
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe sustainable synthesis of levetiracetam by an enzymatic dynamic kinetic resolution and an ex-cell anodic oxidation†
Sebastian
Arndt‡
a,
Birgit
Grill‡
b,
Helmut
Schwab
bc,
Georg
Steinkellner
bd,
Urška
Pogorevčnik
b,
Dominik
Weis
a,
Alexander M.
Nauth
a,
Karl
Gruber
 b,
Till
Opatz
a,
Kai
Donsbach
e,
Siegfried R.
Waldvogel
*a and
Margit
Winkler
*bc
b,
Till
Opatz
a,
Kai
Donsbach
e,
Siegfried R.
Waldvogel
*a and
Margit
Winkler
*bc
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: waldvogel@uni-mainz.de
bacib – Austrian Centre of Industrial Biotechnology, Krenngasse 37, A-8010 Graz, Austria
cInstitute of Molecular Biotechnology, Graz University of Technology, Petersgasse 14, 8010 Graz, Austria. E-mail: margitwinkler@acib.at
dInnophore GmbH, Am Eisernen Tor 3, 8010 Graz, Austria
ePharmaZell GmbH, Hochstrass-Süd 7, 83064 Raubling, Germany
First published on 8th December 2020
Abstract
Levetiracetam is an active pharmaceutical ingredient widely used to treat epilepsy. We describe a new synthesis of levetiracetam by a dynamic kinetic resolution and a ruthenium-catalysed ex-cell anodic oxidation. For the enzymatic resolution, we tailored a high throughput screening method to identify Comamonas testosteroni nitrile hydratase variants with high (S)-selectivity and activity. Racemic nitrile was applied in a fed-batch reaction and was hydrated to (S)-(pyrrolidine-1-yl)butaneamide. For the subsequent oxidation to levetiracetam, we developed a ligand-free ruthenium-catalysed method at a low catalyst loading. The oxidant was electrochemically generated in 86% yield. This route provides a significantly more sustainable access to levetiracetam than existing routes.
Introduction
Levetiracetam (S)-1 is an active pharmaceutical ingredient (API) used for treatment and prevention of hypoxic and ischemic type aggressions of the central nervous system.1 The formulated form is known as Keppra®. Levetiracetam is applied as a medication for epilepsy6 with a global sale of 770 M€ in 2018.2 Numerous strategies for the synthesis of (S)-1 have been explored as summarised by Kotkar and Sudalai, including asymmetric hydrogenations, kinetic resolutions, deracemisation with chiral auxiliaries and proline-catalysed asymmetric α-aminooxylation of n-butyraldehyde.3 Intramolecular cyclisation and elimination,4 and an asymmetric Strecker reaction were also reported.5 Recently, (S)-1 was approached by Co(I)-catalysed single electron reduction of the respective enamide.6 The most efficient approaches to levetiracetam are analysed in view of their sustainability, which may be expressed by the atom economy (AE)7 and the E-factor (EF) (calculations in ESI chapter 2†).8 Technical approaches towards (S)-1 started with the synthesis of 2-aminobutanamide through a Strecker reaction, which is then followed by a chiral resolution,9 and by alkylation/acylation of the amino group (Scheme 1, I).10 Although the Strecker reaction was highly atom-efficient, the resolution and the alkylation were wasteful and low-yielding. Thus, this route afforded 21% overall yield (Table 1). Another technical approach used 2-halobutanoic acid, which was accessed by α-halogenation. Subsequent nucleophilic substitution, a chiral resolution, and finally an amidation yielded the product (Scheme 1, II).11 The larger step count, harsher reaction conditions and more extensive use of chemicals rendered this route less efficient. Overall, 22% of (S)-1 was obtained at an AE of 5%.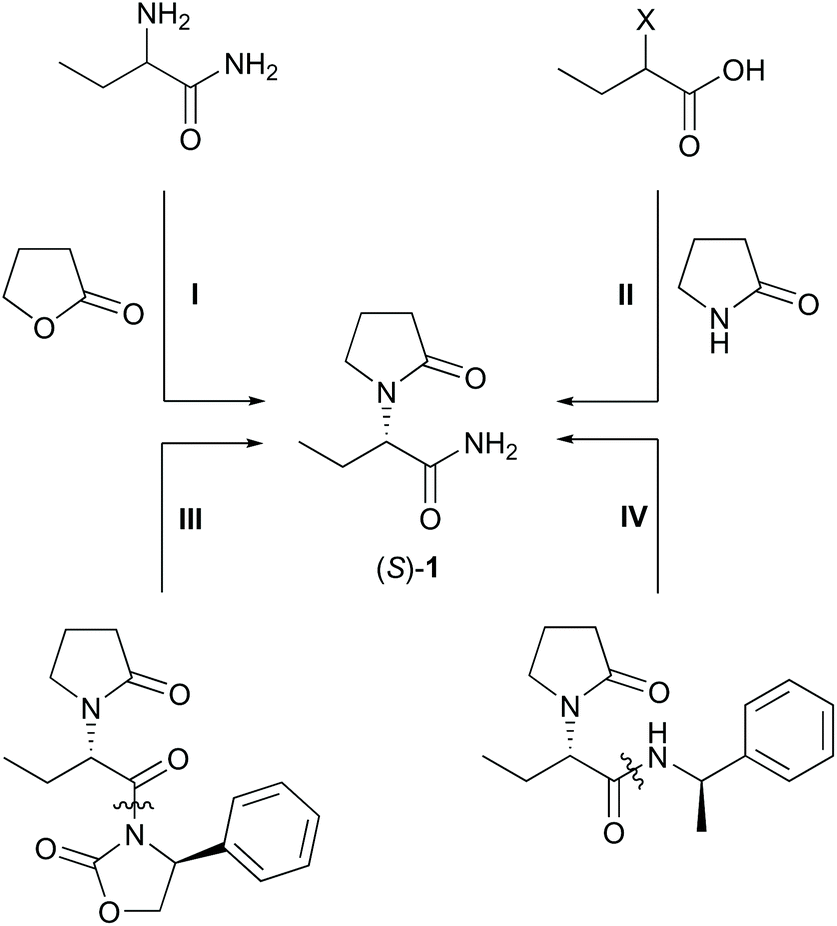 | ||
| Scheme 1 Technical and auxiliary-based approaches to levetiracetam (selected key-intermediates). X = Cl, Br. | ||
| Synthetic route via | AE [%] | EF [kg kg−1] | Overall yield [%] |
|---|---|---|---|
| Comparability might be limited by scale- and optimisation effects, and by the choice of the starting material. TM = transition metal, AE = atom efficiency, EF = E-factor. | |||
| Strecker synthesis | 12 | 52 | 21 |
| α-Halogenation | 5 | 95 | 22 |
| Evans’ auxiliary | 5 | 465 | 37 |
| Sulfinimine auxiliary | 0.8 | 572 | 18 |
| Ugi reaction | 38 | 1544 | 5 |
| Ru catalysis | 3 | 5110 | 25 |
| TM hydrogenation | 39 | 151 | 18 |
| Our approach | 44 | 21 | 44 |
Academic strategies relied upon the use of chiral auxiliaries, such as oxazolidinones (Scheme 1, III) or sulfinimines.12 Those routes gave indeed higher yields, but the removal of the auxiliary and the large number of synthetic steps again resulted in a low overall efficiency. For this reason, those methods are economically prohibitive.
A particularly short method employed the Ugi three-component reaction that, given by the low number of synthetic steps, seemed very efficient at first sight (Scheme 1, IV).13 Revising the resolution,14 however, a very poor overall yield of 5% was obtained accompanied by an uneconomic use of chemicals. Moreover, toxic 2,2,2-trifluoroethanol was used as the solvent. The syntheses of the auxiliaries themselves were not taken into account, which would have impaired the metrics further.15 Catalytic consecutive double-bond migration and oxidative cleavage of (S)-2 under ruthenium catalysis was reported (Scheme 2a), wherein levetiracetam was obtained in 25% over six steps.16 The extensive use of chemicals resulted in by far the lowest sustainability of this route within our comparison. Furthermore, an unacceptable co-solvent according to pharmaceutical guidelines was used (CCl4).17 Bandichhor separately reported the oxidation of (S)-3 and obtained levetiracetam in 42% after amidation of (S)-4.18 Rhodium or cobalt-catalysed approaches were reported by Shevlin and Chirik for the enantioselective hydrogenation of dehydro-levetiracetam (5).6,19–21 Reviewing this pathway,20,21 levetiracetam was obtained in 18% yield, with 39% AE, and with an EF of 151, which still is poor for a technical scale (Scheme 2b). It should be pointed out that N-heterocyclic carbene or chiral phosphine ligands are expensive. Their molar price may be higher than that of the noble transition metal itself. An alternative electrochemical oxidation of (S)-3 was developed by Stahl et al. using a TEMPO-related mediator (Scheme 2a, step II).22 A patent elaborates on an enzymatic approach to levetiracetam and involved an (S)-selective nitrile hydratase (NHase) in a classical enzymatic resolution of (rac)-6, leading to a maximum theoretical yield of 50% after a low yielding two-step chemical synthesis of the oxo-nitrile precursor (Scheme 2c).23
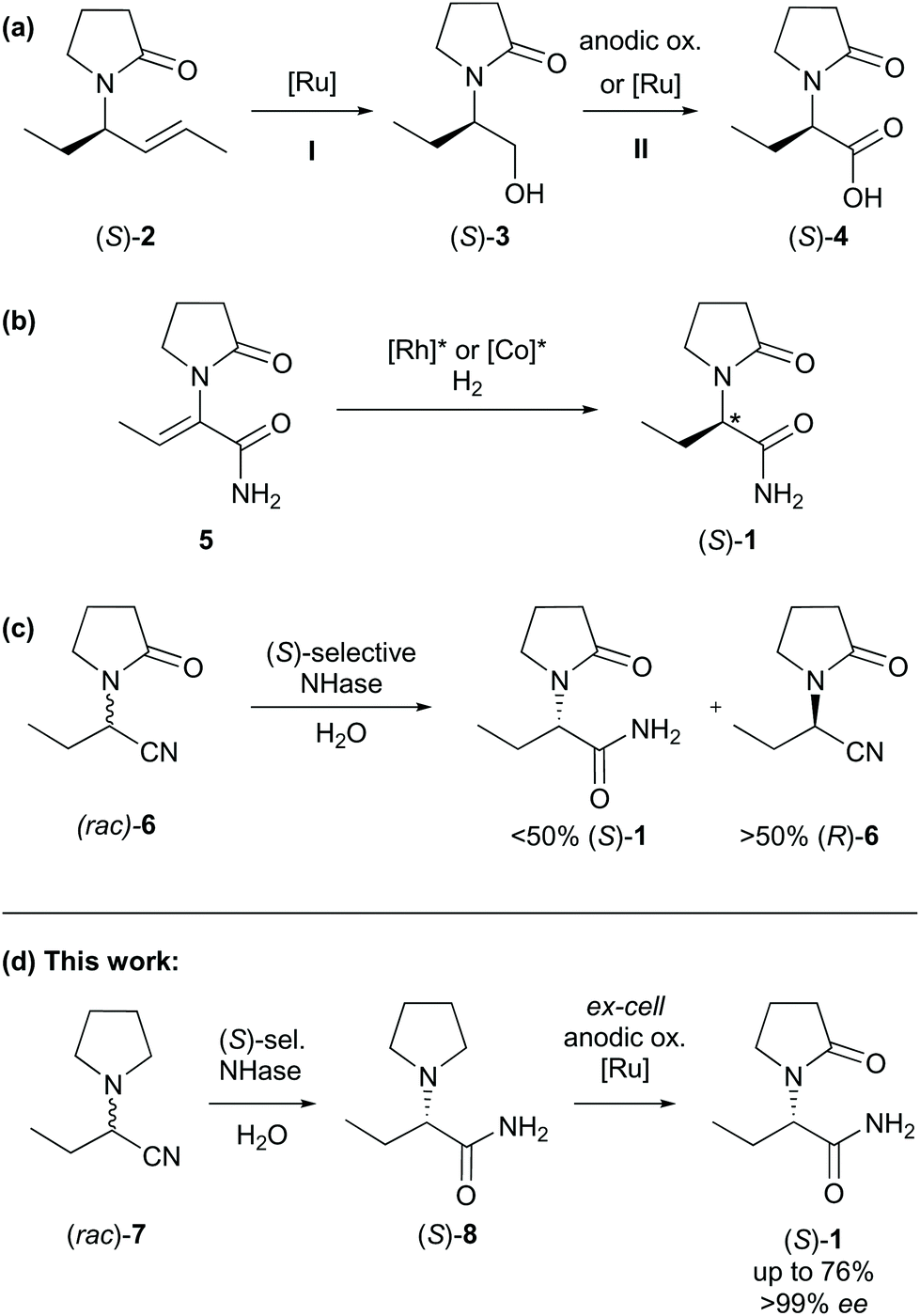 | ||
| Scheme 2 (a–d) Transition metal-catalysed or electrochemical approaches to levetiracetam (selected intermediates). Ox. = oxidation, sel. = selective, ee = enantiomeric excess. | ||
NHases exhibit a broad substrate scope for catalytic enzymatic transformations of nitriles to the corresponding amides.24 The perfectly atom economic reaction proceeds in water – which is both the reagent and solvent, under ambient conditions. Nitrile hydratases are applied on industrial scale e.g. for the production of the bulk chemical acrylamide.25 Likewise, electrolyses are highly atom-efficient, inherently safe, and environmentally benign as they do not require primary oxidants. Electricity is inexpensive, readily available, and sustainable if produced from renewable energy sources.26 Combining these two technologies offers new avenues for sustainable production processes.
In conclusion, technical routes to levetiracetam are more sustainable than ‘academic’ routes, but still suffer from low overall yields. Yet half of the material is lost in a resolution if a recycling of the wrong enantiomer is impossible. Chiral auxiliary-based routes indeed circumvent the resolution, but these methods are accompanied by a large step-count. Unfavourably, the synthesis and the removal of the auxiliary are extremely uneconomic. Catalytic routes ought to feature higher atom efficiencies and a better waste management in theory, but the elaborate syntheses of starting materials, the utilisation of chiral auxiliaries, and the employment of expensive ligands eliminate those advantages. Furthermore, they suffer from high overall costs that impede the application on a technical scale.
The aim of this work was to find both a sustainable and economical route for the synthesis of levetiracetam. From a retro-synthetic point of view, we envisioned a Strecker reaction with the benefit of low-priced and readily available starting materials. As opposed to previous routes, the pyrrolidine moiety shall be directly installed saving one synthetic step. The Strecker reaction is telescoped by an enzymatic dynamic kinetic resolution of 2-(pyrrolidine-1-yl)butanenitrile (rac)-7 to the corresponding amide (S)-8. Finally, we envisioned an electrochemical stereo-conservative and regiospecific oxidation of (S)-8 in position α to the amine (Scheme 2d).
Results and discussion
A highly (S)-selective NHase27 should convert (S)-7 to (S)-8 and we hypothesised that the undesired (R)-enantiomer would disintegrate in aqueous solution and subsequently form (rac)-7 again – the prerequisite for a dynamic kinetic resolution (DKR) to theoretically yield 100% of (S)-8 (Scheme 1d). A DKR similar to that described for (R)-mandelamide28 or 2-aminophenylacetamide29 is not feasible with the racemic oxo-nitrile (rac)-6, since conditions for its racemisation are incompatible with the enzymatic nitrile hydration step.Screening a panel of known and new NHases,30 we identified the thermotolerant cobalt-dependent Comamonas testosteroni NHase (CtNHase) as the most promising enzyme candidate (ESI Table S1†).31 It showed activity from pH 6.0 to 9.5 (ESI Fig. S2†), hydrolysed 7 in the temperature range between 25 and 50 °C (ESI Fig. S3†), and showed the highest (S)-selectivity among the candidates with 89% ee. At 10 mM (rac)-7 substrate concentration the product was detected in 1.1 mM concentration (ESI Table S1†). This small amount of detected product indicated a strong need for a deeper understanding of the limitations of the reaction system.
We investigated potential product inhibition and found that up to 25 mM, no inhibition by (rac)-8 was obvious. Aminonitrile 7 is in equilibrium with its three components propanal, pyrrolidine and hydrocyanic acid in aqueous solution, which is the key requirement for a dynamic kinetic resolution. The equilibrium is pH dependent, with high pH values favouring the formation of 7. On the one hand, the amounts of pyrrolidine, propanal and cyanide in the reaction solution depend on the pH, on the other hand, they also influence the pH. Each of these components may inhibit CtNHase, and especially cyanide has been reported as an NHase inhibitor before.32 Cyanide indeed inhibited CtNHase (ESI Fig. S4†) whereas pyrrolidine and propanal did not (ESI Fig. S5†). In successive optimisation of reaction conditions including increased substrate concentrations, we aimed for minimal cyanide concentrations by pH control and concomitantly adapted buffer capacities in analytical scale reactions. In doing so, product levels could be increased to more than 40% at 50 mM substrate load, and 15% at 200 mM substrate concentration while product ee ranged from 70–90% (ESI Fig. S6†). Time resolved monitoring of 7 hydration showed that the reaction was very fast, but ceased within a short period of time (ESI, Fig. S7†). The reason was not fast deactivation of CtNHase,30,31 but the formation of a side product: propanal and cyanide form 2-hydroxybutanenitrile – a competitive substrate for CtNHase which gives 2-hydroxybutanamide upon hydration, as confirmed by HPLC-MS (ESI Fig. S8†). Formation of (S)-8, enantioselectivity and the substrate selectivity needed to be increased to create a potent biocatalyst suited for industrial application. We therefore focused on CtNHase on the molecular level, docked (S)-7 into the active site of a model (ESI Fig. S11†) and identified amino acid residues within 4 Å around the docked compound as potential targets to shape the active site for improved (S)-7 binding. Putative Cobalt-binding residues, strictly conserved residues in the protein family and Arg52 in the β-subunit (a residue likely involved in proton transfer33) were excluded.
We created site saturation libraries for ten positions: Q93, W120, P126, K131 and R169 of the α-subunit and M34, F37, L48, F51 and Y68 of the β-subunit. To screen for improved variants, a colorimetric assay was established. Specifically, amides can be transacylated by amidases in the presence of hydroxylamine to give hydroxamic acids, which can then be visualised as iron-complex (Scheme 3).34 Using an (S)-selective amidase in the screening assay provides additional selection for (S)-8 and strictly (S)-selective Rhodococcus erythropolis amidase35 (ReAmd) was chosen for this step. The 10 libraries were screened in 96 deep-well plates. As an example, the results for position β-L48 are shown in the ESI (Fig. S12†).
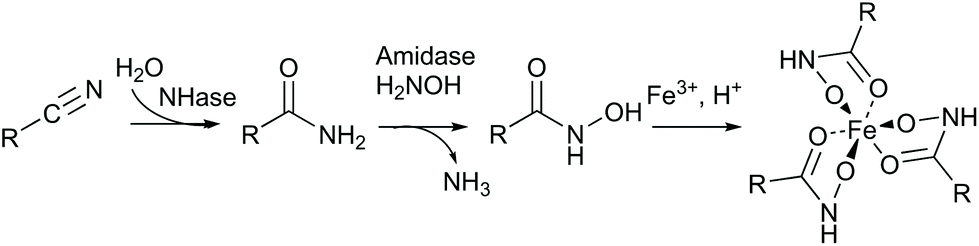 | ||
| Scheme 3 Coupled assay to screen for nitrile hydratase activity. | ||
Cell suspensions of the most promising variants from this screening were used for biotransformation reactions (ESI chapter 5.1 and 5.2†) to simultaneously determine concentration of 8 and the corresponding ee by chiral HPLC. In this re-screening step, false positives generated by potentially formed 2-hydroxybutaneamide were unambiguously eliminated. Whereas variants in the α-subunit showed little effect, several mutants from the β-subunit gave increased formation of 8 and/or ee. Most promising hits were analysed by sequencing (for a selected example see Table S3†). Hydrophobic amino acids instead of Phe in position 51 increased product levels up to twofold with ee of up to 92% at 50 mM of (rac)-7.
Random engineering by error prone PCR was used to complement the rational protein engineering approach.36 To increase the throughput, the above described assay was established on colony level (ESI chapter 8.2†). Colonies are attached to a membrane and separated from their growth medium. First, the membranes are treated with substrate solution. In the second phase they are transferred to a solution of amidase and hydroxylammonium chloride. Finally, the red complex is developed by transfer of the membrane attached colonies to acidic FeSO4 solution. Instead of the full length sequence, four short stretches in CtNHase lining the active site were defined to decrease the library size and therefore reduce the number of variants that must be screened. Random libraries were constructed for each stretch (coloured elements in Fig. 1). Specifically, the regions were amino acid 70–110 (α1) and 120–175 (α2) in the α-, and 30–71 (β1) and 124–170 (β2) in the β-subunit, respectively. The β1 library was based on wild-type CtNHase, whereas the other three libraries were generated on β1 variant βF51L. At least 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 clones per library were screened and 900–1700 variants per library were used for re-screening in the liquid format as described for the site-saturation libraries. Hit positions were evaluated on the 500 μL scale to determine concentration of 8 and ee by chiral HPLC. Library β1 revealed a high number of improved clones, whereby position F51 was found in 16 of 26 sequenced clones, with substitutions to either Ile, Leu or Val (Table 2, entries 3–5) alone or in combination with other amino acid exchanges or silent mutations. Also, substitution of βG54 was found 9 times (entries 6 and 7). The highest ee was found for variant βL48R (96.1%). In contrast, the α1 library consisted of many parents and only pos 110 carried an Ile in 7 of 16 clones (entry 8). This position significantly increased product levels as compared to parent βF51L and slightly the product ee. Similarly, library α2 revealed a substitution of P121 by Ser, Val or Thr beneficial for the formation of 8, whereas ees were typically lower than that of the parent βF51L (entry 9). The hits of library β2 contained mutations in numerous positions, however, only one combination with improved ee was found (entry 10). Associating most impact on CtNHase stretch β1, we screened a focused library that aimed for combinations of the three critical positions 48, 51 and 54, respectively (ESI chapter 9.3†). Substitutions in pos. 48 lead to ees up to 99%, however, product formation were strongly compromised (Table 2, entry 11). Around 60% of amide 8 were obtained (based on rac-7 addition) with βF51V variants combined with Ile, Arg or Val in pos. 54 (entry 12). Finally, combining the most promising mutations from all libraries revealed 10 variants with ees >98.5%. Most of them combined mutations of L48 and G54 (entry 14). Mutations in position 51 alone lead to both, an increase in selectivity and activity, but in combination with other mutations, activity decreased below wild-type level or ees did not reach beyond 94% (data not shown). The best variants and relevant control strains were compared in reactions which were supplemented with 1 equivalent of propanal (Fig. 2) to push the equilibrium towards (rac)-7 formation and compensate for the volatility of propanal. All variants displayed ee >98%. The double mutant αP121T/ βL48R showed two-fold increased product formation in comparison to the wild-type. Three double mutants based on the βL48P exchange reached ee 99.8% or higher. Clearly, position 48 in the β-subunit was responsible for the most significant increase in enantioselectivity, potentially due to improved contact of the amino acid side chain to (S)-7.
000 clones per library were screened and 900–1700 variants per library were used for re-screening in the liquid format as described for the site-saturation libraries. Hit positions were evaluated on the 500 μL scale to determine concentration of 8 and ee by chiral HPLC. Library β1 revealed a high number of improved clones, whereby position F51 was found in 16 of 26 sequenced clones, with substitutions to either Ile, Leu or Val (Table 2, entries 3–5) alone or in combination with other amino acid exchanges or silent mutations. Also, substitution of βG54 was found 9 times (entries 6 and 7). The highest ee was found for variant βL48R (96.1%). In contrast, the α1 library consisted of many parents and only pos 110 carried an Ile in 7 of 16 clones (entry 8). This position significantly increased product levels as compared to parent βF51L and slightly the product ee. Similarly, library α2 revealed a substitution of P121 by Ser, Val or Thr beneficial for the formation of 8, whereas ees were typically lower than that of the parent βF51L (entry 9). The hits of library β2 contained mutations in numerous positions, however, only one combination with improved ee was found (entry 10). Associating most impact on CtNHase stretch β1, we screened a focused library that aimed for combinations of the three critical positions 48, 51 and 54, respectively (ESI chapter 9.3†). Substitutions in pos. 48 lead to ees up to 99%, however, product formation were strongly compromised (Table 2, entry 11). Around 60% of amide 8 were obtained (based on rac-7 addition) with βF51V variants combined with Ile, Arg or Val in pos. 54 (entry 12). Finally, combining the most promising mutations from all libraries revealed 10 variants with ees >98.5%. Most of them combined mutations of L48 and G54 (entry 14). Mutations in position 51 alone lead to both, an increase in selectivity and activity, but in combination with other mutations, activity decreased below wild-type level or ees did not reach beyond 94% (data not shown). The best variants and relevant control strains were compared in reactions which were supplemented with 1 equivalent of propanal (Fig. 2) to push the equilibrium towards (rac)-7 formation and compensate for the volatility of propanal. All variants displayed ee >98%. The double mutant αP121T/ βL48R showed two-fold increased product formation in comparison to the wild-type. Three double mutants based on the βL48P exchange reached ee 99.8% or higher. Clearly, position 48 in the β-subunit was responsible for the most significant increase in enantioselectivity, potentially due to improved contact of the amino acid side chain to (S)-7.
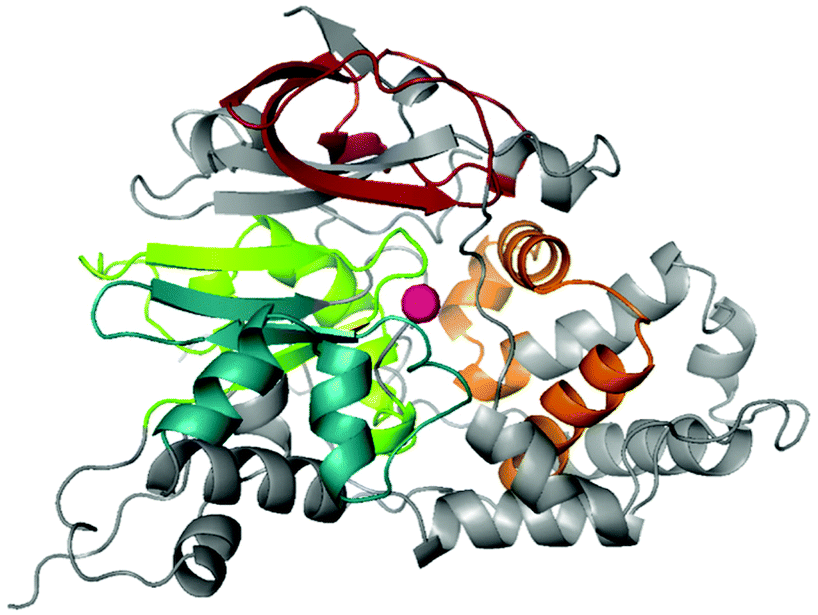 | ||
| Fig. 1 Homology model of CtNHase. The four regions targeted by random mutagenesis are shown in colour. The cobalt ion is displayed as pink sphere, region α1 in blue, α2 in green, β1 in orange and β2 in red. | ||
 | ||
| Fig. 2 Enantioselective hydration of (rac)-7 (100 μM) by CtNHase variants. Reactions were performed in triplicates in Tris-HCl buffer (500 μM) at pH 7.0 in the presence of propanal (100 μM) at 25 °C and 700 rpm for 2 h. | ||
| Entry | CtNHase | Library | Anal. yielda [%] | ee(S) [%] |
|---|---|---|---|---|
| a (rac)-7 (100 mM), resting cells (8.5 mg mL−1), Tris-HCl buffer (500 mM pH 7.0), 25 °C, 700 rpm, 2 h. b Cells cultivated in shake flasks. c Libraries based on mutant βF51L. | ||||
| 1 | Wild-typeb | — | 24 | 85 |
| 2 | βL48R | Random β1 | 26 | 96 |
| 3 | βF51I | Random β1 | 62 | 90 |
| 4 | βF51L | Random β1 | 30 | 91 |
| 5 | βF51V/ βE70L | Random β1 | 50 | 91 |
| 6 | βH53L/ βG54V | Random β1 | 24 | 92 |
| 7 | βN43I/ βG54C | Random β1 | 45 | 90 |
| 8 | αV110I/ βF51L | Random α1c | 57 | 93 |
| 9 | αP121T/ βF51L | Random α2c | 65 | 90 |
| 10 | βF51L/ βH146L/ βF167Y | Random β2c | 50 | 95 |
| 11 | βL48R/ βF51I/ βG54I | β1-Focused | 9 | 98 |
| 12 | βF51V/ βG54V | β1-Focused | 66 | 94 |
| 13 | βP121T/ βL48Rb | Combined | 42 | 98 |
| 14 | βL48R/ βG54Vb | Combined | 31 | 99 |
On the preparative scale, (rac)-7 was hydrated to (S)-8 in pH controlled stirred 50 mL volume fed-batch reactions, using whole cell biocatalysts (ESI chapter 10†). Variant P121T/L48R showed the highest concentration of the desired product on analytical scale and hydrated (rac)-7 (1.3 g substrate in total) to (S)-8 (1.1 g, 95% ee) on a 50 mL scale. The more selective variant L48R/G54V gave (S)-8 in 98% ee (0.78 g at 1.21 g substrate load). The yields of (S)-8 were defined by the degree of cyanohydrin hydration (Scheme 4) and reached 73.3% analytical yield (ESI Fig. S14 and 15†) in case of variant P121T/L48R and 61.7% in case of variant L48R/G54V, respectively.
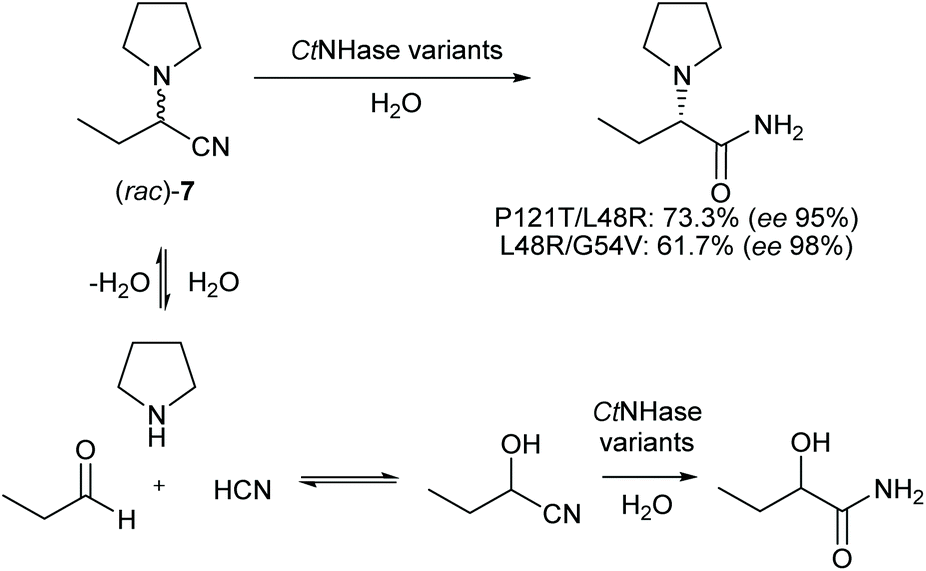 | ||
| Scheme 4 CtNHase mediated dynamic kinetic resolution. | ||
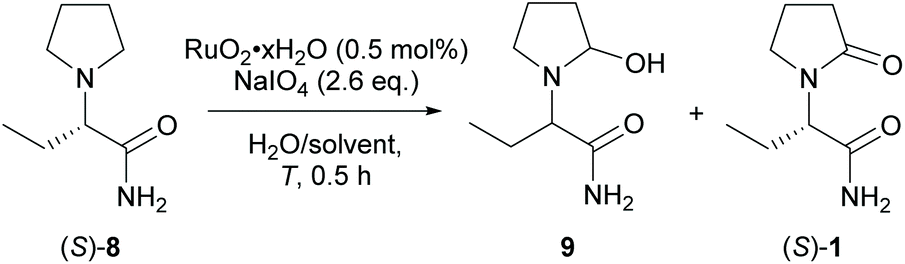
For the final step towards levetiracetam, a direct electrochemical oxidation was envisaged first. Pyrrolidines have been electrolysed in Shono-type oxidations,37 whereby pyrrolidones may be obtained in water as reaction medium.38 Stereo-conservative Shono oxidations have been reported for sulphonamides.39 Cyclic voltammetry indicated an irreversible oxidation of (S)-8 in the desired α-position of the amine. Up to 29% of 1 was determined by GC in optimised batch-electrolyses (detailed in experimental section in ESI, chapter 17†).40 However, chiral HPLC revealed a racemisation of the product during electrolysis. We reasoned that an N-radical intermediate must be formed during the electrolysis causing the racemisation. This prompted us to pursue an ex-cell electrochemical oxidation.
The ex-cell approach was investigated utilising ruthenium catalysts. Oxo ruthenium-catalysed α-oxidation of N-protected pyrrolidines generally feature mild reaction conditions, short reaction times and high selectivity.41 More importantly, RuO4 is known to perform two consecutive concerted oxidation steps that should result in the retention of the absolute configuration.42 Reported favourable conditions are solvent mixtures of CCl4/MeCN/H2O, a low to neutral pH, and low temperatures. Periodate is often used as terminal oxidant.43 The catalysis was performed by dissolving the hydrated ruthenium(IV) oxide (RuO2) and the desired oxidant. The mixture was suspended until the ruthenium tetroxide (RuO4) was formed as indicated by the appearance of a pale-yellow colour. Subsequently, (S)-8 was added as a solid and the reaction progress was monitored by GC. After complete reaction, the yield was determined either by GC using caffeine as an internal standard or the product was isolated by flash column chromatography. Best results were obtained for hydrated RuCl3 or RuO2 pre-catalysts and sodium metaperiodate. The catalytic reactions were performed in mixtures of aqueous and organic solvent such as dimethylformamide, acetonitrile, or acetone (Table 3, entries 2–4). Ethers, benzene, alcohols and tetrachloromethane were not investigated for safety- and regulatory reasons.44 A neutral pH was critical, and an excess of periodate (≥2.6 eq.) as well as a low temperature (0 °C, Table 3, entries 5 and 6) were beneficial. The product (S)-1 was formed in 66% yield and it was isolated in 49% yield and 99.6% ee. A substantial loss of material was observed in the gas chromatograms and in the mass balance after flash column chromatography. To rule out misleading GC data, the intermediate 9 and the potential over-oxidation product (S)-2-(2,5-dioxopyrrolidin-1-yl)butanamide were isolated or synthesised, respectively, but the clear appearance of their GC signals indicated a sufficient GC-stability and response factor. Accordingly, ring opening- and/or polymerisation pathways of the unstable hemi-aminal 9 intermediate were hypothesised.45 To prevent those side reactions, a biphasic solvent system with ethyl acetate/water was investigated similar to the protocols of Hamed et al. and Sashida et al.46 Up to 76% of the product was obtained even at room temperature and at a low amount of organic solvent (Table 3, entry 7), which is favourable from a technical point of view. We reasoned that the RuO4 is transferred into the organic layer where, due to the low water content, the intermediate might be protected from side reactions. The more polar product in turn is transferred to the aqueous phase where it might be protected from over-oxidation. The reaction was scaled up 10-fold obtaining a reproducible yield of 74% [Table 3, entry 8, 1.0 g (S)-8]. The RuO2 was efficiently recovered by filtration over aluminium oxide and the iodate was recovered quantitatively by crystallisation with methanol (up to 96% isolated yield).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | [v%] | T [°C] | GC yield [%] | |||
| 8 |
9a |
1 | ∑ | ||||
| a Relative intensity vs. standard, eq. = equivalents, T = temperature, DMF = dimethylformamide, isol. = isolated yield. Screenings were performed on a 100 mg scale of (S)-8. | |||||||
| 1 | H2O | — | 20 | 5 | 9 | 31 | 45 |
| 2 | DMF | 33 | 20 | 16 | 18 | 37 | 72 |
| 3 | MeCN | 33 | 20 | 1 | 4 | 57 | 62 |
| 4 | Acetone | 33 | 20 | 1 | 4 | 60 | 65 |
| 5 | MeCN | 50 | 0 | 1 | 2 | 65 | 68 |
| 6 | MeCN | 70 | 0 | 0 | 0 | 66 | 66 |
| 7 | EtOAc | 33 | 20 | — | — | 76 | Isol. |
| 8 | EtOAc | 33 | 20 | — | — | 74 | Isol. |
Next, an electrochemical recycling method was developed for the re-oxidation of the iodate generated in the ruthenium catalysis. High-grade periodate is expensive and generates substantial amounts of waste. Therefore, the recycling decreases the E-factor and the costs of the process. The direct oxidation of common iodide was recently published by our group.47 Based on our previous research, the iodate was oxidised in caustic soda. Water is generally considered as an abundant, environmentally benign and non-hazardous solvent.48 A divided electrolysis cell was used equipped with a Nafion membrane, a stainless steel cathode, and a boron-doped diamond (BDD) anode.40 Notably, BDD anodes are innovative and high performance metal-free electrodes, which are considered to be sustainable since they are made from methane.49 In anodic electro-organic synthesis they proved to be superior and technically viable as compared to other electrode materials.50 The conditions were optimised by a statistical screening approach,51 whereby a robust process was found. Optimum conditions were found for an applied charge of Q = 3 F, a current density of j = 10 mA cm−2, and a hydroxide/iodate concentration of 1 M/0.21 M (Table 4, entries 1–3). Paraperiodate (H2IO63−) was obtained in 86% yield and was converted to metaperiodate (IO4−) by acidic recrystallisation (up to 71% isolated yield).52 The electrochemically obtained periodate was tested in the Ru-catalysed oxidation to give reproducible results. Finally, the iodate electrolysis was scaled up in flow electrolysis.53 The electrolysis conditions were adjusted to an increased current density of j = 100 mA cm−2 (Table 4, entries 4–7) and an applied charge of Q = 4 F (Table 4, entries 8–10). The product was obtained in 78% yield, which corresponds to 48 g of paraperiodate.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | V [mL] | Q [F] | j [mA cm−2] | C(NaOH) [M] | LC-PDA yield [%] | ||
| IO3− | IO4− | ∑ | |||||
| BBD = boron-doped diamond, VA = stainless steel, rt = room temperature. Reactions were performed with an initial concentration of iodate of 0.21 M. | |||||||
| 1 | 6 | 3 | 2 | 3 | 20 | 83 | 103 |
| 2 | 6 | 3 | 10 | 1 | 16 | 83 | 99 |
| 3 | 6 | 3 | 10 | 1 | 11 | 86 | 97 |
| 4 | 50 | 3 | 10 | 1 | 20 | 85 | 105 |
| 5 | 50 | 3 | 50 | 1 | 26 | 73 | 99 |
| 6 | 50 | 3 | 100 | 1 | 34 | 63 | 97 |
| 7 | 50 | 3 | 300 | 1 | 63 | 37 | 100 |
| 8 | 500 | 3 | 100 | 1 | 29 | 70 | 99 |
| 9 | 1000 | 3 | 100 | 1 | 37 | 72 | 109 |
| 10 | 1000 | 4 | 100 | 1 | 18 | 78 | 96 |
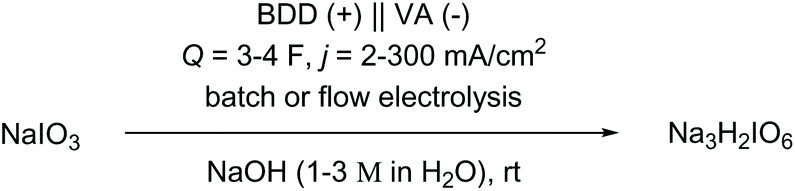
Conclusions
In conclusion, we have developed a sustainable synthetic pathway to levetiracetam that outcompetes the known synthesis routes in terms of atom efficiency, E-factor and overall yield. In the first part, a screening method was designed to enable the screening of tens of thousands of clones for highly (S)-selective NHase variants on colony level and allows us to report the first high throughput engineering study of a NHase. Time consuming chiral HPLC analytics only had to be applied for re-screening purposes of the best variants from rational and random protein engineering libraries. The selectivity of CtNHase for (S)-1versus 2-hydroxybutanenitrile was improved significantly and excellent enantiomeric excess of the desired product was achieved with several CtNHase double mutants. In the second part, a ruthenium-catalysed ex-cell oxidation was developed that provides an economical and sustainable access to levetiracetam. The catalysis was performed without the addition of ligands, by using a low catalyst load of 0.5 mol%, and by using water as “green” main solvent. Levetiracetam was obtained in up to 76% yield with full stereo-retention. In addition, the primary oxidant was electrochemically generated in up to 86%. The recycling process was investigated, which drastically eliminated waste and which formally substituted the primary oxidant periodate by environmentally benign water. BDD anodes feature a superior stability compared to commonly used anode materials for this kind of oxidation. Thus, toxic metal impurities in the terminal oxidant are prevented allowing the use for the synthesis of regulated products. Furthermore, the method might provide synthetic access to other valuable racetams, such as piracetam, brivaracetam, oxiracetam, or nefiracetam.Author contributions
S.A. Conceptualization, Investigation, Formal analysis, Visualization, Writing – original draft, B.G. Investigation, Formal analysis, Visualization, Writing – review & editing; H.S. Conceptualization, Funding acquisition, Writing – review & editing; G.S. Investigation, Visualization, Writing – review & editing, U.P. Investigation, D.W. Investigation, A. M. N. Investigation, Conceptualization, Writing – review & editing, K.G. Supervision, Writing – review & editing, T.O. Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Project administration, K.D. Conceptualization, Resources, Validation, Writing – review & editing; S.R.W. Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing. M.W. Conceptualization, Formal analysis, Data Curation, Funding acquisition, Project administration, Supervision, Writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The skilful technical assistance of Karin Reicher and Gernot A. Strohmeier is gratefully acknowledged. The COMET center acib: Next Generation Bioproduction is funded by BMVIT, BMDW, SFG, Standortagentur Tirol, Government of Lower Austria und Vienna Business Agency in the framework of COMET – Competence Centers for Excellent Technologies. The COMET-Funding Program is managed by the Austrian Research Promotion Agency FFG. UP received funding from the Erasmus+ Program. SRW was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation Wa1276/27-1 and in frame of FOR 2982 – UNODE Wa1276/23-1).References
- V. L. L. Lee, B. K. M. Choo, Y.-S. Chung, U. P. Kundap, Y. Kumari and M. Shaikh, Int. J. Mol. Sci., 2018, 19, 871 CrossRef PubMed.
- UCB, Integrated Annual Report, 2019. https://reports.ucb.com/2019/integrated-annual-report/data-reporting/financial-data.html.
- S. P. Kotkar and A. Sudalai, Tetrahedron Lett., 2006, 47, 6813–6815 CrossRef CAS.
- S. Gibson, H. K. Jacobs and A. S. Gopalan, Tetrahedron Lett., 2011, 52, 887–890 CrossRef CAS PubMed.
- V. Raju, S. Somaiah, S. Sashikanth, E. Laxminarayana and K. Mukkanti, Indian J. Chem., 2014, 53B, 1218–1221 CAS.
- M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888–893 CrossRef CAS PubMed.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- (a) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC; (b) R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC; (c) R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS; (d) K. van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 3 Search PubMed.
- R. He, H. Shao and L. Wang, CN106083642, 2016.
- J. Liu, CN105272897, 2016.
- (a) G. Yang, Y. Chen, G. Zhou, D. Zhu, L. Fu, L. Chen, L. Wang and Y. Yang, CN110590635, 2019; (b) T. Jianye and J. Bufang, CN109503354, 2019; (c) H. Pan, Y. Gong, K. Zhu, Y. Xiao, W. Zhang and P. Wang, CN110831924, 2019; (d) H. Pan, Y. Xiao, W. Zhang and P. Wang, CN110799495, 2019.
- (a) K. Chandra Babu, R. Buchi Reddy, E. Naresh, K. Ram Mohan, G. Madhusudhan and K. Mukkanti, J. Chem., 2013, 2013, 1–6 Search PubMed; (b) K. Chandra Babu, R. Buchi Reddy, K. Mukkanti, K. Suresh, G. Madhusudhan and S. Nigam, J. Chem., 2013, 2013, 1–5 Search PubMed; (c) F. Boschi, P. Camps, M. Comes-Franchini, D. Muñoz-Torrero, A. Ricci and L. Sánchez, Tetrahedron: Asymmetry, 2005, 16, 3739–3745 CrossRef CAS; (d) K. Chandra Babu, R. Buchi Reddy, K. Mukkanti, G. Madhusudhan and P. Srinivasulu, J. Chem. Pharm. Res., 2012, 4, 4988–4994 CAS.
- R. Cioc, L. van Schaepkens Riempst, P. Schuckman, E. Ruijter and R. Orru, Synthesis, 2017, 1664–1674 CAS.
- M. Forcato, F. Massaccesi, L. Cotarca, I. Michieletto and P. Maragni, WO2008/12268, 2008.
- R. A. Sheldon, J. R. Soc., Interface, 2016, 13, 20160087 CrossRef PubMed.
- M. Liniger, Y. Liu and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 13944–13949 CrossRef CAS PubMed.
- (a) European Medicines Agency, ICH guideline Q3D (R1) on elemental impurities, can be found under, https://www.ich.org/page/quality-guidelines; (b) D. R. Abernethy, A. J. Destefano, T. L. Cecil, K. Zaidi and R. L. Williams, Pharm. Res., 2010, 27, 750–755 CrossRef CAS PubMed.
- R. Mylavarapu, R. V. Anand, G. C. M. Kondaiah, L. A. Reddy, G. S. Reddy, A. Roy, A. Bhattacharya, K. Mukkanti and R. Bandichhor, Green Chem. Lett. Rev., 2010, 3, 225–230 CrossRef CAS.
- B. N. Warren and D. S. Davis, US2002042508, 2002.
- C. Ates, J. Surtees, A.-C. Burteau, V. Marmon and E. Cavoy, WO03/014080, 2003.
- J. Surtees, V. Marmon, E. Differding and V. Zimmermann, WO01/64637, 2001.
- M. Rafiee, Z. M. Konz, M. D. Graaf, H. F. Koolman and S. S. Stahl, ACS Catal., 2018, 8, 6738–6744 CrossRef CAS.
- J. L. Tucker, L. Xu, W. Yu, R. W. Scott, L. Zhao and N. Ran, WO2009009117, 2009.
- Z. Cheng, Y. Xia and Z. Zhou, Front. Bioeng. Biotechnol., 2020, 8, 352 CrossRef PubMed.
- Y. Asano, T. Yasuda, Y. Tani and H. Yamada, Agric. Biol. Chem., 1982, 46, 1183–1189 CAS.
- (a) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 ( Angew. Chem. , 2018 , 130 , 6124–6149 ) CrossRef; (b) J. L. Röckl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef; (c) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed; (d) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 ( Angew. Chem. , 2018 , 130 , 5694–5721 ) CrossRef CAS PubMed.
- (a) L. Martínková, N. Klempier, M. Preiml, M. Ovesná, M. Kuzma, V. Mylerová and V. Kren, Can. J. Chem., 2002, 80, 724–727 CrossRef; (b) M. Winkler, L. Martínková, A. C. Knall, S. Krahulec and N. Klempier, Tetrahedron, 2005, 61, 4249–4260 CrossRef CAS.
- S. V. Pawar and G. D. Yadav, Ind. Eng. Chem. Res., 2014, 53, 7986–7991 CrossRef CAS.
- E. Eppinger and A. Stolz, Appl. Microbiol. Biotechnol., 2019, 103, 6737–6746 CrossRef CAS PubMed.
- B. Grill, M. Glänzer, H. Schwab, K. Steiner, D. Pienaar, D. Brady, K. Donsbach and M. Winkler, Molecules, 2020, 25, 2521 CrossRef CAS PubMed.
- K. L. Petrillo, S. Wu, E. C. Hann, F. B. Cooling, A. Ben-Bassat, J. E. Gavagan, R. DiCosimo and M. S. Payne, Appl. Microbiol. Biotechnol., 2005, 67, 664–670 CrossRef CAS PubMed.
- K. Bui, M. Maestracci, A. Thiery, A. Arnaud and P. Galzy, J. Appl. Bacteriol., 1984, 57, 183–190 CrossRef CAS.
- K. H. Hopmann, Inorg. Chem., 2014, 53, 2760–2762 CrossRef CAS PubMed.
- Y.-C. He, C.-L. Ma, J.-H. Xu and L. Zhou, Appl. Microbiol. Biotechnol., 2011, 89, 817–823 CrossRef CAS PubMed.
- Z. Xue, Y. Chao, D. Wang, M. Wang and S. Qian, J. Ind. Microbiol. Biotechnol., 2011, 38, 1931–1938 CrossRef CAS PubMed.
- (a) Z. Shao and F. H. Arnold, Curr. Opin. Struct. Biol., 1996, 6, 513–518 CrossRef CAS PubMed; (b) U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- (a) S. Suga, M. Okajima and J.-i. Yoshida, Tetrahedron Lett., 2001, 42, 2173–2176 CrossRef CAS; (b) T. Shono, Y. Matsumura, K. Uchida, K. Tsubata and A. Makino, J. Org. Chem., 1984, 49, 300–304 CrossRef CAS; (c) T. Shono, Tetrahedron, 1984, 40, 811–850 CrossRef CAS; (d) T. Shono, Y. Matsumura and K. Tsubata, J. Am. Chem. Soc., 1981, 103, 1172–1176 CrossRef CAS; (e) T. Shono, H. Hamaguchi and Y. Matsumura, J. Am. Chem. Soc., 1975, 97, 4264–4268 CrossRef CAS; (f) T. Shono, Y. Matsumura and K. Tsubata, Tetrahedron Lett., 1981, 22, 3249–3252 CrossRef CAS.
- (a) F. Wang, M. Rafiee and S. S. Stahl, Angew. Chem., Int. Ed., 2018, 57, 6686–6690 ( Angew. Chem. , 2018 , 130 , 6796–6800 ) CrossRef CAS PubMed.
- T. Shono, Y. Matsumura, K. Tsubata, K. Uchida, T. Kanazawa and K. Tsuda, J. Org. Chem., 1984, 49, 3711–3716 CrossRef CAS.
- C. Gütz, B. Klöckner and S. R. Waldvogel, Org. Process Res. Dev., 2016, 20, 26–32 CrossRef.
- (a) S.-I. Murahashi and D. Zhang, Chem. Soc. Rev., 2008, 37, 1490–1501 RSC; (b) M. Zhou and R. H. Crabtree, Chem. Soc. Rev., 2011, 40, 1875–1884 RSC.
- (a) B. Plietker, Synthesis, 2005, 2453–2472 CrossRef CAS; (b) J. M. Bakke and A. E. Frøhaug, J. Phys. Org. Chem., 1996, 9, 310–318 CrossRef CAS.
- (a) J.-Y. Lu, M. Riedrich, M. Mikyna and H.-D. Arndt, Angew. Chem., 2009, 121, 8281–8284 ( Angew. Chem. Int. Ed. , 2009 , 48 , 8137–8140 ) CrossRef; (b) K.-i. Tanaka and H. Sawanishi, Tetrahedron: Asymmetry, 1998, 9, 71–77 CrossRef CAS; (c) X. Zhang, A. C. Schmitt and W. Jiang, Tetrahedron Lett., 2001, 42, 5335–5338 CrossRef CAS; (d) P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936–3938 CrossRef CAS; (e) T. K. M. Shing, V. W.-F. Tai and E. K. W. Tam, Angew. Chem., 1994, 106, 2408–2409 ( Angew. Chem. Int. Ed. , 1994 , 33 , 2312–2313 ) CrossRef CAS; (f) T. K. M. Shing, E. K. W. Tam, V. W.-F. Tai, I. H. F. Chung and Q. Jiang, Chem. – Eur. J., 1996, 2, 50–57 CrossRef CAS.
- C. Djerassi and R. R. Engle, J. Am. Chem. Soc., 1953, 75, 3838–3840 CrossRef CAS.
- (a) H. Möhrle and J. Berlitz, Pharmazie, 2008, 63, 7–13 Search PubMed; (b) R. Ito, N. Umezawa and T. Higuchi, J. Am. Chem. Soc., 2005, 127, 834–835 CrossRef CAS; (c) M. Masui, S. Hara and S. Ozaki, Chem. Pharm. Bull., 1986, 34, 975–979 CrossRef CAS; (d) S. Yoshifuji, Y. Arakawa and Y. Nitta, Chem. Pharm. Bull., 1987, 35, 357–363 CrossRef CAS.
- (a) R. B. Hamed, L. Henry, J. R. Gomez-Castellanos, J. Mecinović, C. Ducho, J. L. Sorensen, T. D. W. Claridge and C. J. Schofield, J. Am. Chem. Soc., 2012, 134, 471–479 CrossRef CAS PubMed; (b) M. Kaname, S. Yoshifuji and H. Sashida, Tetrahedron Lett., 2008, 49, 2786–2788 CrossRef CAS.
- (a) S. Arndt, D. Weis, K. Donsbach and S. R. Waldvogel, Angew. Chem., Int. Ed., 2020, 59, 8036–8041 ( Angew. Chem. , 2020 , 132 , 8112–8118 ) CrossRef CAS PubMed; (b) L. J. J. Janssen and M. H. A. Blijlevens, Electrochim. Acta, 2003, 48, 3959–3964 CrossRef CAS; (c) L. J. J. Janssen, NL1013348, 2001; (d) P. S. T. Lehmann, DE10258652, 2004.
- (a) H. C. Hailes, Org. Process Res. Dev., 2007, 11, 114–120 CrossRef CAS; (b) C.-J. Li, Acc. Chem. Res., 2002, 35, 533–538 CrossRef CAS; (c) C.-J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS PubMed.
- (a) S. Lips and S. R. Waldvogel, ChemElectroChem, 2019, 6, 1649–1660 CrossRef CAS; (b) S. R. Waldvogel and B. Elsler, Electrochim. Acta, 2012, 82, 434–443 CrossRef CAS; (c) S. R. Waldvogel, S. Mentizi and A. Kirste, Top. Curr. Chem., 2012, 320, 1–31 CAS; (d) N. Yang, S. Yu, J. V. Macpherson, Y. Einaga, H. Zhao, G. Zhao, G. M. Swain and X. Jiang, Chem. Soc. Rev., 2019, 48, 157–204 RSC; (e) J. H. T. Luong, K. B. Male and J. D. Glennon, Analyst, 2009, 134, 1965–1979 RSC; (f) B. Gleede, T. Yamamoto, K. Nakahara, A. Botz, T. Graßl, R. Neuber, T. Matthée, Y. Einaga, W. Schuhmann and S. R. Waldvogel, ChemElectroChem, 2019, 6, 2771–2776 CrossRef CAS; (g) Y. Einaga, Bull. Chem. Soc. Jpn., 2018, 91, 1752–1762 CrossRef CAS.
- (a) B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2014, 53, 5210–5213 ( Angew. Chem. , 2014 , 126 , 5311–5314 ) CrossRef CAS; (b) S. Lips, A. Wiebe, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., 2016, 128, 11031–11035 ( Angew. Chem. Int. Ed. , 2016 , 55 , 10872–10876 ) CrossRef; (c) A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., 2017, 129, 14920–14925 ( Angew. Chem. Int. Ed. , 2017 , 56 , 14727–14731 ) CrossRef; (d) A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 11801–11805 ( Angew. Chem. , 2016 , 128 , 11979–11983 ) CrossRef CAS PubMed; (e) A. Wiebe, B. Riehl, S. Lips, R. Franke and S. R. Waldvogel, Sci. Adv., 2017, 3, eaao3920 CrossRef PubMed.
- (a) R. Möckel, E. Babaoglu and G. Hilt, Chem. – Eur. J., 2018, 24, 15781–15785 CrossRef PubMed; (b) P. M. Murray, F. Bellany, L. Benhamou, D.-K. Bučar, A. B. Tabor and T. D. Sheppard, Org. Biomol. Chem., 2016, 14, 2373–2384 RSC; (c) M. Santi, J. Seitz, R. Cicala, T. Hardwick, N. Ahmed and T. Wirth, Chem. – Eur. J., 2019, 25, 16230–16235 CrossRef CAS PubMed; (d) S. A. Weissman and N. G. Anderson, Org. Process Res. Dev., 2015, 19, 1605–1633 CrossRef CAS.
- (a) C. L. Mehltretter and C. S. Wise, US2989371, 1961; (b) H. H. Willard and R. R. Ralston, Trans. Electrochem. Soc., 1932, 62, 239 CrossRef.
- M. Selt, R. Franke and S. R. Waldvogel, Org. Process Res. Dev., 2020, 24, 2347–2355 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03358h |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |