
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[24]Crown-8-modified carbon nanotubes for templating metal deposition and active materials for pseudocapacitors†
Ruitao
Zhou‡
a,
Fung-Kit
Tang‡
a,
Raymond Wai-Yin
Sun
b,
Man Kin
Tse
b,
Yujie
Chen
b,
Albert Sun Chi
Chan
b,
Shengzhou
Chen
 c,
Xunjin
Zhu
*a and
Ken Cham-Fai
Leung
*a
c,
Xunjin
Zhu
*a and
Ken Cham-Fai
Leung
*a
aDepartment of Chemistry, The Hong Kong Baptist University, Kowloon Tong, Kowloon, Hong Kong SAR, P. R. China. E-mail: xjzhu@hkbu.edu.hk; cfleung@hkbu.edu.hk
bGuangzhou Lee & Man Technology Company Limited, 8 Huanshi Avenue, Nansha, Guangzhou, P. R. China
cSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou, P. R. China
First published on 26th October 2020
Abstract
A uniform nickel film could be coated on crown ether-modified carbon nanotubes (c-CNTs) by electroless deposition. The products CNT@Ni and c-CNT@Ni were oxidized and compressed into CNT@Ni@(NiOH)2 and c-CNT@Ni@(NiOH)2 discs, respectively. The areal and volumetric capacitances of CNT@Ni@(NiOH)2 and c-CNT@Ni@(NiOH)2 were compared and c-CNT@Ni@(NiOH)2 showed a lower resistance.
A hybrid between the lithium ion battery1 and the supercapacitor is known as a pseudocapacitor, which has a high theoretical specific capacitance. Most of them are made of metal oxides but their reported capacitances are often much lower than their theoretical values because of two reasons: (1) the low conductivity of metal oxides and (2) the low ion diffusion rates of electrolyte inside the metal oxides during the charge–discharge process. To improve the ion diffusion and electron transmission in the pseudocapacitor, one strategy is shortening the electron and ion transfer pathway by synthesizing metal oxides at the nanoscale, such as nanoparticles2 and nanoplates.3 Nanoarchitectured metal oxides can be combined mechanically with current collectors to form electrodes, but the contact interface has a high resistance and the resulting specific capacitance is not satisfactory for application. Another method to improve the contact between metal oxides and current collectors is synthesizing the metal oxides on the current collectors directly. Carbon fabrics and metal foams with a large specific surface area have been widely used as both substrates and current collectors.4 However, the thickness of the metal oxide layer is on the nanoscale and the mass loading of metal oxides on current collectors is low (usually less than 1 mg cm−2). The areal and volumetric capacitances of electrodes are more important parameters in the evaluation of electrochemical performance of the active materials than the specific capacitances.
In order to synthesize low cost electrodes with both a high areal capacitance and volumetric capacitance, herein, nanoporous nickel with a Ni(OH)2 shell and nanoporous nickel–iron with an Fe2O3 shell were synthesized. Nanoporous metal networks were fabricated by stacking metal nanotubes together and fusing them at high temperature, while the metal nanotubes were synthesized by coating Ni and Fe on CNTs and crown ether-modified CNTs (c-CNTs) through an electroless deposition (ELD) process. The resulting surface oxidized nanoporous metal network can function as a high performance freestanding electrode itself. As compared to the reported metal oxide based electrode materials, for example, CNT@Ni(OH)2,5 CNT@Ni@Ni2(CO3)(OH)26 nanotubes and Ni fiber@Ni(OH)2,7 this work presented nanoporous architectures with high conductivities and large specific surface areas. Hence, high areal and volumetric capacitances can be achieved using our new CNT@Ni@Ni(OH)2, c-CNT@Ni@Ni(OH)2 and CNT@Ni–Fe@Fe2O3 materials.
There are several factors that determine the capacitances of materials: mass loading, resistance (contact between the active materials and current collectors), scalability and the nanoporous structures. A modified ELD method was employed to deposit nickel on the substrate of multi-walled carbon nanotubes (MWCNTs).8 It is also worth noting that MWCNTs are readily available commercially in large quantities with relatively inexpensive prices, therfore the cost of production can be further reduced for industrial application.9
The synthesis of the amine-functionalized crown ether (DB24C8-CH2NH2) was achieved through a new synthetic pathway (Fig. 1B). Starting with diol 1, ditosylation yielded ditosylate 2 in 67% yield. Then, ditosylate 2 was macrocyclized with 3,4-dihydroxybenzonitrile using a cesium ion template to yield macrocycle 3 in 68% yield. The cyano group of macrocycle 3 was reduced into the product DB24C8-CH2NH2 by LiAlH4 in 41% yield (Fig. S1–S6, ESI†). Then, the surface of the CNTs was oxidized in acidic medium, and both crown ether modified CNTs (c-CNTs) and pristine oxidized CNTs were used as substrates to initiate ELD of Ni using a silver nitrate catalyst (Fig. 1A and Fig. S7, ESI†). The carboxyl groups on CNTs were activated by SOCl2 and then subsequently reacted with the crown ether DB24C8-CH2NH2 to yield c-CNTs through amide bonds. The crown ether modification on the CNTs was further confirmed by XPS analysis (Fig. S8, ESI†).
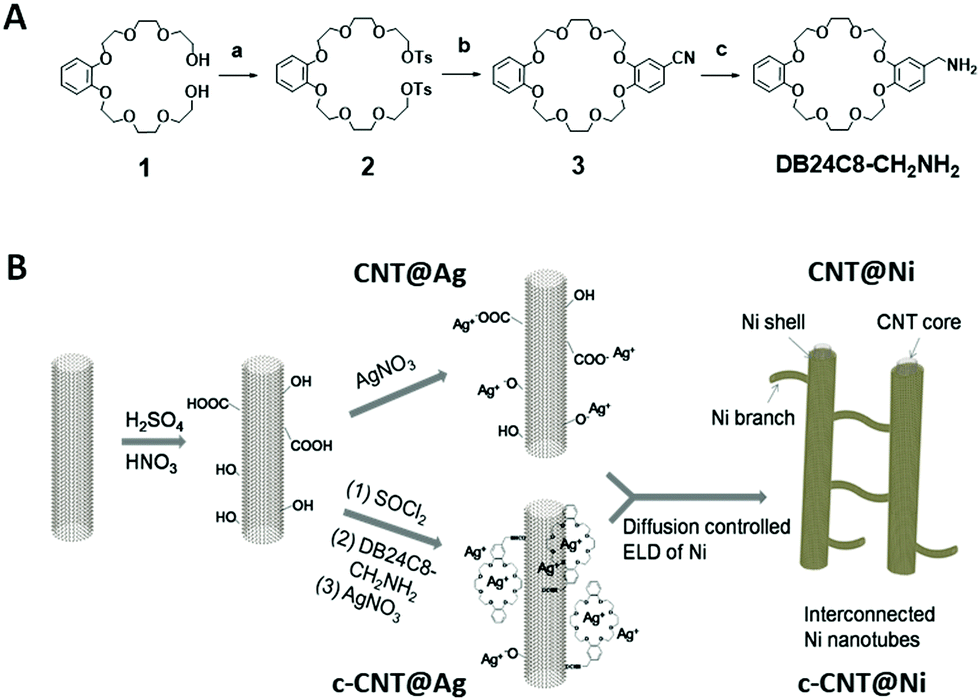 | ||
| Fig. 1 (A) Synthetic scheme of dibenzo[24]crown-8-CH2NH2 (DB24C8-CH2NH2), conditions: (a) TsCl, NEt3, DMAP, r.t., CH2Cl2, 24 h, 67%; (b) 3,4-dihydroxybenzonitrile, Cs2CO3, K2CO3, MeCN, reflux, 24 h, 68%; (c) LiAlH4, THF, reflux, overnight, 41%. (B) Synthetic process of interconnected CNT@Ni and crown ether-based c-CNT@Ni via CNT@Ag and c-CNT@Ag by electroless deposition (ELD). | ||
During the ELD process, Ag+ acts as the catalytic site for Ni adhesion, and the use of a crown ether as an adsorbent for Ag+ ions has been reported,10 hence c-CNTs could provide anchoring points for Ag+ and a more uniform metal coating could be formed. The adsorption of Ag+ on c-CNTs was confirmed by EDX characterization (Fig. S9, ESI†). For the pristine CNTs, the absorption of silver ions was characterized by XPS; two peaks with the binding energy of 368.4 eV and 374.6 eV were observed, corresponding to Ag 3d5/2 and Ag 3d3/2 respectively (Fig. S11a, ESI†). Also, Ag+ has a lower catalytic rate for ELD compared to that of palladium,11 which is advantageous for controlling the mass loading and morphology of nickel deposited on the CNTs, such as by tuning the amount of reducing agent dimethylamine borane (DMAB) and reaction time. The homogeneous deposition of nickel in the bulk solution was diminished with the use of Ag+ and c-CNTs.
The ELD process was carried out with the assistance of ultrasonication (45 kHz) for dispersion. Through the adjustment of the concentration of the reducing agent (DMAB) and pH of the solution, nickel with different morphological structures was synthesized on the CNTs, such as separated nickel islands, connected nickel islands, continuous nickel films and interconnected CNT@Ni with side nickel branches (Fig. 2a–d). Notably, with crown ether modification, the nickel coating on c-CNT@Ni is more evenly distributed and smoother than that of CNT@Ni (Fig. 2e–g), and interconnected branches were arranged in a more uniform way (Fig. 2d and h). Without ultrasound, the branches could not be formed to connect the CNT@Ni and only a thicker metal deposit was observed (Fig. S10, ESI†). This is possibly due to the interference of diffusion rates of Ni2+ for the growth of branches during the ELD process. The nickel coating on CNT@Ni was characterized by XPS (Fig. S11b, ESI†) and XRD (Fig. S11c, ESI†). The resulting CNT@Ni and c-CNT@Ni were tested as positive electrodes in aqueous NaOH electrolyte. Subsequently, the Ni surface was oxidized in situ to form the active Ni(OH)2 materials on the electrodes after the first charging–discharging process (Fig. S7, ESI†).
 | ||
| Fig. 2 TEM images of (a) nickel islands on CNTs; (b) connected Ni islands on CNTs; (c) continuous Ni film on CNTs; (d) interconnected CNT@Ni with side branches; (e–h) interconnected c-CNT@Ni with side branches. | ||
For electrochemical studies, the CNT@Ni material was then fabricated into the area of a 1 cm2 disc as a working electrode. A piece of nickel cloth was fabricated using a reported method12 and directly used as the current collector. The CNT@Ni film had a high specific surface area of 61 m2 g−1 based on the Brunauer–Emmett–Teller analysis, and such a highly porous structure can facilitate the ion transfer between the electrolyte and the electrode (Fig. S12a and b, ESI†). The internal resistance of the porous CNT@Ni film is about 2.4 Ω, which is estimated from the x-intercept of the electrochemical impedance spectroscopy (EIS) curve (Fig. S11d, ESI†). By galvanostatic charge–discharge (GCD) testing, the areal capacitances of the electrode were found to be 34.2, 34.0, 33.9, 32.6 and 22.5 F cm−2 at the current density of 5, 7.5, 10, 15 and 20 mA cm−2, respectively (Fig. S12f, ESI†). The presence of plateaus in the discharging curves of GCD and redox peak pairs in cyclic voltammetry (CV) curves suggest that the capacitance of the porous CNT@Ni film follows a faradaic mechanism (Fig. S12e and f, ESI†). The faradaic capacitance of the electrode originated from the nickel hydroxide layer formed on the surface of CNT@Ni during the charge–discharge process, because Ni can be oxidized into Ni(OH)2 in alkaline solution.13
The oxidation of CNT@Ni was also characterized by XRD (Fig. S11c, ESI†). In the VSM test (Fig. S11d and S13a, ESI†), the magnetic moments of the electrodes decreased after electrochemical testing because of a gradual oxidation on the Ni layer. The oxidation of Ni was fast in the first charge–discharge cycle, and 5.6% Ni was converted into Ni(OH)2. After optimization, the CNT@Ni freestanding electrode was shaped in a round plate (inset of Fig. 3c) with a thickness of 0.253 mm whereas CNT@Ni has a high density of 2.97 g cm−3.
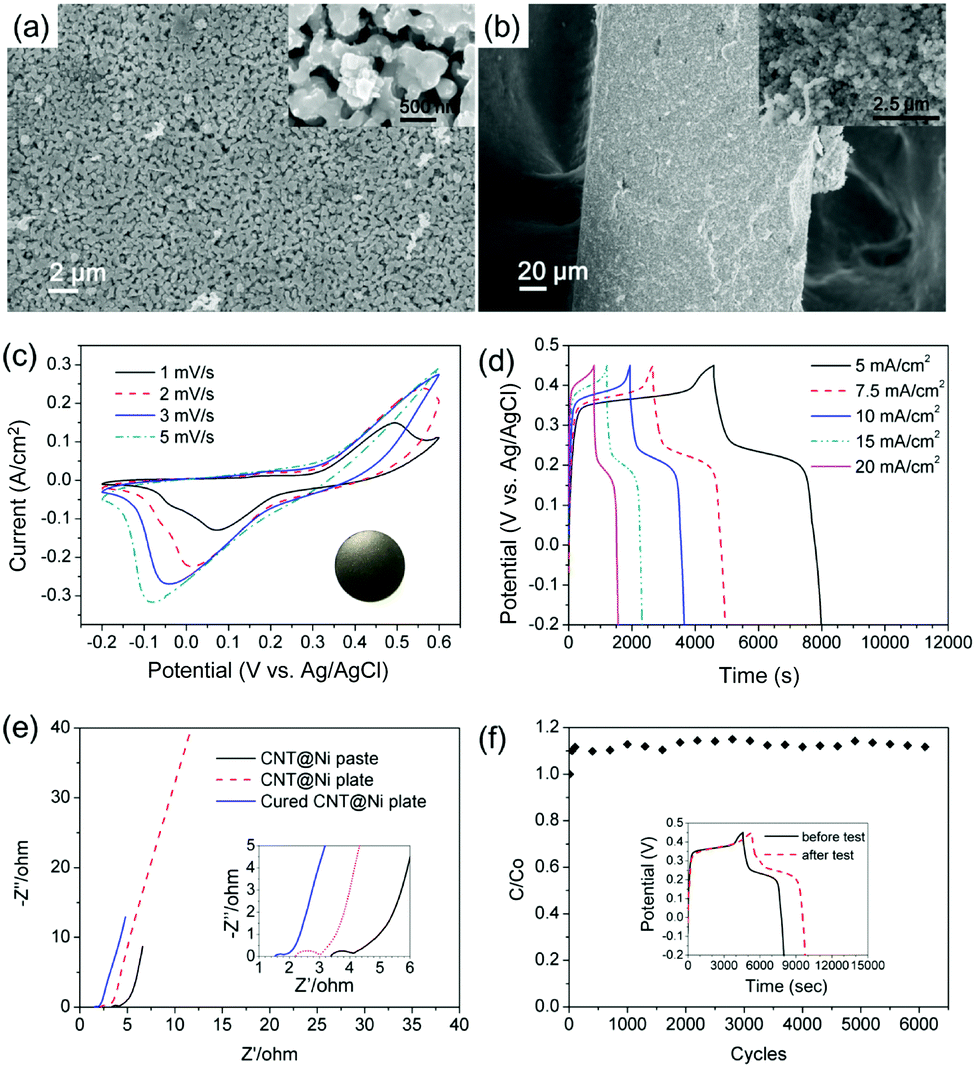 | ||
| Fig. 3 (a and b) SEM images of the CNT@Ni plate (a, front surface; b, section), (c) CV curves of the CNT@Ni plate (inset: photo of a CNT@Ni plate), (d) GCD curves of the c-CNT@Ni plate, (e) electrochemical impedance spectra of CNT@Ni; (f) long cycle test of CNT@Ni by cyclic voltammetry at 50 mV s−1 (inset: GCD curves at 5 mA cm−2 before and after the long cycle test). | ||
Compared to the CNT@Ni film fabricated by filtration, the optimized CNT@Ni plate had smaller pores with sizes of around several hundred nanometers (Fig. 3a and b). In the GCD test, the areal capacitances of the CNT@Ni electrode were 27.7, 28.9, 30.1, 31.5 and 31.2 F cm−2 at the current density of 5, 7.5, 10, 15 and 20 mA cm−2, respectively (Fig. 3d). The increase in both capacitance and current densities could result from an increased IR drop and the non-linear charging of the electrodes. With regards to the thickness and weight of the electrode, the capacitances of the electrode based on volume and weight at 20 mA cm−2 are 1232 F cm−3 and 414.5 F g−1, respectively. Compared to the CNT@Ni film fabricated by filtration, the resistance of the electrode decreased from 3.4 Ω to 2.2 Ω according to EIS characterization (Fig. 3e). The Ni(OH)2 layer was fully accessible to OH−, and the specific capacitance of Ni(OH)2 was 1765 F g−1 at the current density of 40 mA cm−2. However, the CNT@Ni film is stacked loosely by compression, and the contact part between CNT@Ni could be oxidized and result in an increased internal resistance (Fig. S12d, ESI†).
For long testing cycles, the continuous oxidation of the Ni layer caused damage to the contact surface, leading to internal resistance (Fig. S14, ESI†). For further optimization, the electrode was cured at 350 °C in nitrogen for 20 min to fuse the contact between CNT@Ni to give a connected 3-D metal network. The inner resistance of the electrode further decreased to 1.5 Ω (Fig. 3e), indicating that the contact between CNT@Ni was improved by the curing process. The cured electrode was tested up to 6000 cycles using a continuous cyclic voltammetry test at 50 mV s−1 (Fig. 3f). The capacitance of the electrode can be maintained at 117% of the original capacitance after testing for 6000 cycles. An additional GCD test was performed before and after the long cycle test (inset of Fig. 3f), and the capacitance of the electrode by GCD at 5 mA cm−2 was increased by 27%, and this phenomenon was consistent with that of the reported Ni@NiO core–shell electrodes fabricated with activated nickel foam.14
We next fabricated a freestanding electrode with c-CNT@Ni, and similar optimization strategies and electrochemical measurements were carried out via GCD, CV and EIS spectroscopies (Fig. 3d and Fig. S15, ESI†). The areal capacitances of the c-CNT@Ni electrode plate were 22.0, 22.3, 22.5, 22.9 and 23.3 mF cm−2 at the current density of 5, 7.5, 10, 15 and 20 mA cm−2, respectively. With regard to the plate electrode with the thickness of 0.243 mm, c-CNT@Ni has a volumetric capacitance of 960 F cm−3 at the current density of 20 mA cm−2. The measured areal and volumetric capacitances of c-CNT@Ni are similar to those of CNT@Ni, but the inner resistance and charge transfer resistance of c-CNT@Ni are 1.6 Ω, and 0.6 Ω, respectively, via EIS (Fig. S15c, ESI†), hence giving a better energy output efficiency. A similar synthetic strategy can also be applied to nanocarbon materials with different morphologies such as graphene oxide (GO) to give nickel-coated graphene oxide (GO@Ni) and nickel-coated GO–CNT hybrid materials (CNT–GO@Ni) (Fig. S16 and S17, ESI†). In comparison to the reported electrodes with high areal capacitance, including HAB-MOF negative electrodes (23 F cm−2 & 760 F cm−3),15 MnO2/rGO@Ni(OH)2 positive electrodes (17.8 F cm−2),16 and CNT@PPy@MnO2 positive electrodes (16.1 F cm−2),17 both the areal and volumetric capacitances of the electrodes have been greatly improved to higher values in this work.
To match the positive electrodes of CNT@Ni@Ni(OH)2 with a remarkable high areal capacitance, CNT@Ni–Fe@Fe2O3 nanoporous networks were synthesized as negative electrodes. Through the sequential deposition of Ni and Fe on CNTs, CNT@Ni–Fe was synthesized, and CNT@Ni–Fe@Fe2O3 was obtained by the oxidation of CNT@Ni–Fe in hydrogen peroxide. The sequential deposition of Ni and Fe on CNTs was characterized by EDX (Fig. S18, ESI†) while the oxidation of the Fe2O3 layer on CNT@Ni–Fe was confirmed by XRD (Fig. S19, ESI†). The TEM image of CNT@Ni–Fe showed that Ni and Fe were uniformly coated on the CNTs (Fig. 4a). CNT@Ni–Fe@Fe2O3 freestanding electrodes (thickness 0.277 mm) showed high areal and volumetric capacitances via the GCD test (Fig. 4b) of 11.8 F cm−2 and 524 F cm−3, respectively.
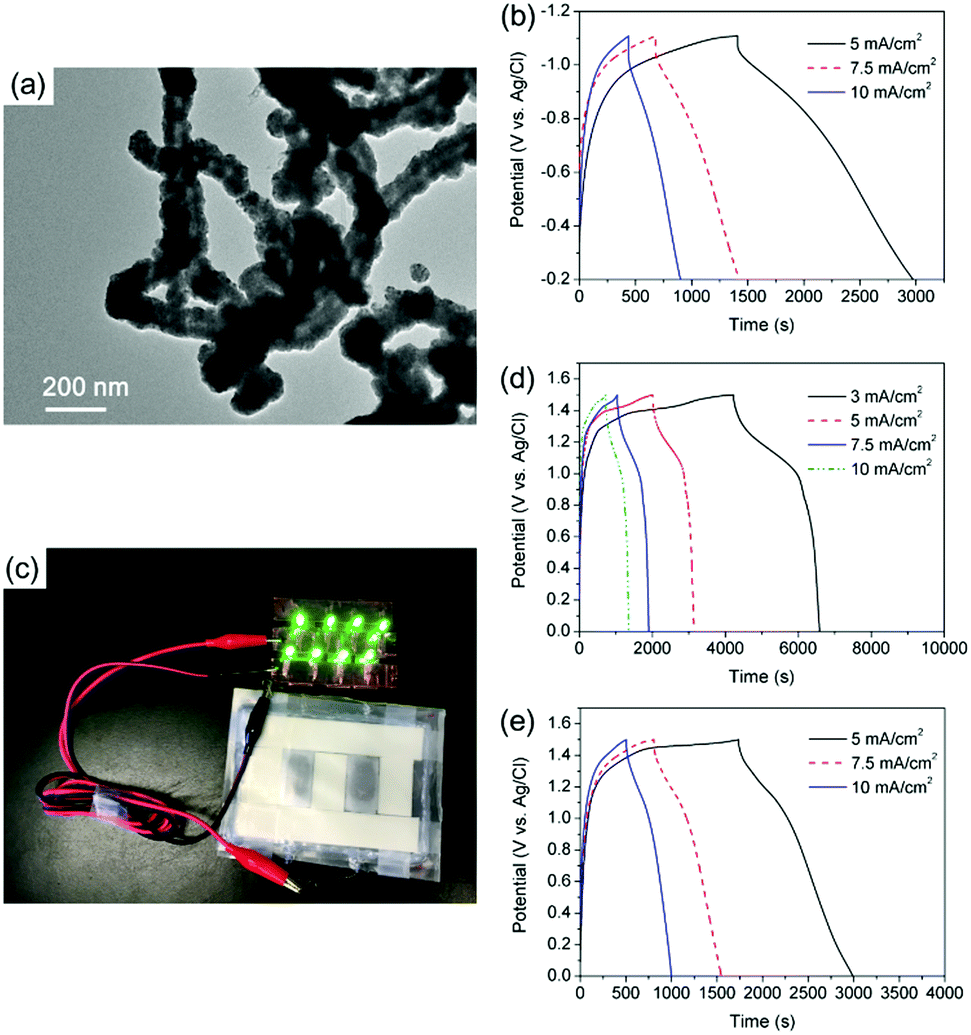 | ||
| Fig. 4 (a) TEM image of CNT@Ni–Fe, (b) GCD curves of CNT@Ni–Fe@Fe2O3 freestanding electrodes, (c) eight LEDs powered by two serially connected flexible supercapacitors, (d) GCD curves of a supercapacitor assembled by CNT@Ni(OH)2 and CNT@Ni–Fe@Fe2O3, and (e) GCD curves of a supercapacitor assembled by c-CNT@Ni(OH)2 and CNT@Ni–Fe@Fe2O3. | ||
An asymmetric supercapacitor was fabricated using CNT@Ni@Ni(OH)2 as the positive electrode and CNT@Ni–Fe@Fe2O3 as the negative electrode (Fig. S20, ESI†) with a balanced mass ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, according to the principle of conservation of charge (Q+ = Q−). From the GCD curves in Fig. 4d, the areal and volumetric capacitances of the device based on the two electrodes were 4.8 F cm−2 and 172 F cm−3, respectively. Calculated by the integration of GCD discharge curves (Fig. 4d), the device had an energy density of 12 W h kg−1 (35 mW h cm−3) based on the overall electrodes. For comparison, another asymmetric supercapacitor was assembled using c-CNT@Ni@Ni(OH)2 as the positive electrode. From the GCD discharge curves (Fig. 4e), the supercapacitor had a capacitance of 4.24 F cm−2 at the current density of 5 mA cm−2, which corresponded to an energy density of 8.2 W h kg−1 (23.7 W h cm−3). As a demonstration, a light-emitting diode (LED) array was powered by two series of flexible connected supercapacitors (Fig. 4c).
:5, according to the principle of conservation of charge (Q+ = Q−). From the GCD curves in Fig. 4d, the areal and volumetric capacitances of the device based on the two electrodes were 4.8 F cm−2 and 172 F cm−3, respectively. Calculated by the integration of GCD discharge curves (Fig. 4d), the device had an energy density of 12 W h kg−1 (35 mW h cm−3) based on the overall electrodes. For comparison, another asymmetric supercapacitor was assembled using c-CNT@Ni@Ni(OH)2 as the positive electrode. From the GCD discharge curves (Fig. 4e), the supercapacitor had a capacitance of 4.24 F cm−2 at the current density of 5 mA cm−2, which corresponded to an energy density of 8.2 W h kg−1 (23.7 W h cm−3). As a demonstration, a light-emitting diode (LED) array was powered by two series of flexible connected supercapacitors (Fig. 4c).
Conclusions
The crown ether DB24C8-CH2NH2 was grafted onto CNTs to improve the absorption of Ag+ for uniform Ni deposition on CNTs. The ELD protocol was modified to control the metal deposition morphology of interconnected CNT@Ni and c-CNT@Ni. The electrodes based on the crown ether-modified CNTs (c-CNT@Ni@(NiOH)2) had lower inner resistance and charge transfer resistance. Asymmetric supercapacitors were assembled using c-CNT@Ni@Ni(OH)2 and CNT@Ni–Fe@Fe2O3 as positive and negative electrodes. The nanoporous metal networks with oxide/hydroxide shells reported in this work showed promising properties in achieving metal deposition and electrochemical performances (Table 1).| Samples | Electrode type | Areal capacitancea | Volumetric capacitancea | Energy density | Internal resistance |
|---|---|---|---|---|---|
| a Measured at the current density of 20 mA cm−2. | |||||
| CNT@Ni@Ni(OH)2 | Positive | 31.2 F cm−2 | 1232 F cm−3 | N/A | 2.2 Ω |
| CNT-GO@Ni@Ni(OH)2 | Positive | 63.65 F cm−2 | 1140 F cm−3 | N/A | N/A |
| c-CNT@Ni@Ni(OH)2 | Positive | 23.3 F cm−2 | 960 F cm−3 | N/A | 1.6 Ω |
| CNT@Ni–Fe@Fe2O3 | Negative | 11.8 F cm−2 | 524 F cm−3 | N/A | N/A |
| Assembled with CNT@Ni@Ni(OH)2 and CNT@Ni–Fe@Fe2O3 | Asymmetric | 4.8 F cm−2 | 172 F cm−3 | 12 W h kg−1 (35 mW h cm−3) | N/A |
| Assembled with c-CNT@Ni@Ni(OH)2 and CNT@Ni–Fe@Fe2O3 | Asymmetric | 4.24 F cm−2 | N/A | 8.2 W h kg−1 (23.7 W h cm−3) | N/A |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Guangdong Province Zhu Jiang Talents Plan (2016ZT06C090), the Guangzhou City Talents Plan (CYLJTD-201609), and the Nansha District Research Project (2016GJ011). This work was partially supported by The Hong Kong Baptist University.Notes and references
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS.
- Y. Zou, C. Cai, C. Xiang, P. Huang, H. Chu, Z. She, F. Xu, L. Sun and H.-B. Kraatz, Electrochim. Acta, 2018, 261, 537–547 CrossRef CAS.
- X. Zhang, W. Shi, J. Zhu, W. Zhao, J. Ma, S. Mhaisalkar, T. L. Maria, Y. Yang, H. Zhang, H. H. Hng and Q. Yan, Nano Res., 2010, 3, 643–652 CrossRef CAS.
- G. W. Yang, C. L. Xu and H. L. Li, Chem. Commun., 2008, 6537–6539 RSC.
- S.-M. Bak, K.-H. Kim, C.-W. Lee and K.-B. Kim, J. Mater. Chem., 2011, 21, 1984–1990 RSC.
- S. Chen, Q. Wu, M. Wen, C. Wang, Q. Wu, J. Wen, M. Zhu and Y. Wang, J. Phys. Chem. C, 2017, 121, 9719–9728 CrossRef CAS.
- S. Xu, X. Li, Z. Yang, T. Wang, W. Jiang, C. Yang, S. Wang, N. Hu, H. Wei and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 27868–27876 CrossRef CAS.
- R. Zhou, Y. Fu, K.-A. Chao and C.-H. Cheng, Renewable Energy, 2019, 135, 1445–1451 CrossRef CAS.
- R. Quintero, D. Y. Kim, K. Hasegawa, Y. Yamada, A. Yamada and S. Noda, RSC Adv., 2014, 4, 8230–8237 RSC.
- M. Hong, X. Wang, W. You, Z. Zhuang and Y. Yu, Chem. Eng., 2017, 313, 1278–1287 CrossRef CAS.
- Y. Yu, C. Yan and Z. Zheng, Adv. Mater., 2014, 26, 5508–5516 CrossRef CAS.
- R. Guo, Y. Yu, Z. Xie, X. Liu, X. Zhou, Y. Gao, Z. Liu, F. Zhou, Y. Yang and Z. Zheng, Adv. Mater., 2013, 25, 3343–3350 CrossRef CAS.
- A. Seghiouer, J. Chevalet, A. Barhoun and F. Lantelme, J. Electroanal. Chem., 1998, 442, 113–123 CrossRef CAS.
- M. Yu, W. Wang, C. Li, T. Zhai, X. Lu and Y. Tong, NPG Asia Mater., 2014, 6, e129–e129 CrossRef CAS.
- D. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. Huang, M. Lee, L. Shaw, S. Chen, A. A. Yakovenko, A. Kulkarni, J. Xiao, K. Fredrickson, J. B. Tok, X. Zou, Y. Cui and Z. Bao, Nat. Energy, 2018, 3, 30–36 CrossRef CAS.
- S. Min, C. Zhao, Z. Zhang, K. Wang, G. Chen, X. Qian and Z. Guo, RSC Adv., 2015, 5, 62571–62576 RSC.
- P. Li, Y. Yang, E. Shi, Q. Shen, Y. Shang, S. Wu, J. Wei, K. Wang, H. Zhu, Q. Yuan, A. Cao and D. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 5228–5234 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, microscopic analysis, electrochemical analysis, NMR spectra. See DOI: 10.1039/d0ma00701c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |