
Coupling liquid chromatography and inductively coupled plasma-mass spectrometry with ultrasonic nebulization for chromium speciation in rice†
Bo-Hao
Chen
a and
Shiuh-Jen
Jiang
 *ab
*ab
aDepartment of Chemistry, National Sun Yat-sen University, Kaohsiung 80424, Taiwan
bDepartment of Medical Laboratory Science and Biotechnology, Kaohsiung Medical University, Kaohsiung 80708, Taiwan. E-mail: sjjiang@faculty.nsysu.edu.tw; Fax: +886-7-5253908
First published on 30th October 2020
Abstract
In this study a procedure based on liquid chromatography (LC) and dynamic reaction cell inductively coupled plasma mass spectrometry (DRC ICP-MS), coupled with ultrasonic nebulization, for chromium speciation in rice was reported. Chromium (Cr) containing species, including Cr(III) and Cr(VI), were well separated by reversed phase HPLC with a C18 column as the stationary phase and a solution containing 0.5 mmol L−1 tetrabutyl ammonium phosphate (TBAP), 0.1 mmol L−1 EDTA and 2% (v/v) methanol (pH 6.9) as the mobile phase using isocratic elution. The chromatographic separation was complete in 4.5 min. Ammonia gas was employed as the reactive gas in the DRC to minimize spectral interference due to 40Ar12C+ and 35Cl16OH+ and 40Ar13C+ and 35Cl18O+ ions on the signal of 52Cr+ and 53Cr+, respectively. The detection limits were in the range 0.011–0.012 ng Cr mL−1. To verify the accuracy, the developed procedure was applied on a certified reference material (NIST SRM 1573a Tomato Leaves) wherein the sum of the concentrations of individual species agreed with the total certified concentration of Cr. Species were extracted using microwave heating with 1% (v/v) HF and 2 mmol L−1 EDTA. After ensuring the accuracy, this procedure has been applied to determine various Cr compounds present in rice and rice cereal. Supernatants were injected directly into the LC-ICP-MS system after dilution. The spike recovery was in the range of 95–105% and precision between sample replicates was better than 5% for all determinations. The LC-ICP-MS results showed satisfactory agreement with the total Cr concentrations obtained by ICP-MS analysis. Both Cr(III) and Cr(VI) were present in the samples analyzed. An unknown Cr compound was found in all the rice samples analyzed.
Introduction
Rice is the most important food of the developing world and the staple food of more than half of the world's population and is a major food crop especially in the Asian region. Rice is rich in nutrients, vitamins and minerals and also is an excellent source of complex carbohydrates. Due to the high consumption, there is a huge concern regarding the intake of toxic heavy metals, through rice, even at ppb levels. Cr is one of the heavy metals that need regular monitoring in rice and also other edible materials as one of its species, Cr(VI), is a known carcinogen.1–3 This leads to the need for the development of a reliable analytical methodology for the determination of Cr(VI) instead of total Cr in different types of rice. The objective is achieved with chromatography in combination with element specific detection.ICP-MS is an elemental analysis methodology capable of detecting most of the elements in the periodic table at milligram to nanogram levels per liter. It is used in the analysis of samples for environmental monitoring, geochemical analysis, characterization of metallurgical compounds and pharmaceuticals, and also clinical research. Cr speciation using liquid chromatography (LC)4–11 and capillary electrophoresis (CE)12,13 coupled with ICP-MS has been reported for a variety of samples. Coupling of LC with ICP-MS is attractive due to the ease of operation, simplicity of interfacing between LC and ICP-MS, availability of isotope ratio information and specificity of the signal intensity of the determined element.14 The determination of Cr by quadrupole ICP-MS is typically compromised by isobaric interference from 40Ar12C+ and 35Cl16OH+ on 52Cr+; 40Ar12CH+ and 37Cl16O+ on 53Cr+. These interferents are reported to be alleviated by employing the dynamic reaction cell (DRC) method with O2 and/or NH3 as the reaction gas.2,15–17
Ultrasonic nebulizers (USNs) with desolvation are employed in ICP based procedures to improve sensitivity and also avoid a high solvent loading. An ultrasonic nebulizer improves the nebulization efficiency compared to a pneumatic nebulizer. The desolvation system of an USN removes most of the water and solvents used for LC separation that might have negative effects on the ICP-MS process. The aims of the present work are to develop a rapid and accurate procedure for the speciation analysis of Cr(III) and Cr(VI) in rice using liquid chromatography DRC-ICP-MS. Herein, optimization of the operating conditions for the LC-USN-DRC-ICP-MS system and its analytical figures of merit when used for the speciation analysis of Cr in rice and rice cereal are presented. An effective microwave-assisted extraction procedure for the extraction of Cr compounds from rice samples has also been presented.
Experimental
Apparatus and conditions
An ELAN 6100 DRC II ICP-MS apparatus (PerkinElmer SCIEX, Concord, ON, Canada) equipped with an USN (Cetac U-5000AT+, Omaha, NB, USA) was used for the optimization studies and also for the speciation analysis of the Cr compounds. The ICP and USN conditions were varied to maximize the signal-to-background (S/B) ratio of Cr while continuously aspirating a solution of 25 ng mL−1 Cr from the mobile-phase selected for the chromatographic separations. The optimized ICP-MS operating conditions are listed in Table 1.| ICP-MS instrument | PerkinElmer SCIEX ELAN 6100 DRC II |
| ICP parameters | |
| RF power | 1300 W |
| Plasma gas flow rate | 15 L min−1 |
| Auxiliary gas flow rate | 1.1 L min−1 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Mass spectrometer settings | |
| Scan mode | Peak hopping |
| Dwell time | 50 ms |
| Sweeps/reading | 21 |
| Reading/replicate | 200 |
| Replicates | 1 |
| Isotopes monitored | 50Cr, 52Cr, and 53Cr |
|
|
| HPLC system | |
| Pump | Hitachi Model CM-5110 LC pump |
| Injector | Rheodyne model 7725i |
| Stationary phase | PerkinElmer C18 reversed phase column, 5 μm diameter particles, 4.6 mm i.d. × 150 mm length |
| Mobile phase | 0.5 mmol L−1 TBAP, 0.1 mmol L−1 EDTA, 2% (v/v) methanol, pH 6.9 |
| Flow rate | 1.0 mL min−1 |
| Sample loop | 100 μL |
|
|
| Ultrasonic nebulizer conditions | |
| Desolvation temperature | 120 °C |
| Condenser temperature | −4 °C |
| Carrier gas flow rate | 1.1 L min−1 |
|
|
| DRC parameters | |
| Reaction gas | NH3 |
| Reaction gas flow rate | 0.6 mL min−1 |
| Rpq | 0.6 |
| Rpa | 0 |
| Axial field voltage | 50 V |
The HPLC system is equipped with a HPLC pump (Hitachi, Model CM-5110), an injector (Rheodyne 7725i) and a reversed phase HPLC column (PerkinElmer, Univ L-RP18, 5 μm diam. particles, 4.6 mm i.d. × 150 mm length). The samples were loaded with a syringe into a 100 μL sample loop. All separations were performed at room temperature under isocratic conditions. The LC conditions were selected to maximize the resolution while lowering the retention times (Table 1). Polytetrafluoroethylene (PTFE) tubing was used to connect the column outlet to the USN of the ICP-MS system.
Chemicals
All chemicals used were of analytical-reagent grade. All solutions were prepared using de-ionized water (resistivity: 18.2 MΩcm) obtained from a Milli-Q water purification system (Millipore, Bedford, MA, USA). Methanol, acetic acid, NH4OH and Suprapur nitric acid (65%) were purchased from Merck (Darmstadt, Germany). Tetra-n-butyl ammonium phosphate (TBAP) was from TCI (Tokyo, Japan). The disodium salt of ethylenediamine tetraacetic acid (EDTA) was obtained from Fisher (Fair Lawn, NJ, USA). Cr(III) chloride was procured from Alfa Chemicals (Danvers, MA, USA). Potassium dichromate(VI) [K2Cr2O7] was obtained from Merck. Cr(III) and Cr(VI) stock standard solutions were prepared using CrCl3·6H2O and K2Cr2O7 in 0.2 mol L−1 EDTA and deionized water, respectively. Ar gas used was provided by Hsin E Li Gases Co., Ltd (Kaohsiung, Taiwan). NH3 gas used was of 99.999% purity (Toyo gas, Taiwan).Sample preparation and extraction
The applicability of the developed procedure to real samples was examined on four rice samples purchased locally (3 polished rice samples and 1 rice cereal sample). The rice samples obtained were ground by using a Retsch MM2000 mixer mill and sieved to 150 μm by using a Retsch VE1000 sieving machine (Retsch, Haan, Germany) to ensure homogeneity. The accuracy of the developed procedure was determined by analyzing a standard reference material, NIST SRM 1573a Tomato Leaves (National Institute of Standards and Technology, Gaithersburg, MD, USA), whose total Cr content was certified.A CEM MARS 6 (Matthews, NC, USA) microwave digester equipped with temperature and pressure sensors was used for the extraction and digestion of Cr species from rice samples. Samples of SRM 1573a tomato leaves and rice (0.3 g each) were weighed into 15 mL polyethylene centrifuge tubes and then the extraction solution (5 mL) containing 0.5 mmol L−1 TBAP, 2% (v/v) methanol, 2 mmol L−1 EDTA, and 1% (v/v) HF was added. The tubes were inserted into a 250 mL beaker (Pyrex glass) containing water (120 mL) and exposed to microwave heating. The microwave system was programmed to maintain the temperature at 90 °C for 50 min; the ramp time was set at 10 min. After microwave heating, the solutions were cooled and centrifuged (3743g, 10 min) directly (MIKRO 22R, Hettich, Germany). The supernatants of different rice samples and SRM 1573a Tomato Leaves were diluted 0 to 12-fold (12 fold for the SRM, 2 fold for Rice 1 and 2, and no further dilution for Rice 3 and Rice cereal) with the mobile phase, filtered through a PVDF filter (Pall; porosity: 0.22 μm) and then injected for LC-USN-DRC-ICP-MS analysis. The concentrations of Cr species were determined through external calibration based on peak areas.
The spike recoveries of the individual compounds were determined by spiking the samples with suitable amounts of Cr(III) and Cr(VI), and then extracting them with the extraction solution. The Cr(III) and Cr(VI) standards were spiked at a level of 1 μg g−1 each in the SRM 1573a tomato leaves; 0.217 and 0.15 μg g−1 in Rice 1; 0.083 and 0.067 μg g−1 in Rice 2; 0.020 and 0.0067 μg g−1 in Rice 3; and 0.083 and 0.020 μg g−1 in Rice cereal. The supernatant solutions were filtered through a PVDF filter before LC separation. The concentration of Cr species was determined by an external calibration procedure based on the peak area. Recoveries were then calculated against the theoretical concentrations.
The extraction efficiency of the Cr compounds was verified through comparison of the total Cr concentrations in the extracts with the certified values and/or the values obtained using the total dissolution procedure. For total dissolution, the samples were digested as follows. To the sample (ca. 0.2 g) taken in a PTFE vessel HNO3 (3 mL) was added. The closed vessel was heated at 150 °C for 20 min, 180 °C for 20 min, and 200 °C for 20 min. The power of the microwave was set at 900 W. The digest was diluted to 5 mL. After suitable dilution the total concentration of Cr in the sample was analyzed through pneumatic nebulization DRC ICP-MS (internal standard: Rh, 1 ng mL−1).
Results and discussion
Selection of LC conditions
As reported in our previous paper,18 the species Cr(VI) is anionic at pH 6–7 and Cr(III) is converted to anionic species by complexing with EDTA. The anionic species are separated using a C8 reversed phase column with TBAP as the ion pairing reagent. To obtain better resolution with reproducible peaks, in this study a C18 column was employed. Several parameters including the concentrations of methanol, EDTA, and TBAP in the mobile phase affected the separation of Cr(III)–EDTA and Cr(VI). The background at Cr major masses m/z 52 and m/z 53 was too high when ICP-MS was operated in standard mode. An ultrasonic nebulizer was employed as the sample introduction device to improve the S/B of Cr compounds. For the following experiments, the carrier gas flow rate, the desolvation tube temperature, and the condenser temperature of the ultrasonic nebulizer were set at 1.3 L min−1, 120 °C, and −2 °C, respectively, as suggested by the supplier. Due to the high background at m/z 52, the ion signals of 50Cr+ and 53Cr+ were used to optimize the LC conditions.The effect of the concentration of EDTA in the mobile phase on the chromatogram was studied at 0.1, 0.15 and 0.2 mmol L−1 at pH 6.9. As shown in Fig. S1 (ESI†), the retention time of Cr(VI) decreased with the EDTA concentration, which could be due to the increase in the ionic strength of the mobile phase. For better separation of Cr(VI) and Cr(III)–EDTA, 0.1 mmol L−1 EDTA was selected. The influence of the concentration of ion pairing reagent TBAP at 0.25, 0.5 and 0.75 mmol L−1, on the chromatogram is shown in Fig. S2 (ESI†). The retention time of Cr(VI) and Cr(III)–EDTA increased with the increase in the TBAP concentration. A TBAP concentration of 0.5 mmol L−1 was considered optimum. The concentration of methanol in the mobile phase also influences the chromatogram and hence the effect of the methanol concentration in the mobile phase on the chromatogram, at 1, 2, and 3% (v/v), was studied. The retention times of Cr(VI) and Cr(III)–EDTA decreased slightly with the methanol concentration. Furthermore better S/B could also be obtained when 2% methanol was added. In the following experiments, 2% (v/v) methanol was added to the mobile phase. The LC operating conditions used in this work are listed in Table 1.
Optimization of USN and DRC ICP-MS conditions
The desolvation tube temperature and the condenser temperature are important operating parameters for the USN. The experimental results showed that the desolvation tube temperature of the USN did not affect the S/B of 53Cr+ and 50Cr+ significantly when the temperature was greater than 120 °C. However, as shown in Fig. S3 (ESI†), the S/B of 53Cr+ and 50Cr+ increased with the decrease in the condenser temperature. To ensure better ion signal USN parameters the carrier gas flow rate, the desolvation tube temperature, and the condenser temperature were set at 1.1 L min−1, 120 °C, and −4 °C, respectively.The background at Cr major masses m/z 52 and m/z 53 was too high to report the better detection limit when ICP-MS was operated in standard mode. Quadrupole ICP-MS instruments equipped with a reaction cell and/or collision cell are known to reduce polyatomic ion interference in the presence of a suitable reaction gas.19,20 Both NH3 and O2 reaction gases are known to lower the background at Cr masses when used as reaction gases in the DRC.2,15,16 In this study, NH3 was selected due to marginally superior estimated detection limits (EDLs). The optimization of DRC conditions has been carried out by continuous introduction of 3 ng mL−1 Cr solution using conventional pneumatic nebulization. The mobile phase containing 300 ng mL−1 Cl− was treated as the blank for this experiment. Fig. S4 (ESI†) shows the effect of the NH3 gas flow rate on the signal of 52Cr+ and 53Cr+, and the blank signals at m/z 52 and 53, respectively. The results indicated that background due to 40Ar12C+, 35Cl16OH+ and 40Ar12CH+, and 37Cl16O+ has been suppressed effectively when NH3 was used as the reaction gas. As shown in Fig. S4,† the blank signal at m/z 52 was suppressed significantly when the NH3 gas flow rate was greater than 0.5 mL min−1. The estimated detection limit (EDL) was determined based on the concentration necessary to yield a net signal equal to three-times the square root of the background. Better detection of 52Cr+ and 53Cr+ could be obtained when the NH3 flow rate was 0.6 mL min−1. Although it did not have a significant effect on the EDL when the rejection parameter q (Rpq) was varied between 0.25 and 0.6, a Rpq value of 0.6 was selected based on the lower background for 52Cr+ and 53Cr+. The rejection parameter a (Rpa) value was set at 0 in this study and the axial field voltage (AFV) was optimized to be 50 V. The DRC conditions used in this work are listed in Table 1.
Analytical performance of HPLC-ICP-MS
Fig. 1(a) and (b) present the chromatograms obtained, under the optimized conditions, for solutions containing 50 ng Cr mL−1 and 5 ng Cr mL−1 each of Cr(III) and Cr(VI), in standard and DRC mode, respectively. The chromatographic separation was complete in 4.5 min. A significant suppression of the background count rates and improvement in the detection power were observed upon proceeding from the standard to the DRC operating mode. Table 1 lists the optimized operating conditions for the HPLC-USN-DRC-ICP-MS analysis. | ||
| Fig. 1 Typical mass-selective chromatogram for Cr(VI) and Cr(III)–EDTA: (a) with standard mode, each Cr species was present at 50 ng Cr mL−1; and (b) with DRC mode, each Cr species was present at 5 ng Cr mL−1. No reaction gas was used in the standard mode. Other LC conditions are given in Table 1. | ||
Under the optimized conditions, the following parameters have been computed. The relative standard deviation of the peak areas was less than 1.9% and the repeatability of retention time was better than 3% for five consecutive injections at 5 ng mL−1 (as the element) each of the two Cr species. Calibration curves (five points) based on the peak height and peak area, were linear with the correlation coefficient (r2) better than 0.9996 for each species in the range studied (0.1–10 ng mL−1). The detection limit was estimated from the peak height versus concentration plot and based on the concentration (as the element) necessary to yield a net signal equal to three times the standard deviation of the baseline fluctuation of the chromatogram. The LC-ICP-MS detection limit obtained in this study was 0.011 and 0.012 ng mL−1 for Cr(III) and Cr(VI), respectively. These results are compared with those of other similar techniques in Table 2. Overall, the detection limits obtained in this work are better than previous results with similar techniques.5,8,16–18,21–26 The lower limit of detection of Cr could be due to the use of the USN as the sample introduction device in this work. A slightly better detection limit could be obtained for 53Cr+ compared to 52Cr+. Hence, in this study, 53Cr+ was used for quantification work.
| Reaction/collision gas | Separation time | Cr(III) | Cr(VI) | Real sample | Reference |
|---|---|---|---|---|---|
| a Limit of quantification. | |||||
| NH3 | 4.5 min | 0.011 | 0.012 | Rice and cereal | This work |
| NH3 | 4 min | 0.1 | 0.1 | Drinking and tap water | 20 |
| NH3 | 7 min | — | 0.11a(mg kg−1) | Cosmetic products | 4 |
| NH3 | 3 min | 0.05 | 0.05 | Oil | 17 |
| He | 7 min | 0.05 | 0.08 | Water | 22 |
| NH3 | 3 min | 0.094 | 0.10 | Water | 15 |
| NH3 | 5 min | 0.05 | 0.05 | Soil and leaves | 7 |
| CH4 | 3 min | 0.5 | 0.6 | Air | 23 |
| He | 7 min | 0.01 | 0.07 | Dairy and cereal products | 24 |
| H2 | 12 min | 1 | 1 | Rice | 21 |
| He | 8 min | 0.08a (μg g−1) | 0.08a (μg g−1) | Dietary supplements | 25 |
Extraction of Cr from rice
Since EDTA was used as the complexing agent in the mobile phase, in this study, the mobile phase with various concentrations of EDTA in the range 0.5–10 mmol L−1 was tested as the extracting reagent. The extraction efficiency increased till 2 mmol L−1 with marginal improvement beyond that when applied on Rice 1. For better recovery, 2 mmol L−1 EDTA was selected. Nevertheless the extraction efficiency was less than 3% when only EDTA was used as the extracting reagent. To improve extraction efficiency, the effect of addition of other acids, including 0.5% (v/v) CH3COOH, HCl, HNO3 and HF, in the extracting solution was studied. As shown in Fig. 2, the addition of HF could improve extraction efficiency significantly. As shown in Fig. S5 (ESI†), extraction efficiency increased with the HF concentration. However, as reported in a previous study,18 Cr(VI) could be converted to Cr(III) during extraction when too high a concentration of HF was used as the extractant. To study the stability of Cr species, a mixture of Cr(VI) and Cr(III) standard solution without the rice extract was heated using microwaves. From the experiments it was found that the Cr(VI) was converted to Cr(III) when the HF concentration was greater than 1% (v/v) (ESI, Fig. S6†). To avoid species transformation, 1.0% (v/v) HF was selected in this study. The effect of the extraction time on extraction efficiency was also studied at 30, 40, 50 and 60 minutes. The extraction efficiency did not change significantly when the extraction time was longer than 50 min; 50 minutes was selected. Thus, for the extraction of Cr species from rice samples, microwave power was used with the extraction solution comprising 0.5 mmol L−1 TBAP, 2% (v/v) methanol, 2 mmol L−1 EDTA and 1% (v/v) HF at 90 °C for 50 min. The total concentrations of Cr in the extracts were determined using pneumatic nebulization DRC ICP-MS. Table 3 reveals that the extraction efficiency was greater than 97% for all determinations.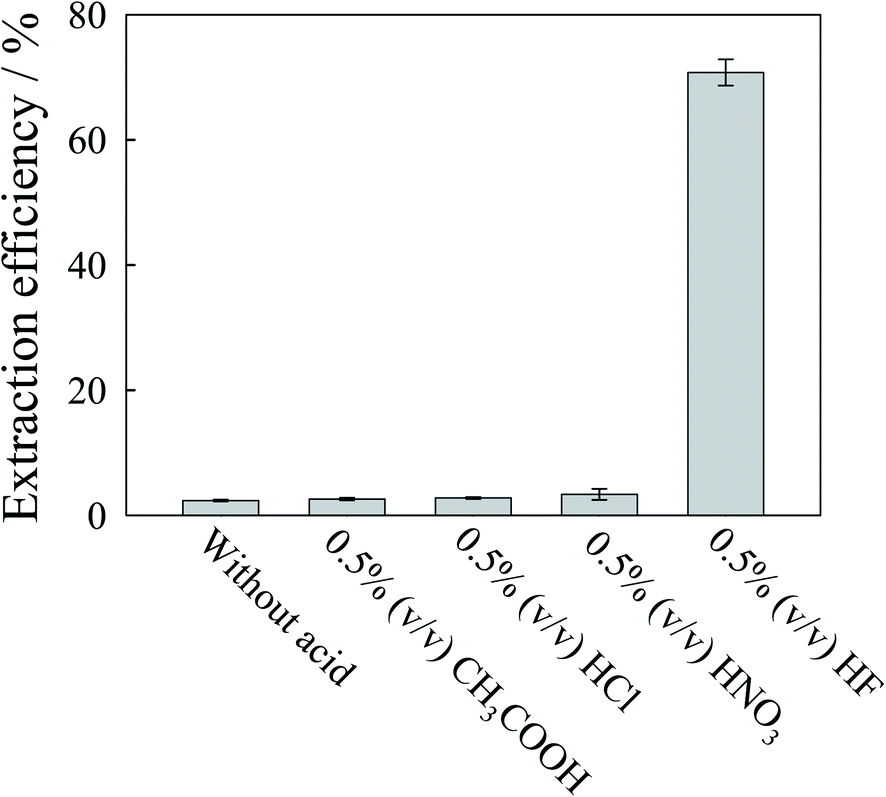 | ||
| Fig. 2 Effect of the extraction reagent on extraction efficiency. 0.3 g of Rice 1 extracted with 5 mL of various extraction reagents. The extraction solutions contained 2 mmol L−1 EDTA and various acids in the mobile phase. Microwave-assisted extraction conditions: 90 °C and 30 min. | ||
| Sample and compound | Concentration found/ng g−1 | Spike recovery/% |
|---|---|---|
| a Values are means of three measurements ± standard deviation. b Column recovery; the value in parentheses is the percentage of the sum of the species to the extracted concentration. c Extraction efficiency; the value in parentheses is the percentage of the extracted concentration to the digested concentration. d NIST certified values. | ||
| NIST SRM 1573a | ||
| Cr(III) | 950 ± 40 | 102 ± 3 |
| Cr(IV) | 1030 ± 40 | 96 ± 2 |
| Sum of Cr species | 1980 ± 60b (102%) | |
| Total Cr (extract) | 1950 ± 70c (100%) | |
| Total Cr (digestion) | 1950 ± 70 | |
| Certifiedd | 1990 ± 60 | |
|
||
| Rice 1 | ||
| Unknown | 52.7 ± 2.1 | |
| Cr(III) | 212 ± 6.7 | 105 ± 4 |
| Cr(IV) | 135 ± 4.4 | 95 ± 2 |
| Sum of Cr species | 400 ± 8.3b (103%) | |
| Total Cr (extract) | 388 ± 11c (98%) | |
| Total Cr (digestion) | 396 ± 13 | |
|
||
| Rice 2 | ||
| Unknown | 14.3 ± 0.7 | |
| Cr(III) | 73.3 ± 2.7 | 105 ± 3 |
| Cr(IV) | 56.4 ± 2.2 | 96 ± 3 |
| Sum of Cr species | 144 ± 3.5b (101%) | |
| Total Cr (extract) | 143 ± 4.1c (97%) | |
| Total Cr (digestion) | 148 ± 4.5 | |
|
||
| Rice 3 | ||
| Unknown | 11.1 ± 0.5 | |
| Cr(III) | 19.9 ± 0.8 | 101 ± 2 |
| Cr(IV) | 6.37 ± 0.3 | 99 ± 1 |
| Sum of Cr species | 37.4 ± 1.0b (98%) | |
| Total Cr (extract) | 38.3 ± 1.3c (98%) | |
| Total Cr (digestion) | 39.2 ± 1.1 | |
|
||
| Rice cereal | ||
| Unknown | 16.9 ± 0.7 | |
| Cr(III) | 79.3 ± 2.9 | 104 ± 4 |
| Cr(IV) | 21.4 ± 0.8 | 96 ± 3 |
| Sum of Cr species | 118 ± 3.1b (98%) | |
| Total Cr (extract) | 120 ± 3.5c (99%) | |
| Total Cr (digestion) | 121 ± 3.9 | |
Sample analysis
In order to prove that the developed chromatographic procedure for the speciation of Cr is suitable for practical analysis, rice and rice cereal samples were analyzed. The accuracy of the procedure has been validated by comparing the sum of the concentration obtained for individual species, using the present procedure, with total concentration of Cr certified in NIST SRM 1573a tomato leaves reference samples. An aliquot of 100 μL was used for the speciation of Cr using the LC USN-DRC ICP-MS procedure. The typical mass selective chromatograms (52Cr+ and 53Cr+) obtained for the NIST SRM 1573a tomato leaves reference sample are shown in Fig. 3. As shown, both Cr(III) and Cr(VI) are present in this leaf sample. To achieve better reproducibility, the peak areas of the elution peaks were used for the quantitative evaluations. The recoveries listed in Table 3 were determined by spiking the tomato leaf reference sample with 1 μg g−1 each of Cr(III) and Cr(VI). The amount of Cr species present in the sample was quantified by an external calibration procedure; the results are listed in Table 3. As shown, the spike recovery was in the range of 96–102% for Cr species in the tomato leaf reference sample. The LC-ICP-MS results are compared with the total concentration of Cr in the tomato leaf reference sample. The LC-USN-DRC-ICP-MS results showed satisfactory agreement with the certified total Cr concentration. | ||
| Fig. 3 Typical mass-selective chromatograms of (a) NIST SRM 1573a Tomato Leaves and (b) the SRM 1573a Tomato Leaves spiked with 1 μg g−1 each of the Cr standards. The extract was diluted 12-fold before separation. The total dilution factor for the injected solution was 200. | ||
As the results for the SRM are satisfactory, this procedure was applied for the speciation analysis of Cr in three polished rice samples and a rice cereal sample. The typical mass selective chromatograms (52Cr+ and 53Cr+) obtained for Rice 1 and Rice 3 are shown in Fig. 4 and Fig. 5, respectively. As shown, both Cr(III) and Cr(VI) are present in these samples. There was an unidentified compound near a retention time of 100 s in all rice and rice cereal samples analyzed. To ensure that the unidentified peak is due to Cr, the ratio of the peak area of m/z 52 to m/z 53 for all the chromatographic peaks was computed. The ratio was in the range 8.32–8.52 and there was no significant difference between the peak area ratios of these peaks according to the Student t-test for a confidence level of 95%. It proves that all the chromatographic peaks shown in the chromatogram do correspond to Cr. The concentration of the compound whose standard was not available was estimated against the sensitivity of the nearest peak. The results of rice and rice cereal are also listed in Table 3. As shown, the spike recovery was in the range of 95–105% for Cr species in rice and rice cereal. A slightly lower recovery of Cr(VI) was observed possibly due to the reduction of Cr(VI) to Cr(III) during extraction. With a lower concentration of HF as the extractant, the reduction could be minimized; however the extraction efficiency of Cr might be degraded. As shown in Table 3, the HPLC-USN-DRC-ICP-MS results were compared with the total concentrations of Cr in the extracts and also in completely digested samples. The results are in satisfactory agreement with the total concentrations. Moreover the Cr(VI) results obtained by the current procedure were in good agreement with the results reported in a previous paper.11 With the current procedure, not only the amount of Cr(VI) was determined, but the concentrations of Cr(III) and an unknown Cr containing compound were also determined. Hence, the current procedure offers complete speciation of Cr in rice.
 | ||
| Fig. 4 Typical chromatograms of (a) reagent blank, (b) the extract of Rice 1, (c) Rice 1 spiked with 217 ng g−1 Cr(III) and 150 ng g−1 Cr(VI). The extract was diluted 2-fold before separation. The total dilution factor for the injected solution was 33.3. The concentration of Cr(VI) in (b) was 4.05 ng mL−1. | ||
 | ||
| Fig. 5 Typical chromatograms of the (a) reagent blank, (b) the extract of Rice 3, and (c) the Rice 3 spiked with 20 ng g−1 Cr(III) and 0.67 ng g−1 Cr(VI). The total dilution factor for the injected solution was 16.7. The concentration of Cr(VI) in (b) was 0.38 ng mL−1. | ||
The precision between sample replicates, of different experiments including sample pretreatment, separation and determination (n = 3), was better than 5% for all determinations. This experiment indicated that the Cr compounds in rice and rice cereal could be readily quantified by the proposed HPLC-USN-DRC-ICP-MS procedure.
Conclusions
The advantages of coupling reversed phase liquid chromatography with DRC-ICP-MS, using ultrasonic nebulization, for the speciation of Cr were presented. With this system, the limits of detection for the Cr were determined to be in the ranges 0.011–0.012 ng Cr mL−1. Using this approach, the concentrations of various Cr compounds in samples such as rice and rice cereal could be quantified. Both Cr(III) and Cr(VI) were present in the samples analyzed. An unknown Cr compound was also detected. The system could provide a sensitive separation technique for chromium speciation in biological samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a grant from the Ministry of Science and Technology (MOST) of the Republic of China under Contracts 107-2113-M-110-010.References
- C.-Y. Hsieh, M.-H. Tsai, D. K. Ryan and O. C. Pancorbo, Sci. Total Environ., 2004, 320, 37–50 CrossRef CAS.
- A. A. Ambushe, R. I. McCrindle and C. M. E. McCrindle, J. Anal. At. Spectrom., 2009, 24, 502–507 RSC.
- C. Kukusamude, P. Sricharoen, N. Limchoowong and S. Kongsri, Food Chem., 2021, 324, 127402 CrossRef.
- A. F. Roig-Navarro, Y. Martinez-Bravo, F. J. López and F. Hernández, J. Chromatogr. A, 2001, 912, 319–327 CrossRef CAS.
- F. Petrucci and O. Senofonte, Anal. Methods, 2015, 7, 5269–5274 RSC.
- R. E. Wolf, M. J. Morrison and M. B. Goldhaber, J. Anal. At. Spectrom., 2007, 22, 1051–1060 RSC.
- N. Kaewkhomdee, S. Mounicou, J. Szpunar, R. Lobinski and J. Shiowatana, Anal. Bioanal. Chem., 2010, 396, 1355–1364 CrossRef CAS.
- C.-Y. Kuo, S.-J. Jiang and A. C. Sahayam, J. Anal. At. Spectrom., 2007, 22, 636–641 RSC.
- J. Scancar and R. Milacic, J. Anal. At. Spectrom., 2014, 29, 427–443 RSC.
- B. Markiewicz, I. Komorowicz, A. Sajnoj, M. Balter and D. Baralkiewicz, Talanta, 2015, 132, 814–828 CrossRef CAS.
- B.-H. Chen, S.-J. Jiang and A. C. Sahayam, Food Chem., 2020, 324, 126698 CrossRef CAS.
- C.-F. Yeh and S.-J. Jiang, J. Chromatogr. A, 2004, 1029, 255–261 CrossRef CAS.
- N. Kovachev, M. Á. Aguirre, M. Hidalgo, K. Simitchiev, V. Stefanova, V. Kmetov and A. Canals, Microchem. J., 2014, 117, 27–33 CrossRef CAS.
- M. Wang, W. Feng, J. Shi, F. Zhang, B. Wang, M. Zhu, B. Li, Y. Zhao and Z. Chai, Talanta, 2007, 71, 2034–2039 CrossRef CAS.
- D. Nixon, K. R. Neubauer, S. J. Eckdahl, J. A. Butz and M. F. Burritt, Spectrochim. Acta Part B, 2002, 57, 951–966 CrossRef.
- D. Barałkiewicz, B. Pikosz, M. Belter and M. Marcinkowska, Accred. Qual. Assur., 2013, 18, 391–401 CrossRef.
- Y.-L. Chang and S.-J. Jiang, J. Anal. At. Spectrom., 2001, 16, 858–862 RSC.
- Y.-A. Lin, S.-J. Jiang, A. C. Sahayam and Y.-L. Huang, Microchem. J., 2016, 128, 274–278 CrossRef CAS.
- H.-W. Hsu, S.-J. Jiang and A. C. Sahayam, Talanta, 2013, 117, 268–272 CrossRef.
- M.-L. Lin and S.-J. Jiang, J. Anal. At. Spectrom., 2011, 26, 1813–1818 RSC.
- S. Catalani, J. Fostinelli, M. E. Gilberti and P. Apostoli, Int. J. Mass Spectrom., 2015, 387, 31–37 CrossRef CAS.
- N. W. Sadiq and D. Beauchemin, Anal. Chem., 2017, 89, 13299–13304 CrossRef CAS.
- J. Sun, L. Ma, Z. Yang and L. Wang, J. Sep. Sci., 2014, 37, 1944–1950 CrossRef CAS.
- M. Stanislawska, B. Janasik and W. Wasowicz, Talanta, 2013, 117, 14–19 CrossRef CAS.
- F. Hernandez, F. Seby, S. Millour, L. Noel and T. Guerin, Food Chem., 2017, 214, 339–346 CrossRef CAS.
- N. Unceta, M. Astorkia, Z. Abrego, A. Gomez-Caballero, M. A. Goicolea and R. J. Barrio, Talanta, 2016, 154, 255–262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ja00356e |
| This journal is © The Royal Society of Chemistry 2021 |