
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-regulated tough elastomers of liquid crystalline inorganic nanosheet/polyurethane nanocomposites†
Toki
Morooka
a,
Yutaka
Ohsedo
 *b,
Riki
Kato
c and
Nobuyoshi
Miyamoto
*a
*b,
Riki
Kato
c and
Nobuyoshi
Miyamoto
*a
aDepartment of Life, Environment and Applied Chemistry, the Graduate School of Engineering, Fukuoka Institute of Technology, 3-30-1, Wajirohigashi, Higashiku, Fukuoka 811-0295, Japan. E-mail: miyamoto@fit.ac.jp
bDepartment of Computer Science and Clothing Environment, Faculty of Human Life and Environment, Nara Women's University, Kitauoyahigashi-machi, Nara 630-8506, Japan. E-mail: ohsedo@cc.nara-wu.ac.jp
cDepartment of Material Science and Production Engineering, the Graduate School of Engineering, Fukuoka Institute of Technology, 3-30-1, Wajirohigashi, Higashiku, Fukuoka 811-0295, Japan
First published on 29th December 2020
Abstract
Liquid crystalline nanosheet/polyurethane composite elastomers with superior mechanical properties were successfully synthesized using the newly developed in situ polymerization technique. A layered clay mineral fluorohectorite was fully exfoliated into single-layer nanosheets and formed a nematic liquid crystalline (LC) state in the N,N′-dimethylformamide/water mixture added with the prepolymer, isocyanate-terminated low-molecular-weight poly(ethylene oxide). The composite elastomer fibers were obtained by injecting this LC mixture into a solution of a four-functional crosslinker molecule, triethylenetetramine, followed by solvent removal. In the composite fibers, the single-layer LC nanosheets were macroscopically aligned along the long axis of the fibers as revealed by polarizing microscope and small angle X-ray scattering. Tensile tests showed that the composite fibers have superior mechanical strength while maintaining a large breaking strain compared to the pristine polyurethane fibers without the nanosheets. The outstanding improvements were due to the physical crosslinks at multiple points between polymer chains and the single-layer nanosheets that are macroscopically ordered.
Introduction
Fabrication of nanocomposites with polymers and inorganic nanoparticles is quite an effective way to achieve extraordinary functions and properties of materials as exemplified by natural nacre, which is a CaCO3/polypeptide nanocomposite exhibiting toughness and light weight. Since the pioneering work on nylon-6/clay nanocomposites,4–6,8,9 inorganic nanosheets or stacked nanosheets obtained from layered crystals have been utilized as excellent nanofillers for nanocomposites due to their large specific surface area, large aspect ratio, low cost, and availability of a variety of materials with desired electric and optical properties.10,11 Their mechanical strength,7,9,12 gas barrier properties,5,13,14 thermal stability,6,7,15 and thermal conductivity16–20 are improved and their electrical and optical properties are also tunable when even a small amount of a layered crystal is composited with a polymer material.However, when a layered crystal is not fully exfoliated into single layers and is not dispersed homogeneously in composites, improvements of the materials' properties are limited; maximized properties along a certain direction and other smart properties are expected when the orientation and spatial distribution of the nanosheets are precisely controlled. Nevertheless, fabrication of such ideal nanocomposites is still very challenging.
In the most conventional synthesis of nanosheet/polymer nanocomposites,21 layered clay mineral powders intercalated with alkylammonium ions, organo-clays, are kneaded in molten polymers. Polymerization of monomers added with organo-clay crystals (in situ intercalative polymerization) is another general method. In these classical methods, however, the layered crystal is usually not fully exfoliated and the control of orientation and spatial distribution of nanosheets is not possible. Layer-by-layer deposition of thin composite films22 is a sophisticated technique to fabricate precisely designed heterostructures; however, only ionic polymers can be used and large scale synthesis is not possible. Doctor-blading,23 spray coating,24 vacuum filtration,25,26 or simply casting27 of polymer-coated nanosheets or polymer/nanosheet solution on a substrate and formation of regulated structures upon solvent evaporation have been also recently reported. In these methods, however, the structure formation depends on the solvent evaporation process so that precise structural control is not straightforward; kinetic control in the complicated situation is necessary in which the intrinsic phase separation of the polymer and nanosheet occurs due to the depletion effect,28 while partial isotropic–nematic transition is driven by the excluded volume effect29 and the viscosity increases.
Considering the above-mentioned situation, for the fabrication of ideal bulk nanocomposite materials with highly regulated structures, splitting of the synthesis process into (1) superstructure formation process of the fillers and (2) fixation processes by in situ polymerization, starting from nanosheet/prepolymer mixture, should be an effective strategy. In line with this strategy, we have been highlighting the structured liquid crystalline colloidal nanosheets since early 2000s.30 Due to the anisotropic shapes of nanosheets, the free rotational motion is restricted and an LC phase is formed when the nanosheet concentration is higher as explained by Onsager's theory.29 By tuning this entropic interaction as well as attractive and repulsive interactions between charged nanosheets, orientational30 and positional31 ordering of nanosheets is precisely controllable on mesoscopic scales in the colloidal state. Furthermore, electric,32 magnetic,33 and shear30 fields are effective to achieve a macroscopic alignment of these mesophases, leading to hierarchical structural control. After a regulated hierarchical superstructure of single-layer nanosheets is obtained, a monomer dissolved in the LC colloid is photo-polymerized to finally obtain a structured nanocomposite gel.34–36 In these gels, not only a great improvement in the mechanical strength but also anisotropic optical and molecular transport properties were demonstrated. However, it has been difficult to apply this synthetic technique to industrially important plastic or elastomer materials mainly composed of hydrophobic polymers because inorganic nanosheets are generally hydrophilic and not dispersed in organic solvents.7,37
In this study, we demonstrate a novel synthetic method to obtain LC single layer nanosheet/polyurethane composite elastomers in which exfoliated nanosheets are embedded, retaining the regulated superstructure of the original LC colloid. The new synthetic method (Fig. 1a) makes use of an isocyanate-terminated low-molecular-weight prepolymer (Fig. 1b) which is soluble in a water/polar organic solvent mixture that is also compatible with the LC nanosheets. The composite elastomer fiber was successfully obtained by extruding the nanosheet/prepolymer mixture into a four-functional crosslinker solution (Fig. 1c and Fig. S1, ESI†). Although, in general, an isocyanate moiety is hydrolyzed easily, it is more reactive with amino groups of the crosslinker triethylenetetramine than water so that the crosslinking reaction predominantly proceeds.38 Due to the shear stress in the extrusion process, the macroscopic orientation of the nanosheets was strongly induced, giving rise to mechanical properties. The present process is also characterized as a new-type of a wet-spinning system where a chemically crosslinked network polymer is obtained in situ in contrast to the conventional system where polymers or nanosheets are physically crosslinked by coaggregation with Ca2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39–41 or solidified by precipitation of a polymer in a poor solvent.
39–41 or solidified by precipitation of a polymer in a poor solvent.
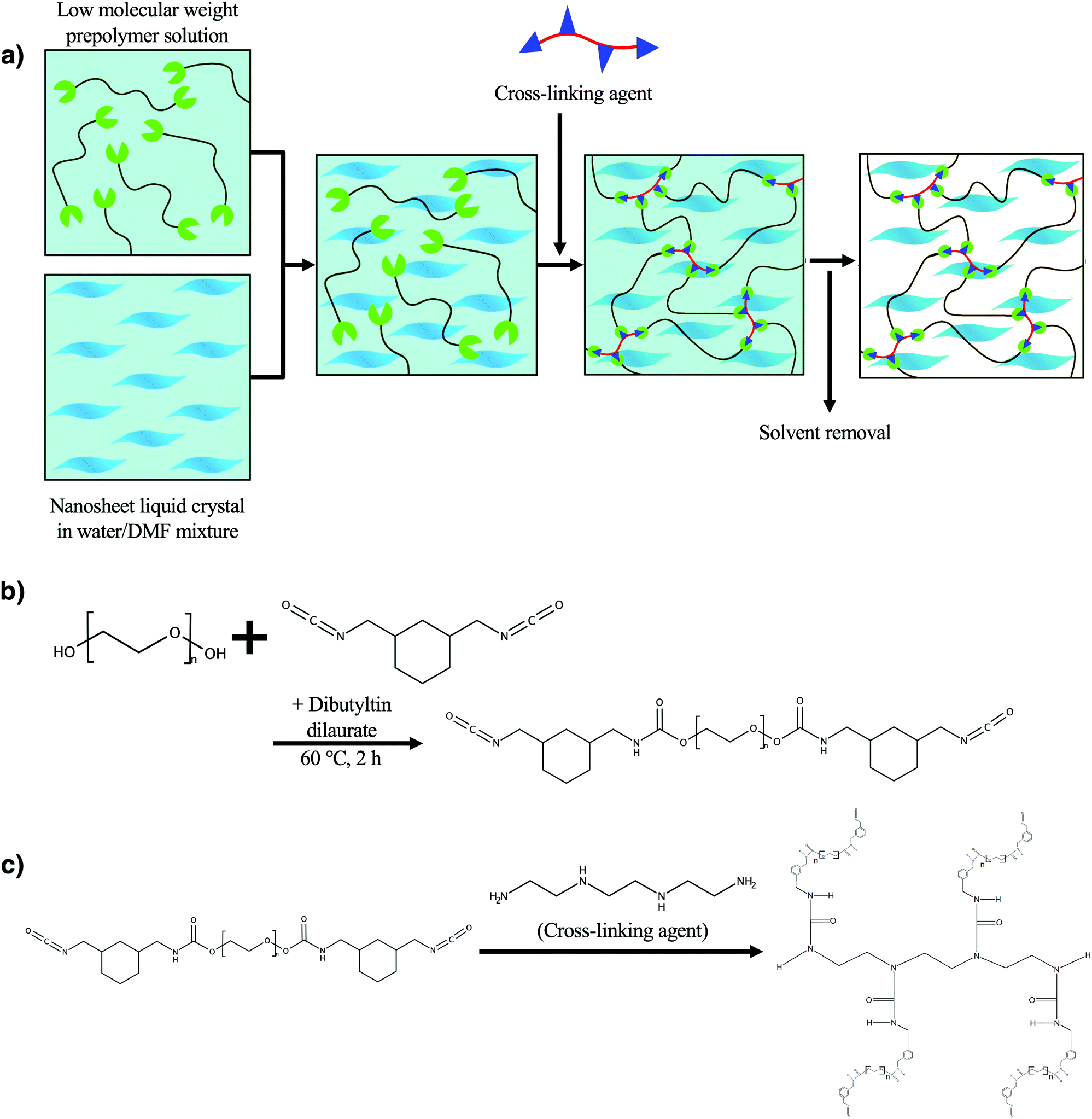 | ||
| Fig. 1 (a) Conceptual scheme of the present study and chemical reaction formula for the synthesis of (b) the prepolymer and (c) the crosslinked polyurethane. | ||
Experimental
Preparation of the nanosheet colloid
The aqueous sol of the synthetic clay mineral fluorohectorite (NHT-B2 SOL) was supplied by Topy Industries. The as-received aqueous sol was centrifuged at 15000 rpm for 1 h to remove impurities and diluted to 1 wt%, according to the previous report.42 Counter cations of the nanosheets were exchanged from Na+ to NH4+ by passing the nanosheet colloid through the column filled with ion exchange resin (Amberlite IR120B, Organo) that was pre-treated with 1 M NH4Cl aq. This aqueous colloid was added with four times volume of N,N-dimethylformamide (DMF, Tokyo Chemical Industry [TCI]) and centrifuged at 15000 rpm for 1 h, followed by removing the supernatant, to finally obtain a concentrated NH4-fluorohectorite nanosheet/water/DMF colloid. During the exfoliation process, mechanical treatment such as ultrasonication was not necessary. Different from van der Waals type layered crystals like BN and MoS2,43,44 it is known that ionic-type layered crystals such as Na-fluorohectorite are spontaneously exfoliated into single layers in water under mild conditions without hard mechanical treatment because of osmotic swelling.42 This was also the case for the present case: NH4-exchanged fluorohectorite in a DMF/water mixture.
Preparation of the nanosheet/polyurethane composite fiber
Polyurethane composite fibers were synthesized by the following method. First, 1.90 g (1.90 mmol) of poly(ethylene glycol) (PEG, Mw: 1000, Sigma-Aldrich) was placed in a N2-purged flask and dissolved in 4 mL of super dehydrated DMF. Then, 0.784 g (4.00 mmol) of 1,3-bis(isocyanatomethyl)cyclohexane (TCI) and 0.027 g (0.043 mmol) of dibutyltin dilaurate (TCI) were added and stirred at 60 °C for 2 h to obtain the solution of the prepolymer which have isocyanate groups at both terminals of the PEG (Fig. 1b). This prepolymer solution (1.00 g) was added to the nanosheet colloid (0.0941, 0.491, or 1.04 g) and extruded into a DMF solution (3.54 M) of the cross-linking agent triethylenetetramine (TCI) by using a syringe pump at a flow rate of 8 mL min−1 (Fig. S1, ESI†). At this time, the beaker containing the crosslinking agent solution was rotated by a turntable at 70 rpm. The cross-linking reaction spontaneously proceeded (Fig. 1c and Fig. S1, ESI†) upon the extrusion and the swollen cross-linked nanosheet/polyurethane composite fiber was obtained. The fiber was stored in the crosslinker solution for 1 day to allow completion of the crosslinking reaction. After the reaction, the composite fiber was stored in DMF for 1 day to remove the unreacted prepolymer and cross-linking agent and was dried in a vacuum oven at 70 °C for 2 hours to finally obtain nanosheet/polyurethane elastomer composite fiber samples.Characterization
For atomic force microscopy (AFM; AFM5000II, HITACHI) observation of the nanosheets, the nanosheet colloid diluted to 0.001 wt% was added dropwise and dried on a mica substrate. A polarized optical microscope (POM; OLYMPUS BX51) with crossed polarizers and a wave plate (530 nm) was used to observe the nanosheet colloid and the prepolymer solution enclosed in a cell having a thickness of 1 mm. The composite fibers were also observed using the same device. Small-angle X-ray scattering (SAXS) was performed using a Rigaku NANOPIX equipped with a CCD-type two-dimensional detector. For the SAXS observation, the nanosheet colloid and the prepolymer solution were enclosed in a glass capillary having a thickness of 0.01 mm and an optical path length of 2 mm, while the composite fiber was measured without a capillary. Field emission scanning electron microscopy (SEM) observation and energy dispersive X-ray analysis (EDS) were performed on a JEOL JSM-7100F with acceleration voltages of 1 and 10 kV, respectively. The thermogravimetric-differential thermal analysis (TG-DTA) of the composite fiber was carried out with a Rigaku TG8120 with a temperature increase rate of 2 °C min−1. The tensile test of the composite polymer was measured with a Shimadzu EZ-L at a tensile speed of 100 mm min−1.Results and discussion
The nanosheets were completely exfoliated in a DMF/water mixture (8:2) and an LC phase was formed even in the presence of the prepolymer. In the AFM image (Fig. S2, ESI†), many nanosheets with a thickness of 1 nm are observed and no stacked nanosheets or aggregates are found, confirming the complete exfoliation into single layers. Based on the AFM images, the average particle size of the nanosheets was evaluated to be around 2.5 μm. In the POM observation (Fig. S3a, ESI†), aggregated particles were not observed, while permanent birefringence and optical textures were observed, indicating that the nanosheets form an LC phase that occurs only when the degree of exfoliation is very high. Even after the addition of the prepolymer, good dispersion and the liquid crystal phase of the nanosheets were maintained (Fig. S3b, ESI†). In contrast, no birefringence was observed in the prepolymer solution without nanosheets (Fig. S3c, ESI†).
As shown in Fig. 2A, long fibers of the composite gel and composite elastomer were obtained continuously. The synthesized composite gel fiber (left image of Fig. 2A) was transparent and tough enough to be held with a tweezer, while it turned white and turbid after the evaporation of the solvent (right side image of Fig. 2A). The change in the sample appearance by solvent evaporation is explained by the formation of macroscopic wrinkles on the fiber surface as well as the formation of sub-micrometer scale heterogeneous domains of polyurethane/fluorohectorite intercalation compounds as described later.
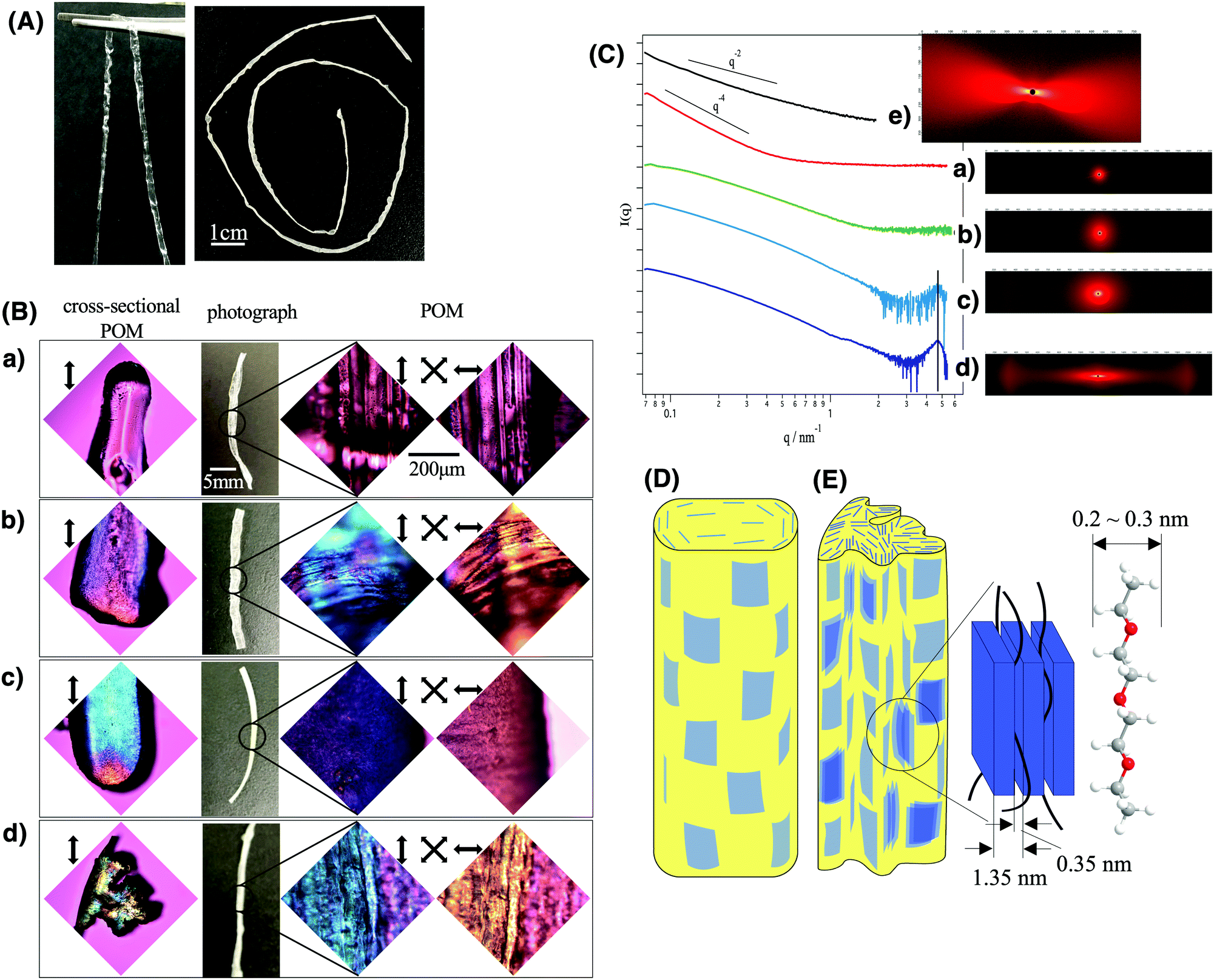 | ||
| Fig. 2 The structural characterization of the (a) pristine polyurethane fiber without nanosheets and nanosheet/polyurethane nanocomposite fibers loaded with (b) 1, (c) 5, and (d) 10 wt% of the nanosheet. (A) Shows the photographs of the composite gel fiber before (left) and after (right) drying. (B) shows the photographs and POM images of the dried samples. (C) shows the SAXS patterns (I–q profiles on the left and 2D patterns on the right) of the (a) pristine polyurethane, (b)–(d) nanocomposites and (e) the nanosheet (2.2 wt%)/water/DMF colloidal sol; the profiles (b)–(d) were obtained after subtraction of the pristine polyurethane scattering (a) as the background. (D) and (E) are the schematic structures of the nanosheet/polyurethane nanocomposite fibers containing smaller and larger amount of the nanosheets, respectively. | ||
In the POM images of the composite fibers (right side images in Fig. 2B), blue and yellow interference colors were observed when the long axis direction of the fiber is parallel and perpendicular to the wave plate, respectively. No aggregated particles were observed. This indicates that the nanosheets maintained a well-dispersed state and the superstructures of the original LC phase without aggregation even after being embedded in polyurethane and that the nanosheets are oriented along the long axis of the fiber. To further characterize the structure, the cross-section of the fibers was observed using a POM (left images of Fig. 2B). According to this observation, the nanosheets are oriented along the outer surface of the fiber. In the sample with 10 wt% of nanosheets, several domains with different orientational directions were formed inside the fiber.
SEM observations of the composite elastomer fiber further confirm that the nanosheets are well-dispersed in the polymer matrix without aggregation with the alignment flat along the fiber surface. In the cross-sectional SEM image (Fig. S4a–c, ESI†), rough surfaces with thin streaks of several μm length are observed. The corresponding EDS mapping image (Fig. S4e and f, ESI†) shows that Si due to nanosheets and C due to the polymer are uniformly distributed without any distinct texture, confirming that the nanosheets are well-dispersed in the polymer matrix without aggregation. The side surface of the composite fiber (Fig. S4d, ESI†) is covered with a platy object with a size of several μm. In contrast, in the case of elastomers without the nanosheets (Fig. S5, ESI†), flatter cross-sections and side surfaces are observed.
SAXS results (Fig. 2C) also revealed that the nanosheets maintained ideal dispersion and orientation in the obtained composite fiber, whereas they partly formed intercalation compounds with the polymer chains when the nanosheet concentration is high. In the nanosheet/polyurethane composites (Fig. 2C(b–d)) as well as LC colloids (Fig. 2C(e)), an anisotropic 2D SAXS pattern was observed and the anisotropy increased with increasing nanosheet concentration in contrast to the pristine polyurethane fiber without the nanosheets (Fig. 2C(a)) that show isotropic scattering. Note that the scattering due to nanosheets is much stronger than that due to polymers so that the scattering pattern of the composites gives information about the nanosheet superstructure but not about the polymer structure. These results suggest that the degree of orientation of the nanosheets increases as the concentration increases. In the I(q) vs. q scattering profiles, broad peaks were found in the wide-angle side for the samples with 5 wt% and 10 wt% of nanosheets (Fig. 3d and e). From the peak position (q = 4.6 nm−1), the d value is calculated to be 1.35 nm according to the relationship of d = 2π/q. This value is slightly larger than the reported value (1.23 nm) for Na-fluorohectorite.45 The gallery height between the nanosheet is calculated to be 0.37 nm by subtracting the thickness of the nanosheet (0.98 nm). Since the thickness of the polyurethane chain is estimated to be 0.2–0.3 nm, it is considered that the polyurethane chains are intercalated between the nanosheets. Furthermore, the slope of the profiles (Fig. 2C(b–e)) in the range of 0.1 < q < 1 was −2, which is ascribed to the form factor of the exfoliated nanosheets. Thus, most of the nanosheets retain a good dispersed state without aggregation even when compounded in the polyurethane.
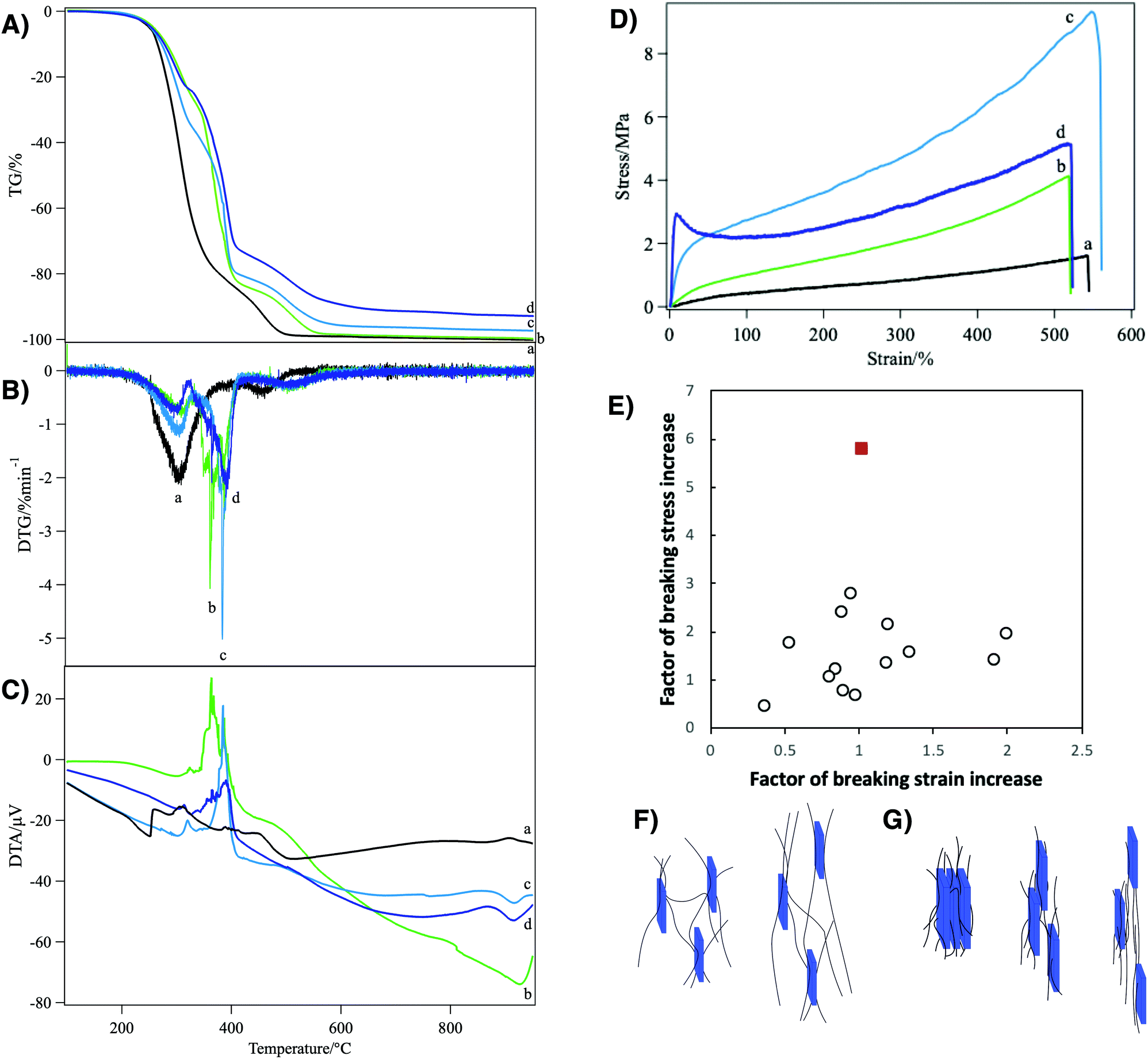 | ||
| Fig. 3 (A) TG, (B) DTG, (C) DTA, and (D) tensile stress–strain curves of the polyurethane nanocomposites loaded with (a) 0, (b) 1, (c) 5, and (d) 10 wt% of nanosheets. (E) Shows the comparison of the mechanical reinforcement factors of the present system (red filled square) with the clay/polymer composite elastomers reported in the literatures1–7 (open circles). The schematic representations of the reinforcement mechanism by (F) weak and (G) strong physical crosslinks in the nanosheet/polyurethane fiber are also shown. | ||
Considering the above-mentioned results, the schematic structures are deduced. The nanosheets in nematic order with a weak positional order are macroscopically aligned along the long axis of the fiber and maintain a fully exfoliated state. While they are mostly dispersed as single layers at low concentrations (Fig. 2D), part of the nanosheets form layered compounds intercalating polyurethane chains at higher nanosheet concentrations (Fig. 2E).
The composite elastomer showed improved thermal stability as characterized by TG-DTA (Fig. 3A and B). The pristine polyurethane without nanosheets showed two-step weight loss characterized by the two DTG peaks at around 300 °C and 460 °C accompanied by exothermal DTA peaks. In the present system, that is a network polymer crosslinked with an n-functional crosslinker, one PEG chain ideally has two terminal groups and 2/n crosslinker molecules. Thus, the weight ratio of the PEG chain to the total polymer weight, wPEG, is calculated as wPEG = MPEG/[MPEG + 2Mter + (2/n)Mcro], where MPEG, Mter and Mcr are the molecular weights of the PEG chain (1000), terminal group (196), and crosslinker (146), respectively. With n = 4, wPEG is calculated as 68% and this value roughly corresponds to the weight loss at the first DTG peak (82%). Thus, DTG peaks at 300 °C and 460 °C are ascribed to the combustion of PEG and other part of the polymer network, respectively. In the nanosheet/polyurethane nanocomposites, a new DTG peak appeared at the intermediate temperature (360–390 °C), replacing the original peak at 300 °C. This new peak can be ascribed to the combustion of the PEG moiety interacting with the nanosheets because the DTA peak as well as the DTG peak shifted to higher temperatures as the nanosheet concentration increased. Thus, it is probable that the presence of the nanosheets in the polyurethane matrix is effective to increase the combustion temperature due to heat absorption by the nanosheets as well as lowering of polymer chain motion through nanosheet–polymer interactions. Meanwhile, the residue weight at 900 °C corresponds to the weight of nanosheets in the present composite elastomer. This value increased with the increase of the loading amount of the nanosheet: 0.0, 0.2, 2.7, and 7.1 wt% for the samples loaded with 0, 1, 5 and 10 wt%. The slight difference between the measured value and the loaded amount is in the range of measurement error (±3 wt%), while it is also possible that a part of the loaded nanosheet escaped into the solution phase during the crosslinking process.
Due to the regulated structure and good dispersion of the nanosheets, the present nanocomposite elastomers showed outstanding mechanical properties as revealed by tensile tests (Fig. 3D). Pristine polyurethane fiber showed an elastic modulus of 0.0053 MPa and a stress–strain (S–S) curve typical of rubber materials,46 while it was broken at a stress of 1.6 MPa and a strain of 542%. With 1 wt% of the nanosheets, the elastic modulus, breaking stress, and breaking strain increased to 0.018 MPa, 4.1 MPa, and 519%, respectively. As the nanosheet concentration increased to 5 wt%, the elastic modulus increased to 0.14 MPa, while the breaking stress was 9.3 MPa and the breaking strain was almost unchanged (548%). With 10 wt% of the nanosheets, the S–S curve shows a yielding point at a strain of 8.4%, followed by stress relaxation. The elastic modulus and breaking stress were largely increased to 0.69 MPa and 5.2 MPa, respectively, while the breaking strain remained similar to other cases (520%). Thus, it is confirmed that the elastic modulus increases as the nanosheet concentration increases, while the breaking stress showed the maximum at a nanosheet concentration of 5 wt%.
Compared with previously reported clay/polymer nanocomposite materials, the above-mentioned mechanical reinforcement in the present system is outstanding. In the present system, the increase of the breaking stress and elastic modulus reach up to 5.9- and 26.8-fold, respectively. In addition, this mechanical improvement was achieved without a decrease of breaking strain. In contrast, in usual polymer/clay composites, the elastic modulus increases with the increase of clay loading whereas the breaking strain and breaking stress significantly decrease. This means that the material becomes harder but more brittle.11 In some good cases of nanocomposite elastomers,1,4–7,47,48 the breaking strain only slightly decreases or even increases; however, as shown in Fig. 3E, the factor of increase of breaking stress is only around 2 or lower, which is much smaller than in the present system.
The mechanical reinforcement in the present nanocomposite elastomer is explained by weaker and stronger physical crosslinks between the polyurethane chains and the nanosheets. Due to the lack of chemically modifiable group such as –OH on the surface of the clay mineral nanosheets, the formation of covalent chemical bonds is not plausible. Because the reinforcement is by physical crosslinks, not by covalent bonds, the crosslinks can slide and form again and again, avoiding reduction of breaking strain. The weaker crosslink is due to the hydrogen bonds between negatively charged nanosheets and polar polyurethane chains49 (Fig. 3F). Although the interaction is weak, very large surface area of the single layer nanosheets and the interactions at multiple points on the polymer chain and nanosheet surface make the physical crosslinks very effective. The mechanical strength improvement at lower nanosheet loading is mainly explained by this model. The stronger one is due to the formation of an intercalation compound in which polyurethane chains are sandwiched between nanosheets (Fig. 3G). It is considered that the steep increase of the tensile stress in the low strain region (Fig. 3D(d)) of the sample with 10 wt% nanosheets is due to the breaking of this stronger physical crosslink. After these strong physical crosslinks are mostly broken at the yielding point, the reinforcement by the weaker physical crosslink only works. Furthermore, the reinforcement by these physical crosslinks is maximized by the macroscopic ordering of the nanosheets. If the nanosheet plane were perpendicular to the stretching direction, that is the long axis of the fiber, the polymer chains should have been disturbed by the nanosheets so that the reinforcement effect is reduced.
Conclusions
Single-layer nanosheets that form an LC phase with highly regulated superstructures were successfully composited with a polyurethane elastomer by using the newly developed in situ polymerization technique for the first time. The LC nanosheet/polyurethane composite fibers showed superior thermal and mechanical properties due to good exfoliation and the regulated superstructure of the nanosheets originated form the LC phase. Since the present nanocomposite elastomer is synthesized by simple contact of the two solutions, production of thin films or small spherical particles as well as fibers would be possible. Due to the superior properties of the elastomer, a wide range of industrial applications in medical materials, smart fabrics, flexible electronics, and dielectric elastomer-based power generation devices are expected.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Research Center for Materials and Energy Devices of Fukuoka Institute of Technology (FIT-ME) (Strategic Research Foundation Grant-Aided Project for Private University [#S1511036L] from MEXT), KAKENHI (#24104005 and #15K05657), Network Joint Research Center for Materials and Devices (#20191350), and the Electronics Research Laboratory of Fukuoka Institute of Technology.References
- M. Pramanik, S. K. Srivastava, B. K. Samantaray and A. K. Bhowmick, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2065–2072 CrossRef CAS.
- Z. Bao, C. Flanigan, L. Beyer and J. Tao, J. Appl. Polym. Sci., 2015, 132, 41521 Search PubMed.
- Q. Zhang, Q. Wang and Y. Chen, J. Appl. Polym. Sci., 2013, 3930–3936 CAS.
- Z. Gu, G. Song, W. Liu, P. Li, L. Gao, H. Li and X. Hu, Appl. Clay Sci., 2009, 46, 241–244 CrossRef CAS.
- J. Y. Kim, W. C. Jung, K. Y. Park and K. D. Suh, J. Appl. Polym. Sci., 2003, 89, 3130–3136 CrossRef CAS.
- Y. Xu, W. J. Brittain, R. A. Vaia and G. Price, Polymer, 2006, 47, 4564–4570 CrossRef CAS.
- S. M. Liff, N. Kumar and G. H. McKinley, Nat. Mater., 2007, 6, 76–83 CrossRef CAS.
- A. Usuki, Y. Kojima, M. Kawasumi, A. Okada, Y. Fukushima, T. Kurauchi and O. Kamigaito, J. Mater. Res., 1993, 8, 1179–1184 CrossRef CAS.
- Y. Kojima, A. Usuki, M. Kawasumi, A. Okada, Y. Fukushima, T. Kurauchi and O. Kamigaito, J. Mater. Res., 1993, 8, 1185–1189 CrossRef CAS.
- P. D. Michael Alexandre, Mater. Sci. Eng., A, 2000, 28, 1–63 CrossRef.
- S. Pavlidou and C. D. Papaspyrides, Prog. Polym. Sci., 2008, 33, 1119–1198 CrossRef CAS.
- S. Gaidukov, R. D. Maksimov, U. Cabulis, E. Plume and A. Stunda-Zujeva, Mech. Compos. Mater., 2013, 49, 333–344 CrossRef CAS.
- T. Ebina and F. Mizukami, Adv. Mater., 2007, 19, 2450–2453 CrossRef CAS.
- M. W. Moller, T. Lunkenbein, H. Kalo, M. Schieder, D. A. Kunz and J. Breu, Adv. Mater., 2010, 22, 5245–5249 CrossRef.
- J. W. Gilman, C. L. Jackson, A. B. Morgan, R. H. Jr., E. M. E. P. G. M. Wuthenow, D. Hilton and S. H. Phillips, Chem. Mater., 2000, 12, 1866–1873 CrossRef CAS.
- Z. Kuang, Y. Chen, Y. Lu, L. Liu, S. Hu, S. Wen, Y. Mao and L. Zhang, Small, 2015, 11, 1655–1659 CrossRef CAS.
- Z. Zhu, C. Li, E. Songfeng, L. Xie, R. Geng, C.-T. Lin, L. Li and Y. Yao, Compos. Sci. Technol., 2019, 170, 93–100 CrossRef CAS.
- C. Yu, J. Zhang, Z. Li, W. Tian, L. Wang, J. Luo, Q. Li, X. Fan and Y. Yao, Composites, Part A, 2017, 98, 25–31 CrossRef CAS.
- J. Zhang, X. Wang, C. Yu, Q. Li, Z. Li, C. Li, H. Lu, Q. Zhang, J. Zhao, M. Hu and Y. Yao, Compos. Sci. Technol., 2017, 149, 41–47 CrossRef CAS.
- X. Wu, Z. Yang, W. Kuang, Z. Tang and B. Guo, Composites, Part A, 2017, 94, 77–85 CrossRef CAS.
- N. T. Skipper, A. K. Soper, J. D. C. McConnell and K. Refson, Chem. Phys. Lett., 1990, 166, 141–145 CrossRef CAS.
- Z. Tang, N. A. Kotov, S. Magonov and B. Ozturk, Nat. Mater., 2003, 2, 413–418 CrossRef CAS.
- E. S. Tsurko, P. Feicht, F. Nehm, K. Ament, S. Rosenfeldt, I. Pietsch, K. Roschmann, H. Kalo and J. Breu, Macromolecules, 2017, 50, 4344–4350 CrossRef CAS.
- M. Wong, R. Ishige, K. L. White, P. Li, D. Kim, R. Krishnamoorti, R. Gunther, T. Higuchi, H. Jinnai, A. Takahara, R. Nishimura and H. J. Sue, Nat. Commun., 2014, 5, 3589–3600 CrossRef.
- T. Verho, M. Karesoja, P. Das, L. Martikainen, R. Lund, A. Alegria, A. Walther and O. Ikkala, Adv. Mater., 2013, 25, 5055–5059 CrossRef CAS.
- H. B. Yao, Z. H. Tan, H. Y. Fang and S. H. Yu, Angew. Chem., Int. Ed., 2010, 49, 10127–10131 CrossRef CAS.
- P. Das, J. M. Malho, K. Rahimi, F. H. Schacher, B. Wang, D. E. Demco and A. Walther, Nat. Commun., 2015, 6, 5967–5980 CrossRef.
- F. M. van der Kooij, M. Vogel and H. N. W. Lekkerkerker, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 5397–5402 CrossRef CAS.
- L. Onsager, Ann. N. Y. Acad. Sci., 1949, 51, 627–659 CrossRef CAS.
- N. Miyamoto and T. Nakato, Adv. Mater., 2002, 14, 1267–1270 CrossRef CAS.
- J. C. P. Gabriel, F. Camerel, B. J. Lemaire, H. Desvaux, P. Davidson, W. Michael and P. Batail, Nature, 2001, 413, 504–508 CrossRef CAS.
- D. Ivan, P. Erwan, D. Partrick, A. Krassimira, B. Isabelle, B. Cristophe and J. M. Laurent, J. Phys. Chem. B, 2011, 115, 7751–7765 CrossRef.
- L. J. Michot, I. Bihannic, S. Maddi, S. S. Funari, C. Baravian, P. Levitz and P. Davidson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16101–16104 CrossRef CAS.
- N. Miyamoto, M. Shintate, S. Ikeda, Y. Hoshida, Y. Yamauchi, R. Motokawa and M. Annaka, Chem. Commun., 2013, 49, 1082–1084 RSC.
- T. Inadomi, S. Ikeda, Y. Okumura, H. Kikuchi and N. Miyamoto, Macromol. Rapid Commun., 2014, 35, 1741–1746 CrossRef CAS.
- M. Shintate, T. Inadomi, S. Yamamoto, Y. Kuboyama, Y. Ohsedo, T. Arimura, T. Nakazumi, Y. Hara and N. Miyamoto, J. Phys. Chem. B, 2018, 122, 2957–2961 CrossRef CAS.
- C. A. Harper, Handbook of Plastics, Elastomers, and Composite, McGraw Hill Companies, Inc., United States of America, 4th edn, 2002 Search PubMed.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS.
- Z. Xu and C. Gao, Nat. Commun., 2011, 2, 571–579 CrossRef.
- D. Kiriya, R. Kawano, H. Onoe and S. Takeuchi, Angew. Chem., Int. Ed., 2012, 51, 7942–7947 CrossRef CAS.
- X. Hu, Z. Xu, Z. Liu and C. Gao, Sci. Rep., 2013, 3, 2374–2381 CrossRef.
- N. Miyamoto, H. Iijima, H. Ohkubo and Y. Yamauchi, Chem. Commun., 2010, 46, 4166–4168 RSC.
- Y. Yao, Z. Lin, Z. Li, X. Song, K.-S. Moon and C.-P. Wong, J. Mater. Chem., 2012, 22 Search PubMed.
- Y. Yao, L. Tolentino, Z. Yang, X. Song, W. Zhang, Y. Chen and C.-P. Wong, Adv. Funct. Mater., 2013, 23, 3577–3583 CrossRef CAS.
- H. Kalo, M. W. Möller, M. Ziadeh, D. Dolejš and J. Breu, Appl. Clay Sci., 2010, 48, 39–45 CrossRef CAS.
- C. Hepburn, Polyurethane elastomers, Elsevier Science Publishers Ltd, 2nd edn, 1992 Search PubMed.
- Z. Bao, C. Flanigan, L. Beyer and J. Tao, J. Appl. Polym. Sci., 2015, 132, 41521 Search PubMed.
- S. B. Bae, C. K. Kim, Y. C. Kim and I. J. Chung, J. Appl. Polym. Sci., 2013, 129, 3089–3095 CrossRef CAS.
- S. Ahmed and F. R. Jones, J. Mater. Sci., 1990, 25, 4933–4942 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/d0ma00768d |
| This journal is © The Royal Society of Chemistry 2021 |