
Nature inspired singlet oxygen generation to access α-amino carbonyl compounds via 1,2-acyl migration†
Waldemar
Schilling
a,
Yu
Zhang
ab,
Prakash Kumar
Sahoo
b,
Samir Kumar
Sarkar
 a,
Sivaraman
Gandhi
c,
Herbert W.
Roesky
a and
Shoubhik
Das
*b
a,
Sivaraman
Gandhi
c,
Herbert W.
Roesky
a and
Shoubhik
Das
*b
aDepartment of Chemistry, Georg-August-Universität Göttingen, Tammanstraße 2-4, 37077 Göttingen, Germany
bDepartment, of Chemistry, Univeriteit Antwerpen, Groenenborgerlaan 171, 2020 Antwerpen, Belgium. E-mail: shoubhik.das@uantwerpen.be
cDepartment of Chemistry, Gandhigram Rural Institute, Gandhigra, 624032, Tamilnadu, India
First published on 23rd November 2020
Abstract
We have discovered chlorophyll catalyzed 1,2-acyl migration reactions to achieve α-amino carbonyl compounds directly from the enaminones. In general, singlet oxygen is generated during photosynthesis in the photosystem II center. This singlet oxygen can readily react with the unsaturated double bonds present in biomolecules. This reactivity intrigued us to apply this concept towards unsaturated enaminones and others to achieve highly valuable compounds. Indeed, this photosensitizer is very cheap, commercially available, main group metal based and provided excellent efficiency for singlet oxygen mediated chemistry by achieving high turnover number (TON) > 300 with a high turnover frequency (TOF) of 50 h−1. Finally, a combination of DFT calculations and detailed mechanistic experiments provided the exact role of the photosensitizer and clear insights into the reaction.
Introduction
Singlet oxygen is a reactive oxygen species (ROS) and is generated via energy transfer to the molecular oxygen during photosynthesis in the photosystem II reaction center.1–3 Chlorophyll after absorbing solar energy reaches the short-lived (∼10–8 s) singlet excited state (1Chl*). Part of this energy is converted into chemical energy via the photosynthetic electron transport chain (PETC), known as photochemical quenching. The rest of the energy stored in 1Chl* can be depleted as heat or as fluorescence or can convert itself to a relatively more stable (∼10–3 s) triplet excited state (3Chl*) via intersystem crossing. This triplet excited state (3Chl*) can easily react with the molecular oxygen generated from the water splitting reaction during photosynthesis and generate highly reactive singlet oxygen species. Due to the strong oxidant nature of this species, other biomolecules such as proteins, carotenoids, lipids etc. can undergo photooxidative reactions via reacting with the unsaturated bonds present in these biomolecules, known as oxidative stress in plants.4,5α-Amino carbonyl compounds are widely represented among pharmaceutically active compounds, peptides, and complex natural products.6–9
The most straightforward way to access α-amino carbonyl compounds is the electrophilic α-amination reaction, which directly introduces a nitrogen atom to the α-position of CH-acidic carbonyl substrates (Fig. 1).10–20
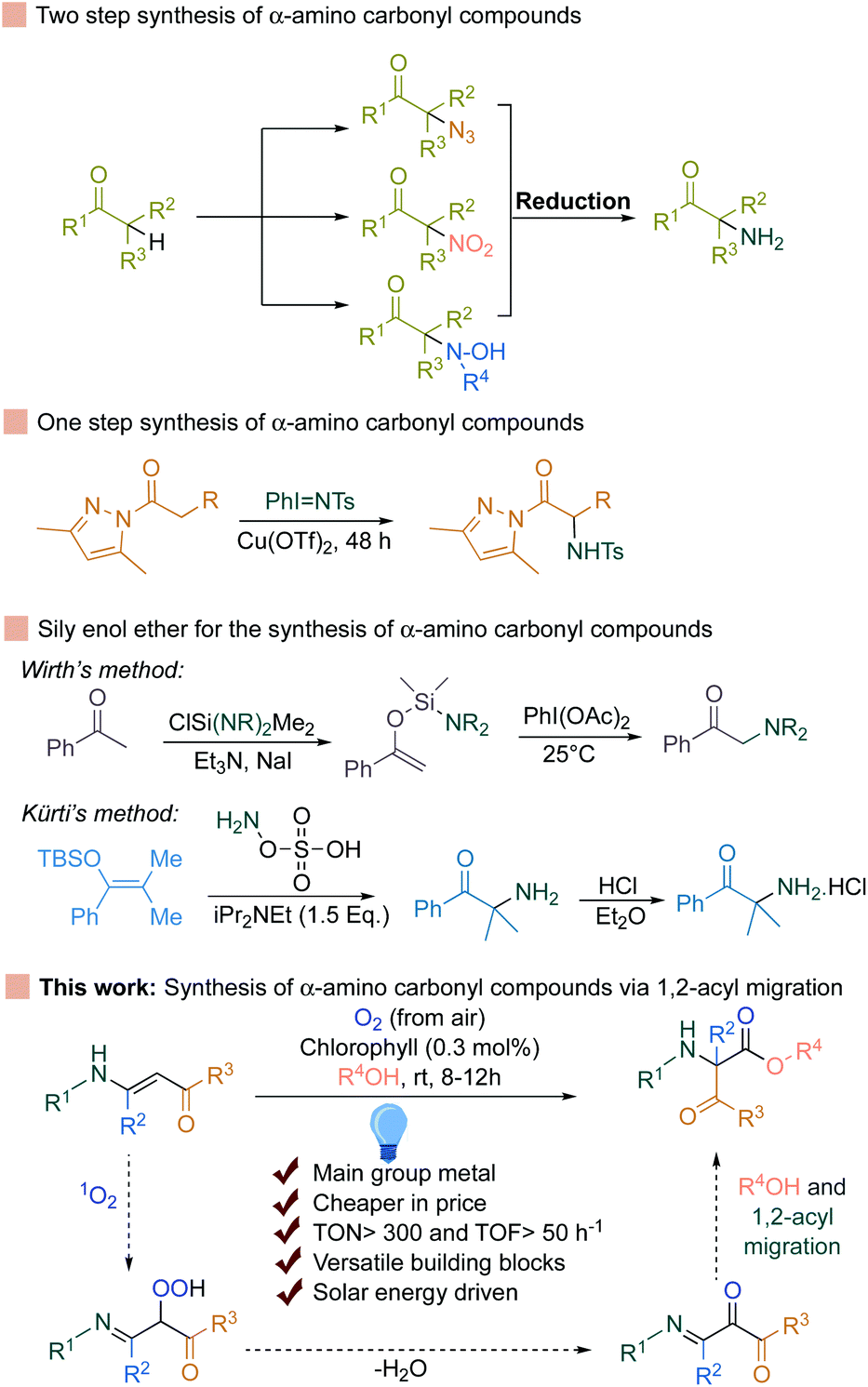 | ||
| Fig. 1 Different synthetic methods for the synthesis of α-amino carbonyl compounds. | ||
This strategy relies on trapping the enolates from the corresponding ketones with an electrophilic nitrogen source. Therefore, the formation of stable enolates from the respective ketones is the prerequisite. To avoid this, excellent achievements have been made by the group of Kürti and Wirth for the selective transformation of silyl enol ether to the corresponding α-amino carbonyls (Fig. 1).21,22
Very recently, concomitant 1,2-acyl migration and the cleavage of C–C bonds to achieve valuable products have also become a strong priority in organic synthesis.23–42 However, in most of the cases, strained cyclic ketone or the presence of a directing group is necessary. To avoid these, Wei et al. recently developed a 1,2-acyl migration reaction followed by olefin isomerization to achieve α,β-unsaturated ketones.43 However, an expensive Ni-complex and strict anhydrous conditions are required to achieve a high yield of the products. In contrast, enaminones can undergo 1,2-acyl migration reactions in the presence of Ru and Pt-based expensive transition metal based photosensitizers (Ru-catalyst: 88 Eur per g, Pt-catalyst: >200 Eur per g) along with a stoichiometric amount of base to access the α-amino carbonyl compounds.44,45 Alternatively, transition metal-free compounds such as main group metal-based complexes provide an interesting avenue for further research and appeal towards the broader synthetic applications.46–59 Intrigued by all this information, we rationalized that if singlet oxygen can be generated using chlorophyll as a photosensitizer and can be applied to the unsaturated double bonds in enaminones, it should generate the corresponding hydroperoxide by reacting with the double bond (Fig. 1). This hydroperoxide can further undergo dehydration and 1,2-acyl migration to the desired α-amino carbonyl compounds. Indeed, chlorophyll is a very cheap (2.13 Eur per g), commercially available, and main group metal (Mg) based porphyrin complex. In fact, the use of natural pigments is a well-known procedure for the generation of singlet oxygen in photochemistry, which was pioneered by Günther Otto Schenck in the 1940s by utilizing chlorophyll for the synthesis of organic compounds.60–69 Based on the early advances for the utilization of singlet oxygen, the Schenck ene reaction and others were developed for a variety of different compounds.70–76
Results and discussion
At the outset of our reaction, (E)-4-(phenylamino)pent-3-en-2-one (1a) was chosen as a model substrate to optimize the reaction conditions (Table S1†). At first, different amounts of chlorophyll were applied using methanol as the solvent under an aerobic atmosphere and under the irradiation of 20 W blue LED for 6 h. Gratifyingly, 54% of the desired methyl 2-methyl-3-oxo-2-(phenylamino)butanoate (1b) was achieved using 0.3 mol% of the catalyst loading and 2 mL of methanol as a solvent (entries 1–3). Further dilution of the solvent increased the product yield to 92% with an excellent turn over frequency (TOF) of 51 h−1 (entries 4–7). However, further increasing the catalyst loading or dilution of the reaction solution did not improve the TOF of the reaction. Comparison with other well-known singlet oxygen generating photosensitizers such as rose bengal, methylene blue, eosin Y and others clearly showed that chlorophyll was the superior catalyst under these conditions (Fig. 2). Furthermore, the investigation of different solvents also did not improve the reaction yield (entries 12–14).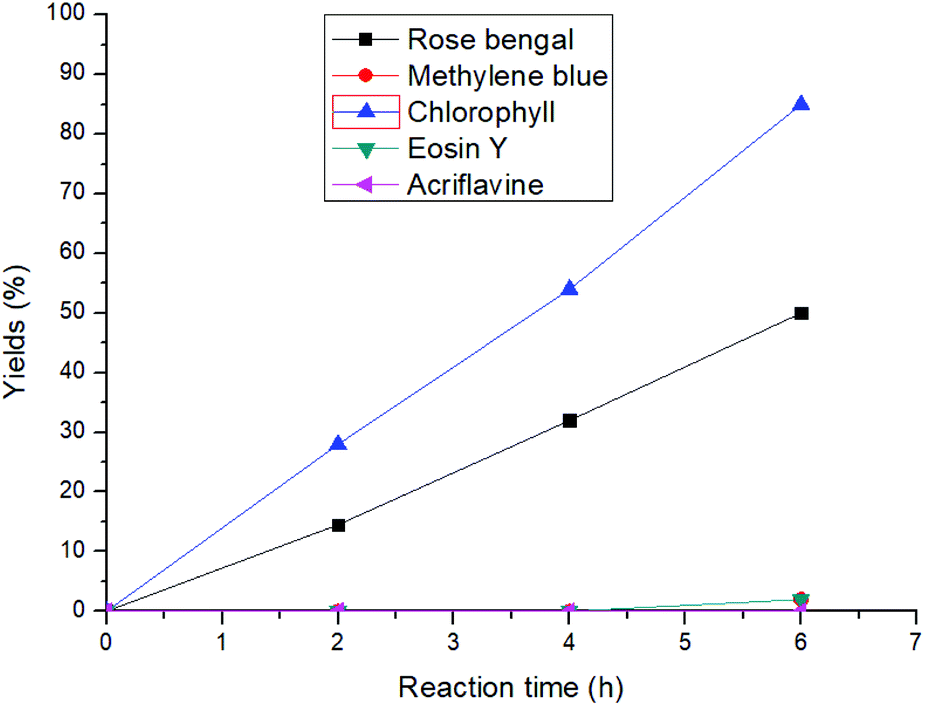 | ||
| Fig. 2 Comparison of different photosensitizers with chlorophyll for the 1,2-acyl migration reaction. | ||
With these optimized reaction conditions in hand, the scope of the 1,2-acyl migration reactions was explored to observe the potential of the photosensitizer (Scheme 1; entries 1b–15b). At first, different alcohols were utilized to change the esterification residue of the newly generated α-amino carbonyl products via 1,2-acyl migration. It should be noted that upon increasing the chain length of the alcohols, the inclusion of branch alcohols was well tolerated and yielded the corresponding products with an excellent TON of 297 (Scheme 1; entries 1b–6b). We observed that with the increase in chain length, the reaction speed decreased slightly. This effect was assumed to be correlated with the higher viscosity of the reaction solution and thereby was slowing the overall reaction speed. In the case of benzyl alcohol, the oxidation of benzyl alcohol to benzaldehyde and benzoic acid was observed but that did not perturb the desired reaction due to the presence of large excess of the benzyl alcohol (Scheme 1; entry 6b). Additionally, all of these reactions exhibited TOF > 20 h−1 which is quite high for the photocatalytic organic synthesis.
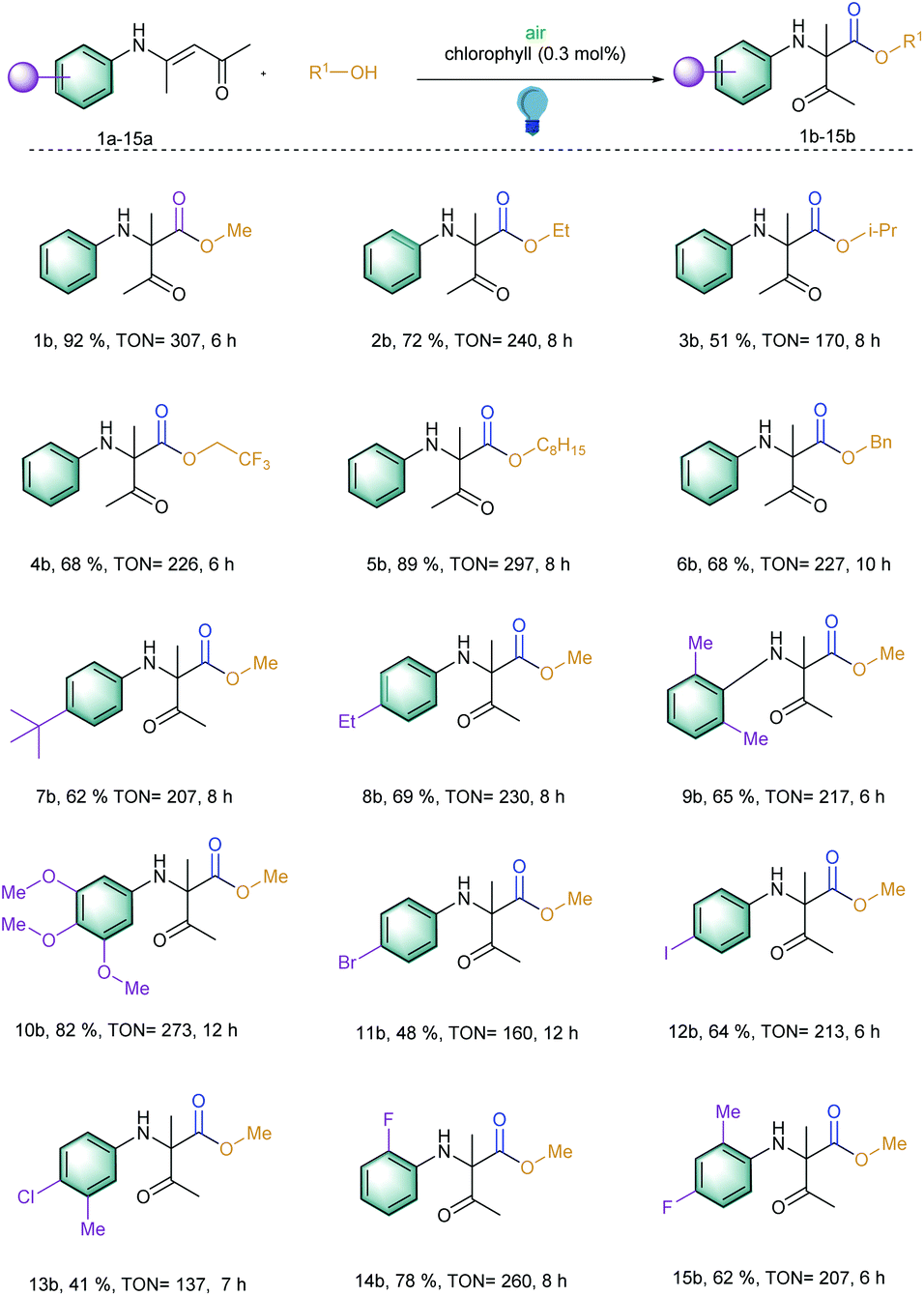 | ||
| Scheme 1 Substrate scope for the 1,2-acyl migration reactions. Reaction conditions: Substrate (0.25 mmol), chlorophyll (0.3 mol%), alcohol (3.5 mL), irradiation with blue LED at 460 nm for given time, at room temperature. | ||
Later, our interest to observe the different substitution patterns at the aromatic ring revealed that a wide variety of aromatic rings could be tolerated and could generate high TON for this 1,2-acyl migration reaction (Scheme 1; entries 7–15b). For example, the reaction exhibited moderate to excellent TON (up to 273) when the aromatic ring contained alkyl (7–9b), methoxy groups (10b) and halides (12b, 14–15b). Additionally, ortho substitution in the aromatic ring with respect to the amino group did not hamper the migration (9b).
Inspired by these above results, we became further interested to explore the 1,2 migration reactions of the longer alkyl chain in the carbonyl part and also on the α-carbon part of the enaminones. To our delight, a pentanone moiety was migrated and generated the corresponding α-amino carbonyl compound with a TON of 237 (Scheme 2, entry 16b). Further extension of the alkyl chain next to the amino group at α-carbon in the starting material led to the TON of 270 (Scheme 2, entry 17b). Interestingly, benzylic amine-substituted amines also exhibited an excellent TON of 237 (Scheme 2, entry 18b). However, changing the aromatic amines to aliphatic amines led to the decomposition of the starting materials under the photocatalytic conditions.
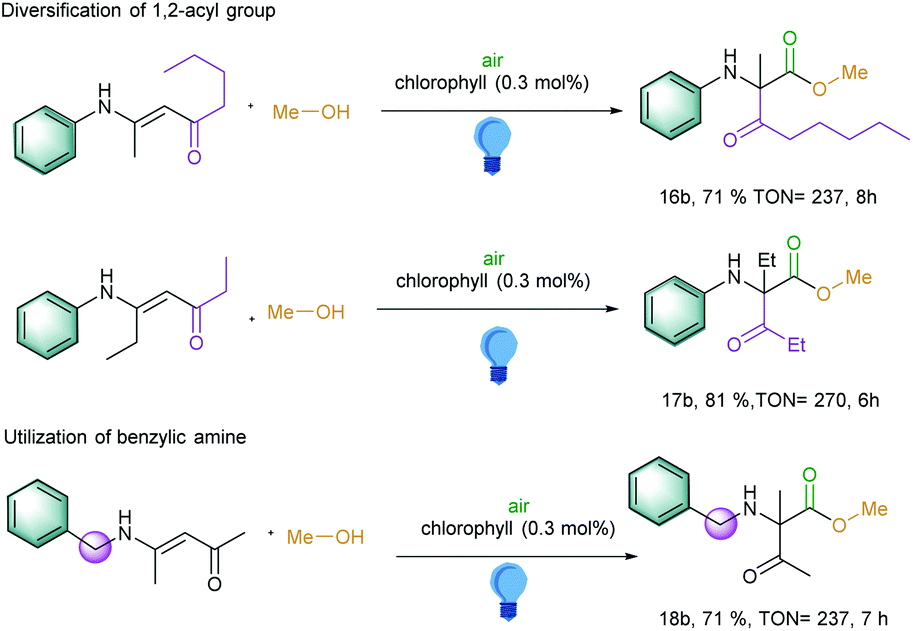 | ||
| Scheme 2 Substrate scope for the 1,2-acyl migration reaction with longer alkyl and amine chain in the starting material. Reaction conditions: Substrate (0.25 mmol), chlorophyll (0.3 mol%), alcohol (3.5 mL), irradiation with blue LED at 460 nm for given time, at room temperature. | ||
Later, an investigation of the impact of different wavelengths on this reaction revealed that the photosensitizer can also be highly reactive under the irradiation of red LED (low in energy); however, it exhibited less reactivity compared to the blue LED (Fig. 3). The observed yields are in accordance with the measured absorbance spectrum of the utilized photosensitizer. These results intrigued us to apply this reaction under the irradiation of direct sunlight. Indeed, sunlight is renewable and available throughout the entire world. The average intensity of the total solar irradiance is about 1366.1 W m−2 which provides roughly 4.3 × 1020 J energy only in 1 h.77–84 Therefore, if it is harvested properly and is utilized in organic synthesis, it can solve many of the sustainable issues. To our delight, the utilization of direct sunlight exhibited up to 82% yields of the products within 5 h (Scheme 3). Additionally, a g-scale reaction under the irradiation of sunlight was also performed efficiently.
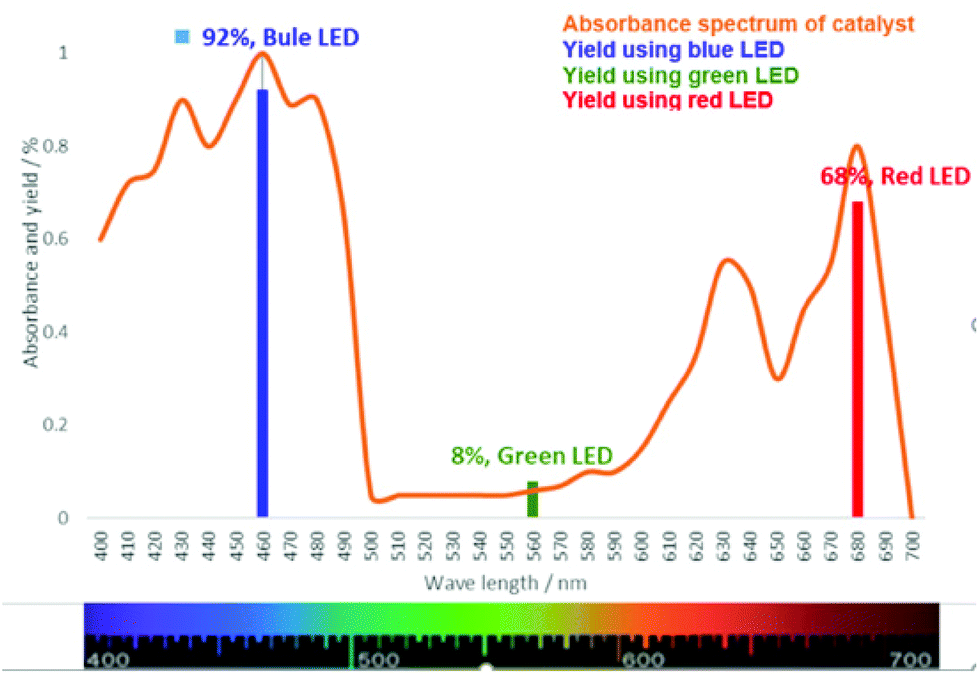 | ||
| Fig. 3 Absorbance spectrum of chlorophyll and the yields of the model substrate at different wavelengths after a 6 h reaction time. | ||
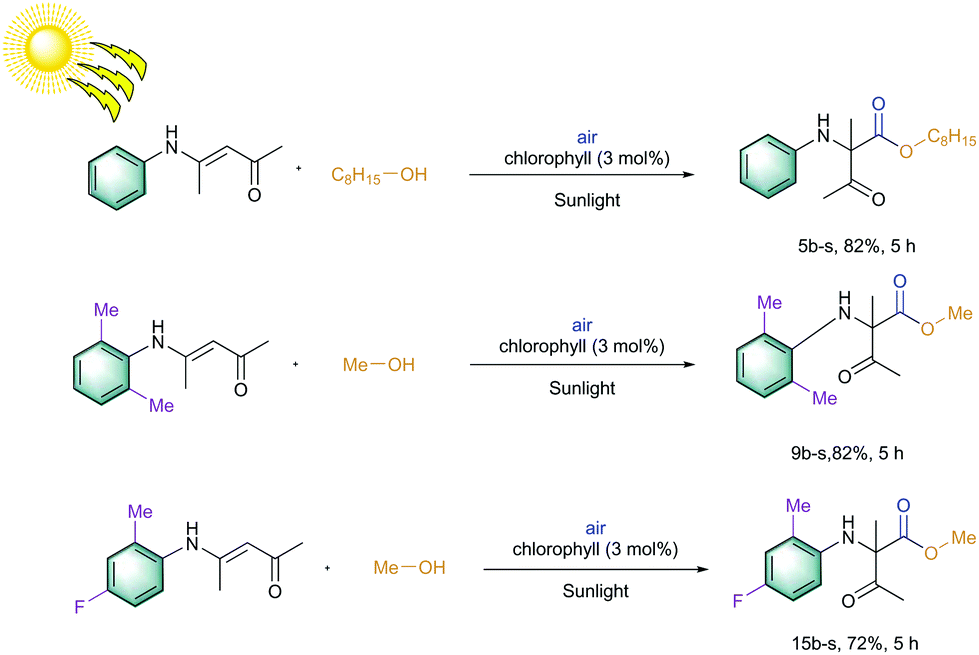 | ||
| Scheme 3 Solar energy mediated 1,2-acyl migration reactions. Reaction conditions: Substrate (0.25 mmol), chlorophyll (0.3 mol%), alcohol (3.5 mL), irradiation with direct sunlight for a given time, at ambient temperature. | ||
After having excellent solar energy driven 1,2-acyl migration reactions, we sought to diversify chlorophyll as a photosensitizer for various singlet oxygen mediated organic transformations (Scheme 4).85–90 For this purpose, we applied this photosensitizer for the oxidative dearomatisation of phenol substrate to achieve para-quinol. To our delight, chlorophyll exhibited an excellent yield of 81% for the formation of the desired product (19b) and this photosensitizer system avoided previously reported transition metals as well as stoichiometric oxidants and reagents.91–95 Furthermore, chlorophyll was applied onto anthracene to obtain endo peroxide (20b) with a yield of 78%.96,97 Furthermore, chlorophyll can selectively cleave both the C–C and C–N bonds in the imidazole moiety to access amide (21b) with a moderate yield of 62%.98–100 Later, our efforts to transform α-terpinene in a single step to ascaridole, an anthelmintic drug (22b), was highly successful with the yield of 83%.101–103
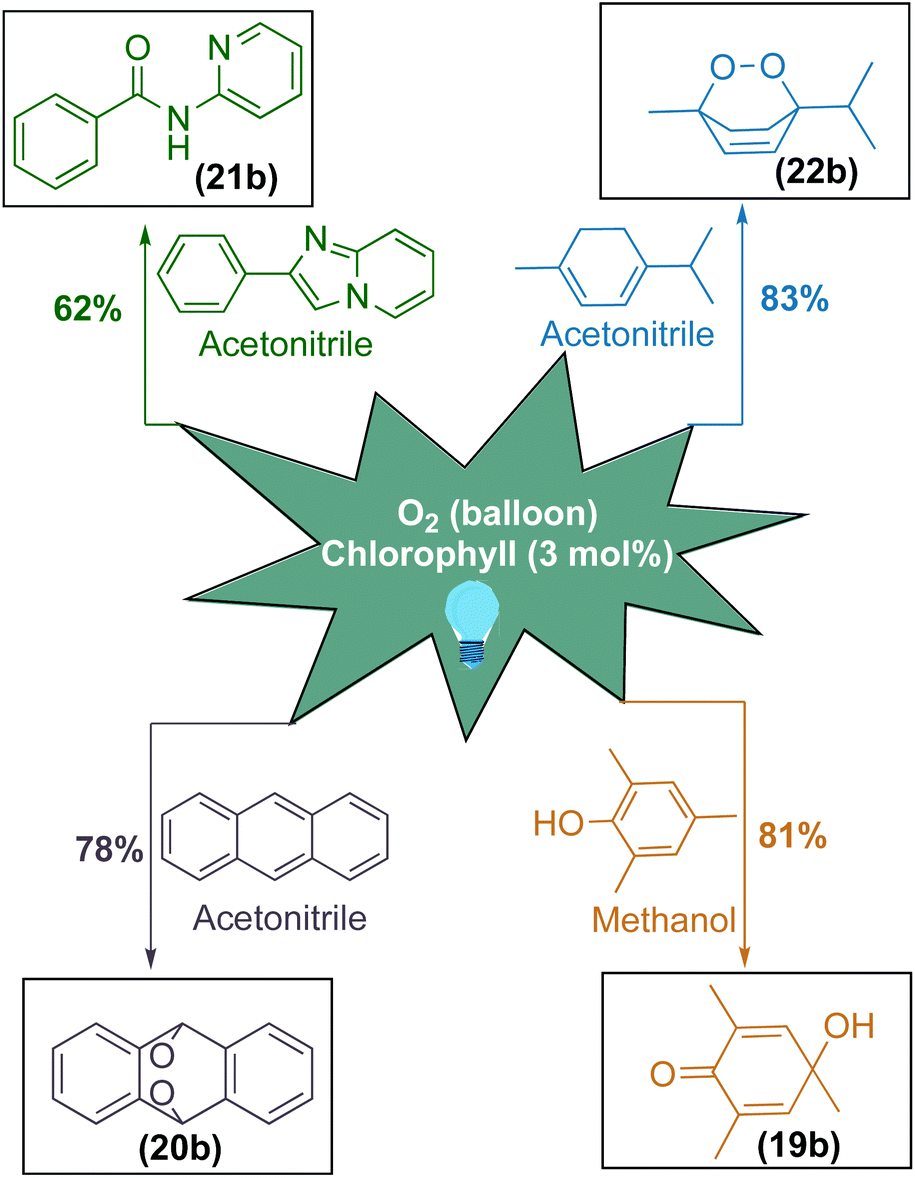 | ||
| Scheme 4 Chlorophyll as a robust photosensitizer for singlet oxygen mediated organic transformations. | ||
Inspired by the generality of this photosensitizer, we sought to gather mechanistic information about the role of the light, oxygen, and the photosensitizer in our reactions (Table 1). According to the control experiments, no product was observed in the absence of light, photosensitizer and oxygen/air. Furthermore, the effect of different quenchers was investigated to figure out the reactive oxygen species and possible intermediates in our reactions (Table 1).104–107 When 2,6-di-tert-butyl-4-methylphenol (BHT) or 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) was added, the yield of the reaction was not changed which proved that the reaction was not following a radical pathway. Further addition of sodium azide in the reaction displayed the presence of singlet oxygen.
| Conditions | Quencher | Quenching | Yield |
|---|---|---|---|
| a STD = standard reaction conditions: 0.3 mol% of photosensitizer, 0.3 mmol of substrate, 3.5 mL MeOH in air, 20 W blue LED, 6 h, rt. | |||
| No oxygen | — | — | 0 |
| No catalyst | — | — | 0 |
| No light | — | — | 2 |
| STD | NaN3 | Singlet oxygen | 0 |
| STD | BHT | Radical | 89 |
| STD | TEMPO | Radical | 87 |
Furthermore, a Stern–Volmer plot revealed that the quenching of the excited state of the chlorophyll was dependent on both the oxygen saturation as well as the concentration of the substrate (Fig. S2–4†).108 Detailed investigation of the absorption and emission spectra with and without the presence of substrate exhibited that both of the spectra underwent significant changes and indicated that the substrate was forming a substrate–catalyst intermediate, which provided access to the fast energy transfer between the catalyst and substrate (Fig. 4). After the addition of the substrate, an additional band was observed with high intensity, which was not present for the pure compounds alone (Fig. 4, left). The quenching of the photosensitizer by oxygen without the presence of the substrate further supported the involvement of singlet oxygen species in the reaction. To further prove the presence of the singlet oxygen species in the reactions, EPR-spin trap (TMP = tetramethyl piperidine) investigations were performed (Fig. 5). Performing the reaction without the oxygen atmosphere revealed no EPR signal which was in strong accordance with the control experiments from Table 1. However, as soon as the reaction was performed under an oxygen atmosphere (with and without the presence of the substrate), typical singlet oxygen signals arrived in the EPR spectra which showed clear proof of the involvement of singlet oxygen in this reaction.109–113
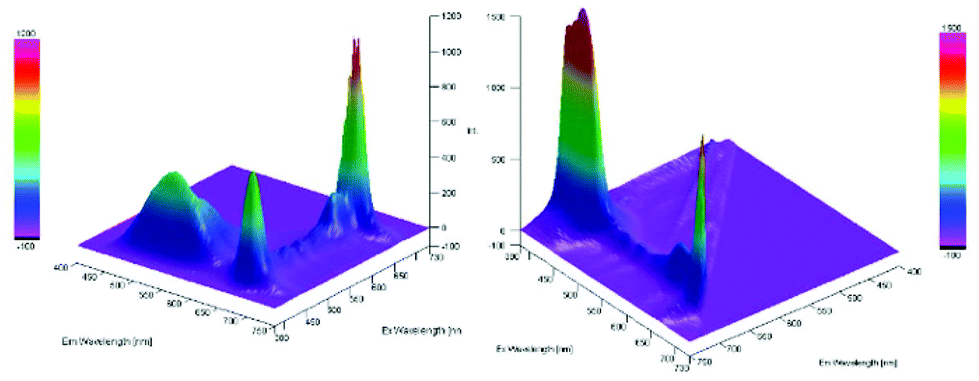 | ||
Fig. 4 3D-absorption and emission spectra for the migration reaction. Left: Substrate–chlorophyll mixture in MeOH (dilution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80). Right: Chlorophyll in MeOH (dilution 1:80). :80). Right: Chlorophyll in MeOH (dilution 1:80). | ||
 | ||
| Fig. 5 Electron spin resonance (EPR) spectrum of the tetramethyl piperidine (TMP) trapped singlet oxygen: (a) reaction mixture with TMP (0.25 mmol) under nitrogen atmosphere; (b) reaction mixture with TMP (0.25 mmol) saturated with air; (c) chlorophyll (0.3 mmol) without the presence of the substrate in methanol and saturated in air. All samples were irradiated for 5 minutes before preparing the EPR sample (0.2 mL). | ||
Further labeling experiments under an 18O2 atmosphere generated a single labeled product and indicated that only one oxygen atom was incorporated from the oxygen atmosphere (see the ESI†). This result combined with the different esterified products from the respective alcohols (Scheme 1) provided evidence that the second oxygen was originated from the alcohol. The oxygen in the acyl group seemed therefore intact and indicated an acyl shift during the reaction to obtain the desired products. Primary kinetic experiments were also performed and a linear time dependence of 16% yield/hour was observed (Fig. S5†). Additionally, ‘on and off’ experiments revealed that under darkness, an additional product (up to a maximum of 8%) was generated (Fig. S6†).
Detailed investigation of the conversion and yield over time in the dark revealed that the conversion was increased during the first 10 min followed by the generation of the product and then further decreased the conversion (Fig. 6). This observation could be explained by assuming two different pathways. The main pathway could activate the substrate and generate singlet oxygen at the same time resulting in a fast reaction. Based on the fact that oxygen was present in high excess and obviously, more singlet oxygen was produced. The singlet oxygen reacted with the substrate and formed a reversible intermediate at a lower rate.
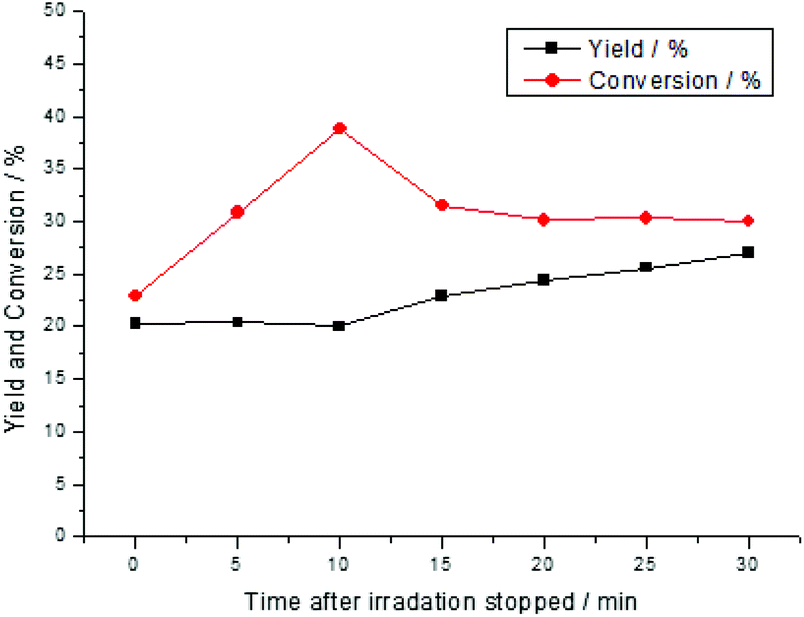 | ||
| Fig. 6 Time-dependent conversion and yield of the reaction after 2 h of the irradiation and samples were taken in the dark phase. | ||
To gain further mechanistic insights into this reaction, computational studies were undertaken to employ the density functional theory (DFT) calculations at the ub3lyp/6-311++g(d,p) level of theory in conjunction with the CPM solvent model for MeOH (Fig. 7 and Scheme 5). All the ground state geometries were optimized followed by a subsequent frequency test to ascertain stationary points. The absorption of a photon upon light irradiation (blue LED = 460 nm) excited the chlorophyll molecule from its ground state to the excited state (chlorophyll*) which interacted with the molecular oxygen. The excitation energy migration from chlorophyll* converted the ground-state oxygen (O2) to singlet oxygen (1O2) and chlorophyll* returned to its ground state by the energy transfer pathway. The frontier molecular orbitals of the O2 bound chlorophyll in the excited state, the highest occupied molecular orbital (HOMO) was spread over the π moiety of the porphyrin ring whereas the lowest unoccupied molecular orbital LUMO resided on O2 alone. It clearly indicated the charge/energy transfer from the porphyrin ring to the oxygen molecule in the excited state (Fig. S7†). The HOMO and LUMO for the chlorophyll catalyst were mainly observed in the π moiety of the porphyrin ring in both the ground and in the excited state (Fig. S8†).
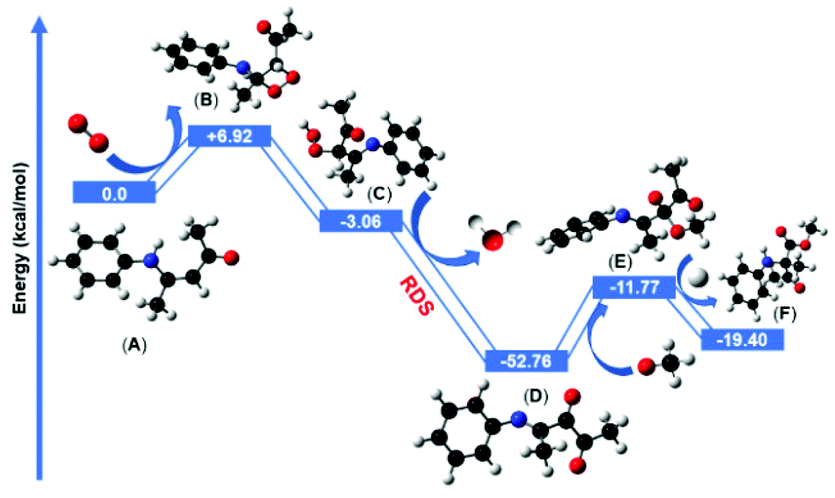 | ||
| Fig. 7 Complete single-point energy profile (relative to the starting material) involving singlet oxygen insertion, dehydration, nucleophilic addition and 1,2-acyl shift steps, calculated at the [ub3lyp/6-311++g(d,p)] level of theory. | ||
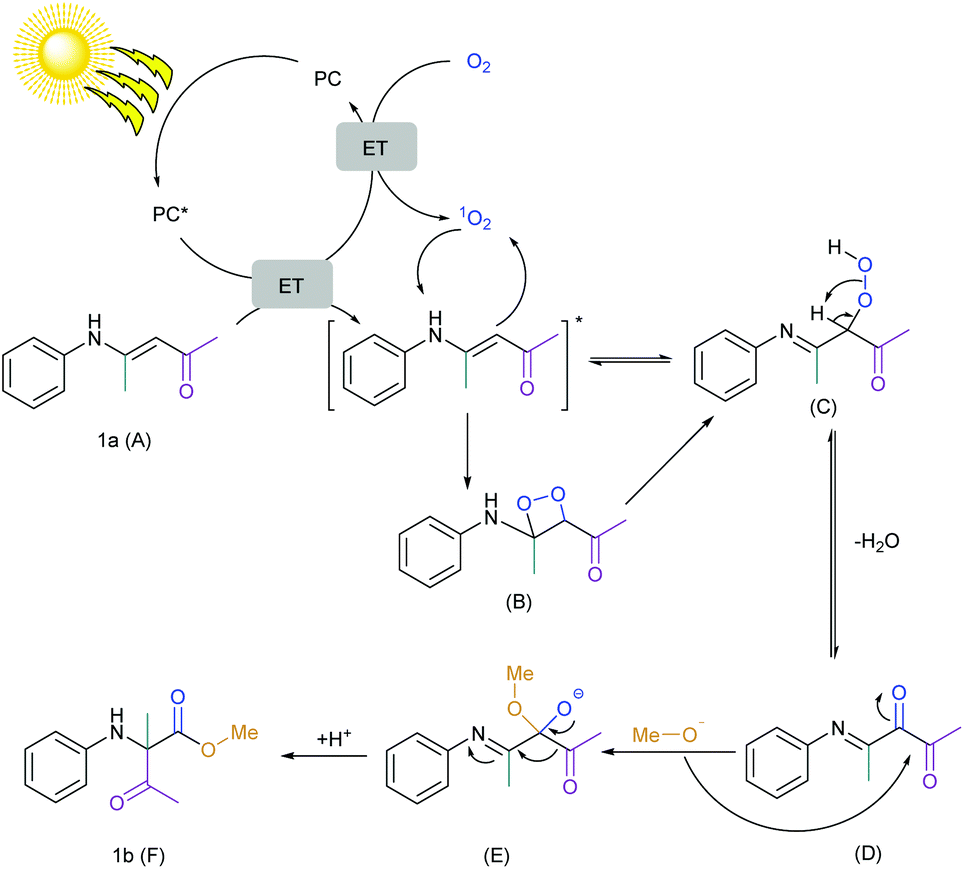 | ||
| Scheme 5 Proposed mechanism for the singlet oxygen mediated 1,2-acyl shift reaction. | ||
The in situ generated reactive 1O2 was then reacted with the electron-rich double bond of enaminone (A) and formed an unstable four-membered dioxetane intermediate (B) which was +6.92 kcal mol−1 uphill in comparison with the starting enaminone substrate (A). The computed energy barrier for this step was +6.92 kcal mol−1 and it is possible to overcome it under the irradiation of blue LED (λ = 460 nm, energy = 2.69 eV). Our model suggested that the four-membered dioxetane intermediate possessed ring strain energy. Thus, the dioxetane intermediate immediately converted to the hydroperoxide intermediate (C) via a ring-opening pathway after abstracting the hydrogen atom from the allylic hydrogen adjacent to the nitrogen atom. The formation of the hydroperoxide intermediate step was favorably exergonic, at −3.06 kcal mol−1, with respect to the four-membered dioxetane intermediate. The hydroperoxide intermediate subsequently underwent a stepwise elimination of H2O to form the dicarbonyl imine intermediate (D). This dehydration step was thermodynamically more favorable and highly exergonic, giving dicarbonyl imine intermediate at −52.76 kcal mol−1 after the removal of H2O molecule which is considered to be the rate determining step. The nucleophilic addition of methanol or MeO− to dicarbonyl imine produced the intermediate (E). Two nucleophilic addition pathways were located and investigated using methanol and MeO− as nucleophiles. Computationally, both of the pathways were feasible, although the barrier height of methanol was 2 kcal mol−1 higher than MeO− for the nucleophilic addition reaction. Proceeding via the more favorable MeO− assisted pathway, the nucleophilic addition reaction was more feasible by −11.77 kcal mol−1 to yield the intermediate (E). The subsequent intramolecular 1,2-acyl migration insertion of (E) to form the final product (F) was computed to occur by protonation with a barrier of −19.4 kcal mol−1, suggesting that the product formation (F) was facile and exergonic by intramolecular 1,2-acyl migration from the β- to α-position of the nitrogen atom.
Free energy calculations were also carried out on various transition states such as molecular oxygen bound chlorophyll (TS-I), dioxygen diradical (triplet O2 bound chlorophyll (TS-II) and mono oxygen bound catalyst (TS-III) (Fig. S9†) in this reaction. The free energy of activation for the formation of different transition states followed the order of mono oxygen bound catalyst (TS-III) > dioxygen diradical bound catalyst TS-II > dioxygen TS-I with the B3LYP functional sets. The overall activation barrier of the TS-III was 24.3 kcal mol−1, which was 12.2 kcal mol−1 higher than TS-II (Fig. S10†). Intrinsic reaction coordinate (IRC) calculations clearly predicted that the conversion of molecular oxygen to the singlet oxygen took place via the dioxygen diradical pathway by energy transfer (ET).114
Combining all this information, the mechanism of this reaction was finally proposed (Scheme 5). At first, chlorophyll was excited by light and the excited state of the chlorophyll underwent (a) an energy transfer (ET) to oxygen to form singlet oxygen which was clear from the EPR spectroscopy and DFT calculations and (b) an energy transfer (ET) to the substrate which was clear from the Stern–Volmer plot. Literature reports also suggested that the oxidation potential of the substrate (1a) (E1a.+/1a) is +1.24 V vs. SCE and is quite higher than the potential of the excited chlorophyll (+0.94 V vs. SCE).44,45,115,116 Therefore, chlorophyll in the excited state could not undergo single electron transfer (SET) with the substrate and ET was more preferable. The substrate was reacted with singlet oxygen to form the peroxo intermediate via the dioxetane intermediate and formed a reversible diketone intermediate after the dehydration. The labeling experiment confirmed the single labeled oxygen provided by the oxygen atmosphere. Therefore, the dicarbonyl compound was irreversibly attacked by the methoxide anion and finally, the 1,2-acyl shift generated the desired product.
Conclusions
In conclusion, we have demonstrated an efficient strategy for the generation of singlet oxygen using a commercially available plant-based chlorophyll under direct sunlight and applied this strategy in organic synthesis. This photosensitizer provided easy access to α-amino carbonyl products, quaternary amino acids, and many others without using transition metal and with high efficiency. Furthermore, detailed mechanistic studies clearly exhibited the different roles of the photosensitizer and oxygen in this reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Fonds der Chemischen Industrie (FCI, Liebig-Fellowship to S. D.), the Chinese Scholarship Council (CSC funding to Y. Z.) and the Humboldt fellowship (support to S. S.) for the financial support. We also thank Shreya Unone for synthesizing some of the starting materials and Daniel Reimer for the scientific discussions.Notes and references
- V. Dogra and C. Kim, Front. Plant Sci., 2020, 10, 1640 CrossRef.
- A. A. Ryan and M. O. Senge, Photochem. Photobiol. Sci., 2015, 14, 638 CrossRef CAS PubMed.
- I. Pibiri, S. Buscemi, A. P. Piccionello and A. Pace, ChemPhotoChem, 2018, 2, 535 CrossRef CAS.
- G. R. Buettner, Arch. Biochem. Biophys., 1993, 300, 535 CrossRef CAS PubMed.
- M. Burke, E. J. Land, D. J. McGarvey and T. G. Truscott, J. Photochem. Photobiol., B, 2000, 59, 132 CrossRef CAS.
- P. C. Meltzer, D. Butler, J. R. Deschamps and B. K. Madras, J. Med. Chem., 2006, 49, 1420 CrossRef CAS PubMed.
- F. I. Carroll, B. E. Blough, P. Abraham, A. C. Mills, J. A. Holleman, S. A. Wolckenhauer, A. M. Decker, A. Landavazo, K. T. McElroy, H. A. Navarro, M. B. Gatch and M. J. Forster, J. Med. Chem., 2009, 52, 6768 CrossRef CAS PubMed.
- C. Bouteiller, J. Becerril-Ortega, P. Marchand, O. Nicole, L. Barré, A. Buisson and C. Perrio, Org. Biomol. Chem., 2010, 8, 1111 RSC.
- M. C. Myers, J. Wang, J. A. Lera, J.-k. Bang, T. Hara, S. Saito, G. P. Zambetti and D. H. Appella, J. Am. Chem. Soc., 2005, 127, 6152 CrossRef CAS PubMed.
- Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356 CrossRef CAS PubMed.
- Z.-Q. Zhang, T. Chen and F.-M. Zhang, Org. Lett., 2017, 19, 1124 CrossRef CAS PubMed.
- N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5360 CrossRef CAS PubMed.
- N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 1080 CrossRef CAS PubMed.
- D. Sandoval, C. P. Frazier, A. Bugarin and J. R. de Alaniz, J. Am. Chem. Soc., 2012, 134, 18948 CrossRef CAS PubMed.
- C. Xu, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2014, 53, 4149 CrossRef CAS PubMed.
- B. Zhao, H. Du and Y. Shi, J. Am. Chem. Soc., 2008, 130, 7220 CrossRef CAS PubMed.
- R. W. Evans, J. R. Zbieg, S. Zhu, W. Li and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074 CrossRef CAS PubMed.
- D. J. Fischer, G. L. Burnett, R. Velasco and J. R. de Alaniz, J. Am. Chem. Soc., 2015, 137, 11614 CrossRef PubMed.
- K. Tokumasu, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2016, 138, 2664 CrossRef CAS PubMed.
- J. A. Smulik and E. Vedejs, Org. Lett., 2003, 5, 4187 CrossRef CAS PubMed.
- P. Mizar and T. Wirth, Angew. Chem., Int. Ed., 2014, 53, 5993 CrossRef CAS PubMed.
- Z. Zhou, Q.-Q. Cheng and L. Kürti, J. Am. Chem. Soc., 2019, 141, 2242 CrossRef CAS PubMed.
- M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC.
- F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed.
- L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed.
- Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640 CrossRef CAS PubMed.
- F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078 RSC.
- T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed.
- P.-H. Chen and G. Dong, Chem. – Eur. J., 2016, 22, 18290 CrossRef CAS PubMed.
- P.-H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340 CrossRef CAS PubMed.
- L. Liu, N. Ishida and M. Murakami, Angew. Chem., Int. Ed., 2012, 51, 2485 CrossRef CAS PubMed.
- T. Xu, H. M. Ko, N. A. Savage and G. Dong, J. Am. Chem. Soc., 2012, 134, 20005 CrossRef CAS PubMed.
- H. M. Ko and G. Dong, Nat. Chem., 2014, 6, 739 CrossRef CAS PubMed.
- P.-H. Chen, T. Xu and G. Dong, Angew. Chem., Int. Ed., 2014, 53, 1674 CrossRef CAS PubMed.
- L. Souillart, E. Parker and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 3001 CrossRef CAS PubMed.
- L. Souillart and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 9640 CrossRef CAS PubMed.
- X. Zhou and G. Dong, J. Am. Chem. Soc., 2015, 137, 13715 CrossRef CAS PubMed.
- X. Zhou, H. M. Ko and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 13867 CrossRef CAS PubMed.
- L. Deng, T. Xu, H. Li and G. Dong, J. Am. Chem. Soc., 2016, 138, 369 CrossRef CAS PubMed.
- T. Sun, Y. Zhang, B. Qiu, Y. Wang, Y. Qin, G. Dong and T. Xu, Angew. Chem., Int. Ed., 2018, 57, 2859 CrossRef CAS PubMed.
- A. M. Dreis and C. J. Douglas, J. Am. Chem. Soc., 2009, 131, 412 CrossRef CAS PubMed.
- Z.-Q. Rong, H. N. Lim and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 475 CrossRef CAS PubMed.
- C. Jiang, H. Lu, W.-H. Xu, H. Wu, T.-Y. Yu, P.-F. Xu and H. Wei, ACS Catal., 2020, 10, 1947 CrossRef CAS.
- W. Fan and P. Li, Angew. Chem., Int. Ed., 2014, 53, 12201 CrossRef CAS PubMed.
- Q.-Y. Meng, T. Lei, L.-M. Zhao, C.-J. Wu, J.-J. Zhong, X.-W. Gao, C.-H. Tung and L.-Z. Wu, Org. Lett., 2014, 16, 5968 CrossRef CAS PubMed.
- S. Harder, Chem. Rev., 2010, 110, 3852 CrossRef CAS.
- C. D. Martin, M. Soleihavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020 RSC.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972 RSC.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434 CrossRef CAS PubMed.
- L. J. Hounjet and D. W. Stephan, Org. Process Res. Dev., 2014, 18, 385 CrossRef CAS.
- S. He, F. Ni, Y. Ji, L. Wang, Y. Wen, H. Bai, G. Liu, Y. Zhang, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2018, 57, 16114 CrossRef CAS PubMed.
- L. C. Wilkins and R. L. Melen, Coord. Chem. Rev., 2016, 324, 123 CrossRef CAS.
- D. Riemer, B. Mandaviya, W. Schilling, A. C. Götz, T. Kühl, M. Finger and S. Das, ACS Catal., 2018, 8, 3030 CrossRef CAS.
- Y. Zhang, W. Schilling, D. Riemer and S. Das, Nat. Protoc., 2020, 15, 822 CrossRef CAS PubMed.
- Y. Zhang, W. Schilling and S. Das, ChemSusChem, 2019, 12, 2898 CrossRef CAS PubMed.
- D. Riemer, W. Schilling, A. Goetz, Y. Zhang, S. Gehrke, I. Tkach, O. Hollóczki and S. Das, ACS Catal., 2018, 8, 11679 CrossRef CAS.
- P. Hirapara, D. Riemer, N. Hazra, J. Gajera, M. Finger and S. Das, Green Chem., 2017, 19, 5356 RSC.
- F. D. Bobbink, S. Das and P. J. Dyson, Nat. Protoc., 2017, 12, 417 CrossRef CAS PubMed.
- D. Riemer, P. Hirapara and S. Das, ChemSusChem, 2016, 9, 1916 CrossRef CAS.
- M. Oelgemöller, Chem. Rev., 2016, 116, 9664 CrossRef PubMed.
- K.-H. Funken, Nachr. Chem., Tech. Lab., 1992, 40, 793 CrossRef CAS.
- A. Kriefer-Liszkay, J. Exp. Bot., 2005, 56, 337 CrossRef PubMed.
- J. Park, D. Feng, S. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 430 CrossRef CAS PubMed.
- A. Krieger-Öiszkay, C. Fufezan and A. Trebst, Photosynth. Res., 2008, 98, 551 CrossRef PubMed.
- M. Trytek, E. Janik, W. Maksymiec, J. Fiedurek, A. Lipke and M. Majdan, J. Photochem. Photobiol., A, 2011, 223, 14 CrossRef CAS.
- A. Y. Rybkin, A. Y. Belik, O. A. Kraevaya, E. A. Khakina, A. V. Zhilenkov, N. S. Goryachev, D. Volyniuk, J. V. Grazulevicius, P. A. Troshin and A. I. Kotelnikov, Dyes Pigm., 2019, 160, 457 CrossRef CAS.
- A. Harriman, B. G. Maiya, T. Murai, G. Hemmi and J. L. Sessler, J. Chem. Soc., Chem. Commun., 1989, 314 RSC.
- P. Klán and J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice, Wiley, Chichester, 2009 Search PubMed.
- K. Schaffner, Angew. Chem., Int. Ed., 2003, 42, 2932 CrossRef CAS.
- M. Prein and W. Adam, Angew. Chem., Int. Ed., Angew. Chem., Int. Ed., 1996, 35, 477 CrossRef CAS.
- H.-G. Bruenker and W. Adam, J. Am. Chem. Soc., 1995, 117, 3976 CrossRef CAS.
- C. S. Foote and E. L. Clennan, Properties and Reactions of Singlet Dioxygen, in Active Oxygen in Chemistry, ed. C. S. Foote, J. S. Valentine, A. Greenberg and J. F. Liebman, Structure Energetics and Reactivity in Chemistry Series, Springer, Dordrecht, 1995, vol. 2 Search PubMed.
- W. Adam, H.-G. Brünker, A. S. Kumar, E.-M. Peters, K. Peters, U. Schneider and H. G. von Schnering, J. Am. Chem. Soc., 1996, 118, 1899 CrossRef CAS.
- K. Gollnick, Adv. Photochem., 1968, 6, 1 CAS.
- C. S. Foote, Acc. Chem. Res., 1968, 1, 104 CrossRef CAS.
- G. Ohloff, Pure Appl. Chem., 1975, 43, 481 CAS.
- Y. Zhang, N. Hatami, N. S. Lange, E. Ronge, W. Schilling, C. Jooss and S. Das, Green Chem., 2020, 22, 4516 RSC.
- D. Cambié, F. Zhao, V. Hessel, M. G. Debije and T. Noël, Angew. Chem., Int. Ed., 2017, 56, 1050 CrossRef.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772 CrossRef CAS.
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L.-Z. Wu, C.-H. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528 CrossRef CAS PubMed.
- J. Nowotny, Energy Environ. Sci., 2008, 1, 565 RSC.
- J. Gong, C. Li and M. R. Wasielewski, Chem. Soc. Rev., 2019, 48, 1862 RSC.
- D. Cambie, J. Dobbelaar, P. Rient, J. Vanderspikken, C. Shen, P. H. Seeberger, K. Gilmore, M. G. Debije and T. Noël, Angew. Chem., Int. Ed., 2019, 58, 14374 CrossRef CAS PubMed.
- A. Lee, “BASF eyes wind and solar stake for ‘world-first’ chemicals plant” can be found under https://www.rechargenews.com/transition/basf-eyes-wind-and-solar-stake-for-world-first-chemicals-plant/2-1-693920, 2019.
- P. Bayer and A. J. von Wangelin, Green Chem., 2020, 22, 2359 RSC.
- J. Dad'ová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620 CrossRef.
- P. Bayer, R. Pérez-Ruiz and A. J. von Wangelin, ChemPhotoChem, 2018, 2, 559 CrossRef CAS.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC.
- M. Zheng, I. Ghosh, B. König and X. Wang, ChemCatChem, 2019, 11, 703 CrossRef CAS.
- D. Petzold, P. Singh, F. Almqvist and D. König, Angew. Chem., Int. Ed., 2019, 58, 8577 CrossRef CAS.
- M. C. Carreno, M. Gonzalez-Lopez and A. Urbano, Angew. Chem., Int. Ed., 2006, 45, 2737 CrossRef CAS.
- M. Uyanik, N. Sasakura, M. Kuwahata, Y. Ejima and K. Isihara, Chem. Lett., 2015, 44, 381 CrossRef CAS.
- K. Miyamoto, Y. Yokota, T. Suefuji, K. Yamaguchi, T. Ozawa and M. Ochiai, Chem. – Eur. J., 2014, 20, 5447 CrossRef CAS PubMed.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, Angew. Chem., Int. Ed., 2004, 44, 310 CrossRef CAS PubMed.
- Y.-F. Liang, K. Wu, Z. Liu, X. Wang, Y. Liang, C. Liu and N. Jiao, Sci. China: Chem., 2015, 58, 1334 CrossRef CAS.
- V. Martinez-Agramunt and E. Peris, Inorg. Chem., 2019, 58, 11836 CrossRef CAS PubMed.
- T. Uchikura, M. Oshima, M. Kawasaki, K. Takahashi and N. Iwasawa, Angew. Chem., Int. Ed., 2020, 59, 7403 CrossRef CAS PubMed.
- K. Yan, D. Yang, W. Wei, G. Li, M. Sun, Q. Zhang, L. Tian and H. Wang, RSC Adv., 2015, 5, 100102 RSC.
- R. Deshidi, M. A. Rizvi and B. A. Shah, RSC Adv., 2015, 5, 90521 RSC.
- Q. Shen, S. Shekhar, J. P. Stambuli and J. F. Hartwig, Angew. Chem., Int. Ed., 2005, 44, 1371 CrossRef CAS PubMed.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2007, 129, 6916 CrossRef CAS PubMed.
- C. Y. Park, Y. J. Kim, H. J. Lim, J. H. Park, M. J. Kim, S. W. Seo and C. P. Park, RSC Adv., 2015, 5, 4233 RSC.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158 RSC.
- J. Kollmann, Y. Zhang, W. Schilling, T. Zhang, D. Riemer and S. Das, Green Chem., 2019, 21, 1916 RSC.
- W. Schilling, Y. Zhang, D. Riemer and S. Das, Chem. – Eur. J., 2020, 26, 390 CrossRef CAS PubMed.
- Y. Zhang, D. Riemer, J. Kollmann and S. Das, ACS Catal., 2018, 8, 6659 CrossRef CAS.
- W. Schilling, D. Riemer, Y. Zhang, N. Hatami and S. Das, ACS Catal., 2018, 8, 5425 CrossRef CAS.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803 RSC.
- É. Hideg, C. Spetea and I. Vass, Biochim. Biophys. Acta, 1994, 1186, 143 CrossRef.
- É. Hideg, C. Spetea and I. Vass, Photosynth. Res., 1994, 39, 191 CrossRef PubMed.
- É. Hideg and I. Vass, Photochem. Photobiol., 1995, 62, 949 CrossRef.
- C. Fufezan, A. W. Rutherford and A. Krieger-Liszkay, FEBS Lett., 2002, 532, 407 CrossRef CAS PubMed.
- X.-H. Li, L.-Z. Wu, L.-P. Zhang, C.-H. Tung and C.-M. Che, Chem. Commun., 2001, 2280 RSC.
- W. T. Borden, R. Hoffmann, T. Stuyver and B. Chen, J. Am. Chem. Soc., 2017, 139, 9010 CrossRef CAS PubMed.
- H. Ishikita, B. Loll, J. Biesiadka, W. Saenger and E.-W. Knapp, Biochemistry, 2005, 44, 4118 CrossRef CAS PubMed.
- M. Kobayashi, S. Ohashi, K. Iwamoto, Y. Shiraiwa, Y. Kato and T. Watanabe, Biochim. Biophys. Acta, Bioenerg., 2007, 6, 596 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, analytical data for compounds, optimization studies and additional mechanistic experiments. See DOI: 10.1039/d0gc03555f |
| This journal is © The Royal Society of Chemistry 2021 |