
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSample dependent performance of aqueous copper hexacyanoferrate/zinc batteries†
Solveig
Kjeldgaard
ab,
Marnix
Wagemaker
 c,
Bo Brummerstedt
Iversen
b and
Anders
Bentien
*a
c,
Bo Brummerstedt
Iversen
b and
Anders
Bentien
*a
aDepartment of Biological and Chemical Engineering, Aarhus University, Denmark. E-mail: bentien@bce.au.dk
bDepartment of Chemistry, Aarhus University, Denmark
cDepartment of Radiation Science & Technology, Technical University Delft, The Netherlands
First published on 22nd February 2021
Abstract
In this work, we investigate factors affecting the capacity retention of aqueous copper hexacyanoferrate/Zn batteries. We compare the performance of four different copper hexacyanoferrate (CuHCF) samples as cathode in CuHCF/Zn cells with Na2SO4 and ZnSO4 based electrolytes at different pH. In Na2SO4 electrolyte, the capacity retention is improved at lower pH, whereas cells with ZnSO4 electrolyte perform similarly at pH 2 and 4. Finally, we show that the performance of CuHCF/Zn cells is highly dependent on the CuHCF sample, underlining the necessity for detailed synthesis description and thorough structural characterization of electrode materials.
Introduction
There is a global drive to replace fossil fuels with solar and wind powered renewable energy sources. The major challenge of incorporating a larger proportion of energy from renewable energy sources in the grid is the inherent intermittent nature of these, and the transition therefore relies on the expansion of stationary energy storage. For stationary energy storage, energy density is less important, while cost and lifetime are main drivers. Here, aqueous based rechargeable batteries are interesting for a number of reasons: (i) raw material sourcing can be decoupled from automotive batteries. (ii) Aqueous electrolytes have a high ionic conductivity compared to organic electrolytes, enabling thicker electrodes, less complex cell design and lower production cost. (iii) Aqueous based electrolytes have higher safety and lower environmental impact. Nonetheless, the number of elements that are redox active within the electrochemical window of water as well as being abundant and low cost is limited, and include metals such as Cu, Mn, Fe, Zn, Ni, Ti and Cr.1,2Zinc anodes are attractive due to the high elemental abundance and low cost of zinc. Furthermore, zinc is compatible with aqueous electrolytes, as opposed to metals such as magnesium, calcium and aluminum. Zinc has a high volumetric energy density of 5855 mA h cm−3 and a low electrode potential of −0.76 V vs. SHE in aqueous solutions. Many research efforts have been invested in the development of aqueous rechargeable Zn-ion batteries. However, it remains a challenge to identify a suitable cathode material with high rate capability and good cycle life. Rechargeable MnO2/Zn batteries have been investigated intensively in both alkaline3 and neutral/mildly acidic4–6 electrolyte. Other cathode materials like vanadium oxide7 have also been investigated, while in particular Prussian Blue Analogues8–14 appear promising.
Prussian Blue Analogues (PBA) are a large family of transition metal hexacyanometallates with the general structural formula AxP[R(CN)6]1−y where A is an insertion ion, often potassium or sodium, P and R are transition metals and y is the number of [R(CN)6]3−/4− vacancies. Generally, PBA belong to cubic space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m,15–20 although for PBA with very high A content (A: Na, K), also rhombohedral21–24 and monoclinic25,26 structures are often reported. The cubic unit cell structure is shown in Fig. 1.
m,15–20 although for PBA with very high A content (A: Na, K), also rhombohedral21–24 and monoclinic25,26 structures are often reported. The cubic unit cell structure is shown in Fig. 1.
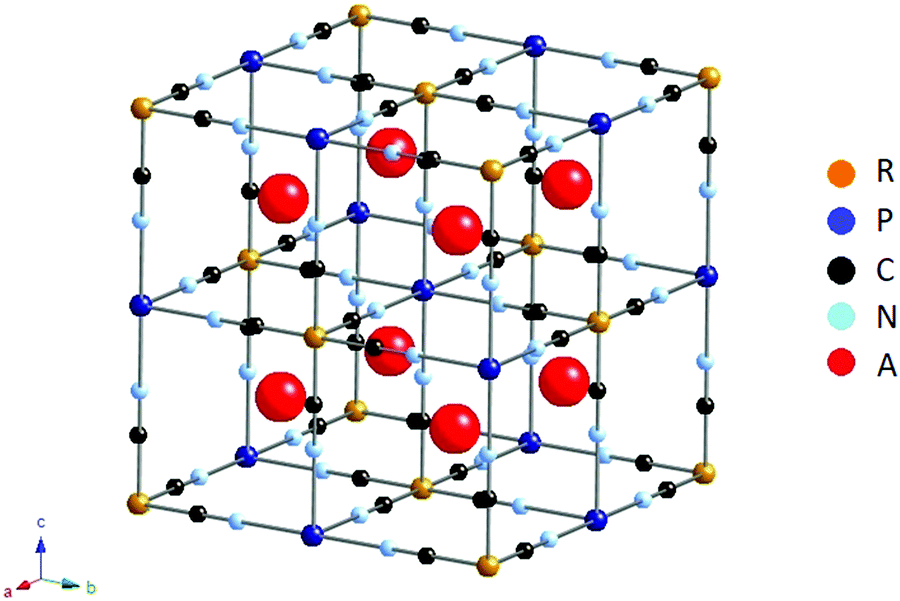 | ||
| Fig. 1 Idealized structure of PBA AxP[R(CN)6]1−y with y = 0 (no vacancies), cubic space group Fmm. Figure made using CrystalMaker. | ||
PBA have a cage-like structure with wide channels allowing for insertion of even large intercalation ions, and PBAs have proved to be able to intercalate even multivalent ions reversibly with fast kinetics in aqueous solution.27 The open framework structure of PBA ensures rapid ionic conduction, enabling remarkably high rate capability.28,29 PBA can be prepared by a simple, scalable and low cost co-precipitation synthesis made from abundant and non-toxic elements.
For aqueous batteries, especially copper hexacyanoferrate (CuHCF) has been considered as cathode material due to its high redox potential that is just below the oxygen evolution potential. Aqueous CuHCF/Zn batteries have a high cell potential of approximately 1.7 V, which in practice is an almost full utilization of the electrochemical window of water. A commonly reported stoichiometry for CuHCF is CuII[FeIII(CN)6]2/3 with 1/3 [Fe(CN)6]3− vacancies, which is likely due to charge balancing of the framework. This stoichiometry is prevalent also for PBA with other transition metals in similar oxidation state.30,31
The existence of [Fe(CN)6]3−/4− vacancies can possibly increase the ionic conduction, and may play a crucial role in the diffusion pathway, especially for larger insertion ions. In a perfect lattice, the ions are conducted through the 〈100〉 channels. A vacancy gives an effective diameter of approximately 8.5 Å, which is larger than the distance between neighboring R sites.32,33 Therefore, adjacent vacancies can connect to form channels, offering an alternative diffusion route. Assuming a random vacancy distribution, the percolation threshold for the face centered cubic (fcc) sublattice of the R site is approximately 0.2,34 however, as shown by Simonov et al., vacancy distribution in PBA tend to be non-random.33 For PBA with composition [PIIRIII(CN)6]2/3, such as CuHCF, the number of vacancies is high enough that the vacancies form an interconnected network regardless of the vacancy distribution being random or ordered. Studies have shown that whereas small cations are conducted through the 〈100〉 channels, larger cations move through vacant [Fe(CN)6]3−/4− positions, implying that vacancies play an important role in ion conduction.35,36
The most commonly reported electrolyte salt for PBA/Zn cells is ZnSO4,8,10–12,14,36,37 but also sodium based electrolyte salts such as Na2SO49,12,13 are commonly reported. Previous studies have reported low cycle life due to Zn-ion poisoning of the cathode, and Na-ion based electrolyte has been proposed as a way to minimize Zn-ion poisoning.13 Nonetheless, in fully discharged PBA/Zn cells, Zn2+ is dissolved in the electrolyte and if this concentration needs to be significantly lower than that of the Na+, this will be the limiting factor for the cell capacity. For this reason, the approach of using sodium intercalation in PBA/Zn cells is a severe limitation of further technological development of PBA/Zn based cells.
PBA/Zn cells often use slightly acidic electrolyte. Zinc dendrite formation is more pronounced in alkaline electrolyte than in neutral electrolytes,7,38,39 and also PBA are often used with acidic electrolyte. In a study from 1992, Stilwell et al. investigated the factors affecting the stability of Prussian Blue films, reporting a remarkable increase in film stability in acidic electrolyte, achieving lifetimes of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at pH 2–3.40 It has previously been reported that PBA are unstable in alkaline electrolyte; several studies report conversion of NiHCF into nickel oxides at pH > 8,41–44 and similar degradation is expected for other PBA. To the best of our knowledge, there are no comprehensive studies exploring the optimal pH of the electrolyte.
000 cycles at pH 2–3.40 It has previously been reported that PBA are unstable in alkaline electrolyte; several studies report conversion of NiHCF into nickel oxides at pH > 8,41–44 and similar degradation is expected for other PBA. To the best of our knowledge, there are no comprehensive studies exploring the optimal pH of the electrolyte.
In literature, the reported capacity retention of CuHCF/Zn cells with ZnSO4 based electrolyte varies considerably. The capacity retention of CuHCF/Zn cells can be improved by using a lower concentration of ZnSO4,45 and also the C-rate can affect capacity retention; Kasiri et al. observe a phase transition of CuHCF after 200 cycles at 1C, and after 600 cycles at 5C.45 However, the difference in capacity retention of CuHCF/Zn cells may also be due to differences in the CuHCF structure. PBA provide a large structural versatility, which can be expected to have a large impact on the electrochemical performance.
In the present study, we investigate factors affecting the electrochemical performance of aqueous CuHCF/Zn cells. The voltage range has a large impact on the capacity retention of CuHCF/Zn cells, with the best capacity retention achieved with voltage range 1.4–2.0 V. We test CuHCF/Zn cells in ZnSO4 and Na2SO4 based electrolytes at different pH. The performance of CuHCF/Zn cells with Na2SO4 based electrolyte is highly dependent on pH, with better performance at lower pH, which is not the case for CuHCF/Zn cells with ZnSO4 based electrolyte. Decreased capacity retention of cells with Na2SO4 based electrolyte at higher pH is due to increased PBA dissolution. PBA dissolution should be understood as dissolution into M2+ and [Fe(CN)6]3−/4−, and is not related to cyanide release. Studies have proved that the cyanide release is low across a wide pH range,46 with the cyanide release being at its minimum at pH 5 (20.01 μg g−1) and maximum at pH 1 (47.47 μg g−1).47 Contrary to previous reports, the capacity retention is not improved by using Na2SO4 based electrolyte rather than ZnSO4 based electrolyte. Finally, we show that CuHCF/Zn cell performance is highly dependent on the CuHCF sample, and large differences in performance are observed even for CuHCF samples that are almost indistinguishable with state-of-the-art analytical methods.
Experimental
Synthesis
The synthesis and structural characterization of CuHCF has been described in detail previously.48 Briefly, CuHCF was prepared by co-precipitation using programmable syringe pumps. The two precursor solutions were added simultaneously to a flask containing 50 mL of water with a flow rate of 4 mL min−1. Following precipitation, samples were left in the mother liquor for a variable period. Finally, the precipitate was washed with water and dried. An overview of synthesis details for all samples in the current study is included in Table 1. Details on structural characterization including ICP-OES, PXRD, TEM and Mössbauer spectroscopy can be found elsewhere.48| Sample | Reagents | Conc. (M) | Flow rate | Atm | T (°C) | Aging |
|---|---|---|---|---|---|---|
| C | CuSO4 | 0.1 | 4 mL min−1 | N2 | 70 | 2 h heat, |
| K3Fe(CN)6 | 0.1 | 2 h stir | ||||
| G | CuSO4 | 0.1 | 4 mL min−1 | — | 70 | 2 h heat, |
| K3Fe(CN)6 | 0.1 | 2 h stir | ||||
| KCl | 2.5 | |||||
| I | CuSO4 | 0.1 | 4 mL min−1 | — | 70 | 2 h heat, |
| K3Fe(CN)6 | 0.15 | 2 h stir | ||||
| K | CuCl2 | 0.1 | 1 mL min−1 | N2 | RT | 18 h stir, |
| K4Fe(CN)6 | 0.1 | H2O2 | ||||
We test four different CuHCF samples with varying synthesis parameters: samples C, G, I and K. The sample names correspond with naming from our previous structural study.48 Sample C was prepared by co-precipitation of 0.1 M CuSO4 and 0.1 M K3Fe(CN)6. In synthesis G, KCl was added to the 50 mL water, making a 2.5 M solution. Samples C, G and K were made from equimolar precursor solutions, and sample I was made with an excess of K3Fe(CN)6. Sample K was prepared from 0.1 M CuCl2 and 0.1 M K4Fe(CN)6, initially yielding CuIIFeII(CN)6 and subsequently oxidized to CuIIFeIII(CN)6 using excess hydrogen peroxide.
Electrode preparation
Powdered CuHCF was mixed with carbon black (Super C45, Timcal) and polyvinylidene fluoride (PVDF) with a dry weight ratio of 70:20:10. N-Methyl-2-pyrrolidone (NMP) was added to obtain a slurry that was stirred for two hours, and subsequently spread onto carbon paper and dried in a vacuum oven at 70 °C overnight. Circular electrodes with a mass loading of approximately 5–10 mg active material were punched out.
Electrochemical characterization
Cells were cycled galvanostatically using coin cells type CR2032 with Zn foil anode (Alfa Aesar, 0.25 mm thick, 99.98%) and glass fiber separator (Whatman GF/D). The electrolytes were 1 M ZnSO4 and 1 M Na2SO4, with pH 2 and 4 adjusted by addition of H2SO4. The volume of electrolyte added was approximately 0.14 mL. All cells were cycled with a current rate of 50 mA g−1, corresponding to approximately 1C.Results and discussion
All CuHCF samples belong to cubic space group Fmm, the idealized structure (no vacancies, y = 0) is seen in Fig. 1. The stoichiometry and particle size of the different samples is reported in Table 2.
| Sample | Stoichiometry | Size (PXRD) [nm] | Size (TEM) [nm] |
|---|---|---|---|
| C | K0.24Cu[Fe(CN)6]0.69 | 33.0 | 30 |
| G | K0.18Cu[Fe(CN)6]0.69 | 53.3 | 80 |
| I | K0.10Cu[Fe(CN)6]0.68 | 37.3 | 48 |
| K | K0.86Cu[Fe(CN)6]0.72 | 31.9 | 24 |
The theoretical capacity for anhydrous KCuFe(CN)6 with no vacancies is 85 mA h g−1. The CuHCF samples in the current study have approximately 1/3 [Fe(CN)6]3− vacancies, which are occupied by water. Water can also be present in the interstitial (A) sites, and the exact water content is dependent on temperature and humidity. The calculated theoretical capacity is 57 mA h g−1 based on a composition of KCu[Fe(CN)6]2/3·4H2O, which agrees well with previous studies on CuHCF.16
Effect of voltage range on capacity retention
Choosing the upper and lower cut-off voltage for charging and discharging is a trade-off between several parameters. Utilization of the full capacity of the battery normally requires high and low cut-off voltages, nonetheless, high cut-off voltages may lead to unwanted side reactions like hydrogen and oxygen evolution. Additionally, attempts to utilize the full capacity may also lead to structural stress on the cathode material as intercalation ions are fully extracted/inserted by full charge/discharge.CuHCF/Zn cells with 1 M ZnSO4 electrolyte at pH 4 were cycled using voltage ranges 1.4–2.0 V, 1.4–2.05 V, 0.8–1.9 V and 1.4–1.9 V. Fig. 2 shows capacity/voltage plots, while Fig. 3 shows the discharge capacity as a function of cycle number.
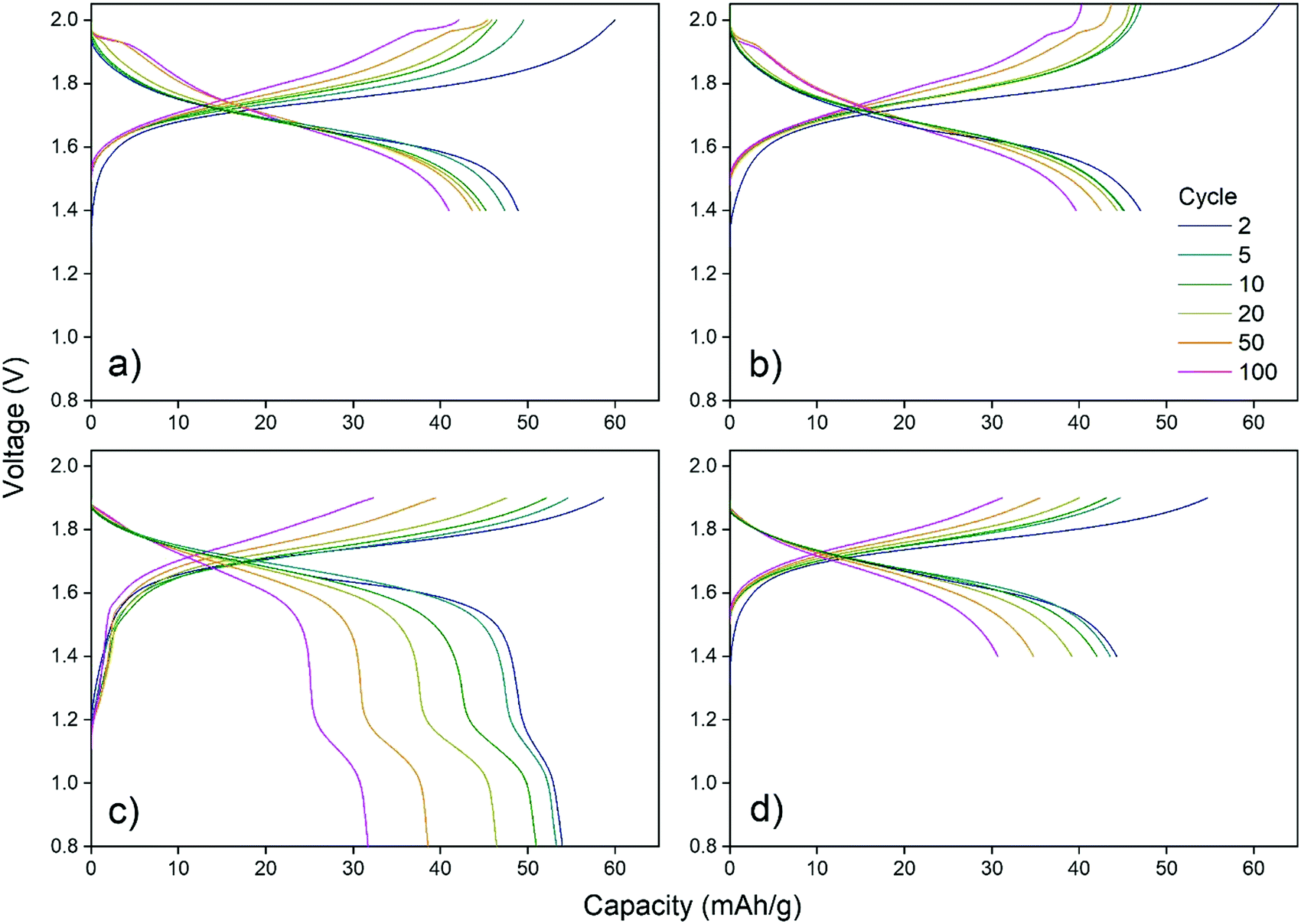 | ||
| Fig. 2 Effect of voltage ranges 1.4–2.0 V, 1.4–2.05 V, 0.8–1.9 V and 1.4–1.9 V on CuHCF/Zn cells with 1 M ZnSO4 electrolyte at pH 4. CuHCF is sample K. | ||
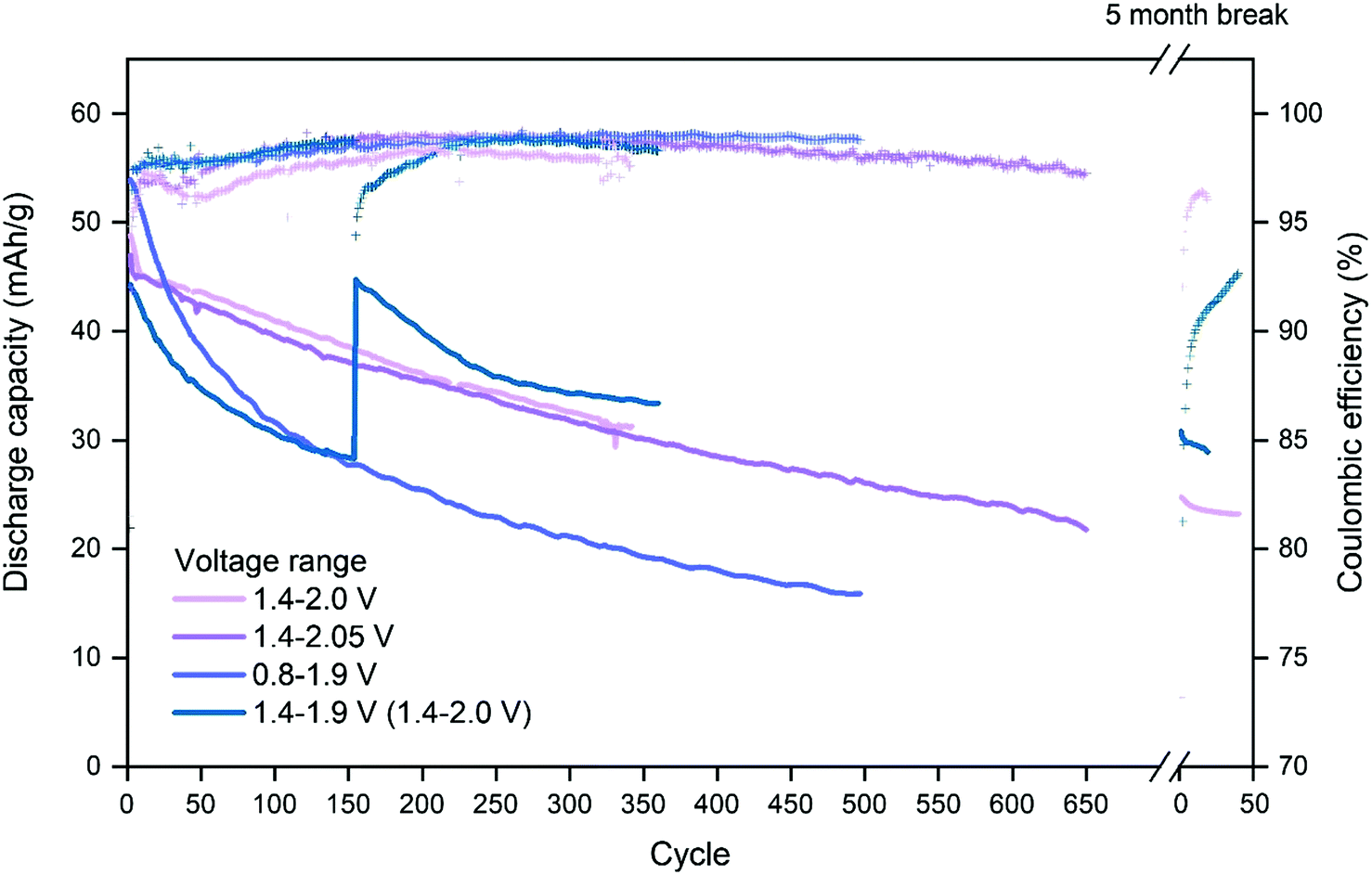 | ||
| Fig. 3 Effect of voltage range on the cycle life of CuHCF/Zn cells with 1 M ZnSO4 electrolyte at pH 4. CuHCF is sample K. After 154 cycles the cut-off voltage of the cell cycled in voltage range 1.4–1.9 V is increased to 2.0 V, resulting in an increase in capacity. | ||
From Fig. 2c and d it is seen that the charging plateau is not yet completed at 1.9 V, which becomes even more pronounced for higher cycle numbers. When the maximum cutoff voltage is increased to 2.0 V (Fig. 2a) or 2.05 V (Fig. 2b) the cells appear to reach full charge. When the lower cutoff voltage is set to 0.8 V, another discharge plateau is observed around 1.0 V to 1.2 V, which gets more pronounced with higher cycle number (Fig. 2c). When the lower cutoff voltage is limited to 1.4 V (Fig. 2a, b and d), the initial discharge capacity is lower, but the capacity retention is improved. The additional plateau at approximately 1.1 V is most likely due to reduction of Cu(II) to Cu(I). Although copper is generally considered to be electrochemically inactive in CuHCF, reduction of Cu(II) to Cu(I) at low potentials has been reported.49,50 X-Ray absorption spectroscopy (XAS) of CuHCF on both metal centers show copper redox activity.50 Cu(I) is observed even in the pristine sample, which has also been reported in other studies using X-ray absorption near edge structure (XANES).51,52
Fig. 3 shows the discharge capacity and coulombic efficiency as a function of cycle number. The capacity retention for the cell with voltage range 0.8–1.9 V is the poorest. Ferro/ferri-cyanide ([Fe(CN)6]4− and [Fe(CN)6]3−) do not form insoluble PBA with monovalent ions, including Cu(I),53 indicating that reduction of copper may result in increased solubility. Therefore, if the plateau at 1.1 V is due to reduction of copper, the poor capacity retention of cells discharged below 1.4 V may be caused by dissolution of CuHCF into the electrolyte.
The cell with the cut-off voltages 1.4 V and 1.9 V has the lowest initial capacity, which is limited by the upper cut-off voltage. Once it is allowed to charge to 2.0 V, the capacity reaches the same level as other cells charged to 2.0 V, see Fig. 3. The cells with voltage ranges 1.4–2.0 V and 1.4–2.05 V have a similar capacity retention which is higher than for the cells charged to only 1.9 V. Still, for cells charged to 2.0 V and 2.05 V, a new plateau at high state-of-charge appears at approximately 1.95 V after 50 cycles, and will be discussed later.
The coulombic efficiency is relatively high and increases slowly with cycle number. By cycle 150, all cells have an efficiency above 98%, except the cell with voltage range 1.4–2.0 V. The cell cycled with voltage range 1.4–2.05 V has a high coulombic efficiency similar to the ones with a cutoff voltage of 1.9 V, which shows that there is no significant gas evolution in the cell, and charging voltages up to 2.05 V appears safe.
After cycling for ∼350 cycles, corresponding to approximately three weeks of cycling, the cell with cut-off voltage 1.4–2.0 is stopped and for five months after which the cycling is resumed with the same conditions. It is observed that the discharge capacity is similar to the capacity prior to the 5 months break, indicating that the decrease in capacity is an effect of cycling rather than an effect of time.
In Table 3, the capacity retention of cells with different voltage ranges are reported. The as-synthesized material is not fully charged, why the discharge capacity of the first cycle is low, and the discharge capacity of cycle two is therefore used as initial capacity.
| Voltage range [V] | Initial capacity | Capacity retention | Capacity retention |
|---|---|---|---|
| Cycle 2 [mA h g−1] | Cycle 50 [mA h g−1 (%)] | Cycle 100 [mA h g−1 (%)] | |
| 1.4–2.0 | 49 | 44 (90%) | 41 (84%) |
| 1.4–2.05 | 47 | 42 (89%) | 40 (85%) |
| 0.8–1.9 | 54 | 39 (72%) | 32 (59%) |
| 1.4–1.9 | 44 | 35 (80%) | 31 (70%) |
Effects of pH and electrolyte composition
Four different CuHCF samples (C, G, I and K) are investigated as cathode material in CuHCF/Zn cells with 1 M Na2SO4 electrolyte at pH 1, 2 and 4, and 1 M ZnSO4 electrolyte at pH 2 and 4. All cells are cycled in the voltage range 1.4–2.0 V with a current of 50 mA g−1. Fig. 4 shows capacity/voltage plots, while the discharge capacity and coulombic efficiency as function of the cycle number is shown in Fig. 5. The capacity retention of the different cells is summarized in Table 4.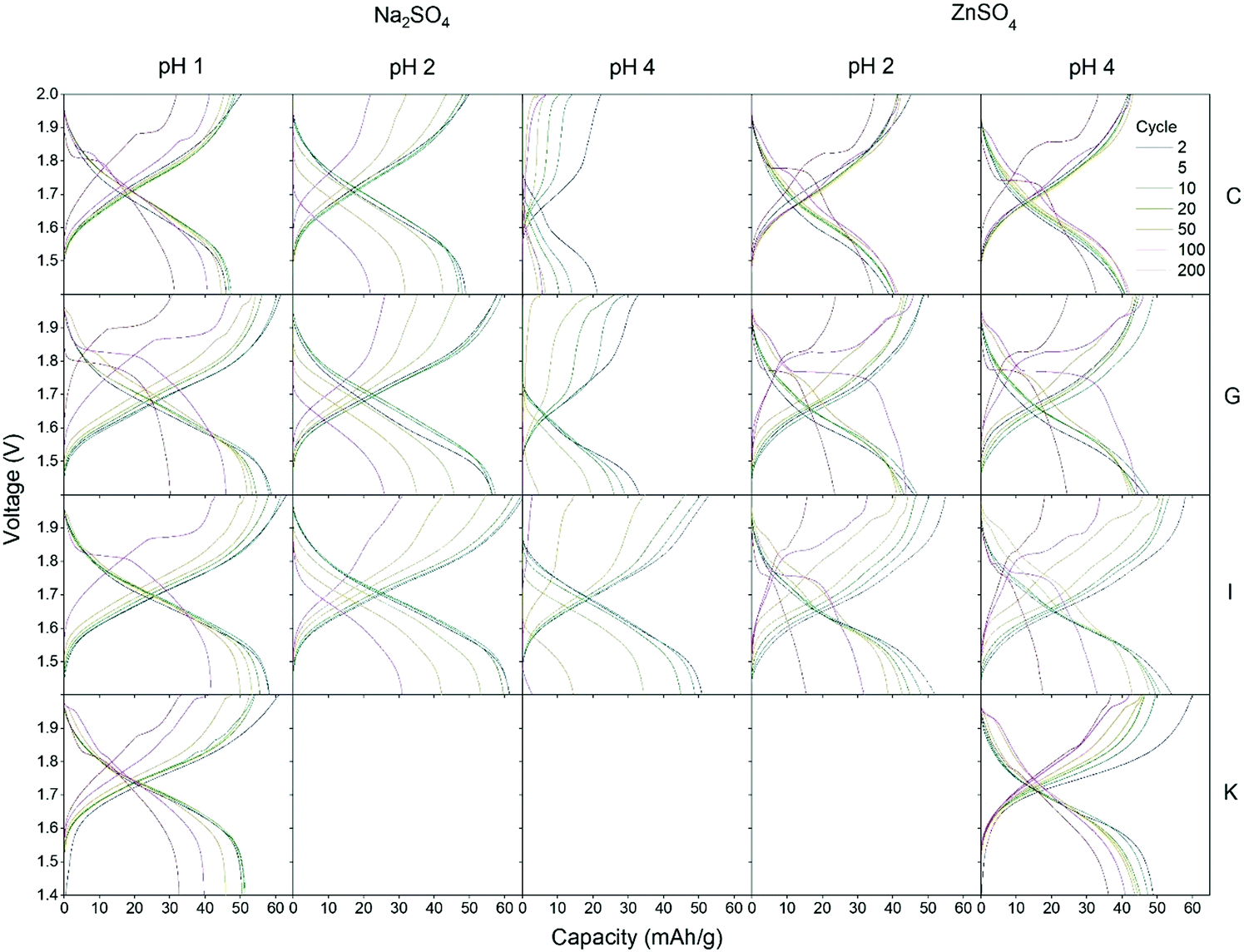 | ||
| Fig. 4 Capacity/voltage plots for the four different CuHCF samples C, G, I and K as cathode material in CuHCF/Zn cells with Na2SO4 electrolyte at pH 1, 2 and 4, and ZnSO4 electrolyte at pH 2 and 4. Cells are cycled in the voltage range 1.4–2.0 V with a current rate of 50 mA g−1. | ||
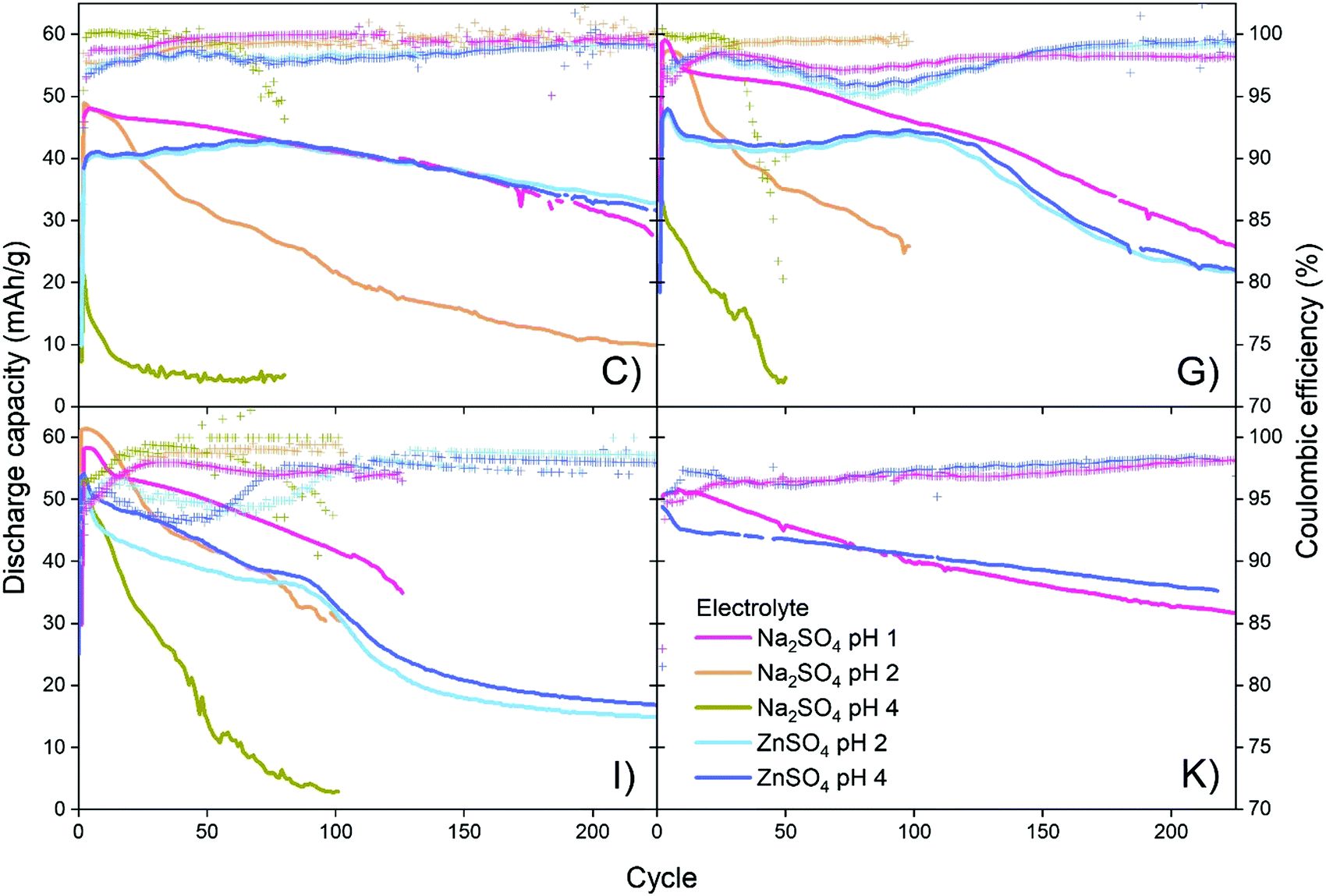 | ||
| Fig. 5 Discharge capacity and coulombic efficiency as a function of cycle for the four different CuHCF samples C, G, I and K as cathode material in CuHCF/Zn cells with Na2SO4 electrolyte at pH 1, 2 and 4, and ZnSO4 electrolyte at pH 2 and 4. Cells are cycled in the voltage range 1.4–2.0 V with a current rate of 50 mA g−1. | ||
| Sample | Initial capacity [mA h g−1] | Capacity retention [mA h g−1 (%)] | ||
|---|---|---|---|---|
| Cycle 50 | Cycle 100 | Cycle 200 | ||
| a Cycle 98. | ||||
| Sample C Na2SO4 pH 1 | 46 | 45 (98%) | 41 (89%) | 32 (70%) |
| Sample C Na2SO4 pH 2 | 49 | 32 (65%) | 22 (45%) | — |
| Sample C Na2SO4 pH 4 | 21 | 4 (19%) | — | — |
| Sample C ZnSO4 pH 2 | 39 | 42 (108%) | 41 (105%) | 34 (87%) |
| Sample C ZnSO4 pH 4 | 38 | 42 (111%) | 42 (111%) | 33 (87%) |
| Sample G Na2SO4 pH 1 | 59 | 52 (88%) | 46 (78%) | 30 (51%) |
| Sample G Na2SO4 pH 2 | 57 | 35 (61%) | 26 (46%)a | — |
| Sample G Na2SO4 pH 4 | 33 | 5 (15%) | — | — |
| Sample G ZnSO4 pH 2 | 46 | 41 (89%) | 44 (96%) | 24 (52%) |
| Sample G ZnSO4 pH 4 | 46 | 42 (91%) | 44 (96%) | 24 (52%) |
| Sample I Na2SO4 pH 1 | 58 | 50 (86%) | 42 (72%) | — |
| Sample I Na2SO4 pH 2 | 61 | 42 (69%) | 31 (51%) | — |
| Sample I Na2SO4 pH 4 | 51 | 15 (29%) | 3 (6%) | — |
| Sample I ZnSO4 pH 2 | 51 | 39 (76%) | 32 (63%) | 16 (31%) |
| Sample I ZnSO4 pH 4 | 54 | 43 (80%) | 33 (61%) | 18 (33%) |
| Sample K Na2SO4 pH 1 | 50 | 46 (92%) | 40 (80%) | 33 (66%) |
| Sample K ZnSO4 pH 4 | 49 | 44 (90%) | 41 (84%) | 36 (73%) |
For cells tested in Na2SO4 based electrolyte, variation in pH has a large effect, and the capacity retention is significantly improved at lower pH. All samples C, G, I and K have a very low capacity retention of maximum 29% after 50 cycles (see Table 4) in Na2SO4 electrolyte at pH 4. At pH 2, the capacity retention reaches a maximum of 69% after 50 cycles, and at pH 1 all cells achieve a high capacity retention ranging from 86% to 98% after 50 cycles. The decrease in capacity of CuHCF/Zn cells with Na2SO4 based electrolyte at higher pH is likely due to dissolution of copper hexacyanoferrate. Dissolution of PBA in aqueous Na2SO4 electrolyte has previously been observed: Zhang et al. detected free [Fe(CN)6]3− by UV-vis spectroscopy after cycling ZnHCF in aqueous Na2SO4 and K2SO4 electrolytes.12 It should be noted that contrary to the results obtained here, a previous study on NiHCF in Na2SO4 electrolyte report similar performance at neutral pH and at pH 2.41
Lowering the pH of the Na2SO4 electrolyte improves the capacity retention, indicating that dissociation is limited by lowering the pH. At low pH copper exists as Cu2+, whereas at higher pH copper precipitates as Cu(OH)2. Precipitation of Cu(OH)2 drives the equilibrium towards further dissolution of CuHCF, as shown in Scheme 1.
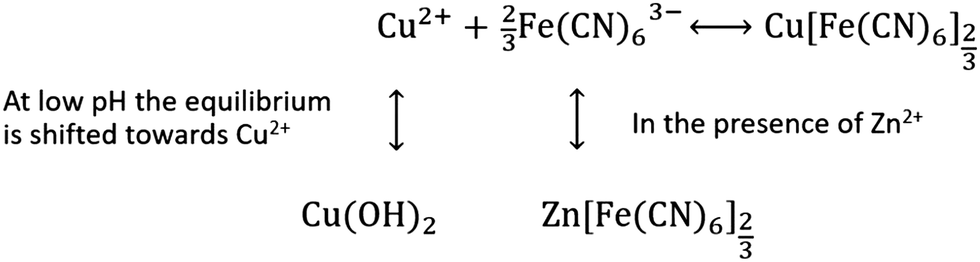 | ||
| Scheme 1 Proposed mechanism for capacity loss of CuHCF/Zn cells. | ||
For cells with ZnSO4 based electrolyte there are no significant differences between pH 2 and pH 4, and the capacity retention is significantly better than for corresponding cells with Na2SO4 based electrolyte. It seems likely that a degree of PBA dissolution occurs in both ZnSO4 and Na2SO4 electrolyte, however in the presence of M2+ ions such as Zn2+, free [Fe(CN)6]3− would immediately precipitate as zinc hexacyanoferrate (ZnHCF).
For cells with Na2SO4 based electrolyte, the concentration of Zn2+ is low and dependent on the state of charge. When the cell is fully charged, the Zn2+ concentration in the electrolyte is zero, while when fully discharged, the concentration of Zn2+ is estimated to be maximum 0.1 M, see calculation in ESI.† Due to the fact that the cell does not reach equilibrium during charge and discharge, there will be a Zn2+ concentration gradient with the highest concentrations towards the anode side. We suggest that the poor capacity retention of CuHCF/Zn cells with Na2SO4 based electrolyte may be explained by diffusion of [Fe(CN)6]3− to the separator or anode where it precipitates as ZnHCF and becomes electrochemically inaccessible. For cells with ZnSO4 based electrolyte, Zn2+ is present at both the cathode and anode at all times, and ZnHCF is primarily precipitated at the cathode where it remains electrochemically accessible, explaining the improved capacity retention compared to cells with Na2SO4 electrolyte.
With reference to the model in Scheme 1, it is somewhat surprising that the CuHCF/Zn cells with Na2SO4 at pH 1 develop a plateau at 1.85 V indicating formation of ZnHCF. It is speculated that this could be explained by the low pH value, which increases the reaction rate between H+ and Zn to form H2 and Zn2+ that diffuses to the cathode side to form ZnHCF. This is supported by the fact that the CuHCF/Zn cells with Na2SO4 electrolyte at pH 1 fail after a maximum of 300 cycles, which could be a consequence of H2 evolution, eventually leading to loss of electrical/ionic contact in the cell.
In Fig. 4 it is seen that most of the CuHCF/Zn cells with ZnSO4 electrolyte develop a new plateau at ∼1.95 V after 20–50 cycles, as was also observed for cells charged to 2.0 V and 2.05 V in Fig. 2. After approximately 50–100 cycles, another plateau at ∼1.85 V develops for CuHCF/Zn cells based on samples C, G and I with ZnSO4 electrolyte. The appearance of the plateau at 1.85 V coincides with an apparent increase in the cell voltage. Additionally, in Fig. 5 a kink is seen for samples C, G and I around cycle 75–100 in the capacity versus cycle number. This is followed by a faster capacity decrease, while the coulombic efficiency increases. The appearance of the new plateau is likely to be related to formation of ZnHCF, which has a standard potential that is approximately 0.15 V higher than that of CuHCF,54 in agreement with the apparent increase in cell voltage. Formation of M-HCF during cycling of PBA in aqueous M2+ electrolyte is supported by previous studies.45,55 For the sample K cell with ZnSO4 electrolyte, a plateau at 1.95 V appears after 20–50 cycles. However the plateau at 1.85 V has not developed after 200 cycles but it is observed after 400 cycles (see Fig. S3, ESI†).
Besides CuHCF dissolution, other models for ZnHCF formation have been proposed. One suggested mechanism proposes ZnHCF nucleation at vacant [Fe(CN)6]3−/4− positions within the CuHCF framework, resulting in a mixed copper/zinc hexacyanoferrate.45 Another possible mechanism explains the formation of mixed hexacyanoferrates as a distortion of the PBA lattice caused by the presence of M2+ in the interstitial (A) sites, which allows substitution of lattice transition metals.56 In the present study, we find no systematic differences between CuHCF/Zn cells (samples C, G and I) with ZnSO4 electrolyte at pH 2 and 4. If ZnHCF forms by nucleating at vacant [Fe(CN)6]3−/4− sites, minimizing vacancies may prevent/decrease ZnHCF formation. Cycling of an almost vacancy free MnHCF/Zn cell with ZnSO4 electrolyte proves that low-vacancy PBA can indeed intercalate Zn-ions, see ESI† (Fig. S6). If on the other hand transition metal substitution is due to lattice distortion caused by M2+ insertion, the number of vacancies should have no effect on the extent of ZnHCF formation. Depending on the mechanism for ZnHCF formation, different strategies could be attempted to limit ZnHCF formation. In a study of CoHCF with 1 M Na2SO4 electrolyte, the capacity retention was improved by addition of 1 wt% CoSO4 to the electrolyte,57 probably because the existence of Co2+ ions in the electrolyte allows free [Fe(CN)6]3− to precipitate with Co2+ as CoHCF. By analogy, M-HCF/Zn cells may benefit from addition of MSO4 to the Na2SO4 electrolyte, however in the case of CuHCF, addition of CuSO4 would lead to copper electroplating on the anode side due to the higher redox potential of the Cu/Cu2+ redox pair compared to Zn/Zn2+.
Previous studies have reported that decreasing the Zn2+ concentration in the electrolyte increases the cycle life of CuHCF/Zn cells.13,45 In a study comparing CuHCF/Zn cells in ZnSO4 and Na2SO4 based electrolytes, the best cycle life was achieved with 1 M Na2SO4 electrolyte acidified with 0.01 M H2SO4.13 In the present study, the capacity retention is not improved by using Na2SO4 electrolyte compared to ZnSO4 electrolyte. It should be noted that the capacity retentions reported in different studies are not directly comparable due to differences in electrolyte concentration and different current rates.
Sample dependence
As noted previously, the plateau at approximately 1.85 V develops much later for cells with sample K cathode than for cells with samples C, G and I (Fig. 4). After 200 cycles, the capacity of CuHCF/Zn cells with ZnSO4 electrolyte based on samples G and I has decreased dramatically, while sample C and sample K retain a capacity retention of 87% and 73%, respectively. The capacity retention of CuHCF/Zn cells seems closely linked to the development of the new plateau at 1.85 V, related to formation of ZnHCF. After 200 cycles, only the cell with sample K cathode has not developed a plateau at 1.85 V in ZnSO4 electrolyte. The CuHCF/Zn cell with ZnSO4 electrolyte at pH 4 with sample C and sample K both have a high capacity retention, however, after approximately 75 cycles, the capacity starts decreasing more rapidly for sample C. After 140 cycles the capacity of sample K is higher than that of sample C, see Fig. S1 (ESI†).It has previously been reported that PBA with a smaller particle size have a higher initial capacity,58–60 while a larger particle size improves capacity retention.60 In the present case, we do not see any clear correlation between performance and particle size. With an average particle size of 53 nm, sample G has the largest particle size, whereas the other samples are similar in size (32–37 nm).
As mentioned previously, it is known that larger cations such as Zn2+ can migrate through vacant [Fe(CN)6]3−/4− positions.35,36 Therefore, the number of vacancies could potentially be important for the electrochemical performance. The difference in the number of vacancies for CuHCF samples is at the edge of the experimental resolution. Nonetheless, the number of vacancies in sample K is slightly lower than that of samples C, G and I. This could possibly be a result of the synthesis from Fe(II) and not Fe(III) as for the other samples. In addition to the number of vacancies, also the vacancy distribution could affect electrochemical performance. As shown by Simonov et al., PBA tend to have a non-random vacancy ordering.33 Vacancy ordering may be affected by synthesis parameters, and could therefore be different for the samples included here.
It remains an interesting question what exactly causes the difference in performance, but here we conclude that performance of CuHCF/Zn cells is highly dependent on the CuHCF sample, and very different performance is observed even for relatively similar samples.
Conclusion
For CuHCF/Zn cells with ZnSO4 based electrolyte, there is negligible difference in performance at pH 2 and 4. In Na2SO4 based electrolyte, the performance is improved significantly at lower pH, which is likely due to decreased dissolution at lower pH. PBA dissolution is likely to occur also in ZnSO4 based electrolyte, but due to the presence of Zn2+ ions in the electrolyte, free [Fe(CN)6]3−/4− react immediately and precipitate as ZnHCF. Contrary to previous reports, the capacity retention is not improved by using Na2SO4 electrolyte rather than ZnSO4 electrolyte.After extended cycling, all CuHCF/Zn cells develop a new plateau in the voltage profile at approximately 1.85 V, which is attributed to the formation of a new ZnHCF phase. For CuHCF/Zn cells with sample K cathode, the plateau appears much later than for the others, resulting in superior capacity retention. The synthesis of sample K differs from the others in three ways: (1) whereas samples C, G and I were prepared in the oxidized form (CuII[FeIII(CN)6]1−y), sample K was initially prepared in the reduced form (CuII[FeII(CN)6]1−y) and was subsequently oxidized using excess hydrogen peroxide. (2) For samples C, G and I a CuSO4 precursor was used, whereas for sample K the precursor was CuCl2. (3) The flow rate for precursor addition was 1 mL min−1 for sample K and 4 mL min−1 for samples C, G and I. Either of these differences could potentially cause the different performance in CuHCF/Zn cells, and invites further study.
CuHCF samples have surprisingly different performance. Structural analysis does not reveal significant differences to account for the different performance of CuHCF samples C, G, I and K. As shown by Simonov et al., PBA exhibit non-random vacancy ordering, which may be affected by synthesis conditions,33 and studies of PBA linking vacancy ordering and electrochemical performance could potentially reveal information about the mechanisms for the different properties of apparently similar samples.
Conflicts of interest
There are no conflicts to declare.References
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933 RSC
.
- London Metal Exchange, https://www.lme.com/en-GB/Metals/Non-ferrous#tabIndex=0.
- M. Huang, M. Li, C. Niu, Q. Li and L. Mai, Adv. Funct. Mater., 2019, 29, 1807847 CrossRef
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS
- J. K. Seo, J. Shin, H. Chung, P. Y. Meng, X. Wang and Y. S. Meng, J. Phys. Chem. C, 2018, 122, 11177–11185 CrossRef CAS
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS
- Z. Jia, B. Wang and Y. Wang, Mater. Chem. Phys., 2015, 149–150, 601–606 CrossRef CAS
- L.-P. Wang, P.-F. Wang, T.-S. Wang, Y.-X. Yin, Y.-G. Guo and C.-R. Wang, J. Power Sources, 2017, 355, 18–22 CrossRef
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Sci. Rep., 2015, 5, 18263 CrossRef CAS
- R. Trócoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef
- T. Gupta, A. Kim, S. Phadke, S. Biswas, T. Luong, B. J. Hertzberg, M. Chamoun, K. Evans-Lutterodt and D. A. Steingart, J. Power Sources, 2016, 305, 22–29 CrossRef CAS
- K. Lu, B. Song, J. Zhang and H. Ma, J. Power Sources, 2016, 321, 257–263 CrossRef CAS
- J. F. Keggin and F. D. Miles, Nature, 1936, 137, 577–578 CrossRef CAS
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 550 CrossRef
- X. Wu, Y. Luo, M. Sun, J. Qian, Y. Cao, X. Ai and H. Yang, Nano Energy, 2015, 13, 117–123 CrossRef CAS
- Y. Yang, E. Liu, X. Yan, C. Ma, W. Wen, X.-Z. Liao and Z.-F. Ma, J. Electrochem. Soc., 2016, 163, A2117–A2123 CrossRef CAS
- D. Yang, J. Xu, X.-Z. Liao, Y.-S. He, H. Liu and Z.-F. Ma, Chem. Commun., 2014, 50, 13377–13380 RSC
- R. Chen, Y. Huang, M. Xie, Z. Wang, Y. Ye, L. Li and F. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 31669–31676 CrossRef CAS
- C. Li, R. Zang, P. Li, Z. Man, S. Wang, X. Li, Y. Wu, S. Liu and G. Wang, Chem. – Asian J., 2018, 13, 342–349 CrossRef CAS
- X. Wu, C. Wu, C. Wei, L. Hu, J. Qian, Y. Cao, X. Ai, J. Wang and H. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 5393–5399 CrossRef CAS
- Y. You, X. Wu, Y. Yin and Y. Guo, Energy Environ. Sci., 2014, 7, 1643–1647 RSC
- Z. Ji, B. Han, H. Liang, C. Zhou, Q. Gao, K. Xia and J. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 33619–33625 CrossRef CAS
- H.-W. Lee, R. Y. Wang, M. Pasta, S. Woo Lee, N. Liu and Y. Cui, Nat. Commun., 2014, 5, 5280 CrossRef CAS
- Y. Moritomo, Y. Kurihara, T. Matsuda and J. Kim, J. Phys. Soc. Jpn., 2011, 80, 103601 CrossRef
- R. Y. Wang, B. Shyam, K. H. Stone, J. N. Weker, M. Pasta, H. Lee, M. F. Toney and Y. Cui, Adv. Energy Mater., 2015, 5, 1401869 CrossRef
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 3, 1149 CrossRef
- A. Zhou, Z. Xu, H. Gao, L. Xue, J. Li and J. B. Goodenough, Small, 2019, 15, 1902420 CrossRef CAS
- H. L. B. Boström, I. E. Collings, A. B. Cairns, C. P. Romao and A. L. Goodwin, Dalton Trans., 2019, 48, 1647–1655 RSC
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506–6507 CrossRef CAS
- C. P. Krap, J. Balmaseda, L. F. del Castillo, B. Zamora and E. Reguera, Energy Fuels, 2010, 24, 581–589 CrossRef CAS
- A. Simonov, T. De Baerdemaeker, H. L. B. Boström, M. L. Ríos Gómez, H. J. Gray, D. Chernyshov, A. Bosak, H.-B. Bürgi and A. L. Goodwin, Nature, 2020, 578, 256–260 CrossRef CAS
- S. C. van der Marck, Int. J. Mod. Phys. C, 1998, 09, 529–540 CrossRef
- Y. Moritomo, K. Igarashi, J. Kim and H. Tanaka, Appl. Phys. Express, 2009, 2, 085001 CrossRef
- V. Renman, D. O. Ojwang, M. Valvo, C. P. Gómez, T. Gustafsson and G. Svensson, J. Power Sources, 2017, 369, 146–153 CrossRef CAS
- J. Lim, G. Kasiri, R. Sahu, K. Schweinar, K. Hengge, D. Raabe, F. La Mantia and C. Scheu, Chem. – Eur. J., 2020, 26, 4917–4922 CrossRef CAS
- F. W. Thomas Goh, Z. Liu, T. S. A. Hor, J. Zhang, X. Ge, Y. Zong, A. Yu and W. Khoo, J. Electrochem. Soc., 2014, 161, A2080–A2086 CrossRef
- J. Ming, J. Guo, C. Xia, W. Wang and H. N. Alshareef, Mater. Sci. Eng., R, 2019, 135, 58–84 CrossRef
- D. E. Stilwell, K. H. Park and M. H. Miles, J. Appl. Electrochem., 1992, 22, 325–331 CrossRef CAS
- P. Marzak, J. Yun, A. Dorsel, A. Kriele, R. Gilles, O. Schneider and A. S. Bandarenka, J. Phys. Chem. C, 2018, 122, 8760–8768 CrossRef CAS
- S. Berchmans, H. Gomathi and G. P. Rao, J. Electroanal. Chem., 1995, 394, 267–270 CrossRef
- S.-M. Chen, C.-Y. Liou and R. Thangamuthu, Electroanalysis, 2007, 19, 2457–2464 CrossRef CAS
- X. Zhou, S. Wang, Z. Wang and M. Jiang, Fresenius’ J. Anal. Chem., 1993, 345, 424–427 CrossRef CAS
- G. Kasiri, R. Trócoli, A. Bani Hashemi and F. La Mantia, Electrochim. Acta, 2016, 222, 74–83 CrossRef CAS
- Y. Yang, C. Brownell, N. Sadrieh, J. May, A. Del Grosso, D. Place, E. Leutzinger, E. Duffy, R. He, F. Houn, R. Lyon and P. Faustino, Clin. Toxicol., 2007, 45, 776–781 CrossRef CAS
- A. Mohammad, Y. Yang, M. A. Khan and P. J. Faustino, Clin. Toxicol., 2015, 53, 102–107 CrossRef CAS
- S. Kjeldgaard, I. Dugulan, A. Mamakhel, M. Wagemaker, B. B. Iversen and A. Bentien, R. Soc. Open Sci., 2021, 8, 201779 CrossRef
- O. Makowski, J. Stroka, P. J. Kulesza, M. A. Malik and Z. Galus, J. Electroanal. Chem., 2002, 532, 157–164 CrossRef CAS
- A. Mullaliu, G. Aquilanti, P. Conti, J. R. Plaisier, M. Fehse, L. Stievano and M. Giorgetti, J. Phys. Chem. C, 2018, 122, 15868–15877 CrossRef CAS
- M. Giorgetti, L. Guadagnini, D. Tonelli, M. Minicucci and G. Aquilanti, Phys. Chem. Chem. Phys., 2012, 14, 5527 RSC
- J. Chaboy, A. Muñoz-Páez, F. Carrera, P. Merkling and E. S. Marcos, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 1–7 CrossRef
- M. D. Adams and J. H. Kyle, Conf. Pap. Minprex 2000.
- M. Jayalakshmi and F. Scholz, J. Power Sources, 2000, 91, 217–223 CrossRef CAS
- A. Dostal, B. Meyer, F. Scholz, U. Schroeder, A. M. Bond, F. Marken and S. J. Shaw, J. Phys. Chem., 1995, 99, 2096–2103 CrossRef CAS
- N. F. Zakharchuk, N. Naumov, R. Stösser, U. Schröder, F. Scholz and H. Mehner, J. Solid State Electrochem., 1999, 3, 264–276 CrossRef CAS
- T. Shao, C. Li, C. Liu, W. Deng, W. Wang, M. Xue and R. Li, J. Mater. Chem. A, 2019, 7, 1749–1755 RSC
- G. He and L. F. Nazar, ACS Energy Lett., 2017, 2, 1122–1127 CrossRef CAS
- P. Padigi, J. Thiebes, M. Swan, G. Goncher, D. Evans and R. Solanki, Electrochim. Acta, 2015, 166, 32–39 CrossRef CAS
- M. Fiore, K. Hurlbutt, S. Wheeler, I. Capone, J. Fawdon, R. Ruffo and M. Pasta, Chem. Mater., 2020, 32(18), 7653–7661 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00949k |
| This journal is © The Royal Society of Chemistry 2021 |