
Visible light-mediated metal-free double bond deuteration of substituted phenylalkenes†
Roman
Iakovenko
 and
Jan
Hlaváč
*
and
Jan
Hlaváč
*
Department of Organic Chemistry, Faculty of Science, Palacký University, 17. listopadu 12, 771 46 Olomouc, Czech Republic. E-mail: jan.hlavac@upol.cz
First published on 14th November 2020
Abstract
Various bromophenylalkenes were reductively photodebrominated by using 1,3-dimethyl-2-phenyl-1H-benzo-[d]imidazoline (DMBI) and 9,10-dicyanoanthracene. With deuterated DMBI analogs (the most effective was DMBI-d11), satisfactory to excellent isotopic yields were obtained. DMBI-d11 could also be regenerated from the reaction mixtures with a recovery rate of up to 50%. The combination of the photodebromination reaction with conventional methods for bromoalkene synthesis enables sequential monodeuteration of a double bond without the necessity of a metal catalyst.
Introduction
Deuteration of organic compounds is of considerable significance in numerous areas of chemistry and chemical biology. Deuterated compounds have become an essential tool for reaction mechanism studies1–3 or structure elucidation.4–6 Deuterium incorporation may be highly beneficial also for drug discovery due to the inhibition of drug metabolism. It also enables “evergreening” of expired patents on previously discovered drugs.7–9As deuterated synthetic building blocks are usually costly, their implementation in an extended reaction sequence is inefficient for preparation of compounds on a larger scale, and also, complex natural compounds eligible for isotopic enrichment do not allow the use of a deuterated precursor. The post-synthetic transformation is the only feasible way of deuterium implementation in these cases.
One of the relevant groups deserving attention is the phenylvinyl fragment, which can be found in many approved drugs, such as entacapone, rilpivirine and cyclobenzaprine, natural compounds including flavones, chalcones, and stilbenoids or more complex compounds such as semisynthetic lysergic acid derivatives, salvianolic acid A, etc. (Fig. 1). Although there are various methods for its deuteration, they have drawbacks including low selectivity, isotopic yield, and configuration stability, and toxicity of heavy metal catalysts or influence of highly reactive species on functional groups. These facts force chemists to study alternative deuteration procedures.
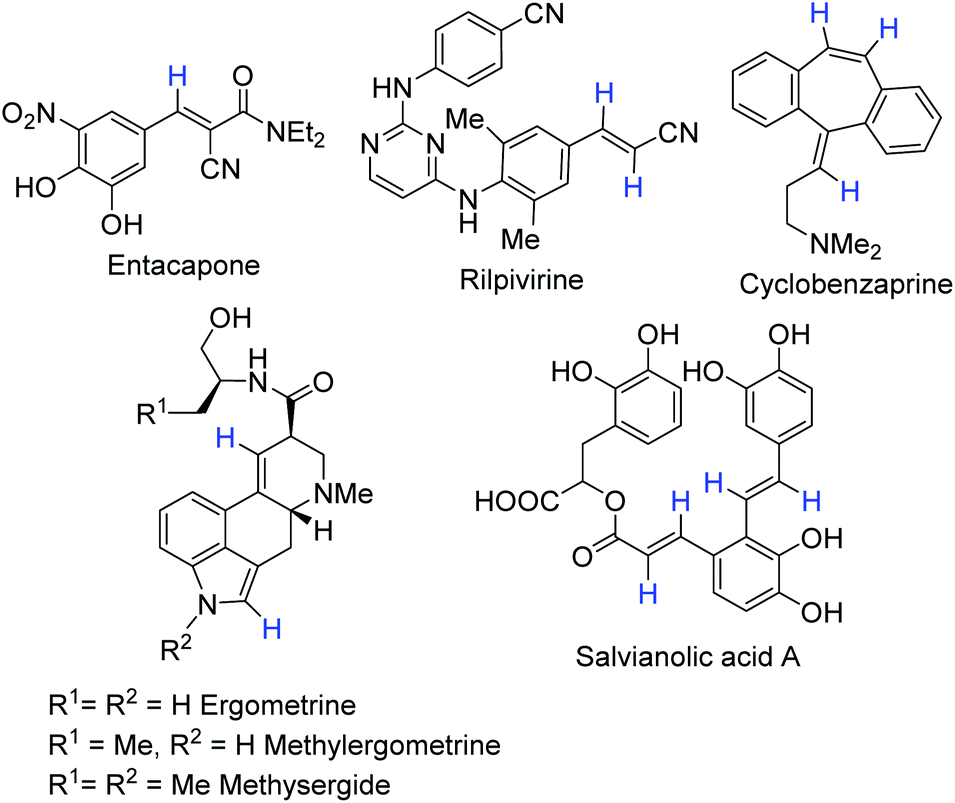 | ||
| Fig. 1 Examples of approved drugs and natural compounds with phenylvinyl moiety. | ||
Recently published methods include direct H/D exchange or replacement of various reactive groups. Deuterium can be transferred from deuterated solvents under transition-metal catalysis10–12 or by hydrolysis of organolithium compounds.13–16
The use of metallocatalysts leads to the problem of contamination of the products resulting in a risk of the final product toxicity as well as contamination of sewage water. Formation of organolithium compounds is suitable only for alkenes with a sufficiently acidic vinyl hydrogen atom and could also affect other functional groups in the molecule. A metal-free H/D exchange of hydrogen atoms at the phenylvinyl double bond using N-heterocyclic carbenes17–20 was described a few times. The disadvantages of this method include mainly low D-enrichment and use of strong bases. In the above papers the phenylvinyl deuteration is usually applied for mechanistic studies or structure confirmation. No systematic study of such deuteration has been published.
Other methods of this double bond deuteration are based on the reduction of different groups, mainly halogen. The systematic study of halogen/deuterium exchange including few examples of phenylvinyl moiety was performed by Kuriyama et al., who used the Pd/N-heterocyclic carbene complexes.21 Although this reaction affords high yields and deuterium enrichment, the use of strong bases and possible interaction of the palladium catalyst with different functional groups can limit its application to other substrates. Palladium as well as many other transition metals are also known to have numerous adverse health effects22,23 and their concentration in e.g. medical substances is strictly regulated.24 A metal-free dehalogenation method for haloarylalkenes was also described,25 but it required the use of hem-diiodoalkene.
In the last decade significant attention was paid to reductive dehalogenation under photoredox catalysis; however no systematic study of deuteration has ever been published. The published dehalogenation of vinyl iodides including phenylvinyl derivatives26 used iridium-based photocatalysts. Although transition-metal photocatalysts can be replaced by different organic dyes with similar excited state reduction potential,27–29 no studies have been performed for deuteration of a carbon–carbon double bond.
Herein we report a simple metal-free procedure for deuterodebromination of a phenylvinyl double bond. This method can also be implemented in a reaction sequence enabling the H/D exchange on a double bond. This selective highly efficient deuterium implementation method can afford cheap and selective isotopic enrichment of such compounds for biological studies or improvement of the pharmacological properties thereof.
Results and discussion
Optimization of the photoreduction system
Before the necessary synthesis of the intended D-donors, we performed the initial reactivity testing with their commercially available non-deuterated analogs (Scheme 1).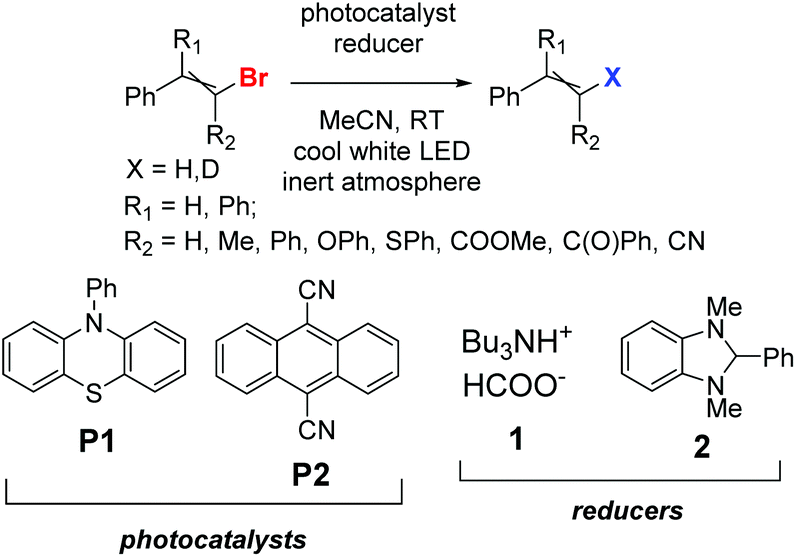 | ||
| Scheme 1 Variety of screened substrates and reagents. | ||
First, we applied the published conditions for the bromoarene reduction catalyzed by 10-phenylphenothiazine (P1) using a 380 nm LED light source.27 We subjected three bromoalkenes β-bromostyrene (A-Br), 2-bromo-1,1-diphenylethylene (B-Br), and bromostilbene (C-Br) to the reaction. According to the described conditions, we used tributylamine formate (1) as a photoactivated H-donor (Table 1, entries 1, 4, and 8).
| # | Substrate (Z/E ratio) | Photocatalyst | Reducing agent | Conv. (NMR) |
|---|---|---|---|---|
| Conditions – Acetonitrile, 5 mol% of photocatalyst, reaction time 16 h.a Light source: 380 nm LED, 13 W.b Light source: cool white LED, 13 W. | ||||
| 1 |
|
P1 | 5 eq. 1 | 8% |
| 2 | P1 | 2 eq. 2 | 28% | |
| 3 | P2 | 2 eq. 2 | 65% | |
| 4 |
|
P1 | 5 eq. 1 | 7% |
| 5 | P1 | 2 eq. 2 | 73% | |
| 6 | P2 | 2 eq. Bu3N | 19% | |
| 7 | P2 | 2 eq. 2 | 96% | |
| 8 |
|
P1 | 5 eq. 1 | 58% |
| 9 | P1 | 2 eq. 2 | 80% | |
| 10 | P2 | 2 eq. Bu3N | 25% | |
| 11 | P2 | 2 eq. 2 | >99% | |
The reactions led to moderate results, and only the conversion of bromostilbene (C-Br) was significant. So, we switched to another reducing agent (H-donor), 1,3-dimethyl-2-phenyl-benzo[d]imidazoline (2), which is widely known for its hydrogen atom donating properties30,31 and is also capable of reductive coupling reactions.32 The conversions with the reducing agent 2 were much higher (Table 1, entries 2, 5, and 9).
When 9,10-dicyanoanthracene (P2) was used as a photocatalyst in combination with cool white LED irradiation similar to the previously described aryl halide transformation,28 the conversion significantly increased. First, P2 was used with tributylamine, as the authors did, with moderate success (Table 1, entries 6 and 10). The pairing of P2 with reducing agent 2 (Table 1, entries 3, 7, and 11) afforded the highest conversions of all three substrates. Additional optimization data for the reducing agent and light source are presented in the ESI (Tables S2 and S3†). (3-Butyl-1-methyl-1H-imidazol-3-ium-2-yl)borane as an alias metal-free borohydride was also tested either alone as a reducer (Table S2,† entries 3 and 9) or with a catalytic amount of 2 (entries 13–16) or 1,3-dimethyl-2-phenylbenzimidazolium iodide [3]I (entry 17), but without success.
The practical setup of the reaction is illustrated in Fig. S1.† A photoreactor can be easily constructed from a commercially available laboratory washing bottle and LED strips and is suitable for carrying out up to six analytical-scale reactions.
Having found the suitable photocatalytic system (P2/2/cool white LED), we tested the reduction of several other bromoalkenes (Table 2), trying to analyse the effects of different substituents on the reaction outcome. Optimal conditions for low-reactive substrates, such as β-bromostyrene A-Br, 2-bromo-1-phenylpropene D-Br, or 1-bromo-1,2,2-triphenylethylene E-Br, include the addition of two portions of P2 in 16-hour intervals (entries 1, 4, and 5). The reactions of bromoalkenes F-Br and G-Br (entries 7 and 8) proceeded successfully in longer reaction time. The reactions of bromophenylalkenes B-Br, C-Br, G-Br, H-Br, I-Br, and J-Br proceeded quantitatively (entries 3 and 7–10). α-Bromostyrene was also tested as a substrate, but it did not undergo reduction with the P2&2 system.
|
|
||||||
|---|---|---|---|---|---|---|
| # | Br-Alkene | Z-/E- | R1 | R2 | Conv. (NMR) | Z-/E- |
| a Two 16 h intervals and two 5 mol% portions of P2. b Reaction time, 24 h. | ||||||
| 1 | A-Br | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 :5 |
H | H | 77%a | — |
| 2 | B-Br | — | Ph | H | 96% | — |
| 3 | C-Br | 1:13 |
H | Ph | >99% | 5.7:1 |
| 4 | D-Br | 1:7.3 |
H | Me | 85%a | 1:1.06 |
| 5 | E-Br | — | Ph | Ph | 70%a | — |
| 6 | F-Br | Z- | H | OPh | 88%b | 1:32 |
| 7 | G-Br | 1:1 |
H | SPh | >99%b | 1:1.1 |
| 8 | H-Br | 1:1 |
H | COOMe | >99% | 1:1.1 |
| 9 | I-Br | Z- | H | C(O)Ph | >99% | 1:19 |
| 10 | J-Br | 1.7:1 |
H | CN | >99% | 1.8:1 |
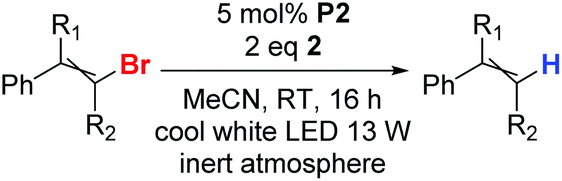
As the P2&2 system exhibited outstanding efficiency, we tried to study the influence of halogen atoms on reactivity. Therefore, we tested a series of chloro-, bromo- and iododerivatives of the selected alkenes (Table 3).
|
|
||||
|---|---|---|---|---|
| # | Structure | Halogen (Z/E) | Time | Conv. (Z/E) |
| Conditions – Acetonitrile, 5 mol% P2, 2 eq. 2, 13 W cool white LED. | ||||
| 1 | A- | Cl (E-) | 16 h | 15% |
| 2 | Br (1:5) |
65% | ||
| 3 | I (E-) | 100% | ||
| 4 | B- | Cl | 50% | |
| 5 | Br | 96% | ||
| 6 | C- | Cl (E-) | 4 h | 35% (1.8:1) |
| 7 | Br (1:13.3) |
56% (4.5:1) |
||
| 8 | I (E-) | 100% (11.5:1) |
||
| 9 | H- | Cl (3.5:1) |
16% (1.3:1) |
|
| 10 | Br (1:1) |
100% (1.2:1) |
||
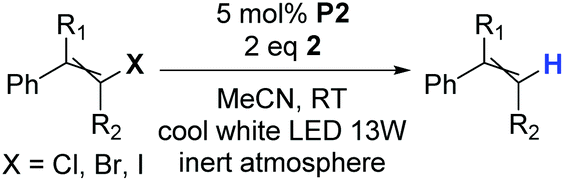
As we expected, chloroalkenes had lower conversions and iodoalkenes had higher conversions into alkenes than their bromo analogs. As the alkene iodination methods are not widely available, and iodoalkenes suffer from low stability, the deuteration process using bromoalkenes seems to be the best compromise.
Arylbromides were unstable towards the P2/2 reducing system – under similar conditions as shown in Table 2, e.g. p-bromoacetophenone was debrominated by 64%, and dimethyl 2-bromoterephthalate was debrominated completely (see ESI, Fig. 27 and 28†). So the aryl halides will probably not remain untouched in the reaction conditions.
A plausible reaction mechanism is presented in Scheme 2. The excited molecule of photocatalyst P2* oxidizes imidazoline 2, affording radical cation of 2(˙+) – a powerful hydrogen atom donor. The excited radical anion of [P2(˙−)]* donates an electron to bromoalkene yielding a vinyl radical, which quickly abstracts hydrogen from 2(˙+). It is worth mentioning that such an interpretation might be incomplete, since an experiment without P2 revealed that photoreduction still proceeded upon irradiation, but at a much slower rate (Table S2,† entries 10 and 11 in comparison to entry 5). So, imidazoline 2 could work, to some extent, as a photoreducing agent as well. Also, the bromoalkene after its single-electron reduction can dissociate to some extent into an alkenyl radical or anion. The formation of the alkenyl anion can explain the stereoselectivity in some cases or high deuterium enrichment in the case of methanol-d4 use.
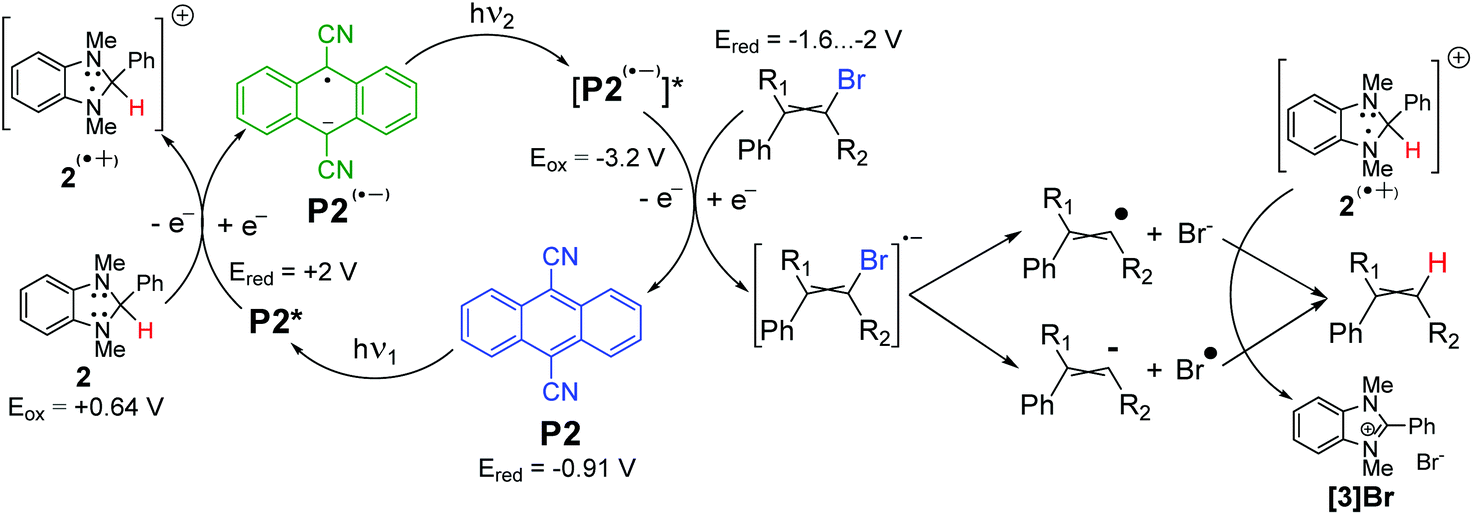 | ||
| Scheme 2 Plausible phenylbromoalkene photoreduction mechanism. Electrode potentials of bromoalkenes,33,34 reducer 235 and photocatalyst P236 are recalculated vs. SCE. | ||
The formation of the alkenyl radical can be responsible for the abstraction of deuterium from the aprotic solvents or incomplete deuteration, when methanol-d4 is used as the solvent (see later).
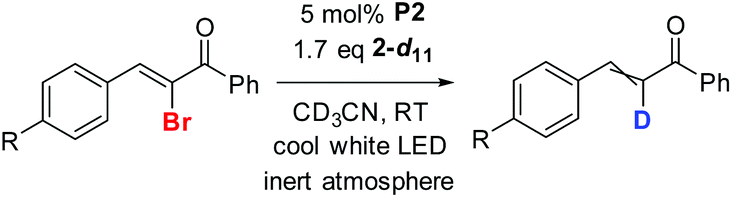
At the beginning of the study of the deuteration process according to Scheme 1 (X = D), we tested the efficiency of deuterated analogs of reducer 2 in various solvents. We used 2-bromo-1,1-diphenylethylene (B-Br) as a model substrate, and synthesized deuterated derivatives 2-d and 2-d7 (see Fig. 2 and ESI†).
 | ||
| Fig. 2 Structure of reducers for Br/D exchange (see Scheme 2). | ||
From the reaction with reducing agent 2 in CD3CN it was shown that migration of deuterium from the solvent is less than 4% (Table S4,† entry 2). Additionally, the kinetic isotope effect of B-Br reduction by 2 in CD3CN was determined to be 2.61 from the competitive reaction experiment (see ESI†).
When monodeuterated reducer 2-d was used in non-deuterated acetonitrile, the D-enrichment was significantly higher and was further increased by the application of acetonitrile-d3 (Table S4,† entries 4 and 5). The application of reducer 2-d7 in acetonitrile and CD3CN significantly increased the conversion, and the isotopic enrichment was almost 90% in the deuterated solvent (Table S4,† entries 6 and 8). The deuterodebromination of B-Br by 2-d7 was also tested without the addition of P2, and despite the deuterium enrichment ratio of 92.6%, the reaction rate was much slower, and after 48 h of irradiation the substrate was converted only by 75% (Table S2,† entry 7).
Then, a comparative study of deuterodebromination of B-Br by 2-d7 in other deuterated solvents was performed (Table S3,† entries 9–12). Comparing to CD3CN, no other solvent had better reaction conversion and D incorporation at the same time.
Some solvents such as acetone-d6 or methanol-d4 afforded higher D-enrichment (Table S4,† entries 9 and 12), while DMSO-d6 led to higher conversion (Table S4,† entry 10).
Results of preparative deuterodebromination experiments
After that, 2-d11 was synthesized, starting from aniline-d5 (see ESI†) and used in preparative deuterodebrominations of ten bromophenylalkenes (Table 4). By comparing the reduction of six substrates with 2-d7 and 2-d11 (Table S5†), it was found that the more highly deuterated reducing agent provided comparable or higher D-enrichment, and similar or higher conversion and yield. In cases where the bromine atom was sterically hindered (E-Br and I-Br), the use of 2-d11 proved to be more advantageous.| # | Substrate (Z-/E-) | Conv.a (NMR) | D content | Time | Yielda (Z-/E-) | [3-d10]Br recovery |
|---|---|---|---|---|---|---|
| For structures of the bromo derivatives see Table 1. Conditions – CD3CN, 5 mol% P2, 1.7 eq. 2-d11, 13 W cool white LED.a Conversion is calculated from the NMR spectra of crude reaction mixtures. The yield is calculated after isolation and purification.b Three 24 h intervals and three 5 mol% portions of P2.c D-Alkene was isolated as a mixture with the bromoalkene starting compound (see ESI† for details).d Two 24 h intervals and two 5 mol% portions of P2 (compound A-Br was purchased from Sigma-Aldrich, Inc; for syntheses of other compounds see ESI†). | ||||||
| 1 |
A-Br (1:5) |
66% | 90% | 72 hb | 58% (2.85:1)c |
37% |
| 2 | B-Br | 99% | 93% | 48 h | 51% | 49% |
| 3 |
C-Br (1:13) |
>99% | 96% | 48 h | 64% (49:1) |
52% |
| 4 |
D-Br (1:7.3) |
60% | 77% (E) | 72 hb | 56% (1.7:1)c |
32% |
| 86% (Z) | ||||||
| 5 | E-Br | 53% | 77% | 72 hb | 45%c | 40% |
| 6 | F-Br (Z-) | 95% | 89% | 48 hd | 94% (E-) | 41% |
| 7 | G-Br (Z-) | >99% | 67% (E) | 48 hd | 84% (1:1) |
55% |
| 77% (Z) | ||||||
| 8 |
H-Br (1:1) |
>99% | 89% (E) | 24 h | 86% (1.3:1) |
41% |
| 96% (Z) | ||||||
| 9 | I-Br (Z-) | >99% | 78% | 24 h | 81% (E-) | 55% |
| 10 |
J-Br (1.7:1) |
>99% | 97% (E and Z) | 24 h | 92% (1.78:1) |
41% |
However, as the higher deuteration of 2 also decreased the reaction rate, we had to consider longer reaction times for reactions with 2-d11. The reactions were monitored every 24 hours for a maximum of three days.
The preparative yields varied between 45 and 94%. In some cases, it was not possible to fully separate the product from the starting compound (Table 4, entries 1, 4, and 5). E/Z-Preference was conserved in most cases during the reaction except for the derivatives A-Br, G-Br, and J-Br.
A scale-up experiment was also performed. The results, yield, reagent and solvent regeneration ratios, are exemplified in Scheme 3.
 | ||
| Scheme 3 High-loading deuterodebromination experiment. | ||
The precipitate of benzimidazolium bromide [3-d10]Br formed during the reduction could be collected and reduced allowing for partial regeneration of the reducer. Thus, up to half of the used amount of 2-d11 can be regenerated.
In order to analyse the influence of substituents in the phenyl ring on the reaction outcome, a series of deuterodebrominations of substituted α-bromochalcones was also performed (Table 5).
|
|
||||||
|---|---|---|---|---|---|---|
| # | Substrate (pure Z-) | R | Conv. (NMR) | D content | Yield (Z-/E-) | [3-d10]Br recovery |
| Conditions – CD3CN, 5 mol% P2, 1.7 eq. 2-d11, 13 W cool white LED, 24 h.a Yield considering partial regeneration of the starting compound.b Time of reaction, 18 h. | ||||||
| 1 | K-Br | NO2 | 87% | 65% (E) | 85%a (1.7:1)b |
55% |
| 89% (Z) | ||||||
| 2 | L-Br | CN | 75% | 71% (E) | 89%a (9:1) |
40% |
| 59% (Z) | ||||||
| 3 | M-Br | MeO | >99% | 87% | 68% (E-) | 53% |
| 4 | N-Br | Me2N | >99% | 85% | 24% (E-) | 60% |
This experiment results in several implications of substituent influence. Firstly, the electron-deficient substrates (K-Br and L-Br) reacted significantly slower and the reaction had to be stopped before completion, because holding it until full conversion led to partial decomposition of the reaction product. Although both substrates were used as Z-isomers, the products were formed as a mixture of E- and Z-chalcones, which could be caused by photoisomerisation of the substrate before the reduction due to lower reactivity.
Secondly, the substrate N-Br despite its excellent conversion into the product was isolated only with a yield as low as 24%, which could be caused by partial decomposition of the reaction product as well as by interaction with silica gel during chromatography.
Regenerated 2-d11 was also tested with the same six substrates that were used for the comparison of 2-d7 and 2-d11 (Table 6). The screening revealed that, as in the case of 2-d7, only with sterically hindered substrates (E-Br and I-Br) there was a great difference in D-enrichment, and so in those cases the use of recovered reducing agent may be less justified.
| # | Substrate (Z-/E-) | Time | Conv. (NMR) | D content | Differenceb | [3-d10]Br recovery |
|---|---|---|---|---|---|---|
| Conditions – CD3CN, 5 mol% P2, 1.7 eq. 2-d11, 13 W cool white LED.a Three 24 h intervals and three 5 mol% portions of P2.b Decrease in %D with recovered 2-d11 compared to the reaction with a new one. | ||||||
| 1 | B-Br | 48 h | 98% | 87% | 6 | 37% |
| 2 | C-Br | 48 h | >99% | 89% | 7 | 41% |
| 3 | E-Br | 72 ha | >99% | 60% | 17 | 49% |
| 4 | H-Br | 24 h | >99% | 87% (E) | 2 (E) | 52% |
| 95% (Z) | 1 (Z) | |||||
| 5 | I-Br | 24 h | >99% | 58% | 20 | 49% |
| 6 | J-Br | 24 h | >99% | 93% (E) | 4 (E) | 60% |
| 97% (Z) | ≈1 (Z) | |||||
Additionally, the NMR spectra of recovered 2-d11 revealed that it had decreased deuterium enrichment in benzimidazoline aromatic positions – by 5.3 and 2.3 percent points for positions 4 and 5, respectively (see Fig. 3). This could serve as evidence that reactive species, which are formed after debromination of E-Br or I-Br, can sometimes abstract hydrogen/deuterium competitively from the aromatic positions of reducing agent 2 due to steric hindrance.
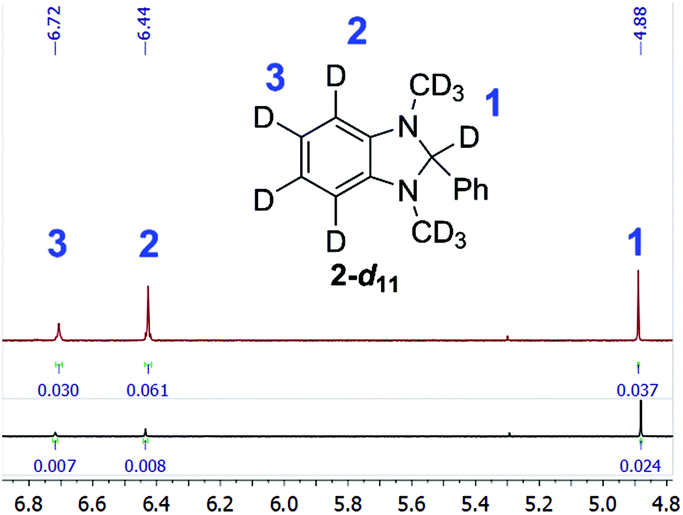 | ||
Fig. 3 Compared fragments of the 1H NMR spectra of the new and 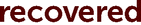 2-d11 (the integrals are normalised to phenyl protons). 2-d11 (the integrals are normalised to phenyl protons). | ||
That fact could, in turn, explain the dramatic decrease in the enrichment and isolated yield in the reactions of E-Br and I-Br with both 2-d7 and recovered 2-d11. Also, in cases where two stereoisomeric D-alkenes were formed, the deuterium content in Z-deuteroalkene was usually higher, and this can also be explained by steric hindrance, because the hydrogen atom at position 2 in the molecule of the reducing agent, being the most electropositive, is not the most sterically accessible one.
Economic aspects of 2-d7/2-d11 selection
We also made some calculations to compare the costs of preparation and implementation of deuterated imidazolines (see ESI,† p. 19). The major part of the cost being the deuterated reagents, and considering the lowest prices found by us for multigram-scale quantities, we estimated the cost of preparation to be 29.5$ per g for 2-d7 and 67.5$ per g for 2-d11. The regeneration procedure lowers these prices by 20–30%, although it requires additional amounts of NaBD4 and CD3OD.Unfortunately, for 2-d11 the recovered reagent cannot be used in reactions with sterically hindered substrates with the same efficiency as a new one (see Table 6, entries 3 and 5), and so in these cases after regeneration 2-d11 should be used with more reactive bromoalkenes. The reagent 2-d7 can be recovered without loss of its properties.
Considering the deuterodebromination of E-Br, the cost of 2-d7 and CD3CN used is 29.3$/1 mmol of obtained D-alkene, and for 2-d11 it is 59.2$/1 mmol of D-alkene. So, the first 56% of deuteration in 1 mmol of E-d is worth 0.52$ per one percent point of deuteration, and if it is needed to achieve higher enrichment, the last 21%D will come at a price of 1.42$ per one percent point. So, the average cost of deuteration of 1 mmol of E-Br up to 77%D will be 0.77$ per percent point of D.
For more reactive substrates, it is possible to greatly cut the costs using 2-d7. Thus, the deuterodebromination of H-Br would consume 20.45$ per mmol of H-d worth of deuterated reagents and solvents, or 0.22$ per percent point of D-enrichment (for details, see ESI,† p. 19). The cost could be further lowered by increasing the regeneration ratio of acetonitrile-d3, which is easier to achieve at higher scales.
So, for non-complicated and non-sterically hindered substrates the reagent of choice is 2-d7 in most cases, since the additional 1–5 percent points of enrichment, which 2-d11 could afford, would be exceptionally costly.
Environmental aspects of the reaction
The deuterodebromination reaction can be considered environmentally benign. The solvent, acetonitrile-d3, can be easily regenerated and corresponds to the green chemistry principles. There is a limited amount of data about the toxicity of photocatalyst P2, but a functionalized dicyanoanthracene compound exhibited very low in vitro cytotoxicity.37 The reducing agent 2 is reported to have a quite high LD50 value of 97 mg kg−1 in mice,38 but it gets converted into salt [3]Br with around 50% recovery ability.By implementation of recovery procedures, the toxic waste of the reaction can be limited to minimum amounts.
Synthesis of bromoalkenes
The bromoalkene starting compounds can be prepared by conventional methods from appropriate alkenes. We demonstrated this possibility for all derivatives by using the methods published in the literature.39–44 It was not possible to obtain only the derivatives F-Br and G-Br in sufficient yield or purity and so they were synthesized via substitution reactions (see ESI† for details).Conclusions
A new metal-free method for light-mediated reductive debromination of phenylvinyl derivatives using (poly)deuterated benzimidazoline derivatives has been developed. It is possible to perform the reaction efficiently with simple and cheap equipment. It allows the synthesis of deuterated derivatives of various substitution patterns, usually with excellent conversion and very high isotopic yield. The reducing agent can be regenerated with a recovery rate of up to 50%. For some substrate types, it is possible to implement a sequential bromination–deuterodebromination process. The developed methodology thus offers a new environmentally benign possibility of Br/D or H/D exchange on a double bond without the need for any special instrumental equipment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Ministry of Education, Youth, and Sports (project IGA_PrF_2020_012).Notes and references
- A. Kohen and H. H. Limbach, Isotope effects in chemistry and biology, CRC Press, 1st edn, 2005 Search PubMed.
- E. Voutyritsa and C. G. Kokotos, Angew. Chem., 2020, 59, 1735–1741 CrossRef CAS.
- E. Voutyritsa, A. Theodorou, M. G. Kokotou and C. G. Kokotos, Green Chem., 2017, 19, 1291–1298 RSC.
- F. W. McLafferty and F. Turecek, Interpretation of Mass-Spectra, Univ. Science Books, Sausalito, CA, USA, 4th edn, 1993 Search PubMed.
- A. Adejare and P. W. Brown, Anal. Chem., 1997, 69, 1525–1529 CrossRef CAS.
- S. Tittebrandt, M. Edelson-Averbukh, B. Spengler and W. D. Lehmann, Angew. Chem., Int. Ed., 2013, 52, 8973–8975 CrossRef CAS.
- E. M. Russak and E. M. Bednarczyk, Ann. Pharmacother., 2019, 53, 211–216 CrossRef CAS.
- G. S. Timmins, Expert Opin. Ther. Pat., 2017, 27, 1353–1361 CrossRef CAS.
- S. Cargnin, M. Serafini and T. Pirali, Future Med. Chem., 2019, 11, 2039–2042 CrossRef CAS.
- M. Hatano, T. Nishimura and H. Yorimitsu, Org. Lett., 2016, 18, 3674–3677 CrossRef CAS.
- J.-F. Li, Z.-Z. Wei, Y.-Q. Wang and M. Ye, Green Chem., 2017, 19, 4498–4502 RSC.
- A. Seoane, N. Casanova, N. Quiñones, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 7607–7610 CrossRef CAS.
- T. Zhu, Z. Li, F. Xiao and W.-L. Duan, Tetrahedron Lett., 2018, 59, 3238–3241 CrossRef CAS.
- P. J. Smith, D. A. J. Crowe and K. C. Westaway, Can. J. Chem., 2001, 79, 1145–1152 CrossRef CAS.
- R. Robiette and J. Pospíšil, Eur. J. Org. Chem., 2013, 836–840 CrossRef CAS.
- S. M. Korneev and D. E. Kaufmann, Synthesis, 2002, 491–496 CrossRef CAS.
- X. Zhang, Q. Chen, R. Song, J. Xu, W. Tian, S. Li, Z. Jin and Y. R. Chi, ACS Catal., 2020, 10, 5475–5482 CrossRef CAS.
- X.-Y. Chen, L.-H. Sun and S. Ye, Chem. – Eur. J., 2013, 19, 4441–4445 CrossRef CAS.
- J. Wu, C. Zhao and J. Wang, J. Am. Chem. Soc., 2016, 138, 4706–4709 CrossRef CAS.
- X.-S. Li, L.-L. Zhao, X.-K. Wang, L. Cao, X.-Q. Shi, R. Zhang and J. Qi, Org. Lett., 2017, 19, 3943–3946 CrossRef CAS.
- M. Kuriyama, G. Yano, H. Kiba, T. Morimoto, K. Yamamoto, Y. Demizu and O. Onomura, Org. Process Res. Dev., 2019, 23, 1552–1557 CrossRef CAS.
- I. Iavicoli, L. Fontana and A. Bergamaschi, in Encyclopedia of Environmental Health, Elsevier, 2011, pp. 307–314 Search PubMed.
- J. Kielhorn, C. Melber, D. Keller and I. Mangelsdorf, Int. J. Hyg. Environ. Health, 2002, 205, 417–432 CrossRef CAS.
- Guideline for elemental impurities Q3D (R1) by European Medicines Agency.
- A. Jayaraman and S. Lee, Org. Lett., 2019, 21, 7923–7927 CrossRef CAS.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS.
- E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- M. Neumeier, D. Sampedro, M. Májek, V. A. de la Peña O'Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem. – Eur. J., 2018, 24, 105–108 CrossRef CAS.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS.
- K. Kunnen, G. Nikonov and L. Yunnikova, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2014, pp. 1–3 Search PubMed.
- E. Hasegawa, T. Ohta, S. Tsuji, K. Mori, K. Uchida, T. Miura, T. Ikoma, E. Tayama, H. Iwamoto, S. Y. Takizawa and S. Murata, Tetrahedron, 2015, 71, 5494–5505 CrossRef CAS.
- T. Igarashi, E. Tayama, H. Iwamoto and E. Hasegawa, Tetrahedron Lett., 2013, 54, 6874–6877 CrossRef CAS.
- L. L. Miller and E. Riekena, J. Org. Chem., 1969, 34, 3359–3362 CrossRef CAS.
- T. V. Magdesieva, I. I. Kukhareva, E. N. Shaposhnikova, G. A. Artamkina, I. P. Beletskaya and K. P. Butin, J. Organomet. Chem., 1996, 526, 51–58 CrossRef CAS.
- E. Hasegawa, T. Seida, N. Chiba, T. Takahashi and H. Ikeda, J. Org. Chem., 2005, 70, 9632–9635 CrossRef CAS.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS.
- H.-K. Wang, S. L. Morris-Natschke and K.-H. Lee, Med. Res. Rev., 1997, 17, 367–425 CrossRef CAS.
- G. N. Krutovskikh, G. F. Gornaeva, K. M. Krivozheiko, M. Z. Girshovich, L. P. Varmanyan and A. V. El'tsov, Chem. Pharm. J., 1980, 14, 130–133 Search PubMed.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Chem. – Eur. J., 2012, 18, 2931–2937 CrossRef CAS.
- T. P. M. Goumans, K. van Alem and G. Lodder, Eur. J. Org. Chem., 2008, 2008, 435–443 CrossRef.
- X. Yang, J. Wu, X. Mao, T. F. Jamison and T. A. Hatton, Chem. Commun., 2014, 50, 3245–3248 RSC.
- J. B. Hendrickson and S. M. Schwartzman, Tetrahedron Lett., 1975, 16, 277–280 CrossRef.
- K. Jouvin, A. Coste, A. Bayle, F. Legrand, G. Karthikeyan, K. Tadiparthi and G. Evano, Organometallics, 2012, 31, 7933–7947 CrossRef CAS.
- K. Kobayashi and T. Nogi, Heterocycles, 2016, 92, 1810 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, product characterization, KIE calculation, reaction optimization data. See DOI: 10.1039/d0gc03081c |
| This journal is © The Royal Society of Chemistry 2021 |