
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical exfoliation of a graphite electrode in 1-ethyl-3-methylimidazolium chloride-[EMIM]+Cl−–AlCl3 ionic liquid and its electrocatalytic application
Gengan
Saravanan
 *ab,
Sankaranarayanan
Sanjay
c,
R. M.
Gnanamuthu
d and
Mutyala
Sankararao
e
*ab,
Sankaranarayanan
Sanjay
c,
R. M.
Gnanamuthu
d and
Mutyala
Sankararao
e
aDepartment of Chemistry, Koneru Lakshmaiah Education Foundation, Hyderabad-500075, Telangana, India. E-mail: saravanan3che@gmail.com; saravanan@klh.edu.in
bYonsei University, 50 Yonsei-ro, Sinchon-dong, Seodaemun-gu, Seoul-120749, South Korea
cDepartment of ECE, Koneru Lakshmaiah Education Foundation, Hyderabad-500075, Telangana, India
dBatteries and Metal-finishing Research Laboratory (+BMRL-), Department of Chemistry, SRM Institute of Science and Technology, Chennai, Tamil Nadu, India
eDepartment of Chemistry, Koneru Lakshmaiah Education Foundation, Vaddeswaram, Guntur, Andhra Pradesh-522502, India
First published on 2nd February 2021
Abstract
The electrochemical exfoliation of graphite using 1-ethyl-3-methylimidazolium chloride-[EMIM]Cl–AlCl3 ionic liquid (IL) has been anticipated to be an environmentally benign and economically viable graphene production technique based on the intercalation of AlCl4− ions into a graphite anode in an IL mixture. An equimolar mixture of the two solids AlCl3− and EMIMCl− becomes a liquid at room temperature via the acid–base reaction to form AlCl4− and EMIM+ ions. This liquid is not only an electrolyte, but also a source of electroactive species for intercalation into graphite. The crystallographic properties of graphite and physicochemical properties of the IL enable better intercalation of AlCl4− (2.95 Å) into the graphite layer (lattice distance 3.3 Å). The results clearly demonstrate that graphene is produced within the graphite anode in AlCl3–EMIM+Cl−. The IL is significantly important in terms of its cost efficiency, wide electrochemical window, and high exfoliation performance for the production of graphene. The as-prepared few-layer graphene was drop-casted on a pre-polished glassy carbon electrode (GCE), followed by the electrodeposition of Pt dendritic nanoparticles, for application in the electrooxidation of glucose.
1. Introduction
Graphene, a single-atom framework with a 2D sp2-hybridized carbon hexagonal sheet structure, has great potential applications in energy conversion and storage, electrocatalysis, sensing, and electronics.1–4 Graphene has a large surface area, good thermal conductivity, high electron mobility, and good optical transmittance.5–8 Although there are various methods to synthesize graphene sheets, it remains a challenge for researchers to supply top-quality graphene sheets via eco-friendly and economically practicable processes.Several graphene preparation methods have been developed since its discovery.9 Among them, the mechanical exfoliation and epitaxial growth of graphene provide high-quality material for fundamental research, but only in limited quantities.10 Chemical vapor deposition (CVD) using catalytic metal substrates such as Ni or Cu produces large-area high-quality graphene.11 The major obstacles to the cost-effective industrial-scale production of CVD-grown graphene are the requirements for high temperature, a sacrificial metal, and multistep transfer processes onto the desired substrates. Chemical exfoliation of graphite based on Hummers’ method is an appealing route to produce solution-processable graphene oxide (GO) at the bulk scale, but thermal or chemical reduction is required to partially restore the electronic properties of graphene.12 Several other methods have been developed to overcome these limitations, such as solvent- and/or surfactant-assisted liquid-phase exfoliation,13 electrochemical expansion,14 and the formation of graphite-intercalating compounds. The electrochemical exfoliation of graphite has been performed mainly in aqueous acids (H2SO4 or H3PO4),15 and the formation of graphene with a small lateral size and a significant amount of oxygen-containing functional groups cannot be avoided due to the overoxidation of graphite. ILs have better electrochemical properties to supply graphene sheets from graphite via the exfoliation method; Coleman et al. reviewed exfoliation to obtain nanotubes and graphene in solvent media.16–18
In the chemical synthesis of graphene, harsh oxidizers and powerful reducing agents are normally used, and many researchers have reported the exfoliation of graphite ionic conductive electrolytes using DC techniques.19–21 The production of graphene using chemical methods in aqueous electrolytes suffers from low electrical conductivity, a narrow electrochemical window, and the limitation of water electrolysis. Since ILs are thermally stable, have wide electrochemical windows, and are liquid at room temperature, various combinations of very large organic cations and relatively bulky inorganic counter-ions have been used as electrolytes for graphite exfoliation.22
In this study, we systematically investigate the exfoliation mechanism at a graphite anode via the intercalation of AlCl4− ions under a constant potential of 7 V and successfully obtain few-layered graphene. Dendritic Pt nanoparticles are electrochemically deposited on a prepolished glassy carbon electrode (GCE) substrate onto which a graphene and Nafion suspension has previously been drop-casted and dried overnight. The resulting electrode has been studied as an electrocatalyst for the electrooxidation of D-glucose.
2. Experimental section
2.1. Electrochemical exfoliation
The schematic in Fig. 1(A) illustrates the electrochemical exfoliation of graphite into graphene in the ionic liquid [EMIM]Cl–AlCl3 followed by the electrodeposition of Pt dendritic nanoparticles onto a graphene-modified GCE for the electrooxidation of glucose. A high-purity graphite rod with a diameter of 5 mm and length of 4 cm (Graphite Store) exposed to the IL was used as the anode, and a platinum wire with a diameter of 2 mm and length of 4.5 cm was used as the counter-electrode. The IL [EMIM]Cl–AlCl3 was prepared by the slow addition of a particular weight of [EMIM]Cl (99%, Merck) to an equimolar amount of AlCl3 (99%, Merck) at room temperature. The IL mixture was continuously stirred using a magnetic bar for 3 h to ensure homogeneity. For the exfoliation of graphite, a constant voltage of 7 V was applied (Powertron, PIPL-0630PR, DC power supply); the current was within ±5–10 mA of 80 mA throughout the course of the experiments. After a brief induction period, the colorless IL became dark yellow-brown; the graphite anode then swelled, and the expanded pieces discharged from the anode into the IL formed a black slurry, as shown in Fig. 1B. The exfoliation process was stopped after 3 h. The product was diluted with a water:ethanol mixture and centrifuged at a speed of 1200 rpm to obtain pure graphene. The dispersion was dialyzed with Millipore water using a dialysis bag with a molecular weight cutoff (MWCO) of 500 Da.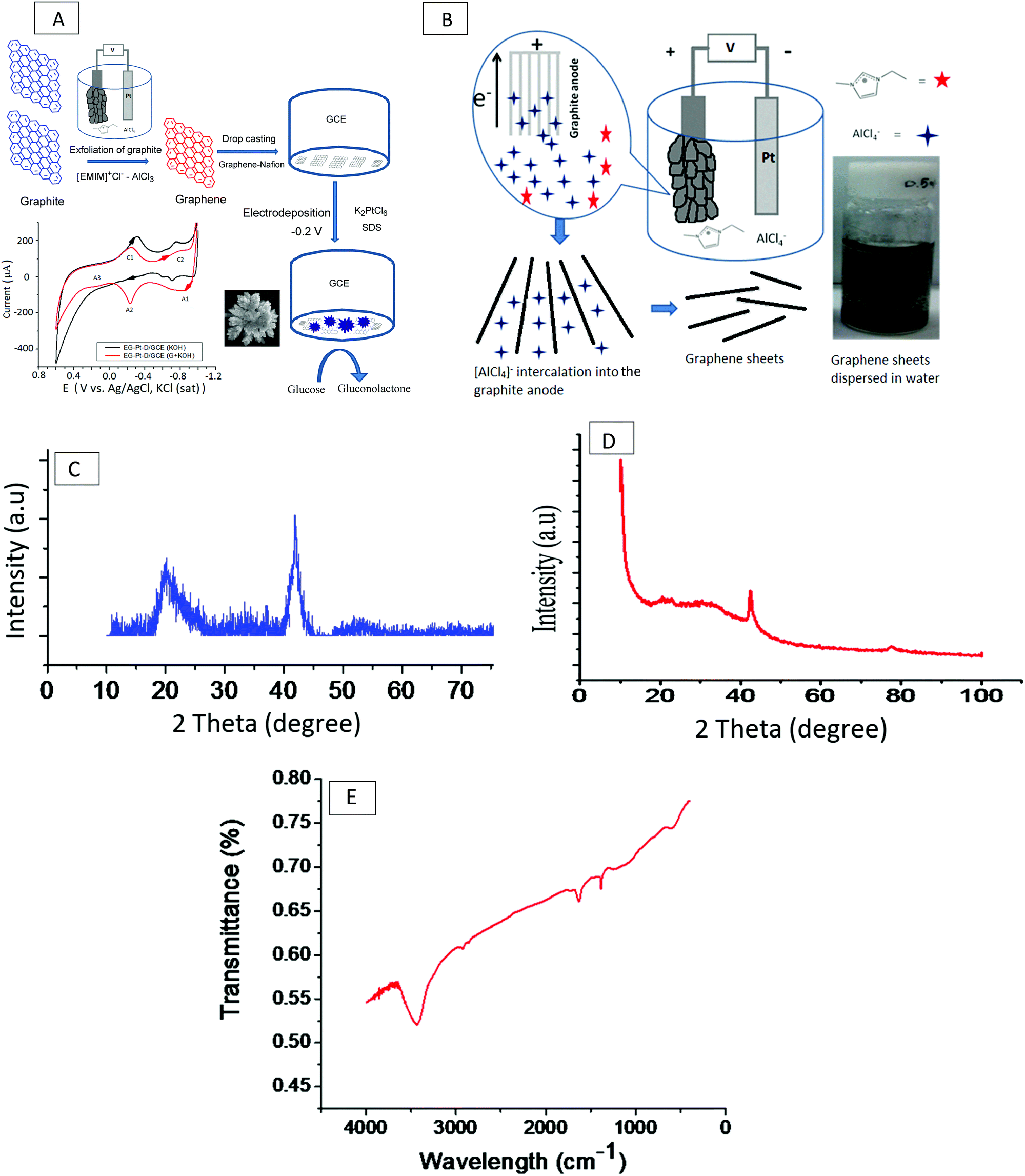 | ||
| Fig. 1 (A) Schematic illustration of the electrochemical exfoliation of graphite to graphene in the IL [EMIM]Cl–AlCl3, followed by drop-casting graphene on a GCE and subsequent electrodeposition of Pt dendritic nanoparticles in 1.0 mM K2PtCl6 and 1 mM SDS to modify an electrode for the electrooxidation of glucose. (B) Scheme illustrating the intercalation of AlCl4− ions into the graphite anode under a constant potential of 7 V for 3 h. (C) XRD pattern of graphene. Peaks correspond to the (002) plane of graphite at 2θ = 23° and the (100) crystal plane of graphene. (D) XRD pattern showing the peaks of the electrodeposited Pt dendritic nanoparticles at 42° and 68° for Pt (111) and (220), respectively. (E) FT-IR spectrum of the graphene electrochemically exfoliated from graphite in the IL [EMIM]Cl–AlCl3. | ||
2.2. Preparation of exfoliated graphene anchored Pt-dendritic nanoparticle modified glassy carbon electrodes (EG–Pt-D NPs/GCE)
Approximately 1 mg of graphene was first dispersed in 0.2 ml of a 0.5 wt% Nafion (Sigma Aldrich) solution, the mixture was stirred to obtain a uniform Nafion–graphene suspension, and a glassy carbon electrode (GCE) was covered with 5 μL of the Nafion–graphene suspension and allowed to dry in air overnight. Electrochemical experiments were performed using a PARSTAT 2273 (Princeton Applied Research, US) electrochemical analyzer with a conventional three-electrode cell, in which platinum foil and Ag/AgCl (saturated KCl) were the auxiliary electrode and reference electrode, respectively. The working electrode was a modified GCE with a diameter of 3 mm (ALS, Tokyo, Japan). Before modification, the surface of the GCE was polished using alumina slurries (0.1 and 0.05 μm) to achieve a mirror finish. The scheme in Fig. 1(B) depicts the intercalation of AlCl4− ions into the graphite anode at a constant potential of 7 V throughout the electrochemical cell. The previously prepared EG/GCE was used as the working electrode for the electrodeposition of Pt dendritic nanoparticles at −0.2 V in an electrolyte consisting of 1.0 mM K2PtCl6 and 1 mM SDS for 400 s. After electrodeposition, the Pt electrode was washed with Millipore Milli-Q water several times and dried at 100 °C for 1 min. Assuming a 100% current efficiency, the Pt loading on the EG/GCE was determined by the charge integrated during the Pt deposition.23 The charge for the deposition of Pt was 0.0316 C, which corresponded to a Pt loading of 16.04 μg.2.3. Characterization of exfoliated graphene (EG) and electrodeposited Pt dendritic nanoparticles (EG–Pt-D/GCE)
The electrochemical experiments were carried out in a conventional three-electrode cell using a PARSTAT 2273 (Princeton Applied Research, US) electrochemical analyzer. The FT-IR spectra were obtained using a PerkinElmer Spectrum GX FT-IR system with pure KBr as the background. The X-ray diffraction pattern of the crystal phases was measured using a Phillips diffractometer over the 2θ range of 10 to 100 degrees using CuKα radiation (λ = 0.154 nm). A Raman spectrum was collected using a confocal Raman spectrometer (WiTec GmbH CRM 200) with a 514.5 nm Nd:YAG laser as a light-weight source. The microstructural morphology of EG and EG–Pt-D/GCE was observed using a scanning electron microscope equipped with energy dispersive X-ray spectroscopy (Hitachi, 3000H). The thickness, width and height of EG and the Pt dendritic nanoparticle topography were studied using atomic force microscopy (AFM, Agilent Technologies, 5500 series) and transmission electron microscopy (TEM, Tecnai 20 G2, FEI make).3. Results and discussion
3.1. Surface morphology of EG and Pt dendritic nanoparticles
Fig. 1C shows the typical XRD pattern of the resulting graphene nanosheets, which exhibit a structure intermediate between the crystalline and amorphous structures. This is evidenced by the reappearance of the C (002) diffraction line at a 2θ value of 23°, which appears as a wider low-intensity peak due to the out-of-plane graphitic reflections that occur at an equivalent position to that of the reflection from bulk graphite;24 the peak at a 2θ value of ∼43° corresponds to the (100) crystal plane of graphene. The in-plane crystallite size (L) is often expressed in terms of the in-plane periodicity peak at a 2θ value of ∼23° as L = 1.84λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ. Similarly, the average crystallite width D is often calculated using the well-known Debye–Scherer equation as D = 0.89λ/βcosθ. The crystalline nature of the Pt dendritic nanoparticles electrodeposited on graphene was confirmed by the XRD pattern, as shown in Fig. 1D. The pattern of EG–Pt-D/GCE exhibits peaks at 42° and 68°, which correspond to Pt (111) and (220), respectively.25–27 The number of graphene layers (n) per domain is often calculated from the XRD peak broadening by combining the Debye–Scherrer and Bragg equations to obtain n = (D/d) + 1; the calculated average number of graphene layers for the exfoliated graphene was 26.27.28Fig. 1E shows the FT-IR spectrum of the graphene electrochemically exfoliated in the IL [EMIM]Cl–AlCl3. An O–H stretching peak due to water absorbed in the ionic liquid can be observed at 3751 cm−1, the peak at 2919 cm−1 corresponds to C–H (stretching), and the aromatic C
cosθ. Similarly, the average crystallite width D is often calculated using the well-known Debye–Scherer equation as D = 0.89λ/βcosθ. The crystalline nature of the Pt dendritic nanoparticles electrodeposited on graphene was confirmed by the XRD pattern, as shown in Fig. 1D. The pattern of EG–Pt-D/GCE exhibits peaks at 42° and 68°, which correspond to Pt (111) and (220), respectively.25–27 The number of graphene layers (n) per domain is often calculated from the XRD peak broadening by combining the Debye–Scherrer and Bragg equations to obtain n = (D/d) + 1; the calculated average number of graphene layers for the exfoliated graphene was 26.27.28Fig. 1E shows the FT-IR spectrum of the graphene electrochemically exfoliated in the IL [EMIM]Cl–AlCl3. An O–H stretching peak due to water absorbed in the ionic liquid can be observed at 3751 cm−1, the peak at 2919 cm−1 corresponds to C–H (stretching), and the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak at ∼1637 cm−1 confirms the hexagonal structure and corresponds to the skeletal vibration of the graphitic backbone, consistent with the literature.29
C peak at ∼1637 cm−1 confirms the hexagonal structure and corresponds to the skeletal vibration of the graphitic backbone, consistent with the literature.29
The confocal Raman features at 1341, 11591, 2696 and 2919 cm−1 are the D, G, 2D and S3 bands, respectively. The G band corresponds to the first-order scattering of the E2g optical mode of the sp2 domain; in contrast, the D-peak is due to the disordered regions containing sp3 carbons with associated out-of-plane vibrations. The 2D band is considered to be an overtone of the D peak, whereas S3 is attributed to the mixture of D and G peaks. The 2D band is a characteristic peak of the graphene structure, and the ratio of the intensity of the 2D and G bands, I2D/IG, depends on the number of graphene layers.30 An I2D/IG ratio of ∼2–3 corresponds to monolayer graphene, 2 > I2D/IG > 1 to bilayer graphene, and I2D/IG < 1 to multilayer graphene. Based on the I2D/IG ratio observed in its Raman spectrum, the graphene exfoliated from graphite in the IL is multilayer, since its I2D/IG value is 0.8, which is smaller than 1.
Fig. 2B shows the EDX spectra of the exfoliated graphene modified GCE with electrodeposited Pt dendritic nanoparticles. Based on the EDX data, the C, O and Pt contents are 73.05%, 8.74% and 18.21% respectively, with a low calculated O/C ratio of 0.1196. This result is consistent with the FT-IR data, which shows the presence of a small quantity of absorbed water in the ionic liquid. Fig. 2C shows the surface topography of the as-obtained EG anchored Pt dendritic nanoparticles. AFM has the advantage of being able to probe minute details related to graphene thickness, and horizontal cross-section analysis gives information about the width and height of the Pt nanoparticles. The thickness of the exfoliated graphene prepared in the ionic liquid ranges from 1 to 2.3 nm, corresponding to single-layer to few-layer sheets, and the Pt dendritic nanoparticles have a width of 200 nm and heights from 2.6 to 10 nm.
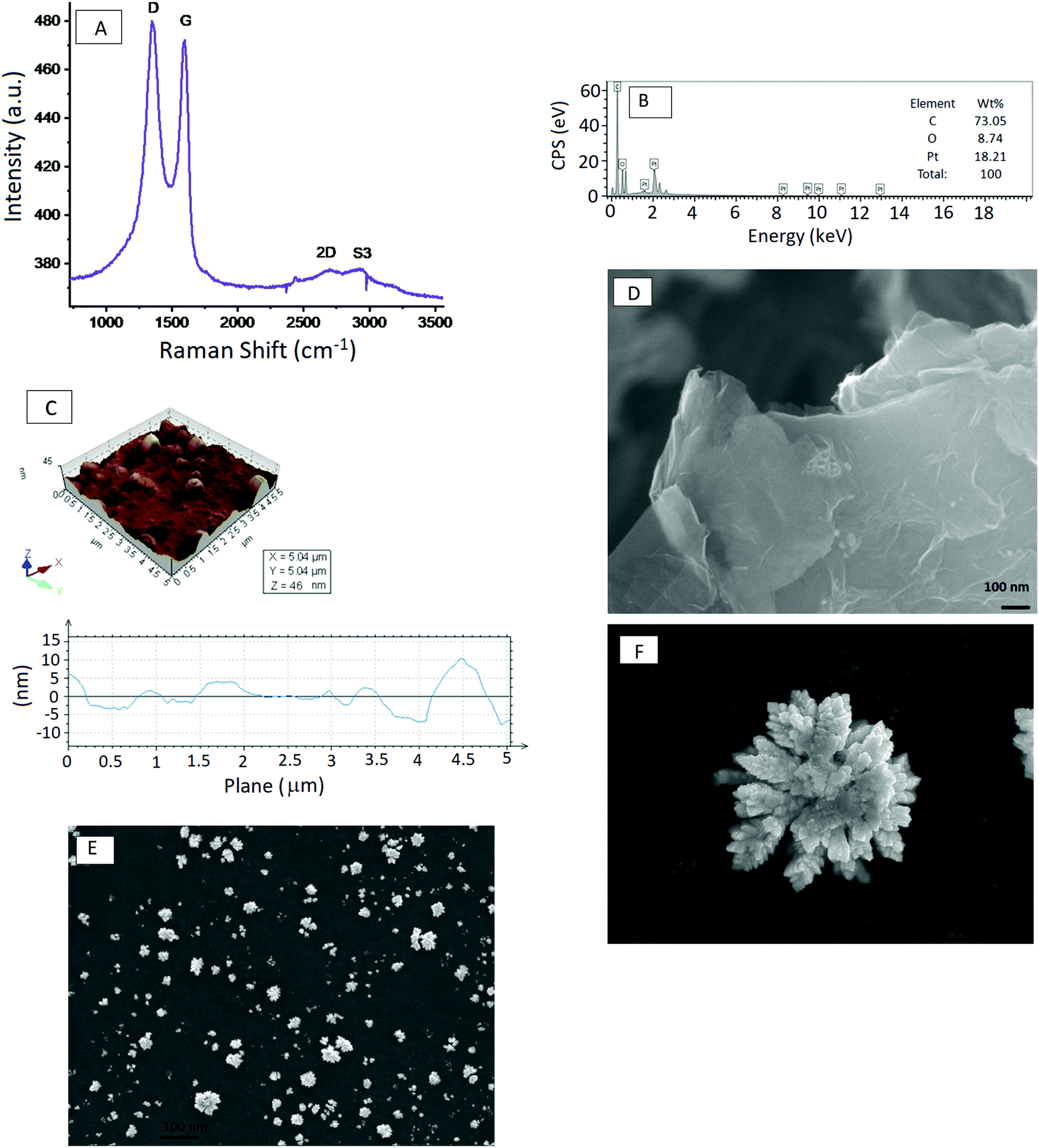 | ||
| Fig. 2 (A) Raman spectrum of the exfoliated graphene nanosheets showing the D, G, 2D and S3 bands of a highly disordered state. (B) EDS spectrum of the exfoliated graphene supported Pt dendritic nanoparticles. (C) Surface topography of the as-obtained EG anchored Pt dendritic nanoparticles. (D) SEM image of the exfoliated graphene sheet. (E) Low-magnification SEM image of the electrodeposited Pt dendritic nanoparticles. (F) High-magnification SEM image of the electrodeposited Pt dendritic nanoparticles. | ||
The surface morphologies of EG and EG–Pt-D/GCE were characterized using scanning electron microscopy (SEM) and TEM, as shown in Fig. 2D–F. The results revealed that the electrochemically produced EG nanosheets were well dispersed with a characteristic wrinkled morphology. The TEM image of the EG sheets shown in Fig. 3A demonstrate a folded silk-like morphology with abundant wrinkles and crumpling, which would prevent them from closely restacking face-to-face when packed or compressed onto an electrode with a high area. The EG–Pt-D/GCE was also observed using SEM. It has been noted that the deposition potential and SDS affect the morphology and therefore the density of Pt nuclei at −0.2 V. Fig. 3B shows that the Pt dendritic nanoparticles are uniform and reside on the surface of EG sheets, which prevents them from aggregating and restacking. The typical Pt dendritic nanoparticle size estimated from the TEM image is 60 ± 5–8 nm.
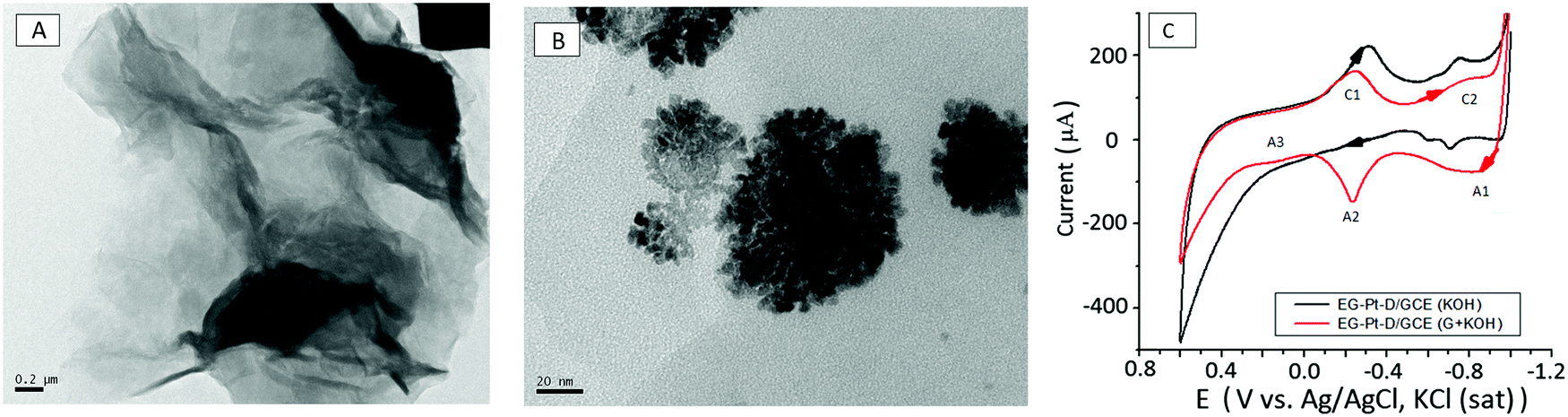 | ||
| Fig. 3 (A) TEM image of graphene nanosheets showing the crumpled morphology of the single-layer graphene sheets. (B) TEM images of Pt dendritic nanoparticles electrodeposited at −0.2 V in a 1.0 mM K2PtCl6 and 1 mM SDS electrolyte solution. (C) Cyclic voltammograms of EG–Pt-D/GCE with 0.5 M KOH at a scan rate of 50 mV s−1 in the presence and absence of 0.05 M glucose. | ||
3.2. Electrochemical intercalation
Wu and associates reported that a planar configuration has been proposed for the intercalated AlCl4− anion, which is unusual, considering that the anion in the IL has both a tetrahedral and planar configuration. The diffusion barrier for the AlCl4− anion in graphite was calculated to be as low as 0.023 eV.31 Graphite contains a high concentration (∼4.2%) of C vacancies with a formation energy of above 7 eV,32,33 consistent with the electrochemical reaction formula proposed by Lin et al.34 Charged AlCl4− anions are intercalated into the bulk graphite, and one electron moves far from the external circuit. The Al–Cl bond lengths and bond angles are distorted as a result of interaction with the graphite layers, which press the AlCl4− tetrahedron from the c-axis direction, and the shape of the anion therefore becomes planar. The planar AlCl4− ion geometry within the graphite is actively more comfortable than the tetrahedral one, and more stable with an energy difference of 0.81 eV. The reason for this is that the intercalation of the AlCl4− cluster might increase the d-spacing between two graphene layers in graphite from 6.025 Å to 8.162 Å after exfoliation; therefore, the planar AlCl4− ion is more favorable when inserted into the graphite.35 The diffusion of the AlCl4− cluster in bulk graphite is must faster than that of Li–graphite intercalation compounds. Thus, it is expected that the movement of the AlCl4− cluster might be instantaneous at room temperature, consistent with transition state theory, which indicates that the AlCl4− cluster can diffuse instantaneously within the bulk graphite. Moreover, the adsorption and diffusion of the AlCl4− cluster on the graphite surface are also relevant to the electrochemistry.In Al batteries, aluminium foil serves as the anode material and graphite serves as the cathode material. Conventional Li-ion or Na-ion batteries can only transfer one unit charge in one redox couple, while Al-ion batteries can transfer three charges in one redox couple (Al3+/Al), and can therefore attain more gravimetric capacity and volumetric capacity than Li-ion or Na-ion batteries. The electrolyte is a mixture of AlCl3 and 1-ethyl-3-methylimidazolium chloride ([EMIm]Cl) in a 1.3:1 molar ratio. In the discharge process, Al metal is oxidized and liberated from the Al foil to form Al3+. In the electrochemical exfoliation of graphite under the influence of an electric field, however, the Al ions move to the cathode side, and Al3+ and AlCl3 coordination anions [AlaClb]− concurrently intercalate into the graphite layers, forming AlxCly. The intercalated AlxCly and neighboring graphite layers interact with one another via van der Waals forces. In the charge process, the opposite electrochemical reactions occur. In the ionic liquid, the coordination ions present are AlCl4− or Al2Cl7−, so the intercalated coordination ion [AlaClb]− might be AlCl4−, Al2Cl7−, or a mixture of these. An equilibrium should exist among the concentrations of Al3+, AlCl4− and Al2Cl7− in the electrolyte to maintain the intercalation and deintercalation process, which requires further study.36–38
3.3. Electrooxidation of glucose using EG–Pt-D/GCE
The electrocatalytic activity of the materials was examined by cyclic voltammetry using the Fe(CN)63−/4− redox couple. The effective surface area of the electrodes was estimated by cyclic voltammetry using 10 mM Fe(CN)63−/4− in 1 M KCl solution. The electroactive surface area was estimated according to the Randles–Sevcik equation, ip = 2.69 × 105n3/2ACD1/2ν1/2, where ip, n, A, C, D, and v are the peak current, number of electrons involved in the reaction, electroactive surface area, concentration of the reactant, diffusion coefficient of the reactant species and scan rate, respectively. The redox reaction of Fe(CN)63−/4− involves only one-electron transfer (n = 1), and the calculated electroactive surface areas of GCE, EG-GCE and Pt-EG-GCE are 3.1 × 10−2, 6.6 × 10−2 and 7.09 × 10−2, respectively. Cyclic voltammograms of EG–Pt-D/GCE in 0.5 M KOH at a scan rate of 50 mV s−1 in the presence and absence of 0.05 M glucose are shown in Fig. 3C. In the absence of glucose, the electrooxidative current through the EG–Pt-D/GCE electrode is nearly negligible. Cyclic voltammetry of the EG–Pt-D/GCE electrode in KOH with potential sweeps from −0.9 V to 0.6 V indicate that at a positive potential, the buildup of the intermediate on the electrode surface inhibits the additional adsorption of glucose, leading to a decrease in peak current at −0.649 V (A1). Platinum–OH surface species begin to form in KOH electrolyte, which can be viewed as the formation of an equivalent somewhat bound species during oxidation. The chemical adsorption of glucose related to the O1 oxidation peak takes place at the OH-adsorbed EG–Pt-D/GCE site. During a continuous positive sweep, reasonably bound poisoning intermediates formed during the reaction in the potential region from −0.648 to 0.218 V are oxidized within the butterfly region at −0.178 V (A2). Consistent with the literature,39–41 the oxidation of glucose on EG–Pt-D/GCE in alkaline solution involves reaction between the adsorbed organic species and therefore the platinum–OH layer. The happenstance of the onset of this peak and therefore the butterfly peak supports the view that it is due to the formation of the Pt–OH layer. Within the hydrogen adsorption and double-layer region, the adsorbed dehydrated intermediates are often further oxidized to weakly adsorbed gluconate,42 the configuration of which depends on the potential at which the weakly adsorbed gluconate species can desorb to form gluconate during a continuous sweep in the positive direction. Given the lower electrooxidation peak current (A3) at 0.216 V, this is often probably partly due to better coverage of the intermediate, which may appreciably reduce the adsorption of OH-species in this potential region. In the −0.65 to 0.216 V region, the adsorbed dehydrogenated intermediate formed lead by oxidation to d-gluconolactone could be formed on the surface and followed by desorption of lactone occurs slowly to gluconate by hydrolysis. The decrease in current after the third current peak (A3) might be due to the formation of thick platinum oxide, which subsequently also inhibits the oxidation of glucose during the negative potential scan. The cathodic current peaks C1 and C2 appear, and therefore the cathodic current peak at −0.283 V should originate from to the reduction of platinum oxide, and that at −0.782 V could be due to the reduction of the oxidized products of glucose or the desorption of the oxidation products. This peak is only apparent in the presence of glucose.43,444. Conclusion
We have investigated the successful exfoliation of graphite into graphene in an IL by an electrochemical method using graphite as the anode. This process is highly efficient and eco-friendly, and represents an excellent candidate as an alternative to the conventional Hummers’ method. This approach is more advantageous in terms of the excellent exfoliation caused by the effective intercalation of AlCl4− anions within the graphitic layers, the wide electrochemical window of the IL, and the greater efficiency of the process. The as-prepared graphene obtained using the exfoliation method was characterized by XRD, FT-IR, Raman, SEM, EDS, AFM and TEM analysis. A suspension of the exfoliated graphene with Nafion was cast onto a GCE, onto which Pt dendritic nanoparticles were then electrodeposited (EG–Pt-D/GCE). The EG–Pt-D/GCE modified electrode was successfully used for the electrochemical oxidation of glucose.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors wish to express their sincere thanks to the President, Vice-president, Director, and Dean of KLEF, Telangana, Hyderabad, for constant encouragement. Gengan Saravanan also thanks the Director of CECRI-CSIR, Karaikudi-6, for the execution of the cyclic voltammetry experiments, SEM, FT-IR, EDS, AFM, TEM and XRD characterization and permission to publish these results.References
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686, 10.1039/C1CS15078B.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286, DOI:10.1038/nature04969.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200, DOI:10.1038/nature04233.
- B. M. Goh, Y. Wang, M. V. Reddy, Y. L. Ding, L. Lu, C. Bunker and K. P. Loh, ACS Appl. Mater. Interfaces, 2014, 6, 9835–9841, DOI:10.1021/acsami.7b03306.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk and J. R. Potts, Adv. Mater., 2010, 22(35), 3906–3924, DOI:10.1002/adma.201001068.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan and F. Miao, Superior thermal conductivity of single-layer graphene, Nano Lett., 2008, 8(3), 902–907, DOI:10.1021/nl0731872.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg and J. Hone, Solid State Commun., 2008, 146(9–10), 351–355, DOI:10.1016/j.ssc.2008.02.024.
- X. Li, Y. Zhu, W. Cai, M. Borysiak, B. Han and D. Chen, Nano Lett., 2009, 9(12), 4359–4363, DOI:10.1021/nl902623y.
- P. W. Sutter, J. I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406, DOI:10.1038/nmat2166.
- Y. Lee, S. Bae, H. Jang, S. Jang, S. E. Zhu, S. H. Sim, Y. I. Song, B. H. Hong and J. H. Ahn, Nano Lett., 2010, 10, 490–493, DOI:10.1021/nl903272n.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274, DOI:10.1038/nnano.2008.83.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun’ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568, DOI:10.1038/nnano.2008.215.
- J. N. Wang, K. K. Manga, Q. L. Bao and K. P. Loh, J. Am. Chem. Soc., 2011, 133(23), 8888–8891, DOI:10.1021/ja203725d.
- C. J. Shih, A. Vijayaraghavan, R. Krishnan, R. Sharma, J. Han, M. H. Ham, Z. Jin, S. C. Lin, G. L. C. Paulus, N. F. Reuel, Q. H. Wang, D. Blankschtein and M. S. Strano, Nat. Nanotechnol., 2011, 6(7), 439–445, DOI:10.1016/j.matpr.2017.11.173.
- J. L. Liu, H. P. Yang, S. G. Zhen, C. K. Poh, A. Chaurasia, J. S. Luo, X. Y. Wu, E. K. L. Yeow, N. G. Sahoo, J. Y. Lin and Z. X. Shen, RSC Adv., 2013, 3, 11745–11750, 10.1039/C3RA41366G.
- Y. Fu, J. Zhang, H. Liu, W. Hiscox and Y. Gu, J. Mater. Chem. A, 2012, 1, 2663–2674, 10.1039/C2TA00353H.
- W. Du, X. Jiang and L. Zhu, J. Mater. Chem. A, 2013, 203(1), 10592–10606 RSC.
- J. N. Coleman, Adv. Funct. Mater., 2010, 19, 3680–3695, DOI:10.1002/adfm.200901640.
- K. Parvez, R. Li, S. R. Puniredd, Y. Hernandez, F. Hinkel and S. Wang, ACS Nano, 2013, 7(4), 3598–3606, DOI:10.1021/nn400576v.
- J. Wang, K. K. Manga, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2011, 133(23), 8888–8891, DOI:10.1021/ja203725d.
- N. Liu, F. Luo, H. Wu, Y. Liu, C. Zhang and J. Chen, Adv. Funct. Mater., 2008, 18(10), 1518–1525, DOI:10.1002/adfm.200700797.
- G. Imperato, B. König and C. Chiappe, Eur. J. Org. Chem., 2007,(7), 1049–1058, DOI:10.1002/ejoc.200600435.
- K. S. Subrahmanyam, S. R. C. Vivekchand, A. Govindaraj and C. N. R. Rao, J. Mater. Chem., 2008, 18, 1517–1523, 10.1039/B716536F.
- J. Yan, J. Liu, Z. Fan, T. Wei and L. Zhang, Carbon, 2012, 50, 2179–2188, DOI:10.1016/j.carbon.2012.01.028.
- G. Saravanan and S. Mohan, Appl. Surf. Sci., 2016, 386(15), 96–102, DOI:10.1016/j.apsusc.2016.05.152.
- G. Saravanan and S. Mohan, Anal. Methods, 2020, 12, 3617–3625, 10.1039/D0AY00021C.
- R. Sharma and N. Chadha Parveen Saini, Indian J. Pure Appl. Phys., 2017, 55, 625–629 Search PubMed.
- A. Reina, X. T. Jia, J. Ho, D. Nezich, H. B. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, J. Nanosci. Lett., 2009, 9(1), 30–35, DOI:10.1021/nl801827v.
- R. Bakhshandeh and A. Shafekhani, Ultrasonic waves and temperature efects on graphene structure fabricated by electrochemical exfoliation method, Mater. Chem. Phys., 2018, 212, 95–102, DOI:10.1016/j.matchemphys.2018.03.004.
- X. Dong, P. Wang, W. Fang, C. Y. Su, Y. H. Chen, L. J. Li, W. Huang and P. Chen, J. Mater. Chem., 2011, 49, 3672–3678, 10.1039/C2JM33286H.
- M. S. Wu, B. Xu, L. Q. Chen and C. Y. Ouyang, Electrochim. Acta, 2016, 195, 158–165, DOI:10.1016/j.electacta.2016.11.039.
- A. A. El-Barbary, R. H. Telling, C. P. Ewels, M. I. Heggie and P. R. Briddon, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 144107–144114, DOI:10.1103/PhysRevB.68.144107.
- M. Y. Lehtinen, A. S. Foster and R. M. Nieminen, New J. Phys., 2004, 6, 68–83 CrossRef.
- M. C. Lin, M. Gong, B. G. Lu, Y. P. Wu, D. Y. Wan, M. Y. Guan, M. Angell, C. X. Chen, J. Yang, B. J. Hwang and H. J. Dai, Nature, 2015, 520, 324–328, DOI:10.1038/nature14340.
- M. L. Agiorgousis, Y.-Y. Sun and S. Zhang, ACS Energy Lett., 2017, 20117(2), 689–693 CrossRef.
- W. Wang, B. Jiang, W. Xiong, H. Sun, Z. Lin, L. Hu, J. Tu, J. Hou, H. Zhu and S. Jiao, Sci. Rep., 2013, 3383–3389, DOI:10.1038/srep03383.
- H. Sun, W. Wang, Z. Yu, Y. Yuan, S. Wang and S. Jiao, Chem. Commun., 2015, 51, 11892–11895, 10.1039/C5CC00542F.
- Z. Yu, S. Jiao, S. Li, X. Chen, W.-L. Song, T. Teng, J. Tu, H.-S. Chen, G. Zhang and D.-N. Fang, Adv. Funct. Mater., 2019, 29(1), 180799–180809 CrossRef.
- Y. Song, S. Jiao, J. Tu, J. Wang, Y. Liu, H. Jiao, X. Mao, Z. Guo and D. J. Fray, J. Mater. Chem. A, 2017, 5, 1282–1291, 10.1039/C6TA09829K.
- Y. B. Vasil'ev, O. A. Khazova and N. N. Nikolaeva, J. Electroanal. Chem., 1985, 196, 127–144, DOI:10.1016/0022-0728(85)85085-3.
- R. R. Adzic, F. Feddrix, B. Z. Nikolić and E. Yeager, J. Electroanal. Chem., 1992, 341(1–2), 287–306, DOI:10.1016/0022-0728(92)80489-Q.
- C. A. Apblett, D. Ingersoll, S. Sarangapani, M. Kelly and P. Atanassov, J. Electrochem. Soc., 2009, 157(1), B86–B89, DOI:10.1007/s11244-015-0499-1.
- D. Zhang, K. Zhang, Y. L. Yao, X. H. Xia and H. Y. Chen, Langmuir, 2004, 20, 7303–7307, DOI:10.1021/la049667f.
- C. E. Banks, A. Crossley, C. Salter, S. J. Wilkins and R. G. Compton, Angew. Chem., Int. Ed., 2006, 45, 2533–2537, DOI:10.1002/anie.200600033.
| This journal is © The Royal Society of Chemistry 2021 |