
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of alkyl group regiochemistry and backbone fluorination on the packing and transistor performance of N-cyanoimine functionalised indacenodithiophenes†
Thomas
Hodsden
a,
Karl J.
Thorley
b,
Aniruddha
Basu
 c,
Andrew J. P.
White
a,
Changsheng
Wang
d,
William
Mitchell
d,
Florian
Glöcklhofer
a,
Thomas D.
Anthopoulos
c and
Martin
Heeney
*a
c,
Andrew J. P.
White
a,
Changsheng
Wang
d,
William
Mitchell
d,
Florian
Glöcklhofer
a,
Thomas D.
Anthopoulos
c and
Martin
Heeney
*a
aDepartment of Chemistry and Centre for Processable Electronics, White City Campus, Imperial College London, London, W12 0BZ, UK. E-mail: m.heeney@imperial.ac.uk
bDepartment of Chemistry, University of Kentucky, Lexington, KY 40506-0055, USA
cKing Abdullah University of Science and Technology (KAUST), KAUST Solar Center (KSC), Thuwal 23955-6900, Kingdom of Saudi Arabia
dPerformance Materials Division, Advanced Technologies, Merck Chemicals Ltd, Chilworth Technical Centre, University Parkway, Southampton, SO16 7QD, UK
First published on 3rd February 2021
Abstract
The synthesis of two novel n-type molecular organic semiconductors based on a fluorinated indacenodithiophene core in combination with an electron withdrawing N-cyanoimine group is reported, and the influence of the regiochemistry of the solubilizing sidechain is investigated. The N-cyanoimine is confirmed to be a strongly electron accepting group, which in combination with the core fluorination resulted in high electron affinities for both materials. Single crystal analysis demonstrated that whilst both materials arrange in ordered slipped stacks with close π–π stacking distances (∼3.40 Å), significant differences in electron transfer integrals for the two regioisomers were observed, relating to differences in relative molecular displacement along the π-stacking direction. Organic thin-film transistors fabricated via blade-coating displayed electron mobility up to 0.13 cm2 V−1 s−1 for the isomer with the larger transfer integral.
Introduction
Conjugated organic semiconductors (OSCs) are one of the key enabling materials for the predicted next generation of printed electronic devices.1,2 Extensive research effort has resulted in the development of a number of promising solution processable materials. However, progress in the development of p-type (or hole transporting) materials has far outstripped that of n-type (or electron transporting) materials in terms of charge carrier mobility, scalability and stability.3–6 New n-type materials are required for use in a number of applications, from the development of CMOS-type circuits with low power consumption to electron-transporting interlayers in hybrid photovoltaic devices.7–10In designing such n-type materials, it is important that the OSC has a high electron affinity to facilitate electron injection and charge transport. This is usually expressed in terms of the energy level of the lowest unoccupied molecular orbital (LUMO), which should be sufficiently low-lying to enable charge injection from common electrode materials. This is typically achieved by the functionalization of an aromatic core with a number of strongly electron withdrawing groups, like nitriles, carbonyl, (di)imide and halogens.9,11–14 Low lying LUMO levels can also help to improve the ambient stability of the resulting reduced species.15–18 In addition to energetic considerations, it is also important for charge transport that the material has a high degree of solid-state ordering with a strong electronic coupling between adjacent units.19–22
A number of fused aromatic cores have been investigated as potential n-type materials. One particularly interesting class is that based on fused aromatics held rigidly co-planar by bridging (hetero)atoms, often termed ladder-type materials.23,24 The high rigidity and coplanarity helps to reduce reorganization energy during charge transfer and facilitates delocalization of the conjugated system. Within the extensive class of ladder-type materials, we and others, have been particularly interested in 4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene (IDT), in which a central benzene ring is flanked by two thienyl units held co-planar by bridging carbon atoms.25–28 The high symmetry of IDT, coupled with its terminal thienyl groups, tends to afford linear materials with low torsional disorder, which have good p-type transistor performance.
The promising performance of the IDT core has prompted interest in its utilization as an n-type material in transistor devices.27,29,30 Since IDT is inherently electron rich, as a consequence of the two fused thiophene rings, efforts have focussed on the functionalization of IDT with various electron withdrawing groups, either in the terminal or bridgehead positions.29,31–34 We recently reported that the functionalization of the bridgehead position of IDT with the strongly electron withdrawing dicyanomethylene group together with simultaneous fluorination of the central benzene ring resulted in a material with a low lying LUMO and promising n-type performance.35 Unusually, we found that this material demonstrated improved solubility compared to the analogous non-fluorinated material, which appeared to result from the steric interactions between the fluorine and the dicyanomethylene group leading to a bowing of the structure.
In this report, we aimed to reduce the molecular distortion whilst retaining the deep LUMO level by combining central core fluorination with the addition of a less sterically demanding electron withdrawing group in the bridgehead positions of the IDT. One such group is the N-cyanoimine group (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-CN), which has been relatively unexplored in the context of n-type materials.36–38 Recently, Rault-Berthelot and co-workers reported that IDT functionalised with N-cyanoimine exhibited a very similar LUMO level to IDT functionalized with dicyanomethylene, thereby demonstrating its strongly electron-withdrawing nature.39 Building upon these results, we hereby report the synthesis and characterization of two novel fluorinated IDT derivatives (diFIDT-di(N(CN))) containing the N-cyanoimine group. In addition, we also investigate how changing the regiochemistry of the required solubilizing hexyl group from the alpha (2,7-) to the beta (3,8-) positions of the IDT core influences solid-state packing and device performance.
N-CN), which has been relatively unexplored in the context of n-type materials.36–38 Recently, Rault-Berthelot and co-workers reported that IDT functionalised with N-cyanoimine exhibited a very similar LUMO level to IDT functionalized with dicyanomethylene, thereby demonstrating its strongly electron-withdrawing nature.39 Building upon these results, we hereby report the synthesis and characterization of two novel fluorinated IDT derivatives (diFIDT-di(N(CN))) containing the N-cyanoimine group. In addition, we also investigate how changing the regiochemistry of the required solubilizing hexyl group from the alpha (2,7-) to the beta (3,8-) positions of the IDT core influences solid-state packing and device performance.
Results and discussion
Design and synthesis
As a starting point we examined the predicted effect on backbone planarity of the N-cyanoimine group by performing molecular modelling using DFT with a B3LYP level of theory and 6-311+G(d,p) basis set. The N-cyanoimine group is asymmetric and therefore three stereoisomers could possibly be formed by its attachment to the IDT bridging positions. DFT calculations were performed to establish the ground state conformation and HOMO/LUMO energy levels of each isomer (Fig. S1, ESI†). The results indicated that the structure is planar in all three isomers, suggesting that unlike dicyanomethylene, N-cyanoimine would not introduce any undesired steric interactions with the fluorinated core. The Z,Z-isomer was predicted to be the most stable, as indicated by a lower HOMO energy level. These calculations also predicted a low-lying LUMO energy level (−4.25 eV), indicating suitability for n-type applications.2,7-Dihexyl-diFIDT-di(N(CN)) and 3,8-dihexyl-diFIDT-di(N(CN)) were synthesised following a modified literature procedure (Scheme 1).36,40 The starting 2,7-dihexyl-diFIDT-di(O) was synthesised following our recently reported route,35 and the isomeric 3,8-dihexyl-diFIDT-di(O) was prepared by a modification of the route reported to the non-fluorinated analogue.32 Thus, Stille cross-coupling of diethyl 2,5-dibromo-3,6-difluoroterephthalate ([1]) with trimethyl(4-hexylthiophen-2-yl)stannane ([2]) using tetrakis(triphenylphosphine)palladium(0) afforded diethyl 2,5-difluoro-3,6-bis(4-hexylthiophen-2-yl)terephthalate ([3]) in high yield (83%) after purification by silica gel chromatography. Saponification with KOH in ethanol afforded a quantitative conversion to the diacid ([4]), which was subsequently converted to the acid chloride by treatment with SOCl2 in DCM in the presence of a catalytic amount of DMF. After solvent removal, the resulting water-sensitive material was used directly without purification. Ring closure was achieved by a Lewis acid-promoted intramolecular Friedel–Crafts acylation with AlCl3 to afford the diketone, 3,8-dihexyl-5,10-difluoro-s-indaceno[1,2-b:5,6-b′]dithiophene-4,9-dione, 3,8-dihexyl-diFIDT-di(O) (78%).
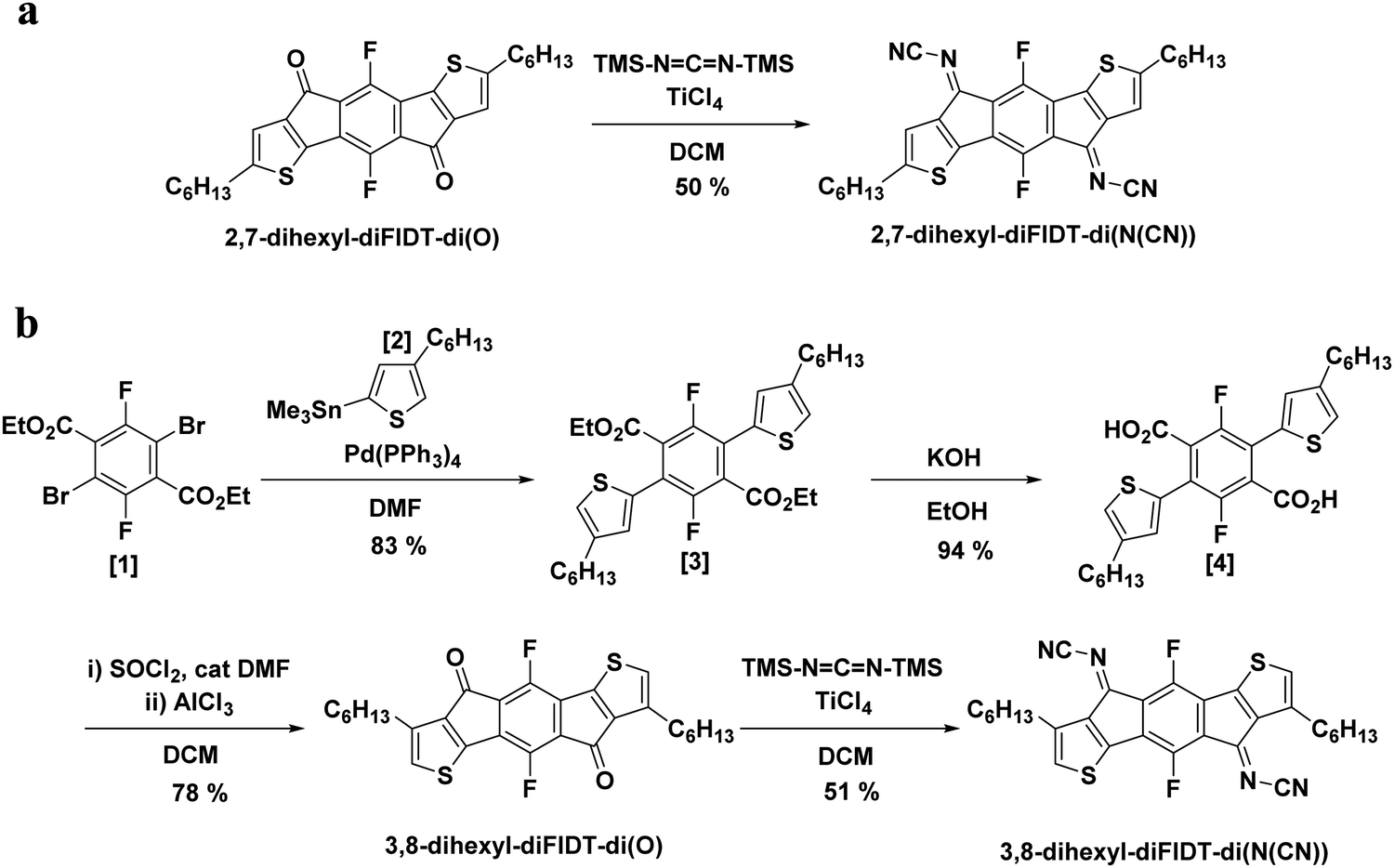 | ||
| Scheme 1 Synthesis of (a) 2,7-dihexyl-diFIDT-di(N(CN)) and (b) 3,8-dihexyl-diFIDT-di(N(CN)). | ||
It is noted that limiting the reaction time of the Friedel–Crafts acylation for the 3,8-dihexyl-diFIDT-di(O) to 4 h was crucial in avoiding the formation of unwanted side-products. NMR and mass spectrometric analysis suggested that the side-products were a mixture of higher molecular weight dimer/oligomer structures (Fig. S12 and S13, ESI†). It is well-known that thiophene can polymerise in the presence of a strong Lewis acid (e.g. AlCl3), hence it is feasible that these side-products arise due to the presence of a reactive α-position on the thienyl units in this isomer.40 This is supported by the fact that no side-products were observed for 2,7-dihexyl-diFIDT-di(O), where the α-positions are occupied by alkyl chains.
The respective diketone starting materials were reacted with TiCl4 and bis(trimethylsilyl)carbodiimide in a Knoevenagel-type condensation reaction at room temperature to give 2,7-dihexyl-diFIDT-di(N(CN)) (50%) and 3,8-dihexyl-diFIDT-di(N(CN)) (51%) respectively after purification by reprecipitation. A single Z,Z-isomer was confirmed to be formed in both cases, in agreement with the DFT calculations, by a combination of 1H NMR, 19F NMR, 13C NMR, IR, HRMS and single crystal XRD.
Molecular organisation
Single crystals of 2,7-dihexyl-diFIDT-di(N(CN)) and 3,8-dihexyl-diFIDT-di(N(CN)) were grown via the solvent vapour exchange method using chlorobenzene/methanol mixtures. The crystal structure for 2,7-dihexyl-IDT-di(N(CN)) was downloaded from the Cambridge Crystallographic Data Centre for comparison.2,7-Dihexyl-diFIDT-di(N(CN)) crystallised in the monoclinic P21/n space group. The IDT core was completely coplanar, in agreement with the DFT calculations (Fig. 1a). However the N-cyanoimine groups are slightly bent out-of-plane with the IDT (2.3°) core. This distortion is more pronounced than that present in non-fluorinated, 2,7-dihexyl-IDT-di(N(CN)) (1.3°) (Fig. 1c). However, this distortion is less pronounced than the corresponding dicyanomethylene-bridged analogue previously reported (7.5°), which in agreement with the smaller size of the N-cyanoimine group versus dicyanomethylene. The hexyl chains on the thienyl α-position lie outside the plane with a dihedral angle of 88°. A single N⋯H (2.61 Å) short contact is observed between stacks, along with intramolecular C⋯C (3.20 Å) and N⋯F (2.96 Å) interactions (Fig. 1b). It is interesting to find that fluorination does not lead to specific fluorine-based interactions being introduced.
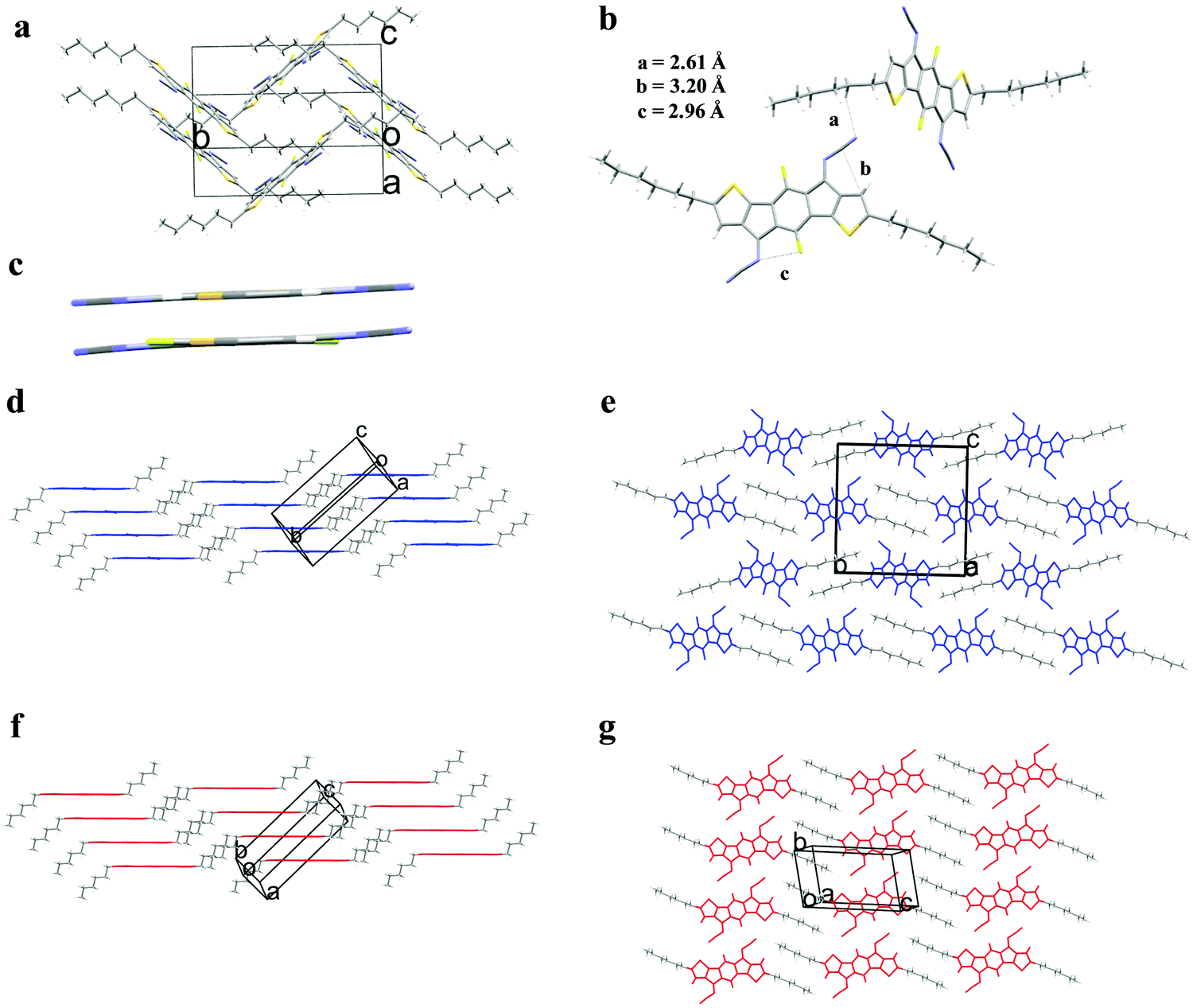 | ||
| Fig. 1 (a) Unit cell of 2,7-dihexyl-diFIDT-di(N(CN)); (b) inter- and intramolecular short contacts; (c) backbone planarity of 2,7-dihexyl-IDT-di(N(CN)) (top) versus2,7-dihexyl-diFIDT-di(N(CN)) (bottom), with alkyl chains removed for clarity (viewed down the molecular short axis); (d and e) Crystal packing motifs of 2,7-dihexyl-FIDT-di(N(CN)); (f and g) crystal packing motifs of 2,7-dihexyl-diIDT-di(N(CN)). | ||
A planar slipped π–π stacking arrangement is formed along the a axis, however adjacent stacks (along the molecular short axis) are arranged almost perpendicular to each other (85.3°) with poor inter-stack registry. Alkyl chain interdigitation is observed between stacks along the long axis (Fig. 1d and e). This is in contrast to the non-fluorinated analogue 2,7-dihexyl-IDT-di(N(CN)), which displays a planar slipped π–π stacking motif without such tilting (Fig. 1f and g). This change in packing motif upon core fluorination is similar to that observed for the dicyanomethylene-containing analogue, 2,7-dihexyl-diFIDT-di(C(CN)2), reported in our previous study.35 There is co-facial packing of the conjugated core within a stack, along the a axis, with an interplanar stacking distance of 3.41 Å and a slipping distance of 3.24 Å (angle 43.6°) (Fig. S22, ESI†). These values are similar to 2,7-dihexyl-IDT-di(N(CN)) (interplanar stacking distance of 3.41 Å, slipping distance of 3.51 Å).
One key factor affecting charge transport through organic crystals is the electronic coupling between frontier molecular orbitals on neighbouring units, known as the transfer integral. A larger value indicates better overlap between frontier molecular orbitals on neighbouring units (LUMO–LUMO in the case of electron transport) and therefore the potential for more efficient charge transfer. 2,7-Dihexyl-diFIDT-di(N(CN)) displays a one-dimensional (1D) charge transfer pathway, with an electron transfer integral of 106 meV for the main π-stack. This value is high, and larger than that calculated for 2,7-dihexyl-IDT-di(N(CN)) (87 meV), indicating that core fluorination is expected to be beneficial for charge transfer.
To gain insight into the intermolecular interaction energies, symmetry adapted perturbation theory (SAPT0/jun-cc-pvdz) calculations were performed. SAPT0 analysis allows the total interaction energies between molecular pairs extracted from the crystal structures to be decomposed into the respective non-covalent interactions. Analysis was performed on molecular pairs of 2,7-dihexyl-IDT-di(N(CN)) and 2,7-dihexyl-diFIDT-di(N(CN)) from their respective crystal packing motifs. In addition, the 2,7-dihexyl-IDT-di(N(CN)) molecules in their crystal packing were altered by substitution of the central protons for fluorine atoms, thus leading to the hypothetical 2,7-dihexyl-diFIDT-di(N(CN)) molecules in the 2,7-dihexyl-IDT-di(N(CN)) packing. Similarly, 2,7-dihexyl-IDT-di(N(CN)) molecules were placed in the 2,7-dihexyl-diFIDT-di((N(CN)) packing. This allowed the specific effect of F substitution to be isolated.
In the 2,7-dihexyl-diFIDT-di(N(CN)) packing, the substitution of F with H makes very little difference to pairwise interaction energies (Fig. 2a). In contrast, there is a significant difference upon substitution of H with F in the 2,7-dihexyl-IDT-di(N(CN)) packing (Fig. 2b). In this packing, the non-fluorinated derivative shows an interaction stabilisation 8.33 kcal mol−1 stronger than the fluorinated one. This difference is the result of a large steric repulsion by the inclusion of fluorine, which is too large to fit into the close packing between the N-cyanoimine groups. The repulsion is to such an extent that the overall interaction becomes slightly positive (ESAPT = +0.20 kcal mol−1). In can be concluded that the main difference in packing of the two compounds originates from the in-plane edge-to-edge interaction in the 2,7-dihexyl-IDT-di(N(CN)) crystal, between molecules in adjacent stacks. 2,7-Dihexyl-diFIDT-di(N(CN)) prefers to adopt the crystal packing where intermolecular interactions involving the fluorine atoms are minimised.
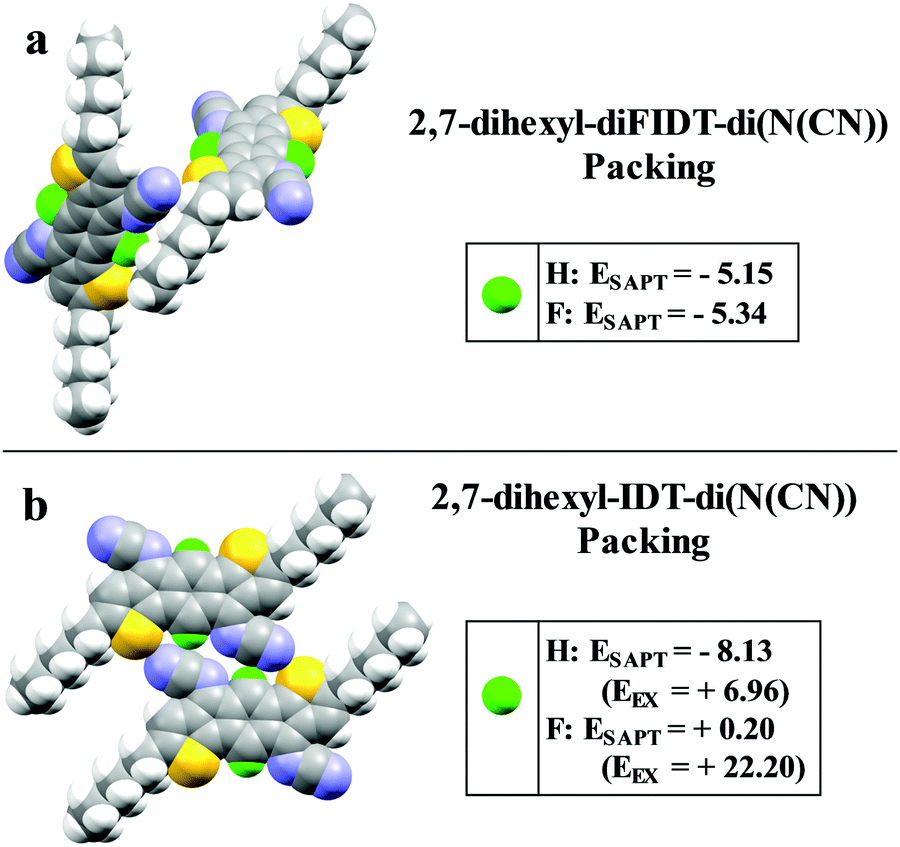 | ||
| Fig. 2 In-plane longitudinal interaction in (a) the 2,7-dihexyl-diFIDT-di(N(CN)) crystal packing and (b) the 2,7-dihexyl-IDT-di(N(CN)) crystal packing. Total SAPT energies (ESAPT) and repulsive exchange energies (EEX) are given for 2,7-dihexyl-IDT-di(N(CN)) and 2,7-dihexyl-diFIDT-di(N(CN)) molecules in each packing motif. All energies in kcal mol−1. Atom colours: yellow = sulfur, green = hydrogen or fluorine, purple = nitrogen. | ||
3,8-Dihexyl-diFIDT-di(N(CN)) crystallised in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The IDT core was completely coplanar, in agreement with the DFT calculations. The hexyl chains on the thienyl β-position lie outside the plane with a dihedral angle of 80° (Fig. 3a). The N-cyanoimine groups are slightly bent out-of-plane with the IDT (13.7°) (Fig. 3c). The bending is greater than that for 2,7-dihexyl-diFIDT-di(N(CN)), indicating increased steric crowding due to the proximity of the alkyl chains in the 3,8-positions. As opposed to 2,7-dihexyl-diFIDT-di(N(CN)), a number of fluorine-based non-covalent interactions do appear to be present in 3,8-dihexyl-diFIDT-di(N(CN)). One intermolecular S⋯F (3.27 Å) short contact was observed, along with several intramolecular interactions, including C⋯H (2.59 Å), N⋯H (2.55 Å) and S⋯F (2.91 Å) (Fig. 3b). These heteroatom interactions lead to a sizeable SAPT interaction energy of −9.6 kcal mol−1, where a combination of electrostatic and dispersion components overcome the repulsive exchange energy at close interatomic distance (Fig. S28, ESI†). Replacing fluorine with a proton weakens the intermolecular stabilisation, mostly due to a decrease in electrostatic energy. It was not feasible to directly compare the 2,7- and 3,8- substituted derivatives by the same methodology due to the large reorganisation required to fit the alkyl chains into the available space.
space group. The IDT core was completely coplanar, in agreement with the DFT calculations. The hexyl chains on the thienyl β-position lie outside the plane with a dihedral angle of 80° (Fig. 3a). The N-cyanoimine groups are slightly bent out-of-plane with the IDT (13.7°) (Fig. 3c). The bending is greater than that for 2,7-dihexyl-diFIDT-di(N(CN)), indicating increased steric crowding due to the proximity of the alkyl chains in the 3,8-positions. As opposed to 2,7-dihexyl-diFIDT-di(N(CN)), a number of fluorine-based non-covalent interactions do appear to be present in 3,8-dihexyl-diFIDT-di(N(CN)). One intermolecular S⋯F (3.27 Å) short contact was observed, along with several intramolecular interactions, including C⋯H (2.59 Å), N⋯H (2.55 Å) and S⋯F (2.91 Å) (Fig. 3b). These heteroatom interactions lead to a sizeable SAPT interaction energy of −9.6 kcal mol−1, where a combination of electrostatic and dispersion components overcome the repulsive exchange energy at close interatomic distance (Fig. S28, ESI†). Replacing fluorine with a proton weakens the intermolecular stabilisation, mostly due to a decrease in electrostatic energy. It was not feasible to directly compare the 2,7- and 3,8- substituted derivatives by the same methodology due to the large reorganisation required to fit the alkyl chains into the available space.
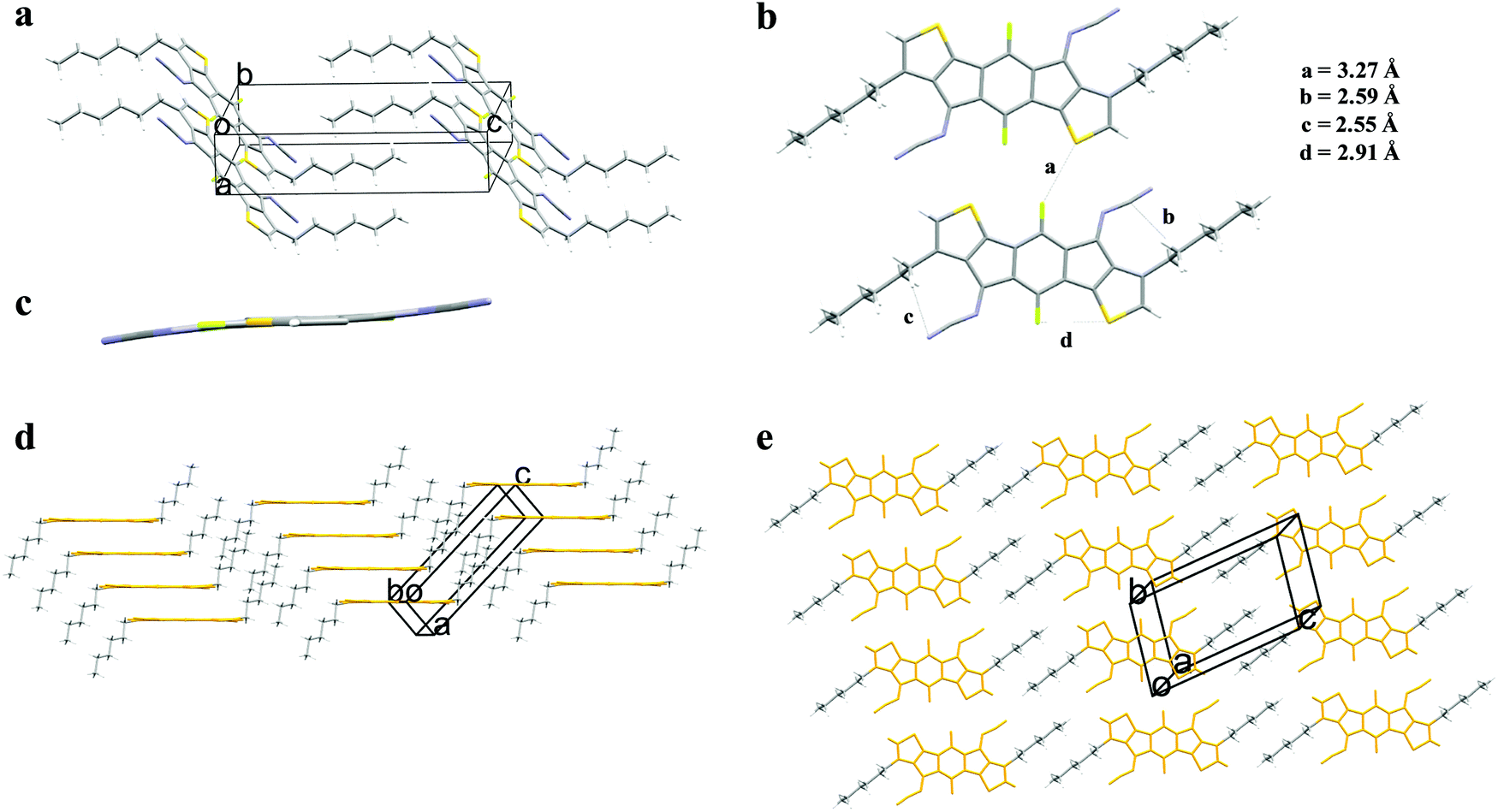 | ||
| Fig. 3 (a) Unit cell of 3,8-dihexyl-diFIDT-di(N(CN)); (b) inter- and intramolecular short contacts; (c) backbone planarity, with alkyl chains removed for clarity (viewed down the molecular short axis); (d and e) crystal packing motif of 3,8-dihexyl-diFIDT-di(N(CN)). | ||
A planar slipped π–π stacking arrangement is formed along the a axis and alkyl chain interdigitation is observed between stacks (Fig. 3d and e). Stacks are arranged co-planar along the short axis. This is contrasting to that observed for 2,7-dihexyl-diFIDT-di(N(CN)), where a perpendicular arrangement is present. Hence, the position of the alkyl chain is shown to have a significant impact on the crystal packing. There is co-facial packing of the conjugated core within a stack, along the a axis, with an interplanar stacking distance of 3.39 Å and a slipping distance of 3.25 Å (angle 43.8°) (Fig. S27, ESI†). These values are almost identical to those present in 2,7-dihexyl-diFIDT-di(N(CN)).
3,8-Dihexyl-diFIDT-di(N(CN)) displays a one-dimensional (1D) charge transfer pathway, with an electron transfer integral of 29 meV for the main π-stack. This is much lower than for 2,7-dihexyl-diFIDT-di(N(CN)) (106 meV), which indicates that there is a poorer LUMO–LUMO interaction. Such a large difference is surprising, considering that the interplanar stacking distance and slipping distance are so similar, but highlights the sensitivity of charge transfer to the relative positioning of the frontier molecular orbitals on neighbouring molecules. This phenomenon has been widely studied, showing that, while the transfer integral generally decreases with increasing displacement, the relative displacement of neighbouring molecules (e.g. along the molecular short- and/or long-axes) is also important.41,42 In this instance, poorer LUMO–LUMO overlap for 3,8-dihexyl-diFIDT-di(N(CN)) (Fig. S29c, ESI†) compared to 2,7-dihexyl-diFIDT-di(N(CN)) (Fig. S29a, ESI†) appears to be a result of a larger displacement along the molecular short-axis (Fig. S29d, ESI†).
Electrochemical properties
Electrochemical studies of both dialkyl-diFIDT-di(N(CN)) analogues were performed by cyclic voltammetry (CV) using a 0.1 M solution of Bu4NPF6 in DCM solutions and a ferrocene (Fc/Fc+) internal standard. Potentials were swept from 0 V to a positive maximum, then swept back to a negative maximum before returning to 0 V. In line with best practice, energy levels were estimated from E1/2 (when reversibility was observed) or Ei (when irreversibility was observed), where E1/2 is the half-wave potential and Ei is the inflection-point potential.43 HOMO = −(E1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ori + 4.8) eV and LUMO = −(E1/2ori + 4.8) eV for oxidation and reduction respectively, with the ferrocene/ferrocenium reference redox system at 4.8 eV below the vacuum level. Electrochemical data for 2,7-dihexyl-IDT-di(N(CN)) was taken from Rault-Berthelot and co-workers, where E1/2 for the first reduction peak was extracted to provide a better comparison.39
ori + 4.8) eV and LUMO = −(E1/2ori + 4.8) eV for oxidation and reduction respectively, with the ferrocene/ferrocenium reference redox system at 4.8 eV below the vacuum level. Electrochemical data for 2,7-dihexyl-IDT-di(N(CN)) was taken from Rault-Berthelot and co-workers, where E1/2 for the first reduction peak was extracted to provide a better comparison.39
No major differences in the cyclic voltammograms were observed between dialkyl-diFIDT-di(N(CN)) analogues in solution, reflecting the negligible impact of alkyl chains on the electrochemical properties. The cyclic voltammograms displayed two chemically reversible reductions along with one or two irreversible oxidations (Fig. 4). These compounds displayed deep LUMO energy levels (−4.16 eV to −4.19 eV), indicating the suitability of N-cyanoimine as an effective EWG. Direct comparison between the LUMO energy levels of 2,7-dihexyl-IDT-di(N(CN)) and 2,7-dihexyl-diFIDT-di(N(CN)) demonstrated a small depression (0.16 eV) due to the introduction of fluorine groups onto the central phenyl moiety.
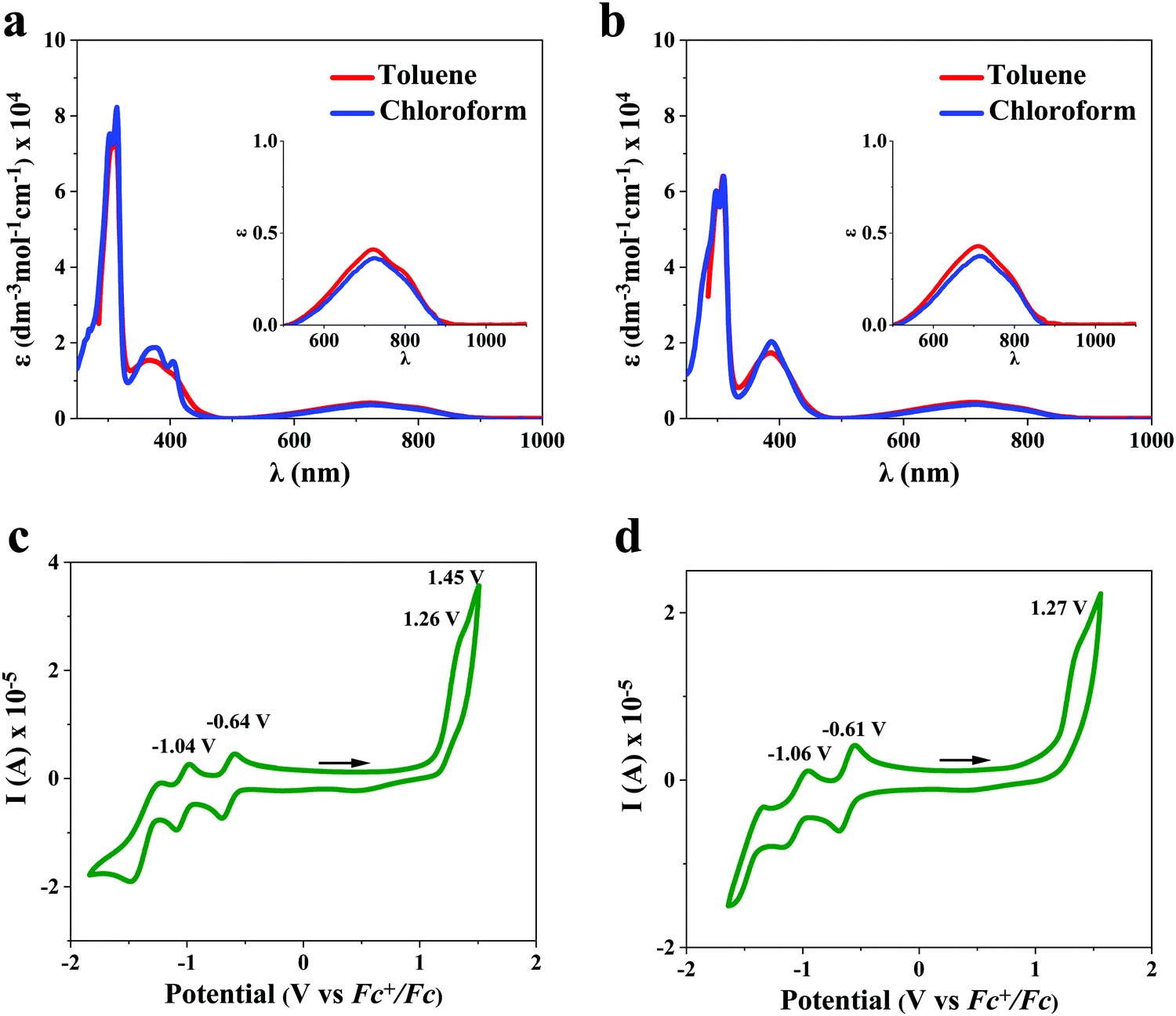 | ||
| Fig. 4 Solution absorption spectra for (a) 2,7-dihexyl-diFIDT-di(N(CN)) and (b) 3,8-dihexyl-diFIDT-di(N(CN)) in toluene and chloroform; cyclic voltammagrams of dialkyl-diFIDT-diN(CN)) analogues in CH2Cl2, (0.1 M solution [n-Bu4N]PF6 as supporting electrolyte), showing corresponding E1/2ori potentials for (c) 2,7-dihexyl-diFIDT-di(N(CN)) and (d) 3,8-dihexyl-diFIDT-di(N(CN)). | ||
UV-vis absorption spectra
The absorption spectra for both dialkyl-diFIDT-di(N(CN)) analogues in solution (toluene and chloroform, 10−5 M) are shown in Fig. 4 and summarised in Table 1. Both dialkyl-diFIDT-di(N(CN)) analogues exhibited intense, well-defined peaks in the high energy region (250–450 nm), and a weak, broad transition at low energy (550–900 nm). On the basis of time-dependent DFT (TD-DFT) calculations, the low energy bands for both materials are assigned to a HOMO/LUMO transition with very weak oscillator strength (f 0.055 and 0.061 for 2,7-dihexyl-diFIDT-di(N(CN)) and 3,8-dihexyl-diFIDT-di(N(CN))), as shown in Fig. S19 (ESI†). The weak oscillator strength arises from the poor overlap between the LUMO, which is centralised on the N-cyanoimine and cyclopentadienyl ring, and the HOMO, located on the IDT core. From the onset of absorption at the longer wavelengths (around 850–900 nm), optical gaps of ∼1.4 eV could be extracted.| OSC | HOMOa [eV] | LUMOa [eV] | λ max (solution) [nm] | T d [°C] | T m [°C] | ΔHm [J g−1] |
|---|---|---|---|---|---|---|
| a Extracted from CV measurements. b Data for this compound taken from Rault-Berthelot et al.25 c Reanalysed as E1/2. d In THF. e In toluene. f In chloroform. g No transitions observed in DSC. | ||||||
| 2,7-Dihexyl-IDT-di(N(CN)) | −5.72 | −4.00c | 711, 322d | 364 | 139 | 25 |
| 2,7-Dihexyl-diFIDT-di(N(CN)) | −6.06 | −4.16 | 719, 365, 312e | 321 | —g | —g |
| 726, 405, 373, 314, 303f | ||||||
| 3,8-Dihexyl-diFIDT-di(N(CN)) | −6.07 | −4.19 | 708, 386, 309e | 334 | 92, 110 | 19, 11 |
| 713, 387, 310, 298f | ||||||
Thermal properties
The thermal behaviour of both dialkyl-diFIDT-di(N(CN)) analogues was investigated by a combination of thermal gravimetric analysis (TGA, Fig. S30, ESI†) and differential scanning calorimetry (DSC, Fig. S31, ESI†). The key data is summarised in Table 1.Both diFIDT-di(N(CN)) analogues exhibited high thermal stabilities (temperature at which 5% mass loss is reached, Td > 320 °C), though a distinct drop in mass (∼1% loss) can be seen at around 250–275 °C. This feature was also present for the non-fluorinated analogue (2,7-dihexyl-IDT-di(N(CN))), as previously reported by Rault-Berthelot et al., though this feature was not explicitly discussed.39 The Td of 364 °C reported for 2,7-dihexyl-IDT-di(N(CN)) was >40 °C higher than for 2,7-dihexyl-diFIDT-di(N(CN)) (Td = 321 °C), indicating that core fluorination was detrimental for the thermal stability. Repositioning of the hexyl chains from 2,7-position to 3,8-position led to a small increase in thermal stability (13 °C).
DSC of both dialkyl-diFIDT-di(N(CN)) analogues were recorded to up to 200 °C. Further heating to 250 °C resulted in degradation and a loss of reversibility, corresponding well with the 1% mass loss in the TGA. 2,7-dihexyl-diFIDT-di(N(CN)) did not display any thermal transitions below 200 °C. The non-fluorinated 2,7-dihexyl-IDT-di(N(CN)) was reported by Rault-Berthelot and co-workers to exhibit a weak crystal-to-mesophase transition in this temperature range.39
Conversely, 3,8-dihexyl-diFIDT-di(N(CN)) displayed two low enthalpy, reproducible endotherms around 100 °C, which appeared stable upon repeated thermal cycling (Fig. 5). Rault-Berthelot and co-workers detail similar transitions for 2,7-dihexyl-IDT-di(N(CN)), identifying the formation of an undefined mesophase.39 Analysis of these transitions in 3,8-dihexyl-diFIDT-di(N(CN)) by polarised optical microscopy (POM) showed the absence of an isotropic phase up to 225 °C, supporting the notion that the isotropic transitions in these OSCs lie beyond the decomposition temperature (∼250 °C). However, the inability to cool the samples from the melt, coupled with the low enthalpy of the transitions, meant that no further information about these thermal transitions could be gleaned from POM.
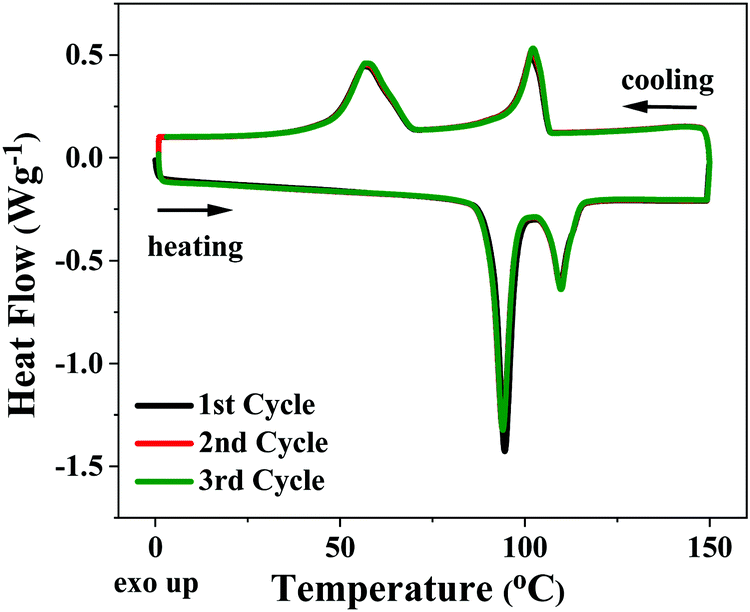 | ||
| Fig. 5 DSC plots (all cycles) for 3,8-dihexyl-diFIDT-di(N(CN)). | ||
Organic field-effect transistors
The charge transport behaviour of both materials was investigated in OFET devices. Previously reported transistors utilizing the non-fluorinated 2,7-dihexyl-IDT-di(N(CN)) were fabricated via vacuum deposition, achieving relatively modest device performance with a charge carrier mobility up to 1.4 × 10−3 cm2 V−1 s−1.39 In our case we found that the solubility of both dialkyl-diFIDT-di(N(CN)) OSCs was sufficient in chlorobenzene to fabricate transistor devices by blade-coating, a potentially scalable technique for solution based devices.35,44 Devices were fabricated in a bottom-contact top-gate (BC – TG) configuration, with the active layer deposited onto the substrate at 100 °C, followed by spin coating of the gate dielectric (CytopTM) and thermal evaporation of the aluminium gate electrode. The key results are summarised in Table 2. All devices exhibited unipolar electron transport, reflecting the role of the deep LUMO level in facilitating electron transport. In addition, the deep HOMO energy level (∼−6.05 eV) imparted a large energy level offset with the Au S/D electrodes, helping to suppress hole injection and possible ambipolar behaviour.| OSC | V T [V] | μ linear [cm2 V−1 s−1] | μ saturated [cm2 V−1 s−1] | I on/Ioff |
|---|---|---|---|---|
| a Annealed at 200 °C. | ||||
| 2,7-Dihexyl-diFIDT-di(N(CN)) | 45.5 | 0.02 | 0.09 (0.13) | 104 |
| 3,8-Dihexyl-diFIDT-di(N(CN)) | 23.7 | 0.01 | 0.03 (0.03) | 103 |
The saturated mobility was extracted from the linear part of the square root of the drain current (Fig. S32a and S33a, ESI†). The absence of any double slope in the plot endorses the reliability of the data,45 although a weak gate voltage-dependence of the saturated mobility above VT suggests some device non-idealities were present (Fig. S32c and S33c, ESI†). Furthermore, the large variations in the threshold voltages suggest the presence of interfacial traps and a strong relation to the local processing conditions during coating.
The best performance was obtained for 2,7-dihexyl-diFIDT-di(N(CN)) devices, annealed at 200 °C, with a saturated electron mobility up to 0.13 cm2 V−1 s−1 (average 0.09 cm2 V−1 s−1 across 8 devices) (Fig. 6). The performance of pristine devices was over an order of magnitude lower (average 0.003 cm2 V−1 s−1). Lower saturated mobilities were achieved with the 3,8-dihexyl-diFIDT-di(N(CN)) (up to 0.03 cm2 V−1 s−1), in agreement with the reduced electron transfer integral calculated from the single crystal packing. In contrast to 2,7-dihexyl-diFIDT-di(N(CN)), no difference in performance was realised between pristine devices and devices annealed at 200 °C for 3,8-dihexyl-diFIDT-di(N(CN)). All devices displayed performance at least an order of magnitude higher than previously reported for the vacuum-processed, non-fluorinated 2,7-dihexyl-IDT-di(N(CN)), highlighting the beneficial role of core fluorination.
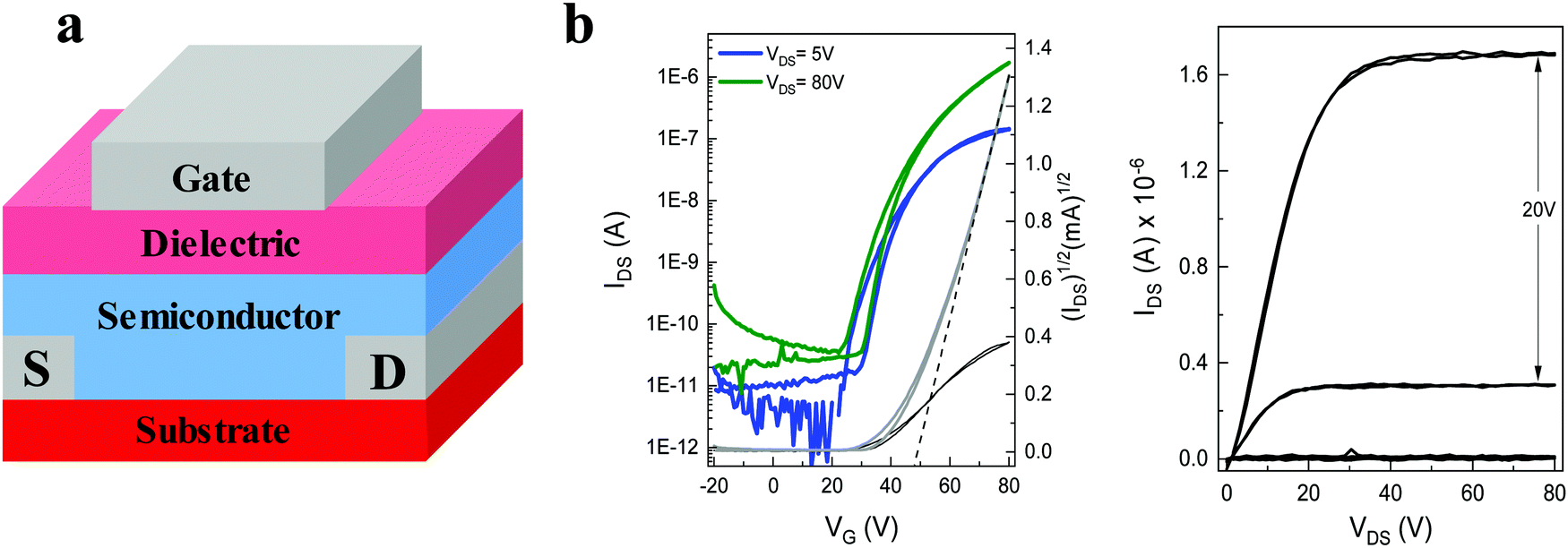 | ||
| Fig. 6 (a) OFET device architecture, where S/D are source/drain electrodes (b) transfer (left) and output (right) curves for blade-coated BC-TG devices of 2,7-dihexyl-diFIDT-di(N(CN)), post-annealed at 200 °C and tested in N2. | ||
Conclusion
Two novel, highly electron deficient semiconductors, 2,7-dihexyl-diFIDT-di(N(CN)) and 3,8-dihexyl-diFIDT-di(N(CN)), were synthesised from the corresponding diketones via a Knoevenagel condensation reaction. The products were isolated by a simple hot reprecipitation. Introduction of the N-cyanoimine groups was confirmed to be effective at stabilising the LUMO (−4.16 eV to −4.19 eV). Single crystal analysis of 2,7-dihexyl-diFIDT-di(N(CN)) and 3,8-dihexyl-diFIDT-di(N(CN)) demonstrated that they arrange into highly ordered planar slipped stacking arrangements with very close π–π stacking distances (∼3.40 Å) and minimal π–π slippage. Despite exhibiting similar stacking distances, a significant difference in electron transfer integrals for the two regioisomers was observed, relating to differences in relative molecular displacement along the π-stacking direction. OFET devices incorporating dialkyl-diFIDT-di(N(CN)) were fabricated via a scalable blade-coating and displayed good unipolar electron mobility, with μsat up to 0.13 cm2 V−1 s−1 for 2,7-dihexyl-diFIDT-di(N(CN)). Lower performance was observed for 3,8-dihexyl-diFIDT-di(N(CN)) regioisomer (up to 0.03 cm2 V−1 s−1), in agreement with the reduced transfer integrals observed. Overall, these results show that the inclusion of the N-cyanoimine groups cause less backbone distortion than the previously investigated dicyanomethylene group. The combination of N-cyanoimine with core fluorination leads to very low lying LUMO levels and appears a useful strategy in the design of n-type materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) (grant EP/L016702/1), the Royal Society and the Wolfson Foundation (for Royal Society Wolfson Fellowship) and King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under award numbers OSR-2019-CRG8-4095.3, and OSR-2020-CRG8-4095.2 for their support. F. G. thanks the Austrian Science Fund (FWF) for funding: J 4463 (project number).References
- X. Guo and A. Facchetti, Nat. Mater., 2020, 19, 922–928 CrossRef CAS.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 201801079 CrossRef.
- J. T. E. Quinn, J. Zhu, X. Li, J. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS.
- H. Jia and T. Lei, J. Mater. Chem. C, 2019, 7, 12809 RSC.
- H. Sun, X. Guo and A. Facchetti, Chem, 2020, 6, 1310–1326 CAS.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2018, 6, 10390–10410 RSC.
- J. Dhar, U. Salzner and S. Patil, J. Mater. Chem. C, 2017, 5, 7404–7430 RSC.
- A. Naibi Lakshminarayana, A. Ong and C. Chi, J. Mater. Chem. C, 2018, 6, 3551–3563 RSC.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. W. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS.
- H. Luo, D. He, Y. Zhang, S. Wang, H. Gao, J. Yan, Y. Cao, Z. Cai, L. Tan, S. Wu, L. Wang and Z. Liu, Org. Lett., 2019, 21, 9734–9737 CrossRef CAS.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS.
- T. D. Anthopoulos, F. B. Kooistra, H. J. Wondergem, D. Kronholm, J. C. Hummelen and D. M. de Leeuw, Adv. Mater., 2006, 18, 1679–1684 CrossRef CAS.
- P. Kafourou, B. Park, J. Luke, L. Tan, J. Panidi, F. Glöcklhofer, J. Kim, T. D. Anthopoulos, J.-S. Kim, K. Lee, S. Kwon and M. Heeney, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202013625.
- D. X. Long, M. Karakawa and Y.-Y. Noh, Phys. Chem. Chem. Phys., 2016, 18, 23904–23909 RSC.
- C. Sutton, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef CAS.
- G. Gryn’ova, K.-H. Lin and C. Corminboeuf, J. Am. Chem. Soc., 2018, 140, 16370–16386 CrossRef.
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS.
- S. Arumugam, I. A. Wright, A. R. Inigo, S. Gambino, C. T. Howells, A. L. Kanibolotsky, P. J. Skabara and I. D. W. Samuel, J. Mater. Chem. C, 2014, 2, 34–39 RSC.
- J. Chen, K. Yang, X. Zhou and X. Guo, Chem. – Asian J., 2018, 13, 2587–2600 CrossRef CAS.
- U. Scherf, J. Mater. Chem., 1999, 9, 1853–1864 RSC.
- J. Kirkpatrick, C. B. Nielsen, W. M. Zhang, H. Bronstein, R. S. Ashraf, M. Heeney and I. McCulloch, Adv. Energy Mater., 2012, 2, 260–265 CrossRef CAS.
- W. M. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS.
- Y. Li, M. Gu, Z. Pan, B. Zhang, X. Yang, J. Gu and Y. Chen, J. Mater. Chem. A, 2017, 5, 10798–10814 RSC.
- A. Wadsworth, H. Chen, K. J. Thorley, C. Cendra, M. Nikolka, H. Bristow, M. Moser, A. Salleo, T. D. Anthopoulos, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2020, 142, 652–664 CrossRef CAS.
- X. Song, N. Gasparini, M. M. Nahid, H. Chen, S. M. Macphee, W. Zhang, V. Norman, C. Zhu, D. Bryant, H. Ade, I. McCulloch and D. Baran, Adv. Funct. Mater., 2018, 28, 1802895 CrossRef.
- A. Casey, Y. Han, Z. Fei, A. J. P. White, T. D. Anthopoulos and M. Heeney, J. Mater. Chem. C, 2015, 3, 265–275 RSC.
- H. Bristow, K. J. Thorley, A. J. P. White, A. Wadsworth, M. Babics, Z. Hamid, W. Zhang, A. F. Paterson, J. Kosco, J. Panidi, T. D. Anthopoulos and I. McCulloch, Adv. Electron. Mater., 2019, 5, 1900344 CrossRef CAS.
- C. Zhao, Y. Zhang and M.-K. Ng, J. Org. Chem., 2007, 72, 6364–6371 CrossRef CAS.
- H. Tian, Y. Deng, F. Pan, L. Huang, D. Yan, Y. Geng and F. Wang, J. Mater. Chem., 2010, 20, 7998–8004 RSC.
- J.-D. Peltier, B. Heinrich, B. Donnio, J. Rault-Berthelot, E. Jacques and C. Poriel, ACS Appl. Mater. Interfaces, 2017, 9, 8219–8232 CrossRef CAS.
- T. Hodsden, K. J. Thorley, J. Panidi, A. Basu, A. V. Marsh, H. Dai, A. J. P. White, C. Wang, W. Mitchell, F. Glöcklhofer, T. D. Anthopoulos and M. Heeney, Adv. Funct. Mater., 2020, 30, 2000325 CrossRef CAS.
- S. Chen, Y. Zhao, A. Bolag, J.-I. Nishida, Y. Liu and Y. Yamashita, ACS Appl. Mater. Interfaces, 2012, 4, 3994–4000 CrossRef CAS.
- K. Takahashi and K. Kobayashi, J. Org. Chem., 2000, 65, 2577–2579 CrossRef CAS.
- A. S. Batsanov, M. R. Bryce, M. A. Coffin, A. Green, R. E. Hester, J. A. K. Howard, I. K. Lednev, N. Martín, A. J. Moore, J. N. Moore, E. Ortí, L. Sánchez, M. Savirón, P. M. Viruela, R. Viruela and T.-Q. Ye, Chem. – Eur. J., 1998, 4, 2580–2592 CrossRef CAS.
- J.-D. Peltier, B. Heinrich, B. Donnio, O. Jeannin, J. Rault-Berthelot, E. Jacques and C. Poriel, J. Mater. Chem. C, 2018, 6, 13197–13210 RSC.
- P. Kovacic and K. N. McFarland, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 1963–1976 CrossRef CAS.
- K. J. Thorley and C. Risko, J. Mater. Chem. C, 2016, 4, 4040–4048 RSC.
- J. L. Brédas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 CrossRef.
- E. M. Espinoza, J. A. Clark, J. Soliman, J. B. Derr, M. Morales and V. I. Vullev, J. Electrochem. Soc., 2019, 166, H3175–H3187 CrossRef CAS.
- A. Basu, M. R. Niazi, A. D. Scaccabarozzi, H. Faber, Z. Fei, D. H. Anjum, A. F. Paterson, O. Boltalina, M. Heeney and T. D. Anthopoulos, J. Mater. Chem. C, 2020, 8, 15368–15376 RSC.
- H. H. Choi, K. Cho, C. D. Frisbie, H. Sirringhaus and V. Podzorov, Nat. Mater., 2018, 17, 2–7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2050898 and 2050899. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00091h |
| This journal is © The Royal Society of Chemistry 2021 |