
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGrowth amplification in ultrahigh-throughput microdroplet screening increases sensitivity of clonal enzyme assays and minimizes phenotypic variation†
Paul Jannis
Zurek
 ab,
Raphaëlle
Hours
ab,
Ursula
Schell
b,
Ahir
Pushpanath
b and
Florian
Hollfelder
*a
ab,
Raphaëlle
Hours
ab,
Ursula
Schell
b,
Ahir
Pushpanath
b and
Florian
Hollfelder
*a
aDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, CB2 1GA Cambridge, UK. E-mail: fh111@cam.ac.uk
bJohnson Matthey Plc, 260 Cambridge Science Park, CB4 0WE Cambridge, UK
First published on 12th November 2020
Abstract
Microfluidic ultrahigh-throughput screening of enzyme activities provides information on libraries with millions of variants in a day. Each individual library member is represented by a recombinant single cell, compartmentalised in an emulsion droplet, in which an activity assay is carried out. Key to the success of this approach is the precision and sensitivity of the assay. Assay quality is most profoundly challenged when initially weak, promiscuous activities are to be enhanced in early rounds of directed evolution or when entirely novel catalysts are to be identified from metagenomic sources. Implementation of measures to widen the dynamic range of clonal assays would increase the chances of finding and generating new biocatalysts. Here, we demonstrate that the assay sensitivity and DNA recovery can be improved by orders of magnitude by growth of initially singly compartmentalised cells in microdroplets. Homogeneous cell growth is achieved by continuous oxygenation and recombinant protein expression is regulated by diffusion of an inducer from the oil phase. Reaction conditions are adjusted by directed droplet coalescence to enable full control of buffer composition and kinetic incubation time, creating level playing field conditions for library selections. The clonal amplification multiplies the product readout because more enzyme is produced per compartment. At the same time, phenotypic variation is reduced by measuring monoclonal populations rather than single cells and recovery efficiency is increased. Consequently, this workflow increases the efficiency of lysate-based microfluidic enzyme assays and will make it easier for protein engineers to identify or evolve new enzymes for applications in synthetic and chemical biology.
Introduction
Enzymes are increasingly used for sustainable chemical synthesis, as they are powerful ‘green’ catalysts providing efficient turnover and remarkable stereo- and enantioselectivity under mild reaction conditions.1,2 If a reaction that does not exist in nature is to be catalysed, there are two principal routes to enzymes with novel activities: either natural enzymes with secondary, promiscuous activities3–5 are fine-tuned to the requisites of industrial processes, often via directed evolution; alternatively, new enzyme functions can either be discovered by functional metagenomics in large samples of natural biodiversity6–10 or introduced by computational design.11–13 In all cases, the initially weak activity of an enzyme is likely to require improvements and directed evolution is usually the method of choice. This artificial Darwinian process consists of repeated cycles of mutagenesis and selection and has led to many successful biocatalysts.14–16 The prospects of directed evolution campaigns, however, improve with the practical ability to explore greater diversity via ever higher throughput screening approaches. In the last decade, compartmentalisation of single library members and miniaturisation of reaction volumes in droplet microfluidics (see Fig. 1) has proven a valuable and efficient tool for enzyme evolution.17,18 Single library members (e.g. cells expressing19 or displaying20 enzyme variants) are co-encapsulated with the substrate and reaction progress is followed in chromogenic or fluorogenic reactions by optical interrogation of droplet-compartmentalised library members. Droplets can be sorted with kHz frequencies, enabling fast screening of tens of millions of variants in a day, in picoliter volumes.19–21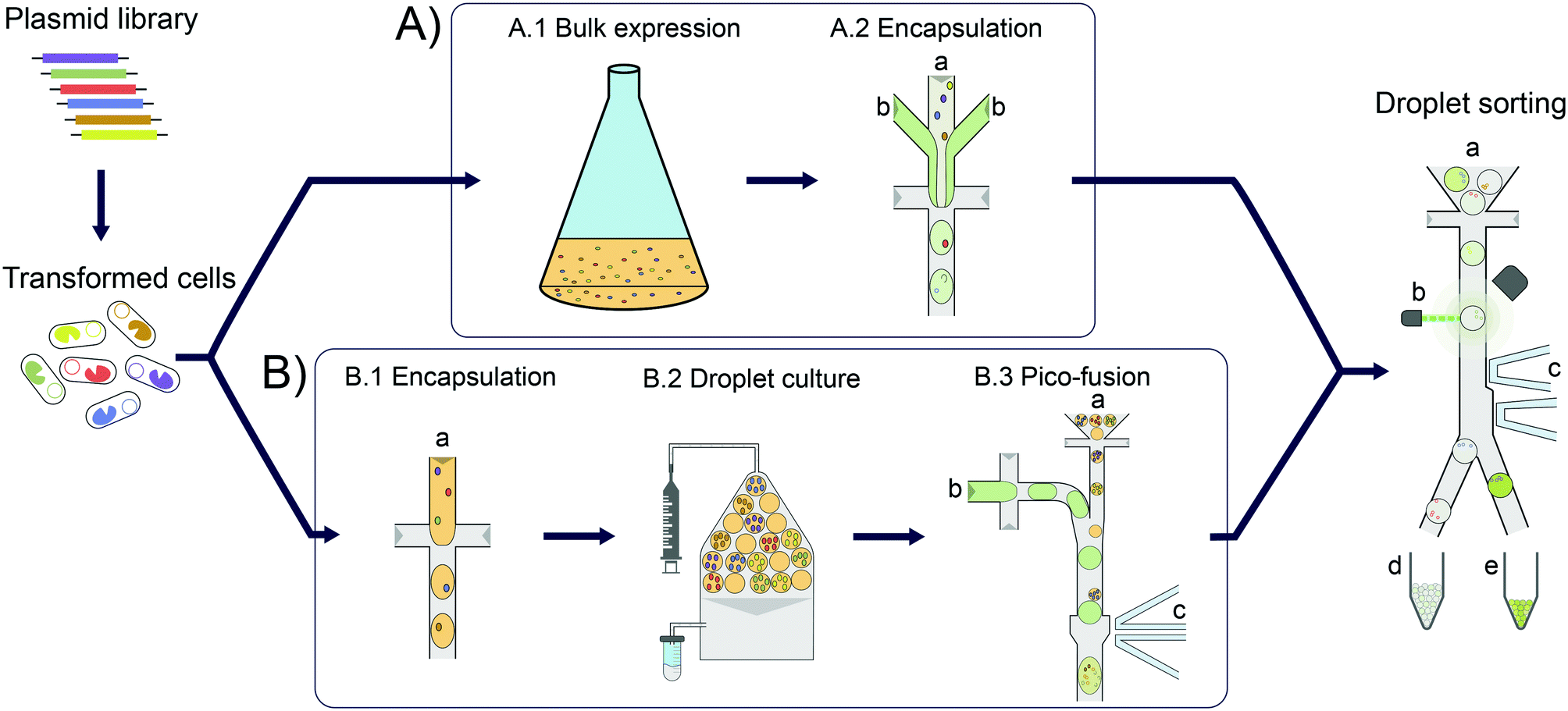 | ||
| Fig. 1 Overview of cell lysate screening after growth amplification in comparison to standard single cell activity assays. In each case, E. coli cells are transformed with a plasmid library for expression of enzyme variants. These cells are then Poisson-distributed in microfluidic droplets, so that the majority of droplets has one or no cell (‘monoclonal droplets’). A) Conventional single cell lysate assay: bulk cell growth and protein expression (A.1). The cell suspension (A.2 a) is then co-encapsulated with substrate and lysis agent (A.2 b) at the point of droplet formation. After allowing time for sufficient enzymatic turnover to reach a detectable signal, the droplets are sorted on a microfluidic chip. B) Clonal amplification in droplets for enzyme lysate assays: to amplify the signal in microfluidic enzyme assays, single cells are encapsulated and grown in droplets (B.1). Droplets are incubated under oil flow (grey) to allow for homogeneous cell growth as well as protein expression (B.2). Substrate and lysis agent (B.3 b) are added to the droplets (B.3 a) in a second step via selective droplet coalescence (B.3) and subsequently sorted on a microfluidic chip. | ||
In addition to maximal throughput, the dynamic range and sensitivity of the assay (including the analytical technologies for measuring product concentrations) are key features relevant for success. Indeed, concentrations of fluorescent reaction product as low 2.5 nM could be detected in a metagenomic screening of hydrolases, corresponding to just ∼2500 molecules of the reaction product fluorescein in 2 pL droplets.6 Consequently, even hits with relatively low activities (kcat/KM ∼50 M−1 s−1) could be identified. This means the sensitivity of droplet screening is increased by several orders of magnitude compared to e.g. conventional plate screening, so that metagenomic hits with inefficient expression and/or low promiscuous activities can be discovered or subsequently evolved in droplets. However, for very low promiscuous activities or enzymes from metagenomic libraries with low expression, detection limits may still preclude catalyst discovery. The superb sensitivity afforded by fluorescence detection is difficult to match when other optical assays that involve detection of product absorbance22 or anisotropy23 are used, where detection limits of 10 μM and 0.1 μM, respectively, have been determined. Other non-optical detection modes have recently been applied to assay enzymatic activity and enabled detection limits in a similar range to the new optical methods, e.g. 1 μM for electrochemical24 and 30 μM for mass spectrometric25 detections. In any case, a crucial condition in all droplet approaches is the necessity to compartmentalise, by Poisson-distribution, just one cell (or gene) per droplet compartment. This renders the droplet monoclonal, i.e. defined by just one unique DNA sequence. On the other hand, while necessary for monoclonality, compartmentalisation of single cells imposes limits on the amount of enzyme produced that is in turn leading to formation of detectable product. A larger enzyme concentration would make it easier to detect reaction product, which means that low promiscuous activity or only weakly expressed enzymes (e.g. in metagenomic libraries) can be identified and become selectable.
Here we establish a general workflow (Fig. 1) that addresses the problem of detection limits in droplet screening by facilitating homogeneous cell growth in droplet compartments, resulting in increased enzyme content per droplet. We show that the individual enzyme performance necessary for successful product detection is lowered ∼10-fold. We validate the workflow by demonstrating its applicability to the conversion of an amino acid dehydrogenase to an amine dehydrogenase biocatalyst, an important class of biocatalysts for the industrial synthesis of chiral amines.26
Results and discussion
Achieving homogeneous cell growth in droplets
First, a suitable device for droplet incubation needed to be prepared that allows for robust storage and convenient handling of emulsions, such as direct re-injection into a sorting device. For this purpose, modified 0.5 mL reaction tubes were used (Fig. 2A and S1†). These droplet incubation chambers can easily be prepared by opening access holes into a standard plastic reaction tube with a biopsy punch and attaching access tubing with high-viscosity plastic glue. The droplets are directly collected from the generation device into such a chamber pre-filled with oil. During incubation the droplets packed at the top of the oil in the chamber, allowing for a convenient and robust way to incubate large emulsion volumes without destabilization by surface effects and for easy re-injection into subsequent microfluidic devices, as necessary for extensive microfluidic screening campaigns.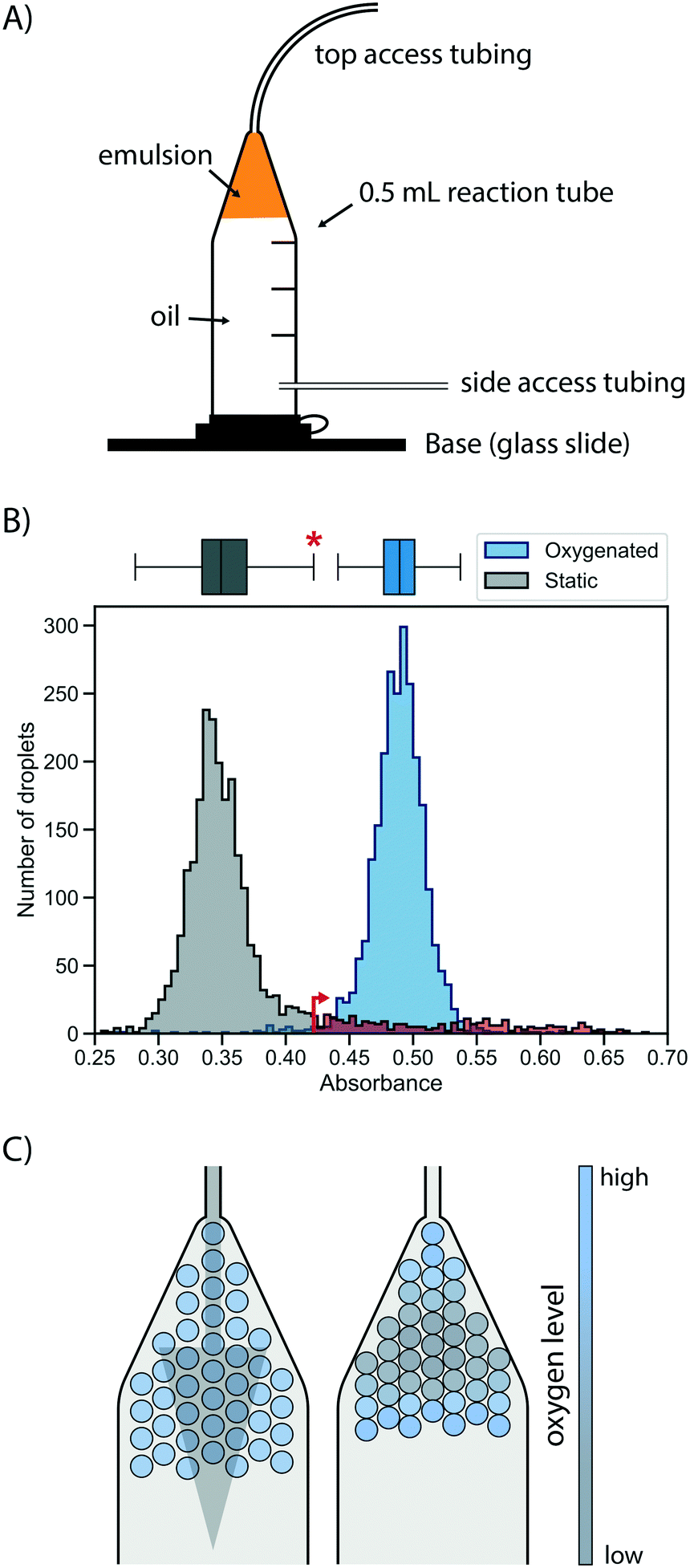 | ||
| Fig. 2 Oxygen supply via the oil phase ensures rapid and homogeneous cell growth in droplets. A) Schematic of a droplet incubation chamber, built from a standard 0.5 mL plastic reaction tube, high viscosity glue and polyethylene tubing. These chambers allow for robust storage and convenient handling of droplet emulsions, which pack on top of the oil phase in the chamber. Droplets can easily be collected, stored, oxygenated and ejected from an incubation chamber. B) Histograms of occupied droplets (n = 2500) starting from singly compartmentalised E. coli that are grown in static (gray) or actively oxygenated (blue, 4 μl min−1 HFE-7500 with 1% RAN surfactant) culture for 16 hours at 37 °C. Cell growth was measured via the absorbance of the live cell stain formazan dye WST-1 (that detects NADH) and analysed in AADS.22 The upper fence of the corresponding boxplot for the static culture (corresponding to the third quartile plus 1.5-fold interquartile range) is marked with an asterisk and an arrow, and the static growth distribution beyond this point is coloured in red, highlighting the strong tail of the distribution. C) Illustrative scenarios for distribution of oxygen throughout a droplet sample during incubation for cell growth. Oxygenation by oil flow (left) leads to a homogeneous distribution, whereas a lack of active oxygenation leads to oxygen depletion in the central, static population (right) with little mixing and exchange. | ||
Next, cell growth was established in these incubation chambers. To test the hypothesis that oxygen availability is key to homogeneous growth, static and oxygenated cell growth in droplets was compared. A live cell stain compatible with absorbance-activated droplet sorting (AADS) was found in WST-1 that was injected into the droplets after growth. WST-1 absorbance changes concomitantly with reduction of NAD+ in a coupled reaction.22 Amplification of cells in droplets was first tested in the most straightforward format, by encapsulating single cells into droplets filled with growth medium and incubation at 37 °C for 16 h without any oxygenation or mixing. Occupied droplets (empty droplets are ignored) incubated under these static growth conditions showed a main peak at low absorbance values with a very evident tail (Fig. 2B), indicating that most droplets show little growth amplification whereas few droplets (showing up in the tail) provide conditions for higher cell growth, so that a mixed population is the result. We sought to establish homogeneity in the droplet population by pushing oil through the incubation chamber, thus providing gentle mixing and oxygen supply. When relatively low oil flow rates for oxygenation (4 μl min−1 per 100 μl of emulsion) were used, homogeneous cell growth could be achieved. To quantify the effect of oxygenation on the growth homogeneity of the population, a robust measure of scale, the interquartile range, was used. This was necessary because of the non-normality of the static growth distribution: droplets with ‘extreme’ growth values, by convention defined as outliers with an absorbance greater than the third quartile plus 1.5-fold interquartile range (0.35% for a reference normal distribution), are abundant in static growth conditions, resulting in 12.1% of the total number of droplets being ‘extreme’ outliers. When oxygenation was applied, the peak of occupied droplets showed a reduction to a mere 1.1% droplets with ‘extreme’ growth, quantifying the beneficial effect of oxygenation on growth homogeneity by removing overly grown outliers.
This situation that static cell growth in droplets, as demonstrated in previous studies,27–30 did not lead to fast growth amplification and homogeneity is unsatisfactory for precise assays: growth homogeneity is crucial for reliable screening, because identical conditions in every droplet are necessary to provide a quantitative readout of enzymatic turnover. The number of cells per droplet determines the enzyme concentration proportionally, thus influencing the enzymatic activity on which selections are based. Previous screening campaigns (e.g. with selections for antimicrobial resistance31 or with initial filtering for cell growth by assessment of light scattering properties in flow cytometry32) were based on simple binary or merely qualitative selections, where different degrees of activity were not differentiated. As a consequence, they may not share the same sensitivity to growth homogeneity as screens for catalytic turnover. A possible reason for the homogeneity in static growth conditions is differential oxygen availability depending on the location of individual droplets in the device. While oxygen can diffuse into the emulsion from the oil, it is also rapidly depleted by the growing cells. As a result, cell growth is not only reduced, but – as the availability of oxygen is greater in droplets close to the interface – heterogeneity is introduced dependent on a droplet's position in the incubation chamber (Fig. 2C). The work of Mahler and colleagues33 has come to a similar conclusion. In their work a custom 3D-printed droplet incubation device was used for oxygenation of cells growing within droplets in a circular set-up, in which a peristaltic pump is used to push oil through an emulsion at high flow rates, to supply a maximal amount of oxygen. Our simple set-up does not yield the cell growth rates comparable to regular shaking flasks achieved by Mahler et al.,33 yet oxygenation with comparatively low oil flow rates provides growth homogeneity. This set-up requires only regular syringe pumps for oil flow and self-made reaction tubes as incubation chambers.
Induction of recombinant protein expression without droplet manipulation
In order to carry out enzyme assays in directed evolution, recombinant protein expression must be induced for each member of a gene library (coding for enzyme mutants or a metagenomic collection of proteins). Many protein expression systems rely on the addition of an external inducer after an initial growth phase, such as the T7 expression system34,35 present in the pET vector family. Here, recombinant expression is usually induced indirectly by the non-hydrolysable lactose analog IPTG (inducing expression of the T7 RNA polymerase via a lactose promoter, which then expresses the gene of interest via the T7 promoter), allowing control over the timing and extent of the induction.36 In a single cell lysate assay, protein expression can be induced in bulk culture before encapsulation. By contrast, when cell growth is performed for signal amplification, protein expression must be induced after encapsulation and growth in each individual droplet compartment. This poses a challenge to conventional induction systems, as an additional step is required to add the inducer, increasing workload and potentially endangering droplet stability. Therefore, a protein expression system offering similarly good control over cell count and protein expression efficiency without the need for an additional droplet manipulation step was investigated.Protein expression was studied in a model system by taking advantage of the easy visualisation of the oxygen-independent fluorescent protein iLOV37 with different induction systems in E. coli BL21 (DE3). (i) Firstly, auto-induction of the aforementioned T7 expression system in a medium containing glucose and lactose was investigated, because it promises expression from the T7 promoter without the external addition of an inducer. Glucose inhibits the recombinant protein expression in the early growth phase, but once it is metabolised the remaining lactose induces protein expression.38 This set-up successfully led to protein expression in droplets (Fig. 3). However, finding the concentrations of glucose and lactose required for optimal protein production, as well as incubation times for high expression proved challenging, as conditions from bulk culture were not directly transferable to droplet culture.
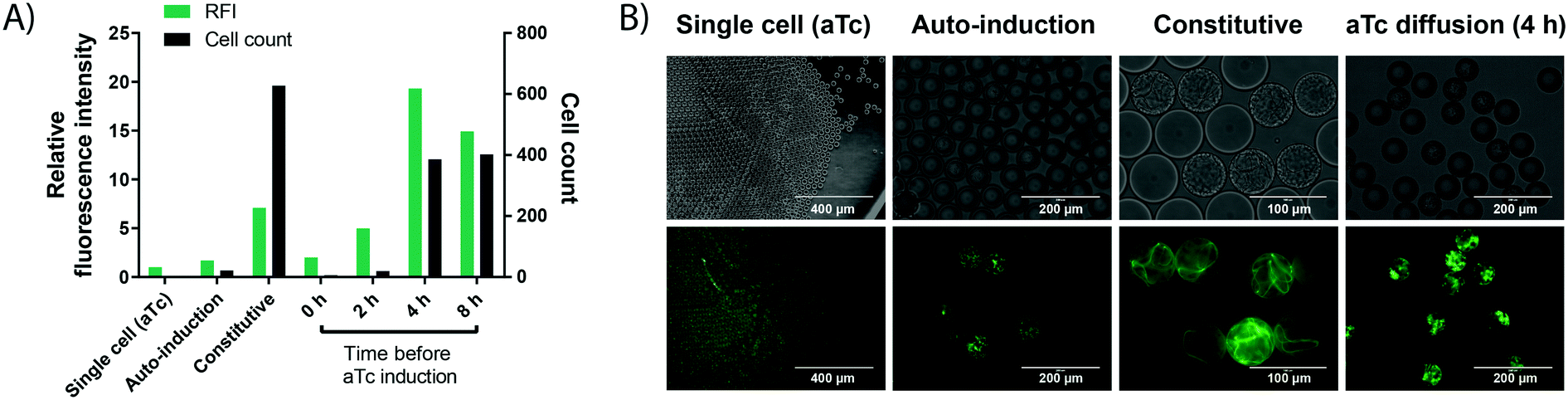 | ||
| Fig. 3 Cell growth in droplets leads to higher protein level in each compartment. Single cells are encapsulated in 50 pL droplets containing growth medium and droplets are incubated for 16 h at 37 °C in incubation chambers. Growth is performed in TB medium, except for auto-induction in ZYP-505238 medium. aTc induction is performed via solubilisation of 400 ng mL−1 of aTc in the oil, followed by exchange of the oil phase at different time points. Cell growth conditions are compared to a conventional single cell control, in which iLOV expression is induced by aTc in bulk, followed by encapsulation of single cells and direct assessment of fluorescence without any growth. A) Cell count (black bars) and fluorescence (green bars) of cells expressing the oxygen-independent fluorescent protein iLOV via different expression systems in droplets. After growth, droplets were de-emulsified with an antistatic gun41 and iLOV fluorescence was measured in a spectrophotometer at 475 nm/510 nm. Cell count was determined by counting colony forming units after plating the de-emulsified droplets on LB-agar. B) Bright field and fluorescence images of cells expressing the oxygen-independent fluorescent protein iLOV via different expression systems in droplets. Strong cell growth visible in the bright field image for constitutive expression. Strong fluorescence is visible for constitutive expression and aTc diffusion, showing the potential of high protein production from few cells via aTc diffusion. | ||
(ii) Secondly, more straightforward constitutive expression was tested. Expression under control of the constitutive lacI promoter showed increased green fluorescence, while the number of cells per droplet increased dramatically. This can be an issue for many enzyme assays, including the one chosen as a model in this study, because of unspecific background activity from cellular components, in this case due to intracellular reducing compounds.39 High background activity reduced the assay sensitivity and introduced undesired variation, making the assay somewhat unreliable.
(iii) Finally, we tested an induction system allowing for strong protein production in droplets from only a few cells, based on the anhydrotetracycline (aTc) inducible promoter. The inducer aTc was barely soluble in the fluorocarbon carrier oil (HFE-7500) and, if delivered from the oil phase, induces protein expression in droplets by diffusion across the phase barrier into the aqueous droplets. We could thus grow cells in droplets as described in the previous section, and at any chosen time point add aTc to the oil used for oxygenation to induce protein expression. A similar approach has recently been applied to regulate the pH inside aqueous droplets during cell cultivation by adding small amounts of acetic acid or diethylamine to the carrier oil phase.40 The experimentally straightforward supply of inducer via diffusion from the oil phase is practically convenient, does not require any additional droplet manipulation steps and allows for control over the total expression strength and final cell count (by controlling the time point of induction) compared to the other two expression methods. Crucially, the inducible promoter provided the basis for strong protein expression from few cells by changing the time point of induction (Fig. 3). For an enzymatic assay (e.g. the detection of WST-1 as a coupled readout for enzymatic activity later in this study), high cellular background prevented the use of 4 h as an induction time point, which shows increased expression strength and cell growth compared to the used 2 h induction time point. However, we expect the achievable improvements in assay sensitivity from cell growth to be even more pronounced in assays with no cellular background, because the onset of stronger cell growth is easily controllable by a freely chosen induction time point. Consequently, this arrangement constitutes a simple and user-friendly way for induction that avoids an additional droplet manipulation step.
Detection and sorting of low enzyme activities
To test the utility of the growth amplification for enzyme assays, we chose the conversion of an amino acid dehydrogenase (AADH) to an amine dehydrogenase (AmDH), an industrially valuable class of enzymes. AADHs can be tuned to accept an amine substrate instead of an amino acid via two active site mutations,42,43 but the rate of this new activity is too low to be conveniently detected. Both activities, however, can in principle be assayed in the same way: NADH produced by the enzyme reaction will reduce WST-1 to its chromatic form, detectable in AADS at 455 nm.22 Indeed, when a well-known AmDH was used as a test case for a novel enzyme or an enzyme having a secondary promiscuous3–5 activity (with therefore low kcat/KM = 0.5 s−1 mM−1;42 corresponding to <1% of the original AADH activity of kcat/KM = 71 s−1 mM−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22), no product was detectable using AADS from droplets containing single cells expressing this enzyme (Fig. 4A). All droplets showed low absorbance, suggesting no detectable activity was apparent over background and single Poisson-distributed cells cannot be distinguished against droplets without a cell. The minimal turnover (determined as approximately 1300 substrate turnovers per enzyme molecule or 10 μM with the formazan-dye coupled absorbent assay described by Gielen et al.22) cannot be reached from a single cell. In cases of low AmDH activity and limited enzyme stability, insufficient product is produced to be detected. This scenario makes such enzymes practically unevolvable, as the absence of detectable signal will thwart all selection efforts.
22), no product was detectable using AADS from droplets containing single cells expressing this enzyme (Fig. 4A). All droplets showed low absorbance, suggesting no detectable activity was apparent over background and single Poisson-distributed cells cannot be distinguished against droplets without a cell. The minimal turnover (determined as approximately 1300 substrate turnovers per enzyme molecule or 10 μM with the formazan-dye coupled absorbent assay described by Gielen et al.22) cannot be reached from a single cell. In cases of low AmDH activity and limited enzyme stability, insufficient product is produced to be detected. This scenario makes such enzymes practically unevolvable, as the absence of detectable signal will thwart all selection efforts.
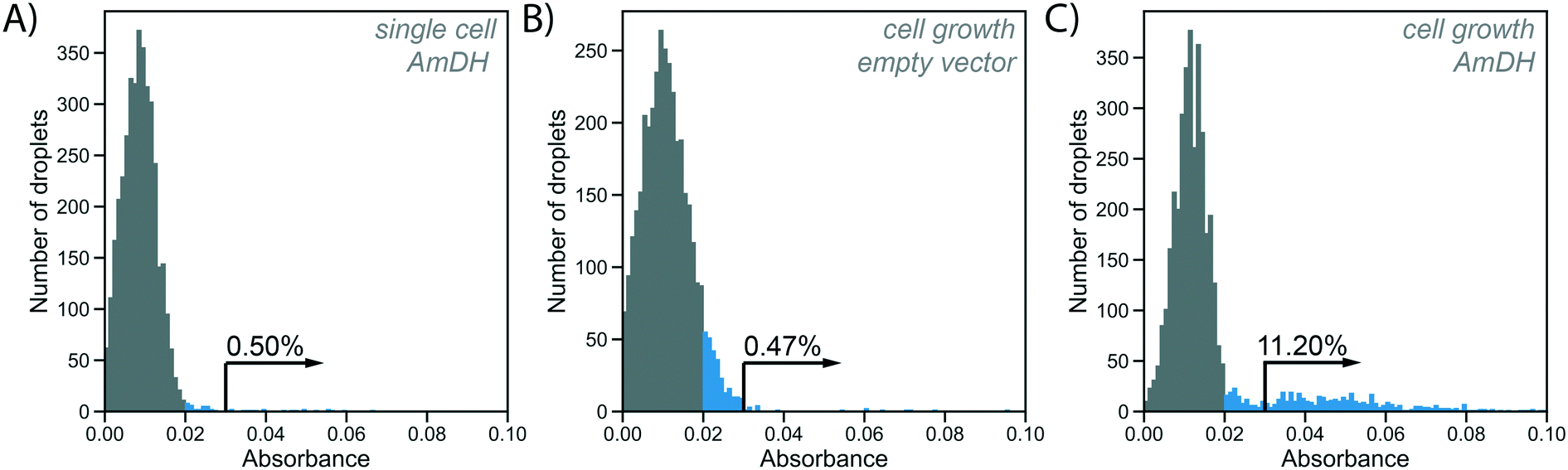 | ||
| Fig. 4 Absorbance of WST-1 as a readout for enzyme activity in droplets with and without cell growth. Histograms show absorbance of 4000 droplets generated at an occupancy of 15%, WST-1 absorbance is measured at 455 nm. Absorbances greater than 0.02 are coloured blue to indicate activity above background in the single cell assay (shown in A). Arrows and percentages represent values of absorbance >0.03, indicating activity above the cell growth background (shown in B), used to detect AmDH activity in enrichment experiment (shown in C). A) Single AmDH expressing cells are directly co-encapsulated with substrate and lysis solution. Absorbance of droplets is measured after 16 h of incubation. Droplets containing cells are not distinguishable from unoccupied droplets. B) Single cells with an empty plasmid (negative control) are encapsulated into 100 pL growth medium droplets. Cell growth is performed at 37 °C for 2 h, followed by oxygenation with inductor oil (400 ng mL−1 aTc in HFE-7500 with 1% RAN surfactant) for 16 h at 20 °C. After cell growth and protein expression, droplets are fused with 200 pL substrate and lysis solution. Absorbance of droplets is measured after 2 h of incubation to allow for enzymatic turnover. C) Single cells harbouring AmDH plasmid are treated in the cell growth workflow as in B). A large peak of unoccupied droplets with low absorbance is visible, as well as a tail of occupied droplets with higher absorbance due to AmDH activity, with 11.2% of all droplets corresponding roughly to the expected droplet occupancy. | ||
In our novel workflow this situation is remedied by growing cells for 2 h at 37 °C, followed by overnight protein expression at 20 °C induced via aTc diffusion for increased protein production. To start the enzymatic reaction, substrate and lysis solution were added to the droplets via selective droplet coalescence (‘pico-fusion’, Fig. S2†) after cell growth. Control of the timing of addition of these reagents, compared to co-encapsulation and incubation with substrate during growth, ensures a level playing field between all clones: the assay reaction starts at the same time for all library members, under identical conditions, minimizing non-enzymatic background reaction signal and avoiding kinetic complications e.g. with the non-linearity of enzymatic time courses. The separate addition of reagent furthermore enables cell lysis to release intracellular enzymes and can be used to create reaction conditions after pico-fusion of a relatively larger volume of buffer that differs from cell growth medium.
Practically, an emulsion of 100 pL droplets with grown cells was fused with 200 pL droplets containing the substrate and lysis agent in the pico-fusion device. An excess of substrate and lysis solution was added to the cell droplet to enable efficient lysis and buffer adjustment. The enzymatic turnover of the substrate was measured by the absorbance of the coupled reaction product in AADS at multiple timepoints. Droplets containing no cells again showed low absorbance, but now there are also droplets with higher absorbance detectable, corresponding to AmDH activity (Fig. 4C). To exclude increased cellular background as the reason for increased absorbance in the cell growth assay, cells not expressing the AmDH but only containing an empty plasmid were used in the cell growth workflow as a control. In this case, empty droplets are not distinguishable from droplets containing cells, although the negative peak is wider, indicating potentially increased background activity (Fig. 4B). Thus, when the cell growth workflow is applied, activity of the AmDH becomes detectable.
To further verify these findings, highly absorbing droplets were sorted from a 1:200 dilution of cells harbouring an AmDH plasmid in cells containing an empty plasmid using the cell growth workflow, resulting in a ∼80-fold enrichment, demonstrating the utility of this workflow to detect enzymes with low activities. However, to make sorting possible, erroneous fusions as well as non-fused droplets must be excluded from the sorting (Fig. S3†). The previously implemented sorting algorithm for AADS employed a simple point-over-threshold comparison,22 which was extended here to true peak detection (see ESI† for the improved Arduino sorting algorithm). This enables the sorting of a specific range of absorbance, as well as implementing a selection based on the signal duration that served as an approximation for correct droplet size.
Quantification of improvements to sensitivity and accuracy
To test whether the improvement in assay sensitivity would affect screening success, an AmDH mutant just active enough to be detected from single cell activity was used, so that a quantitative comparison between conventional and growth enhanced, clonally amplified droplets was possible, yielding an improvement factor. To this end, a computationally stabilized variant of the AADH was generated using the PROSS algorithm.44 The stabilized AADH (AADHmut, Fig. S4†) shows greatly increased soluble expression (soluble fraction = 94%) compared to the non-stabilized parent AADH (soluble fraction = 44%), at a similar level of activity (activity in cell lysate: 77% of the non-stabilized variant). The strong solubilization while maintaining enzyme activity achieved by the PROSS algorithm highlights the success of the combined strategy employed with help of the automated webserver of Goldenzweig et al.:44 using evolutionary precedent to reduce the false positive rate in energy calculations (and prohibiting mutations around the active site) results in strong stabilization with no change of activity. The stabilized AADHmut is turned into an AmDH by active site mutagenesis of residues K66X N262X, in analogy to the reference AmDH.42Single cells expressing this stabilized variant, AmDHmut, were either (i) directly co-encapsulated with substrate and lysis solution in a single cell lysate assay, or (ii) encapsulated in growth medium to enable cell growth and protein expression in droplets followed by addition of substrate and lysis solution via pico-fusion (as described above). Product formation was measured via interrogation of the absorbance of the droplets in AADS at multiple time points. The average detection signal difference of occupied droplets to the peak of unoccupied droplets was plotted in the line graph in Fig. 5A. After 0.5 h, the occupied droplets in the cell growth assay reach an average detection voltage difference to the peak of unoccupied droplets of 3.99 V (Fig. 5C), corresponding to ∼2.5 mM reduced WST-1 indicating conversion of most of the available substrate (3 mM). In contrast, the activity in droplets with single cells start plateauing after 2 h at an average detection voltage difference of 1.32 V (corresponding to ∼1.1 mM reduced WST-1; Fig. 5B). Consequently, using the cell growth assay the initial rate of product formation by AmDHmut is 12.1-fold increased compared to the single cell assay.
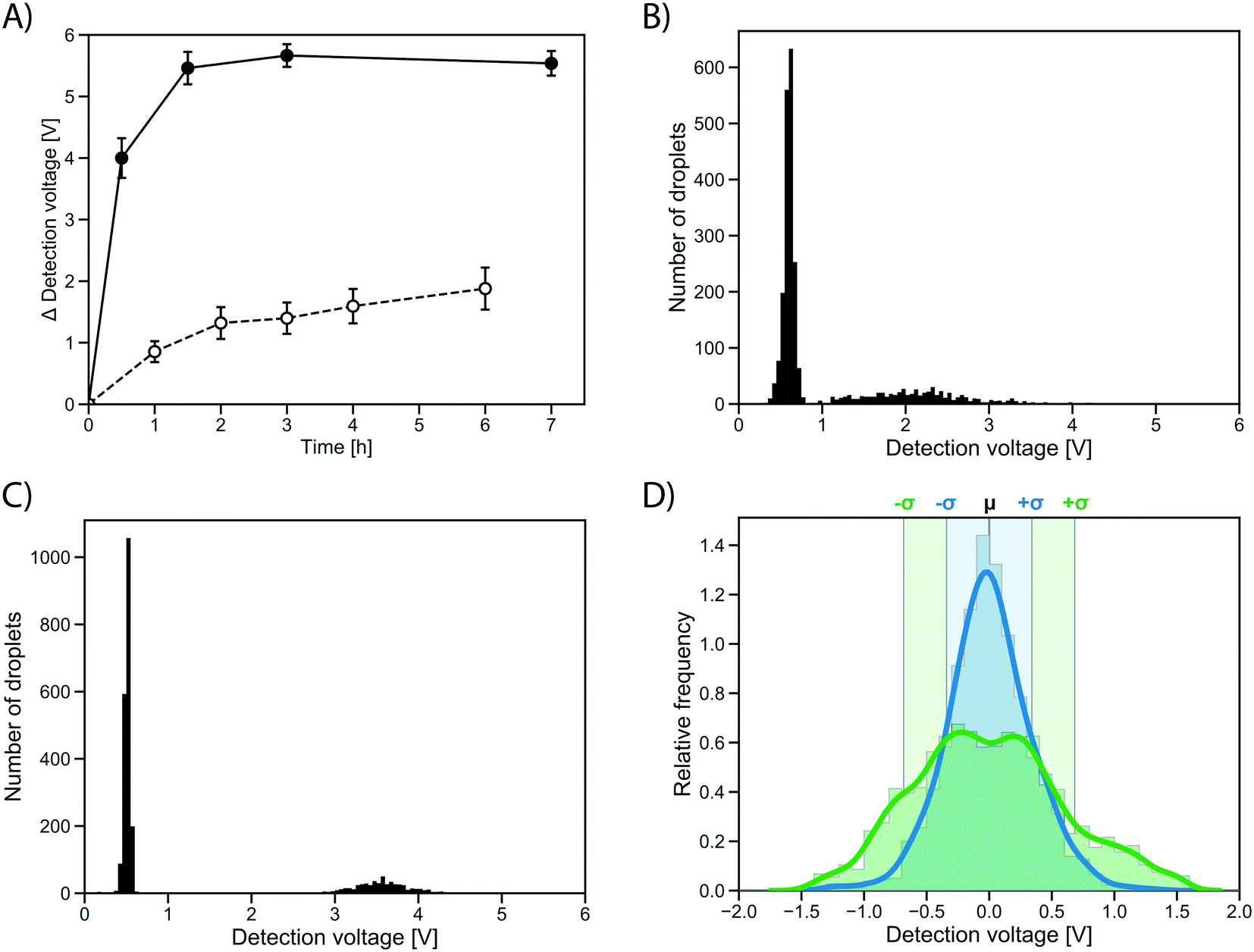 | ||
| Fig. 5 Quantification of growth effects. A) Difference in absorbance detection signal of the peak of occupied droplets and unoccupied droplet peak for a stabilized AmDH (AmDHmut) in single cell (dashed line) and cell growth assay (solid line). Average calculated from 2500 detected droplets, error bars show standard deviation. B) Histogram (n = 2500) of the absorbance of droplets containing single AmDHmut expressing cells co-encapsulated with substrate and lysis solution after 4 h of incubation. A peak of unoccupied droplets with low absorbance can clearly be separated from a peak of droplets containing single cells. C) Histogram (n = 650) of the absorbance of droplets containing approximately 20–30 cells grown in droplets and expressing AmDHmut. Absorbance measured 30 minutes after addition of substrate and lysis agent to the droplets. D) Comparison of variation in the in-droplet cell growth (blue) and single cell workflows (green). The detection signal of the occupied droplet peak was isolated and normalized to have a mean of one. Chosen time points are 4 h for single cell assay (22.7% relative standard deviation, fully shown in panel B) and 30 min for cell growth (10.7% relative standard deviation, fully shown in panel C). Sigma intervals are indicated, corresponding to 68% of all droplets. | ||
The distribution of the measured absorbance values for an identical clone in many droplets made it immediately obvious that the cell growth workflow not only increases the signal strength, but also decreases signal variation over the single cell assay (Fig. 5D). The peaks of occupied droplets showed 5 to 15% relative standard deviation, compared to 20 to 25% for the single cell assay. By comparison, the variation in the assay was at least reduced by half as a consequence of measuring activity of a population of cells rather than a single cell in each droplet (Fig. 5D). When multiple cells are responsible for protein production, the idiosyncratic or stochastic effects of single cells are averaged, resulting in a more reliable and consistent signal.
Conclusion
The new workflow achieves an increase in the detection limit, sensitivity and recovery efficiency of microdroplet-based screening of enzyme libraries and delivers the following:(i) A device for droplet incubation in a densely filled chamber. Emulsions are incubated in modified commercial 0.5 mL reaction tubes requiring no additional equipment, in a much simpler set-up and higher throughput compared to the more complicated devices used previously for cell biological analysis45 or for metagenomic screening.33 Incubation in the modified chamber results in tightly packed emulsions by passively draining the excess oil phase left from droplet generation, providing the large numbers and stability necessary for prolonged screening campaigns. Droplets can easily be withdrawn for analysis or manipulation and returned for incubation under oxygenating conditions. Oxygenation by oil perfusion resulted high growth rates and crucially homogeneous populations.
(ii) Passive delivery of assay component via the oil phase. Supplying each droplet continuously via the oil flow with oxygen leads to faster cell growth and growth homogeneity in all droplets. Supplementing the oil additionally with the inducer aTc switches on protein expression, allowing separate timescales for cell growth and expression (e.g. for proteins that are toxic or assays with high cellular background). By clonal amplification more enzyme molecules are produced per droplet, leading to a higher product signal and easier detection.
(iii) Active delivery of assay components via droplet manipulation. Controlled addition of substrate and lysis reagents by pico-fusion of droplets permits precise timing of the reaction start, so that the optimal ratio of signal generated by the enzymatic reaction to noise of the uncatalysed background reactions can be chosen. This increases the dynamic range for reactions with high chemical backgrounds. Furthermore, the buffer composition for reaction is adjusted from the cell growth medium in this step, widening the scope of detectable reactions and allowing cell lysis to release intracellularly expressed enzymes.
(iv) Clonal amplification multiplies the product readout. Cell growth leads to more expressed enzyme, which, after catalytic turnover, leads to more product generated per droplet compartment. Here, we demonstrated in an absorbance assay in droplets22 that the detection limit (of originally 1300 turnovers per enzyme molecule) could be reduced to ∼100 molecules per enzyme molecule. This improvement was based on the observed 12-fold increase in reaction rate brought about by more available enzyme after expression by multiple cells. It may be possible to grow even larger number of cells to achieve larger amplification factors, potentially resulting in up to 48-fold improvements (based on quantification of GFP expression, see Fig. 3). It will also be possible to grow cells displaying proteins, e.g. using droplet-compartmentalised E. coli autodisplay46 or yeast display,20 instead of cell growth combined with lysis to liberate intracellular protein as demonstrated here.
(v) Increased precision of droplet enzymatic assays. Differential protein expression levels per cell (phenotypic variation) and variability in the time of lysis in single cell assays and have been suggested as sources for droplet-to-droplet variation.47,48 This variation is reduced here by averaging a population of cells, potentially decreasing the number of false positives and allowing the detection of more genuine hits. This idea had been considered before,28,32 but was never quantified and is achieved here for the first time for enzyme activity. This advance is not only a general improvement of the reliability and efficiency of droplet screening, but could also prove useful for applications such as deep mutational scanning, in which quantitative measures of variant frequencies rely on oversampling and rediscovering the same variant multiple times in distinct gates.49
(vi) Enhanced recovery efficiency. Enhanced recovery efficiency is brought about by an increased number of genetic elements after cell multiplication. After sorting, selected droplets are de-emulsified and the plasmid DNA is isolated for subsequent cell transformation. In case of single cell droplet assays, the recovery efficiency is low, as the amount of plasmid from one cell is not always enough to produce a transformant. In the first study on single cell lysate droplet assays, Kintses et al. describe a theoretical single cell recovery efficiency of 87% with an ultra-high copy plasmid.19 In the case of cell growth in droplets, the transformation of sorted DNA usually yielded between 10 and 100 times the number of sorted droplets, depending on the actual cell count within the droplet, thus recovering all sorted variants with an up to 100-fold higher chance even with the medium-high copy aTc-inducible plasmid.
Taken together, this workflow increases the sensitivity of droplet enzymatic assays by several orders of magnitude, making catalyst discovery campaigns more likely to be successful. While an increasing number of catalyst screens in droplets have been successfully implemented,50 some approaches categorically require higher sensitivity to meet success. The successful detection of a weak promiscuous activity that remained undetectable (and thus unselectable) without clonal amplification by cell growth was demonstrated in this work in selections of an AADH for its weaker AmDH activity as a paradigm for adaptive evolution of promiscuous reaction. Likewise, metagenomic screenings will detect larger numbers of catalysts as here the low expression efficiency from metagenomic DNA in heterologous hosts (and often in the absence of a useful promotor) is a typical challenge that ‘hides’ interesting novel enzymes.
Alternative detection modes beyond fluorescence and absorbance, such as electrochemistry24 or mass spectrometry25 would be useful to enlarge the types of assays (i.e. more substrate and reaction types) to be conducted in droplets, but suffer from low sensitivity. For example, it has not been possible to carry out selections in a monoclonal manner using mass spectrometry, likely due to the requirement of very high total turnover numbers owing to the combination of comparably high detection limits (30 μM) and large droplets (25 nL).25 Assuming a similar expression efficiency as seen by Gielen et al., a single cell would provide ∼8 × 105 enzyme molecules to a droplet,22 requiring each enzyme molecule to turn over 22600 or 565000 substrate molecules to reach detection limit in electrochemical24 or mass spectrometric25 detection, respectively. An increased supply of enzyme to the droplet via the workflow presented here may pave the way for better versatility of droplet microfluidics by opening up prospects for these new detection modes.
More generally, reliable recovery of unstable, poorly expressed or inactive enzyme variants and biocatalysts will be especially relevant in bioprospecting of novel biocatalysts in functional metagenomics or for engineering new functions into enzymes in the early stages of directed evolution. An order of magnitude gain in sensitivity, increasing the number of genuine positive (while decreasing false positives by averaging the readout from multiple cells), enhancing the utility of new assay technologies, a 100-fold better recovery and control over assay timing will reveal catalysts that are currently orders of magnitude short of being detectable. The capacity to detect additional catalytic sequences will no doubt enable more adventurous and challenging bioprospecting and protein engineering campaigns in the future and bring ultrahigh throughput droplet microfluidic screening closer to being a mainstay of combinatorial biochemistry and biotechnology.
Experimental
Materials
All chemicals and oligonucleotides were purchased from Sigma-Aldrich unless noted otherwise. Enzymes were purchased from New England Biolabs. The plasmid pASK-IBA36b+ was purchased from IBA lifesciences, the plasmid pRSF-Duet1 from Novagen. The 008-FluoroSurfactant was purchased from RAN biotechnologies.Microfluidic chip fabrication
Microfluidic chips were designed with AutoCAD 2018 and fabricated using standard soft lithography procedures.51 Silicon wafers (3 inch diameter, Siegert Wafer) were coated with photoresist (SU-8 2050, Microchem) and patterned by exposure to UV light through a printed photomask. Polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning) imprints were bonded to glass slides after surface plasma treatment and channels were made hydrophobic by flushing with 1% trichloro(1H,1H,2H,2H-perfluorooctyl)silane in HFE-7500 (3M Novec).Plasmid preparation
The oxygen-independent fluorescent protein iLOV was ordered from Addgene in a pET28a plasmid under control of a T7 promoter (plasmid #63723). The iLOV gene was cloned into pASK-IBA36b+ for aTc inducible expression via Gibson assembly. For constitutive expression, the iLOV gene was cloned to replace the lacI gene in a reduced pRSF-Duet1 plasmid, thus being under control of the placI promoter. The pRSF-Duet1 plasmid was modified before to remove the two original T7 promoter sites by amplification via inverse PCR from T7 terminator to T7 promoter, followed by intramolecular ligation. The Rhodococcus sp. M4 phenylalanine dehydrogenase (Uniprot ID Q59771) with AmDH mutations as determined by Ye et al.42 was ordered as a gene string and cloned into pASK-IBA63b+ for aTc inducible expression. The origin of replication for this plasmid is a medium-high copy ColE1 derivate.Cell growth in droplets and comparison of induction conditions
Droplet incubation chambers were built as described in the ESI.† For cell growth, droplets were generally incubated under oil flow to achieve mixing and oxygenation. HFE-7500 with 1% 008-FluoroSurfactant was pushed through the droplet incubation chamber at 4 μl min−1 per 100 μl of droplet emulsion. To quantify the cell growth in comparison to non-oxygenated incubation, a 15 mM WST-1 solution was added to the droplets via pico-fusion (Fig. S2†). WST-1 detects NADH from live cells and can be measured in AADS as described previously.22 For comparison of induction conditions, iLOV expressing E. coli BL21 (DE3) cells were grown in different expression conditions. Autoinduction was performed by growth in ZYP-5052 medium.38 Growth for all other expression conditions was performed in TB medium (12 g l−1 tryptone, 24 g l−1 yeast extract, 5 g l−1 glycerol, 2.3 g l−1 KH2PO4, 12.5 g l−1 K2HPO4). Induction via aTc was performed by solubilizing 400 ng mL−1 aTc in the carrier oil. After growth and protein expression, droplets were de-emulsified with an antistatic gun41 and iLOV fluorescence was measured in a spectrophotometer at 475 nm/510 nm. Cell count was determined by counting colony forming units after plating the de-emulsified droplets on LB-agar.Enzyme assay from single cell lysate in droplets
Single cell lysate assays are performed similarly to the published droplet assay for phenylalanine dehydrogenases (PheDH), except that a substrate for amine dehydrogenases (AmDH) is used.22 After transformation of E. coli BL21 (DE3), all colonies are washed off the agar plate with 3 mL of LB medium. This stock is used to inoculate 4 mL fresh LB in a sterile culture tube to an optical density at 600 nm (OD600) of 0.8. To this culture, inducer is immediately added (200 ng ml−1 aTc (Acros Organics)) and it is incubated overnight shaking (200 rpm) at 20 °C for protein expression. After expression, the OD600 is measured again and the cells are washed and diluted with encapsulation buffer (25% (v/v) Percoll in 100 mM glycine–KOH pH 9). Dilution is based on the assumption that for E. coli BL21, an OD600 of 1 is equalling approximately 5 × 108 cells per ml, aiming to enable droplet occupancies of 0.15 cells per droplet in order to avoid excess double encapsulations. The cell solution, the substrate and lysis solution (6 mM WST-1 (NBS Biologicals), 6 mM R-1-methyl-3-phenylpropylamine, 2 mM NAD+, 5 μg ml−1 1-methoxy-5-methylphenazinium methyl sulfate (mPMS), 1 μl ml−1 rLysozyme (Merck) and 0.8× CelLytic B in 100 mM glycine–KOH pH 9) and the oil phase (HFE-7500 with 1% 008-FluoroSurfactant) are drawn up into syringes. A conventional droplet generator (80 μm height and 50 μm width at the flow-focusing junction) was used to generate 300 pl droplets at high frequency (>1 kHz) with flow rates of 30 μl min−1 oil and 8 μl min−1 for each of the two aqueous phases. The emulsion is incubated at 22 °C, absorbance is measured and droplets are sorted as described in Gielen et al.22Enzyme assays after cell growth in originally monoclonal droplets
A non-induced cell solution is prepared and diluted as in the early steps of the single cell lysate assay. A conventional droplet generator (50 μm height and 50 μm width at the flow-focusing junction) was used to generate 100 pl droplets at high frequency (>1 kHz) with flow rates of 24 μl min−1 oil (HFE-7500 with 1% 008-FluoroSurfactant) and 8 μl min−1 diluted cell solution. Droplets are collected in an incubation chamber and grown as described above. For induction of protein expression with aTc, the droplets were oxygenated and incubated at 37 °C for 1 h. The oil phase was then changed to inducer oil (HFE-7500 with 1% 008-FluoroSurfactant and 400 ng ml−1 aTc). Inducer oil was pushed for 10 min at 10 μl min−1 from the top to quickly exchange to oil in the emulsion and then used for oxygenation at 4 μl min−1 overnight at 20 °C. After cell growth, the emulsion was injected into the pico-fusion chip (Fig. S2†). Droplets were injected at 1 μl min−1 and spaced with HFE-7500 flowing at 1.5 μl min−1. A flow-focusing junction on the same chip (50 μm width, 50 μm height) was used to generate 200 pl droplets of substrate and lysis solution (4.5 mM WST-1, 4.5 mM R-1-methyl-3-phenylpropylamine, 3 mM NAD+, 7.5 μg ml−1 mPMS, 200 μg ml−1 streptomycin, 2 μl ml−1 rLysozyme and 0.6× CelLytic B in 100 mM glycine–KOH pH 9). If droplets were incubated for sorting, 4 mM tartrazine was added in the substrate and lysis solution as an absorbant offset. Substrate and lysis droplets were generated with an aqueous flow rate of 2 μl min−1 and oil (HFE-7500 with 1% 008-FluoroSurfactant) at 3 μl min−1. The injected droplets with grown cells and the generated substrate droplets are synchronized in the delay channel by slight adjustments to the droplet re-injection flow rate. The smaller cell droplets pack behind the larger substrate droplets in the delay channel, before entering the fusion chamber. An electric field of 400 V and 10 kHz is applied on salt water electrodes52 to facilitate droplet coalescence. Fused droplets are collected and incubated in an incubation chamber before injection into a sorting chip to measure absorbance and select droplets with increased absorbance, as described by Gielen et al.22Verification of activities in plate screening
Identity of sorted variants was confirmed in a secondary screening in 96-well plates. Transformed colonies after sorting were used to inoculate 400 μl LB medium in a 96-well deep well plate. After initial growth overnight at 37 °C, 25 μl of this culture were used to inoculate 425 μl fresh LB medium. After 2 h of growth, 50 μl LB with 10× aTc (2 μg ml−1) was added for induction of protein expression, which was performed at 20 °C overnight. Cells were pelleted (20 min at 3220 × g) and lysed (200 μl 25 mM tris-HC pH 8, 0.1% Triton X-100, 1 μl/30 ml benzonase (Merck)). In a new plate, the detection reaction was started by adding substrate solution (10 mM R-1-methyl-3-phenylpropylamine, 2 mM NAD+ in 100 mM glycine–KOH pH 9) to 20 μl of the cell lysate. Reaction progress was monitored as increase in absorbance at 340 nm for 5 min.Generation of a stabilized AmDH
The PROSS algorithm44 was used to generate stabilized designs of the parental AADH, based on its crystal structure (PDB ID 1C1D). The active site (all residues less than 8 Å away from either substrate L-phenylalanine and NAD+) was excluded from alteration. Six designs were synthesised (Geneart) and tested. The design showing the highest expression strength and activity was chosen for further experiments (ESI† sequence 1). Soluble expression was determined via gel densitometry in ImageJ Fiji after SDS-PAGE separation of the clarified lysate and pellet fraction (Fig. S4A†) of an expression culture grown and lysed as described above. Activity was tested in lysate assays relative to the non-stabilized variant, as described above (Fig. S4B†). The fourth design (Pross 4, ESI† sequence 1) was chosen to be turned into an AmDH as it showed the highest soluble expression strength. AmDHmut was generated from this stabilized AADH by introduction of mutations K66X N262X and screening for AmDH activity in plates, as described above. Variant K66Q N262M with high activity was identified.Author contributions
PJZ, RH and FH designed the research. PJZ performed the experiments. PJZ and FH wrote the paper with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Christian Gylstorff for help with the droplet incubation chambers. We thank Dr. Tomasz Kaminski and the members of the Hollfelder group for helpful comments on the manuscript. This work was funded by the EPSRC and Johnson Matthey (studentship to RH), the European Union's Horizon 2020 research and innovation programme (PJZ via the EU ITN ES-Cat (722610); FH via MetaFluidics (685474)). FH holds an ERC Advanced Investigator award (695669).References
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS.
- N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–573 CrossRef CAS.
- I. Nobeli, A. D. Favia and J. M. Thornton, Nat. Biotechnol., 2009, 27, 157–167 CrossRef CAS.
- U. T. Bornscheuer and R. J. Kazlauskas, Angew. Chem., Int. Ed., 2004, 43, 6032–6040 CrossRef CAS.
- R. J. Kazlauskas, Curr. Opin. Chem. Biol., 2005, 9, 195–201 CrossRef CAS.
- P.-Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvönen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Nat. Commun., 2015, 6, 10008 CrossRef CAS.
- L. Fernández-Arrojo, M.-E. Guazzaroni, N. López-Cortés, A. Beloqui and M. Ferrer, Curr. Opin. Biotechnol., 2010, 21, 725–733 CrossRef.
- P. Lorenz and J. Eck, Nat. Rev. Microbiol., 2005, 3, 510–516 CrossRef CAS.
- T. Uchiyama and K. Miyazaki, Curr. Opin. Biotechnol., 2009, 20, 616–622 CrossRef CAS.
- A. S. Tauzin, M. R. Pereira, L. D. Van Vliet, P.-Y. Colin, E. Laville, J. Esque, S. Laguerre, B. Henrissat, N. Terrapon, V. Lombard, M. Leclerc, J. Doré, F. Hollfelder and G. Potocki-Veronese, Microbiome, 2020, 8, 141 CrossRef.
- A. Zanghellini, Curr. Opin. Biotechnol., 2014, 29, 132–138 CrossRef CAS.
- L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Röthlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, C. F. Barbas, D. Hilvert, K. N. Houk, B. L. Stoddard and D. Baker, Science, 2008, 319, 1387–1391 CrossRef CAS.
- D. Röthlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, S. Albeck, K. N. Houk, D. S. Tawfik and D. Baker, Nature, 2008, 453, 190–195 CrossRef.
- J. L. Porter, R. A. Rusli and D. L. Ollis, ChemBioChem, 2016, 17, 197–203 CrossRef CAS.
- C. Zeymer and D. Hilvert, Annu. Rev. Biochem., 2018, 87, 131–157 CrossRef CAS.
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866–876 CrossRef CAS.
- P.-Y. Colin, A. Zinchenko and F. Hollfelder, Curr. Opin. Struct. Biol., 2015, 33, 42–51 CrossRef CAS.
- S. Neun, P. J. Zurek, T. S. Kaminski and F. Hollfelder, Methods Enzymol., 2020, 643, 317–343 Search PubMed.
- B. Kintses, C. Hein, M. F. Mohamed, M. Fischlechner, F. Courtois, C. Lainé and F. Hollfelder, Chem. Biol., 2012, 19, 1001–1009 CrossRef CAS.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS.
- P. Mair, F. Gielen and F. Hollfelder, Curr. Opin. Chem. Biol., 2017, 37, 137–144 CrossRef CAS.
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7383–E7389 CrossRef CAS.
- F. Gielen, M. Butz, E. J. Rees, M. Erdelyi, T. Moschetti, M. Hyvönen, J. B. Edel, C. F. Kaminski and F. Hollfelder, Anal. Chem., 2017, 89, 1092–1101 CrossRef CAS.
- H. Goto, Y. Kanai, A. Yotsui, S. Shimokihara, S. Shitara, R. Oyobiki, K. Fujiwara, T. Watanabe, Y. Einaga, Y. Matsumoto, N. Miki and N. Doi, Lab Chip, 2020, 20, 852–861 RSC.
- D. A. Holland-Moritz, M. K. Wismer, B. F. Mann, I. Farasat, P. Devine, E. D. Guetschow, I. Mangion, C. J. Welch, J. C. Moore, S. Sun and R. T. Kennedy, Angew. Chem., 2020, 132, 4500–4507 CrossRef.
- G. Grogan, Curr. Opin. Chem. Biol., 2018, 43, 15–22 CrossRef CAS.
- B. L. Wang, A. Ghaderi, H. Zhou, J. Agresti, D. A. Weitz, G. R. Fink and G. Stephanopoulos, Nat. Biotechnol., 2014, 32, 473–478 CrossRef CAS.
- T. Beneyton, S. Thomas, A. D. Griffiths, J.-M. Nicaud, A. Drevelle and T. Rossignol, Microb. Cell Fact., 2017, 16, 18 CrossRef.
- P. Hammar, S. A. Angermayr, S. L. Sjostrom, J. van der Meer, K. J. Hellingwerf, E. P. Hudson and H. N. Joensson, Biotechnol. Biofuels, 2015, 8, 193 CrossRef.
- S. Siedler, N. K. Khatri, A. Zsohár, I. Kjærbølling, M. Vogt, P. Hammar, C. F. Nielsen, J. Marienhagen, M. O. A. Sommer and H. N. Joensson, ACS Synth. Biol., 2017, 6, 1860–1869 CrossRef.
- X. Liu, R. E. Painter, K. Enesa, D. Holmes, G. Whyte, C. G. Garlisi, F. J. Monsma, M. Rehak, F. F. Craig and C. A. Smith, Lab Chip, 2016, 16, 1636–1643 RSC.
- J. M. Duarte, I. Barbier and Y. Schaerli, ACS Synth. Biol., 2017, 6, 1988–1995 CrossRef CAS.
- L. Mahler, M. Tovar, T. Weber, S. Brandes, M. M. Rudolph, J. Ehgartner, T. Mayr, M. T. Figge, M. Roth and E. Zang, RSC Adv., 2015, 5, 101871–101878 RSC.
- F. W. Studier and B. A. Moffatt, J. Mol. Biol., 1986, 189, 113–130 CrossRef CAS.
- J. W. Dubendorf and F. W. Studier, J. Mol. Biol., 1991, 219, 45–59 CrossRef.
- G. L. Rosano and E. A. Ceccarelli, Front. Microbiol., 2014, 5, 172 Search PubMed.
- S. Chapman, C. Faulkner, E. Kaiserli, C. Garcia-Mata, E. I. Savenkov, A. G. Roberts, K. J. Oparka and J. M. Christie, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20038–20043 CrossRef CAS.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS.
- R. Huang, H. Chen, D. M. Upp, J. C. Lewis and Y.-H. P. J. Zhang, ACS Catal., 2019, 9, 11709–11719 CrossRef CAS.
- M. Tovar, L. Mahler, S. Buchheim, M. Roth and M. A. Rosenbaum, Microb. Cell Fact., 2020, 19, 16 CrossRef CAS.
- M. Karbaschi, P. Shahi and A. R. Abate, Biomicrofluidics, 2017, 11, 044107 CrossRef.
- L. J. Ye, H. H. Toh, Y. Yang, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2015, 5, 1119–1122 CrossRef CAS.
- M. J. Abrahamson, J. W. Wong and A. S. Bommarius, Adv. Synth. Catal., 2013, 355, 1780–1786 CrossRef CAS.
- A. Goldenzweig, M. Goldsmith, S. E. Hill, O. Gertman, P. Laurino, Y. Ashani, O. Dym, T. Unger, S. Albeck, J. Prilusky, R. L. Lieberman, A. Aharoni, I. Silman, J. L. Sussman, D. S. Tawfik and S. J. Fleishman, Mol. Cell, 2016, 63, 337–346 CrossRef CAS.
- H. Kleine-Brüggeney, L. D. van Vliet, C. Mulas, F. Gielen, C. C. Agley, J. C. R. Silva, A. Smith, K. Chalut and F. Hollfelder, Small, 2019, 15, 1804576 CrossRef.
- B. van Loo, M. Heberlein, P. Mair, A. Zinchenko, J. Schüürmann, B. D. G. Eenink, J. M. Holstein, C. Dilkaute, J. Jose, F. Hollfelder and E. Bornberg-Bauer, ACS Synth. Biol., 2019, 8, 2690–2700 CrossRef CAS.
- M. B. Elowitz, A. J. Levine, E. D. Siggia and P. S. Swain, Science, 2002, 297, 1183–1186 CrossRef CAS.
- A. Raj and A. van Oudenaarden, Cell, 2008, 135, 216–226 CrossRef CAS.
- N. Peterman and E. Levine, BMC Genomics, 2016, 17, 206 CrossRef.
- S. Neun, P. J. Zurek, T. S. Kaminski and F. Hollfelder, Methods Enzymol., 2020, 643, 317–343 Search PubMed.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- A. Sciambi and A. R. Abate, Lab Chip, 2014, 14, 2605–2609 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00830c |
| This journal is © The Royal Society of Chemistry 2021 |