of equilibration of the HPM suspensions with atmospheric CO2 at 400 ppmv would have only a minor effect on Pb speciation and solubility. Specifically, in the absence of atmospheric CO2, the predominant species of dissolved Pb at equilibrium with HPM at pH 6.7 in 0.1 mM phosphate with 0.05 M NaNO3 are predicted to be free Pb2+ (69.9%), PbNO3+ (23.3%), PbOH+ (4.90%) and PbHPO04 (0.54%). Assuming equilibrium of the same system with 400 ppmv CO2 only slightly alters soluble Pb speciation to 68.7% free Pb2+, 22.9% PbNO3+, 4.82% PbOH+ and 0.53% PbHPO04. Furthermore, Visual MINTEQ predicts that equilibration with CO2 at pH 6.7 would only slightly raise Pb solubility from 8.40 × 10−8 to 8.52 × 10−8. Thus, the aqueous suspensions investigated in this study were allowed to equilibrate with atmospheric CO2.
To confirm accuracy and reproducibility of ICP-OES measurement of dissolved Pb at low Pb concentrations over the wide range of Na phosphate concentrations present in the experimental solutions, Pb standards at 100 μg L−1 were prepared from a certified Pb(NO3)2 stock solution. These were acidified with 2.0 mL ultrapure concentrated HNO3 added per liter. The solutions were spiked with appropriate quantities of 0.10 M Na2HPO4 solution to create triplicate 100 μg L−1 Pb standards ranging from 0 to 10 mM Na phosphate. The final pH of all solutions were measured and found to be 1.65 or lower. The Pb standard solutions containing 0, 0.10 mM, 1.0 mM, and 10 mM Na2HPO4, were determined by ICP-OES, to contain 113 ± 6, 110 ± 0, 103 ± 6, and 97 ± 6 μg Pb L−1, respectively, indicating good accuracy and precision with only a small apparent matrix effect of relatively high concentrations of phosphate and Na on Pb measurement by ICP-OES.
Results and discussion
The measured pH, free Pb2+ activities, and solubilities of Pb and phosphate in filtered solutions after equilibration of the Na phosphate solutions with HPM for 8, 15 and 30 days are summarized in Table 1. These results are further analyzed and discussed in the following sections.| Solution property | Added PO4 (mM) | Equilibration time (days) | ||
|---|---|---|---|---|
| 8 | 15 | 30 | ||
| pH | 0.0 | 5.91 ± 0.27 | 5.86 ± 0.11 | 6.04 ± 0.08 |
| 0.1 | 6.69 ± 0.03 | 6.71 ± 0.09 | 6.77 ± 0.07 | |
| 1.0 | 6.49 ± 0.07 | 5.67 ± 0.05 | 5.86 ± 0.04 | |
| 10 | 6.80 ± 0.11 | 5.70 ± 0.06 | 5.80 ± 0.03 | |
| 50 | 6.85 ± 0.01 | 6.15 ± 0.01 | 6.03 ± 0.01 | |
| Dissolved PO4 (μM) | 0.0 | 18.4 ± 1.2 | 25.0 ± 9.0 | 20.4 ± 2.2 |
| 0.1 | 135 ± 4.7 | 135 ± 2.3 | 143 ± 6.1 | |
| 1.0 | 1288 ± 61.3 | 1288 ± 60.1 | 1343 ± 76.1 | |
| 10 | 12404 ± 44.2 |
12375 ± 172 |
12476 ± 365 |
|
| 50 | 55760 ± 293 |
55246 ± 157 |
52286 ± 1383 |
|
| Dissolved Pb (μM) | 0.0 | 2.58 ± 0.26 | 2.28 ± 0.16 | 1.44 ± 1.03 |
| 0.1 | 0.18 ± 0.06 | 0.048 ± 0.00 | 0.121 ± 0.034 | |
| 1.0 | 0.17 ± 0.10 | 0.20 ± 0.13 | 0.05 ± 0.02 | |
| 10 | 0.23 ± 0.25 | 0.11 ± 0.10 | 0.13 ± 0.10 | |
| 50 | 0.16 ± 0.05 | 0.15 ± 0.01 | 0.11 ± 0.06 | |
| −log(Pb2+) | 0.0 | 6.61 ± 0.09 | 6.23 ± 0.09 | 6.48 ± 0.22 |
| 0.1 | 6.98 ± 0.04 | 6.91 ± 0.06 | 7.72 ± 0.17 | |
| 1.0 | 8.66 ± 0.04 | 8.59 ± 0.06 | 8.60 ± 0.14 | |
| 10 | 9.34 ± 0.01 | 9.18 ± 0.01 | 9.17 ± 0.04 | |
| 50 | 9.3 ± 0.04 | 9.05 ± 0.03 | 9.13 ± 0.07 | |
Dissolved Pb at equilibrium with OH-pyromorphite
Equilibration of the crystalline HPM in aqueous solutions containing a wide range of dissolved phosphate (from 0.01 to 50 mM) had the effect of reducing dissolved Pb at higher phosphate levels, as summarized in Table 1 and displayed in Fig. 2. The relatively large variability in measured dissolved Pb observed here is commonly seen in equilibrated aqueous media where Pb precipitation is limiting solubility.6,31 A possible reason for the variability which is seen only at the lowest dissolved phosphate concentrations, is that the addition of phosphate may have changed the surface charge of HPM by anion adsorption.5 If the HPM suspension without added phosphate was in a more dispersed colloidal state due to surface charge, some of the smaller HPM crystallites may have passed through the paper filters and caused poorer reproducibility and an overestimate of dissolved Pb. A recent study has shown that the aggregation of ultrafine Pb phosphate particles is an important factor in allowing filtration of water to trap particles with much smaller diameters than the nominal filter pore size.32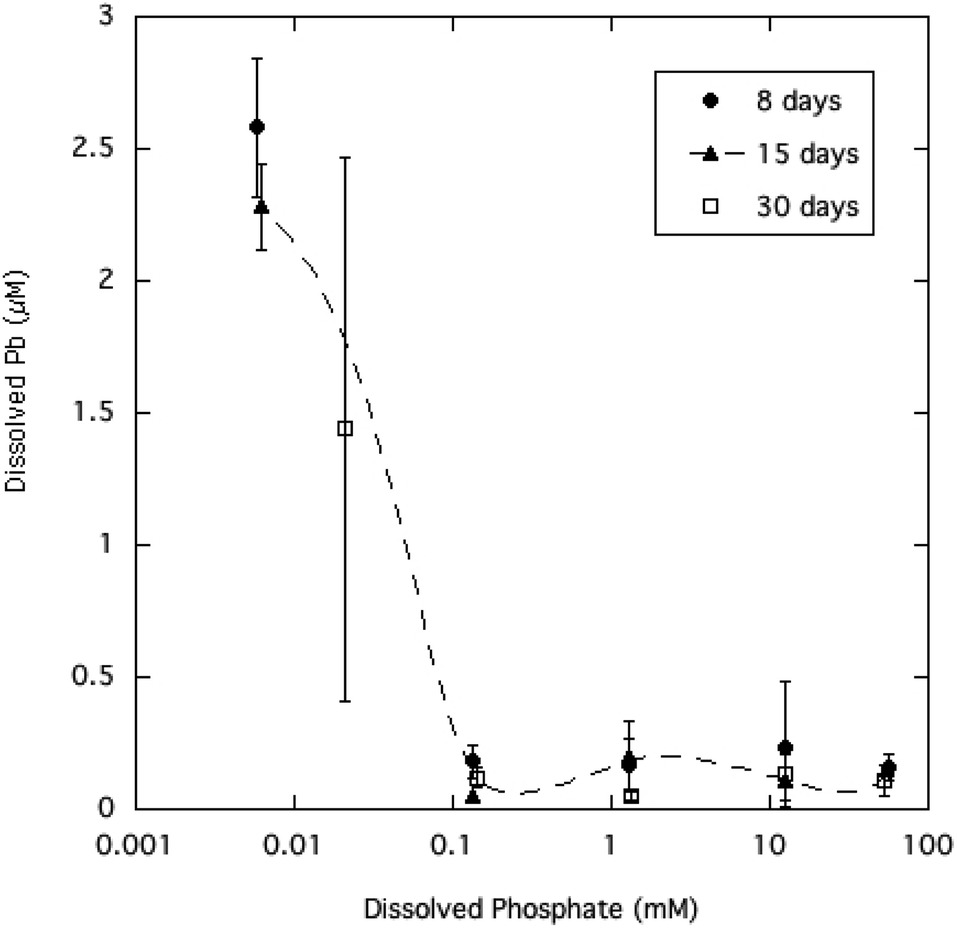 | ||
| Fig. 2 Dissolved Pb concentration as a function of soluble phosphate in aqueous HPM suspensions equilibrated from 8 to 30 days. | ||
Equilibration times ranging from 8 to 30 days did not significantly affect average dissolved Pb, indicating that the microcrystalline mineral had reached steady state with the solution phase. The lowered dissolved Pb at higher phosphate is qualitatively consistent with expectation from the principle of mass action, as higher phosphate in solution inhibits dissolution of the Pb phosphate mineral phase. However, as Fig. 2 shows, increasing dissolved phosphate above 0.1 mM (about 3 mg P L−1) had no significant effect in reducing Pb solubility beyond the initial decrease. The pH of the aqueous suspensions of HPM were generally in the range of 5.7 to 6.8 (Table 1), approaching 7.0 at the higher phosphate levels as the added Na2HPO4 added alkalinity to the solutions. Longer reaction times (15 and 30 days) with sample agitation caused the pH in most cases to decrease to about 5.7–6.0 as slow equilibration with CO2 in the atmosphere neutralized excess alkalinity.
The mole ratio of dissolved Pb to phosphate was determined for the equilibrated OH-pyromorphite suspensions without added phosphate in aqueous NaNO3 at 8, 15 and 30 days equilibration and found to average 0.53 ± 0.26, 0.28 ± 0.25 and 0.25 ± 0.36, respectively, well below the ratio of 1.67 that would be expected for stoichiometric dissolution. There was no statistically significant effect of equilibration time on the average Pb/P ratio in solution, and the overall mean value of 0.355 ± 0.29 indicates a high degree of non-stoichiometric mineral dissolution favoring release of phosphate over Pb to solution. Zhu et al.21 also observed the Pb/P mole ratio to be well below the stoichiometric value of 1.67 when HPM was equilibrated in pure water at pH 5.6. Incongruent dissolution is a well-known characteristic of pyromorphite and other minerals of the apatite group.15 The systems studied here had pH close to neutral, but dissolution under more strongly acidic conditions has been shown to favor release of Pb relative to phosphate, with mole ratios indicative of stoichiometric dissolution at least in the early stages of mineral dissolution.11
Pb2+ ion activity at equilibrium with OH-pyromorphite
The activity of free Pb2+ ions in aqueous solutions at equilibrium with HPM as measured by ion selective electrode (ISE) was lowered with increasing dissolved phosphate concentrations, as shown in Table 1 and Fig. 3. As solution phosphate was increased from about 0.02 mM to more than 50 mM, free Pb2+ activity decreased progressively by approximately 2.7 log units. This observation is qualitatively consistent with a shift in the solid-solution equilibrium in favor of greater precipitation of Pb at higher dissolved phosphate activity. However, as shown in Fig. 4, dissolved Pb did not decrease in response to added phosphate to the extent that free Pb2+ activity did, reaching a near-constant solubility at 0.1 mM dissolved phosphate and higher. At high dissolved phosphate, free Pb2+ activity was more than 2 log units lower than dissolved Pb, a result that may be explained by increased ion-pair formation between Pb2+ and the phosphate anion. This large difference between free Pb2+ activity and Pb solubility was not predicted by the Visual MINTEQ program as shown in Fig. 4. This simulation predicted the difference between Pb activity and solubility to be much smaller than was actually measured and virtually unaffected by dissolved phosphate concentrations. As discussed below, underestimation of the stability constants of soluble Pb-orthophosphate ion pairs could account for this discrepancy between model and measurement.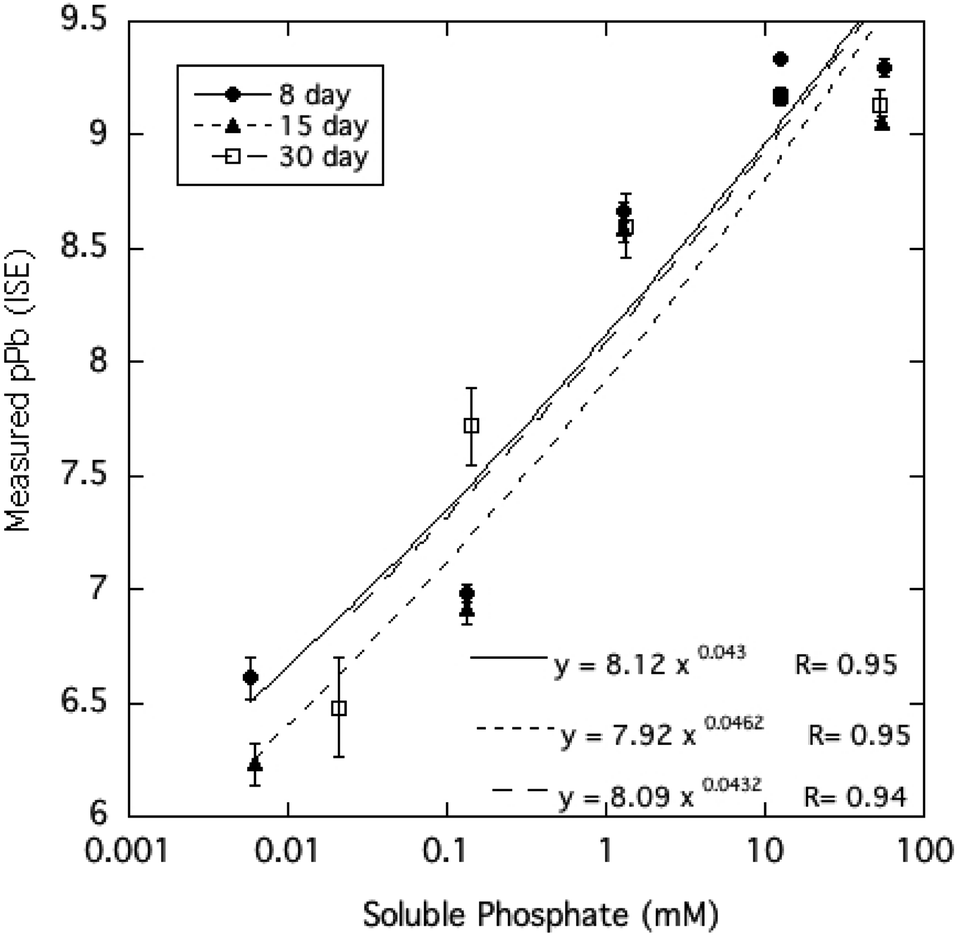 | ||
| Fig. 3 Pb2+ activity measured by ion-selective electrode as a function of soluble phosphate in aqueous HPM suspensions equilibrated from 8 to 30 days. | ||
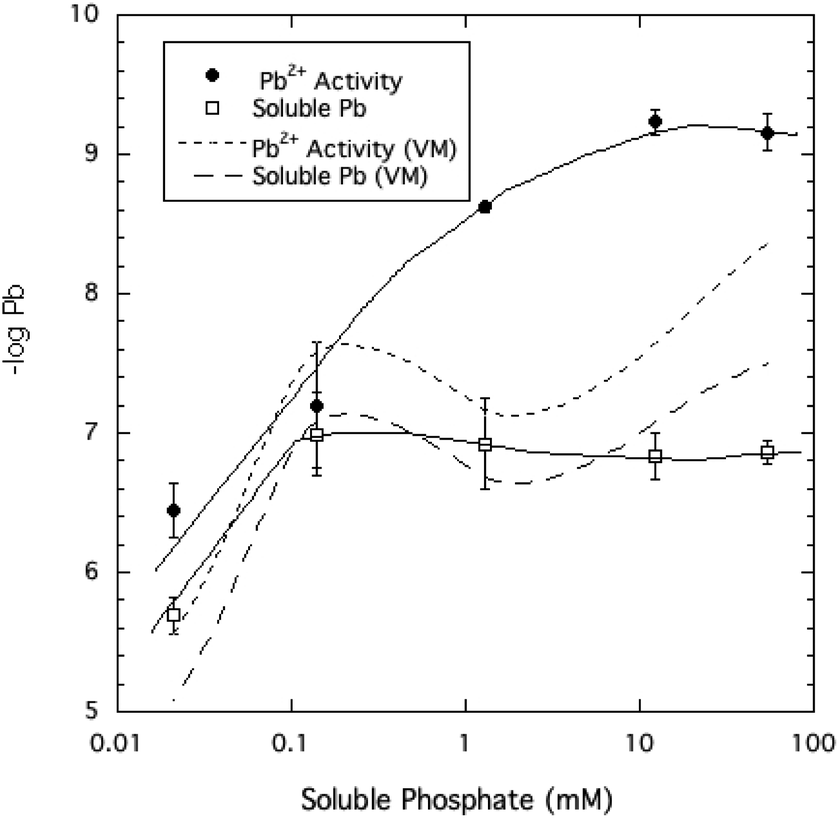 | ||
| Fig. 4 Pb2+ ion activity and soluble Pb measured and modeled (by Visual MINTEQ, VM) as a function of soluble phosphate at equilibrium with crystalline HPM. | ||
The dominant species of Pb expected to be in solution over the range of phosphate tested in the experiment are shown in Fig. 5 based on predictions of the Visual MINTEQ chemical equilibrium model. At soluble phosphate concentrations of 1 mM and lower, Pb-phosphate ion pairs are predicted to be minor soluble species, as the free Pb2+ ion (>60%) and PbNO3+ ion pair (<30%) are the prevalent species. The model indicates that very high phosphate concentrations (>1 mM) result in the PbHPO04 ion pair becoming the dominant soluble form of Pb. The Pb-phosphate ion pairing favored at high phosphate levels can at least qualitatively explains the large difference between dissolved Pb and free Pb2+ concentrations.
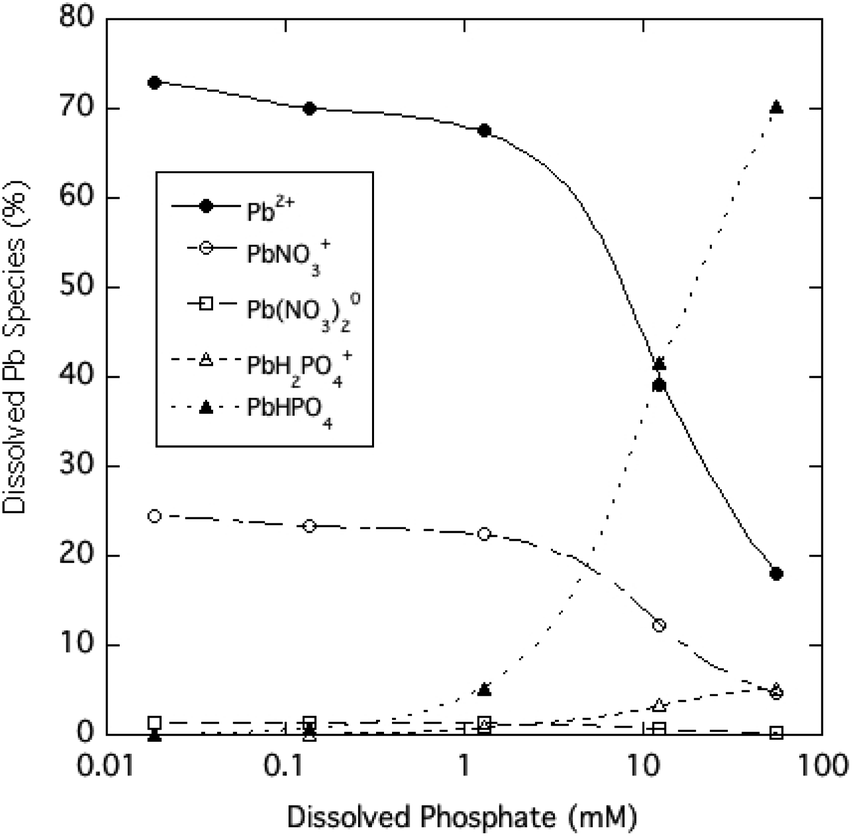 | ||
| Fig. 5 Estimated distribution of dissolved Pb species as a function of soluble phosphate for aqueous HPM suspensions (calculated using Visual MINTEQ). | ||
In order to determine whether the Pb speciation in solution predicted by Visual MINTEQ quantitatively accounts for the large effect of high dissolved phosphate in lowering free Pb2+ activity as measured by the ISE, Pb2+ activity was estimated separately by entering the measured dissolved Pb, phosphate, nitrate, and pH into the Visual MINTEQ speciation program. Fig. 6 compares these two independent determinations of Pb2+ ion activity, revealing a large discrepancy at the highest phosphate levels, with the ISE-determined Pb2+ activity being smaller than the Visual MINTEQ-calculated estimates by as much as 2 orders of magnitude. One reasonable explanation for the discrepancy in these estimates of free Pb2+ (Fig. 6), which becomes greater as soluble phosphate increases, is that the stability constants of the Pb-phosphate ion pairs in the database of the Visual MINTEQ speciation program underestimate the true values. Although no direct measurement of formation constants for Pb-orthophosphate ion pairs have been reported to the knowledge of the authors, research has shown that Pb2+ ions bond selectively and strongly with phosphate groups of phosphate monoesters and nucleic acids.23,33,34 It therefore is reasonable to conclude that Pb2+ bonds much more strongly than alkaline earth metal ions with orthophosphate.
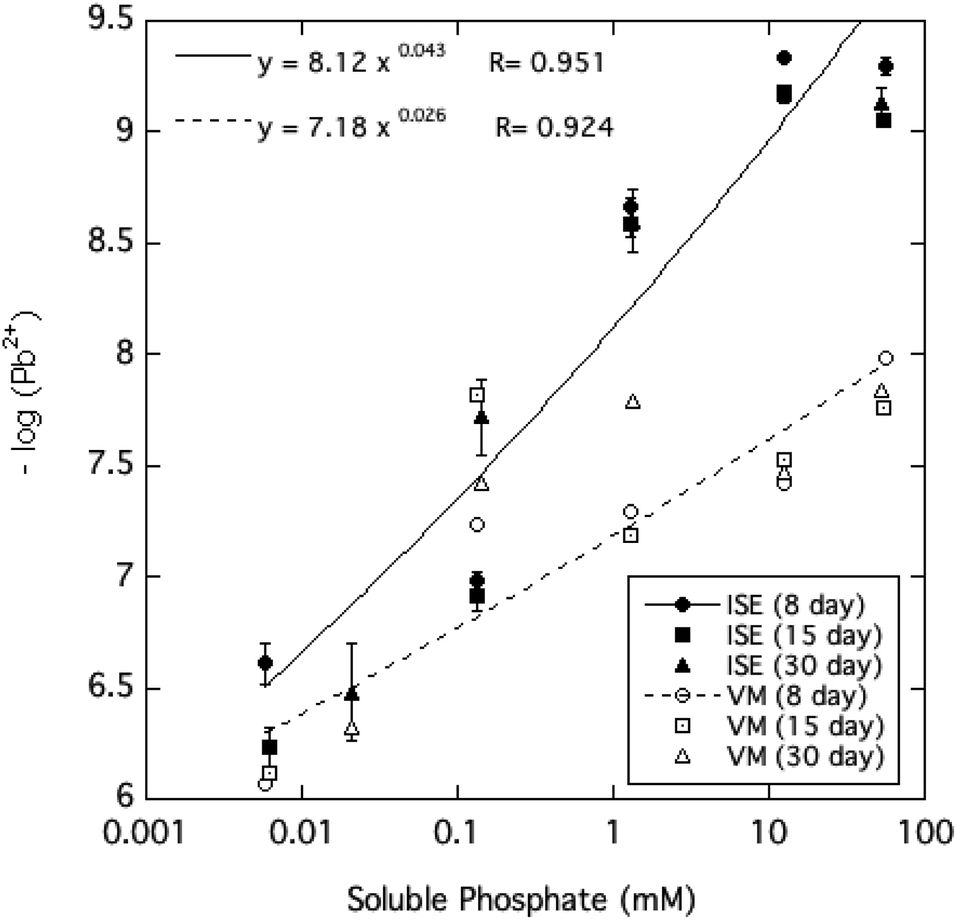 | ||
| Fig. 6 Comparison of Pb2+ activities measured by ion-selective electrode (ISE) and calculated using Visual MINTEQ (VM) as a function of soluble phosphate in aqueous HPM suspensions equilibrated from 8 to 30 days. | ||
Ion activity products (IAPs) of solution phases at equilibrium with OH-pyromorphite
The ion activity products of HPM, given in eqn (4), were calculated by two approaches based on direct and indirect estimation of the free Pb2+ ion activities as described in the Methods section. These are plotted in Fig. 7 as a function of solution pH for all of the equilibration times and phosphate concentrations tested. Despite the considerable variability of estimated logIAP values over the range of pH, phosphate concentrations and reaction times, it is clear from Fig. 7 that the IAPs estimated using the ISE to determine Pb2+ ion activity were generally lower than those estimated using an indirect determination of Pb2+ ion activity. This suggests that the indirect determination using Visual MINTEQ to speciate dissolved Pb may have underestimated ion pair formation of Pb with phosphate as explained earlier, and therefore overestimated free Pb2+ activity.
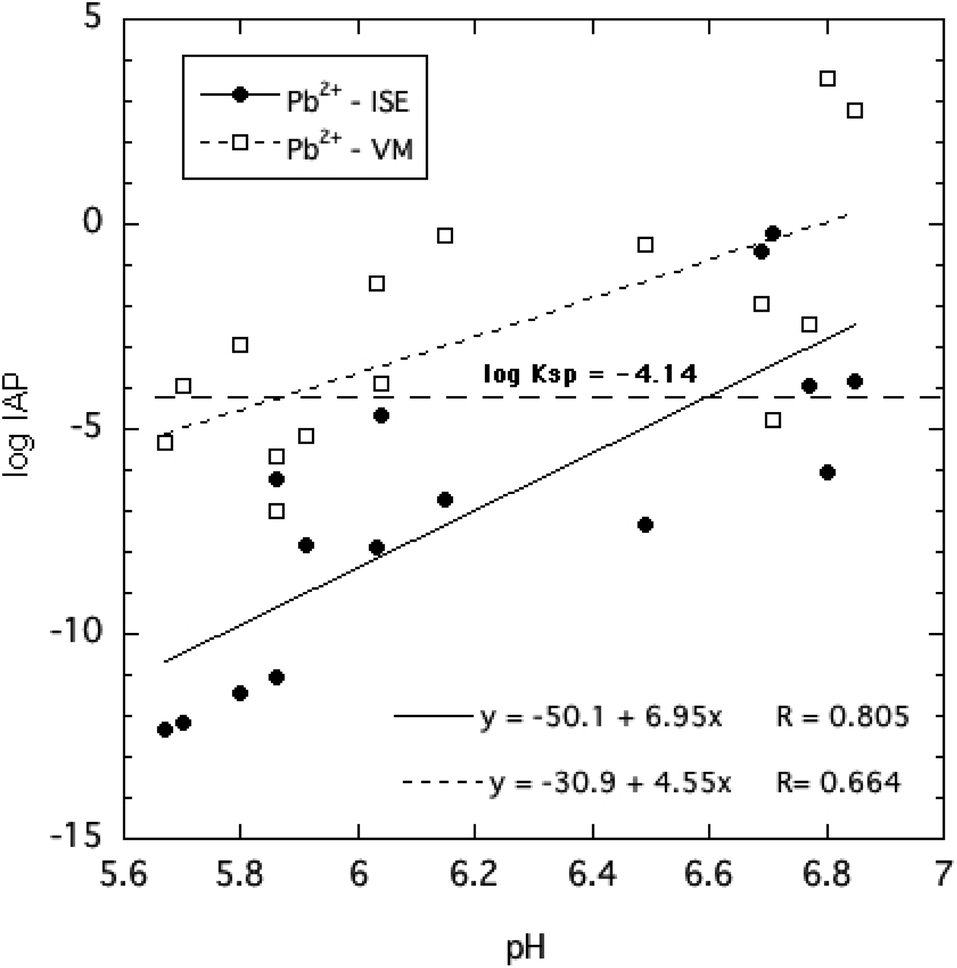 | ||
| Fig. 7 Comparison of ion activity products (IAP) of all solutions in equilibrium with HPM suspensions over a wide range of soluble phosphate concentrations, calculated from ion-selective electrode (ISE) measurement of Pb2+ activities, pH and dissolved phosphate. | ||
Both estimates of logIAP trended toward less negative values at higher pH, implying a shift in mineral solubility depending on pH. Most of the measured logIAPs deviated by several log units from the widely reported KSP of −4.14. The results strongly suggest that crystalline HPM does not display reversible dissolution–precipitation behavior expected for minerals that dissolve congruently. Furthermore, the assumption that the solubility behavior of this mineral can be described by a single invariant solubility product applicable over a range of solution conditions including variable pH and dissolved phosphate concentrations must be questioned. Nevertheless, as little as 3 mg L−1 of P in the form of soluble phosphate added to aqueous solutions equilibrated with excess HPM effectively reduced free Pb2+ ion activity and lowered dissolved Pb below 40 μg L−1.
Conclusions
The solubility of Pb in aqueous solutions equilibrated with microcrystalline HPM was decreased by the addition of 0.1 mM phosphate. Higher added phosphate concentrations up to 50 mM had no significant further effect in lowering solubility, but free Pb2+ ion activity decreased from about 10−6.5 to 10−9 as soluble phosphate was increased in this range. The lowered Pb2+ activity is accounted for by inhibited dissolution of HPM and increased Pb2+-phosphate ion pair formation in solution at higher solution concentrations of phosphate. The much greater Pb solubilities compared to free Pb2+ ion concentrations at high phosphate concentrations are attributed largely to PbHPO04 and PbH2PO4+ ion pair formation. Measurement of the ion activity products (IAP) of the solutions at equilibrium with HPM gave highly variable results that were sensitive to pH and generally not consistent with the reported solubility product of this mineral. It is concluded that a meaningful and constant solubility product of HPM cannot be established because of the incongruent dissolution behavior of this mineral in the near-neutral pH range.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge research funding from the National Institute of Food and Agriculture (Hatch project NYC-125400) awarded to M. B. M.References
- M. A. Chappell and K. G. Scheckel, Pyromorphite formation and stability after quick lime neutralization in the presence of soil and clay sorbents, Environ. Chem., 2007, 4, 109–113 CrossRef CAS.
- J. D. Hopwood, G. R. Derrick, D. R. Brown, C. D. Newman, J. Haley, R. Kershaw and M. Collinge, The identification and synthesis of lead apatite minerals formed in lead water pipes, J. Chem., 2016, 2016, 1–11 CrossRef.
- S. J. Traina and V. Laperche, Contaminant bioavailability in soils, sediments, and aquatic environments, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3365–3371 CrossRef CAS.
- J. D. Noel, Y. Wang and D. E. Giammar, Effect of water chemistry on the dissolution rate of the lead corrosion product hydrocerrusite, Water Res., 2014, 54, 237–246 CrossRef CAS.
- J. Zhao, D. E. Giammar, J. D. Pasteris, C. Dai, Y. Bae and Y. Hu, Formation and aggregation of lead phosphate particles: implications for lead immobilization in water supply systems, Environ. Sci. Technol., 2018, 52, 12612–12623 CrossRef CAS.
- Y. Bae, J. D. Pasteris and D. E. Giammar, Impact of orthophosphate on lead release from pipe scale in high pH, low alkalinity water, Water Res., 2020, 177, 115764 CrossRef CAS.
- Y. Zhu, B. Huang, Z. Zhu, H. Liu, Y. Huang, Z. Zhao and M. Liang, Characterization, dissolution and solubility of the hydroxypyromorphite-hydroxyapatite solid solution [(PbxCa1-x)5(PO4)3OH] at 25 °C and pH 2–9, Geochem. Trans., 2016, 17, 2 CrossRef.
- J. O. Nriagu, Lead orthophosphate. I. Solubility and hydrolysis of secondary lead orthophosphate, Inorg. Chem., 1972, 11, 2499–2503 CrossRef CAS.
- J. O. Nriagu, Lead orthophosphates-II. Stability of chloropyromorphite at 25 °C, Geochim. Cosmochim. Acta, 1973, 37, 367–377 CrossRef CAS.
- J. P. Gustafsson, Visual MINTEQ 3.1, 2018 Search PubMed.
- J. Topolska, M. Manecki, T. Bajda, O. Borkiewicz and P. Budzewski, Solubility of pyromorphite Pb5(PO4)3Cl at 5-65 °C and its experimentally determined thermodynamic parameters, J. Chem. Thermodyn., 2016, 98, 282–287 CrossRef CAS.
- J. Flis, M. Manecki and T. Bajda, Solubility of pyromorphite Pb5(PO4)3Cl- mimetite Pb5(AsO4)3Cl solid solution series, Geochim. Cosmochim. Acta, 2011, 75, 1858–1868 CrossRef CAS.
- C. E. Martinez, A. R. Jacobson and M. B. McBride, Lead phosphate minerals: Solubility and dissolution by model and natural ligands, Environ. Sci. Technol., 2004, 38, 5584–5590 CrossRef CAS.
- L. Xie and D. E. Giammar, Equilibrium solubility and dissolution rate of the lead phosphate chloropyromorphite, Environ. Sci. Technol., 2007, 41, 8050–8055 CrossRef CAS.
- H. B. Pan and B. W. Darvell, Calcium phosphate solubility: the need for re-evaluation, Cryst. Growth Des., 2009, 9, 639–645 CrossRef CAS.
- E. Atlas and R. M. Pytkowicz, Solubility behavior of apatites in seawater, Limnol. Oceanogr., 1977, 22, 290–300 CrossRef CAS.
- M. B. McBride, S. E. Kelch, M. P. Schmidt, S. Sherpa, C. E. Martinez and L. Aristilde, Oxalate-enhanced solubility of lead (Pb) in the presence of phosphate: pH control on mineral precipitation, Environ. Sci.: Processes Impacts, 2019, 21, 738–747 RSC.
- S. Sauvé, M. McBride and W. Hendershot, Lead phosphate solubility in water and soil suspensions, Environ. Sci. Technol., 1998, 32, 388–393 CrossRef.
- P. V. Winger, P. J. Lasier and B. P. Jackson, The influence of extraction procedure on ion concentrations in sediment pore water, Arch. Environ. Contam. Toxicol., 1998, 35, 8–13 CrossRef CAS.
- L. Weltje, W. den Hollander and H. T. Wolterbeek, Adsorption of metals to membrane filters in view of their speciation in nutrient solution, Environ. Toxicol. Chem., 2003, 22, 265–271 CrossRef CAS.
- Y. Zhu, Z. Zhu, X. Zhao, Y. Liang and Y. Huang, Characterization, dissolution and solubility of lead hydroxypyromorphite [Pb5(PO4)3OH] at 25-45 °C, J. Chem., 2015, 2015, 1–10 Search PubMed.
- C. W. Childs, A potentiometric study of equilibria in aqueous divalent metal orthophosphate solutions, Inorg. Chem., 1970, 9, 2465–2469 CrossRef CAS.
- C. P. Da Costa and H. Sigel, Stabilities of complexes formed between Pb(II) and simple phosphonate or phosphate monoester ligands including some pyrimidine-nucleoside 5’-monophosphates (CPM2-, UMP2-, dTMP2-), J. Biol. Inorg Chem., 1999, 4, 508–514 CrossRef CAS.
- A. Saha, N. Saha, L.-N. Ji, J. Zhao, F. Gregán, S. A. A. Sajadi, B. Song and H. Sigel, Stability of metal ion complexes formed with methyl phosphate and hydrogen phosphate, J. Biol. Inorg Chem., 1996, 1, 231–238 CrossRef CAS.
- W. L. Lindsay, Chemical Equilibria in Soils, J. Wiley and Sons, New York, 1979 Search PubMed.
- R. M. Smith and A. E. Martell, Critical Stability Constants, Springer, New York, 1976, vol. 4: Inorganic Complexes Search PubMed.
- M. Xu, J. P. Larentzos, M. Roshdy, L. J. Criscenti and H. C. Allen, Aqueous divalent metal-nitrate interactions: hydration versus ion pairing, Phys. Chem. Chem. Phys., 2008, 10, 4793–4801 RSC.
- F. Alkan, T. Small, S. Bai, A. Dominowski and C. Dybowski, Ion pairing in H2O and D2O solutions of lead nitrate, as determined with 207Pb NMR spectroscopy, J. Struct. Chem., 2016, 57, 369–375 CrossRef CAS.
- S. A. Miller, Investigation of lead solubility and orthophosphate addition in high pH low DIC water, M.S. Thesis, University of Cincinnati, 2014.
- Y. Xiong, L. Kirkes, T. Westfall and R. Roselle, Experimental determination of solubilities of lead oxalate (PbC2O4(cr)) in a NaCl medium to high ionic strengths, and the importance of lead oxalate in low temperature environments, Chem. Geol., 2013, 342, 128–137 CrossRef CAS.
- D. Marani, G. Macchi and M. Pagano, Lead precipitation in the presence of sulfate and carbonate: testing of thermodynamic predictions, Water Res., 1995, 29, 1085–1092 CrossRef CAS.
- D. A. Lytle, M. R. Schock, C. Formal, C. Bennett-Stamper, S. Harmon, M. N. Nadagouda, D. Williams, M. K. DeSantis, J. Tully and M. Pham, Lead particle size fractionation and identification in Newark, New Jersey's drinking water, Environ. Sci. Technol., 2020, 54, 13672–13679 CrossRef.
- V. G. Kanellis and C. G. dos Remedios, A review of heavy metal cation binding to deoxyribonucleic acids for the creation of chemical sensors, Biophys. Rev., 2018, 10, 1401–1414 CrossRef CAS.
- R. S. Brown, B. E. Hingerty, J. C. Dewan and A. Klug, Pb(II)-catalysed cleavage of the sugar-phosphate backbone of yeast tRNAPhe – implications for lead toxicity and self-splicing RNA, Nature, 1983, 303, 543–546 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |