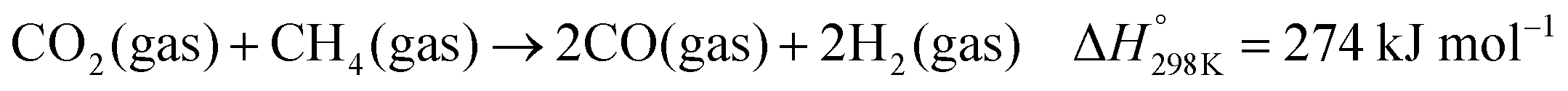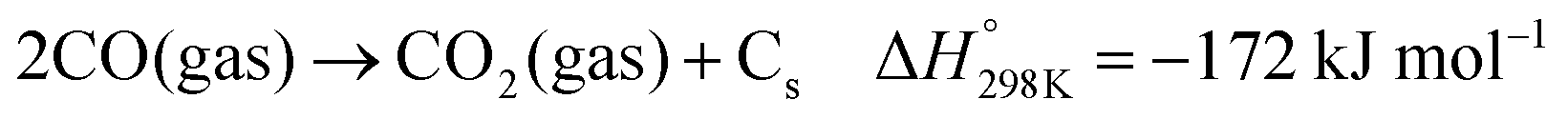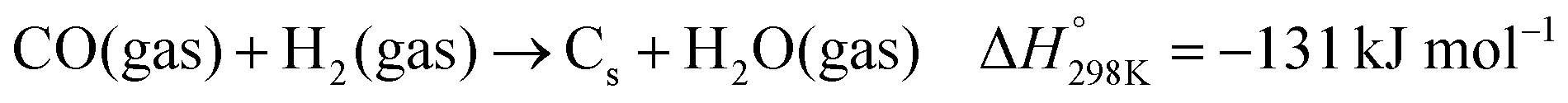DOI:
10.1039/D0MA00917B
(Paper)
Mater. Adv., 2021,
2, 1715-1730
Silicon oxycarbonitride ceramic containing nickel nanoparticles: from design to catalytic application†
Received
23rd November 2020
, Accepted 24th January 2021
First published on 25th January 2021
Abstract
Nickel-containing silicon oxycarbonitride ceramic nanocomposites are synthesized from hydrous nickel acetate and poly(vinyl)silazane (Durazane 1800) or perhydropolysilazane NN120-20 (A) (PHPS). A room temperature chemical reaction results in Ni-containing polysilazane precursors which are transformed into ceramic nanocomposites with nickel nanoparticles (2–4 nm) upon pyrolysis at elevated temperatures (700–1100 °C) under an argon atmosphere. The ceramic nanocomposites derived from the Durazane 1800-Ni precursor by the thermolysis process at 700 and 900 °C manifest a microporous structure with a BET specific surface area of ∼361 and ∼232 m2 g−1, respectively. In contrast, all pyrolyzed samples derived from the PHPS-Ni precursor exhibit a nonporous structure. The Ni/SiOCN ceramic nanocomposites – tested in a plug-flow fixed-bed reactor – display significant catalytic activity in dry methane reforming to syngas. The highest CH4 reaction rate of 0.18 mol min−1 gNi−1 is observed at 800 °C for the sample derived from the PHPS-Ni precursor by pyrolysis at 900 °C. All these make the materials developed in this work, i.e. nickel nanoparticles in situ formed in the SiOCN ceramic matrix, as promising candidates for heterogeneous catalysis.
Introduction
Metal nanoparticles can be used for a wide range of applications varying from spintronics to electrocatalysis and heterogeneous catalysis.1–5 However, the low chemical stability and sintering of metal nanoparticle catalysts under operating conditions limit their practical applications. These problems have been solved by embedding these metal nanoparticles into ceramic supports such as silica,6 alumina,7 zirconia,8 and other metal oxides.9–14 Although these supports are commercially available, they display several drawbacks, such as poor thermal conductivity and chemical reactivity at high temperatures. Silicon-based polymer-derived ceramics (PDCs), which are well known for their excellent thermomechanical properties, high thermal stability, and high resistance towards corrosion and oxidation under extreme environmental conditions,15–17 might be a promising candidate for replacing these commercial supports. Micro-mesoporous PDCs with high surface areas have been reported as promising catalytic supports.18–20 For instance, metal-containing (e.g. Co and Ni) SiOC nanocomposites with hierarchical porosity, derived from polysiloxane polymers, show high catalytic activity for CO2 methanation and Fischer–Tropsch synthesis.18,20–24 Similarly, some transition metal-containing (e.g. Cu, Ru, Pd, and Ir) SiCN nanocomposites have been reported as heterogeneous catalysts for liquid-phase oxidation,25 hydrogeneration,26,27 and sustainable synthesis.28
Silicon-based PDCs have been synthesized in single- and multi-phase as well as nanocomposite structures by thermal pyrolysis of organosilicon polymer precursors denoted as preceramic polymers.29–31 Metal-containing PDC nanocomposites have been commonly prepared by direct mixing of preceramic polymers with metal/metal oxide powders, followed by thermolysis under a controlled atmosphere.32–35 However, this approach might lead to pore blocking effects, heterogeneous distributions, and agglomeration of the metal nanoparticles in the ceramic supports. Therefore, the in situ formation of metal nanoparticles during the synthesis of the microporous/mesoporous PDC matrix has been alternatively applied. This approach can be achieved by the thermolysis of (i) commercially available metallopolymers or (ii) organosilicon polymers modified by metallorganic compounds. Metallopolymers are usually pyrolyzed at relatively low temperatures. For instance, uniform metallic α-Fe nanoparticles with an average size of 4 nm have been in situ formed in the SiC matrix during the thermolysis of poly(ferrocenylsilanes) (PFSs) at 700 °C under a nitrogen atmosphere.36 However, this synthesis approach is greatly restricted by the variety of available metallopolymers. In contrast, the chemical modification of preceramic polymers with metallorganic compounds has been widely used because most of the organosilicon polymers contain functional groups, such as Si–H, N–H, and –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, which can easily react with metallorganic compounds to obtain metal-modified polymer precursors. Metal carbonyls,37 metal alkoxides,38 metal acetylacetonates,39 and bis(cyclopentadienyl) metal dichlorides (Cp2MCl2)40 are a few examples of metallorganic compounds that can react with preceramic polymers. Nevertheless, some metallorganic compounds are extremely toxic, and harsh reaction conditions are usually required. Recently, Motz et al. have developed a molecular approach to incorporate transition metals (e.g. Fe, Ni, Cu, and Pt) into the PDC matrix by the chemical modification of polysilazane HTT1800 with aminopyridinato metal complexes.25,41–43In situ formation of nickel nanoparticles during the reaction of the polysilazane HTT1800 polymer with trans-[bis(2-aminoethanol-N,O)diacetato-nickel(II)] in an ice bath under an argon atmosphere appeared to be a quite complex process due to the complicated and time-consuming synthesis of the nickel(II)-containing complex.44,45
CH2, which can easily react with metallorganic compounds to obtain metal-modified polymer precursors. Metal carbonyls,37 metal alkoxides,38 metal acetylacetonates,39 and bis(cyclopentadienyl) metal dichlorides (Cp2MCl2)40 are a few examples of metallorganic compounds that can react with preceramic polymers. Nevertheless, some metallorganic compounds are extremely toxic, and harsh reaction conditions are usually required. Recently, Motz et al. have developed a molecular approach to incorporate transition metals (e.g. Fe, Ni, Cu, and Pt) into the PDC matrix by the chemical modification of polysilazane HTT1800 with aminopyridinato metal complexes.25,41–43In situ formation of nickel nanoparticles during the reaction of the polysilazane HTT1800 polymer with trans-[bis(2-aminoethanol-N,O)diacetato-nickel(II)] in an ice bath under an argon atmosphere appeared to be a quite complex process due to the complicated and time-consuming synthesis of the nickel(II)-containing complex.44,45
In our previous work, we designed and successfully synthesized several M/SiOCN nanocomposites (M = Mn, Fe, Co, Cu, Zn, and Ag) by chemical reaction of poly(vinyl)silazane (Durazane 1800) with metal acetates followed by pyrolysis of as-derived precursors.46 Most of the metal nanoparticles were in situ formed in the synthesized precursors during the reaction at room temperature. Transition metals (Fe and Co) catalyzed the cross-linking of polymer precursors, which in turn increased the final ceramic yield. Compared with previous approaches, our synthesis method appeared to be advantageous, owing to the very simple reaction conditions (in an ice bath under an argon atmosphere), the use of rather inexpensive poly(vinyl)silazane (Durazane 1800), and easily accessible commercial metal acetate materials. Accordingly, in the present work, we extend our approach to synthesize Ni-containing precursors by reacting nickel(II) acetate tetrahydrate with either poly(vinyl)silazane (Durazane 1800) or carbon-free perhydropolysilazane NN120-20 (A) (PHPS). These two polymers are chosen for the present study because they possess different molecular structures, which allows us to study the influence of the hydrocarbon side groups, such as methyl and vinyl groups of Durazane 1800 on the chemical compositions, microstructures and catalytic properties of the resulting ceramic nanocomposites. We mainly focus on the clarification of the reaction mechanism between nickel(II) acetate tetrahydrate and polysilazanes through comprehensive characterizationof the synthesized Ni-containing precursors. In addition, the polymer-to-ceramic transformation process, phase compositions, and porosity development in the pyrolyzed ceramic nanocomposites are investigated. The catalytic performance of obtained Ni/SiOCN ceramic nanocomposites (porous SiOCN ceramic matrix-supported very fine Ni nanoparticles) towards dry reforming of methane (DRM) with carbon dioxide is evaluated for the first time. As compared with previous studies on Ni-based catalysts,11,12,47–53 the obtained Ni/SiOCN catalysts show remarkable catalytic activity towards the DRM process even though the catalyst configuration was not optimized for this particular reaction. Thus, the in situ formation of nickel nanoparticles in the PDC matrix can be a promising approach to prepare heterogeneous catalysts for the DRM process.
Experimental methods
Materials
Nickel(II) acetate tetrahydrate (Ni(Ac)2·4H2O, ≥99.9) and anhydrous tetrahydrofuran (THF, ≥99.99%) were obtained from Sigma-Aldrich. Poly(vinyl)silazane Durazane 1800 and NN120-20 (A) that is a 20 wt% solution of perhydropolysilazane (PHPS) in di-n-butyl ether were purchased from durXtreme GmbH, Germany. Argon gas used during both the reaction and pyrolysis steps was used as received (>99.999%).
Synthesis of Ni-containing polysilazane precursors
All synthesis reactions were carried out according to the following procedures reported recently:46 4 g Durazane 1800 or 20 g NN120-20 (A) (i.e. 4 g PHPS) was first dissolved in 50 mL anhydrous THF and then added into a dropping funnel. Similarly, 1.6 g Ni(Ac)2·4H2O was dispersed in 50 mL anhydrous THF in another flask. The flask and dropping funnel were then assembled in a glovebox without allowing both reactants to be mixed. The reaction assembly was immediately removed from the glovebox. The flask containing nickel acetate tetrahydrate solution was connected to Schlenk-lines and submerged in an ice bath (−6 °C). Polymer and nickel acetate tetrahydrate were then mixed in the flask by opening the valve of the dropping funnel under slow argon flow. The synthesis process continued under slow argon flow and was stirred for 24 h until the end of gas liberation. Finally, the THF solvent was removed under vacuum for at least 5 h, and the synthesized Ni-containing precursors were collected in the glovebox. The synthesized Ni-containing precursors derived from Durazane 1800 and PHPS were denoted as Du1800-Ni and PHPS-Ni precursors, respectively.
Synthesis of Ni/SiOCN ceramic nanocomposites
Ceramic nanocomposites were obtained by the thermolysis of the synthesized Du1800-Ni and PHPS-Ni precursors in a tube furnace using a quartz Schlenk tube under flowing argon (approximately 40 mL min−1) at three different pyrolysis temperatures (700, 900, and 1100 °C). In a typical pyrolysis route, the obtained Ni-containing precursors were heated from room temperature to 250 °C with a rate of 50 °C h−1, held for 2 h at 250 °C, heated from 250 °C to a final pyrolysis temperature (700, 900 or 1100 °C) with a rate of 50 °C h−1 and held for 3 h at this temperature and finally cooled to room temperature with a rate of 180 °C h−1. The ceramic nanocomposites obtained by pyrolyzing the Du1800-Ni precursor at 700, 900, and 1100 °C are labeled as 700Ar-Du1800-Ni, 900Ar-Du1800-Ni, and 1100Ar-Du1800-Ni, respectively, while, the samples obtained at the same temperatures but from the PHPS-Ni precursor are denoted as 700Ar-PHPS-Ni, 900Ar-PHPS-Ni and 1100Ar-PHPS-Ni, respectively. The schematic of the synthesis of Ni/SiOCN ceramic nanocomposites is shown in Fig. S1 (ESI†).
Catalytic tests
Catalytic tests were performed using a plug-flow fixed-bed reactor. For a given test, 25 mg of catalyst was diluted using quartz (washed and calcined, 200–800 μm particle size, from Merck) to a total volume of 0.5 cm3 and implemented inside a quartz tubular reactor (4 mm inner diameter, 400 mm of length). The reactor was mounted inside a Reetz GmbH oven, and a k-type thermocouple placed at the packed-bed height next to the reactor wall allowed for temperature control. Gas flow was controlled with different mass flow controllers (MFCs) by Bronkhorst High-Tech B.V. CH4 (purity of 99.95%), CO2 (purity of 99.995%), H2 (purity of 99.999%), and N2 (99.999%) gases were purchased from Air Liquide. All the catalytic tests were performed at reactant ratios of CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2:N2 = 1:1:3 and GHSV = 120000 N mL h−1 gcat−1. Heating to 500 °C and cooling after catalysis were performed under the N2 flow of 30 N mL min−1 and 20 °C min−1. Before testing, the catalysts were in situ reduced at 500 °C using a 10% H2:N2 (v/v) of 30 N mL min−1 for 1 hour. Gas characterization was performed using a 7890A gas chromatograph (GC) from Agilent Technologies equipped with FID and TCD detectors. Prior to testing, the GC was calibrated using calibration gas bottles purchased from Deuste Gas Solutions. To ensure reproducibility, the back pressure of the GC was controlled using a Swagelok K-series pressure regulator. A carbon balance was performed to all data points and was always above 95%.
:CO2:N2 = 1:1:3 and GHSV = 120000 N mL h−1 gcat−1. Heating to 500 °C and cooling after catalysis were performed under the N2 flow of 30 N mL min−1 and 20 °C min−1. Before testing, the catalysts were in situ reduced at 500 °C using a 10% H2:N2 (v/v) of 30 N mL min−1 for 1 hour. Gas characterization was performed using a 7890A gas chromatograph (GC) from Agilent Technologies equipped with FID and TCD detectors. Prior to testing, the GC was calibrated using calibration gas bottles purchased from Deuste Gas Solutions. To ensure reproducibility, the back pressure of the GC was controlled using a Swagelok K-series pressure regulator. A carbon balance was performed to all data points and was always above 95%.
Characterization methods
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) measurements of the synthesized Ni-containing precursors and pyrolyzed samples were measured from 550 cm−1 to 4000 cm−1 in the glovebox using Nicolet iS5 (Thermo Fisher Scientific, USA) and Specac Golden Gate (Waltham, MA, USA) with a diamond plate. X-ray photoelectron spectroscopy (XPS) was performed on an ESCALAB 250Xi (Thermo Fisher Scientific, USA). The size of the X-ray spot on the sample was 100 μm. The samples for X-ray photoelectron measurement were prepared by sprinkling a small amount of sample powders onto the surface of sticky carbon conductive tape on the sample holder in the glovebox. All XPS spectra were calibrated using the C 1s core line with a binding energy of 284.8 eV. The released gases during the synthesis were determined using a gas chromatography system (Agilent 7820A, USA) equipped with a thermal conductivity detector. Synchrotron X-ray diffraction (XRD) measurements were carried out at the beamline 12.2.2 of the Advanced Light Source (Lawrence National Berkeley Laboratory, California, USA) using monochromatic synchrotron radiation with λ = 0.495 Å (25 keV/30 μm spot size) in the angle-dispersive transmission mode using an RDI flat panel detector with the dark image and strain correction. The patterns were measured on the sample powder placed in 0.7 mm capillaries. For the in situ synchrotron XRD experiments, about 1 mg of the catalysts powder was heated in a 0.7 mm outer diameter quartz capillary (Hilgenberg GmbH, Germany) under quasi flowing conditions (CH4:CO2:N2 = 1:1:3 and 1:2:3). The gases were injected through a 0.5 mm outer diameter tungsten tube. The capillary is heated at a 10 °C min−1 heating rate in an infrared heated SiC tube furnace as described elsewhere.54,55 The patterns were recorded using a PerkinElmer flat panel detector (XRD 1621, dark image, and strain correction). A NIST 660b LaB6 standard was used to calibrate the measurement. Elemental analysis (for Si and Ni) was performed with inductively coupled plasma optical emission spectroscopy (ICP-OES) in a Horiba Scientific ICP Ultima2 (Horiba, Japan). The powder samples were digested in an aqueous suspension with the addition of HNO3 and HF at 200 °C for 5 h in an autoclave. The polymer to ceramic conversion was investigated in an argon atmosphere with a heating rate of 5 °C min−1 with thermal gravimetric analysis (TGA) on a STA 409 PC LUXX (Netzsch, Germany) coupled with a mass spectrometer OMNi Star GSD 320 (Pfeiffer Vacuum, Germany). Solid-state nuclear magnetic resonance (NMR) spectra were recorded with a Bruker Avance 400 MHz spectrometer operating at 100.56 MHz for 13C, 79.44 MHz for 29Si, and 399.88 MHz for 1H. 1H–13C and 1H–29Si cross-polarization magic angle spinning (CP-MAS) NMR experiments were carried out at a MAS rate of 10 kHz using a 4 mm MAS HX double-resonance probe. The 1H π/2 pulse length was 3.1 μs, and two pulse phase modulation (TPPM) heteronuclear dipolar decoupling was used during acquisition. The spectra were measured using a contact time of 2.0 ms and a recycle delay of 2 s. All 13C and 29Si spectra are referenced to those of external tetramethylsilane (TMS) at 0 ppm for 13C and 29Si using adamantane and tetrakis(trimethylsilyl)silane (TKS) as secondary references, respectively. The finely ground sample powders were inserted into the NMR rotor with a funnel in the glovebox. Transmission electron microscopy (TEM) images and energy-dispersive X-ray spectroscopy (EDX) spectra were obtained using an FEI Tecnai G2 20 S-TWIN equipped with a LaB6-source at 200 keV acceleration voltage (FEI, USA). The samples for TEM analysis were prepared in the glovebox by dispersion in anhydrous THF, followed by dropping them onto a copper grid covered with a carbon film. Particle size distribution was calculated from TEM images using the Nano measurer software by quantifying 100 particles. X-ray absorption near-edge spectroscopy (XANES) was performed in the transmission mode at beamline KMC-2 of the BESSY-II synchrotron light source at Berlin, Germany, equipped with a graded Si–Ge(111) double crystal monochromator to characterize the oxidation states of Ni in the synthesized Ni-containing precursors.56 The samples were prepared in the form of powder applied onto adhesive Kapton tape. High harmonics were rejected by detuning the monochromator so that the intensity of the beam on the samples was 65% of the maximum possible intensity. Reference spectra were simultaneously measured for energy calibration. X-ray absorption fine structure (XAFS) measurements were carried out with a novel self-developed wavelength-dispersive spectrometer in von Hámos geometry to characterize the oxidation states of Ni in the pyrolyzed Ni/SiOCN ceramic nanocomposites.57 The spectrometer was equipped with a microfocus X-ray tube, a curved Highly Annealed Pyrolytic Graphite mosaic crystal, and a hybrid photon counting CMOS detector with 512 × 1030 pixel and a pixel size of 75 μm × 75 μm. The tube was operated with a high voltage of 16 kV and a current of 200 μA. The references (Ni foil, Ni2Si, and NiO) were prepared as powders on scotch tape while the samples were prepared as wax-pellets (with Hoechst wax C), due to their lower concentration of Ni, with 13 mm pellet diameter. All references and samples were constantly moved during the measurements to minimize the effects of local thickness inhomogeneity. The beam size on the samples was around 3 mm × 3 mm. The gathered spectral range covered the Ni K absorption edge at 8332 eV. The EXAFS evaluation was performed by using the Demeter software.58 Nitrogen sorption measurements were carried out using a QuadraSorb Station 4 apparatus (Quantachrome, USA). Isotherms were recorded at 77 K after degassing under vacuum at 150 °C for 12 h before the actual measurement. The surface area was calculated using Brunauer–Emmett–Teller (BET) calculations. All nitrogen sorption data were analyzed using the Quantachrome/QuadraWin software version 5.05.
Results and discussion
Chemical structure and formation mechanisms of the synthesized Ni-containing polysilazane precursors
As displayed in Fig. 1, the ATR-FTIR spectrum of Durazane 1800 shows the characteristic absorption bands of poly(vinyl)silazane: the bands attributed to –Si–NH–Si– groups such as N–H stretching at 3382 cm−1 and the vibration of the NH unit bridging two silicon atoms at 1162 cm−1. The bands related to vinyl silyl groups (CH2CH–Si–) are also observed as C–H vibrations at 3042 and 3006 cm−1, CC stretching at 1590 cm−1, and scissoring of terminal methylene at 1402 cm−1. The strong absorption band assigned to Si–H vibration appears at 2116 cm−1. The main characteristic band of Si–CH3 groups is located at 1255 cm−1 with two bands at 2953 and 2896 cm−1 corresponding to the C–H stretching.44,59 Similarly, the ATR-FTIR spectrum of PHPS shows the bands associated with N–H (3382 cm−1), Si–H (2146 cm−1) and Si–NH–Si (1170 cm−1). A remarkable decrease in the intensity of absorption bands corresponding to Si–H and –NH– (in –Si–NH–Si–) groups is observed in the ATR-FTIR spectra of synthesized Du1800-Ni and PHPS-Ni precursors. Moreover, new bands related to the CO vibration (1712 cm−1) and acetate group (1561, 1414, and 1368 cm−1) are detected in the spectra of both precursors. The absorption bands corresponding to CO vibration can be mainly attributed to the formed Si–OOCCH3 in the synthesized precursors. These results suggest the successful chemical reaction between the nickel acetate tetrahydrate with both polymers.46 Additionally, for the synthesized Du1800-Ni precursor, the absorption bands at 3042 and 3006 cm−1 that were attributed to the vinyl groups vanished, which can be explained by a hydrosilylation reaction between Si–H and –Si–CHCH2. Although the hydrosilylation reaction usually takes place at T > 120 °C in pure polysilazanes,60,61 previous works showed that the presence of inorganic catalysts such as transition metals or metal complexes could remarkably lower this reaction temperature.44,46
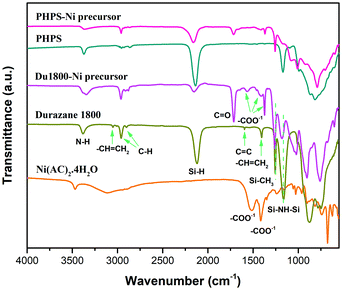 |
| | Fig. 1 The ATR-FTIR spectra of Du1800-Ni and PHPS-Ni precursors along with reactants Ni(Ac)2·4H2O, Durazane 1800, and PHPS. | |
The chemical structure of the Du1800-Ni precursor was further confirmed by 13C{1H} and 29Si{1H} CPMAS NMR characterization (Fig. 2). As displayed in Fig. 2a, the 13C {1H} CPMAS NMR spectrum of the Du1800-Ni precursor shows the characteristic signal shift of the CH3COO– group with a chemical shift at 20 ppm (–CH3) and 167 ppm (CO), which are not observed for Durazane 1800 (Fig. S4, ESI†). Moreover, the NMR signals corresponding to vinyl groups (–CHCH2–), which are found between 128 and 142 ppm in the spectrum of Durazane 1800, vanish in the synthesized precursor.62 These results confirm the chemical reaction between nickel acetate tetrahydrate and polymer as well as the hydrosilylation reaction between –CH2CH2– and Si–H groups, which agrees with ATR-FTIR results. The NMR signals between −10 and 4 ppm in the spectra of Durazane 1800 and Du1800-Ni precursor can be assigned to the carbon atoms of aliphatic groups bonded to a silicon atom (Si–CH3). These results are also in accordance with the 29Si {1H} CPMAS NMR spectrum (Fig. 2b), which shows the presence of Si(CH3)(CH2CH2)N2 and Si(CH3)(H)N2 units in the synthesized Du1800-Ni precursor at −8 and −25 ppm, respectively.63,64 The weak signal observed at −35 ppm can be assigned to the residue Si(CHCH2)N3 units in the Du1800-Ni precursor.63,65,66 The signals between −40 and −60 ppm might be owed to Si–OOCCH3.67
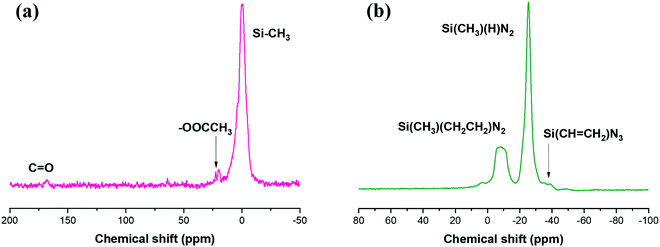 |
| | Fig. 2 The (a) 13C {1H} CPMAS and (b) 29Si {1H} CPMAS NMR spectra of the Du1800-Ni precursor. | |
The oxidation states of incorporated nickel on the surface of synthesized Ni-containing precursors were investigated by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 3, XPS Ni 2p spectra of Du1800-Ni and PHPS-Ni precursors can be deconvoluted into two doublet peaks characteristic of 2p1/2 and 2p3/2 spin–orbit splitting. The doublet peak observed at 852.7 and 869.9 eV can be assigned to metallic Ni0, while the doublet at 855.5 and 872.7 eV can be attributed to Ni2+. The broad peaks at 861.1 and 877.0 eV are satellite peaks corresponding to Ni 2p3/2 and 2p1/2, respectively, which is in good agreement with previous reports on metallic Ni and NiO compounds.68–70 These results suggest the in situ formation of metallic Ni during the reaction between nickel acetate tetrahydrate and poly(vinyl)silazane Durazane 1800 or PHPS polymers. Most probably, the surface of metallic Ni nanoparticles formed in both precursors (as indicated by TEM, see below) is oxidized to Ni2+.
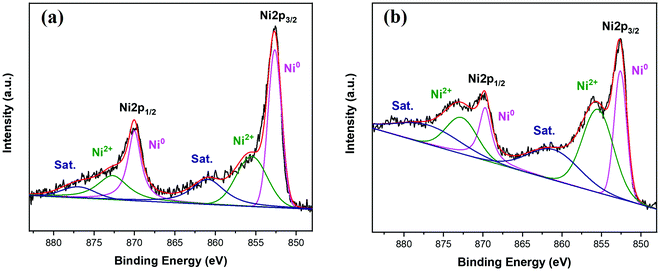 |
| | Fig. 3 The Ni 2p X-ray photoelectron spectra of (a) Du1800-Ni and (b) PHPS-Ni precursors. Sat. denotes the satellite peaks. | |
The oxidation states of nickel in the synthesized Ni-containing precursors were further confirmed by X-ray absorption near-edge spectra (XANES) characterization. Fig. 4a shows the normalized Ni K-edge XANES spectra of Du1800-Ni and PHPS-Ni precursors as well as those of Ni compounds used as reference materials. The first derivative spectra of the XANES are shown in Fig. 4b. The pre-edge peak, which depends on the oxidation states of the 3d transition element,71,72 in the synthesized Du1800-Ni and PHPS-Ni precursors is located at energies between those of reference Ni0 foil (8333 eV) and Ni2+(Ac)2·4H2O/Ni2+O (8331 eV) references. This indicates that nickel oxidation states in the synthesized precursors are between Ni0 and Ni2+, which is consistent with the XPS results (Fig. 3). Nanocrystalline nickel particles in the synthesized Du1800-Ni precursor is also confirmed by the high-resolution XRD patterns (Fig. S6a, ESI† and Fig. 13a). The high-resolution XRD pattern of the Du1800-Ni precursor manifests clear reflections of the metallic nickel phase (ICDD-PDF-00-004-0850), while that of the PHPS-Ni precursor exhibits an amorphous structure (Fig. S6b, ESI,† and Fig. 13b).
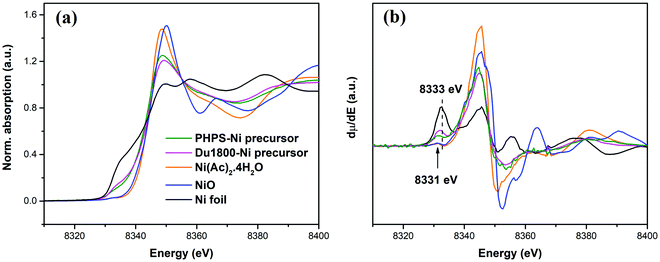 |
| | Fig. 4 (a) The normalized Ni K-edge XANES spectra and (b) the corresponding first derivative of Du1800-Ni and PHPS-Ni precursors as well as reference materials (Ni0 foil, Ni2+O, and Ni2+(Ac)2·4H2O). | |
As revealed by the TEM characterization, the Du1800-Ni precursor consists of homogeneously distributed nickel nanoparticles (Fig. 5a) with an average size of ∼2.4 nm (Fig. 5c) in an amorphous polymer matrix. In contrast, although nickel nanoparticles are not observed in the PHPS-Ni precursor (Fig. 5b), EDX analysis shows the presence of the nickel element in the amorphous polymer matrix (Fig. 5d). This result can be due to the too tiny size of formed nickel nanoparticles to be observed, which agrees with high-resolution XRD results (see the next section). The particle size of nickel in this sample is found to be ∼2.5 nm even after pyrolyzing at 700 °C in an argon atmosphere (see the next section). The incorporation of metallic Ni and Ni2+-compounds in the PHPS-Ni precursor is also confirmed by XPS (Fig. 3) and XANES (Fig. 4) results.
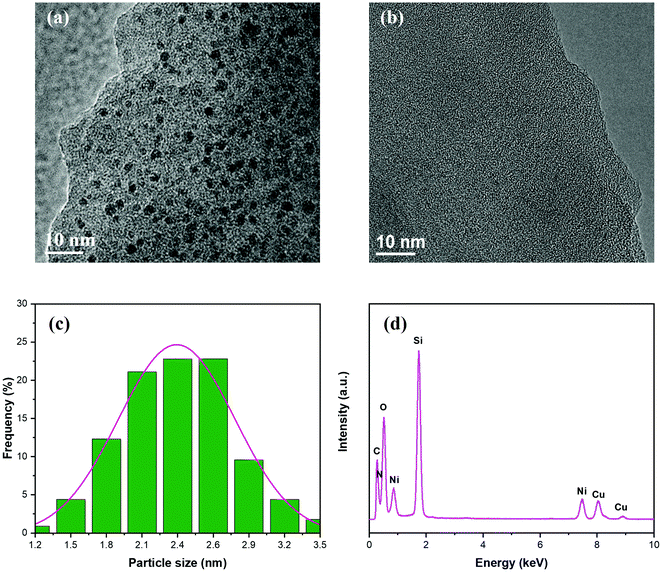 |
| | Fig. 5 TEM images of (a) Du1800-Ni and (b) PHPS-Ni precursors. (c) The particle size distribution of formed nickel nanoparticles in the Du1800-Ni precursor. (d) EDX spectrum of the PHPS-Ni precursor. | |
According to ATR-FTIR, 13C {1H} and 29Si {1H} CPMAS NMR, XPS, XANES, XRD, and TEM characterization, the mechanisms involved in the reaction of Ni(Ac)2·4H2O with poly(vinyl)silazane Durazane 1800 can be explained as follows. As shown in Fig. 6, the acetate groups of Ni(Ac)2·4H2O react first with Si–H groups of Durazane 1800, causing the formation of acetosilyl groups (CH3COO–Si–), Ni nanoparticles, and H2 gas. The in situ formation of metallic Ni nanoparticles in this step might be due to electron transfer from negatively charged hydrogen in –Si–H to Ni2+ in the Ni(II) acetate that results in Ni2+ reduction with in situ formation of metallic Ni nanoparticles, H2 evolution, and formation of acetic acid. Then the formed acetic acid might react with –Si–H and –Si–NH–Si– groups of Durazane 1800, releasing gaseous H2 and NH3, respectively. The release of gaseous H2 and NH3 during the reaction is confirmed by gas chromatography (Fig. S5, ESI†), and HCl titration, respectively. Moreover, gaseous bubbles and changes in solution color are observed during this chemical reaction (Fig. S2, ESI†). Our finding is in line with recently proposed mechanisms for the endothermic reaction of polysilazanes with carboxy end groups of the unsaturated polyesters.73 This proposed reaction mechanism also agrees with our previous report on the mechanism involved in the reaction of polysilazane HTT1800 with trans-[bis(2-aminoethanol-N,O)diacetato-nickel(II)].44 As PHPS consists also of active groups such as –Si–H and –Si–NH–Si– (Fig. S3, ESI†), similar reaction mechanisms might be involved in its reaction with Ni(Ac)2·4H2O. However, a hydrosilylation reaction between –Si–H and –Si–CHCH2 groups in Durazane 1800 might take place under these reaction conditions, forming carbosilane bonds (–Si–CH2–CH2–Si–) in the synthesized Du1800-Ni precursor.
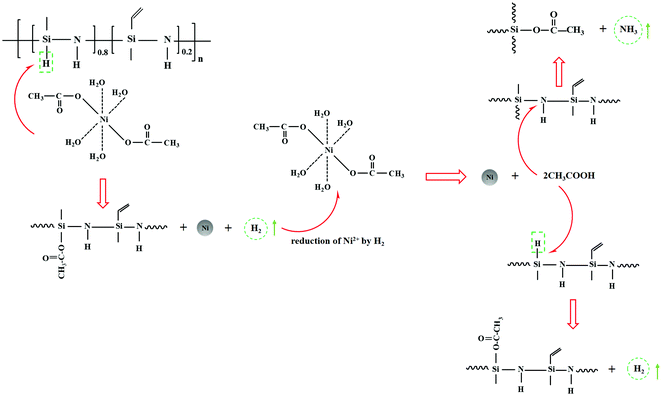 |
| | Fig. 6 Schematic of possible mechanisms of reaction between poly(vinyl)silazane Durazane 1800 and Ni(Ac)2·4H2O that leads to the formation of metallic nickel nanoparticles. | |
The transformation of Du1800-Ni and PHPS-Ni precursors into Ni/SiOCN ceramic nanocomposites
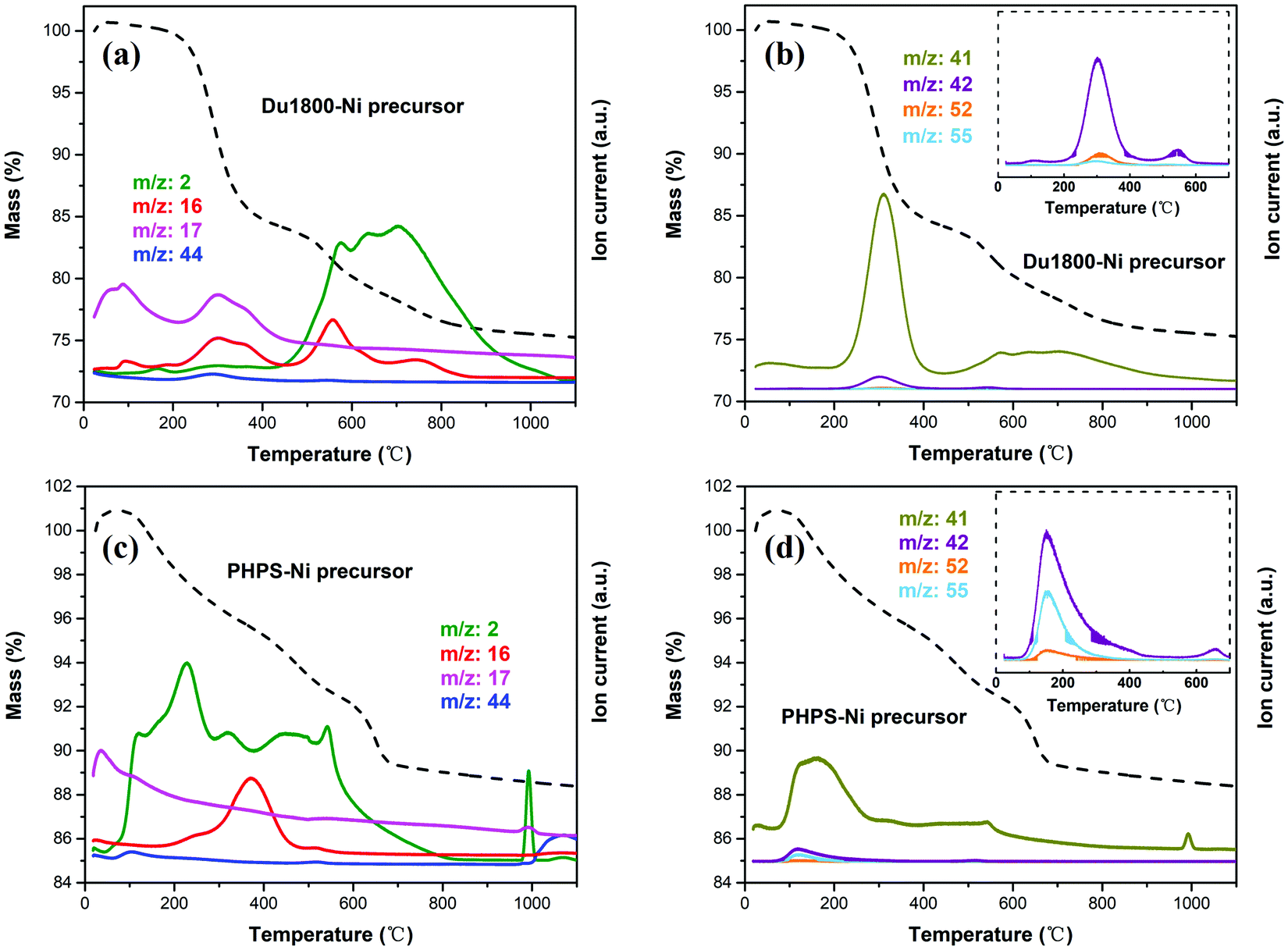
The pyrolysis of Du1800-Ni (Fig. 7a and b) and PHPS-Ni (Fig. 7c and d) precursors is accompanied by the evolution of gaseous H2 (m/z = 2), CH4 (m/z = 16), NH3 (m/z = 17) and CO2 (m/z = 44) as well as the distillation of oligomer fragments (m/z = 41, 42, 52, 55). For the Du1800-Ni precursor, the NH3 evolution corresponds to the transamination reaction and CH4 evolution originates from the decomposition of Si–CH3, Si–OOCCH3, or the cleavage of C–C bonds, both cause the weight loss of ∼17% at T < 500 °C. Since the PHPS polymer does not contain carbon in its backbone chain, CH4 evolution should be attributed to the decomposition of side groups (i.e. –Si–OOCCH3 units) in the PHPS-Ni precursor. This suggestion is confirmed by detecting CHCO+ (m/z = 41) and CH2CO+ (m/z = 42) ions in the same temperature range (see Fig. 7b and d). A high amount of H2 is released during the cross-linking of the PHPS-Ni precursor and the weight loss is ∼7% at T < 500 °C, which is much lower than that observed for the Du1800-Ni precursor. The CH4 evolution and H2 evolution due to the dehydrocoupling reaction of Si–H/Si–H and Si–H/N–H groups are responsible for the weight loss of ∼8 and 5% at T > 500 °C for Du1800-Ni and PHPS-Ni precursors, respectively. These results differ significantly from those reported for the pyrolysis of unmodified Durazane 1800 and PHPS polymers.46,74 For instance, the gaseous NH3 and H2 evolved below 200 °C for Du1800-Ni and PHPS-Ni precursors were not observed during the thermolysis of unmodified polymers. These results can be explained by the desorption of gaseous NH3 and H2 that are formed during the synthesis process (see Fig. 6) and adsorbed on the surface of the Ni-containing precursors. Besides, the hydrocarbon oligomers (m/z = 52 and 55) detected in the temperature range of 200–400 °C are not observed during the pyrolysis of unmodified polymers. This result suggests that the in situ formed metallic nickel nanoparticles in the precursors might catalyze the formation of these hydrocarbons. In addition, the cross-linking and polymer–ceramic transformation processes are completed at 900 and 700 °C for Durazane 1800 and PHPS polymers, respectively. In contrast, for Du1800-Ni and PHPS-Ni precursors, the weight losses are observed with the evolution of H2 up to 1100 °C. This finding can be explained by the activation of dehydrogenation reactions by nickel nanoparticles. Finally, Du1800-Ni and PHPS-Ni precursors show a ceramic yield of ∼75 and 88%, respectively, which is much higher than reported values for unmodified Durazane 1800 (∼66%) and PHPS (∼80%) polymers.46,74 These results can be explained by the increased cross-linking degree of the polymer upon chemical modification with nickel acetate tetrahydrate (see Fig. 6).
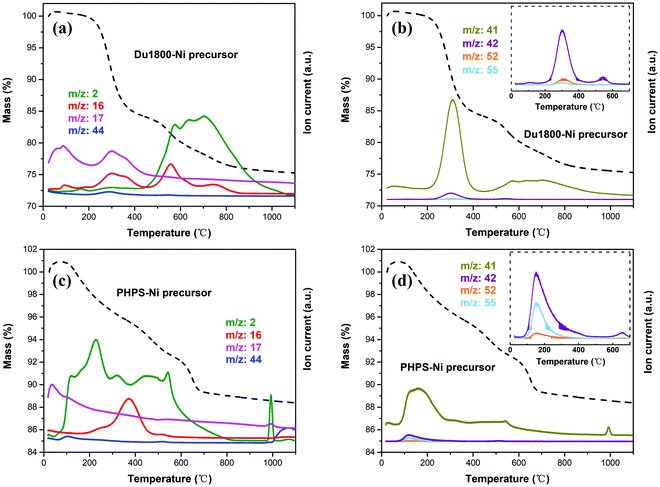 |
| | Fig. 7 Results of simultaneous thermal gravimetric (dashed lines) and mass spectrometry analysis of (a and b) Du1800-Ni and (c and d) PHPS-Ni precursors. | |
The cross-linking and polymer–ceramic transformation processes of Du1800-Ni and PHPS-Ni precursors were further confirmed by ATR-FTIR characterization. Fig. 8 shows the ex situ ATR-FTIR spectra of ceramic nanocomposites derived from Du1800-Ni and PHPS-Ni precursors by pyrolyzing at different temperatures under an argon atmosphere for 3 h. The intensity of absorption bands corresponding to N–H (3343 cm−1), C–H (2960 cm−1), Si–H (2152 cm−1), CO (1712 cm−1), and –COO−1 (1610–1330 cm−1) gradually decreases with increasing pyrolysis temperature. For the Du1800-Ni precursor that pyrolyzed at 900 and 1100 °C, broad absorption bands attributed to Si–O (1026 cm−1), Si–N (904 cm−1), and Si–C (766 cm−1) are observed, suggesting the formation of SiOCN ceramic. As shown in Fig. 8b, the formation of SiOCN in the PHPS-Ni precursor occurs at lower pyrolysis temperature (700 °C) than that of the Du1800-Ni precursor (900 °C), which agrees with the results of TG-MS analysis (Fig. 7).
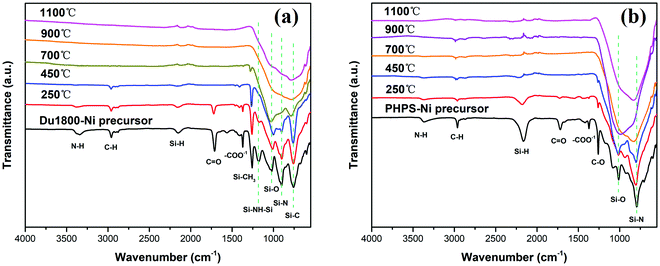 |
| | Fig. 8 ATR-FTIR spectra of (a) Du1800-Ni and (b) PHPS-Ni precursors thermally treated at 250 °C, 450 °C, 700 °C, 900 °C, and 1100 °C under an argon atmosphere for 3 h. | |
Chemical composition and structure of Ni/SiOCN ceramic nanocomposites
In the next step, the chemical and phase compositions of Ni/SiOCN ceramic nanocomposites obtained by the pyrolysis of synthesized Ni-containing precursors at 700, 900, and 1100 °C under an argon atmosphere for 3 h were characterized using ICP-OES, XRD, XAFS, and TEM techniques. As shown in Table S1 (ESI†), the weight ratio Si/Ni in the pyrolyzed Du1800-Ni and PHPS-Ni samples is 4.1 ± 0.1 and 6.3 ± 0.1, respectively, regardless of the pyrolysis temperatures. In contrast, the analysis of the X-ray photoelectron spectra survey reveals that the surface composition (especially C, O, and N amount) varies significantly with the pyrolysis temperature (see Tables S2 and S3, ESI†). These results are in good agreement with TG-MS analysis, which indicates the continuous evolution of gaseous species that in turn causes composition changes during the pyrolysis of precursors up to 1100 °C.
As shown in Fig. S6a (ESI†), the high-resolution XRD pattern of the 700Ar-Du1800-Ni sample obtained by pyrolysis of the Du1800-Ni precursor at 700 °C under an argon atmosphere reveals the presence of graphitic carbon (ICDD-PDF-03-065-6212) and nanocrystalline metallic nickel (ICDD-PDF-00-004-0850). The latter reacts partially with the SiOCN matrix at higher pyrolysis temperatures to form the nickel silicide Ni2Si (ICDD-PDF-00-048-1339) phase in 900Ar-Du1800-Ni and 1100Ar-Du1800-Ni samples. In addition to the Ni2Si phase, crystalline SiC (ICDD-PDF-00-049-1428) is formed in the 1100Ar-Du1800-Ni sample. Although the formation of SiC in polymer-derived ceramics usually takes place at temperatures higher than 1400 °C,75 previous works reported that manganese and iron could act as catalysts and lower its formation temperature to 1100 °C.46 For 700Ar-PHPS-Ni and 900Ar-PHPS-Ni samples, only nanocrystalline metallic nickel is detected in their XRD patterns, while nickel silicide Ni2Si (ICDD-PDF-00-048-1339) and NiSi (ICDD-PDF-00-038-0844) phases are formed in the 1100Ar-PHPS-Ni sample by increasing the pyrolysis temperature to 1100 °C (Fig. S6b, ESI†). The XRD reflections corresponding to the metallic Ni phase in PHPS-Ni samples are much broader than those of the same phase in Du1800-Ni samples, suggesting the smaller crystallite sizes of Ni in PHPS-Ni samples. These results are in good agreement with the TEM characterization, as discussed below.
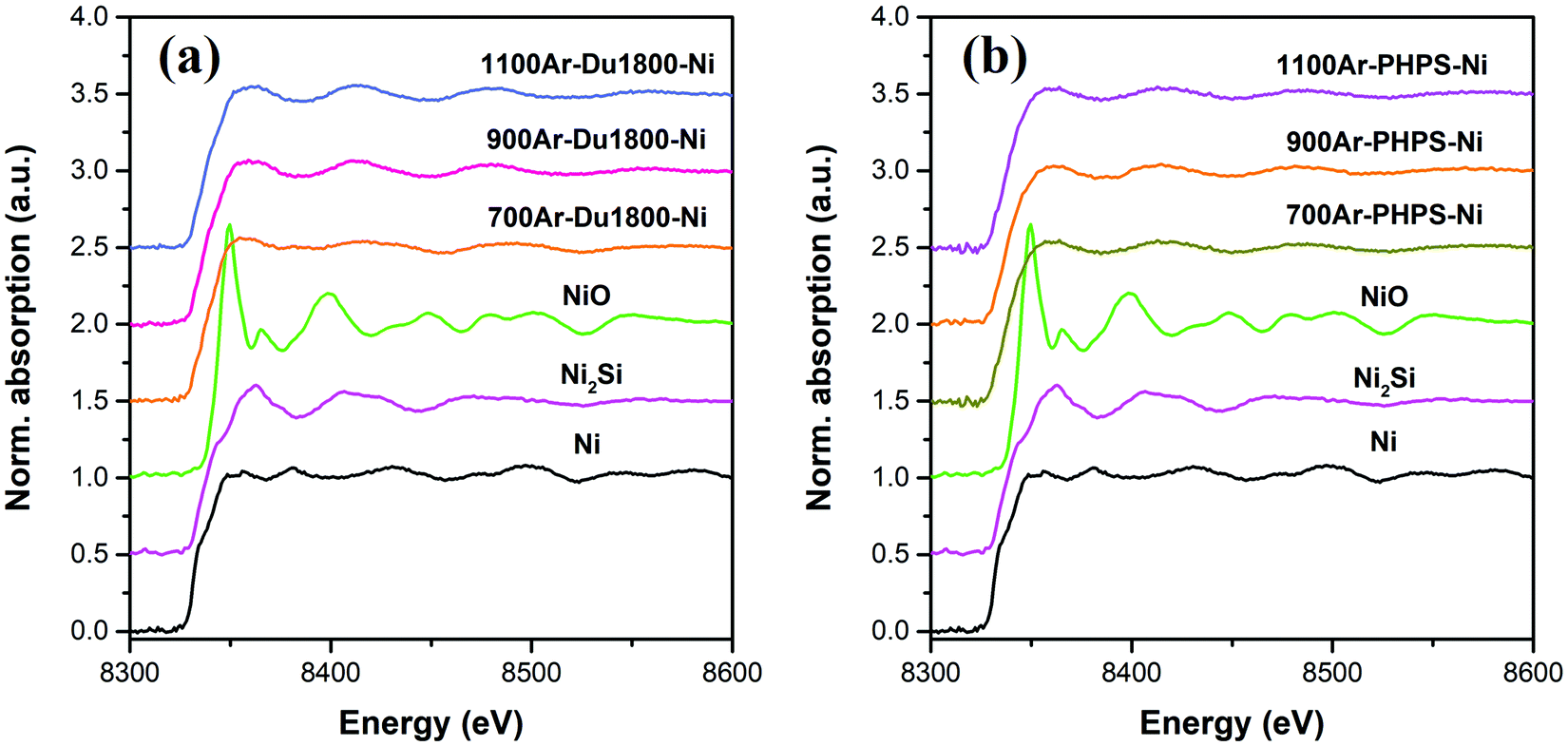
X-ray absorption fine structure (XAFS) measurements were conducted to obtain further information about phase compositions in the pyrolyzed samples. The Ni K-edge XAFS spectra of pyrolyzed samples, as well as nickel compounds (Ni, Ni2Si, and NiO), used as reference materials, are shown in Fig. 9. As shown in Fig. S7 (ESI†), the linear combination fitting (LCF) of normalized Ni K-edge XAFS spectra of pyrolyzed samples with those reference materials confirms the presence of both metallic Ni and Ni2Si phases in all samples. The weight ratio Ni2Si/Ni in pyrolyzed Du1800-Ni and PHPS-Ni samples is found to increase with increasing pyrolyzing temperature, which agrees with the XRD results. Additionally, small weight fractions of the NiO phase (≤12 wt%) are found in all samples. No NiO phase is detected by XRD characterization, which might be due to its low crystallinity or amorphous structure.
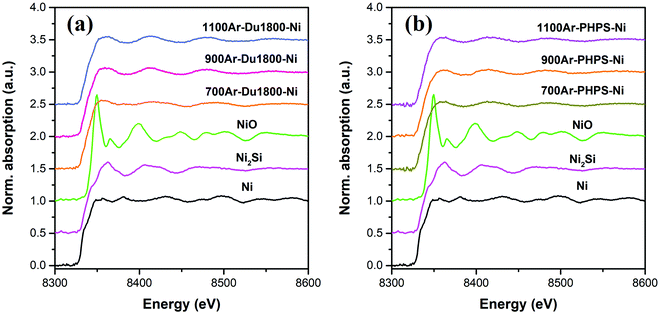 |
| | Fig. 9 The Ni K-edge X-ray absorption fine structure (XAFS) spectra of Ni/SiOCN samples derived from (a) Du1800-Ni and (b) PHPS-Ni precursors at different pyrolysis temperatures as well as reference materials (Ni, Ni2Si, and NiO). | |
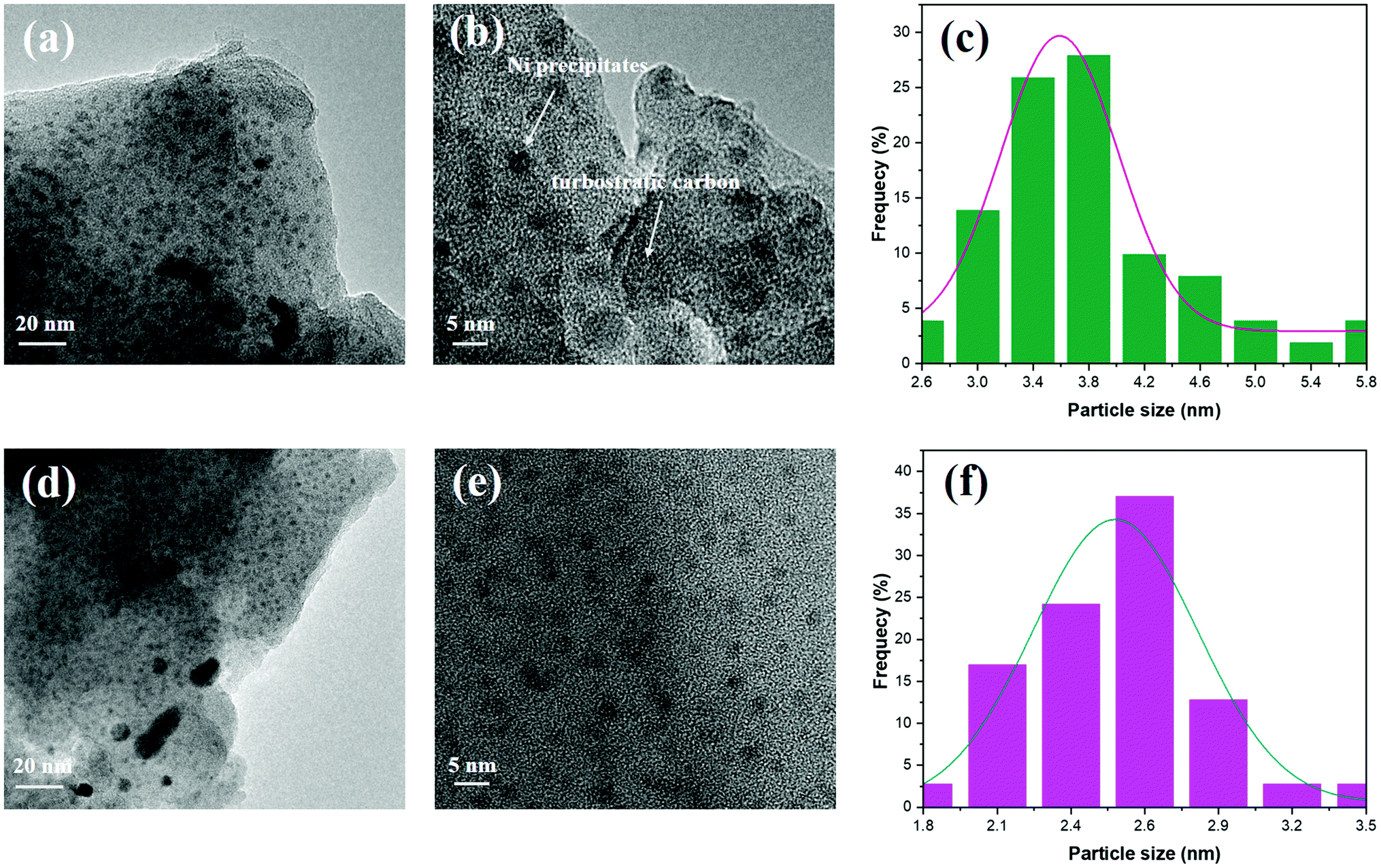
The results of TEM characterization of 700Ar-Du1800-Ni (Fig. 10a and b) and 700Ar-PHPS-Ni (Fig. 10d and e) samples, as well as EDX mapping of the 1100Ar-Du1800-Ni sample (Fig. S9, ESI†) confirm the homogeneous distribution of Ni nanoparticles in the amorphous SiOCN matrix. The average size of the nanoparticles in 700Ar-Du1800-Ni and 700Ar-PHPS-Ni samples is around 3.6 and 2.5 nm, respectively (Fig. 10c and f). Turbostratic carbon is observed only in the 700Ar-Du1800-Ni sample (Fig. 10b), which agrees with XRD results (Fig. S6a, ESI†). Raman spectra also confirm the formation of turbostratic carbon, which is characterized by the D and G bands,76 in all pyrolyzed Du1800-Ni samples (Fig. S8, ESI†). In contrast, for those samples derived from the PHPS-Ni precursor, both D and G peaks are not observed. Typically, the free carbon phase in polymer-derived ceramics has not yet been found upon thermolysis between 700 and 900 °C, and higher pyrolysis temperatures are usually required to initiate the formation and precipitation of turbostratic carbon.77–81 However, the presence of transition metal catalysts such as Ni and Fe in PDCs can greatly lower its formation temperature.43,82 In addition, no turbostratic carbon is observed in the 700Ar-PHPS-Ni sample, which can be explained by the carbon-free backbone chain of the PHPS polymer.
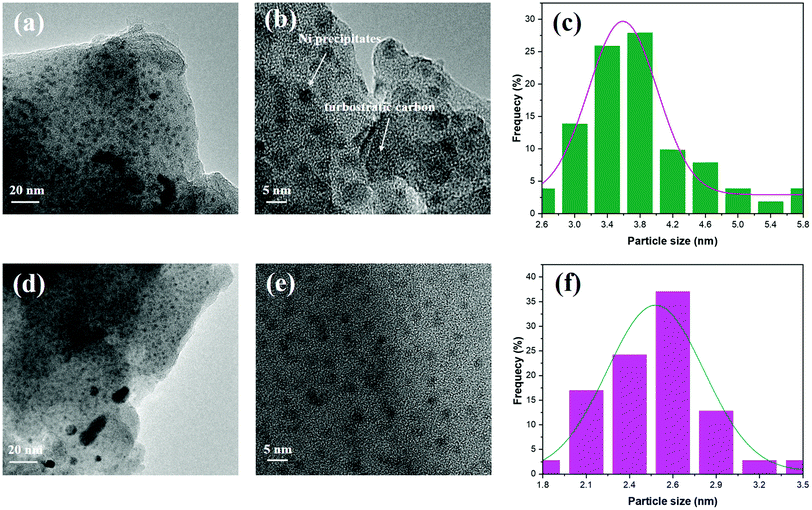 |
| | Fig. 10 TEM images of (a and b) 700Ar-Du1800-Ni and (d and e) 700Ar-PHPS-Ni samples. Their particle size distribution is shown in (c) and (f), respectively. | |
Porosity and surface area of Ni/SiOCN ceramic nanocomposites
Fig. 11 shows nitrogen physisorption isotherms of Ni/SiOCN ceramic nanocomposites derived from Du1800-Ni and PHPS-Ni precursors at different pyrolysis temperatures. 700Ar-Du1800-Ni and 900Ar-Du1800-Ni samples derived from the Du1800-Ni precursor exhibit type I isotherms corresponding to microporous materials, while the 1100Ar-Du1800-Ni sample shows a nonporous structure. Accordingly, 700Ar-Du1800-Ni and 900Ar-Du1800-Ni samples possess a BET specific surface area of ∼361 and ∼232 m2 g−1, respectively, which are much higher than that of the 1100Ar-Du1800-Ni sample (6.8 m2 g−1). Interestingly, all Ni/SiOCN ceramic nanocomposites derived from PHPS-Ni precursor manifest nonporous structures with a low BET specific surface area of ∼9, ∼7.5, and ∼7 m2 g−1 for 700Ar-PHPS-Ni, 900Ar-PHPS-Ni, and 1100Ar-PHPS-Ni, respectively. The nucleation and growth of micropores in PDCs can be explained by the amount and type of gaseous species released during the polymer-to-ceramic transformation process such as H2, CH4, NH3, and other hydrocarbons.83 For instance, the PHPS polymer has no hydrocarbon units in its backbone chain (Fig. S3, ESI†). Therefore, the thermolysis of the PHPS-Ni precursor results in the evolution of few gaseous species such as H2 and NH3 in addition to a small amount of gaseous CH4 from the decomposition of acetate groups. Consequently, all Ni/SiOCN ceramic nanocomposites derived from the PHPS-Ni precursor show nonporous structures with a low surface area. In contrast, Durazane 1800 is a hydrocarbon-rich polymer, and its thermolysis results in the release of several gaseous hydrocarbons. Therefore, micropores are formed in 700Ar-Du1800-Ni and 900Ar-Du1800-Ni samples during the pyrolysis of the Du1800-Ni precursor at 700–900 °C. However, the generated micropores are not stable and will collapse with increasing thermolysis temperature. Thus, the 1100Ar-Du1800-Ni sample exhibits a nonporous structure. Moreover, our previous work showed that the in situ formed metallic nickel nanoparticles could also play a role in the formation and stability of these micropores in the PDCs during thermolysis.44 As discussed above, nickel nanoparticles can catalyze the formation of hydrocarbon species and reduce the critical barrier for the formation of gaseous species during the polymer-to-ceramic transformation, which facilitates the heterogeneous nucleation of pores due to the release of small gas molecules such as H2, CH4, and NH3. In this way, micropores are prone to be formed instead of macropores and abnormal pore growth. In addition, higher cross-linking of the precursors through the formation of carbosilane units can enhance the pore stability during the pyrolysis. Moreover, nickel nanoparticles lower the formation temperature of turbostratic carbon by some hundred degrees. Accordingly, the formed nickel nanoparticles and turbostratic carbon with higher mechanical stability than that of the neighboring microporous matrix behave as nanofillers and reinforce the microporous structure, subsequently increasing the stability of micropores in the entire matrix of the sample.
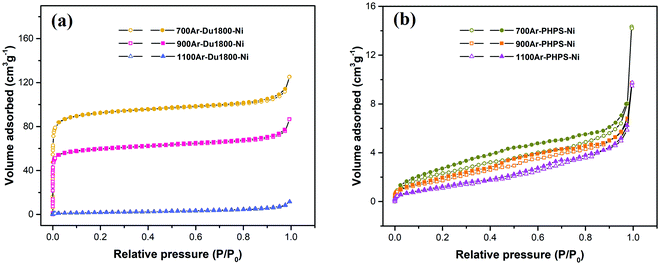 |
| | Fig. 11 Nitrogen physisorption isotherms of Ni/SiOCN ceramic nanocomposites derived from (a) Du1800-Ni and (b) PHPS-Ni precursors (open and filled symbols indicate the adsorption and desorption stage, respectively). | |
Catalytic properties of Ni/SiOCN ceramic nanocomposites in dry reforming of methane
Dry reforming of methane (DRM) with carbon dioxide is a promising process to convert the two main greenhouse gases into CO/H2 mixtures with a ratio close to unity (eqn (1)).84 Typical deviation from the unity H2:CO ratio and higher CO2 reaction rates compared to CH4 are due to the occurrence of the reverse water gas shift (RWGS) reaction (eqn (2)). The DRM process has been chosen as a representative catalytic system containing supported nickel nanoparticles that perform under harsh conditions.| | 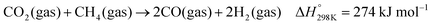 | (1) |
| | 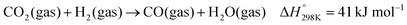 | (2) |
The deactivation of catalysts that are mainly caused by nickel particle sintering (the decrease of active surface area) and potentially via coke formation can contribute to the decrease of reaction rates at a given temperature. Surface carbon formation resulting from CO disproportionation or CH4 decomposition (eqn (3)–(5)) has been claimed to deactivate the catalyst or also to be involved in the reaction mechanism,53,85,86 although an excess carbon can plug the reactor.87 Another option can be the transformation of the active metal nickel nanoparticles to their silicide phases,88 which displays a diminished activity toward syngas formation. The occurrence of the different potential deactivation mechanisms will be later discussed using post-catalytic XRD, XPS, and TEM data.| | 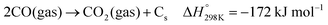 | (3) |
| | 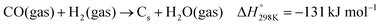 | (4) |
| | 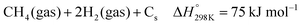 | (5) |
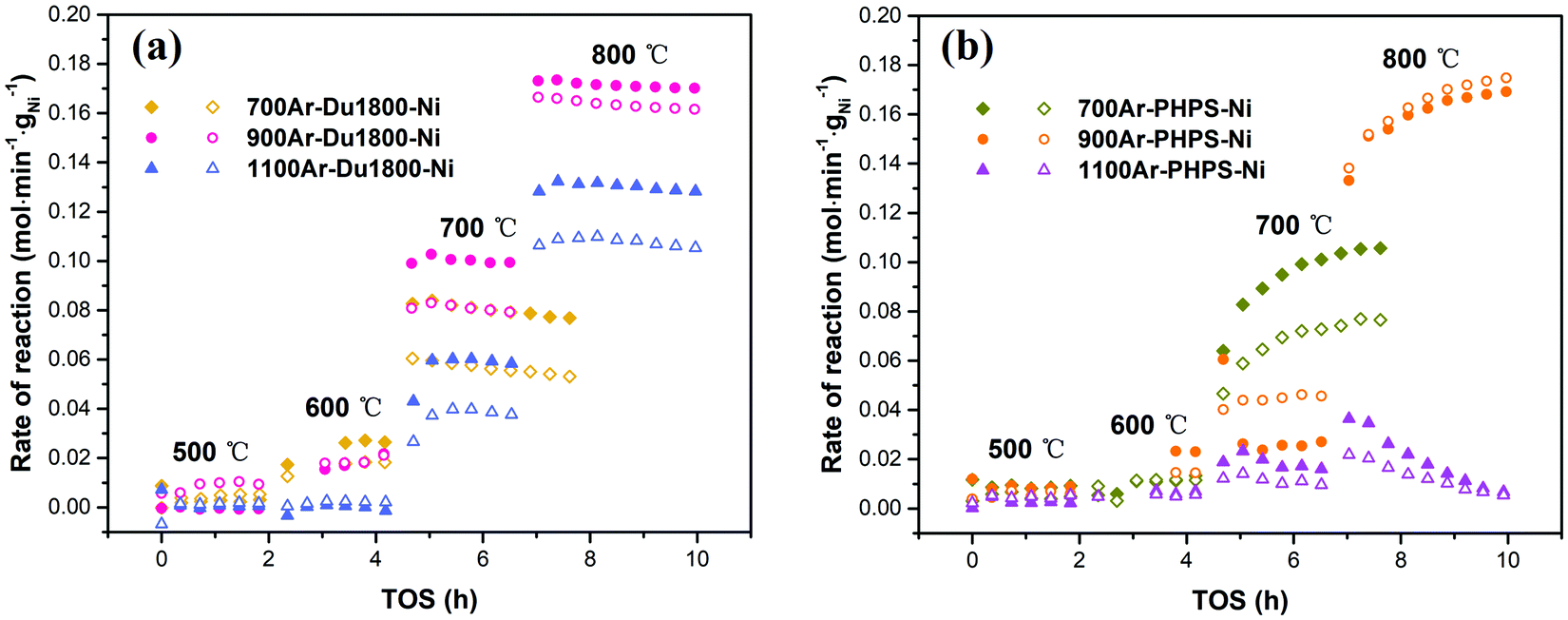
The catalytic properties of Ni/SiOCN ceramic nanocomposites derived from Du1800-Ni and PHPS-Ni precursors are studied in the DRM process in a plug-flow fixed bed-reactor at reactant ratios of CH4:CO2:N2 = 1:1:3 and GHSV = 120000 N mL h−1 gcat−1, as shown in Fig. 12. All samples show low or no activity towards DRM at 500 °C, which is most possibly due to the low detection limit of the GC. At higher temperatures such as 600 °C, 700 °C, and 800 °C, all samples show activity for the DRM process. The reaction rates increase with temperature due to the endothermic nature of the DRM reaction. For all catalytic tests, the reaction rate of CO2 is always higher than that of CH4. This can be attributed to the occurrence of the reverse water gas shift (RWGS) reaction (eqn (2)), which further reacts to CO2 and produces the observed deviation from the unity of the H2:CO ratio. However, the CO2 reaction rate is a little lower than that of CH4 for catalyst 900Ar-PHPS-Ni, which is caused by more injection of CO2 than expected into the reactor. For the samples derived from the Du1800-Ni precursor, all show very close reaction rates when measured at 500 and 600 °C. However, with increasing test temperature, catalyst 900Ar-Du1800-Ni shows much better performance, and the CH4 reaction rate at 700 and 800 °C is around 0.08 and 0.16 mol min−1 gNi−1, respectively (Fig. 12a). Moreover, all the Du1800-Ni precursor derived samples display remarkable stability at all temperatures tested, suggesting a soft deactivation effect. All catalysts derived from the PHPS-Ni precursor show very low reaction rates at 500 °C and 600 °C (Fig. 12b). The reaction rate is greatly increased when the test is conducted at 700 °C except for catalyst 1100Ar-PHPS-Ni, which displays a moderate increase in terms of the reaction rate and a strong deactivation effect. In contrast, the catalysts pyrolyzed at 700 and 900 °C show an increase in the reaction rate at higher testing temperatures. This activation phenomenon might be explained by changes in the crystallite/particle size, a phase transition, and/or the removal of turbostratic carbon caused by oxidation with CO2 or H2O. Surface carbon oxidation can also occur viaeqn (4). Although H2O is not introduced as a reactant, it can be produced by the RWGS and oxidize carbon to produce CO and H2. Our HRTEM investigations on PHPS-Ni catalysts showed that the metallic Ni nanoparticles are embedded in carbon onions, which might reduce their catalytic activity. Removal of carbon can clean the catalyst surface, exposing more nickel to reaction and thereby increasing reaction rates.53 The 900Ar-PHPS-Ni catalyst exhibits the best performance among all samples with the highest CH4 reaction rate of 0.18 mol min−1 gNi−1 at 800 °C. The reason for the differences in the performance of these samples can be explained by differences in the catalyst configuration, which includes the surface area (Fig. 11), initial nickel nanoparticle size (Fig. 10), and chemical compositions (Fig. 9 and Fig. S6, ESI†).
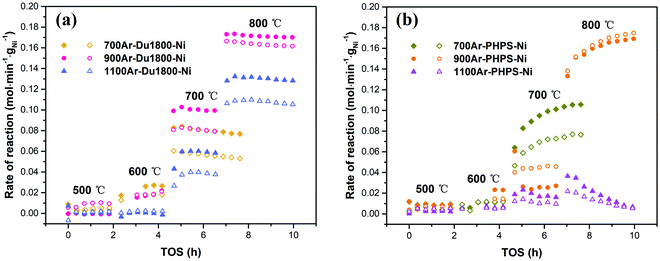 |
| | Fig. 12 CO2 and CH4 reaction rates for the tests at 500 °C, 600 °C, 700 °C, and 800 °C, using a plug-flow fixed-bed reactor at reactant ratios of CH4:CO2:N2 = 1:1:3 and GHSV = 120000 N mL h−1 gcat−1 (open and filled symbols represent CH4 and CO2 reaction rate, respectively). | |
The catalytic performance (reaction rate of CH4) in our work is summarized in Table S4 (ESI†). As compared with other studies (Table S5, ESI†),47–52 our prepared Ni/SiOCN catalysts show remarkable catalytic activity towards the DRM process even though the catalyst configuration was not optimized for this particular reaction. This demonstrates that the in situ formation of nickel nanoparticles in the PDC matrix is a promising approach to prepare heterogeneous catalysts for the DRM process. Catalyst design and optimization can further improve the performance of this type of catalyst for the DRM and other catalytic processes that occur on supported nickel nanoparticles.
To understand the catalytic performance, post-catalytic characterization is required on the spent catalysts. However, the spent catalysts recovered from the plug-flow fixed-bed reactor after the catalytic tests contain quartz powder to dilute the catalysts and ensure homogeneous temperature and pressure across the catalytic bed. These impurities in the spent catalysts might add difficulties during the preparation of the samples for post characterization as well as the interpretation of the results of characterization. Therefore, the catalytic tests were repeated without using any powder for dilution in a recirculating batch reactor initially containing a CH4:CO2:Ar = 1:1:8 mixture at 1000 mbar of pressure, as detailed in the ESI.† All samples showed activity towards the DRM and the production of syngas as demonstrated by the conversion of CO2 and the detection of CO and H2 in the MS spectra (Fig. S10, ESI†), therefore allowing the use of the spent catalysts for further characterization. The high-resolution XRD patterns of the catalysts before and after DRM tests are shown in Fig. 13. For each given catalyst, the patterns of PHPS-Ni catalysts before and after DRM generally reveal no noteworthy phase transitions during catalysis. In contrast, a new set of XRD reflections corresponding to the NiSi phase are observed in the patterns of 900Ar-Du1800-Ni and 1100Ar-Du1800-Ni catalysts after the DRM test. Moreover, the full width at half maximum (FWHM) of the Ni(111) reflection belonging to each sample is slightly decreased after the DRM test, suggesting the slight increase in the crystallite size of metallic nickel in the catalysts. The increase in the crystallite size of metallic Ni (i.e., crystallinity) in the 900Ar-Du1800-Ni catalyst under different DRM conditions is also confirmed by in situ synchrotron XRD experiments (Fig. S11, ESI†). As shown in Fig. S11a (ESI†), the XRD reflections corresponding to the metallic Ni phase become more intense and sharp with time during the pre-reduction step in a H2 atmosphere at 600 °C as well as under DRM reactions at 600 and 800 °C. In contrast, no remarkable structural changes are observed for the 1100Ar-PHPS-Ni sample during the pre-reduction step and DRM reactions (see Fig. S11b, ESI†), which agrees with the ex situ XRD results. This might be due to the low activity of this sample towards DRM (see Fig. 12b). The formation of nickel silicide and the sintering of nickel particles can result, to some degree, in catalyst deactivation.53,88 This deactivation is soft for the time on stream tested as proved by the catalytic tests previously. Surface carbon formation resulting from CO disproportionation or CH4 decomposition (eqn (3)–(5)) can be another reason for the deactivation of the catalyst.53,85,86 However, the formation of surface carbon cannot be confirmed from XRD results because all Du1800-Ni group samples contain graphitic carbon before interaction with the DRM reactants. Similarly, the PHPS-Ni group samples display no graphitic reflections before or after DRM, suggesting that (if formed) surface carbon resulting from eqn (3)–(5) is amorphous or below the technique's detection limit.
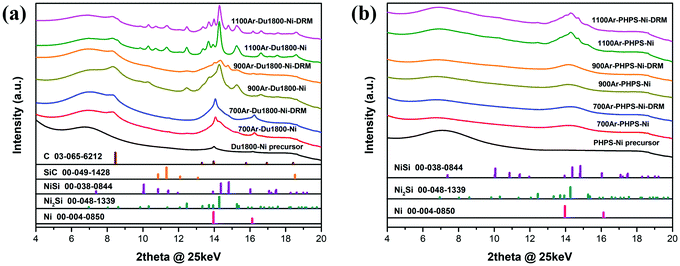 |
| | Fig. 13 Room-temperature high-resolution XRD patterns of (a) Du1800-Ni and (b) PHPS-Ni precursors and their derived ceramic nanocomposites after pyrolysis at 700 °C, 900 °C, and 1100 °C before and after DRM. Reference material patterns are Ni (ICDD-PDF-00-004-0850), Ni2Si (ICDD-PDF-00-048-1339), NiSi (ICDD-PDF-00-038-0844), SiC (ICDD-PDF-00-049-1428) and C (ICDD-PDF-03-065-6212). | |
The Ni 2p X-ray photoelectron spectra of all samples before and after the DRM test are shown in Fig. S12 (ESI†). The peaks at 855.8 eV and 873.4 eV that are associated with Ni2+ disappeared after the DRM test, which suggests the reduction of the NiO phase in the samples to metallic Ni or nickel silicide under DRM conditions. This change is logical if we consider the reductive nature of the effluent gas composition after DRM. As shown in Fig. S13 (ESI†), the C 1s X-ray photoelectron spectra of all samples before and after DRM can be fitted with four peaks at 284.1, 284.8, 286.3, and 288.6 eV, which can be assigned to C–Si, C–C, C–N, and C–O bonds, respectively.89 For Ni/SiOCN samples derived from the Du1800-Ni precursor, the main peak in the spectra is due to C–C bonds in graphite or amorphous carbon, while the main peak in the spectra of PHPS-Ni samples is assigned to C–Si bonds. Moreover, the analysis of the spectrum survey (Tables S2 and S3, ESI†) reveals that the amount of carbon on the surface of Du1800-Ni samples is much higher than that on the surface of PHPS-Ni samples and increases after DRM. This result points out the formation of surface carbon from eqn (3)–(5). For the PHPS-Ni samples (except for the sample pyrolyzed at 700 °C), the carbon content on the surface of the catalysts after DRM slightly increases. These results complement XRD analysis and suggest that the carbon content on the surface of catalysts increases after DRM. No remarkable increase in the amount of graphitic carbon can be observed by XRD, suggesting that the formed carbon might be amorphous or in a low amount.
Fig. 14 shows the TEM characterization performed on the catalysts after DRM-atmosphere exposure. Several multiwalled sp2-carbon nanotubes are formed on the surface of the spent 900Ar-Du1800-Ni catalyst, produced from CH4 decomposition or CO disproportionation. In contrast, a small number of carbon nanotubes are formed on the surface of the spent 1100Ar-PHPS-Ni catalyst. The presence of surface carbon is not necessarily linked to catalyst deactivation, as under specific conditions surface carbon can act as an intermediate toward the generation of syngas by oxidation with CO2.53 Therefore, the presence of nanotubes on the 900Ar-Du1800-Ni samples does not necessarily point to higher deactivation but can be a result of an increased CH4 cracking activity, which produces the necessary intermediate for the reaction with CO2. This fact has been supported by an increased reaction rate for this type of catalyst, as shown in Fig. 12a. TEM characterization further complements XRD and XPS data and highlights that carbon formation is a surface phenomenon mainly occurring on the surface of Du1800-Ni samples. The relatively small amount of graphitic carbon results in no observable bulk changes for the Du1800-Ni samples but a significant increase of the surface carbon concentration, as proved by XPS. For these catalysts, the formation of carbon nanotubes does not lead to catalyst deactivation as verified by remarkably stable reaction rates of the Du1800-samples for the duration of the catalytic tests. All synthesized samples have proved their activity for CH4 and CO2 conversion to produce syngas, and we anticipate their applicability to other catalytic reactions performed on supported nickel nanoparticles.
 |
| | Fig. 14 TEM images of the (a and b) 900Ar-Du1800-Ni and (c and d) 1100Ar-PHPS-Ni catalysts after the DRM test in a recirculating batch reactor. | |
Conclusions
Ni-Containing polysilazane Du1800-Ni and PHPS-Ni precursors have been synthesized through the chemical reaction of Ni(Ac)2·4H2O with poly(vinyl)silazane (Durazane 1800) and perhydropolysilazane NN120-20 (A) (PHPS), respectively. Nickel nanoparticles are in situ formed during the reaction between Ni(Ac)2·4H2O and –Si–H groups of the polymers. The formed Ni nanoparticles in Ni-containing precursors are stable after pyrolyzing at 700 °C under an argon atmosphere, but they react with the SiOCN matrix at higher pyrolysis temperatures (≥900 °C) to form the nickel silicide (Ni2Si) phase. The ceramic nanocomposites derived from the Du1800-Ni precursor by pyrolyzing at 700 and 900 °C show a high BET specific surface area of ∼361 and ∼232 m2 g−1, respectively, while all the pyrolyzed samples derived from the PHPS-Ni precursor exhibit a nonporous structure. Catalytic tests for the dry reforming of methane process reveals that all samples are active towards syngas formation, but the samples pyrolyzed at 900 °C show the best performance. Demonstrated by its applicability to the dry reforming of methane reaction, in situ formation of nickel nanoparticles in PDCs shows great potential as a preparation method for heterogeneous catalysts.
Author contributions
J. Wang synthesized the materials, carried out ATR-FTIR characterization, analyzed and discussed the experimental data, prepared and drew the images and graphics for the publication and wrote the manuscript. A. Gili and R. Schomäcker assisted in the catalytic tests in a plug-flow fixed-bed reactor; A. Gili conducted the collection of the synchrotron XRD patterns. M. Grünbacher and S. Penner carried out the catalytic tests in the recirculating batch reactor. S. Praetz and C. Schlesiger were involved in the XAFS characterization and result analysis. J. D. Epping supported the solid NMR characterization. O. Görke helped with Raman and EDX characterization. G. Schuck carried out the XANES characterization. A. Gurlo and M. F. Bekheet supervised this research project, and contributed to the experimental design and data interpretation.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We would like to thank Harald Link for collecting ICP-OES data, Heinz Sap for TG-MS, Maria Unterweger for XPS, and Christina Eichenauer for nitrogen sorption. We thank Sören Selve and Jan Ron Justin Simke for performing TEM characterization. Jun Wang acknowledges the financial support from the China Scholarship Council (201604910900) and DAAD STIBET-grant for degree completion awarded by Technische Universität Berlin. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. Albert Gili thanks the Unifying Systems in Catalysis (UniSysCat), which is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1-390540038.
References
- V. Pareek, A. Bhargava, R. Gupta, N. Jain and J. Panwar, Adv. Sci., Eng. Med., 2017, 9, 527–544 CrossRef CAS.
- M. J. Ndolomingo, N. Bingwa and R. Meijboom, J. Mater. Sci., 2020, 55, 6195–6241 CrossRef CAS.
-
A. J. Shnoudeh, I. Hamad, R. W. Abdo, L. Qadumii, A. Y. Jaber, H. S. Surchi and S. Z. Alkelany, in Biomaterials and Bionanotechnology, ed. R. K. Tekade, Academic Press, 2019, pp. 527–612 Search PubMed.
- H. H. El-Maghrabi, A. A. Nada, S. Roualdes and M. F. Bekheet, Int. J. Hydrogen Energy, 2020, 45, 32000–32011 CrossRef CAS.
- H. H. El-Maghrabi, A. A. Nada, M. F. Bekheet, S. Roualdes, W. Riedel, I. Iatsunskyi, E. Coy, A. Gurlo and M. Bechelany, J. Colloid Interface Sci., 2020, 587, 457–466 CrossRef.
- R. N. Baig and R. S. Varma, ACS Sustainable Chem. Eng., 2013, 1, 805–809 CrossRef.
- J. H. Kang, L. D. Menard, R. G. Nuzzo and A. I. Frenkel, J. Am. Chem. Soc., 2006, 128, 12068–12069 CrossRef CAS.
- H. S. Whang, M. S. Choi, J. Lim, C. Kim, I. Heo, T.-S. Chang and H. Lee, Catal. Today, 2017, 293, 122–128 CrossRef.
- Z. Jin, M. Xiao, Z. Bao, P. Wang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 6406–6410 CrossRef CAS.
- M. F. Bekheet, M. Grünbacher, L. Schlicker, A. Gili, A. Doran, J. D. Epping, A. Gurlo, B. Klötzer and S. Penner, CrystEngComm, 2019, 21, 145–154 RSC.
- M. F. Bekheet, P. Delir Kheyrollahi Nezhad, N. Bonmassar, L. Schlicker, A. Gili, S. Praetz, A. Gurlo, A. Doran, Y. Gao, M. Heggen, A. Niaei, A. Farzi, S. Schwarz, J. Bernardi, B. Klötzer and S. Penner, ACS Catal., 2021, 11, 43–59 CrossRef CAS.
- N. Bonmassar, M. F. Bekheet, L. Schlicker, A. Gili, A. Gurlo, A. Doran, Y. Gao, M. Heggen, J. Bernardi, B. Klötzer and S. Penner, ACS Catal., 2020, 10, 1102–1112 CrossRef CAS.
- A. Gili, L. Schlicker, M. F. Bekheet, O. Görke, D. Kober, U. Simon, P. Littlewood, R. Schomäcker, A. Doran, D. Gaissmaier, T. Jacob, S. Selve and A. Gurlo, ACS Catal., 2019, 9, 6999–7011 CrossRef CAS.
- L. Schlicker, M. F. Bekheet, A. Gili, A. Doran, A. Gurlo, K. Ploner, T. Schachinger and S. Penner, J. Solid State Chem., 2018, 266, 93–99 CrossRef CAS.
- R. Riedel, A. Kienzle, W. Dressler, L. Ruwisch, J. Bill and F. Aldinger, Nature, 1996, 382, 796–798 CrossRef CAS.
- A. Müller, J. Peng, H. J. Seifert, J. Bill and F. Aldinger, Chem. Mater., 2002, 14, 3406–3412 CrossRef.
- S. Fu, M. Zhu and Y. Zhu, J. Adv. Ceram., 2019, 8, 457–478 CrossRef CAS.
- A. Lale, M. Schmidt, M. D. Mallmann, A. V. A. Bezerra, E. D. Acosta, R. A. F. Machado, U. B. Demirci and S. Bernard, Surf. Coat. Technol., 2018, 350, 569–586 CrossRef CAS.
- S. Schwarz, M. Friedrich, G. Motz and R. Kempe, Z. Anorg. Allg. Chem., 2015, 641, 2266–2271 CrossRef CAS.
- D. Schumacher, M. Wilhelm and K. Rezwan, J. Am. Ceram. Soc., 2020, 103, 2991–3001 CrossRef CAS.
- M. Schubert, M. Wilhelm, S. Bragulla, C. Sun, S. Neumann, T. M. Gesing, P. Pfeifer, K. Rezwan and M. Bäumer, Catal. Lett., 2017, 147, 472–482 CrossRef CAS.
- H. P. Macedo, R. L. Medeiros, J. Ilsemann, D. M. Melo, K. Rezwan and M. Wilhelm, Microporous Mesoporous Mater., 2019, 278, 156–166 CrossRef CAS.
- M. Adam, S. Kocanis, T. Fey, M. Wilhelm and G. Grathwohl, J. Eur. Ceram. Soc., 2014, 34, 1715–1725 CrossRef CAS.
- M. Wilhelm, M. Adam, M. Baeumer and G. Grathwohl, Adv. Eng. Mater., 2008, 10, 241–245 CrossRef CAS.
- G. Glatz, T. Schmalz, T. Kraus, F. Haarmann, G. Motz and R. Kempe, Chem. – Eur. J., 2010, 16, 4231–4238 CrossRef CAS.
- D. Forberg, T. Schwob, M. Zaheer, M. Friedrich, N. Miyajima and R. Kempe, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- D. Forberg, T. Schwob and R. Kempe, Nat. Commun., 2018, 9, 1–7 CrossRef CAS.
- D. Forberg, J. Obenauf, M. Friedrich, S.-M. Hühne, W. Mader, G. Motz and R. Kempe, Catal. Sci. Technol., 2014, 4, 4188–4192 RSC.
- M. Birot, J.-P. Pillot and J. Dunogues, Chem. Rev., 1995, 95, 1443–1477 CrossRef CAS.
- P. Colombo, G. Mera, R. Riedel and G. D. Soraru, J. Am. Ceram. Soc., 2010, 93, 1805–1837 CAS.
- P. Greil, Adv. Eng. Mater., 2000, 2, 339–348 CrossRef CAS.
- P. Greil, J. Am. Ceram. Soc., 1995, 78, 835–848 CrossRef CAS.
- A. Francis, E. Ionescu, C. Fasel and R. Riedel, Inorg. Chem., 2009, 48, 10078–10083 CrossRef CAS.
- M. Hojamberdiev, R. M. Prasad, K. Morita, M. A. Schiavon and R. Riedel, Microporous Mesoporous Mater., 2012, 151, 330–338 CrossRef CAS.
- Y. Wang, Y. Feng, X. Guo, Y. Liu and H. Gong, RSC Adv., 2017, 7, 46215–46220 RSC.
- K. Kulbaba, A. Cheng, A. Bartole, S. Greenberg, R. Resendes, N. Coombs, A. Safa-Sefat, J. E. Greedan, H. D. Stöver and G. A. Ozin, J. Am. Chem. Soc., 2002, 124, 12522–12534 CrossRef CAS.
- X. Chen, Z. Su, L. Zhang, M. Tang, Y. Yu, L. Zhang and L. Chen, J. Am. Ceram. Soc., 2010, 93, 89–95 CrossRef CAS.
- E. Ionescu, C. Linck, C. Fasel, M. Müller, H. J. Kleebe and R. Riedel, J. Am. Ceram. Soc., 2010, 93, 241–250 CrossRef CAS.
- T. Ishikawa, S. Kajii, K. Matsunaga, T. Hogami, Y. Kohtoku and T. Nagasawa, Science, 1998, 282, 1295–1297 CrossRef CAS.
- P. Amoros, D. Beltran, C. Guillem and J. Latorre, Chem. Mater., 2002, 14, 1585–1590 CrossRef CAS.
- G. Hahn, J. K. Ewert, C. Denner, D. Tilgner and R. Kempe, ChemCatChem, 2016, 8, 2461–2465 CrossRef CAS.
- S. M. Sachau, M. Zaheer, A. Lale, M. Friedrich, C. E. Denner, U. B. Demirci, S. Bernard, G. Motz and R. Kempe, Chem. – Eur. J., 2016, 22, 15508–15512 CrossRef CAS.
- T. Schmalz, T. Kraus, M. Günthner, C. Liebscher, U. Glatzel, R. Kempe and G. Motz, Carbon, 2011, 49, 3065–3072 CrossRef CAS.
- M. Seifollahi Bazarjani, H.-J. Kleebe, M. M. Müller, C. Fasel, M. Baghaie Yazdi, A. Gurlo and R. Riedel, Chem. Mater., 2011, 23, 4112–4123 CrossRef.
- M. Seifollahi Bazarjani, M. M. Müller, H.-J. Kleebe, Y. Jüttke, I. Voigt, M. Baghaie Yazdi, L. Alff, R. Riedel and A. Gurlo, ACS Appl. Mater. Interfaces, 2014, 6, 12270–12278 CrossRef CAS.
- J. Wang, V. Schölch, O. Görke, G. Schuck, X. Wang, G. Shao, S. Schorr, M. F. Bekheet and A. Gurlo, Open Ceram., 2020, 1, 100001 CrossRef.
- X. Li, D. Li, H. Tian, L. Zeng, Z.-J. Zhao and J. Gong, Appl. Catal., B, 2017, 202, 683–694 CrossRef CAS.
- Z. Li, L. Mo, Y. Kathiraser and S. Kawi, ACS Catal., 2014, 4, 1526–1536 CrossRef CAS.
- T. Xie, L. Shi, J. Zhang and D. Zhang, Chem. Commun., 2014, 50, 7250–7253 RSC.
- J. Zhang and F. Li, Appl. Catal., B, 2015, 176–177, 513–521 CrossRef CAS.
- I. Luisetto, S. Tuti and E. Di Bartolomeo, Int. J. Hydrogen Energy, 2012, 37, 15992–15999 CrossRef CAS.
- K. Mette, S. Kühl, H. Düdder, K. Kähler, A. Tarasov, M. Muhler and M. Behrens, ChemCatChem, 2014, 6, 100–104 CrossRef CAS.
- A. Gili, L. Schlicker, M. F. Bekheet, O. Görke, S. Penner, M. Grünbacher, T. Götsch, P. Littlewood, T. J. Marks and P. C. Stair, ACS Catal., 2018, 8, 8739–8750 CrossRef CAS.
- A. Doran, L. Schlicker, C. Beavers, S. Bhat, M. Bekheet and A. Gurlo, Rev. Sci. Instrum., 2017, 88, 013903 CrossRef CAS.
- L. Schlicker, A. Doran, P. Schneppmüller, A. Gili, M. Czasny, S. Penner and A. Gurlo, Rev. Sci. Instrum., 2018, 89, 033904 CrossRef.
- D. M. Többens and S. Zander, J. Large-Scale Res. Facil., 2016, 2, 49 CrossRef.
- C. Schlesiger, L. Anklamm, H. Stiel, W. Malzer and B. Kanngießer, J. Anal. At. Spectrom., 2015, 30, 1080–1085 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- J. Yuan, S. Hapis, H. Breitzke, Y. Xu, C. Fasel, H.-J. Kleebe, G. Buntkowsky, R. Riedel and E. Ionescu, Inorg. Chem., 2014, 53, 10443–10455 CrossRef CAS.
- Y. D. Blum, D. B. MacQueen and H.-J. Kleebe, J. Eur. Ceram. Soc., 2005, 25, 143–149 CrossRef CAS.
- G. D. Soraru, F. Dalcanale, R. Campostrini, A. Gaston, Y. Blum, S. Carturan and P. R. Aravind, J. Mater. Chem., 2012, 22, 7676–7680 RSC.
- J. Bill, J. Seitz, G. Thurn, J. Dürr, J. Canel, B. Janos, A. Jalowiecki, D. Sauter, S. Schempp and H. Lamparter, Phys. Status Solidi A, 1998, 166, 269–296 CrossRef CAS.
- Y. L. Li, E. Kroke, R. Riedel, C. Fasel, C. Gervais and F. Babonneau, Appl. Organomet. Chem., 2001, 15, 820–832 CrossRef CAS.
- J. Seitz, J. Bill, N. Egger and F. Aldinger, J. Eur. Ceram. Soc., 1996, 16, 885–891 CrossRef CAS.
- M. Weinmann, T. W. Kamphowe, J. Schuhmacher, K. Müller and F. Aldinger, Chem. Mater., 2000, 12, 2112–2122 CrossRef CAS.
- S. Traßl, D. Suttor, G. Motz, E. Rössler and G. Ziegler, J. Eur. Ceram. Soc., 2000, 20, 215–225 CrossRef.
- M. Zaheer, C. D. Keenan, J. Hermannsdörfer, E. Roessler, G. n. Motz, J. r. Senker and R. Kempe, Chem. Mater., 2012, 24, 3952–3963 CrossRef CAS.
- J. C. De Jesus, I. González, A. Quevedo and T. Puerta, J. Mol. Catal. A: Chem., 2005, 228, 283–291 CrossRef CAS.
- J. Zhang, J. Tu, X. Xia, Y. Qiao and Y. Lu, Sol. Energy Mater. Sol. Cells, 2009, 93, 1840–1845 CrossRef CAS.
- Y. Cao, L. Nyborg and U. Jelvestam, Surf. Interface Anal., 2009, 41, 471–483 CrossRef CAS.
- M. F. Bekheet, L. Dubrovinsky and A. Gurlo, J. Solid State Chem., 2015, 230, 301–308 CrossRef CAS.
- M. F. Bekheet, L. Schlicker, A. Doran, K. Siemensmeyer and A. Gurlo, Dalton Trans., 2018, 47, 2727–2738 RSC.
- S. Steffen, M. Bauer, D. Decker and F. Richter, J. Appl. Polym. Sci., 2014, 131, 40375 CrossRef.
- M. Sokri, M. Nazri, Y. Daiko, Z. Mouline, S. Honda and Y. Iwamoto, Inorganics, 2016, 4, 5 CrossRef.
- N. Soltani, U. Simon, A. Bahrami, X. Wang, S. Selve, J. D. Epping, M. I. Pech-Canul, M. F. Bekheet and A. Gurlo, J. Eur. Ceram. Soc., 2017, 37, 4809–4820 CrossRef CAS.
- G. Mera, A. Navrotsky, S. Sen, H.-J. Kleebe and R. Riedel, J. Mater. Chem. A, 2013, 1, 3826–3836 RSC.
- H. J. Kleebe, C. Turquat and G. D. Sorarù, J. Am. Ceram. Soc., 2001, 84, 1073–1080 CrossRef CAS.
- G. Mera, R. Riedel, F. Poli and K. Müller, J. Eur. Ceram. Soc., 2009, 29, 2873–2883 CrossRef CAS.
- H. J. Kleebe, Phys. Status Solidi A, 1998, 166, 297–313 CrossRef CAS.
- C. Turquat, H. J. Kleebe, G. Gregori, S. Walter and G. D. Sorarù, J. Am. Ceram. Soc., 2001, 84, 2189–2196 CrossRef CAS.
- G. Ziegler, H.-J. Kleebe, G. Motz, H. Müller, S. Traßl and W. Weibelzahl, Mater. Chem. Phys., 1999, 61, 55–63 CrossRef CAS.
- M. Scheffler, P. Greil, A. Berger, E. Pippel and J. Woltersdorf, Mater. Chem. Phys., 2004, 84, 131–139 CrossRef CAS.
- C. Schitco, M. S. Bazarjani, R. Riedel and A. Gurlo, J. Mater. Chem. A, 2015, 3, 805–818 RSC.
- M. Bradford and M. Vannice, Catal. Rev., 1999, 41, 1–42 CrossRef CAS.
- J. Wei and E. Iglesia, J. Catal., 2004, 225, 116–127 CrossRef CAS.
- J. Wei and E. Iglesia, J. Catal., 2004, 224, 370–383 CrossRef CAS.
- P. Djinović, I. G. O. Črnivec, B. Erjavec and A. Pintar, Appl. Catal., B, 2012, 125, 259–270 CrossRef.
- C. Hoffmann, P. Plate, A. Steinbrück and S. Kaskel, Catal. Sci. Technol., 2015, 5, 4174–4183 RSC.
- N. B. Laidani and G. D. Sorarù, J. Mater. Res., 2015, 30, 770–781 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00917b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Christopher
Schlesiger
c,
Christopher
Schlesiger