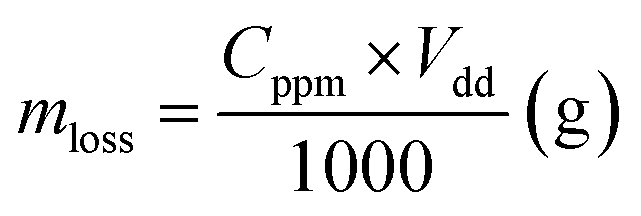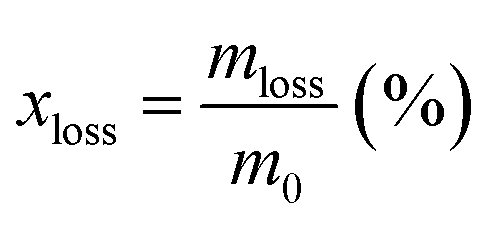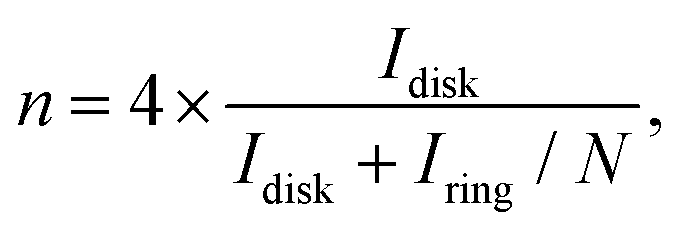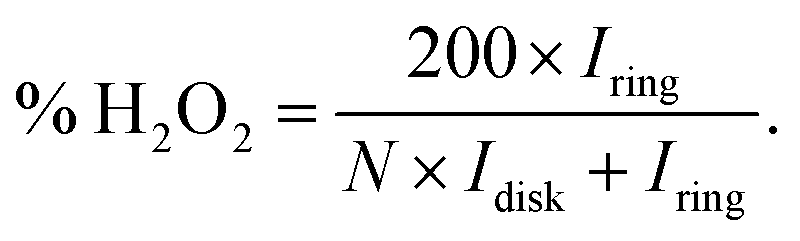DOI:
10.1039/D0MA00718H
(Paper)
Mater. Adv., 2021,
2, 322-335
N-Doped few-layer graphene encapsulated Pt-based bimetallic nanoparticles via solution plasma as an efficient oxygen catalyst for the oxygen reduction reaction†
Received
18th September 2020
, Accepted 11th November 2020
First published on 16th November 2020
Abstract
N-Doped few-layer graphene encapsulated Pt-based bimetallic nanoparticles were successfully synthesized via the solution plasma (SP) method. This synthesis strategy can be achieved in one pot by using only pure dimethylformamide (DMF) as the reaction solution at room temperature and atmospheric pressure. The structural analyses exhibited a fine core–shell structured nanoparticle (2–4 nm), which had Pt and Pt-based bimetallic nanoparticles as a core and N-doped few-layer graphene (NFG), having 2–4 layers, as a shell. Pt-based bimetallic cores (i.e., PtAu, PtAg, and PtPd) were varied depending on the metal electrodes used in the SP process. The electrocatalytic activity toward the oxygen reduction reaction (ORR) of the obtained samples in an acidic solution was found to be acceptable, while possessing excellent durability, compared to those of the commercial carbon-supported Pt catalyst. The effective ORR activity was possibly attributed to the synergistic effect of the Pt-based bimetallic core and N-doped graphene shell which contained high amounts of pyridinic-N and quaternary-N. The results in a single fuel cell test expressed the superiority of our SP-derived sample in the fuel cell performances to commercial Pt/C. This study not only proposed potential alternative ORR catalysts with acceptable electrocatalytic activity and high durability but also provided guidelines for the facile synthesis of carbon-based core–shell nanostructured materials with low chemical use via the SP process.
1. Introduction
Nowadays, the world is facing the depletion of fossil fuel resources and the effect of pollutants from the utilization of fossil fuel. Therefore, finding alternative sustainable energy resources has drawn much attention in both academic and industrial fields. Fuel cells have been extensively studied in order to replace conventional energy conversion devices using fossil fuel.1 Notably, the proton exchange membrane fuel cell (PEMFC) has been presented as one of the most promising technologies because it can convert chemical energy to electrical energy without causing pollution.2 A PEMFC generally uses hydrogen gas (H2) and oxygen gas (O2) as fuels. H2 molecules are oxidized on the anode releasing electrons, which flow through an external circuit to the cathode where O2 molecules are reduced, the rate-determining step influencing overall performance.3 To overcome this problem, the cathodic catalysts have been developed to enhance the oxygen reduction reaction (ORR). Platinum (Pt) has been proposed as the most effective cathodic catalyst because it can effectively promote the ORR at low temperatures.4 According to the availability of Pt, the design of using a combination of Pt and other metals in ORR catalyst has been recently underlined, such as Pt-based bimetallic nanoparticles or alloys.5–7 The electrocatalytic activity of Pt-based alloys was found to be improved relating to their strain effects. The presence of other metal atoms can lead to strain in the structure of Pt. The strain effects usually influence the electronic charge redistribution on the surface of catalysts, which can alter the binding and releasing ability of the surface with the reactants, intermediates, and products of ORR.8 Even though Pt and Pt-based bimetallic catalysts have exhibited excellent ORR activity, the improvement of their durability and stability is still one of the concerning issues. Providing diversity in the architectural design of the electrocatalyst is an effective approach to improve the electrocatalyst capability in various aspects, not only electrocatalytic performance but also durability and stability, for instance, using metal–organic frameworks to overcome several limitations according to the synergistic effects of the selected materials.9,10 Currently, carbon materials, which have excellent electronic conductivity, high thermal and chemical stability, and low cost,11 are being widely used as supports for Pt and Pt-based catalysts, for example, Pt3Au/C and Pt3Pd/C, which exhibited higher ORR activity compared to Pt/C.12–14 Seeking a new design and the synthetic route to obtain an outstanding ORR catalyst is still challenging, especially in terms of durability and stability improvement. Besides, it is necessary to meet the economic feasibility and environmental concerns to make sustainable development.
Graphene is one of the important candidate materials for several applications, due to its promising properties, including exceptionally high chemical stability.15 It has been investigated and used as a protective coating for metals in a variety of sizes, including at the nanoscale.16,17 However, applying the graphene layer, which contains only sp2 hybridized carbon–carbon bonding,18 may change the surface chemistry of the catalysts and lower their catalytic activity. One of the alternatives to overcome this problem is doping heteroatoms into the carbon structure because it provides asymmetrical spin and charge densities to the surface which help to enhance the adsorption of O2.19,20 Hence, when Pt-based bimetallic nanoparticles are encapsulated by N-doped graphene, it should enhance their durability and boost their ORR activity.
The synthesis of carbon-encapsulated metal nanoparticles can be conducted via several methods, such as chemical vapour deposition and thermal treatment.21,22 So far, these methods usually need high temperatures and a multi-step process, which includes the synthesis of metal nanoparticles before the coating of carbon layers. Recently, the liquid-phase plasma technology, the so-called solution plasma (SP) technology, has been reported that can simply synthesize bimetallic nanoparticles and carbon doped with heteroatoms under room temperature and pressure conditions.23,24 In the SP process, plasma can be generated between electrodes submerged in a solution by using a bipolar-pulsed power supply. Some molecules in the solution near the plasma can be transformed into highly reactive species, which can rapidly react with other surrounding highly reactive species or molecules to form products.23 The obtained products depend on the type of solution used. Therefore, the synthesis of N-doped graphene can be done in one step by using an organic liquid containing nitrogen via SP.25 Moreover, SP was reported to have the potential to synthesize bimetallic materials by using different types of metal electrodes. During the SP process, the rapid bombardment of energetic ions was found to occur at the tip of these metal electrodes, leading to the erosion of the metal electrode, diffusion of metal ions, and formation of metal clusters.24 According to these phenomena, Pt-based bimetallic nanoparticles should be rapidly synthesized and simultaneously encapsulated in N-doped graphene via the SP at room temperature and atmospheric pressure with low chemical use.
In this work, we addressed a one-step synthesis of Pt-based bimetallic nanoparticles, i.e., PtAu, PtAg, and PtPd, encapsulated in N-doped few-layer graphene (NFG) via the SP using a pair of Pt and Au, Ag, or Pd electrodes. The synthesis was done at room temperature and atmospheric pressure without the addition of metal catalysts. Only DMF was used as the NFG precursor. The obtained products were characterized using high-resolution transmission electron microscopy (HR-TEM), X-ray powder diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and Raman spectroscopy to confirm the formation of Pt-based bimetallic nanoparticles encapsulated in NFG. Their utilization as an ORR electrocatalyst for operating under an acidic environment in comparison with the commercial Pt/C catalyst was also investigated and discussed in detail, including their electrocatalytic activity and durability.
2. Experimental section
2.1 Materials
Dimethylformamide (DMF, 99.5%), acetone (>99.5%), hydrofluoric acid (HF, 46.0–48.0%), hydrochloric acid (HCl, 35.0–37.0%) and 0.5 M sulfuric acid (H2SO4, 99.5%) were purchased from Kanto Chemical Co., Inc., Japan. Platinum nanoparticles (particle size <50 nm), Nafion® D-521 solution (5 wt% in a mixture of lower aliphatic alcohols and water), 20 wt% platinum on a Vulcan XC-72 (20% Pt/C), and standard solutions of platinum, palladium, gold, and silver of concentration 1000 ppm were purchased from Sigma Aldrich, Germany. 1.0 mm diameter wires of platinum (Pt, 99.98%), palladium (Pd, 99.95%), gold (Au, 99.95%), and silver (Ag, 99.99%) were purchased from Nilaco Co., Japan.
2.2 Solution plasma (SP) synthesis
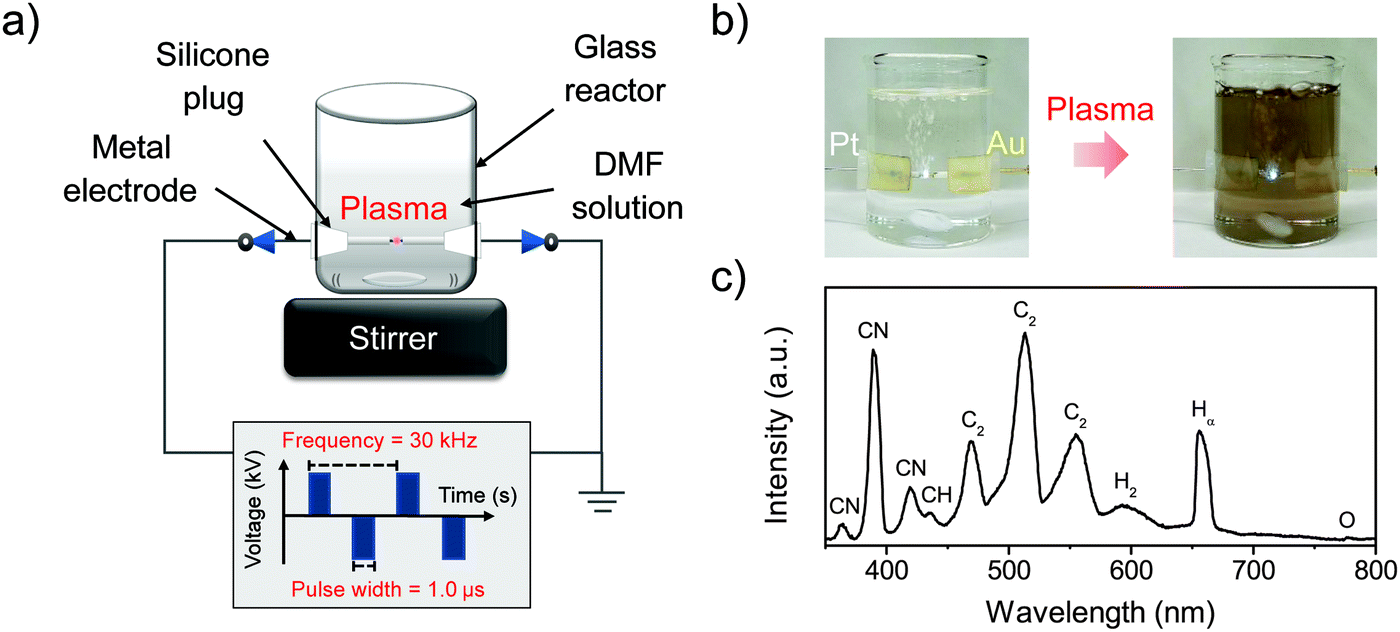
Pt-based bimetallic nanoparticles coated with NFG were synthesized via SP using Pt wire with Au, Ag, or Pd wire as an opposite electrode. In the SP process, a pair of electrodes shielded with insulating ceramic tubes were plugged into a glass reactor, and the tips of the electrodes were placed at the center of the reactor with a gap distance of 1.0 mm, as shown in Fig. 1. Plasma discharge was generated and maintained between the two electrodes submerged under 100 ml of DMF by using a bipolar pulsed power supply (Kurita Seisakusho Co., Ltd, Japan). Plasma operating conditions were fixed at a pulse width of 1.0 μs and a repetition frequency of 30 kHz. During the plasma discharge, black solid particles were continuously generated. After 60 min of the reaction, the black solid particles were separated from the resulting mixture and washed with acetone by filtration using PTFE 0.1 μm membrane (JVWP04700, Merck). The collected solid samples were dried at 100 °C for 12 h and kept in a dark-bottle in a desiccator before further characterization.
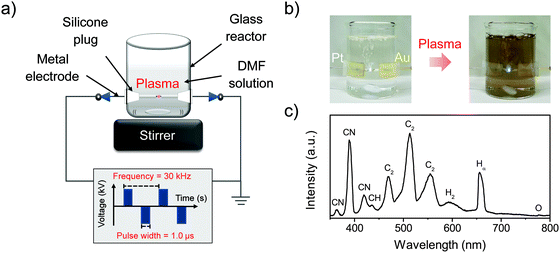 |
| | Fig. 1 Experimental set up of the SP process (a), the SP synthesis of PtAu-NFG (b), and the OES spectra of SP in DMF at 30 kHz (c). | |
2.3 Characterizations
The detection of generated radicals during SP discharge in DMF was done by using optical emission spectroscopy (OES, UV/Vis USB 2000+, Ocean Optics Inc., USA). The morphologies and nanostructures of the synthesized samples were observed via high-resolution transmission electron microscopy (HR-TEM, JEM-2100F, JEOL Ltd, Japan) at an accelerating voltage of 200 kV. The crystal structures were examined via X-ray diffraction (XRD, Rigaku Corp., Japan) with Cu Kα radiation (λ = 0.154 nm) at a scan rate of 4 degrees per minute. Raman spectroscopy (Raman, Leica DM 2500M Ren (RL/TL) microscope, Renishaw Plc., England) was also used to analyze the structural information of carbon-shell with a 532 nm laser excitation source. X-Ray photoelectron spectroscopy (XPS, PHI 5000 Versa Probe II, ULVAC-PHI, Inc., Japan) using a Mg Kα X-ray source was applied to analyze the surface properties and elemental composition of the obtained samples.
2.4 Electrochemical tests
The electrochemical properties of the obtained samples were tested via cyclic voltammetry (CV) and linear sweep voltammetry (LSVs) using three-electrode cells with a potentiostat (Hokuto Denko Inc., HZ500) system. The three electrode cells consisted of a 5 mm diameter glassy carbon disk surrounded by an 8 mm outer diameter platinum ring as a rotating ring-disk electrode (RRDE, HR2-RD1-Pt8/GC5) to be a working electrode, platinum electrode as a counter electrode, and Ag/AgCl as a reference electrode. The commercial Pt/C was used to compare with the SP-derived samples in this work. For preparation of the ink containing Pt/C, 5.0 mg of Pt/C was suspended in a mixture of 0.5 ml of isopropanol and 50 μl of Nafion® solution under sonification until a homogeneous dispersion was obtained. Whereas, the ink containing SP-derived samples was prepared by using a mixture of 1.0 mg SP-derived sample and 4.0 mg of carbon black (Carbon Black XC-72, Fuel Cell Earth LLC). 10.0 μl of the obtained homogeneous suspension was dropped onto an RRDE and dried in the air under room temperature. All electrochemical measurements were evaluated in a 0.5 M H2SO4 solution at 50 mV s−1 with potential range from 0.0 V to 1.4 V (V vs. RHE). Before the measurements, N2 gas was flowed into 0.5 M H2SO4 solution for 60 min to remove the dissolved O2 gas. Then, O2 gas was flowed into 0.5 M H2SO4 solution for 30 min to estimate the ORR performance of the obtained samples. A 500-cycle CV measurement was done at the scan rate of 100 mV s−1 from 0.0 V to 1.4 V (V vs. RHE) to explore the stability for long-term use. A chronoamperometric study was also conducted at 0.4 V (V vs. RHE) for 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s at a rotating speed of 1600 rpm. Besides that, the methanol tolerance was tested in a chronoamperometric study with the addition of 3 M methanol (MeOH, >99.8%, Kanto Chemical Co., Inc., Japan) at 0.6 V (V vs. RHE). The procedure is as follows: first, the system was maintained for the first 500 s to ensure that equilibrium was reached. Then the methanol was introduced into the system after 500 s. The relative currents were monitored over time to see the tolerance effect of methanol on the samples. These experiments were also repeated on commercial Pt/C as a control, to make clear comparisons.
000 s at a rotating speed of 1600 rpm. Besides that, the methanol tolerance was tested in a chronoamperometric study with the addition of 3 M methanol (MeOH, >99.8%, Kanto Chemical Co., Inc., Japan) at 0.6 V (V vs. RHE). The procedure is as follows: first, the system was maintained for the first 500 s to ensure that equilibrium was reached. Then the methanol was introduced into the system after 500 s. The relative currents were monitored over time to see the tolerance effect of methanol on the samples. These experiments were also repeated on commercial Pt/C as a control, to make clear comparisons.
2.5 Durability tests
The durability of the synthesized catalysts was examined by acid immersion by applying a static potential. The amount of metal corroded from the synthesized samples was calculated into a mass loss percentage, which is a reverse variation with the durability of the catalysts. The suspension of each 40.0 μl sample, which was prepared as mentioned in the electrochemical test, was dropped onto the glassy carbon electrode and dried at room temperature. The tests were performed in 100 ml of a mixed solution of H2SO4 solution pH 3, 30 ppm HF, and 10 ppm HCl at 80 °C and using three-electrode cells with a static potential of 0.9 V (V vs. RHE) for 100 h. As a reference, the 20% Pt/C was also tested by using the same amount of the synthesized sample. Inductively coupled plasma atomic emission spectroscopy (ICP-OES, SPS7800, Hitachi Co., Japan) was applied for detecting the metal concentration in the collected solution. As a result, the mass loss amount and mass loss percentage can be determined from the equation as follows:
Mass loss amount:
| | 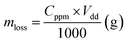 | (1) |
Mass loss percentage:
| | 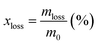 | (2) |
where
Cppm is the metal concentration in the collected solution (ppm),
Vdd is the volume of the tested solution (l), and
m0 is the mass of the tested sample (g).
2.6 Single-cell test
The current density–voltage and current density–power density data were recorded on the membrane electrolyte assembly (MEA) installed in a single cell of a PEMFC. For the fabrication of the cathode, 20.0 mg of commercial Pt/C, or a mixture of 4.0 mg of the sample and 16.0 mg of carbon black was sonicated in 900 μl of isopropanol and 253 μl of 5 wt% Nafion for 1 h. For the anode, the way to prepare the ink was the same as that for the cathode but using 20.0 mg of commercial Pt/C. The catalyst ink was dropped on the Teflon plate with the catalyst loading at 0.2 mg cm−2, then it was dried under room temperature and pressure conditions. After drying, the prepared Teflon plates (cathode and anode with 1 cm2 area) were pressed at 150 °C and 2 MPa, onto a Nafion® membrane NRE-212, and held for 3 min. The single fuel cell measurements were tested under a humidified atmosphere at 80 °C with the flow rate of H2 and O2 at 200 cm3 min−1 for the anode and cathode flow channels, respectively. A potentio-galvanostat (VMP-300, Biologic) with a back pressure of 0.2 MPa was used to monitor the current, voltage, and power of the fuel cell generated over time. Electrochemical impedance spectroscopy (EIS) was also used in the single-cell test under the same test conditions. The sweeping frequencies were ranged from 10 kHz to 10 mHz with 10 points per decade. The amplitude of the AC signal was fixed at 10 mV to measure the steady-state current at 0.7 V and 0.6 V.
3. Results and discussion
In this study, the samples obtained from the plasma discharge in DMF solution using a pair of electrodes, which were Pt–Au, Pt–Ag, Pt–Pd, and Pt alone, were assigned to PtAu-NFG, PtAg-NFG, PtPd-NFG, and Pt-NFG respectively. During the plasma discharge, there were color changes of solutions which were yellow-black, yellow-grey, dark grey, and dark yellow, when different pairs of electrodes of Pt–Au, Pt–Ag, Pt–Pd, and only Pt, respectively, were used. Fig. 1(b) illustrates the digital image of the synthesis of PtAu-NFG by the SP. The formation of black particles in the SP system was suggested to be carbon material. They were produced through the decomposition and recombination of reactive species (i.e., C2, CH, H, and CN radicals) generated from organic compounds during SP discharge.26 These radicals were confirmed by using OES spectrometry, as shown in Fig. 1(c). A similar phenomenon was also reported in the previous study, which used the nitrogen-containing organic liquid as a precursor to form N-doped carbon nanoparticles.27 Besides, the bimetallic nanoparticles could be formed from the erosion of metal electrodes via the SP, resulting in individual metal atoms, which could be further combined and alloyed with other surrounding metal atoms.28 Therefore, the color change of solution was related to the suspension of carbon and metal particles formed during the SP process.
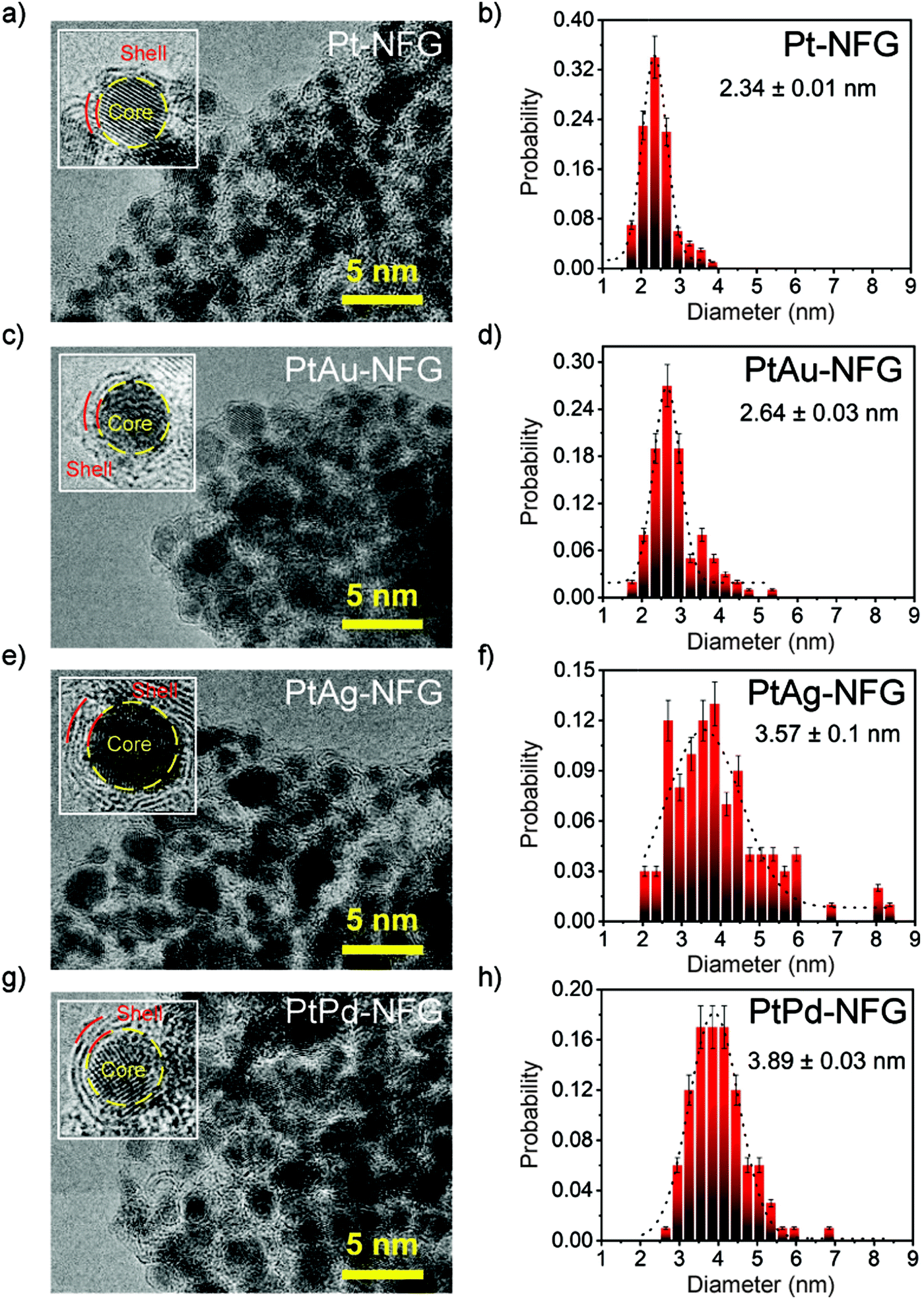
According to HR-TEM images (Fig. 2), the obtained particles in all systems were found to be agglomerated particles having a narrow size distribution with an average size of 2–4 nm. Moreover, morphological structures of the obtained samples were found to be core–shell structures with clear layers. Enlarged-view images of each sample revealed the presence of thin layers, ranging from 2 to 4 layers. These thin layers were found to closely coat the core which was confirmed to be Pt alone for Pt-NFG, and the coexistence of Pt and Au, Ag, or Pd for PtAu-NFG, PtAg-NFG, and PtPd-NFG, respectively, by using EDS measurements (Fig. S1, S2 and Table S1, ESI†). The lattice fringes of thin layers revealed double layers with a d-spacing of approximately 0.34 nm, which corresponded to that of few-layer graphene that was previously reported.29 This coating has been reported to help in stabilizing the structure of the metal core, which enhances the durability of the metal catalyst.30 Besides, it was also reported that the heteroatom-containing carbon-shell enhanced the electrical conductivity on metals, leading to the promotion of the catalytic activity.30,31
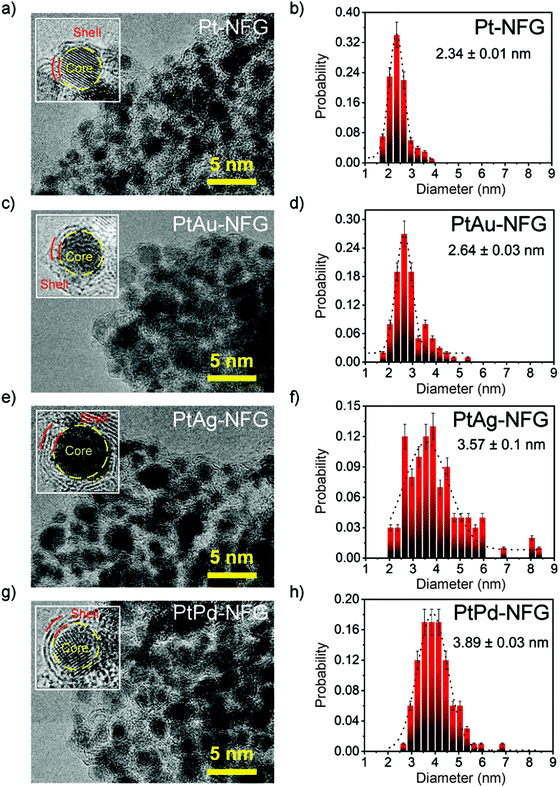 |
| | Fig. 2 HR-TEM images (left) and size distribution results (right) for Pt-NFG (a and b), PtAu-NFG (c and d), PtAg-NFG (e and f), and PtPd-NFG (g and h). | |
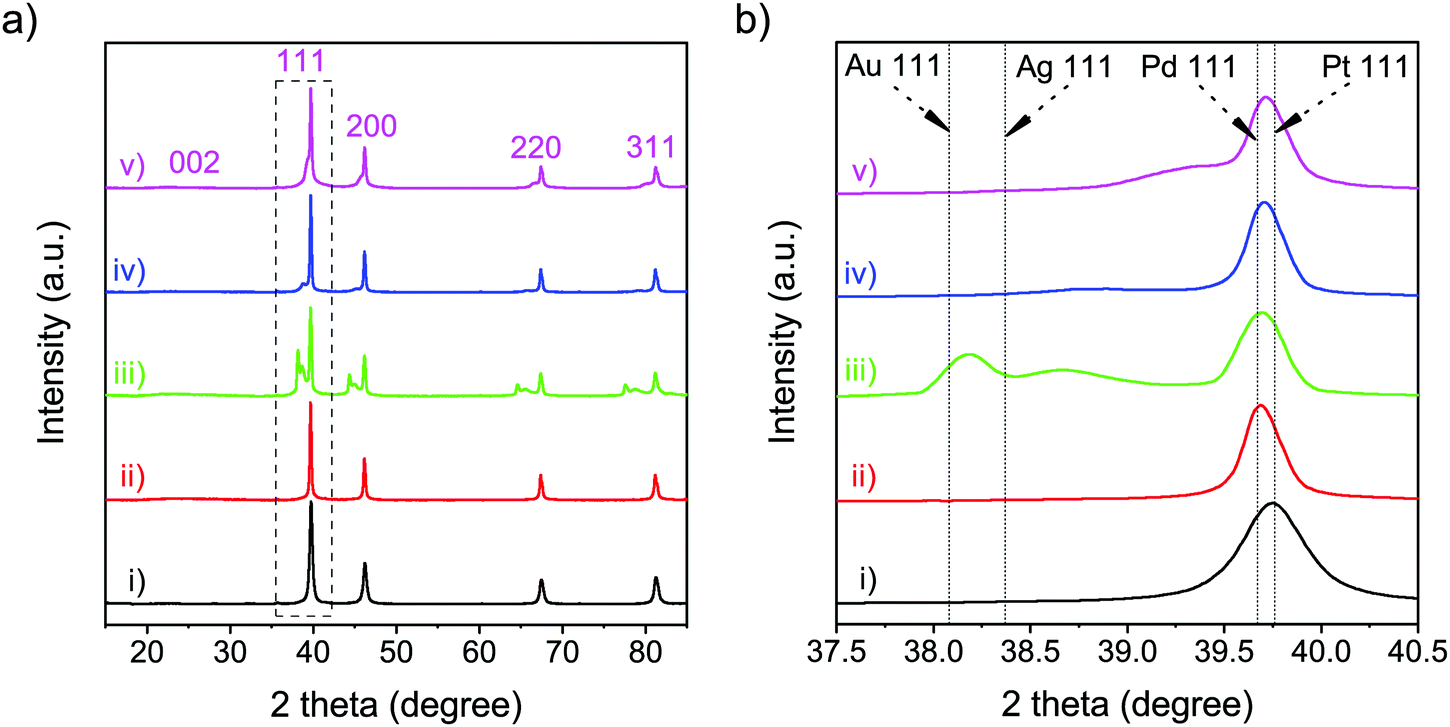
The structural details of the obtained samples were further investigated via XRD and Raman measurements. The XRD patterns of all samples (Fig. 3(a)) showed a broad diffraction peak at 2θ = 23.50°, corresponding to the (002) plane of graphene.32 In addition, XRD analysis was carried out to compare the diffraction patterns of the obtained nanoparticles with that from the standard JCPDS cards of each metal (Table S2, ESI†). In the XRD pattern of Pt-NFG, sharp peaks were exhibited at 2θ = 39.70°, 46.15°, 67.40°, and 81.20°, corresponding to the (111), (200), (220), and (311) planes, which were four characteristic peaks of face-centered cubic (fcc) crystalline Pt. However, according to the enlarged view in Fig. 3(b), it was found that the (111) plane shifted slightly toward the lower 2θ region, compared to that of the pure commercial Pt, which could be caused by a lattice strain due to interaction between the core and the shell.33,34 A similar pattern was also revealed for other obtained samples that have Pt-based bimetallic nanoparticles as a core. For the samples with a bimetallic core (i.e., PtAu-NFG, PtAg-NFG, and PtPd-NFG), additional peaks were found in the XRD patterns. In Fig. 3(b), the XRD pattern of PtAu-NFG showed the a small broadened peak at 2θ = 38.65°, associated with the (111) plane of Au; moreover, the peak between (111) planes of Au and Pt could be observed, which implied the formation of alloyed AuPt.35 Accordingly, in PtAu-NFG, there should be the coexistence of the AuPt alloy as dominant species, and individual Au, which was in agreement with the results of the previous study.28 However, for PtAg-NFG and PtPd-NFG, the (111) planes of Ag and Pd were not observable, but the additional peaks 2θ = 38.85° and 39.35° were revealed, respectively. These additional peaks were found to be at a lower 2θ region, compared to the (111) planes of Pt. This evidence could suggest that Ag and Pd atoms, which have smaller atomic size than Pt, were alloyed with Pt atoms, leading to the change in the lattice structure of the nanoparticles and the appearance of the additional peak in the XRD pattern, compared to that of Pt alone. Therefore, PtAg-NFG and PtPd-NFG should contain Pt and the alloyed PtAg and PtPd, respectively. The change of the geometric structure from alloying can lead to an altered binding energy of O2 on the surface of catalysts, which plays a decisive role in the ORR performance of alloy catalysts.8
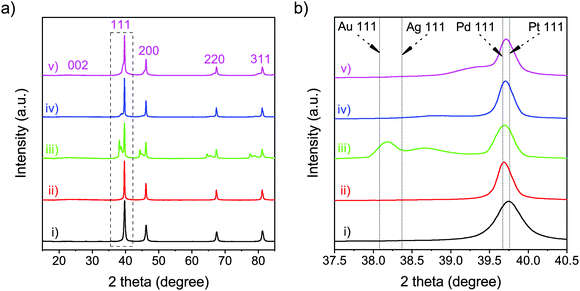 |
| | Fig. 3 XRD patterns (a) and the enlarged view of the XRD peaks corresponding to the (111) plane (b) of the commercial Pt (i), Pt-NFG (ii), PtAu-NFG (iii), PtAg-NFG (iv), and PtPd-NFG (v). | |
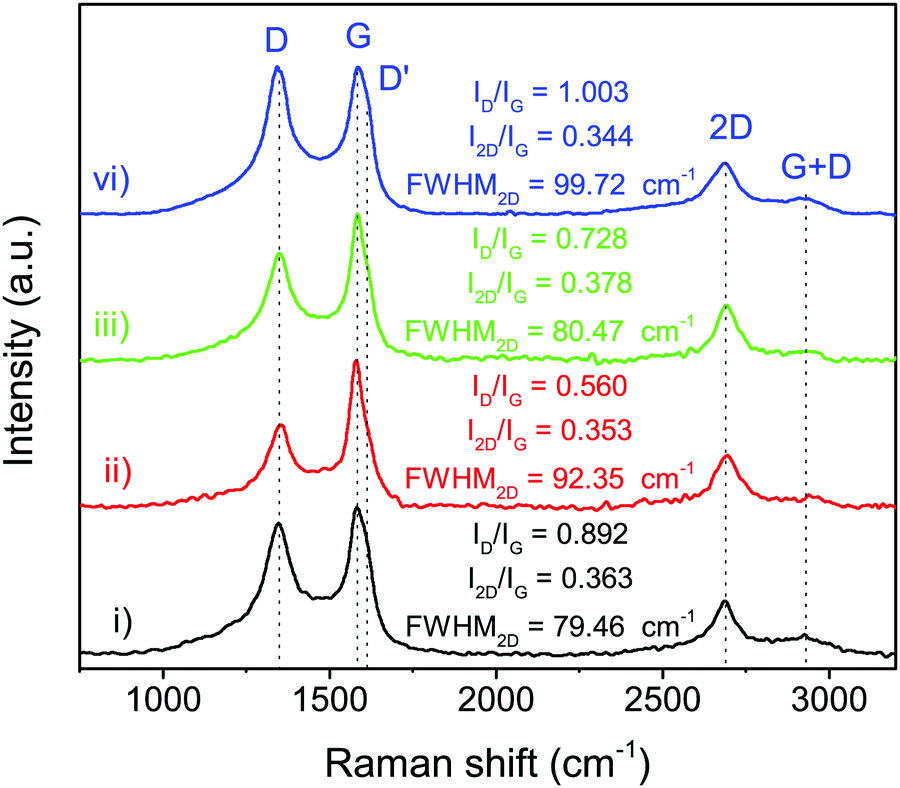
The structural information of N-doped few-layer graphene was further investigated by Raman spectroscopy. As shown in Fig. 4, the Raman spectrum of N-doped few-layer graphene in all obtained samples showed a similar pattern having four main peaks, which are typically found in graphene, at approximately 1348 cm−1, 1580 cm−1, 2675 cm−1, and 2940 cm−1, attributed to the D band, G band, 2D band, and G + D band, respectively.25 The D band usually indicates some defects in the crystal structure, the edge of graphene or disordered structure, while the G band relates to the vibrations of in-plane sp2 phonons of carbon atoms in the graphitic planes.36 The relative intensity ratio of the D band to the G band (ID/IG) is typically employed for the degree of graphitization, or defect ratio in carbon materials.37,38 The ID/IG ratios of the obtained samples were estimated, as shown in Fig. 4. PtPd-NFG showed the relatively high value of ID/IG, indicating the high defect sites. Meanwhile, the value of ID/IG obtained from the Raman spectra of PtAu-NFG was the lowest. As a result, the value of ID/IG seemed to vary depending on the metal nanoparticles in the core. In addition, the 2D band relates to the higher-order carbon structure, which can be employed to indicate the number of layers in graphene.36 The value of I2D/IG can be used to identify monolayer (I2D/IG > 2), bilayer (I2D/IG ≈ 1), and multilayer (I2D/IG < 1) graphene.39 The values of I2D/IG obtained in this work ranged from 0.34 to 0.37, which could suggest that the SP-induced shell was multilayer graphene. Besides, the full width at half maximum (FWHM) of the 2D band, which is generally used for determining the number of single-atomic layers in graphene (i.e., ∼31 cm−1 for bilayer graphene),40 also indicated that the obtained shell was few-layer graphene, which was consistent with the TEM results. The additional peak, referring to the D′ peak, was also observed as a shoulder of the G peak at higher wavenumbers for all obtained samples. The D′ peak was reported as the defect peak caused by doping heteroatoms, such as nitrogen doping.25 Thus, the XRD and Raman results could suggest that the obtained nanoparticles in this work had the shell of the N-doped few-layer graphene coating on the core, which contained a mixture of metal alone and alloys.
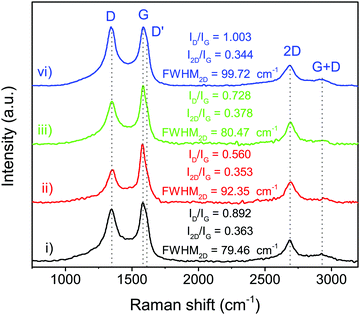 |
| | Fig. 4 Raman spectra of Pt-NFG (i), PtAu-NFG (ii), PtAg-NFG (iii), and PtPd-NFG (iv). | |
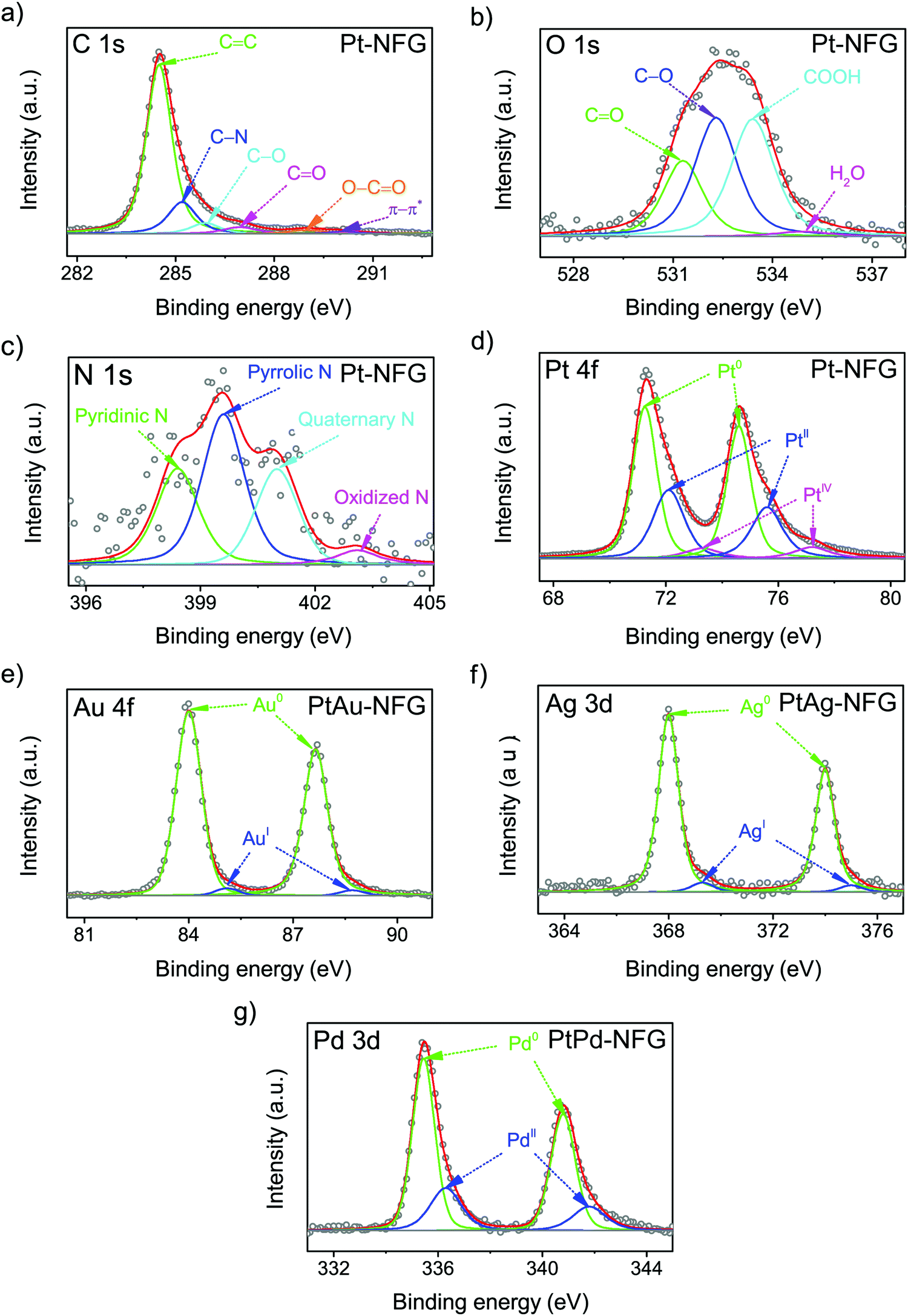
Furthermore, the surface element composition and chemical states of the obtained samples were investigated by XPS, which is sensitive on the 1–10 nm depth scale.41 The result of XPS showed that the obtained atomic compositions were found to belong to not only N-doped graphene layers but also the bimetallic core (Table 1). This could support the suggestion that the shells of the obtained samples were few-layer graphene, which theoretically has the thickness of a single layer equaling 0.345 nm.42 According to Table 1, the presence of nitrogen could be evidence indicating the successful incorporation of nitrogen atoms into the carbon framework and N/C ratios were found to be comparable for all obtained samples. The high-resolution XPS spectra were then deconvoluted to gain a deeper insight into the chemical bonding states of a particular element, as displayed in Fig. 5. The C 1s spectrum, shown in Fig. 5(a), exhibited the characteristic peak centered at 284.3 ± 0.1 eV, corresponding to the sp2 bonding configuration. A long tail at the higher binding energy was composed of several deconvoluted peaks assigned to C–N (285.4 ± 0.2 eV), C–O (285.8 ± 0.1 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (286.6 ± 0.2 eV), O–CO (289.2 ± 0.2 eV), and π–π* interaction (289.9 ± 0.1 eV).43,44 The N 1s spectrum with four components corresponding to pyridinic-N (398.6 ± 0.2 eV), pyrrolic-N (399.5 ± 0.2 eV), quaternary-N (401.3 ± 0.2 eV), and oxidized-N (403.2 ± 0.2 eV) are shown in Fig. 5(c).45 Notably, the pyrrolic-N, pyridinic-N, and quaternary-N significantly contribute to the shape of the N 1s peaks, while oxidized-N was found to be a minor component. This finding indicated that the nitrogen atoms were mostly bonded to neighboring carbon atoms.46 In particular, the presence of pyridinic-N and quaternary-N indicated that it could facilitate the adsorption of O2 and was proven to be active for the ORR.46 Besides, the oxygen contents were also observed in the samples, which were obtained from the oxidation during SP treatment.26 The O/C ratios were significantly high for PtPd-NFG, compared to that of PtAu-NFG, and PtAg-NFG. The oxygen contents could be from not only the oxygen species present at the defect edges, such as CO (531.3 ± 0.1 eV) and –COOH (532.9 ± 0.1 eV) evident in the O 1s peak (Fig. 5(b)) but also could be the result of the formation of metal oxides in the cores. According to the previous study, the reactivity and oxygen-binding for Pt, Pd, Au, and Ag atoms were revealed by density functional theory (DFT) calculations.47 The adsorption and reaction with oxygen were found to be exothermic for Pt and Pd, and favored to form oxides of Pt and Pd, respectively. Meanwhile, Au and, especially, Ag could lead to an endothermic reaction, and it was more difficult for them to form bonds with oxygen. Consistent evidence was also showed in the obtained deconvoluted XPS spectra in this work. The deconvoluted XPS peaks of Pt 4f, Au 4f, Ag 3d, and Pd 3d are exhibited in Fig. 5(d–g). The relative integrated area for each species in the Au 4f and Ag 3d peaks of PtAu-NFG and PtAg-NFG could indicate that the oxide species were rarely found,48,49 compared to that of Pt-NFG and PtPd-NFG. The deconvoluted peaks of Pt 4f and Pd 3d of Pt-NFG and PtPd-NFG could suggest that there should be the presence of PtO and PtO2, and PdO, respectively,50,51 in relatively high amounts. In addition, it was also reported that the presence of oxide species at the core structure could affect the formation of the graphene layer.52 When the oxygen was present on the metal core, it could hinder the formation of larger size graphene flakes, due to diminishing of the graphene nucleation density. Therefore, the metals without the presence of oxides could give the formation of compact graphene. Accordingly, it could explain the relatively high density of defect sites, from the Raman results, in the Pt-NFG and PtPd-NFG samples, which contained a high amount of oxide species on the cores. Moreover, the slight shift of peaks of Pt 4f, Au 4f, Ag 3d, and Pd 3d could be observed. The XPS spectra of Pt 4f of our synthesized samples were found to have two main peaks at 71.2 eV and 74.6 eV which represented the Pt 4f7/2 and Pt 4f5/2 states, respectively.53 These peaks were positively shifted compared to a pure metallic state of Pt on Pt/C which was located at 70.7 eV and 74.2 eV for Pt 4f7/2 and Pt 4f5/2 (Fig. S3, ESI†), respectively. The positive shift was interpreted in previous studies by two major reasons. The first was the effect of the difference in electronegativity between Pt and the carbon shell.54–56 Pt transfers its electron to the carbon shell making itself less electron-dense. Besides, this effect was enhanced by nitrogen atoms doped to the carbon shell where the nitrogen is more electronegative than the carbon atom. Thus, the upshift of binding energy on the Pt 4f state arises. The second was because of the alloying effect on Pt with a secondary metal.57 Generally, the work function of metals descends from Pt > Pd > Au > Ag. The electron transfer must move from the other metal to Pt. This should have a tendency to cause a negative shift in binding energy. However, it upshifts the Fermi level of Pt electronic structures resulting in an opposite downshift of the d-band center, as well as Pt 4f7/2. Therefore, it strengthened the binding energy found in photoelectron measurements. The positive shifts of Pt 4f7/2 could confirm the presence of a bimetallic core for the PtM-NFG samples. Although there was no secondary metal on Pt-NFG, a positive shift was also found.
O (286.6 ± 0.2 eV), O–CO (289.2 ± 0.2 eV), and π–π* interaction (289.9 ± 0.1 eV).43,44 The N 1s spectrum with four components corresponding to pyridinic-N (398.6 ± 0.2 eV), pyrrolic-N (399.5 ± 0.2 eV), quaternary-N (401.3 ± 0.2 eV), and oxidized-N (403.2 ± 0.2 eV) are shown in Fig. 5(c).45 Notably, the pyrrolic-N, pyridinic-N, and quaternary-N significantly contribute to the shape of the N 1s peaks, while oxidized-N was found to be a minor component. This finding indicated that the nitrogen atoms were mostly bonded to neighboring carbon atoms.46 In particular, the presence of pyridinic-N and quaternary-N indicated that it could facilitate the adsorption of O2 and was proven to be active for the ORR.46 Besides, the oxygen contents were also observed in the samples, which were obtained from the oxidation during SP treatment.26 The O/C ratios were significantly high for PtPd-NFG, compared to that of PtAu-NFG, and PtAg-NFG. The oxygen contents could be from not only the oxygen species present at the defect edges, such as CO (531.3 ± 0.1 eV) and –COOH (532.9 ± 0.1 eV) evident in the O 1s peak (Fig. 5(b)) but also could be the result of the formation of metal oxides in the cores. According to the previous study, the reactivity and oxygen-binding for Pt, Pd, Au, and Ag atoms were revealed by density functional theory (DFT) calculations.47 The adsorption and reaction with oxygen were found to be exothermic for Pt and Pd, and favored to form oxides of Pt and Pd, respectively. Meanwhile, Au and, especially, Ag could lead to an endothermic reaction, and it was more difficult for them to form bonds with oxygen. Consistent evidence was also showed in the obtained deconvoluted XPS spectra in this work. The deconvoluted XPS peaks of Pt 4f, Au 4f, Ag 3d, and Pd 3d are exhibited in Fig. 5(d–g). The relative integrated area for each species in the Au 4f and Ag 3d peaks of PtAu-NFG and PtAg-NFG could indicate that the oxide species were rarely found,48,49 compared to that of Pt-NFG and PtPd-NFG. The deconvoluted peaks of Pt 4f and Pd 3d of Pt-NFG and PtPd-NFG could suggest that there should be the presence of PtO and PtO2, and PdO, respectively,50,51 in relatively high amounts. In addition, it was also reported that the presence of oxide species at the core structure could affect the formation of the graphene layer.52 When the oxygen was present on the metal core, it could hinder the formation of larger size graphene flakes, due to diminishing of the graphene nucleation density. Therefore, the metals without the presence of oxides could give the formation of compact graphene. Accordingly, it could explain the relatively high density of defect sites, from the Raman results, in the Pt-NFG and PtPd-NFG samples, which contained a high amount of oxide species on the cores. Moreover, the slight shift of peaks of Pt 4f, Au 4f, Ag 3d, and Pd 3d could be observed. The XPS spectra of Pt 4f of our synthesized samples were found to have two main peaks at 71.2 eV and 74.6 eV which represented the Pt 4f7/2 and Pt 4f5/2 states, respectively.53 These peaks were positively shifted compared to a pure metallic state of Pt on Pt/C which was located at 70.7 eV and 74.2 eV for Pt 4f7/2 and Pt 4f5/2 (Fig. S3, ESI†), respectively. The positive shift was interpreted in previous studies by two major reasons. The first was the effect of the difference in electronegativity between Pt and the carbon shell.54–56 Pt transfers its electron to the carbon shell making itself less electron-dense. Besides, this effect was enhanced by nitrogen atoms doped to the carbon shell where the nitrogen is more electronegative than the carbon atom. Thus, the upshift of binding energy on the Pt 4f state arises. The second was because of the alloying effect on Pt with a secondary metal.57 Generally, the work function of metals descends from Pt > Pd > Au > Ag. The electron transfer must move from the other metal to Pt. This should have a tendency to cause a negative shift in binding energy. However, it upshifts the Fermi level of Pt electronic structures resulting in an opposite downshift of the d-band center, as well as Pt 4f7/2. Therefore, it strengthened the binding energy found in photoelectron measurements. The positive shifts of Pt 4f7/2 could confirm the presence of a bimetallic core for the PtM-NFG samples. Although there was no secondary metal on Pt-NFG, a positive shift was also found.
Table 1 The atomic composition of Pt-NFG, PtAu-NFG, PtAg-NFG, and PtPd-NFG by XPS measurements
| Sample |
Atomic composition (at%) |
Doping ratio |
| C |
N |
O |
Pt |
Ma |
N/C |
O/C |
|
M represented secondary metal in the Pt-based bimetallic catalysts.
|
| Pt-NFG |
83.96 |
1.53 |
5.14 |
9.37 |
0.00 |
0.018 |
0.061 |
| PtAu-NFG |
82.54 |
1.89 |
3.77 |
3.99 |
7.81 |
0.023 |
0.046 |
| PtAg-NFG |
84.82 |
2.87 |
5.80 |
4.60 |
1.91 |
0.034 |
0.068 |
| PtPd-NFG |
73.04 |
1.58 |
17.63 |
3.00 |
4.74 |
0.022 |
0.241 |
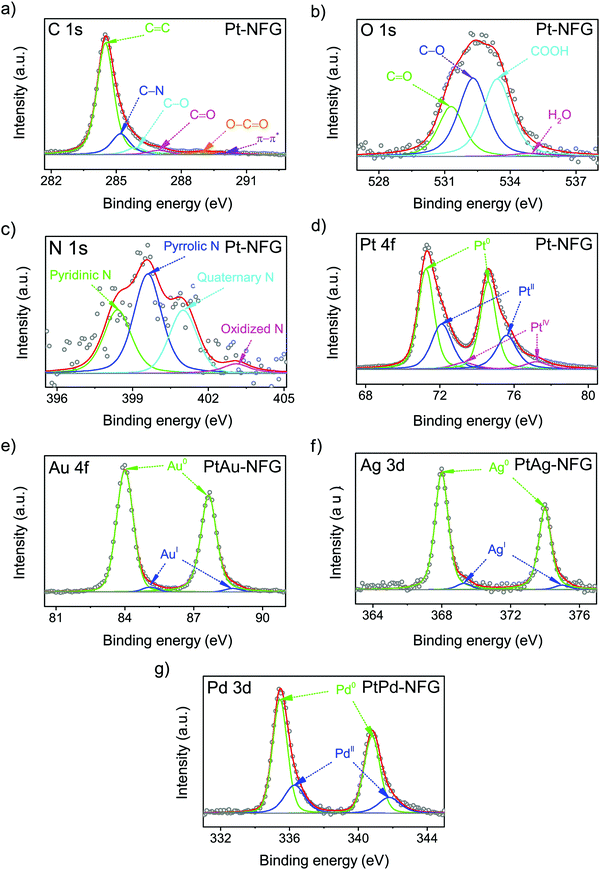 |
| | Fig. 5 Deconvoluted XPS peaks of C 1s (a), O 1s (b), N 1s (c), and Pt 4f of Pt-NFG (d), Au 4f of PtAu-NFG (e), Ag 3d of PtAg-NFG (f), and Pd 3d of PtPd-NFG (g). | |
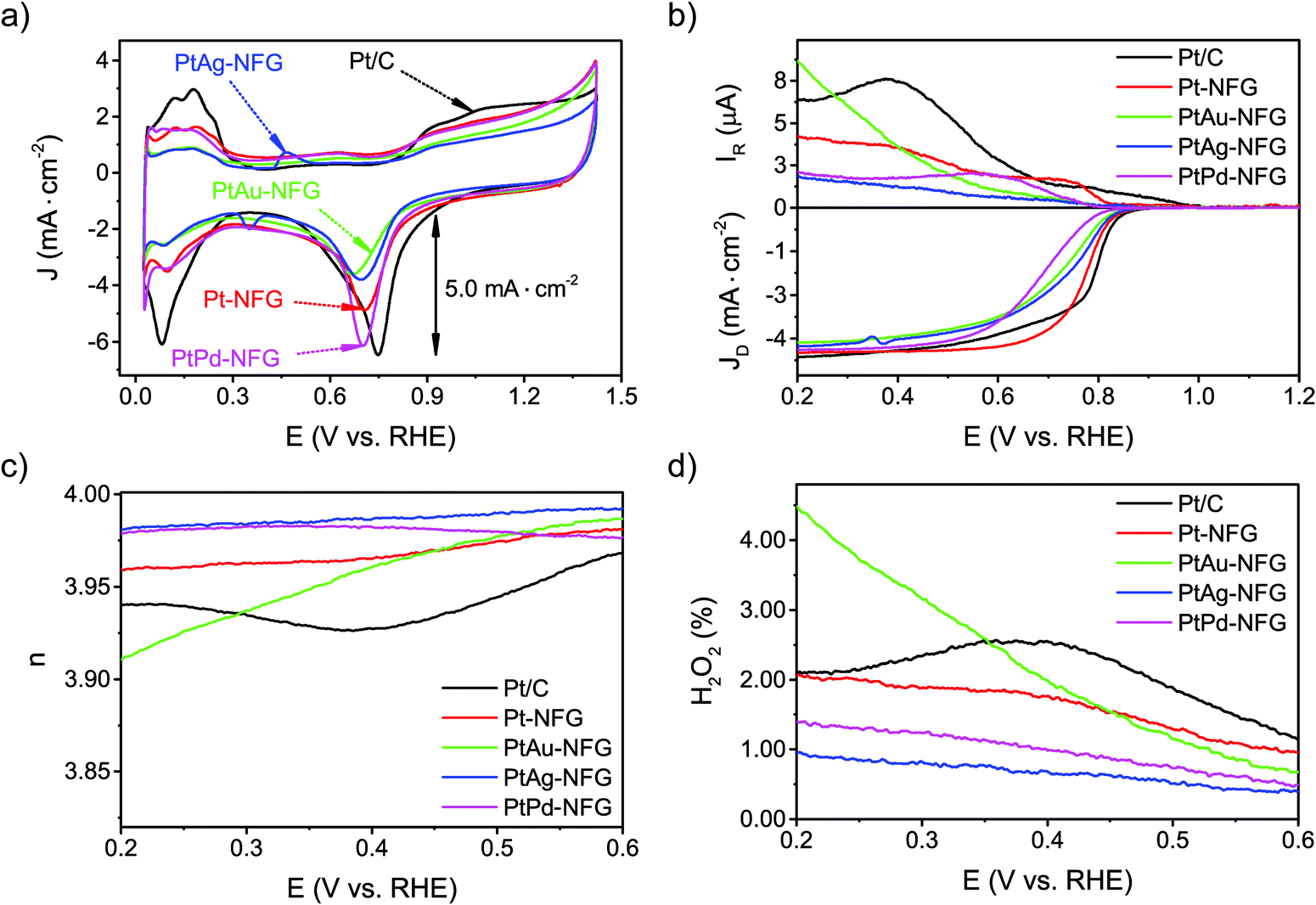
The CV measurements were conducted to understand the ORR activity of the obtained samples. They were performed in 0.5 M H2SO4 in the presence of O2 bubbled in solution with the scan rate of 50 mV s−1 (Fig. 6(a)). The potential between 0.0 and 0.4 V (V vs. RHE) was attributed to the hydrogen region, which can indicate the adsorption and desorption abilities of the catalysts with hydrogen.58 The result pointed out that the obtained NFG-encapsulated Pt or Pt-based bimetallic nanoparticles showed a lower ability to adsorb and desorb hydrogen molecules, compared to the commercial Pt/C. For the oxygen region, at a potential ranging from 0.4 to 1.4 V (V vs. RHE), the CV curves in the O2-saturated solution exhibited well-defined peak potentials, which showed the electrochemical activity toward ORR. The peak potentials of Pt-NFG, PtAu-NFG, PtAg-NFG, and PtPd-NFG appeared at 0.706, 0.702, 0.698, and 0.669 V (V vs. RHE), respectively. As a result, the peak potentials showed negative shifts compared to the commercial Pt/C. For the current density, the PtPd-NFG could provide a comparable value to that obtained from the commercial Pt/C, while other samples showed relatively lower current density. Moreover, the LSV measurements on an RRDE were also conducted at various rotation speeds (Fig. S4, ESI†) to gain more information about the oxygen reduction reaction performance. Fig. 6(b) shows LSV results of all samples obtained in this work at the rotational speed of 1600 rpm with the potential sweep in a negative direction from 1.2 to 0.2 V (V vs. RHE). All samples showed the onset potential values close to 0.880 V (V vs. RHE) which showed that they were comparable in terms of catalytic performance to commercial Pt/C. Also, the Pt-NFG, PtAu-NFG, PtAg-NFG, and PtPd-NFG could give the current density from the disk electrode at 4.314, 4.018, 4.125, and 4.228 mA cm−2, which were similar to that of commercial Pt/C at 4.438 mA cm−2 after reaching the diffusion-controlled region. However, at the kinetic-diffusion mixed region, the vertical section on LSV curves showed different gradients meaning that the diffusion rates of the O2 to react at the surface of the catalysts were different. The steeper gradient leads to a faster ORR rate and vice versa. The half-wave potential (E1/2) was used to explain the rate of ORR.59 The E1/2 sorted in the descending order of samples was as follows: Pt/C > Pt-NFG > PtAg-NFG > PtAu-NFG > PtPd-NFG. The results in Fig. 6(b) show that all SP-derived samples exhibited a slightly slower ORR rate relative to Pt/C. One of the possible explanations for the slower ORR rate that occurred in the kinetic-diffusion mixed region in SP-derived samples could be the hydrophobicity of the NFG shell that hindered the transport of O2 molecules dissolved in an aqueous electrolyte to contact the surface of the catalyst. However, this evidence suggested that the heteroatom-doped few-layer graphene shell can be the key for preserving the ORR activity of the core–shell sample in this study, while the previous study reported that a pristine graphene shell could alter the active surface of the metal-core.18
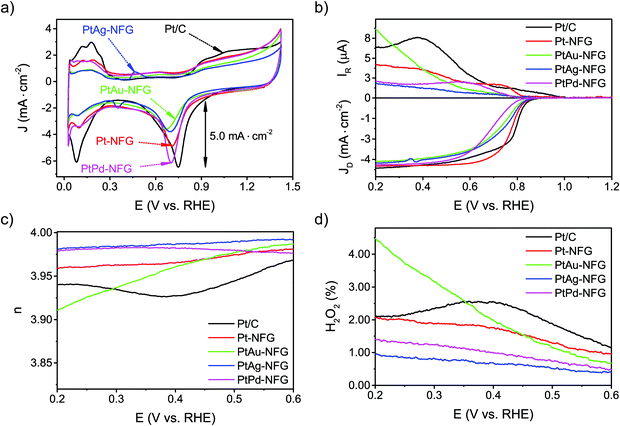 |
| | Fig. 6 CV curve results of measurement with scan rate at 50 mV s−1 in O2-saturated 0.5 M H2SO4 (a). LSV curves (b), electron transfer number (n) values (c), and H2O2 yields (d) of measurement with scan rate at 10 mV s−1 in O2-saturated 0.5 M H2SO4. | |
The possible pathways of the ORR were also evaluated. In general, the ORR can proceed via two major pathways, direct four-electron and two-electron pathways in acidic solutions. The direct four-electron pathway in an acidic electrolyte, which is more favorable for the ORR in fuel cell applications, is shown as follows:
| | | O2 + 4H+ + 4e− → 2H2O (E° = +1.23 V) | (3) |
while the two-electron pathway forms a H
2O
2 intermediate resulting in a two-step reduction, as shown below:
| | | O2 + 2H+ + 2e− → H2O2 (E° = +0.70 V) | (4) |
| | | H2O2 + 2H+ + 2e− → 2H2O (E° = +1.78 V). | (5) |
The formation of hydrogen peroxide (H2O2) and the corresponding electron transfer number (n) per O2 were then discovered using the following equations:
| | 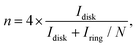 | (6) |
| | 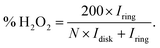 | (7) |
Where
Idisk and
Iring are the disk current and the ring current, respectively.
N is the collection efficiency which was determined to be 0.482 using 0.1 M HClO
4 with a 10 mM K
3Fe(CN)
6 electrolyte. As the result, in
Fig. 6(c), the
n values of the obtained samples in this work derived from the RRDE measurements ranged from 3.92 to 3.98. Notably, the
n values of PtAg-NFG and PtPd-NFG were higher than those of commercial Pt/C in the whole measurement range. A significantly low H
2O
2 (1–4%) was observed for all obtained samples, which was comparable to that of the commercial Pt/C. In the light of the evidence, it could be proposed that the obtained Pt-based bimetallic nanoparticles coated with N-doped few-layer graphene tended to catalyze the ORR through a direct four-electron pathway, similar to the commercial Pt/C. Besides, the
n values were also obtained from the Koutecky–Levich (K–L) plots (Fig. S4(f), ESI
†). The values were consistent with those derived from the RRDE measurements.
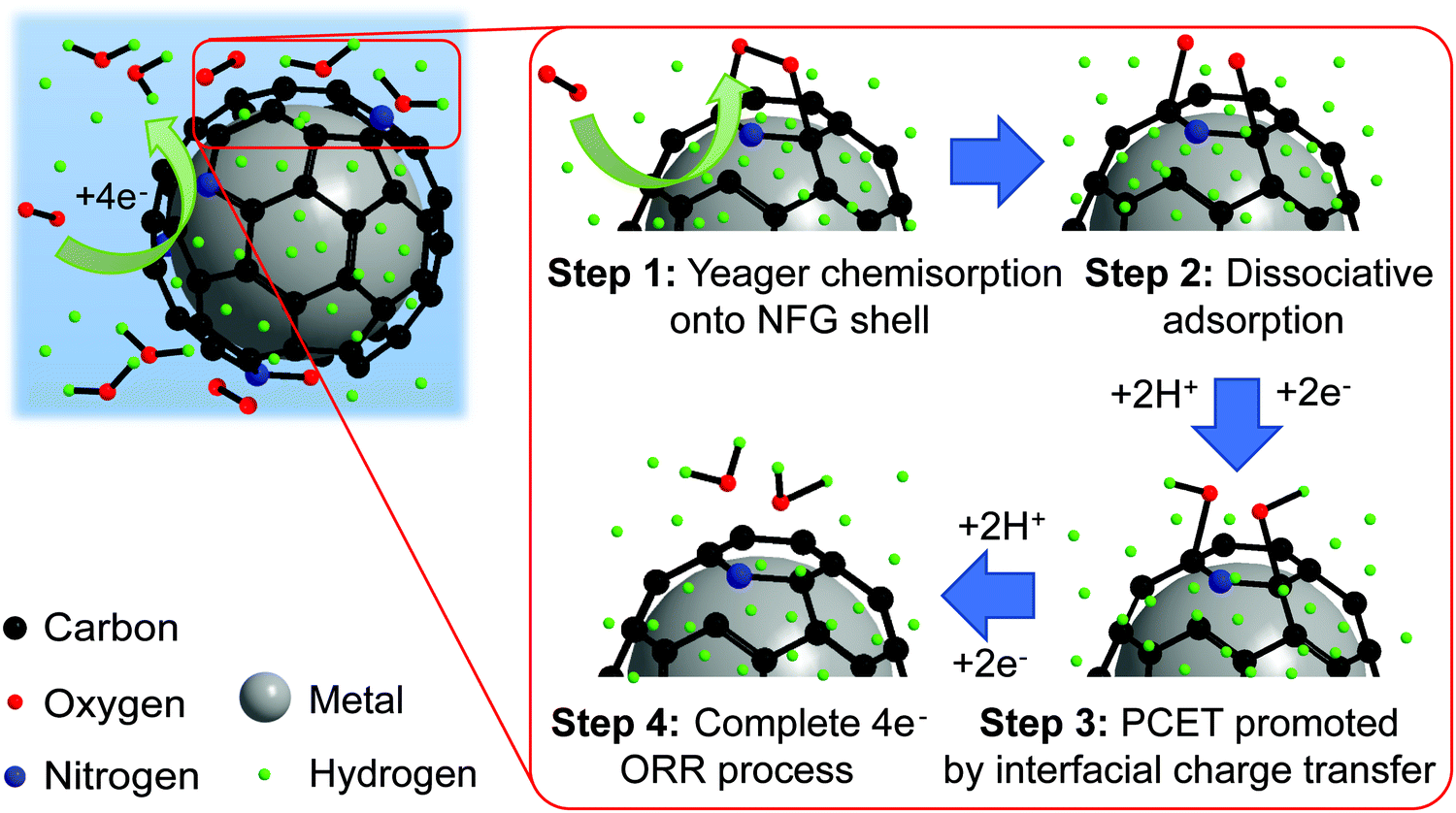
To gain a deeper insight into the shift toward the four-electron route on PtM-NFG samples (M refers to other metals), the possible mechanism of O2 adsorption and ORR of the metal nanoparticles encapsulated in N-doped few-layer graphene is also proposed as shown in Fig. 7. Theoretically, the O2 could be adsorbed on the carbon-based metal-free catalyst via two possible modes, including side-on (Yeager model) and end-on (Pauling model), as shown in Fig. S5 (ESI†).60 Then, both protons and electrons were transferred to the adsorbed oxygen, which was called proton-coupled electron transfer (PCET).61 After PCET, the ORR follows. A higher concentration of protons on the N-doped graphene-shell surface, compared to bare carbon, acts as a driving force to push the ORR toward direct four-electron reduction. Also, the existing nitrogen and oxygen atoms, which have higher electronegativity than a carbon atom, create electropositive sites. These promote the O2 adsorption onto the N-doped graphene surface by decreasing the adsorption barrier.60,62 Typically, the metal-free carbon catalyst, even N-doped carbon, is preferential to the associative adsorption (shown in eqn (S1)–(S5), ESI†) since there was a difficulty in overcoming the energy barrier of dissociative adsorption by breaking the O–O bond.63,64 However, the synergistic effect from the presence of both the nitrogen atom in the carbon framework and the metal core could reduce the activation energy of dissociative adsorption of oxygen, shifting the mechanism towards dissociative adsorption, eqn (S6) (ESI†), through Yeager chemisorption.65 Oxygen atoms can be individually adsorbed and undergo ORR without the formation of H2O2. Therefore, the N-doped few-layer graphene encapsulated Pt-based bimetallic nanoparticles, obtained in this study, showed high selectivity towards direct four-electron reduction. The pieces of evidence obtained in this work prove that the N-doped few-layer graphene not only acted as a protective shell but also collaborated with the Pt-based bimetallic core to efficiently catalyze the desired ORR. Therefore, N-doped few-layer graphene-encapsulated Pt-based bimetallic nanoparticles could be proposed as potential alternative ORR catalysts for electrocatalysis research.
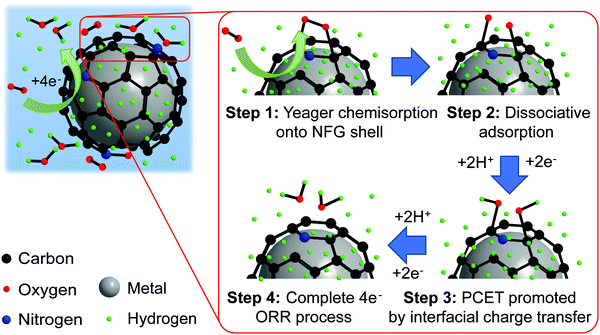 |
| | Fig. 7 Proposed mechanism for ORR and proton transfer of bimetal coated by the NFG core–shell catalyst synthesized by SP. | |
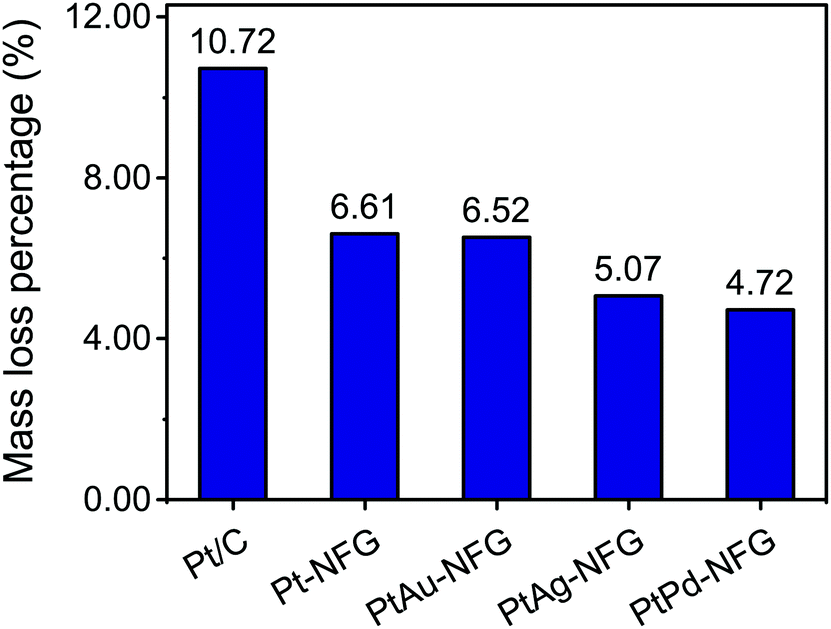
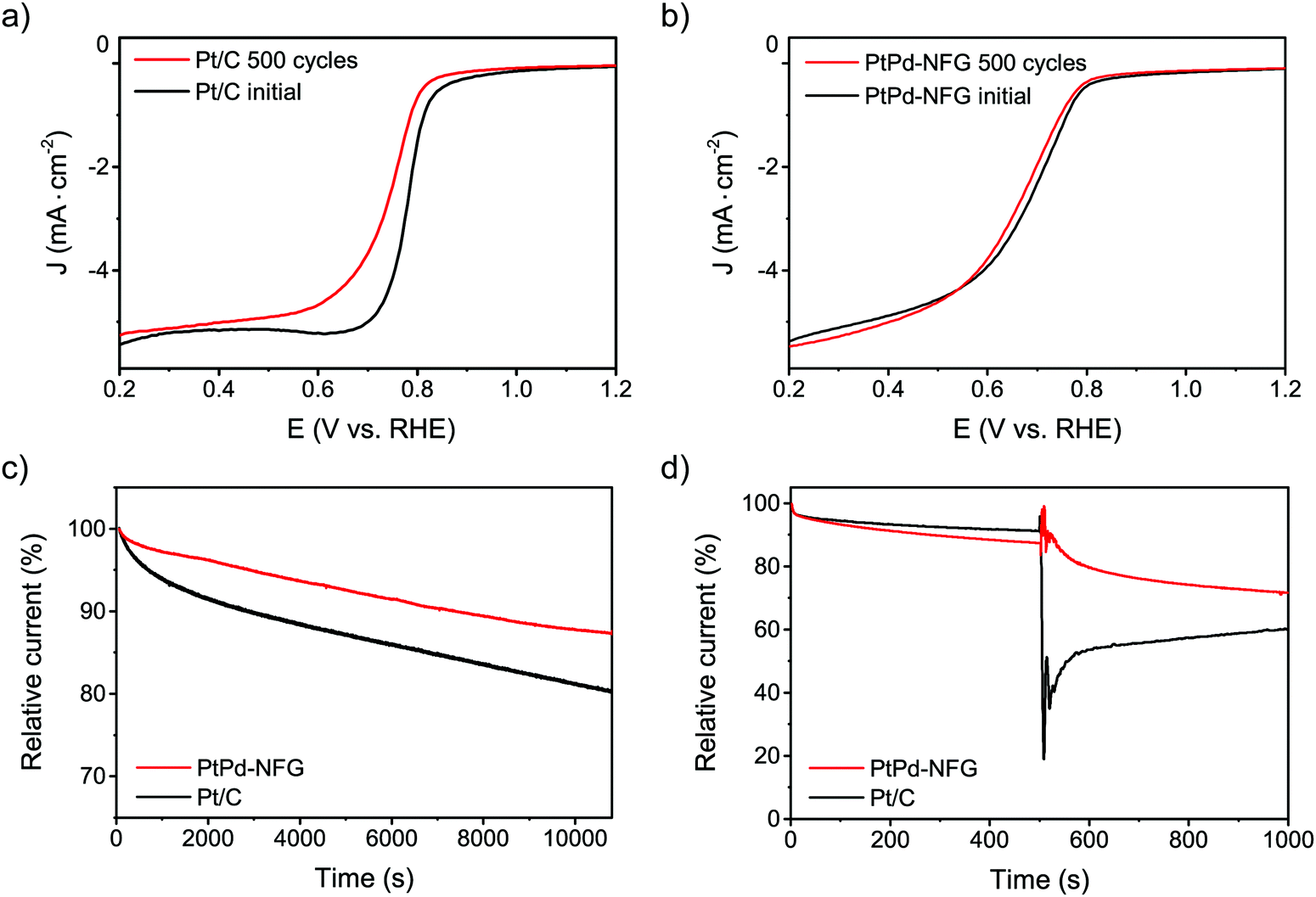
Other than the enhancement of the selectivity toward the four-electron pathway of Pt-based-NFG samples, the protectability of the NFG shell was also demonstrated in this section. Fig. 8 reveals the result of the durability tests using the measurement of metal mass loss based on ICP-OES performed by submerging the samples in the prepared acid medium at 80 °C with applying static potential at 0.9 V (V vs. RHE) for 100 h. The percentages of Pt mass loss from all obtained samples in this work were relatively low compared to that of the commercial Pt/C, indicating that the N-doped few-layer graphene acted as a protective layer to prevent the corrosion of the metal-core. Accordingly, the Pt-bimetallic nanoparticles coated with N-doped few-layer graphene not only showed comparable ORR activity to the commercial Pt/C with high selectivity toward four-electron reduction but also showed higher durability than the commercial Pt/C, especially PtPd-NFG. Therefore, PtPd-NFG was selected to be tested and compared with commercial Pt/C in further investigations. The durability test with different techniques was conducted. Fig. 9(a and b) show the ORR polarization curves for Pt/C and PtPd-NFG at the initial and 500th cycles of CV, respectively. The LSV curve of Pt/C exhibited a significant change in the onset potential and limiting current density between the initial and at the 500th cycle measurements. However, no significant change could be observed in the curve of PtPd-NFG. This implies that PtPd-NFG had better stability over multicycle use. This was in good agreement with the results from the chronoamperometric test, shown in Fig. 9(c). The Pt/C showed a big drop in the current at the start followed by the constant rate of decrease in the relative current after 1000 s while PtPd-NFG had a slight drop at the beginning. In summary, our obtained core–shell catalyst, e.g., PtPd-NFG, revealed higher durability compared to the commercial Pt/C, which was due to the protective N-doped graphene shell. Besides that, the durability and tolerance of the obtained catalyst to the methanol crossover effect in an acidic solution were also investigated. Methanol, which is generally toxic to Pt-based catalysts, was added after 500 s in chronoamperometric measurement, at 0.6 V (V vs. RHE) and with the rotating speed at 1600 rpm at the concentration of 3 M, to test the tolerance of our samples. The result was shown in Fig. 9(d). There was a significant decrease in current on Pt/C, while the PtPd-NFG showed only a slight drop in current. This result confirms that the synthesized core–shell structured catalyst by SP in this work had superior durability and tolerance to the effect of methanol crossover in an acidic solution to commercial Pt/C. From all that we have mentioned, it implies that the NFG shell plays an important role, acting as a protective barrier which improves the chemical stability of the catalyst.
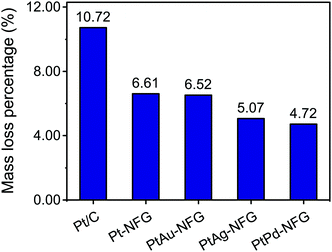 |
| | Fig. 8 Durability test results of Pt/C, Pt-NFG, PtAu-NFG, PtAg-NFG, and PtPd-NFG. | |
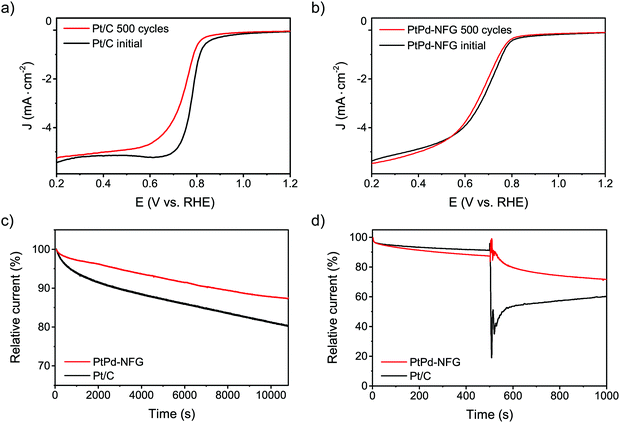 |
| | Fig. 9 LSV curves of Pt/C (a) and PtPd-NFG (b) at the initial and after 500 cycles of CV at 100 mV s−1 in O2-saturated 0.5 M H2SO4. Current–time curves of PtPd-NFG and Pt/C at 0.4 V (V vs. RHE) in O2-saturated 0.5 M H2SO4 at 1600 rpm (c). Chronoamperometric curves of PtPd-NFG and Pt/C at 0.6 V (V vs. RHE) in O2-saturated 0.5 M H2SO4 at 1600 rpm followed by the addition of 3 M methanol (d). | |
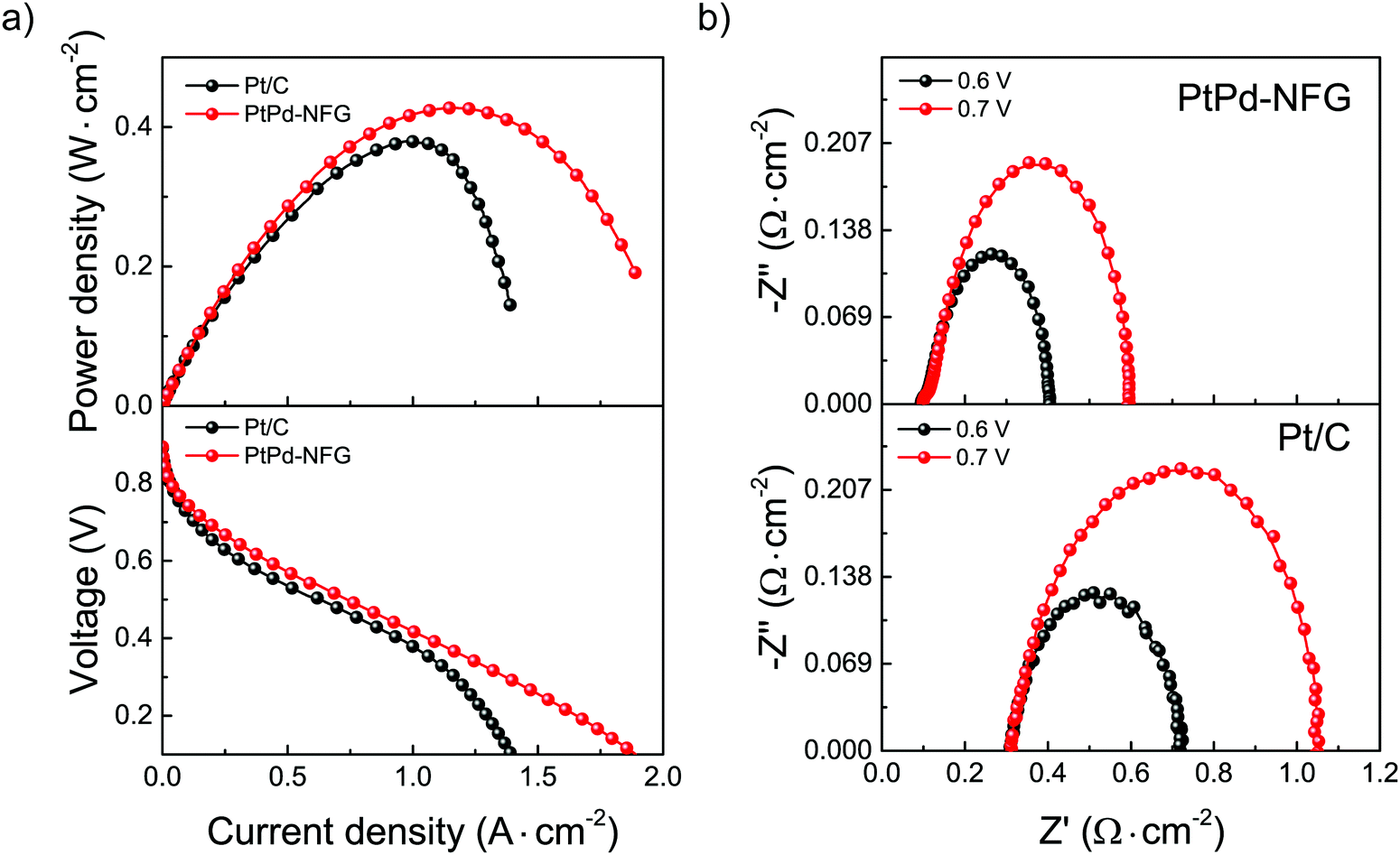
Testing MEA fuel cell performance on an electrocatalyst is one of the important steps to determine the capability for practical purposes. The comparisons of the catalytic performance of PtPd-NFG and commercial Pt/C were evaluated by applying them as a cathode catalyst in a single cell H2/O2 fuel cell at an operating temperature of 80 °C. The current density–voltage–power density (I–V–P) curves of PtPd-NFG and Pt/C are demonstrated in Fig. 10(a). The open-circuit voltage (OCV) values of both catalysts were found to be similar at 0.9 V which means that PtPd-NFG had a comparable activation loss and similar feasibility to start the reaction to commercial Pt/C while the maximum power densities (Pmax) of PtPd-NFG and Pt/C were found to be 427 and 379 mW cm−2, respectively. Moreover, Pt/C showed a rapid drop in current density after the current was reached at 1.2 A cm−2, indicating that the catalytic activity was suddenly diminished due to a blockage of O2 transport by water molecules formed from the ORR since the surface of platinum is hydrophilic.66 On the other hand, our PtPd-NFG exhibited a gradual current density drop over the test period. This indicates a superior catalytic activity. This was due to that the change in surface chemistry for the ORR catalysis from Pt in Pt/C to NFG in PtPd-NFG. A characteristic of NFG is that it is hydrophobic, and therefore it can prevent the occlusion of water molecules generated from ORR facilitating the active sites available for the following reaction.67
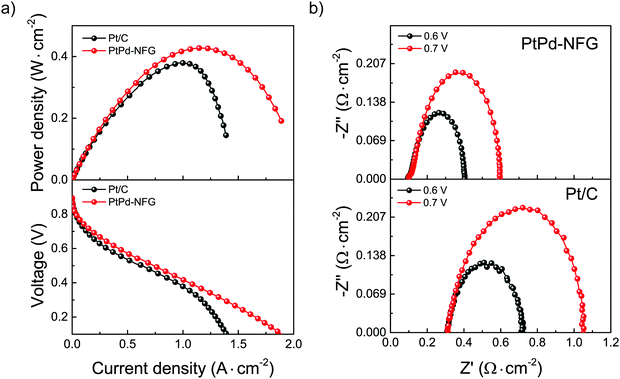 |
| | Fig. 10
I–V–P curves (a) and EIS spectra at OCV conditions (b) of single cell testing of Pt/C and PtPd-NFG at 80 °C. | |
Fig. 10(b) presents the EIS results using Nyquist plots of the MEA fuel cells with different cathodic materials of PtPd-NFG (above) and Pt/C (below) while the anode was fixed to be the same as Pt/C. The tests were used in the same setup (shown in Fig. S6, ESI†) to fit the obtained impedance spectra. There was only one loop for each spectrum which corresponds to a charge transfer resistance (Rct). The Rct of PtPd-NFG and Pt/C were 0.50 Ω cm−2 and 0.74 Ω cm−2 at the constant potential of 0.7 V, respectively. The lower value of Rct on PtPd-NFG implies that the cathode activation loss was smaller than that of Pt/C. It is because the positive interaction between the NFG shell and bimetallic core promotes charge transfer.68 Moreover, total ohmic resistances (RΩ), which can be obtained from the high-frequency intercept at Z′ (x-axis), of PtPd-NFG and Pt/C were 0.09 Ω cm−2 and 0.31 Ω cm−2, respectively. The higher value of Pt/C suggests a greater amount of water adsorbed on the surface that hinders the active sites to cause the reaction69 whereas PtPd-NFG shows a lower RΩ than Pt/C due to the hydrophobicity of the NFG shell that could adsorb water molecules on the surface less than the surface of Pt in Pt/C. Furthermore, the lower value of the total impedance at both 0.6 V and 0.7 V of the fuel cell fabricated with PtPd-NFG compared to those of Pt/C demonstrated that our SP-derived sample was preferable for the overall fuel cell performance.70 From all results we mentioned above, our synthesized sample had promising potential and can be used practically in PEM fuel cell applications.
4. Conclusion
In conclusion, we have rationally designed and presented an efficient strategy to synthesize N-doped few-layer graphene encapsulated Pt-based bimetallic nanoparticles as efficient ORR catalysts. The synthesis was successfully conducted in one pot via solution plasma using only DMF as the reaction solution at room temperature and atmospheric pressure. The metallic core can be tuned by varying electrodes, while various heteroatom doped shells can be prepared by simply changing precursors. N-Doped few-layer graphene played an important role as the protective shell, which helps to improve the durability of the catalyst in comparison to commercial Pt/C. In addition, the incorporation of N atoms in the carbon framework of the graphene shell could collaborate with the Pt-based metallic core, leading to an ORR with high selectivity toward the four-electron pathway which is the desired reaction for fuel cell applications. This may open a great opportunity for the further development to achieve a new high-performance catalyst, as an alternative to the commercial Pt/C catalyst, via a facile and tunable synthesis method, solution plasma method, that can meet the economic feasibility and environmental concerns.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has been financially supported by the Japan Science and Technology Corporation-Strategic International Collaborative Research Program (Grant number: JPMJSC18H1) and the Japan Science and Technology Corporation-Open Innovation Platform with Enterprises, Research Institute, and Academia (Grant number: JPMJOP1843). The author also acknowledges the study support from JICA Technical Cooperation Project for ASEAN University Network/Southeast Asia Engineering Education Development Network (Program number: J-1710176).
References
-
B. Gou, W. Na and B. Diong, Fuel cells: modeling, control, and applications, CRC Press, 2017 Search PubMed.
- S. J. Peighambardoust, S. Rowshanzamir and M. Amjadi, Acc. Chem. Res., 2010, 35, 9349–9384 CAS.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef.
- O. T. Holton and J. W. Stevenson, Platinum Met. Rev., 2013, 57, 259–271 CrossRef.
- M. Ammam and E. B. Easton, J. Power Sources, 2013, 236, 311–320 CrossRef CAS.
- Y. Zhao, J. Liu, Y. Zhao and F. Wang, Phys. Chem. Chem. Phys., 2014, 16, 19298–19306 RSC.
- H. Abe, J. Liu and K. Ariga, Mater. Today, 2016, 19, 12–18 CrossRef CAS.
- M. Shao, J. H. Odell, A. Peles and D. Su, Chem. Commun., 2014, 50, 2173–2176 RSC.
- Y. Fu, P. Richardson, K. Li, H. Yu, B. Yu, S. Donne, E. Kisi and T. Ma, Nano–Micro Lett., 2020, 12, 1–13 CrossRef.
- Y. Fu, K. Li, M. Batmunkh, H. Yu, S. W. Donne, B. Jia and T. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 44830–44839 CrossRef CAS.
- C. Su, Y. Liu, Z. Luo, J.-P. Veder, Y. Zhong and Z. Shao, Chem. Eng. J., 2020, 406, 126883 CrossRef.
- K.-S. Lee, H.-Y. Park, H. C. Ham, S. J. Yoo, H. J. Kim, E. Cho, A. Manthiram and J. H. Jang, J. Phys. Chem. C, 2013, 117, 9164–9170 CrossRef CAS.
- S. Beckord, A. K. Engstfeld, S. Brimaud and R. J. Behm, J. Phys. Chem. C, 2016, 120, 16179–16190 CrossRef CAS.
- Y. Tang, F. Gao, S. Mu, S. Yu and Y. Zhao, Russ. J. Electrochem., 2015, 51, 345–352 CrossRef CAS.
- C. Soldano, A. Mahmood and E. Dujardin, Carbon, 2010, 48, 2127–2150 CrossRef CAS.
- A. Manikandan, L. Lee, Y.-C. Wang, C.-W. Chen, Y.-Z. Chen, H. Medina, J.-Y. Tseng, Z. M. Wang and Y.-L. Chueh, J. Mater. Chem. A, 2017, 5, 13320–13328 RSC.
- Z. Yang, W. Sun, L. Wang, Z. Wan, J. Wang, S. Wang and G. Liu, Corros. Sci., 2020, 175, 108860 CrossRef CAS.
- M. Topsakal, H. Şahin and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155445 CrossRef.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS.
- Q. Lv, W. Si, J. He, L. Sun, C. Zhang, N. Wang, Z. Yang, X. Li, X. Wang and W. Deng, Nat. Commun., 2018, 9, 1–11 CrossRef.
- S. Chen, A. Zehri, Q. Wang, G. Yuan, X. Liu, N. Wang and J. Liu, ChemistryOpen, 2019, 8, 58–63 CrossRef CAS.
- W. Lei, H. M. Barnes, J. Zhang and Z. Cai, Wood Fiber Sci., 2017, 49, 22–32 Search PubMed.
- T. Morishita, T. Ueno, G. Panomsuwan, J. Hieda, A. Yoshida, M. A. Bratescu and N. Saito, Sci. Rep., 2016, 6, 36880 CrossRef CAS.
- X. Hu, X. Shen, O. Takai and N. Saito, J. Alloys Compd., 2013, 552, 351–355 CrossRef CAS.
- S. Chae, G. Panomsuwan, M. A. Bratescu, K. Teshima and N. Saito, ACS Appl. Nano Mater., 2019, 2, 1350–1355 CrossRef CAS.
- C. Chokradjaroen, S. Kato, K. Fujiwara, H. Watanabe, T. Ishii and T. Ishizaki, Sustainable Energy Fuels, 2020, 4, 4570–4580 RSC.
- O. L. Li, S. Chiba, Y. Wada, G. Panomsuwan and T. Ishizaki, J. Mater. Chem. A, 2017, 5, 2073–2082 RSC.
- M. A. Bratescu, O. Takai and N. Saito, J. Alloys Compd., 2013, 562, 74–83 CrossRef CAS.
- H. Calderon, A. Okonkwo, I. Estrada-Guel, V. Hadjiev, F. Alvarez-Ramírez and F. R. Hernández, Adv. Struct. Chem. Imaging, 2016, 2, 1–12 CrossRef.
- Y. Su, Y. Zhu, H. Jiang, J. Shen, X. Yang, W. Zou, J. Chen and C. Li, Nanoscale, 2014, 6, 15080–15089 RSC.
- C. Lv, B. Liang, K. Li, Y. Zhao and H. Sun, Biosens. Bioelectron., 2018, 117, 802–809 CrossRef CAS.
- F. T. Johra, J.-W. Lee and W.-G. Jung, J. Ind. Eng. Chem., 2014, 20, 2883–2887 CrossRef CAS.
- T. Daio, A. Staykov, L. Guo, J. Liu, M. Tanaka, S. M. Lyth and K. Sasaki, Sci. Rep., 2015, 5, 13126 CrossRef CAS.
- E. Westsson, S. Picken and G. Koper, Chem. Commun., 2019, 55, 1338–1341 RSC.
- A. Balkis and A. P. O'Mullane, Mater. Chem. Phys., 2014, 143, 747–753 CrossRef CAS.
- K. Hyun, T. Ueno, O. L. Li and N. Saito, RSC Adv., 2016, 6, 6990–6996 RSC.
- C. H. Choi, S. H. Park and S. I. Woo, J. Mater. Chem., 2012, 22, 12107–12115 RSC.
- G. Panomsuwan, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2015, 3, 9972–9981 RSC.
- P. R. Kidambi, C. Ducati, B. Dlubak, D. Gardiner, R. S. Weatherup, M.-B. Martin, P. Seneor, H. Coles and S. Hofmann, J. Phys. Chem. C, 2012, 116, 22492–22501 CrossRef CAS.
- P. Tan, W. Han, W. Zhao, Z. Wu, K. Chang, H. Wang, Y. Wang, N. Bonini, N. Marzari and N. Pugno, Nat. Mater., 2012, 11, 294–300 CrossRef CAS.
- S. Hajati and S. Tougaard, J. Surf. Anal., 2009, 15, 220–224 CrossRef CAS.
- Y. Zhu, H. Ji, H.-M. Cheng and R. S. Ruoff, Natl. Sci. Rev., 2018, 5, 90–101 CrossRef CAS.
- N. Thongwichit, O. L. H. Li, W. Yaowarat, N. Saito and U. Suriyapraphadilok, Jpn. J. Appl. Phys., 2015, 55, 01AE10 CrossRef.
- J. Kim, J. Chun, S.-G. Kim, H. Ahn and K. C. Roh, J. Electrochem. Sci. Technol., 2017, 8, 338–343 CrossRef CAS.
- D.-w. Kim, O. L. Li and N. Saito, Phys. Chem. Chem. Phys., 2014, 16, 14905–14911 RSC.
- G. Panomsuwan, N. Saito and T. Ishizaki, Carbon, 2016, 98, 411–420 CrossRef CAS.
- L. Xiao and W. F. Schneider, Chem. Phys. Lett., 2010, 484, 231–236 CrossRef CAS.
- N. J. Firet, M. A. Blommaert, T. Burdyny, A. Venugopal, D. Bohra, A. Longo and W. A. Smith, J. Mater. Chem. A, 2019, 7, 2597–2607 RSC.
- J.-P. Sylvestre, S. Poulin, A. V. Kabashin, E. Sacher, M. Meunier and J. H. Luong, J. Phys. Chem. B, 2004, 108, 16864–16869 CrossRef CAS.
- J. H. Kim, J. Y. Cheon, T. J. Shin, J. Y. Park and S. H. Joo, Carbon, 2016, 101, 449–457 CrossRef CAS.
- G. K. Reddy, C. Ling, T. C. Peck and H. Jia, RSC Adv., 2017, 7, 19645–19655 RSC.
- Y. Hao, M. S. Bharathi, L. Wang, Y. Liu, H. Chen, S. Nie, X. Wang, H. Chou, C. Tan, B. Fallahazad, H. Ramanarayan, C. W. Magnuson, E. Tutuc, B. I. Yakobson, K. F. McCarty, Y.-W. Zhang, P. Kim, J. Hone, L. Colombo and R. S. Ruoff, Science, 2013, 342, 720–723 CrossRef CAS.
- C. Detoni, A. R. P. da Silva and M. M. Souza, J. Porous Mater., 2020, 27, 1–11 CrossRef.
- J. Ma, A. Habrioux, Y. Luo, G. Ramos-Sanchez, L. Calvillo, G. Granozzi, P. B. Balbuena and N. Alonso-Vante, J. Mater. Chem. A, 2015, 3, 11891–11904 RSC.
- P. Kannan, T. Maiyalagan, N. G. Sahoo and M. Opallo, J. Mater. Chem. B, 2013, 1, 4655–4666 RSC.
- T. Holme, Y. Zhou, R. Pasquarelli and R. O'Hayre, Phys. Chem. Chem. Phys., 2010, 12, 9461–9468 RSC.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2006, 110, 23489–23496 CrossRef CAS.
- D. Zhan, J. Velmurugan and M. V. Mirkin, J. Am. Chem. Soc., 2009, 131, 14756–14760 CrossRef CAS.
- L. Zhang, L. Fan, P. Yang, M. Li, H. Zhang, Y. Tang, Z. Kang, H. Guo, R. Wang and D. Sun, Mater. Adv., 2020, 15, 2010–2018 RSC.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS.
- E. Odella, S. J. Mora, B. L. Wadsworth, J. J. Goings, M. A. Gervaldo, L. E. Sereno, T. L. Groy, D. Gust, T. A. Moore and G. F. Moore, Chem. Sci., 2020, 11, 3820–3828 RSC.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 125, 389–393 CrossRef.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS.
- Y. Peng and S. Chen, Green Energy Environ., 2018, 3, 335–351 CrossRef.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- Q. Yan, H. Toghiani and H. Causey, J. Power Sources, 2006, 161, 492–502 CrossRef CAS.
- S. Yu, X. Li, S. Liu, J. Hao, Z. Shao and B. Yi, RSC Adv., 2014, 4, 3852–3856 RSC.
- Y. Xue, J. M. Baek, H. Chen, J. Qu and L. Dai, Nanoscale, 2015, 7, 7078–7083 RSC.
- B. Najafi, P. Bonomi, A. Casalegno, F. Rinaldi and A. Baricci, Energies, 2020, 13, 3643 CrossRef CAS.
- B. Y. Kaplan, N. Haghmoradi, E. Biçer, C. Merino and S. A. Gürsel, Int. J. Hydrogen Energy, 2018, 43, 23221–23230 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00718h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Rinyarat
Naraprawatphong
a,
Rinyarat
Naraprawatphong