
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe performance of conjugated polymers as emitters for triplet–triplet annihilation upconversion†
Riley
O’Shea
ab,
Can
Gao
 c,
Tze Cin
Owyong
ab,
Jonathan M.
White
b and
Wallace W. H.
Wong
*ab
c,
Tze Cin
Owyong
ab,
Jonathan M.
White
b and
Wallace W. H.
Wong
*ab
aARC Centre of Excellence in Exciton Science, School of Chemistry, University of Melbourne, Parkville, VIC 3010, Australia. E-mail: wwhwong@unimelb.edu.au
bSchool of Chemistry, Bio21 Institute, University of Melbourne, Parkville, VIC 3010, Australia
cBeijing National Laboratory for Molecular Sciences, Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China
First published on 19th February 2021
Abstract
A series of poly(phenylene–ethynylene) copolymers with various aryl spacer units were synthesized for use as emitters in triplet–triplet annihilation upconversion. The upconversion performance of these conjugated polymers was compared to that of well-known poly(phenylene–vinylene) polymers, MEH-PPV and super yellow, in chloroform solution. The copolymer containing anthracene units outperformed both reference polymers recording a maximum upconversion quantum yield of 0.18%.
Introduction
Triplet–triplet annihilation upconversion (TTA-UC), also known as triplet fusion upconversion, is a photochemical process by which two lower energy photons can be combined to produce one photon of higher energy.1 It sees use in raising the efficiency of solar cells above their thermodynamic limit (the Shockley–Queisser limit).2,3Two chromophores are required in a typical TTA-UC system – a triplet sensitizer and an annihilator/emitter.1 The triplet sensitizer absorbs a photon promoting it to its singlet excited state (Fig. 1a). Intersystem crossing (ISC) leads to the formation of the triplet excited state on the sensitizer. This triplet exciton can then be transferred to an emitter molecule via triplet energy transfer (TET). As the triplet exciton population of emitters builds, two triplet excitons can combine leading to triplet–triplet annihilation (TTA) generating a higher energy singlet exciton. This singlet exciton then undergoes radiative decay releasing a photon that is higher in energy than the photon absorbed by the sensitizer.
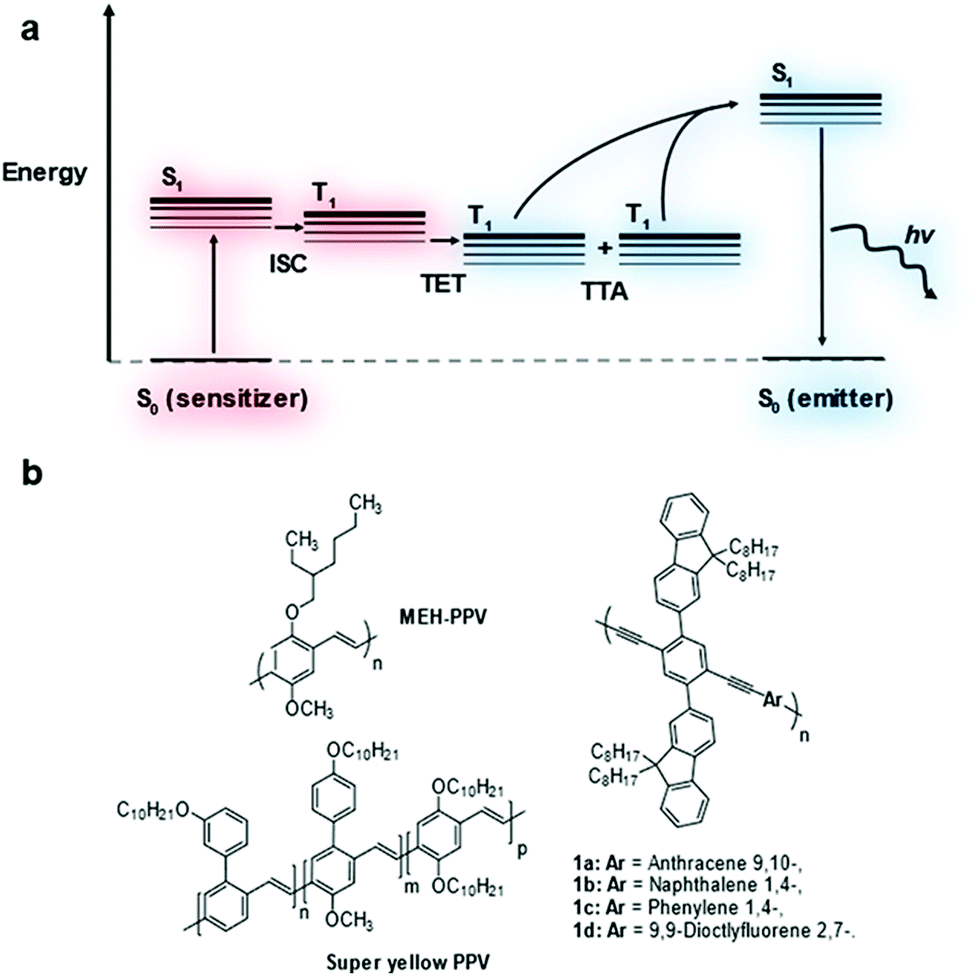 | ||
| Fig. 1 Jablonski diagram depicting the process involved for TTA-UC (a). Structures of poly(phenylene–vinylene)s, MEH-PPV and super yellow, and copolymers based on poly(phenylene–ethynylene) investigated as emitters for TTA-UC (b). | ||
Transition metal complexes are most commonly used as triplet sensitizers but metal chalcogenide quantum dots have also been used.4,5 As for the emitter component, polycyclic aromatic hydrocarbon molecules have been widely investigated.6–9 Much less well-established is the use of conjugated polymers as emitters in TTA-UC.10–14 It was shown in theoretical models that the extended conjugation of these materials leads to improved triplet exciton diffusion which may assist in TTA-UC performance.15 Measurements of TTA-UC systems containing conjugated polymers showed some promise but accurate comparison of TTA-UC efficiency has not been reported.14,16 In this study, the TTA-UC performance of several conjugated polymers in solution was investigated and compared (Fig. 1b).
There are some important parameters to consider when measuring TTA-UC performance.17 The TTA-UC efficiency is measured by the upconversion quantum yield (ΦUC), which is a product of all the quantum yields for photochemical processes involved, given by the equation:
| ΦUC = ΦISCΦTETΦTTAΦPL | (1) |
In measuring ΦUC, a relative measurement is typically preferred due to its simplicity.21 This can be done by comparing the integrated photoluminescence intensity of a standard fluorescent dye with a known ΦPL and that of the upconversion dye pair, eqn (2):
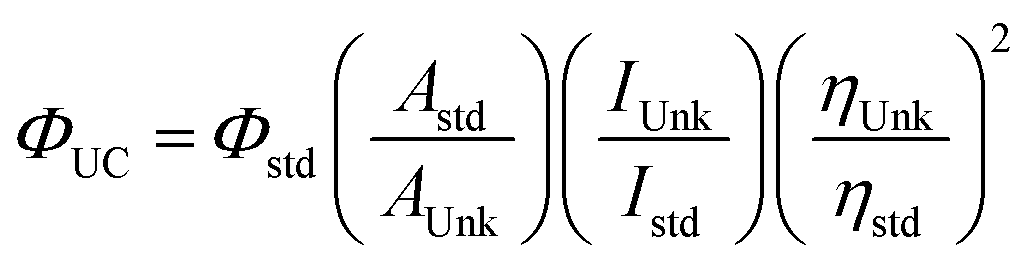 | (2) |
Herein, the ΦUC of solutions containing commercially available poly(phenylene–vinylene)s, MEH-PPV and super yellow, were measured using an absolute quantum yield method with meso-tetraphenyl-tetrabenzoporphine palladium complex (PdTPTBP) as the triplet sensitizer. We then compare the performance of MEH-PPV and super yellow to a series of poly(phenylene–ethynylene) copolymers. The TTA-UC samples were in a similar concentration range in chloroform solution and were optimized for maximum TTA-UC emission.
Results and discussion
A series of copolymers based on poly(phenylene–ethynylene) (PPE) was devised bearing dioctylfluorene sidechains to increase polymer solubility and to reduce molecular interactions that increase the rate of non-radiative decay (Fig. 1). It is well-known that PPEs with alkyl sidechains can show strong molecular association leading to low photoluminescence quantum yield.23 The aryl comonomer unit was varied to fine tune the photophysical properties of the polymer backbone.The bis-ethyne monomer 4 was synthesized by Suzuki coupling followed by treatment with TBAF to remove the TMS protecting groups in 74% yield (Fig. S2, ESI†). The single crystal structure of monomer 4 was obtained by slow evaporation from a chloroform solution. The structure showed a slip stacked packing, with a symmetrical kink in of one of the octyl chains on each of the fluorene units caused by close packing (for further details see Table S1, ESI†). The para-aryldibromide monomers 5a–d were either commercially available or synthesized using literature methods (see ESI† for details).
The polymers were synthesized using Sonogashira polycondensation between monomer 4 and dibromide monomers 5a–d, with reaction yields varying between 41–66% (Table S2, ESI†). The molecular weight of the polymers was found to be fairly low <15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (Table 1, see ESI,† Fig. S7 for GPC traces). Attempts to increase these values proved to be unsuccessful. Neither longer reaction times nor higher temperatures had any effect on the molecular weight. Similarly, no change was noticed when the corresponding para-aryldiiodides were used in place of the para-aryldibromides 5. When using Pd(dppf)Cl2 as the catalyst, a slightly higher molecular weight range was obtained, however the polymers produced were weakly emissive. Similar PPEs reported in prior literature also exhibited low molecular weights with Sonogashira polycondensations.24–26 Higher molecular weights in homopolymers could be obtained by alkyne metathesis,27 however this method is difficult to implement for construction of copolymers. Concerning the polymer properties for TTA-UC, it should be noted that as little as 5 repeat units have been shown to be sufficient for similar photophysical properties as the corresponding PPE.24
000 g mol−1 (Table 1, see ESI,† Fig. S7 for GPC traces). Attempts to increase these values proved to be unsuccessful. Neither longer reaction times nor higher temperatures had any effect on the molecular weight. Similarly, no change was noticed when the corresponding para-aryldiiodides were used in place of the para-aryldibromides 5. When using Pd(dppf)Cl2 as the catalyst, a slightly higher molecular weight range was obtained, however the polymers produced were weakly emissive. Similar PPEs reported in prior literature also exhibited low molecular weights with Sonogashira polycondensations.24–26 Higher molecular weights in homopolymers could be obtained by alkyne metathesis,27 however this method is difficult to implement for construction of copolymers. Concerning the polymer properties for TTA-UC, it should be noted that as little as 5 repeat units have been shown to be sufficient for similar photophysical properties as the corresponding PPE.24
| M w (g mol−1) | Đ | DPa | Abs λmax (nm) | PL λmax (nm) | Φ PL (%) | |
|---|---|---|---|---|---|---|
| a DP = degree of polymerization. b Solution ΦPL measured at 10 μg mL−1 concentration of polymer in chloroform using anthracene as a reference with excitation at 350 nm. | ||||||
| 1a | 11600 |
2.0 | 11 | 321 | 533 | 49.5 ± 0.2 |
| 1b | 5310 | 1.6 | 7 | 319 | 462 | 97.8 ± 2.1 |
| 1c | 7070 | 1.6 | 9 | 320 | 450 | 74.5 ± 1.5 |
| 1d | 5910 | 1.9 | 6 | 323 | 445 | 93.8 ± 2.2 |
The photoluminescence spectrum of the copolymers showed some variation in the peak emission with the most significant difference for anthracene copolymer 1a. The extended π system of the anthracene in 1a led to the most red-shifted UV-Vis absorption edge and photoluminescence (PL) peak maxima in the series (Fig. 2). All copolymers have an absorption maximum at ∼320 nm which can be attributed to the fluorene sidechains.28 Copolymer 1a also showed vibronic features in its absorption and PL spectra reminiscent of the anthracene building block. The ΦPL of the polymers ranged from 50% to 100% (Table 1), with the anthracene derivative 1a being the least emissive and the naphthalene derivative 1b being the most emissive. The most likely reason for the ΦPL variations is the positioning of the fluorene sidechains and their effect on polymer–polymer interactions. Some insights are discussed in the DFT calculations section below. These PLQY values are reminiscent of many PPE copolymers, with solution ΦPL values typically being in excess of 50%, with many of those containing simple alkyl side chains displaying solution ΦPL close to 100%.23,29
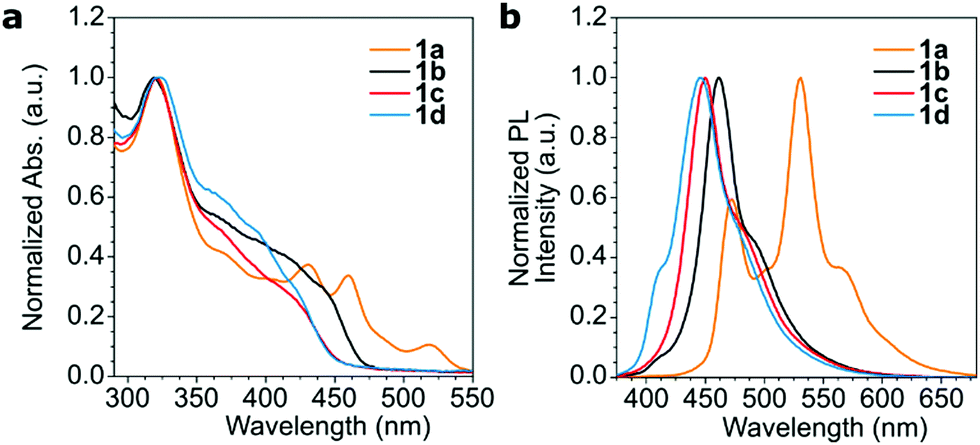 | ||
| Fig. 2 Normalized UV-Vis absorption (a) and photoluminescence (b) spectra of copolymers 1a–d in chloroform solution. Excitation wavelength: 350 nm. | ||
Next, the TTA-UC performance of the polymers were tested with PdTPTBP as the triplet sensitizer. The polymers (0.25 mg mL−1) and the PdTPTBP (7.5 μM) were well-dissolved in chloroform as it was apparent from the absorption spectrum of the samples (Fig. S9, ESI†). A red-to-green upconversion was observed with the anthracene-based PPE 1a with 632 nm excitation (Fig. 3). The upconversion emission intensity was optimized and found to occur using a polymer concentration of 0.25 mg mL−1 with a 7.5 μM concentration of PdTPTBP. Surprisingly, none of the other PPE derivatives showed any upconversion emission under the same conditions. In fact, the polymers 1b–d failed to quench the phosphorescence of PdTPTBP at 800 nm (Fig. 3). This is a clear indication that only 1a has appropriate triplet excited state energy to interact with the PdTPTBP sensitizer.
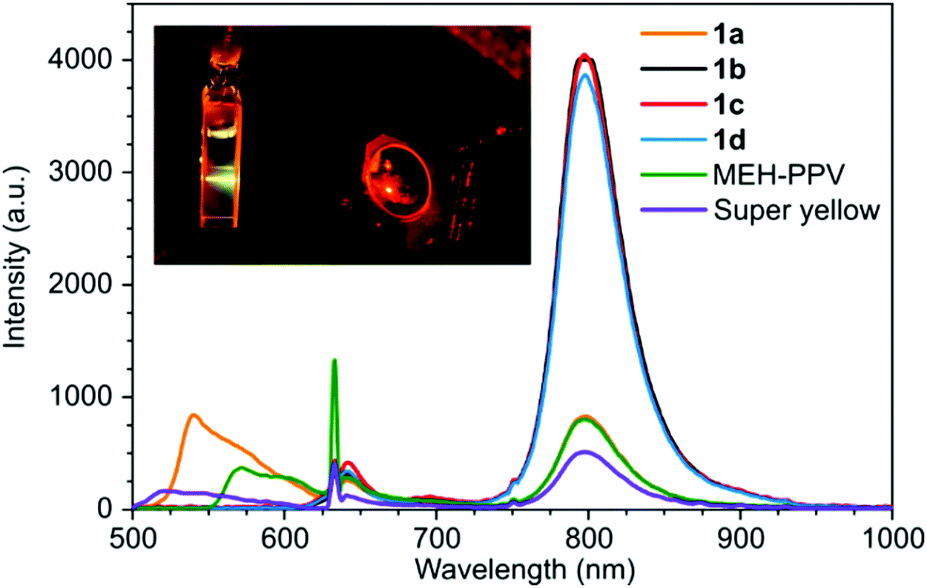 | ||
| Fig. 3 Emission spectrum of samples containing copolymers 1a–d (0.25 mg mL−1), MEH-PPV (0.5 mg mL−1), super yellow PPV (0.5 mg mL−1) with PdTPTBP (7.5 μM) as the sensitizer in chloroform solution under 632 nm excitation. Phosphorescence of PdTPTBP was observed at 800 nm and the upconverted emission was observed at 500–600 nm. The insert is a photo of a cuvette containing copolymer 1a and PdTPTBP showing yellow TTA-UC emission. | ||
The upconverted emission maximum for the sample containing 1a was close to that of the conventional photoluminescence maximum for 1a at 540 nm but the emission profiles were rather different especially with the 600 nm banc pass filter used on the upconverted emission measurement (Fig. S15, ESI†).
These copolymers were then compared to commercially available PPV emitters – MEH-PPV and super yellow – whose ΦUC was determined via an integrating sphere method under nitrogen atmosphere to be 0.039% and 0.029% respectively. These values were determined in chloroform solution with an excitation intensity of 985 mW cm−2 at 632 nm, with an optimized polymer concentration of 0.5 mg mL−1 and PdTPTBP at 7.5 μM. Despite the higher ΦPL of super yellow at 69% compared to 27% for MEH-PPV, its ΦUC is lower than MEH-PPV.30,31 The lower upconversion intensity of super yellow and its substantially higher cost compared to MEH-PPV, makes MEH-PPV preferable as an upconversion standard. These commercial polymers serve as adequate standards, with a large degree of spectral overlap due to their broad photoluminescent emission in the same region displayed by the PPE copolymers.
The ΦUC values were measured as a function of the excitation intensity for 1a and MEH-PPV. A maximum ΦUC value of 0.18% was reached at 8910 mW cm−2 (Fig. 4). The ΦUC of MEH-PPV reached a maximum value of 0.065% at 10500 mW cm−2. These values were also measured relative to super yellow PPV and found to have little to no relative error (Table S3, ESI†). Although these are low values of ΦUC compared to the best solution TTA-UC systems,32 this work showed PPE materials are possible emitters and performed significantly better than both MEH-PPV and super yellow.
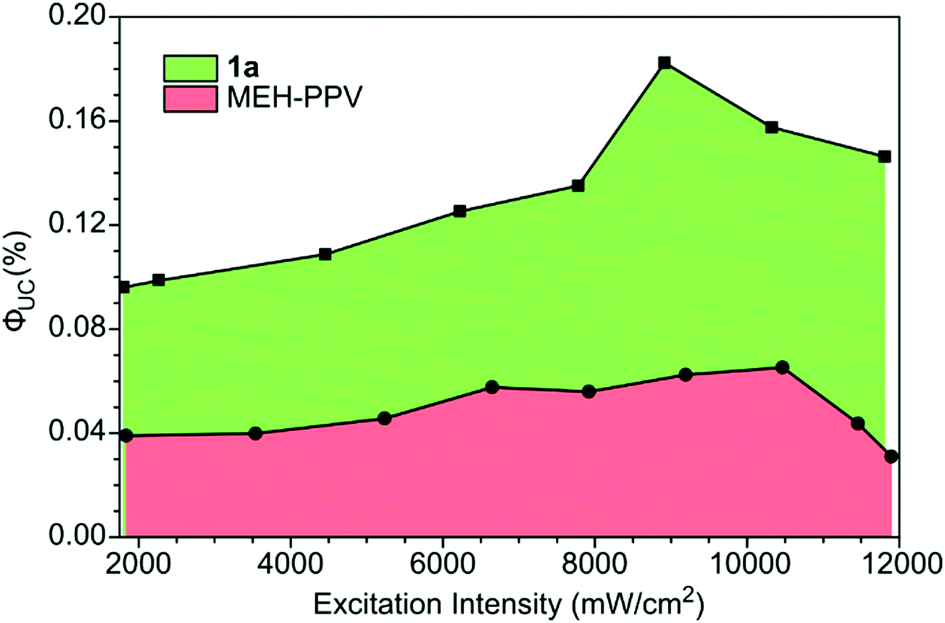 | ||
| Fig. 4 Dependence of upconversion quantum yield on excitation intensity for PPE 1a (0.25 mg mL−1), MEH-PPV (0.5 mg mL−1) with PdTPTBP (7.5 μM) as the sensitizer in chloroform solution under 632 nm excitation. | ||
To gain insight into the effect of various aryl monomer building blocks on the properties of the polymers, density functional theory (DFT) calculations were performed (Fig. 5 and 6). A semi-empirical method (PM6) was used to optimize the geometry of these polymers, then the TD-DFT was performed with B3LYP/6-311G(d,p) to obtain the singlet and triplet excited state energies. As mentioned previously, the difference in the ΦPL values (Table 1) could be related to the positioning of the fluorene sidechains. For the naphthalene and fluorene-based copolymers 1b and 1d, the sidechains of the optimized structures are directed inwards towards the arene units (Fig. 5). This is expected to reduce polymer–polymer backbone interactions leading to higher ΦPL. For 1a and phenylene-based 1c, the fluorene sidechains occupied the same plane, leaving the polymer backbone more exposed.
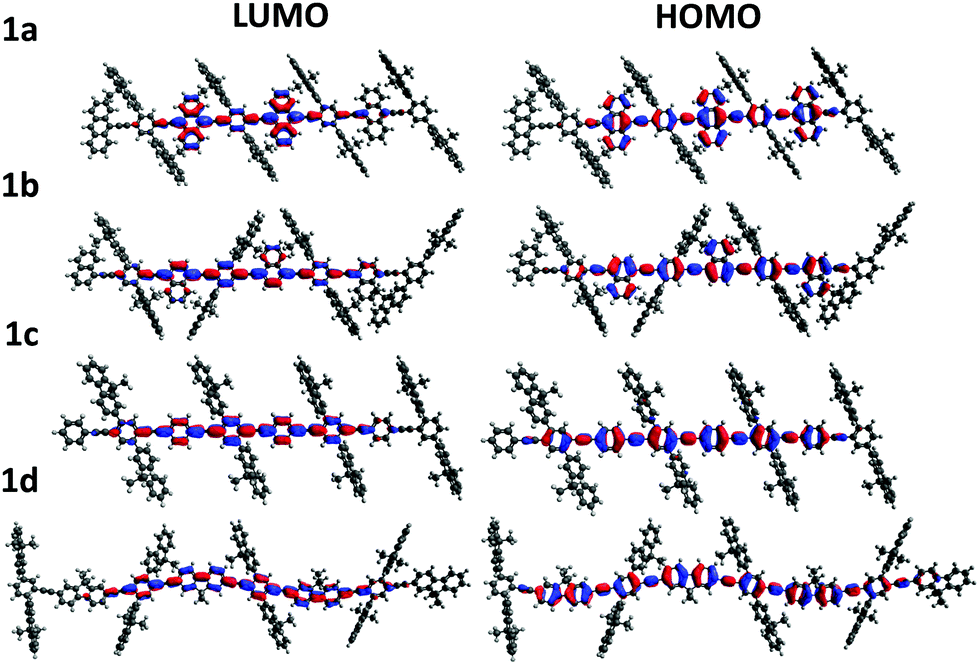 | ||
| Fig. 5 DFT calculations of model oligomers of 1a–d displaying the electron probability distributions for the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) for various aryl comonomer units. | ||
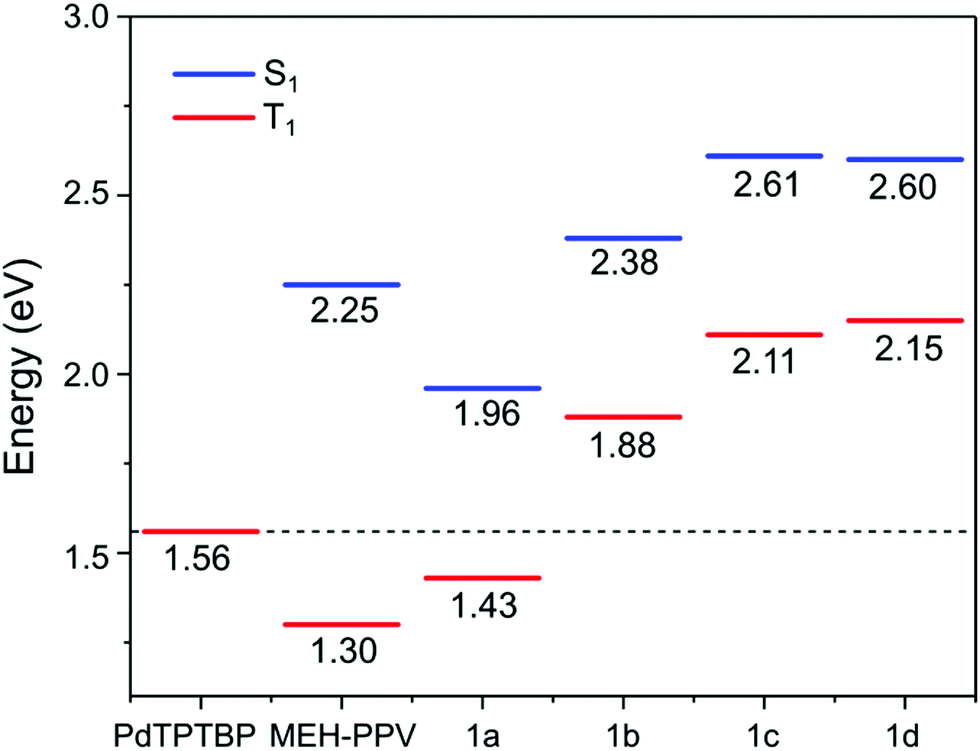 | ||
| Fig. 6 Energy diagram depicting the calculated singlet and triplet excited state energies obtained by DFT of the model oligomers of 1a–d compared to the triplet excited state energy of PdTPTBP. The values for MEH-PPV are taken from ref. 34. | ||
The electron distributions for the HOMO and LUMO of all polymers showed a high level of overlap (Fig. 5). Additionally, for the anthracene polymer, more of the electron distribution was found on the anthracene unit itself than for the other polymers, which mainly reside on the conjugated polymer backbone. This may explain the retained vibronic emission features seen in anthracene-based polymer 1a (Fig. 2).
The calculated singlet (S1) and triplet (T1) energies are shown in Fig. 6. These values are similar to a previous theoretical study on the energy levels of PPEs by Kohler and Beljonne, where T1 of 1.51, 2.40 and 2.25 eV were found for the anthracene, phenylene and dioctylfluorene-based polymers respectively.33 The T1 values of 1a and MEH-PPV, at 1.43 eV and 1.30 eV34 respectively, were lower than T1 of the triplet sensitizer PdTPTBP.35 This indicated that energetically favourable triplet energy transfer from the sensitizer was possible for 1a and MEH-PPV. This agreed with observations in the phosphorescence quenching and TTA-UC emission measurements.
Although alternative triplet sensitizers could theoretically produce upconversion with copolymers 1b–d, no single sensitizer can work with all polymers in this study given the energy level requirements for TTA-UC. Instead, many of the polymers could be excited directly at sufficiently high excitation energies in preference to a sensitizer, resulting in down-converted emission. As the primary aim of this study was to assess the performance of conjugated polymers as emitters for upconversion and MEH-PPV/PdTPTBP was the reference system, only PdTPTBP was used.
Conclusions
In summary, a series of PPE copolymers with a variety of aryl spacer units were synthesized and tested as emitters for TTA upconversion. Commercially available conjugated polymers, MEH-PPV and super yellow PPV, were used as references for ΦUC measurements after their ΦUC values were determined by absolute quantum yield method. The anthracene-based polymer 1a outperformed both references achieving maximum ΦUC of 0.18% with 632 nm excitation and 8910 mW cm−2 intensity. Other copolymers in the series did not quench the phosphorescence of the sensitizer and therefore showed no upconverted emission. DFT calculations indicated only 1a has the appropriate triplet energy for triplet energy transfer from the PdTPTBP sensitizer. We are in the process of exploring the TTA-UC performance of a wider range of conjugated polymers to further assess their viability in photon upconversion applications.Abbreviations
| TTA | Triplet–triplet annihilation |
| UC | Upconversion |
| PPV | Poly(phenylene–vinylene) |
| PPE | Poly(phenylene–ethynylene) |
| DP | Degree of polymerization |
| UV-Vis | Ultraviolet-visible |
| PL | Photoluminescence |
| PdTPTBP | Palladium(II) meso-tetraphenyltetrabenzoporphyrin |
| DFT | Density functional theory |
| TD | Time dependent |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest occupied molecular orbital |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ARC Centre of Excellence in Exciton Science (CE170100026). We thank Prof Kenneth Ghiggino for helpful discussions on photophysical analysis of the upconversion systems.References
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC.
- Y. Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, R. G. C. R. Clady, T. F. Schulze, N. J. Ekins-Daukes, M. J. Crossley, B. Stannowski, K. Lips and T. W. Schmidt, Energy Environ. Sci., 2012, 5, 6953–6959 RSC.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- N. Nishimura, J. R. Allardice, J. Xiao, Q. Gu, V. Gray and A. Rao, Chem. Sci., 2019, 10, 4750–4760 RSC.
- E. M. Gholizadeh, S. K. K. Prasad, Z. L. Teh, T. Ishwara, S. Norman, A. J. Petty, J. H. Cole, S. Cheong, R. D. Tilley, J. E. Anthony, S. Huang and T. W. Schmidt, Nat. Photonics, 2020, 14, 585–590 CrossRef CAS.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS.
- C. Gao, S. K. K. Prasad, B. Zhang, M. Dvořák, M. J. Y. Tayebjee, D. R. McCamey, T. W. Schmidt, T. A. Smith and W. W. H. Wong, J. Phys. Chem. C, 2019, 123, 20181–20187 CrossRef CAS.
- V. Gray, A. Dreos, P. Erhart, B. Albinsson, K. Moth-Poulsen and M. Abrahamsson, Phys. Chem. Chem. Phys., 2017, 19, 10931–10939 RSC.
- T. Singh-Rachford and F. Castellano, J. Phys. Chem. A, 2008, 112, 3550–3556 CrossRef CAS.
- D. Hertel, H. Bässler, R. Guentner and U. Scherf, J. Chem. Phys., 2001, 115, 10007–10013 CrossRef CAS.
- F. Laquai, G. Wegner, C. Im, A. Büsing and S. Heun, J. Chem. Phys., 2005, 123, 074902 CrossRef.
- S. Baluschev, P. E. Keivanidis, G. Wegner, J. Jacob, A. C. Grimsdale, K. Müllen, T. Miteva, A. Yasuda and G. Nelles, Appl. Phys. Lett., 2005, 86, 061904 CrossRef.
- S. Baluschev, J. Jacob, Y. S. Avlasevich, P. E. Keivanidis, T. Miteva, A. Yasuda, G. Nelles, A. C. Grimsdale, K. Müllen and G. Wegner, ChemPhysChem, 2005, 6, 1250–1253 CrossRef CAS.
- V. Jankus, E. W. Snedden, D. W. Bright, V. L. Whittle, J. A. G. Williams and A. Monkman, Adv. Funct. Mater., 2013, 23, 384–393 CrossRef CAS.
- I. Lyskov, E. Trushin, B. Q. Baragiola, T. W. Schmidt, J. H. Cole and S. P. Russo, J. Phys. Chem. C, 2019, 123, 26831–26841 CrossRef CAS.
- Y. Zhou, F. N. Castellano, T. W. Schmidt and K. Hanson, ACS Energy Lett., 2020, 5, 2322–2326 CrossRef CAS.
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC.
- S. M. Bachilo and R. B. Weisman, J. Phys. Chem. A, 2000, 104, 7711–7714 CrossRef CAS.
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC.
- F. Deng, J. Blumhoff and F. N. Castellano, J. Phys. Chem. A, 2013, 117, 4412–4419 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- N. Yanai, K. Suzuki, T. Ogawa, Y. Sasaki, N. Harada and N. Kimizuka, J. Phys. Chem. A, 2019, 123, 10197–10203 CrossRef CAS.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS.
- P. Wautelet, M. Moroni, L. Oswald, J. Le Moigne, A. Pham, J. Y. Bigot and S. Luzzati, Macromolecules, 1996, 29, 446–455 CrossRef CAS.
- J. M. Hong, H. N. Cho, D. Y. Kim and C. Y. Kim, Synth. Met., 1999, 102, 933–934 CrossRef CAS.
- D. Y. Kim, J. M. Hong, J. K. Kim, H. N. Cho and C. Y. Kim, Macromol. Symp., 1999, 143, 221–230 CrossRef CAS.
- L. Kloppenburg, D. Jones and U. H. F. Bunz, Macromolecules, 1999, 32, 4194–4203 CrossRef CAS.
- B.-H. Sohn, K. Kim, D. S. Choi, Y. K. Kim, S. C. Jeoung and J.-I. Jin, Macromolecules, 2002, 35, 2876–2881 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- R. O'Shea and W. W. H. Wong, Polym. Chem., 2020, 11, 2831–2837 RSC.
- E. W. Snedden, L. A. Cury, K. N. Bourdakos and A. P. Monkman, Chem. Phys. Lett., 2010, 490, 76–79 CrossRef CAS.
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS.
- A. Köhler and D. Beljonne, Adv. Funct. Mater., 2004, 14, 11–18 CrossRef.
- P. C. Jha, E. Jansson and H. Ågren, Chem. Phys. Lett., 2006, 424, 23–27 CrossRef CAS.
- R. Tao, J. Zhao, F. Zhong, C. Zhang, W. Yang and K. Xu, Chem. Commun., 2015, 51, 12403–12406 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1992339. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00068c |
| This journal is © The Royal Society of Chemistry 2021 |