
Visible-light-induced denitrogenative phosphorylation of benzotriazinones: a metal- and additive-free method for accessing ortho-phosphorylated benzamide derivatives†
Fushan
Chen
,
Shanshan
Hu
,
Sipei
Li
,
Guo
Tang
 * and
Yufen
Zhao
* and
Yufen
Zhao
Department of Chemistry, College of Chemistry and Chemical Engineering, and the Key Laboratory for Chemical Biology of Fujian Province, Xiamen University, Xiamen, Fujian 361005, China. E-mail: t12g21@xmu.edu.cn
First published on 10th December 2020
Abstract
Metal-free, visible-light-induced denitrogenative phosphorylation of 1,2,3-benzotriazinones was achieved. With the use of eosin Y as a photoredox catalyst, N,N-diisopropylethylamine as a base, CH3CN–H2O as a solvent and sunlight or a blue LED as a light source, a variety of aryl-phosphonates, aryl-phosphinates, and aryl-phosphine oxides were efficiently prepared. In addition, B2pin2 instead of P-nucleophiles as a radical acceptor was also demonstrated. The key advantages of this newly developed method are the clean reaction profile, use of a low-cost organic-dye catalyst, energy efficiency, broad substrate scope, good to excellent yields and large-scale synthetic applicability. The gram-scale synthesised compounds could be isolated in pure form just upon extraction, followed by re-crystallisation; no tedious chromatographic purification was required.
Introduction
Organophosphorus compounds are widely utilised in the fields of pharmaceuticals,1 functional materials2 and agricultural chemistry;3 in particular, aromatic phosphorus compounds act as useful ligands in transition-metal-catalysed reactions.4 Consequently, considerable attention has been paid to the study of aryl-P bond formation, and various strategies for the synthesis of these scaffolds have been established over the past few decades. Pioneered by Hirao,5 the transition-metal-catalysed phosphorylation of different aryl precursors such as arylhalides, aryl boronic acids, aryl mesylates, triarylbismuths, arylnitriles, aryl sulfides, arylsilanes and aroylhydrazides with phosphorus-based nucleophiles has attracted an explosion of interest.6 Although remarkable achievements have been made in this field, high reaction temperature and long reaction time are still needed.Over the past several years, transition-metal-triggered denitrogenative annulation of 1,2,3-benzotriazin-4(3H)-ones with various π-compounds has provided an applicable route to heterocyclic skeletons (Scheme 1A).7 This synthetic strategy has aroused great attention due to the use of benzotriazinones as a versatile and important building block, which can be easily prepared from commercially available methyl anthranilate. Besides, nitrogen is the only byproduct in the reactions. Recently, Yu et al. discovered an iridium-polypyridyl complex catalysed denitrogenative alkyne insertion of 1,2,3-benzotriazinones for the preparation of isoquinolones via a radical mechanism (Scheme 1B).8 Apart from cyclisation products, nickel-catalysed ortho-alkenylated, ortho-arylated and ortho-alkylketonated benzamides were successfully synthesised from benzotriazinones by the research groups of Mannathan,9a Cheng9b and Li (Scheme 1C).9c The Mannathan group also reported an efficient palladium-catalyzed ortho-alkynylation reaction of 1,2,3-benzotriazinones with aromatic terminal alkynes (Scheme 1D).7g Although the use of transition-metal catalysts in the synthesis of fine chemicals and pharmaceutical intermediates has become common in the last few decades, the use of transition metals often entails additional procedures to reduce the content of the residual metals and improve the quality of pharmaceutical ingredients.10
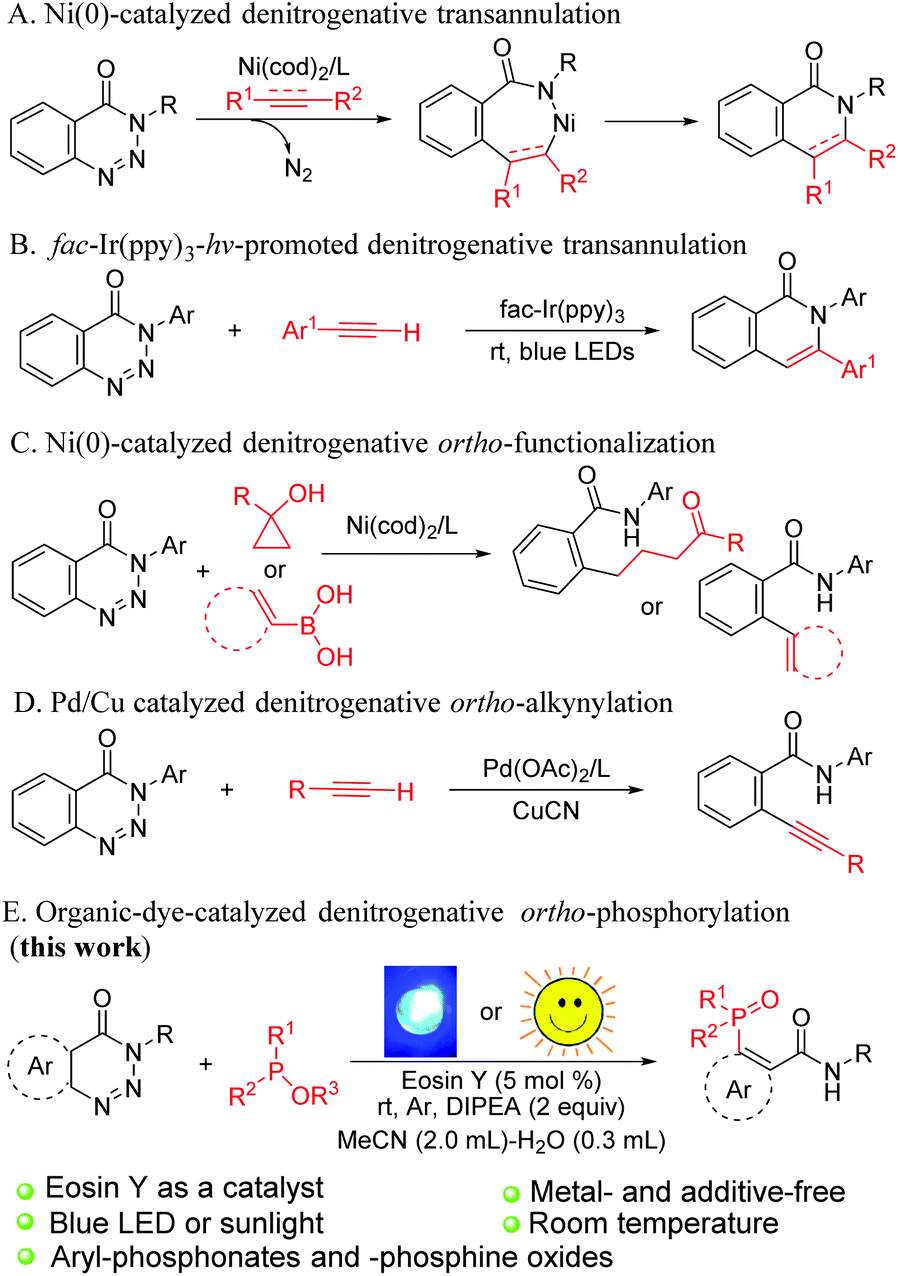 | ||
| Scheme 1 Denitrogenative reactions of benzotriazinones. | ||
Photoredox catalysis has emerged as a powerful and reliable tool for the construction of C–C and C–heteroatom bonds because of its sustainable and economy friendly characteristics. Myriad elegant works of visible-light-induced reaction have been well documented for years.11 From the environmental and economic points of view, organic dye is an attractive alternative to expensive and multi-step prepared iridium and ruthenium polypyridyl complexes.12 Thus, a metal-free photocatalytic system has been documented as a promising approach for modern organic synthesis. However, arylation of phosphorus reagents via a single-electron-transfer (SET) process under environment-friendly and metal-free conditions is relatively scarce.13 Given our ongoing efforts on the exploration of green methods of C–P bond construction,14 herein, we report a convenient and efficient tactic for the synthesis of aromatic phosphorus compounds via organic-dye-catalysed denitrogenative phosphorylation of 1,2,3-benzotriazin-4-(3H)-ones under the irradiation of blue light or sunlight without any metal catalysts or additives (Scheme 1E).
Results and discussion
A preliminary optimization of the reaction conditions was carried out with 1,2,3-benzotriazinone 1a and triethyl phosphite 2a as the model substrates (Table 1). A mixture of both compounds, N,N-diisopropylethylamine (DIPEA) and a photocatalyst was irradiated with LEDs under an argon atmosphere. To our delight, in the presence of fluorescein as the photocatalyst, the reaction in DMSO under irradiation by blue LEDs afforded the desired product 3a in 48% yield (entry 1). Encouraged by this result, other cheap and readily available organic dyes such as rhodamine B, rhodamine 6G, eosin Y, eosin B and Na2eosin Y were next examined (entries 2–6), and eosin Y delivered 3a in a yield of 54% (entry 4). In order to further improve the efficiency of the reaction, many solvents other than DMSO were investigated. 3a was obtained in a similar yield when the reaction was performed in MeCN or water (entries 7 and 8), and other solvents such as DCE, DMF, THF, acetone and EtOH did not provide satisfactory results (entry 9). Pleasingly, the targeted product 3a was detected in 85% yield in the mixed solvent of MeCN (2 mL) and H2O (0.1 mL) at room temperature for 90 min (entry 10). The amount of H2O seemed to significantly affect the reaction (entries 7–10). Furthermore, a 93% yield was achieved with the mixed solvent of MeCN (2 mL) and H2O (0.3 mL) (entry 11). Further increasing the amount of H2O to 0.5 mL, however, resulted in 3a with a slightly lower yield (entry 12). From an environmental and practical point of view, we were intrigued to use abundant sunlight for activating this chemical transformation. To our delight, the reaction under sunlight could be completed within 6 h with a yield of 90% (entry 13). The reaction didn't work without light even at 60 °C (entry 14). Control experiments without DIPEA or a catalyst gave no desired product at all (entries 15 and 16).|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Solvent (mL) | Yieldb (%) |
| a Reaction conditions: The reactions were performed with mixtures of 1a (0.2 mmol), 2a (0.3 mmol), base (0.4 mmol) and the photocatalyst (5 mol%) in the solvent (2.0 mL) at room temperature under irradiation by blue LEDs (30 W) for 90 min. b Yield of 3a, determined by 31P NMR analysis of the crude reaction mixture using nC8H17P(O) (δ: 48 ppm) as the internal standard. c Solvent: DCE, DMF, THF, acetone, and EtOH were used. d The value in parentheses indicates the isolated yield. e The reaction worked with sunlight (25–30 °C). f The reaction was carried out at 60 °C in darkness. g Without DIPEA. | |||
| 1 | Fluorescein | DMSO | 48 |
| 2 | Rhodamine B | DMSO | 40 |
| 3 | Rhodamine 6G | DMSO | 7 |
| 4 | Eosin Y | DMSO | 54 |
| 5 | Eosin B | DMSO | 42 |
| 6 | Na2Eosin Y | DMSO | 32 |
| 7 | Eosin Y | MeCN | 58 |
| 8 | Eosin Y | H2O | 49 |
| 9 | Eosin Y | Solventc | <33 |
| 10 | Eosin Y | MeCN/H2O (0.1 mL) | 85 |
| 11 | Eosin Y | MeCN/H2O (0.3 mL) | 93(91)d |
| 12 | Eosin Y | MeCN/H2O (0.5 mL) | 85 |
| 13e | Eosin Y (sunlight) | MeCN/H2O (0.3 mL) | 90 |
| 14f | Eosin Y (dark) | MeCN/H2O (0.3 mL) | 0 |
| 15g | Eosin Y | MeCN/H2O (0.3 mL) | 0 |
| 16 | — | MeCN/H2O (0.3 mL) | 0 |
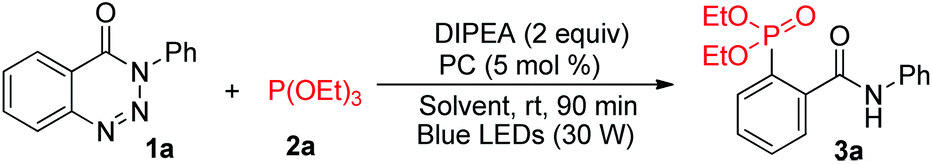
With the optimal reaction conditions identified, the scope of this visible-light-catalysed denitrogenative ring-opening of 1,2,3-benzotriazin-4-(3H)-ones for the synthesis of ortho-phosphorylated aryl amide derivatives was investigated (Table 2). It was found that 1,2,3-benzotriazin-4-(3H)-one substrates possessing various functional groups, such as alkyl, halogens, trifluoromethyl, ester, ether and thioether, at different positions on benzene ring 1 could be employed in this reaction, readily affording the respective products in good to excellent yields (74–91%). Besides, substrates with multisubstituted benzene ring 1 were also well tolerated (3n and 3o). Notably, N-naphthyl, N-pyridyl and N-styrenyl substrates reacted smoothly in the system to give the corresponding phosphorylated products 3p–3s in moderate yields (74–82%). Subsequently, we turned our attention to explore the scope of substituents on benzene ring 2. As expected, both electron-deficient groups (such as –CO2Me, –Cl and –Br) and electron-rich groups (such as –Me and –OMe) on benzene ring 2 did not disturb the efficiency of this transformation, and the expected products (3t–3x) were isolated in yields ranging from 81 to 87%. Significantly, the reaction proceeded well in the presence of a reactive furan ring, leading to thiophene-derived phosphonate 3y in 87% yield. Unfortunately, substrates bearing an aliphatic chain instead of an aryl group at the nitrogen atom were unreactive under the standard conditions and could be almost fully recovered probably due to the instability of the radical anion intermediate formed during the reaction (3z and 3aa). These results clearly revealed that an aryl group at the nitrogen atom was necessary to stabilise the key intermediate, and the electronic and steric factors of the aryl groups had a negligible effect on the coupling reactivity. Furthermore, sunlight as a light source could also effectively promote the reaction. When the reaction tube was put on the window sill under sunlight for 6 hours without stirring, the reaction mixture became turbid, indicating an ongoing reaction and affording the corresponding phosphorylated products 3a–3y in satisfactory yields.
| Reaction conditions: 1 (0.2 mmol), 2a (0.3 mmol), DIPEA (0.4 mmol), eosin Y (5 mol%), MeCN (2.0 mL), H2O (0.3 mL), room temperature, blue LEDs (30 W), stirring under argon for 90 min. Isolated yield. Yields in parentheses were determined by 31P NMR analysis using nC8H17P(O) (δ: 48 ppm) as the internal standard, and the reaction was performed under irradiation by sunlight at 25–30 °C for 6 h. |
|---|
|
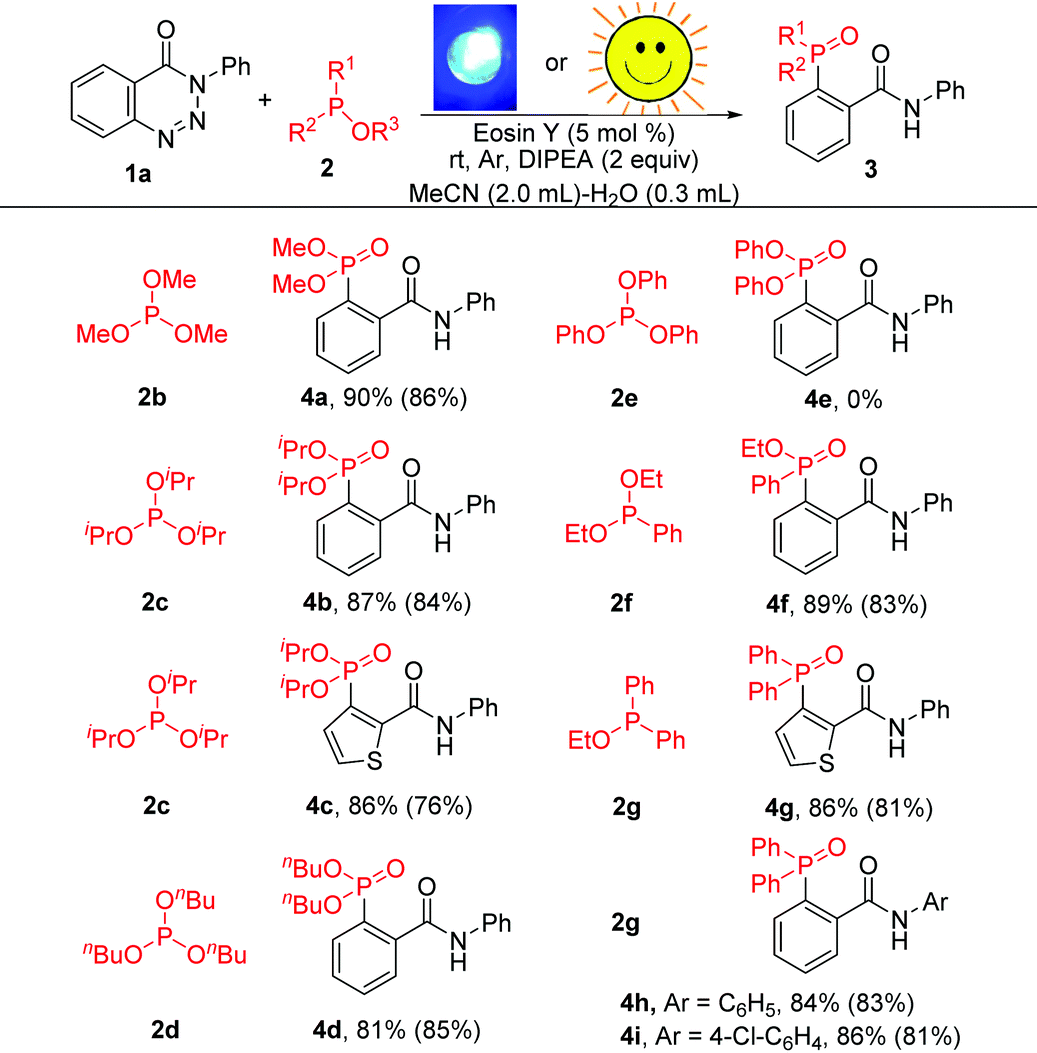
The scope of other phosphites was also examined to further demonstrate the generality of this methodology. As shown in Table 3, apart from 2a, other phosphites such as P(OMe)3, P(OiPr)3 and P(OnBu)3 were all compatible in this reaction providing 4a, 4b, 4c and 4d in yields of 90%, 87%, 86% and 81%, respectively. Unfortunately, the reaction was completely blocked when triphenylphosphite was taken as the substrate. However, benzotriazinones underwent phosphorylation successfully with 2f and 2g to deliver the target products (4f–4i) with excellent reaction efficiency. Triaryl tertiary phosphine oxides (4g–4i), which are important precursors of triarylphosphine ligands,4 could be prepared in high yields under the optimal conditions. Furthermore, the reactions could also be conducted using sunlight as the light source in MeCN–H2O at 25–30 °C and gave the corresponding products in good yields. Obviously, this organic-dye-catalysed cleavage of C–N and N–N bonds provides a facile route for the preparation of various valuable aryl-phosphonates and aryl-phosphine oxides.
| a Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol), DIPEA (0.4 mmol), eosin Y (5 mol%), MeCN (2.0 mL), H2O (0.3 mL), room temperature, blue LEDs (30 W), stirring under argon for 90 min. Isolated yield. Yields in parentheses were determined by 31P NMR analysis using nC8H17P(O) as the internal standard, and the reaction was performed under irradiation by sunlight at 25–30 °C for 6 h. |
|---|
|
To examine the synthetic utility of this transformation, gram-scale experiments were carried out (Scheme 2). Compounds 3a and 4i were obtained with high efficiency in yields of 82% and 78% after prolonging the blue LED irradiation time to 24 h. The pure gram-scale synthesized compounds were isolated just upon extraction, followed by re-crystallization using a petroleum ether/AcOEt mixture as the solvent; no tedious chromatographic purification was required. Furthermore, sunlight could also effectively promote the reaction, affording compound 3a in 78% yield.
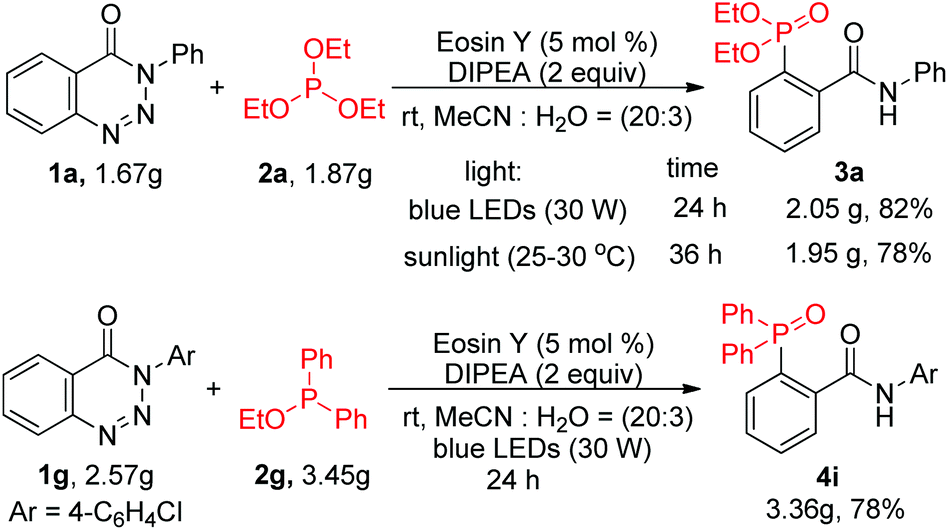 | ||
| Scheme 2 Gram-scale preparation of 3a and 4i. | ||
Furthermore, the luminescence of eosin Y with excitation at 575 nm could be readily quenched by DIPEA (Fig. 1, see the ESI†). This luminescence quenching is most likely due to photoinduced electron transfer.
 | ||
| Fig. 1 Luminescence quenching experiments. | ||
To understand the mechanism, further experiments were conducted (Scheme 3). Only a trace amount of 3a was detected when 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was added to the reaction system as a radical inhibitor. Recently, significant progress has been made in C–B bond formation through a single-electron transfer process.15 Diboron (B2pin2) as a good radical acceptor could be applied to this system to produce the corresponding borylated products 6a and 6b in good yields. These results suggest that this reaction may involve a radical process. Based on the above results and previous reports,8,13c,d,16 a plausible mechanism for the visible-light-promoted denitrogenation of 1,2,3-benzotriazin-4-(3H)-one was proposed. Initially, excited-state eosin Y (Eredp/2 = 0.83 V)17 was generated under the irradiation of visible light, which was then reductively quenched by DIPEA (Eoxp/2 = 0.63 V, see the ESI†). A single-electron transfer between the anion radical eosin Y˙− and benzotriazinone 1 (Eoxp/2 = 2.04 V, see the ESI†) took place to produce the radical anion A with the regeneration of ground-state eosin Y. Intermediate A could undergo an elimination of nitrogen to produce the corresponding aryl radical anion intermediate B, which reacted with phosphite 2 to give the unstable phosphoranyl radical C. The anion species D could be obtained by releasing an alkyl radical from C, followed by the abstraction of a proton from water to give the final desired product 3.
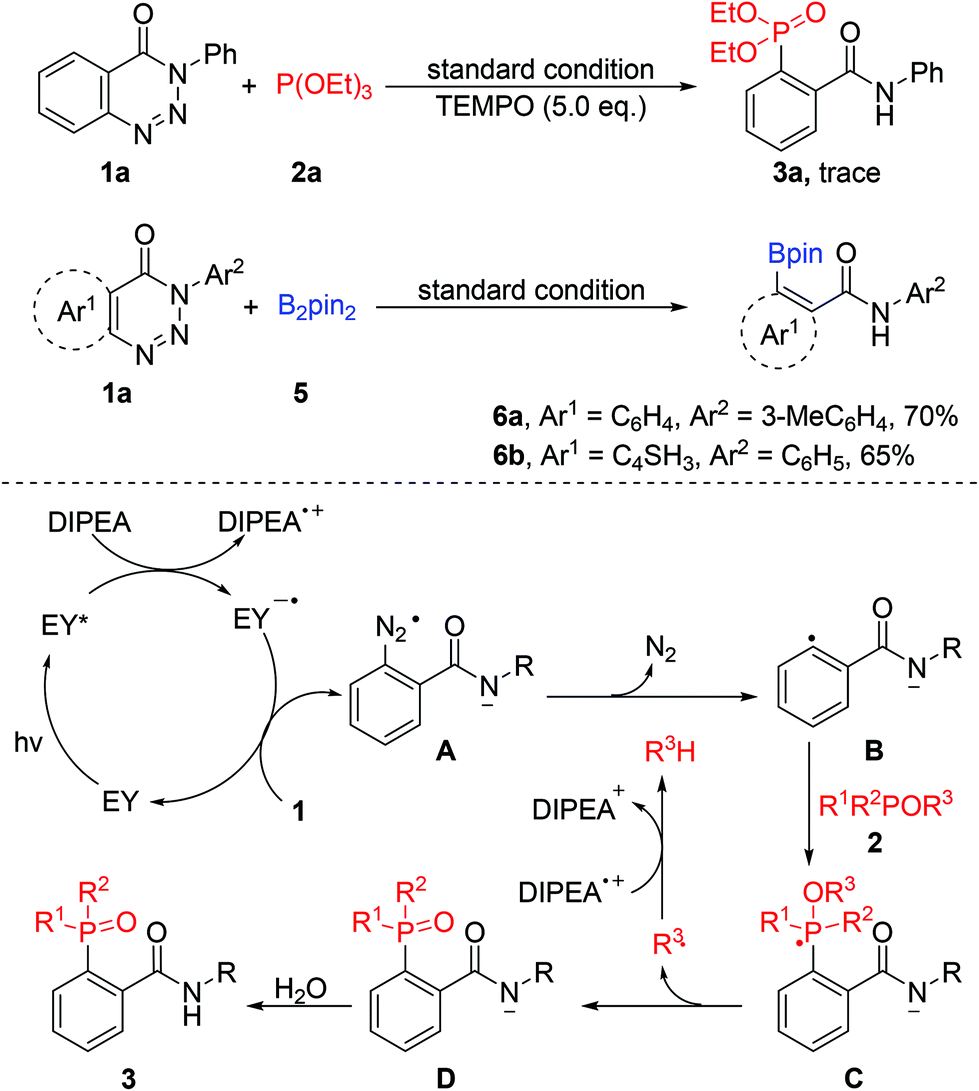 | ||
| Scheme 3 Investigation of the reaction mechanism. | ||
Conclusions
In summary, we have developed an organic-dye-catalysed denitrogenative phosphorylation of 1,2,3-benzotriazin-4-(3H)-ones for the synthesis of aryl-phosphonates, aryl-phosphinates and aryl-phosphine oxides. This method features low-cost organic-dye catalysis, environmentally friendly reaction conditions, broad substrate scope and excellent functional group compatibility. The radical mechanism was verified by control experiments and use of B2pin2 as the radical acceptor. Moreover, this method can be easily adapted to large-scale preparation. Further investigation of the mechanism and application of this reaction is currently underway.Experimental
Organic-dye-catalyzed denitrogenative phosphorylation of benzotriazinones
A Schlenk tube containing benzotriazinone derivatives (1, 0.2 mmol) and eosin Y (0.01 mmol) was evacuated and purged with argon three times. DIPEA (0.4 mmol), phosphorylation reagent (2, 0.3 mmol), H2O (0.3 mL) and CH3CN (2.0 mL) were sequentially added to the system at room temperature. Then the system was stirred at room temperature under the irradiation of 30 W blue LEDs for 90 min (or irradiated by sunlight at 25–30 °C for 6 h). Afterwards, the solvent was removed under vacuum, and the residues were purified by silica gel column chromatography using petroleum ether/ethyl acetate as the eluent (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford the desired phosphorylation products.
:1) to afford the desired phosphorylation products.
Gram-scale preparation 3a
A round flask containing benzotriazinone derivative 1a (7.5 mmol, 1.67 g) and eosin Y (5 mol%, 243 mg) were evacuated and purged with argon three times, DIPEA (2.0 equiv, 3.6 mL), 2a (1.5 equiv, 1.87 g), a mixture of H2O (11 mL) and CH3CN (75 mL) were sequentially added to the system at room temperature. Then the system was stirred at room temperature under the irradiation of 30 W blue LEDs for 24 hours (or irradiated by sunlight at 25–30 °C for 36 h without stirring). The reaction mixture was quenched with potassium carbonate saturated solution to pH = 8.5 and extracted with ethyl acetate (3 × 60 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The residue was recrystallized using a petroleum ether/AcOEt mixture as the solvent to afford the desired product 3a (2.05 g, blue LEDs; 1.95 g, sunlight).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Key Research and Development Program of China (2020YFA0608300) and NSFC (21772163, 21778042).Notes and references
- For selected references, see: (a) The Chemistry of Organophosphorous Compounds, ed. F. H. Hartley, Wiley, New York, 1996 Search PubMed; (b) Phosphorus Chemistry II, in Topics in Current Chemistry, ed. L. Montchamp, Springer, Switzerland, 2015 Search PubMed; (c) C. Schultz, Bioorg. Med. Chem., 2003, 11, 885–898 CrossRef CAS; (d) S. Demkowicz, J. Rachon, M. Daśko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC.
- For selected references, see: (a) P. H. Mutin, G. Guerrero and A. Vioux, J. Mater. Chem., 2005, 15, 3761–3768 RSC; (b) C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef.
- (a) C. Sine, Farm Chemicals Handbook, Meister Publishing Company, Willoughby, OH, USA, 1993 Search PubMed; (b) D. Z. Shi, Chinese Pesticide Dictionary, Chemical Industry Press Co, Lit., Beijing, China, 2008 Search PubMed.
- For selected reviews, see: (a) P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193, 499–556 CrossRef; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef CAS; (c) Y.-M. Li, F.-Y. Kwong, W.-Y. Yu and A. S. C. Chan, Coord. Chem. Rev., 2007, 251, 2119–2144 CrossRef CAS; (d) J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC; (e) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS.
- T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Tetrahedron Lett., 1980, 21, 3595–3598 CrossRef CAS.
- For selected reviews, see: (a) H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151–4202 CrossRef CAS; (b) J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS; (c) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem. (Z), 2018, 376, 1–19 CrossRef; (d) T. Chen and L.-B. Han, Synlett, 2015, 26, 1153–1163 CrossRef CAS; (e) T. Chen, J. S. Zhang and L.-B. Han, Dalton Trans., 2016, 45, 1843–1849 RSC; (f) S. Vander Jeught and C. V. Stevens, Chem. Rev., 2009, 109, 2672–2702 CrossRef CAS; (g) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS.
- For selected references, see: (a) T. Miura, M. Yamauchi and M. Murakami, Org. Lett., 2008, 10, 3085–3088 CrossRef CAS; (b) M. Yamauchi, M. Morimoto, T. Miura and M. Murakami, J. Am. Chem. Soc., 2010, 132, 54–55 CrossRef CAS; (c) T. Miura, M. Morimoto, M. Yamauchi and M. Murakami, J. Org. Chem., 2010, 75, 5359–5362 CrossRef CAS; (d) T. Miura, Y. Nishida, M. Morimoto, M. Yamauchi and M. Murakami, Org. Lett., 2011, 13, 1429–1431 CrossRef CAS; (e) Z.-J. Fang, S.-C. Zhang, Z. Guo, J.-Y. Guo, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2015, 54, 9528–9532 CrossRef CAS; (f) V. H. Thorat, N. S. Upadhyay, M. Murakami and C.-H. Cheng, Adv. Synth. Catal., 2018, 360, 284–289 CrossRef CAS; (g) M. H. Balakrishnan and S. Mannathan, Org. Lett., 2020, 22, 542–546 CrossRef.
- H. Wang and S. Yu, Org. Lett., 2015, 17, 4272–4275 CrossRef CAS.
- (a) M. H. Balakrishnan, K. Sathriyan and S. Mannathan, Org. Lett., 2018, 20, 3815–3818 CrossRef; (b) V. H. Thorat, N. S. Upadhyay and C.-H. Cheng, Adv. Synth. Catal., 2018, 360, 4784–4789 CrossRef CAS; (c) J. Li, Y. Zheng, M. Huang and W. Li, Org. Lett., 2020, 22, 5020–5024 CrossRef CAS.
- For selected reviews, see: (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS; (b) S. Roscales and A. G. Csák, Chem. Soc. Rev., 2014, 43, 8215–8225 RSC; (c) R. Rossi, M. Lessi, C. Manzini, G. Marianetti and F. Bellina, Adv. Synth. Catal., 2015, 357, 3777–3814 CrossRef CAS.
- For selected reviews, see: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS; (d) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00030; (e) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC; (f) X.-P. Ma, C.-M. Nong, Y.-F. Liang, P.-P. Xu, X.-Y. Guo, C. Liang, C.-X. Pan, C.-F. Su and D.-L. Mo, Green Chem., 2020, 22, 3827–3834 RSC.
- For selected reviews, see: (a) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC; (b) D. Prasad and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- (a) K. Luo, Y.-Z. Chen, W.-C. Yang, J. Zhu and L. Wu, Org. Lett., 2016, 18, 452–455 CrossRef CAS; (b) P. Peng, L. Peng, G. Wang, F. Wang, Y. Luo and A. Lei, Org. Chem. Front., 2016, 3, 749–752 RSC; (c) R. S. Shaikh, S. J. S. Düsel and B. König, ACS Catal., 2016, 6, 8410–8414 CrossRef CAS; (d) R. Li, X. Chen, S. Wei, K. Sun, L. Fan, Y. Liu, L. Qu, Y. Zhao and B. Yu, Adv. Synth. Catal., 2018, 360, 4807–4813 CrossRef.
- (a) J. Pan, R. Zhao, J. Guo, D. Ma, Y. Xia, Y. Gao, P. Xu and Y. Zhao, Green Chem., 2019, 21, 792–797 RSC; (b) S. Shi, J. Chen, S. Zhuo, Z. Wu, M. Fang, G. Tang and Y. Zhao, Adv. Synth. Catal., 2019, 361, 3210–3216 CrossRef CAS; (c) J. Wu, D. Ma, G. Tang and Y. Zhao, Org. Lett., 2019, 21, 7674–7678 CrossRef CAS.
- G. Yan, D. Huang and X. Wu, Adv. Synth. Catal., 2018, 360, 1040–1053 CrossRef CAS.
- W. Lecroq, P. Bazille, F. Morlet-Savary, M. Breugst, J. Lalevée, A.-C. Gaumount and S. Lakhdar, Org. Lett., 2018, 20, 4164–4167 CrossRef CAS.
- D.-M. Yan, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378–380 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis, spectral data and NMR spectra of compounds 3a–3y, 4a–4d, 4f–4i, 6a and 6b. See DOI: 10.1039/d0gc03613g |
| This journal is © The Royal Society of Chemistry 2021 |