
Microfluidic assay of antiplatelet agents for inhibition of shear-induced platelet adhesion and activation†
Shekh Mojibur
Rahman
a and
Vladimir
Hlady
 *b
*b
aDepartment of Chemical Engineering, University of Utah, Salt Lake City, UT 84112, USA
bDepartment of Biomedical Engineering, University of Utah, 20 S. 2030 E., Rm. 506B, Salt Lake City, UT 84112, USA. E-mail: vladimir.hlady@utah.edu; Tel: +1 (801) 581 5042
First published on 13th November 2020
Abstract
We have developed a microfluidic system to perfuse whole blood through a flow channel with an upstream stenotic region and a downstream protein capture region. This flow-based system was used to assay how effectively antiplatelet agents suppress shear-induced platelet adhesion and activation downstream of the stenotic region. Microcontact printing was used to covalently attach one of three platelet binding proteins [fibrinogen, collagen, or von Willebrand factor (vWf)] to the surface of the downstream capture region. Whole blood with an antiplatelet agent was transiently exposed to an upstream high wall shear rate (either 4860 s−1 or 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 560 s−1), and subsequently flowed over the downstream capture region where the platelet adhesion was measured. Several antiplatelet agents (acetylsalicylic acid, tirofiban, eptifibatide, anti-vWf, and anti-GPIbα) were evaluated for their efficacy in attenuating downstream adhesion. Following antibody blocking of vWf or GPIbα, downstream platelet activation was also assessed in perfused blood by flow cytometry using two activation markers (active GPIIb/IIIa and P-selectin). Acetylsalicylic acid demonstrated its inability to diminish shear-induced platelet adhesion to all three binding proteins. GPIIb/IIIa inhibitors (tirofiban and eptifibatide) significantly reduced platelet adhesion to fibrinogen. Antibody blocking of vWf or GPIbα effectively diminished platelet adhesion to all three capture proteins as well as platelet activation in perfused blood, indicating an essential role of vWf–GPIbα interaction in mediating shear-induced platelet aggregation.
560 s−1), and subsequently flowed over the downstream capture region where the platelet adhesion was measured. Several antiplatelet agents (acetylsalicylic acid, tirofiban, eptifibatide, anti-vWf, and anti-GPIbα) were evaluated for their efficacy in attenuating downstream adhesion. Following antibody blocking of vWf or GPIbα, downstream platelet activation was also assessed in perfused blood by flow cytometry using two activation markers (active GPIIb/IIIa and P-selectin). Acetylsalicylic acid demonstrated its inability to diminish shear-induced platelet adhesion to all three binding proteins. GPIIb/IIIa inhibitors (tirofiban and eptifibatide) significantly reduced platelet adhesion to fibrinogen. Antibody blocking of vWf or GPIbα effectively diminished platelet adhesion to all three capture proteins as well as platelet activation in perfused blood, indicating an essential role of vWf–GPIbα interaction in mediating shear-induced platelet aggregation.
Introduction
Cardiovascular disease, a leading cause of death for people in the world, is often treated through the surgical introduction of vascular devices such as stents, shunts, catheters, and vascular grafts. Due to intimal fibrous hyperplasia, vascular devices such as synthetic small-diameter vascular grafts often become stenotic (narrow).1–3 Atherosclerosis plaque formation is a common cause of blood vessel stenosis.4,5 In addition to the thrombogenic surface of vascular devices and atherosclerotic plaque rupture, the flow-induced shear force plays a key role in thrombus formation. High shear forces have been shown to trigger intracellular signaling in platelets, leading to activation of their surface receptors such as GPIIb/III and GPIbα.6,7 Following such intracellular signaling, circulating platelets recognize adhesive proteins such as fibrinogen and von Willebrand factor (vWf) via their surface receptors. Following a series of biochemical reactions and morphological changes, platelets become fully activated to form aggregates, ultimately leading to thrombus formation.The platelet homeostatic response of vascular device implantation and atherosclerotic plaque formation often necessitates that the patients remain on antiplatelet therapies to reduce the risk of thrombotic complications such as stroke, embolism, and myocardial infarction. Examples of available prescribed antiplatelet agents are thromboxane A2 (TxA2) inhibitors (e.g., acetylsalicylic acid), GPIIb/IIIa inhibitors (e.g., abciximab, tirofiban, and eptifibatide), and adenosine diphosphate (ADP) receptor P2Y12 inhibitors (e.g., clopidogrel, prasugrel, and cangrelor). Several shear-based instruments are commercially available to assess the effect of antiplatelet agents on platelet function in vitro under high shear flow. However, the majority of these devices are limited in terms of their ability to measure the efficacy of antiplatelet agents in the hemodynamic environment that platelets experience in vivo. For example, platelets are often sheared continuously either in a cone-and-plate analyzer (CPA, Matis Medical, Belgium) or in a platelet function analyzer (PFA-100/200, Siemens, USA) to assess different antiplatelet agents.8–13 However, these devices do not take into account the transient interactions between platelets and increased shear forces, which usually occurs when blood circulates through a stenotic vessel or implanted vascular device. While several studies explored the effect of transiently increased shear forces on local platelet function using microfluidic devices,4,14–18 the ability of antiplatelet agents in suppressing shear-induced platelet aggregation was not thoroughly investigated. Moreover, these studies examined the platelet function at the sites of stenosis while the effects of platelet exposure to elevated shear forces and subsequent downstream events were largely ignored. We have recently developed a microfluidic assay to investigate the effects of transiently elevated upstream shear forces on downstream platelet adhesion and activation and used it to show that transient exposure to increased upstream shear forces primes platelets for enhanced downstream adhesion and activation.19–21
Shear gradients generated by transiently elevated shear forces such as those that occur in stenotic vascular vessels and grafts may exert profound effects on platelet function. There is abundant evidence that shear-induced platelet aggregation plays a critical role in the pathogenesis of acute arterial thrombosis.22,23 Despite the prevalence of antiplatelet agents, their efficacy in suppressing shear-induced platelet aggregation is not fully understood. Thus, pharmacological modulation of platelet function under high shear force is believed to be of clinical importance. Furthermore, the evaluation of antiplatelet agents is crucial to understand the mechanisms of shear-induced platelet aggregation, which may lead to the proper selection of antiplatelet agents for the prevention of thrombotic complications after vascular device implantation and atherosclerotic plaque formation.
Our microfluidic assay is used here to assess the effects of several antiplatelet agents on downstream platelet adhesion following exposure to transiently increased upstream shear forces. Platelets in whole blood with an antiplatelet agent were transiently exposed to an increased shear force (either 4860 s−1 or 11560 s−1) in an upstream stenotic region. Downstream from the stenotic region, one of the three proteins (fibrinogen, collagen, or vWf) was covalently immobilized to serve as a platelet capture agent. We tested several antiplatelet agents: – acetylsalicylic acid (ASA, also known as aspirin) as a TxA2 inhibitor, and – tirofiban or eptifibatide as an inhibitor of the binding between fibrinogen and platelet GPIIb/IIIa. We have also blocked vWf and GPIbα using anti-vWf and anti-GPIbα (AK2), respectively, to inhibit the binding between vWf and GPIbα. Platelet adhesion to a downstream capture agent was characterized by using a phase contrast microscopy. To evaluate the effect of blocking vWf or GPIbα on platelet activation, two activation markers (GPIIb/IIIa and P-selectin) were quantified in perfused blood using flow cytometry.
Method
Flow channel preparation
Each experimental flow channel, containing an upstream stenotic region and a downstream capture region (Fig. 1), was manufactured by placing a silicone gasket (BioPlexus, Ventura, CA, USA) between a microscope slide (VWR International, Radnor, PA, USA) and a Nexterion-H slide (Schott, Tempe, AZ, USA) as previously described.20 Flow channel features were designed using SolidWorks (Dassault Systemes, Waltham, MA, USA) and patterned on the silicone gasket utilizing a laser cutter (VLS3.60, Universal Laser Systems, Scottsdale, AZ, USA). Flow channel dimensions were 1 mm wide, 0.18 mm high, and 70 mm long. Stenotic regions were created 5 mm downstream from the inlet to allow for the development of laminar flow. Both the upstream stenotic region and the downstream capture region were 10 mm in length. Two different transiently increased shear forces (wall shear rates of 4860 s−1 and 11560 s−1) were generated by changing the width of the upstream stenotic region from 1.0 mm to 0.4 and 0.2 mm, respectively. The wall shear rates in the pre- and post-stenotic regions of the flow channels were 1620 s−1, and the control flow channel had no stenotic region. Wall shear rates were calculated by simulating constant blood flow (0.5 mL min−1) through three-dimensional (3-D) rectangular flow channels using COMSOL Multiphysics (COMSOL, Inc., Burlington, MA, USA) according to the previously published protocol.20 Continuity and Navier–Stokes equations were used to derive the wall shear rates. Blood traveling time within the stenotic regions was 86 and 43 milliseconds for 0.4 and 0.2 mm wide stenotic channels, respectively. Reynolds numbers within the stenotic regions were 8.8 and 13.4 for 0.4 and 0.2 mm wide stenotic channels, respectively. Reynolds number within the control flow channel with no stenotic region was 4.3. These Reynolds numbers confirmed the laminar flow within the microchannels. The distance between the upstream stenotic region and the downstream capture region was 25 mm. Following exposure to the upstream stenotic region, blood flowed ∼540 milliseconds to travel this 25 mm distance before entering the downstream capture region.
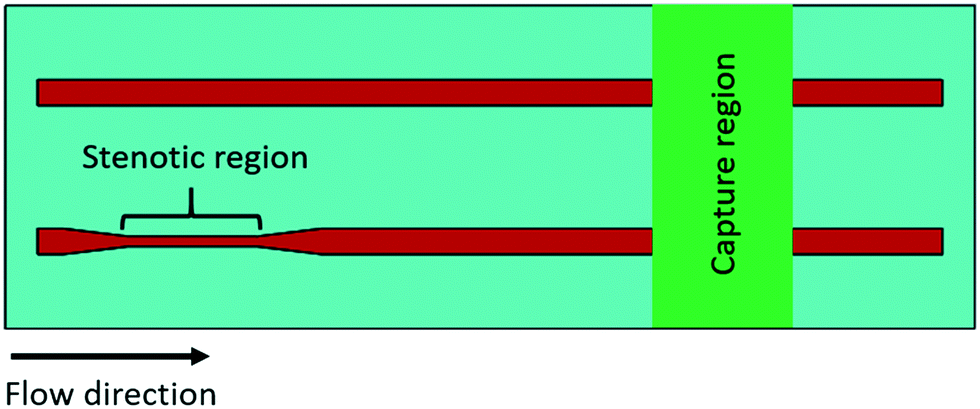 | ||
| Fig. 1 Schematic diagram of the microfluidic flow system. The upstream stenotic region was created by changing the width of the flow channel. The control flow channels had no stenotic region. One of three platelet binding proteins (fibrinogen, collagen, or vWf) was immobilized downstream on a reactive glass substrate by microcontact printing to create a platelet capture region. | ||
The downstream capture region (10 mm in length) was prepared by microcontact printing described in detail elsewhere.24 Fibrinogen and vWf (Haematologic Technologies, Essex Junction, VT, USA), and collagen I (Ibidi, Fitchburg, WI, USA) were covalently immobilized at 100% surface coverage on the surface of Nexterion-H slide. To create the capture region, a flat polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, Midland, MI, USA) stamp was coated with a protein solution for 10 min, subsequently allowed to dry under a stream of nitrogen, and placed in contact with a Nexterion-H slide for 1 h. The Nexterion-H slide contained reactive N-hydroxysuccinimide (NHS) esters, which allowed covalent protein immobilization via their –NH2 moieties.25
Platelet adhesion studies
Flow channels were assembled and connected to a syringe pump (Kent Scientific, Torrington, CT, USA). A solution of human serum albumin (HSA, 1 mg mL−1, Sigma Aldrich, St. Louis, MO, USA) was perfused through each flow channel and incubated for 1 h at room temperature to passivate the nonreacted regions of the Nexterion-H glass surfaces as well as the walls of each flow channel and connecting tubing. Fresh whole blood was drawn via venipuncture from four healthy human donors (36–41% hematocrit, 214–258 × 103 platelets per μL, ∼0.0035 Pa s viscosity). All experiments were performed in compliance with the relevant laws and guidelines approved by the Institutional Review Board of the University of Utah (IRB # 00051506). Study participants were fully informed regarding the purposes of the study and consent was obtained. Blood was drawn into buffered 3.2% (0.105 M) sodium citrate and was immediately treated with Phe-Pro-Arg-chloromethylketone (PPACK, 80 μM, Haematologic Technologies, Essex Junction, VT, USA) to inhibit thrombin-induced coagulation. The percentage of activated platelets in the anticoagulated blood prior to microfluidic experiments was determined by flow cytometry. The expression levels of active GPIIb/IIIa and P-selectin was approximately 2.3% and 2.2%, respectively (for donor variability see ESI†). The blood samples were kept at 37 °C in a water bath and used within 2 h of draw from donors. Currently prescribed antiplatelet agents: ASA, tirofiban, and eptifibatide were purchased from Sigma Aldrich (St. Louis, MO, USA). Sheep anti-human vWf (Haematologic Technologies, Essex Junction, VT, USA) and anti-GPIbα (AK2, Novus Biologicals, Littleton, CO, USA) were tested to assess their efficacy in diminishing downstream platelet adhesion and activation. Prior to perfusion, anticoagulated blood was incubated with each of antiplatelet agents at their recommended blood concentration (i.e., 30 μg mL−1 ASA, 0.33 μg mL−1 tirofiban, 3 μg mL−1 eptifibatide, 10 μg mL−1 anti-vWf, and 20 μg mL−1 AK2) for 30 min at 37 °C. Blood was then perfused through the flow channels at a constant flow rate of 0.5 mL min−1. The flow was sustained for 5 min and 2.5 mL of blood was perfused through each channel, after which the flow channels were rinsed with prewarmed Tyrode's buffer (37 °C, pH 7.4) and fixed in 4% paraformaldehyde solution. The attached cells were imaged using a phase contrast microscope (Diaphot 200, Nikon, Tokyo, Japan) with 40× objective (ELWD, NA 0.55, Nikon, Tokyo, Japan), and the number of adhered platelets was quantified. Total 40 images (10 images for each donor) were analyzed for quantifying platelet adhesion.Platelet activation studies
Flow cytometry was used to measure the expression levels of active GPIIb/IIIa and CD62P (P-selectin) on perfused platelets. Following blood perfusion through a flow channel with a stenotic region but with no capture region, a 5 μL aliquot of blood supernatant was incubated for 20 min with PAC-1 and anti-human CD62P (BD Biosciences, San Jose, CA, USA) to label active GPIIb/IIIa and P-selectin, respectively. Two 5 μL blood aliquots were obtained and labeled prior to perfusion. Platelet activation in one aliquot was achieved by the addition of thrombin before labeling (c = 0.1 unit mL−1, EMD Millipore, Billerica, MA, USA) and another aliquot was left unstimulated to serve as positive and negative activation controls, respectively. Blood was not treated with PPACK for thrombin-stimulated positive control. Following labeling, platelets were fixed in 1% paraformaldehyde and stored at 4 °C. Analysis of 100000 events was conducted on a FACScanto analyzer (BD Biosciences, San Jose, CA, USA).
Statistical analysis
All tests were carried out at least four replicates. Box and whisker plots presented the distribution of platelet adhesion density through their quartiles and outliers. Bar plots were presented as the mean ± standard deviation of platelet activation data. Statistical significance was established using paired t-test at p-values < 0.05. The significance level was presented as *p < 0.05, **p < 0.005, or ***p < 0.0005. Data were analyzed for significance with OriginPro 9.0 software (OriginLab, Northampton, MA, USA).Results
TxA2 inhibitor
For each increased upstream wall shear rate, two samples of anticoagulated whole blood were prepared. One sample was untreated, and another sample was treated with ASA. Fig. 2 shows the downstream platelet adhesion on three capture proteins. While the presence of ASA in the blood resulted in smaller platelet adhesion to fibrinogen and collagen, the number of adhered platelets did not significantly change in the presence of ASA at each upstream wall shear rate (p > 0.05). ASA showed no inhibitory effect on platelet adhesion to the vWf-coated capture region.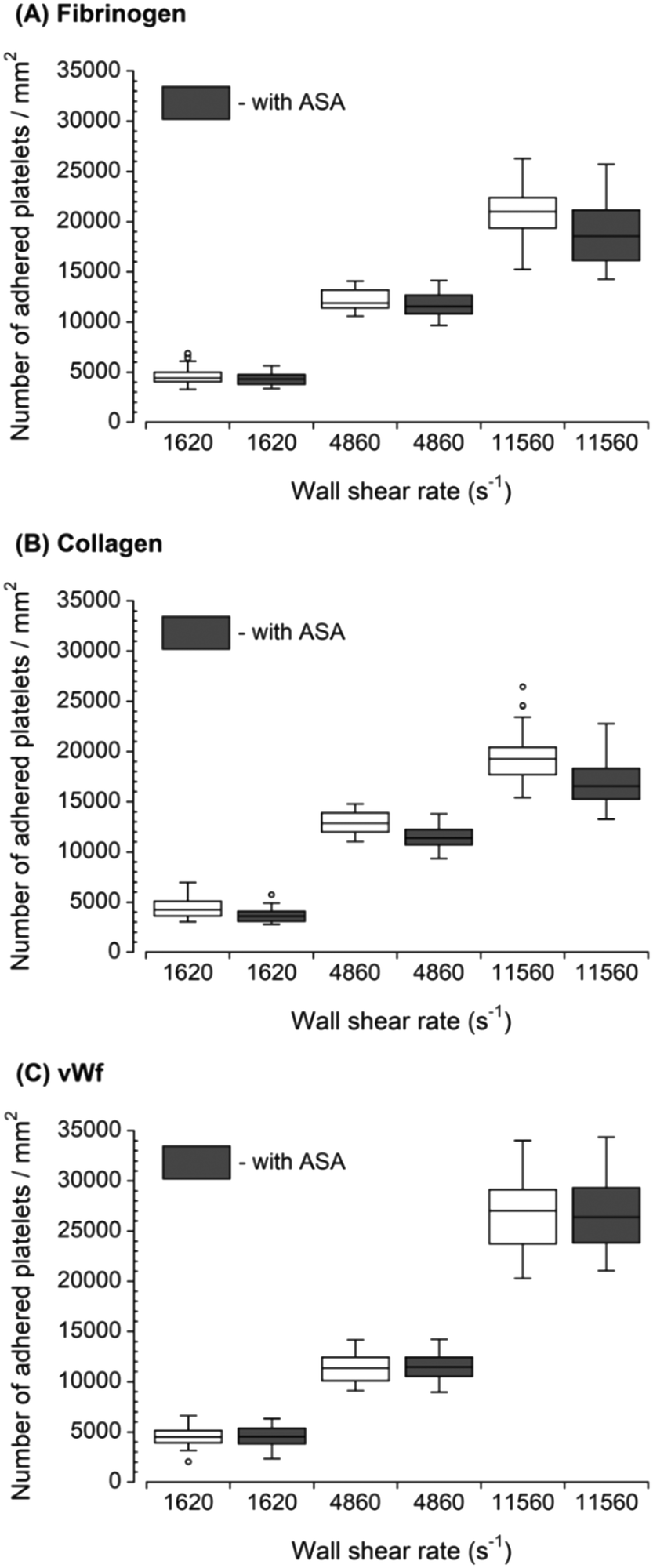 | ||
| Fig. 2 Platelet adhesion to three capture proteins placed downstream in the presence of ASA after transient exposure to a higher upstream wall shear rate. The 1620 s−1 flow channel contained no stenotic region thus serving as a control. The effect of ASA on downstream platelet adhesion was compared to control samples containing no ASA at each wall shear rate (n = 4 and p > 0.05). Total 40 images (10 images from each independent experiment) were analyzed for generating box and whisker plots. Platelet adhesion data for samples containing no ASA were adapted from Rahman and Hlady.20 | ||
GPIIb/IIIa inhibitors
Two GPIIb/IIIa inhibitors: tirofiban (a nonpeptide inhibitor) and eptifibatide (a cyclic heptapeptide), were used to inhibit platelet binding to the fibrinogen capture region after the blood was exposed to transiently increased wall shear rate (Fig. 3). Both eptifibatide and tirofiban at their recommended blood concentration effectively diminished, but did not fully eliminate the number of adhered platelets. The platelet adhesion was statistically significant compared to controls (i.e., samples containing no GPIIb/IIIa inhibitors in the blood) at each upstream wall shear rate. These results illustrated the ability of the assay to assess GPIIb/IIIa inhibitors at different shear conditions. Since tirofiban and eptifibatide are highly specific inhibitors of fibrinogen binding to GPIIb/IIIa receptors, platelet adhesion to collagen and vWf after the treatment of tirofiban and eptifibatide was not analyzed.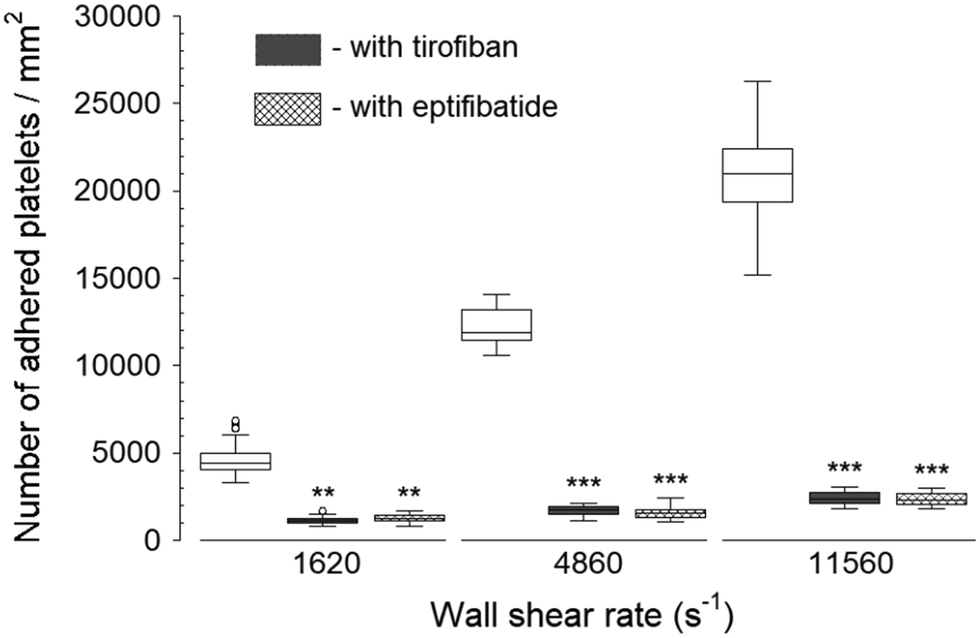 | ||
| Fig. 3 Platelet adhesion to fibrinogen capture region in the presence of GPIIb/IIIa inhibitors after transient exposure to a higher upstream wall shear rate. The 1620 s−1 flow channel contained no stenotic region thus serving as a control. Both tirofiban and eptifibatide significantly diminished the number of adhered platelets compared to control samples containing no GPIIb/IIIa inhibitors at each wall shear rate (n = 4, **p < 0.005, and ***p < 0.0005). Total 40 images (10 images from each independent experiment) were analyzed for generating box and whisker plots. Platelet adhesion data for samples containing no GPIIb/IIIa inhibitors were adapted from Rahman and Hlady.20 | ||
Anti-vWf and anti-GPIb
The inhibition of vWf–GPIbα binding by blocking of vWf or GPIbα prior to blood perfusion significantly diminished the platelet adhesion to all three capture proteins (Fig. 4 and 5). These results implied that antibody blocking of vWf or GPIbα prevented the activation of GPVI, GPIa/IIa, and GPIIb/IIIa that ultimately led to decreased platelet adhesion, not only to vWf but also to fibrinogen and collagen. For the vWf-coated capture region, blocking of vWf showed a relatively lower number of adhered platelets (Fig. 4) compared to blocking of GPIbα (Fig. 5), possibly due to the interactions of anti-vWF with both plasma vWf and surface-bound vWf.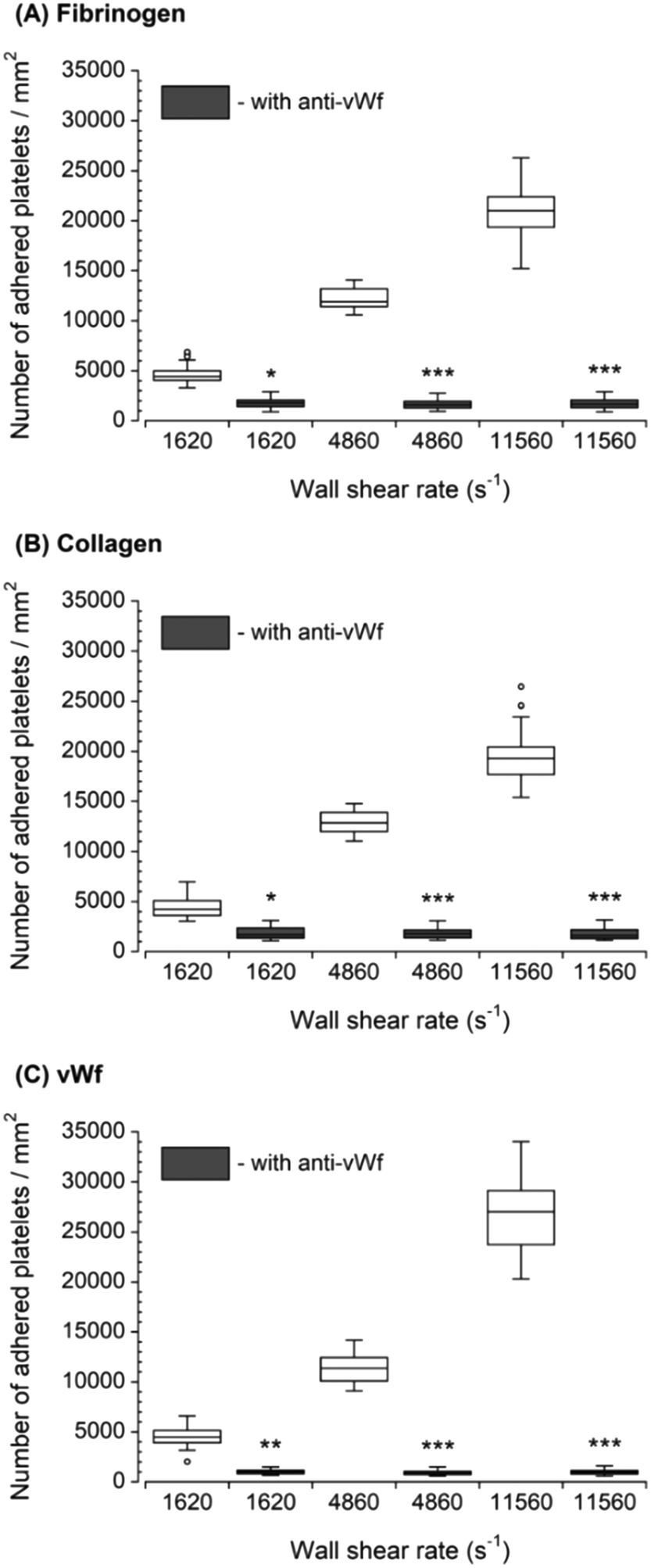 | ||
| Fig. 4 Platelet adhesion to three capture proteins in the presence of anti-vWf after transient exposure to a higher upstream wall shear rate. The 1620 s−1 flow channel contained no stenotic region thus serving as a control. Blocking vWf significantly diminished the number of platelets adhered to all three proteins compared to control samples containing no anti-vWf at each wall shear rate (n = 4, *p < 0.05, **p < 0.005, and ***p < 0.0005). Total 40 images (10 images from each independent experiment) were analyzed for generating box and whisker plots. Platelet adhesion data for samples containing no anti-vWf were adapted from Rahman and Hlady.20 | ||
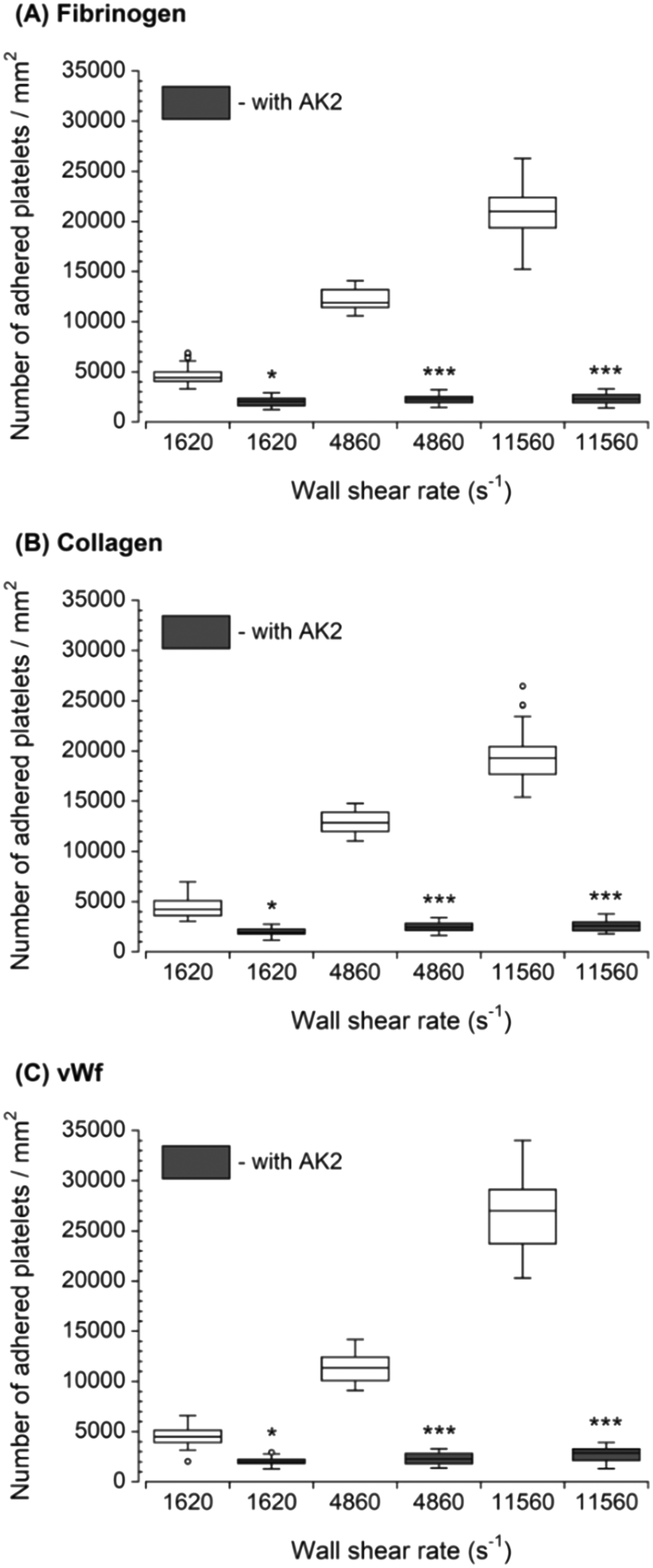 | ||
| Fig. 5 Platelet adhesion to three capture proteins in the presence of anti-GPIbα after transient exposure to a higher upstream wall shear rate. The 1620 s−1 flow channel contained no stenotic region thus serving as a control. Blocking GPIbα significantly diminished the number of platelets adhered to all three proteins compared to control samples containing no anti-GPIbα at each wall shear rate (n = 4, *p < 0.05, and ***p < 0.0005). Total 40 images (10 images from each independent experiment) were analyzed for generating box and whisker plots. Platelet adhesion data for samples containing no AK2 were adapted from Rahman and Hlady.20 | ||
Platelet activation after transient exposure to higher wall shear rates was also assessed quantitatively using flow cytometry. Fig. 6 shows the results of the flow cytometry analysis of whole blood perfusate. We previously found that platelet activation markers (GPIIb/IIIa and P-selectin) were significantly higher when platelets were transiently exposed to wall shear rates ranging from 2975 s−1 to 11560 s−1.20 Approximately 19% and 34% of platelets expressed GPIIb/IIIa at wall shear rates of 4860 s−1 and 11560 s−1, respectively. About 17% and 31% of platelets expressed P-selectin at wall shear rates of 4860 s−1 and 11560 s−1, respectively. Following the blocking of vWf or GPIbα, the present study showed that the expression levels of GPIIb/IIIa and P-selectin were only significantly higher compared to the negative control (i.e., blood with no prior perfusion) after platelets were exposed to 11560 s−1 upstream wall shear rate. However, in comparison to the perfused samples containing no anti-vWf and anti-GPIbα at each higher upstream wall shear rate (either 4860 s−1 or 11560 s−1), blocking of vWf or GPIbα significantly decreased the expression levels of GPIIb/IIIa and P-selectin (p < 0.005).
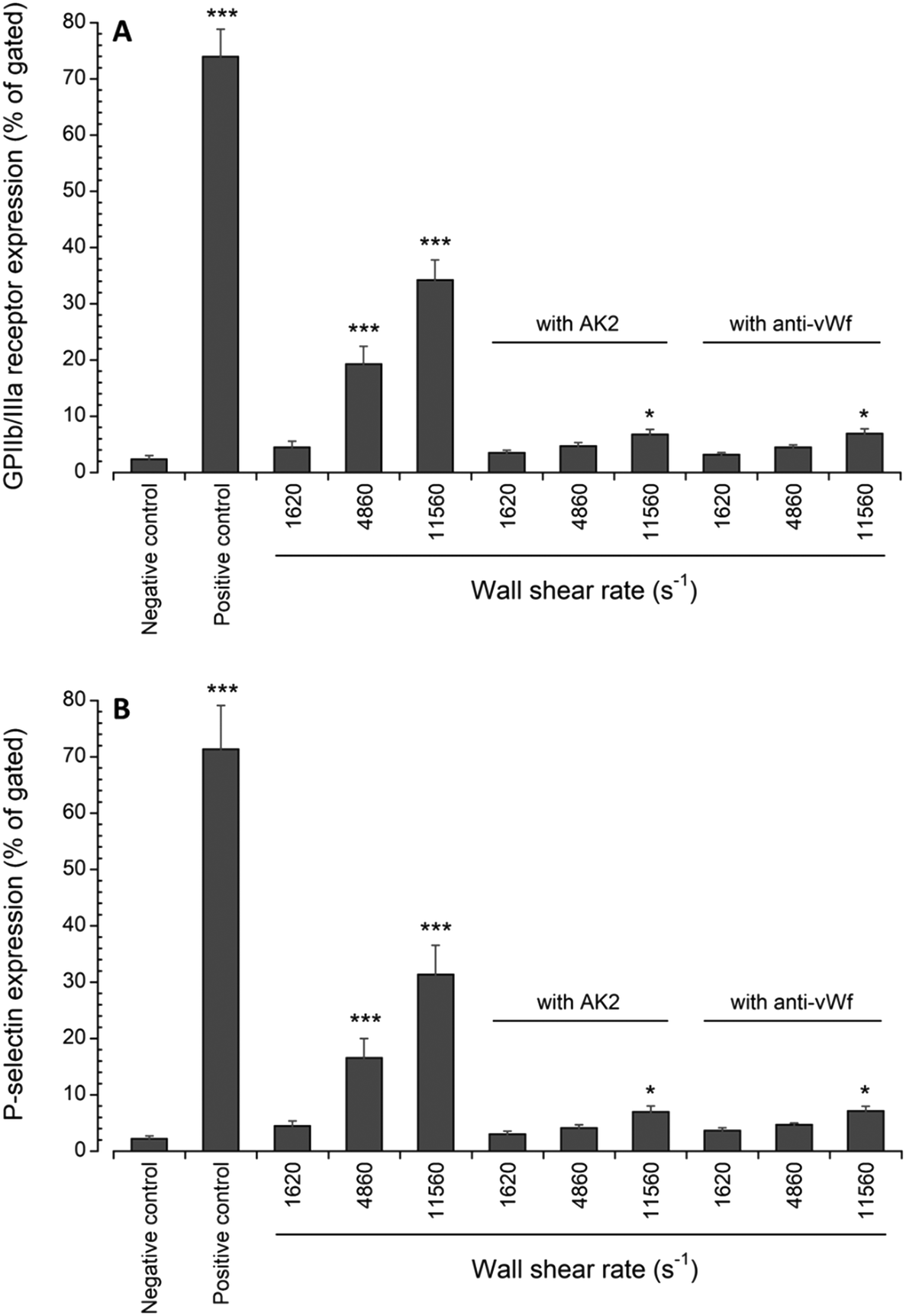 | ||
| Fig. 6 Expression levels of (A) active GPIIb/IIIa and (B) P-selectin in perfused blood samples compared with unstimulated (negative control) samples collected prior to perfusion (n = 4, *p < 0.05, and ***p < 0.0005). Flow cytometry data for samples containing no anti-vWf or AK2 were adapted from Rahman and Hlady.20 | ||
Discussion
Antithrombotic (anticoagulant and antiplatelet) agents are often used during and after the surgical introduction of a vascular device as well as for the treatment of atherosclerotic disease. A large proportion of the patients continue to receive these agents indefinitely due to the increased risk of thrombosis, which causes a considerable loss in quality of life. Shear-induced platelet adhesion and activation is an important mechanism of thrombosis at stenotic arterial regions. Platelet aggregation induced by a high shear force depends on the availability of vWf and the presence of both GPIbα and GPIIb/IIIa on the platelet surface.4,26 Several studies suggested that high shear forces initially promote the interaction between vWf and GPIbα,7,27 which is followed by a series of events such as Ca2+ mobilization,28 the release of serotonin,26,29 actin polymerization and cytoskeletal rearrangements,30,31 protein kinase C expression,32 and GPIIb/IIIa activation.6 The efficacy of antiplatelet agents under high shear conditions is not thoroughly understood. Commercially available platelet function assays often fail to mimic the hemodynamic environment that platelets experience upon passage through a stenotic vascular device or vessel. Following shear-induced intracellular signaling, in vitro evaluation of antiplatelet agents might be helpful to monitor their efficacy. In whole blood flow, platelets are marginated near the vessel wall, as expected from the Fahraeus–Lindqvist and Segre–Silberberg effects.33,34 Such platelet margination strongly depends on the wall shear rates.35,36 Hence, wall shear rates play an important role in influencing platelet function and formation of thrombi. Platelet margination also depends on hematocrit since platelets are smaller and fewer in number than red blood cells (RBCs). Elevated shear rates caused by stenotic regions have been reported to promote platelet margination to maintain the balance between hydrodynamic lift away from the wall and hydrodynamic pair collisions, which ultimately results in more platelets concentrated near the walls as well as increased thickness of the cell-depleted layer (i.e., a near-wall layer of blood plasma absents of RBCs).36,37 In the present study, platelets in whole blood with an antiplatelet agent were exposed to high shear in a stenotic region, and subsequently, downstream platelet response was measured under low shear (Fig. 1). This microfluidic flow system mimicked the transient flow acceleration and deceleration (i.e., fluid shear gradients) around the stenotic region that platelets usually experience in vivo. The downstream effects were measured by studying platelet adhesion and activation on three immobilized platelet binding proteins after treatment with several antiplatelet agents.TxA2 is an agonist released by activated platelets from their phospholipid membrane. TxA2 recruits additional platelets from the circulation to the adhesion site and amplifies platelet activation by working as a signal transducer for more potent agonists such as thrombin and ADP. Activation of the cyclooxygenase-1 (COX-1) results in the production of TxA2. ASA is a potent inhibitor of COX-1, which in turn blocks the production of TxA2 from the activated platelets. The results of the present study suggested that shear-induced platelet aggregation is independent of COX-1, and thus ASA-independent (Fig. 2). These findings supported the previously published observations, which documented that the treatment of platelets with ASA had no statistically significant inhibitory effect on platelet aggregation mediated by high shear forces.14,38–42 Ratnatunga et al. reported that high-dose ASA affects the platelet response to shear forces,43 which was not investigated in the present study. It was also reported that ASA effectively inhibits shear-induced platelet aggregation in dual antiplatelet therapy with dipyridamole and clopidogrel.12,44 However, the inhibitory mechanisms in these cases remain uncertain.
GPIIb/IIIa is a heterodimer complex formed by Ca2+-dependent association of an α-subunit (GPIIb) and a β-subunit (GPIIIa). In the resting platelets, GPIIb/IIIa exhibits low affinity to bind to its ligands (fibrinogen and vWf). Platelet activation induced by the agonist(s) triggers the inside-out signaling pathway for a conformational change in GPIIb/IIIa, leading to high-affinity binding sites for fibrinogen and vWf.45 The binding between fibrinogen and GPIIb/IIIa is one of the final common pathways for platelet aggregation. Tirofiban and eptifibatide inhibit platelet aggregation by preventing the binding between solution fibrinogen and GPIIb/IIIa on the platelet surface by inhibiting the GPIIb/IIIa receptor. We previously demonstrated that upstream wall shear rates of 4860 s−1 and 11560 s−1 significantly activate GPIIb/IIIa on the platelet surface, resulting in increased platelet adhesion to surface-bound fibrinogen downstream.20 We also found that the wall shear rate of 1620 s−1 is capable of increasing GPIIb/IIIa activation; however, the level of activation was statistically insignificant. The platelet adhesion results of the present study showed that tirofiban and eptifibatide effectively prevented the binding between surface-immobilized fibrinogen and platelet GPIIb/IIIa (Fig. 3). The results also showed that tirofiban and eptifibatide achieved similar levels of inhibition. Abciximab (monoclonal antibody) is another commonly prescribed GPIIb/IIIa inhibitor, which was not tested in this study. Neumann et al. reported that abciximab, tirofiban, and eptifibatide achieve similar levels of inhibition of platelet aggregation at their recommended blood concentration.46
Since it has been shown that high shear forces initially promote the binding between vWf and GPIbα,7,27 this binding could exert control over fibrinogen–GPIIb/IIIa, collagen–GPVI, and collagen–GPIa/IIa interactions. However, there is no commercially available prescribed drug to inhibit the binding between vWf and GPIbα. We tested how anti-vWf (polyclonal antibody) and AK2 (monoclonal antibody) inhibit the binding between vWf and GPIbα. It was previously found that anti-vWf and AK2 can effectively block vWf and GPIbα, respectively.47,48 After blocking either vWf or GPIbα, we assessed platelet adhesion on all three immobilized platelet binding proteins to identify the role of vWf–GPIbα interactions in promoting downstream thrombus formation. Antibody blocking of vWf or GPIbα significantly inhibited upstream shear-induced platelet adhesion downstream (Fig. 4 and 5). The results of such blocking supported previous findings,26,49,50 which suggested that vWf binding to GPIbα is an essential event for leading to irreversible platelet adhesion, following by GPIIb/IIIa activation, platelet aggregation, and subsequent thrombus formation.
To determine the effect of blocking vWf or GPIbα on downstream platelet activation, GPIIb/IIIa and P-selectin expression upon the exposure to higher shear forces were analyzed in the perfused blood using flow cytometry. These platelet activation results did not fully mirror the platelet adhesion results. While blocking of vWf or GPIbα significantly diminished platelet adhesion to all three capture proteins for all three upstream wall shear rates, platelet expressions of GPIIb/IIIa and P-selectin were significantly higher after the transient exposure to 11560 s−1 compared to the negative control (Fig. 6). We previously showed that wall shear rates of 4860 s−1 and 11560 s−1 can induce platelet lysis; however, only transient exposure to 11560 s−1 resulted in statistically significant platelet lysis.20 The present study results imply that the blocking of vWf or GPIbα would not prevent platelet activation induced by platelet lysis. Another difference between platelet activation and adhesion results could also be due to flow cytometry protocol which required 20 min of incubation time of perfused blood with platelet activation markers. This incubation time was significantly longer than the time blood spent flowing in the microfluidic channels. Hence, it was not possible to measure the expression levels of GPIIb/IIIa and P-selectin immediately after blood perfusion. Hence, it is possible that the longer incubation time of flow cytometry might have led to an increased expression of GPIIb/IIIa and P-selectin. We need to point out that platelet lysis caused by a high shear rate of 11560 s−1 may have a dual role in mediating downstream platelet adhesion. Platelet lysis may cause the release of procoagulant factors (e.g., ADP), which stimulate further platelet activation and promote the recruitment of additional platelets to the adhesion site. Simultaneously platelet lysis caused by upstream stenosis may result in a reduced number of activated platelets available for adhesion to the downstream capture region.
There are some limitations of the microfluidic system used in the present study. We designed 3-D microchannels to simulate blood flow in stenotic vascular devices or vessels. The use of rectangular microchannels facilitated the immobilization of platelet binding proteins on a reactive glass substrate to create downstream platelet capture regions. However, vascular devices or vessels are cylindrical in vivo. Roughly 6 × 108 platelets were perfused for each single experiment. Flow cytometry analysis determined that approximately 1.14 × 108 and 2.04 × 108 platelets expressed GPIIb/IIIa, of which approximately 1.2 × 105 and 2.1 × 105 platelets adhered to the fibrinogen-coated downstream capture region at wall shear rates of 4860 s−1 and 11560 s−1, respectively. The number of platelets adhered to the downstream capture region was significantly lower compared to the number of activated platelets. These results indicated that a large fraction of platelets activated by upstream stenosis did not adhere to the downstream capture region. However, the results demonstrated that downstream platelet adhesion increased with the increase of the wall shear rate in the upstream stenotic region. Our previous study indicated that the activation of fibrinogen receptor GPIIb/IIIa by transient exposure to high upstream wall shear rates progresses in a time-dependent manner during the downstream flow of platelets.21 As mentioned earlier, platelet activation in perfused blood was measured using flow cytometry, which required 20 min of incubation with platelet activation markers. Because of the limitation of flow cytometry, it was not possible to measure the percent of activated platelets immediately after the blood perfusion. Thus, it was not possible to determine exactly what percentage of activated platelets adhered to the downstream capture region.
Since platelet activation is a time-dependent process, the downstream platelet adhesion could be dependent on the time duration of platelet exposure to upstream elevated shear forces. In this study, blood was perfused through the microchannels at a constant flow rate of 0.5 mL min−1. The width of the stenotic region was selected to create two different elevated shear forces. Thus, the traveling time of platelets through the stenotic region was varied with the width. The results demonstrated that the traveling time decreased with increasing shear forces in the stenotic region, which might have an effect on platelet adhesion to the downstream capture region. It could be possible to expose the platelets to a constant traveling time through the stenotic region by adjusting the length of the stenotic region. Following exposure to upstream stenosis, platelets traveled the distance between the upstream stenotic region and the downstream capture region at a wall shear rate of 1620 s−1. The traveling time for this distance and wall shear rate of 1620 s−1 may have some effects on platelet adhesion to the downstream capture region, which was not evaluated in this study. Non-uniform shear distribution in a stenotic microchannel causes a higher state of platelet priming near the wall, whereas many platelets away from the wall may not be primed. Thus, it is difficult to correlate the number of platelets primed by upstream stenosis with the number of platelets adhered to the downstream capture region.
The evaluation of antiplatelet agents is essential for the management of patients with implanted vascular devices or treated for atherosclerotic disease.51 With the advent of newer antiplatelet agents, the selection of appropriate antiplatelet agents remains challenging. Shear-induced platelet aggregation is a common pathway for thrombus formation.52,53 The results of the present study demonstrated that the microfluidic assay could be applied to assess the efficacy of antiplatelet agents on platelet function following transient exposure to various higher wall shear rates. The assay is a single-pass perfusion system that requires a small volume of blood and permits the study of platelets in the presence of other blood elements. The present assay could be utilized to study the efficacy of ADP receptor inhibitors as ADP is known to contribute to shear-induced platelet aggregation.54 However, the majority of available ADP receptor inhibitors require metabolic activation as they are available as prodrugs, making them challenging for similar in vitro studies. The present assay could also be utilized to study how effective an antiplatelet agent is in suppressing multiple agonist pathways as well as to explore how effective a combination of antiplatelet agents could be in suppressing a single agonist pathway. In the present study, only two elevated shear forces (4860 s−1 and 11560 s−1) were studied. Additional shear forces relevant to mild to severe stenosis could be included for further assessment. Although GPIIb/IIIa is a common receptor for both fibrinogen and vWf, platelet adhesion to vWf in the presence of GPIIb/IIIa inhibitors was not assessed. The assessment of platelet adhesion to vWf in the presence of GPIIb/IIIa inhibitors could provide additional insights into the mechanisms of shear-induced platelet aggregation.
Conclusions
The present study demonstrated the ability of a microfluidic assay to screen several selected antiplatelet agents under different wall shear rates spanning from physiological to pathological level. While ASA showed little or no effect, GPIIb/IIIa inhibitors were found to be very effective against shear-induced platelet adhesion. Antibody blocking of vWf or GPIbα revealed that the binding between vWf and GPIbα is a crucial event for downstream adhesion and activation after exposure to an upstream high shear force. These results have some implications for minimizing the thrombotic risk associated with high shear forces. The results are also relevant to understanding the mechanisms of thrombus formation at the post-stenotic regions of vascular devices or vessels. The findings could help assess the hemocompatibility of biomaterials as well as for the management of patients with atherosclerotic disease.Author contributions
S. Rahman conducted the research, analyzed the data, and prepared the draft of the manuscript. V. Hlady designed the research, interpreted the data, and critically revised the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by the National Institutes of Health (R01 HL126864). The authors would like to thank Dr. A. Fogelson for valuable discussions and Dr. Robert Campbell from Weyrich's lab for providing human blood necessary to complete the experiments.References
- S. H. Swedberg, B. G. Brown, R. Sigley, T. N. Wight, D. Gordon and S. C. Nicholls, Circulation, 1989, 80, 1726–1736 CrossRef CAS.
- A. H. Davis, T. R. Magee, R. N. Baird, E. Sheffield and M. Horrocks, Br. J. Surg., 1992, 79, 1019–1021 CrossRef.
- Y. G. Wilson, A. H. Davies, I. C. Currie, M. Morgan, C. McGrath, R. N. Baird and P. M. Lamont, Eur. J. Vasc. Endovasc. Surg., 1996, 11, 164–169 CrossRef CAS.
- E. Westein, A. D. van der Meer, M. J. Kuijpers, J. P. Frimat, A. van den Berg and J. W. Heemskerk, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1357–1362 CrossRef CAS.
- K. S. Sakariassen, L. Orning and V. T. Turitto, Future Sci. OA, 2015, 1, FSO30 Search PubMed.
- Z. Chen, N. K. Mondal, J. Ding, S. C. Koenig, M. S. Slaughter, B. P. Griffith and Z. J. Wu, Mol. Cell. Biochem., 2015, 409, 93–101 CrossRef CAS.
- W. Deng, Y. Xu, W. Chen, D. S. Paul, A. K. Syed, M. A. Dragovich, X. Liang, P. Zakas, M. C. Berndt, J. D. Paola, J. Ware, F. Lanza, C. B. Doering, W. Bergmeier, X. F. Zhang and R. Li, Nat. Commun., 2016, 7, 12863 CrossRef CAS.
- J. L. Moake, N. A. Turner, N. A. Stathopoulos, L. Nolasco and J. D. Hellums, Blood, 1988, 71, 1366–1374 CrossRef CAS.
- S. K. Kundu, E. J. Heilmann, R. Sio, C. Garcia and R. A. Ostgaard, Clin. Appl. Thromb./Hemostasis, 1996, 2, 241–249 CrossRef.
- N. A. Turner, J. L. Moake and L. V. McIntire, Blood, 2001, 98, 3340–3345 CrossRef CAS.
- J. H. Haga, L. K. Jennings and S. M. Slack, Ann. Biomed. Eng., 2002, 30, 1262–1272 CrossRef.
- T. Nakamura, S. Uchiyama, M. Yamazaki and M. Iwata, Cerebrovasc. Dis., 2002, 14, 234–238 CrossRef CAS.
- J. C. Resendiz, S. Feng, G. Ji, K. A. Francis, M. C. Berndt and M. H. Kroll, Mol. Pharmacol., 2003, 63, 639–645 CrossRef CAS.
- P. A. Holme, U. Orvim, M. J. Hamers, N. O. Solum, F. R. Brosstad, R. M. Barstad and K. S. Sakariassen, Arterioscler., Thromb., Vasc. Biol., 1997, 17, 646–653 CrossRef CAS.
- W. S. Nesbitt, E. Westein, F. J. Tovar-Lopez, E. Tolouei, A. Mitchell, J. Fu, J. Carberry, A. Fouras and S. P. Jackson, Nat. Med., 2009, 15, 665–673 CrossRef CAS.
- M. Li, D. N. Ku and C. R. Forest, Lab Chip, 2012, 12, 1355–1362 RSC.
- T. V. Colace and S. L. Diamond, Arterioscler., Thromb., Vasc. Biol., 2013, 33, 105–113 CrossRef CAS.
- H. Ha and S. J. Lee, Microvasc. Res., 2013, 90, 96–105 CrossRef.
- S. M. Rahman, C. D. Eichinger and V. Hlady, Acta Biomater., 2018, 73, 228–235 CrossRef.
- S. M. Rahman and V. Hlady, Acta Biomater., 2019, 91, 135–143 CrossRef CAS.
- S. Rahman, A. Fogelson and V. Hlady, Colloids Surf., B, 2020, 193, 111118 CrossRef CAS.
- S. Goto, H. Sakai, M. Goto, M. Ono, Y. Ikeda, S. Handa and Z. M. Ruggeri, Circulation, 1999, 99, 608–613 CrossRef CAS.
- N. Ajzenberg, P. Aubry, M. G. Huisse, A. Cachier, W. E. Amara, L. J. Feldman, D. Himbert, D. Baruch, M. C. Guillin and P. G. Steg, J. Am. Coll. Cardiol., 2005, 45, 1753–1756 CrossRef.
- L. E. Corum, C. D. Eichinger, T. W. Hsiao and V. Hlady, Langmuir, 2011, 27, 8316–8322 CrossRef CAS.
- G. M. Harbers, K. Emoto, C. Greef, S. W. Metzger, H. N. Woodward, J. J. Mascali, D. W. Grainger and M. J. Lochhead, Chem. Mater., 2007, 19, 4405–4414 CrossRef CAS.
- D. M. Peterson, N. A. Stathopoulos, T. D. Giorgio, J. D. Hellums and J. L. Moake, Shear-induced platelet aggregation requires von Willebrand factor and platelet membrane glycoproteins Ib and IIb-IIIa, Blood, 1987, 69, 625–628 CrossRef CAS.
- A. J. Reininger, H. F. Heijnen, H. Schumann, H. M. Specht, W. Schramm and Z. M. Ruggeri, Blood, 2006, 107, 3537–3545 CrossRef CAS.
- T. W. Chow, J. D. Hellums, J. L. Moake and M. H. Kroll, Blood, 1992, 80, 113–120 CrossRef CAS.
- G. H. Anderson, J. D. Hellums, J. Moake and C. P. Alfrey, Thromb. Res., 1978, 13, 1039–1047 CrossRef CAS.
- Y. Yuan, S. Kulkarni, P. Ulsemer, S. L. Cranmer, C. L. Yap, W. S. Nesbitt WS, I. Harper, N. Mistry, S. M. Dopheide, S. C. Hughan, D. Williamson, C. de la Salle, H. H. Salem, F. Lanza and S. P. Jackson, J. Biol. Chem., 1999, 274, 36241–36251 CrossRef CAS.
- M. J. Maxwell, S. M. Dopheide, S. J. Turner and S. P. Jackson, Arterioscler., Thromb., Vasc. Biol., 2006, 26, 663–669 CrossRef CAS.
- M. H. Kroll, J. D. Hellums, Z. Guo, W. Durante, K. Razdan, J. K. Hrbolich and A. I. Schafer, J. Biol. Chem., 1993, 268, 3520–3524 CAS.
- R. Fahraeus and T. Lindqvist, Am. J. Physiol., 1931, 96, 562–568 CrossRef CAS.
- G. Segre and A. Silberberg, Nature, 1961, 189, 209–210 CrossRef.
- A. W. Tilles and E. C. Eckstein, Microvasc. Res., 1987, 33, 211–223 CrossRef CAS.
- P. A. Aarts, S. A. van den Broek, G. W. Prins, G. D. Kuiken, J. J. Sixma and R. M. Heethaar, Arteriosclerosis, 1988, 8, 819–824 CAS.
- A. Yazdani and G. E. Karniadakis, Soft Matter, 2016, 12, 4339–4351 RSC.
- J. R. O'Brien and G. P. Salmon, Blood, 1987, 70, 1354–1361 CrossRef.
- J. R. O'Brien, Lancet, 1990, 335, 711–713 CrossRef.
- K. Konstantopoulos, K. K. Wu, M. M. Udden, E. I. Banez, S. J. Shattil and J. D. Hellums, Biorheology, 1995, 32, 73–93 CAS.
- Y. Miyazaki, S. Nomura, T. Miyake, H. Kagawa, C. Kitada, H. Taniguchi, Y. Komiyama, Y. Fujimura, Y. Ikeda and S. Fukuhara, Blood, 1996, 88, 3456–3464 CrossRef CAS.
- E. D. Michos, R. Ardehali, R. S. Blumenthal, R. A. Lange and H. Ardehali, Mayo Clin. Proc., 2006, 81, 518–526 CrossRef CAS.
- C. P. Ratnatunga, S. F. Edmondson, G. M. Rees and I. B. Kovacs, Circulation, 1992, 85, 1077–1082 CrossRef CAS.
- R. R. Makkar, N. L. Eigler, S. Kaul, A. Frimerman, M. Nakamura, P. K. Shah, J. S. Forrester, J. M. Herbert and F. Litvack, Eur. Heart J., 1998, 19, 1538–1546 CrossRef CAS.
- B. Nieswandt, D. Varga-Szabo and M. Elvers, J. Thromb. Haemostasis, 2009, 7, 206–209 CrossRef CAS.
- F. J. Neumann, W. Hochholzer, G. Pogatsa-Murray, A. Schomig and M. Gawaz, J. Am. Coll. Cardiol., 2001, 37, 1323–1328 CrossRef CAS.
- K. M. Dayananda, I. Singh, N. Mondal and S. Neelamegham, Blood, 2010, 116, 3990–3998 CrossRef CAS.
- C. D. Eichinger and V. Hlady, Biointerphases, 2017, 12, 02C406 CrossRef.
- M. Sugimoto, S. Tsuji, M. Kuwahara and H. Matsui, Int. J. Hematol., 1999, 69, 48–53 CAS.
- K. Tomokiyo, Y. Kamikubo, T. Hanada, T. Araki, Y. Nakatomi, Y. Ogata, S. M. Jung, T. Nakagaki and M. Moroi, Blood, 2005, 3, 1078–1084 CrossRef.
- A. Rana, E. Westein, B. Niego and C. E. Hagemeyer, Front. Cardiovasc. Med., 2019, 6, 141 CrossRef CAS.
- D. M. Pushin, T. Y. Salikhova, K. S. Zlobina and G. T. Guria, PLoS One, 2020, 15, e0234501 CrossRef CAS.
- S. Xu, J. Piao, B. Lee, C. Lim and S. Shin, Biosens. Bioelectron., 2020, 165, 112395 CrossRef CAS.
- M. Cattaneo, M. L. Zighetti and R. Lombardi, Br. J. Haematol., 1994, 88, 826–829 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00756k |
| This journal is © The Royal Society of Chemistry 2021 |