
Electro-oxidative C–H alkylation of quinoxalin-2(1H)-ones with organoboron compounds†‡
Kaikai
Niu
a,
Yanke
Hao
a,
Lingyun
Song
a,
Yuxiu
Liu
 a and
Qingmin
Wang
*ab
a and
Qingmin
Wang
*ab
aState Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, People's Republic of China. E-mail: wangqm@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, People's Republic of China
First published on 15th December 2020
Abstract
Radical cleavage of C–B bonds to accomplish C–H functionalization is synthetically appealing but practically challenging. We report herein a mild electro-oxidative method for efficient C–H alkylation of quinoxalin-2(1H)-ones by means of radical addition reactions of alkyl boronic acids and esters and alkyl trifluoroborates to afford C–C coupled products.
Carbon radicals are synthetically useful intermediates in C–C bond-forming reactions,1 and the addition of carbon radicals to heteroarenes would constitute a synthetically useful method for rapid construction of aromatic building blocks.2 Organoboron compounds, which are widely used in organic synthesis3 and can act as bioisosteres in bioactive molecules,4 constitute a readily available, minimally toxic source of stable carbon radicals.5 Therefore, the generation of alkyl radicals by deboration of organoboron compounds under mild conditions is strategically appealing. However, owing to the high oxidation potential of C–B bonds (Scheme 1a),6 their cleavage requires harsh conditions. In the classic methods to accomplish this difficult transformation (Scheme 1b), alkyl trifluoroborates and boronic acids undergo single-electron oxidation by stoichiometric strong oxidants such as AgNO3/K2S2O8,7 Mn(OAc)38 and Cu(OAc)2,9 or O2.10 In addition, alkyl trifluoroborates can undergo single-electron transfer to generate alkyl radicals under catalytic photoredox conditions,11 and alkyl boronic acids and esters can be converted to alkyl radicals by photoredox activation in the presence of a Lewis base catalyst.12
 | ||
| Scheme 1 Approaches for generating radicals from boron compounds. | ||
One attractive alternative to chemical oxidants is electrochemical oxidation, which uses electrons as a clean, renewable reagent (Scheme 2).13 However, anodic generation of alkyl radicals from boronic acid derivatives remains underexplored owing to their high oxidation potentials, which can lead to radical dimerization or to overoxidation to generate carbocations.14 Only a few examples have been reported, and they proceed via indirect pathways involving a redox mediator such as a photoredox catalyst15 or a Mn(OAc)316 catalyst (Scheme 1b).
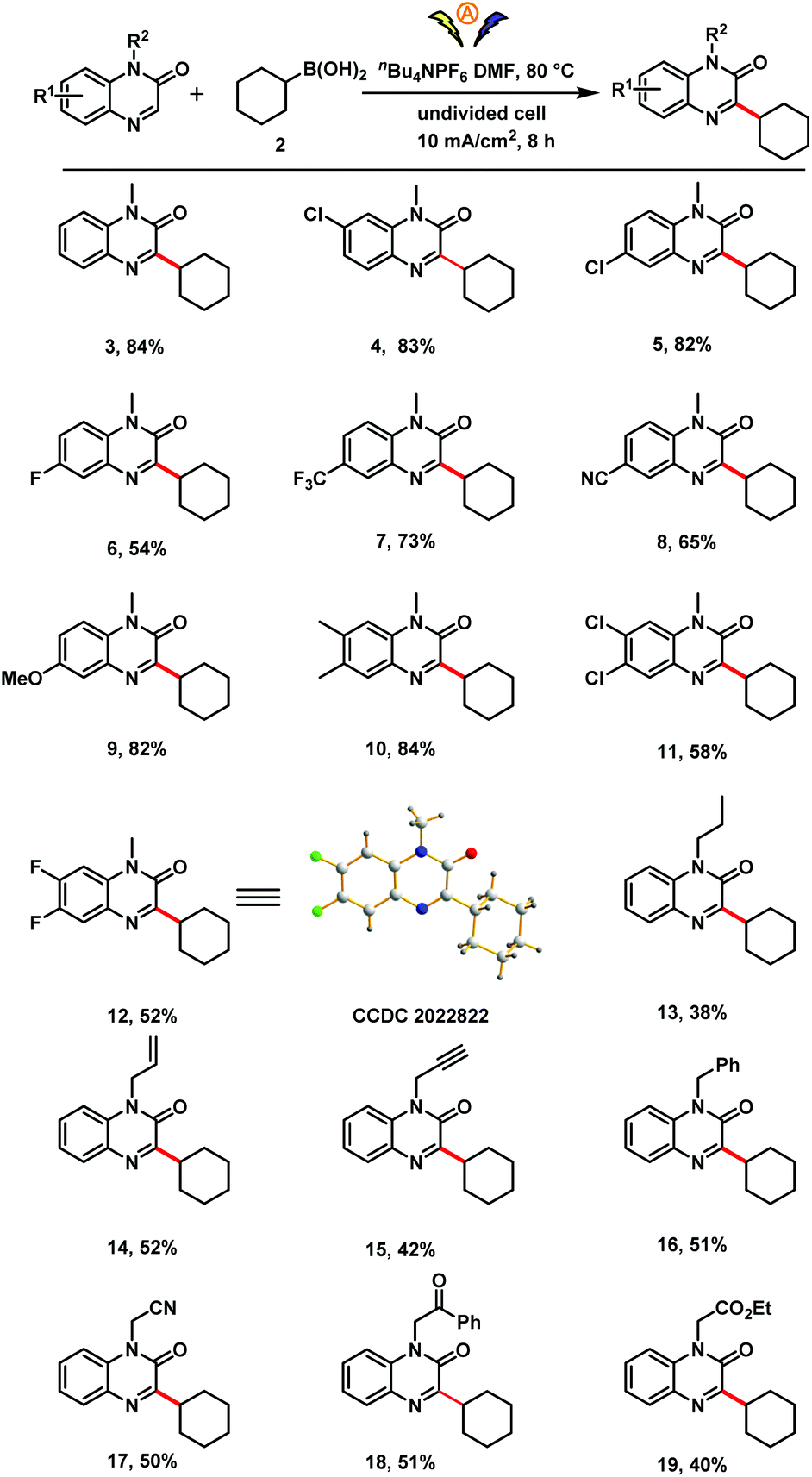 | ||
| Scheme 2 Substrate scope with respect to the quinoxalin-2(1H)-one. Reaction conditions: Quinoxalin-2(1H)-one (0.5 mmol), 2 (1.5 mmol), nBu4NPF6 (2.0 equiv), TFA (4.0 equiv), DMF (5 mL), undivided cell with two graphite electrodes (each 1.0 × 1.0 cm2), 80 °C, 10 mA cm−2, 8 h. Isolated yields are provided. | ||
To our knowledge, the direct use of organoboron compounds as carbon radical sources for electro-oxidative C–H functionalization reactions has not yet been achieved. Herein, as part of our ongoing research on organic radical chemistry,17 we describe a newly developed electrochemical protocol that uses alkyl boronic acids and esters and alkyl trifluoroborates as alkylating agents for C–H alkylation reactions of quinoxalin-2(1H)-ones via direct electrochemical oxidation under mild conditions (Scheme 1c).
We began by carrying out reactions of 1-methylquinoxalin-2(1H)-one (1) and cyclohexylboronic acid (2) as model substrates (Table 1). The optimal conditions involved the use of graphite plates as electrodes, N,N-dimethylformamide (DMF) as the solvent, nBu4NPF6 (2.0 equiv.) as the electrolyte, trifluoroacetic acid (TFA, 4.0 equiv.) as an additive, and a current density of 10 mA cm−2. Under these conditions, desired alkylation product 3 could be obtained in 85% isolated yield after 24 h (entry 1). Control experiments indicated that the acid additive (entry 2), electricity (entry 3), and the electrolyte (entry 4) were critical for this reaction; in their absence, most of the 1 was recovered. When the amount of TFA or 2 was reduced to 2.0 equiv., the yield of 3 decreased (entries 5 and 6). Decreasing the current density to 5 mA cm−2 also lowered the yield (entry 7), whereas increasing it to 20 mA cm−2 had little effect (entry 8). Evaluation of various solvents revealed that DMF was the best choice (entries 9–11). Notably, the reaction time could be shortened to 8 h by increasing the reaction temperature to 80 °C (entry 12). The similarity of the yields under air and Ar atmospheres indicate that the cyclohexylboronic acid was not oxidized by oxygen (compare entries 1 and 13).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
| a Standard reaction conditions: 1 (0.5 mmol), 2 (1.5 mmol), nBu4NPF6 (2.0 equiv.), TFA (4.0 equiv.), DMF (5 mL), undivided cell with two graphite electrodes (each 1.0 × 1.0 cm2), room temperature (r.t.), 10 mA cm−2, 24 h. b Isolated yields are provided. | ||
| 1 | None | 85 |
| 2 | No TFA | Trace |
| 3 | No electricity | 0 |
| 4 | No electrolyte | 0 |
| 5 | 2.0 equiv. TFA | 72 |
| 6 | 2.0 equiv. 2a | 63 |
| 7 | 5 mA cm−2 | 33 |
| 8 | 20 mA cm−2 | 82 |
| 9 | MeCN as solvent | 15 |
| 10 | DMA as solvent | 10 |
| 11 | DCM as solvent | Trace |
| 12 | 80 °C, 8 h | 82 |
| 13 | Under Ar | 84 |

Using the optimized conditions (Table 1, entry 12), we investigated the scope of the reaction with respect to the quinoxalin-2(1H)-one (Scheme 2). Substrates with a variety of substituents on the aromatic ring (methyl, chloro, fluoro, trifluoromethyl, cyano, methoxy) reacted smoothly with 2 to afford corresponding products 4–12 in moderate to good yields. The structure of difluoro-substituted product 12 was unambiguously confirmed by X-ray analysis. Various quinoxalinones bearing reactive functional groups on the nitrogen atom, including propyl, alkenyl, propargyl, benzyl, cyanomethyl, carbomethyl and ethoxycarbonylmethyl were also suitable substrates (13–19).
Next we investigated the scope with respect to the organoboron compound (Scheme 3). A series of acyclic and cyclic primary, secondary, and tertiary alkylboronic acids proved to be suitable substrates for this electro-oxidative C–H alkylation reaction (3, 20–24). In addition, cyclic secondary and tertiary organotrifluoroborates could be used in the reaction (3, 21, 23, 24). To our delight, cyclic and tertiary boronic esters were also compatible with the oxidative radical coupling conditions (3, 24).
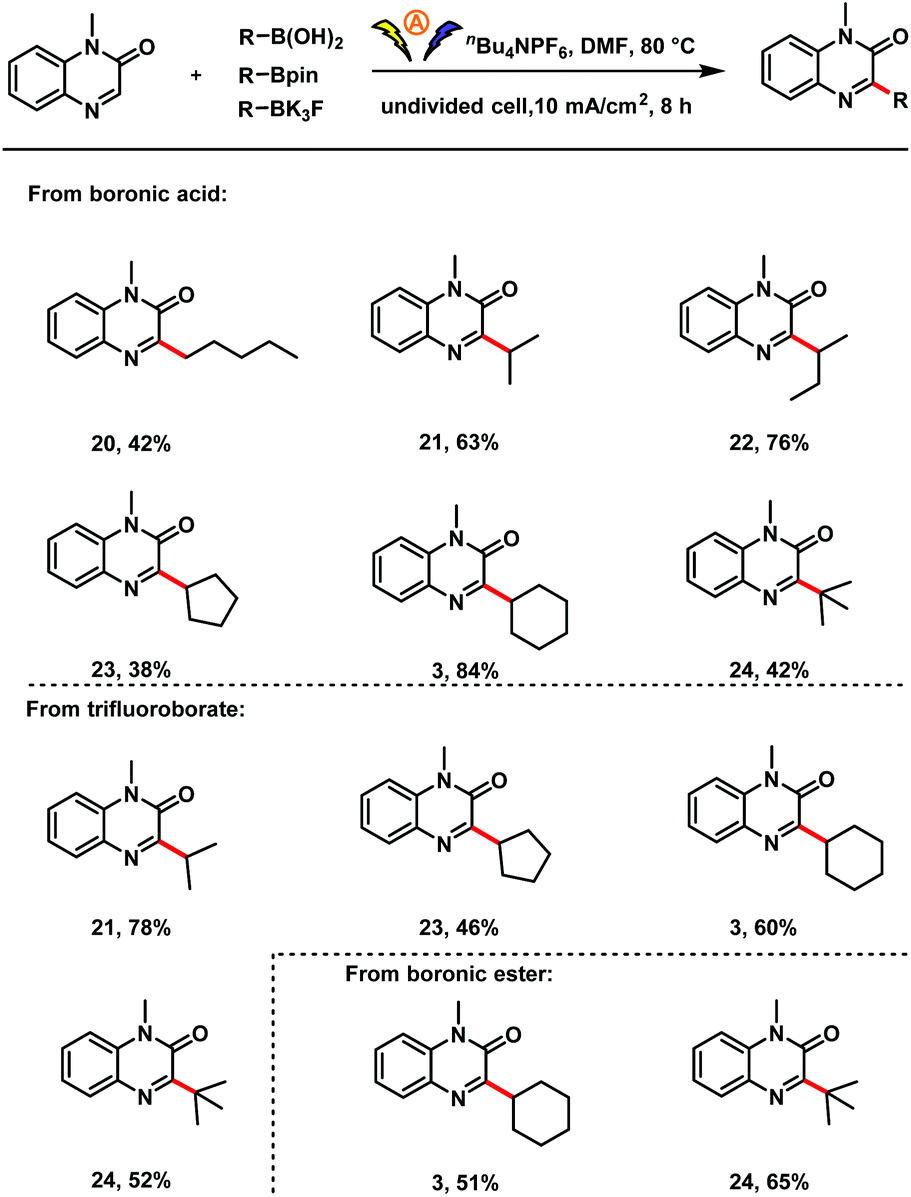 | ||
| Scheme 3 Substrate scope with respect to the organoboron compound. Reaction conditions: 1 (0.5 mmol), organoboron compound (1.5 mmol), nBu4NPF6 (2.0 equiv.), TFA (4.0 equiv.), DMF (5 mL), undivided cell with two graphite electrodes (each 1.0 × 1.0 cm2), 80 °C, 10 mA cm−2, 8 h. Isolated yields are provided. | ||
Heteroaryl motifs are widely exist in natural products, small-molecule drugs, organic materials, and ligands for metal catalysts.18 Preliminary results showed that benzoquinoxalinone (26) and a phenanthridine (27) could also serve as acceptable substrates under the electro-oxidative conditions, suggesting that the protocol may be useful for derivatization of pharmaceuticals (Scheme 4).
 | ||
| Scheme 4 Functionalization of other heteroarene. | ||
To verify the practicality of this electrochemical protocol, we carried out a scaled-up reaction (Scheme 5): reaction of 5.0 mmol of 1-methylquinoxalin-2(1H)-one (1) with boronic acid 2 under the standard conditions afforded 3 in 79% yield. To our delight, the protocol could even be carried out with a battery as a power supply, demonstrating that the reaction could be used to accomplish the desired transformation without the need for a complex DC power device. Furthermore, in an additional test of the protocol's utility, we performed a click reaction of alkylation product 13 to transform the alkyne into a triazole skeleton (25, 97% yield).
 | ||
| Scheme 5 Practicality of the electrochemical method. | ||
The reaction mechanism was elucidated by means of several control experiments (Scheme 6). First, the electro-oxidative C–H alkylation reaction was strongly suppressed by a radical inhibitor, BHT (2,6-di-tert-butyl-4-methylphenol).19 Second when N-phenylmethacrylamide was subjected to electrolysis with boronic acid 2 under the standard conditions, radical relay product 29 was isolated in 8% yield. These results indicate that an alkyl radical was formed.
 | ||
| Scheme 6 Control experiments. | ||
Based on the above-described experimental results, we propose the reaction mechanism outlined in Scheme 7. Single-electron oxidation of the boronic compound at the anode generates an alkyl radical, which reacts with the protonated heteroarene to give radical cation I. Then the radical cation loses a proton to give C-radical intermediate II. Finally, II undergoes single-electron oxidation at the anode to give the product.
 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
In summary, we have developed a protocol for electro-oxidative C–H alkylation reactions of quinoxalin-2(1H)-ones with organoboron compounds. Organoboronic acids, trifluoroborates, and even boronic esters could be converted into alkyl radicals by direct electrochemical oxidation without the need for a metal, an oxidant, or a photoredox reagent. Further studies utilizing alkyl radicals generated from organoboron compounds are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21732002, 22077071) for generous financial support for our programs.Notes and references
- C. Chatgilialoglu and A. Studer, Encyclopedia of Radicals in Chemistry, Biology and Materials, Wiley, Chichester, 2012 Search PubMed.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS.
- (a) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS; (b) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, 1045 CAS; (c) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CrossRef CAS.
- D. B. Diaz and A. K. Yudin, Nat. Chem., 2017, 9, 731–742 CrossRef CAS.
- (a) W. Liu, P. Liu, L. Lv and C. J. Li, Angew. Chem., Int. Ed., 2018, 57, 13499–13503 CrossRef CAS; (b) Y. Cheng, C. Muck-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS.
- J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512–3522 RSC.
- Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. Del Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292–3295 CrossRef CAS.
- A. S. Demir, O. Reis and M. Emrullahoglu, J. Org. Chem., 2003, 68, 578–580 CrossRef CAS.
- G. Sorin, R. Martinez Mallorquin, Y. Contie, A. Baralle, M. Malacria, J. P. Goddard and L. Fensterbank, Angew. Chem., Int. Ed., 2010, 49, 8721–8723 CrossRef CAS.
- L. Zhang and Z. Q. Liu, Org. Lett., 2017, 19, 6594–6597 CrossRef CAS.
- G. X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC.
- F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. Van der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS.
- S. Inagi and T. Fuchigami, Curr. Opin. Electrochem., 2017, 2, 32–37 CrossRef CAS.
- H. Yan, Z. W. Hou and H. C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef CAS.
- Y. Yuan, Y. Zheng, B. Xu, J. Liao, F. Bu, S. Wang, J.-G. Hu and A. Lei, ACS Catal., 2020, 10, 6676–6681 CrossRef CAS.
- (a) J. Dong, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, Sci. Adv., 2019, 5, eaax9955 CrossRef CAS; (b) K. Niu, L. Song, Y. Hao, Y. Liu and Q. Wang, Chem. Commun., 2020, 56, 11673–11676 RSC.
- (a) R. R. Gupta, in Bioactive heterocycles V, Topics in Heterocyclic Chemistry V, ed. R. R. Gupta, Wiley-VCH, Springer, Heidelberg, Hoboken, 2008, vol. 11 Search PubMed; (b) S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef.
- W. Zhao, R. P. Wurz, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 12153–12156 CrossRef CAS.
Footnotes |
| † Dedicated to the 100th anniversary of chemistry at Nankai University. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2022822. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc03892j |
| This journal is © The Royal Society of Chemistry 2021 |