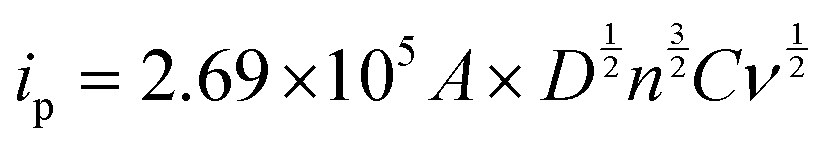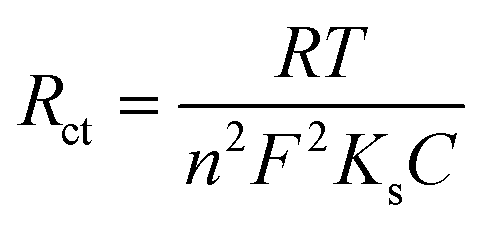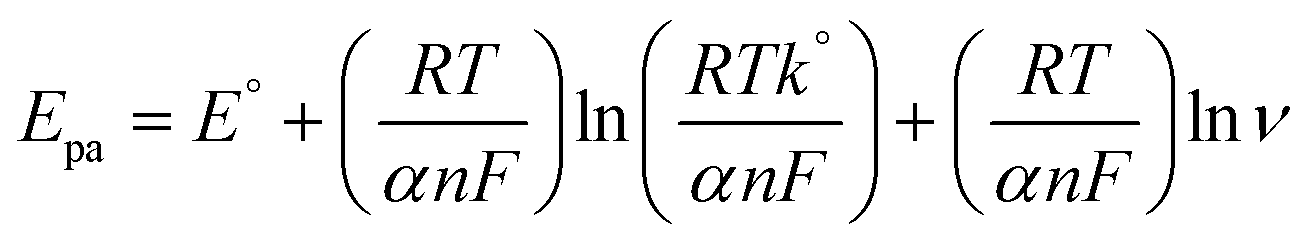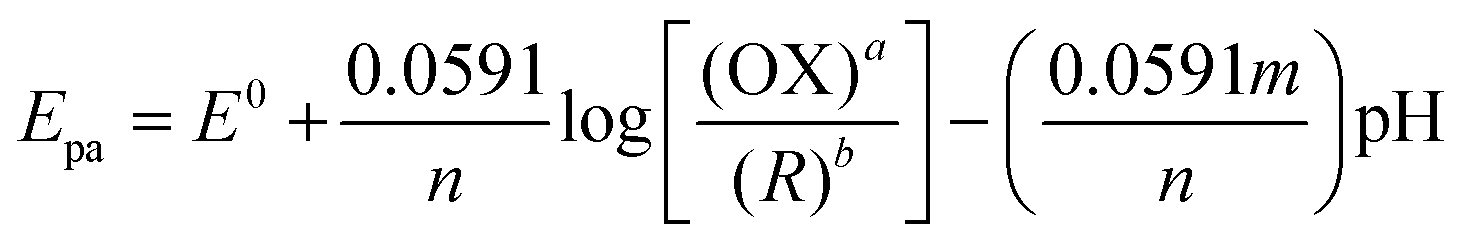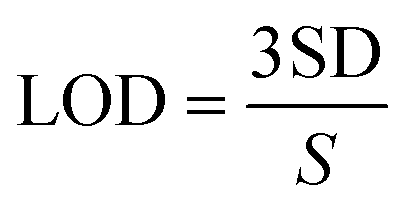DOI:
10.1039/D0MA00974A
(Paper)
Mater. Adv., 2021,
2, 3336-3345
An electrochemically exfoliated graphene/poly(3,4-ethylenedioxythiophene) nanocomposite-based electrochemical sensor for the detection of nicotine†
Received
11th December 2020
, Accepted 31st March 2021
First published on 31st March 2021
Abstract
A novel, simple and ‘green’ electrochemical exfoliation method was used to prepare a graphene (GR) dispersion. The as-obtained GR was characterized using HR-TEM, which confirmed the exfoliation of a few layers of graphene from graphite sheets. Furthermore, the GR film was coated on a glassy carbon electrode (GCE) surface via drop-casting. Then, electro-polymerization was carried out using the GR-modified electrode in acetonitrile solution (ACN) containing 10 μM 3,4-ethylenedioxythiophene (EDOT) and 100 mM lithium perchlorate (LiClO4). The electro-polymerized PEDOT (poly(3,4-ethylenedioxythiophene)) on the GR film-modified electrode was analyzed using FE-SEM and Raman spectroscopy, which confirmed the successful incorporation of PEDOT on the GR surface. The electrochemical characterization of PEDOT/GR/GCE was carried out by electrochemical impedance spectroscopy (EIS) in 0.1 M KCl containing 5 mM [Fe(CN)6]3−/4−. The electrochemical measurements revealed that our proposed PEDOT/GR film exhibited a high active surface area, redox currents and high charge transfer rate (Ks). Under optimized conditions, PEDOT/GR/GCE showed a higher oxidation peak current for nicotine at +0.91 V (vs. Ag/AgCl) in 0.1 M phosphate buffer solution (PBS; pH 7). Furthermore, PEDOT/GR/GCE responded linearly for nicotine detection from 0.5 to 1000 μM with a limit of detection at 47 nM. The selectivity and sensitivity of the electrochemical sensor (PEDOT/GR/GCE) were determined in the presence of important interfering molecules. In addition, the stability and reproducibility of the PEDOT/GR/GCE sensor were tested. Finally, the PEDOT/GR-based sensor was successfully applied to detect nicotine in tobacco samples with high accuracy.
1. Introduction
Nicotine is a toxic metabolite that is present in tobacco leaves, and is a heavily used addictive substance among adults.1–4 As stated by the Centers for Disease Control and Prevention (CDC), about half-million Americans die each year due to exposure to secondhand smoke or smoking.5 Smokers burn tobacco leaves, which causes many problems, such as heart disease, stroke, cancer, and lung disease, and also negatively affects the human brain.6,7 The concentration of nicotine is ∼1–3% in tobacco products, which determines its quality. Therefore, accurate detection of nicotine is important in various samples for toxicology, medicine and tobacco-related research and development.8–10 Traditional methods such as gas chromatography-mass spectrometry (GC-MS),11 high-performance liquid chromatography (HPLC),12,13 radioimmunoassays,14 electro-chemiluminescence (ECL)15 and liquid chromatography-mass spectrometry (LC-MS)3 have been used to analyze the concentration of nicotine. Due to the high cost, extensive sample extraction procedures and sample pretreatment requirements,16 alternative highly selective electrochemical sensors have been prepared.
Recently, detection of nicotine was demonstrated using carbon paste electrode (CPE),17 glassy carbon electrode (GCE)18 and pencil graphite electrode (PGE).1 To further improve the stability, selectivity and sensitivity of sensors, chemically modified electrodes were reported.19–21 Graphene (GR) is a two-dimensional (2D) honeycomb lattice structured carbon-based material with a delocalized electronic network.22–26 GR has exhibited high thermal conductivity, optical transparency, mechanical strength, good flexibility, deep ultraviolet emission, high thermal stability, chemical tolerance (for protective coatings), etc.22,27–29 GR can be prepared by either bottom-up or top-down methods.30–35 Electrochemical exfoliation is a top-down method which can produce high-yield GR nanosheets at low cost.36–39 The as-prepared GR can be used to synthesize nanocomposites with metals, metal oxides,40 conducting polymers (CPs)41 and some other functional materials to improve sensing performance.
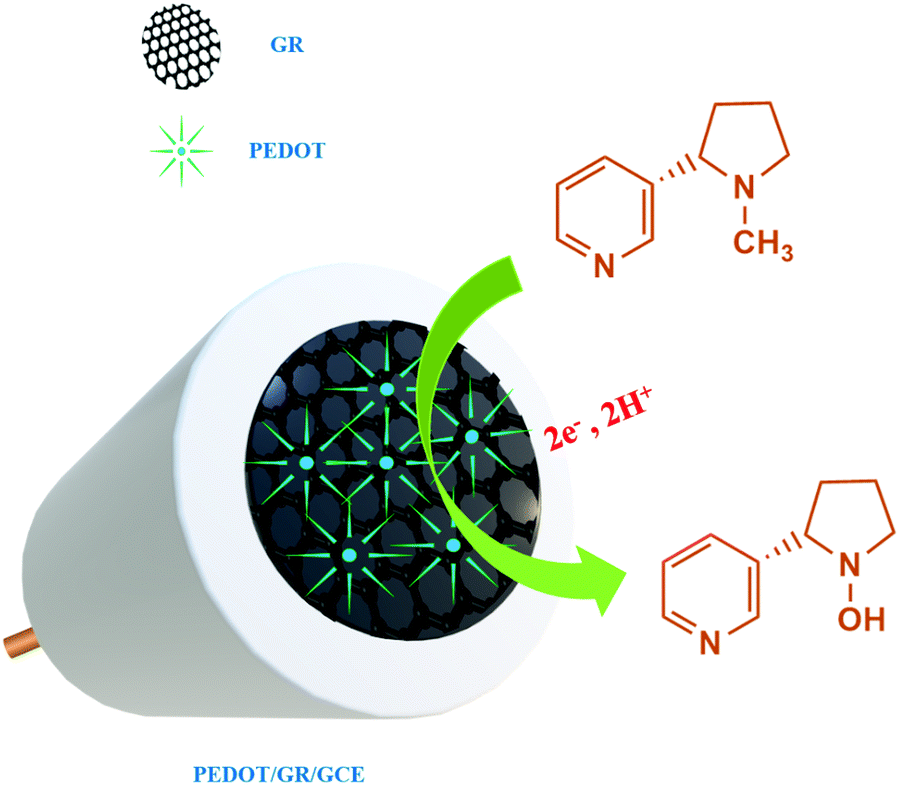
On the other hand, poly(3,4-ethylenedioxythiophene) (PEDOT) is an important π-conjugated polymer which has been widely used in the fabrication of sensors due to its high stability, conductivity, good biocompatibility, fast electron transfer properties and electro-catalytic activity.42,43 PEDOT has also shown anti-fouling properties, which are needed to improve the sensitivity, stability and repeatability of electrochemical sensors.44–46 Therefore, the preparation of PEDOT-based nanocomposites could be useful to fabricate electrochemical sensors for nicotine detection with high sensitivity and selectivity. Hybrid films composed of PEDOT and GR have received great attention due to the synergistic effect between the two components.47–49 In the present work, electrochemical detection of nicotine was demonstrated using a PEDOT/GR composite-modified electrode.
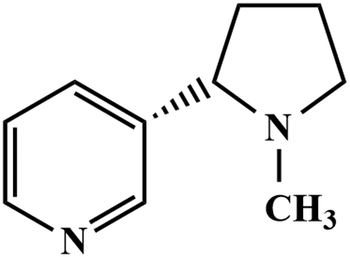
Firstly, GR was prepared by an electrochemical exfoliation method and characterized by HR-TEM and selected area electron diffraction (SAED) techniques. Secondly, PEDOT was electrochemically deposited onto the GR film-modified electrode. The as-prepared PEDOT/GR hybrid film was characterized by Raman spectroscopy and FE-SEM to investigate the successful deposition and surface morphology analysis. The obtained PEDOT/GR nanocomposite-modified electrode (PEDOT/GR/GCE) showed good electro-catalytic activity and high selectivity towards nicotine. Moreover, the stability and selectivity of the PEDOT/GR hybrid film-modified electrode was tested. The linear response range and limit of detection (LOD) of nicotine were also determined. Finally, the PEDOT/GR/GCE sensor was applied to detect nicotine (Scheme 1) in tobacco samples with high selectivity.
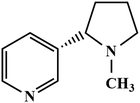 |
| | Scheme 1 Chemical structural formula of nicotine. | |
2. Experimental
2.1 Materials and reagents
Graphite sheets were obtained from Graphite Shop, Inc. (United States). Acetonitrile (AR, ACS), L-ascorbic acid (AR), L-cysteine (CHR), potassium ferrocyanide (AR), potassium ferricyanide (AR) and uric acid (AR) were purchased from Sisco Research Laboratories Pvt. Ltd, India. Dextrose anhydrous, oxalic acid (99.5%, AR/ACS) and sulfuric acid (H2SO4) were purchased from Loba Chemie, India. Melamine (99%) and nicotine (≥99%) were obtained from Sigma-Aldrich, India. Sodium phosphate dibasic heptahydrate (Na2HPO4·7H2O) and sodium dihydrogen phosphate monohydrate (H2NaPO4·H2O) were purchased from Spectrochem Pvt, Ltd, India. Potassium chloride (KCl) was purchased from Fisher Scientific, India. Hydrogen peroxide was obtained from Merck Life Science Pvt, Ltd, India. The phosphate buffer solution (PBS) (pH 7) was used to prepare the standard (0.1 M) solutions, and the pH values were adjusted by utilizing different ratios of NaH2PO4·H2O and Na2HPO4·7H2O. Double distilled (Milli-Q) water was used for the preparation of nicotine stock solutions and stored under dark conditions. Milli-Q water (18.2 MΩ cm) was obtained from a Millipore ultrapure water system. Commercially available cigarettes were purchased from local shops and utilized for real sample preparation and analysis.
2.2 Characterizations
The crystal structure and morphology of GR were characterized by a high-resolution transmission electron microscope (HR-TEM) (JEM-2100 Plus Electron Microscope, Japan). A field emission scanning electron microscope (FESEM-EDS) (Quanta 400 FEG, FEI) was used to analyze the microstructures and morphological features of the PEDOT and PEDOT/GR films. All Raman spectra were recorded using the excitation of a 633 nm laser (LabRAM HR evolution, Horiba, 3.2 mW laser power source) connected to an Olympus imaging microscope (using Labspec6 Raman software). The calibration of the Raman spectrometer was carried out using a silica standard (520.16 cm−1) before measuring the Raman spectra of each sample with 3.2% of the maximum laser power (100 mW). Electrochemical studies such as cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and amperometry were carried out using an electrochemical workstation (CHI-760E, CH Instruments, USA). Ag/AgCl (3 M KCl) and platinum wire were used as the reference and counter electrodes, respectively.
2.3 Preparation of GR
GR was prepared by an electrochemical exfoliation method.50,51 Briefly, graphite sheets (3 cm × 0.5 cm) were used as electrodes. Graphite sheets (∼1.5 cm length of both the anode and cathode) were dipped into 50 mL of 0.1 M PBS (pH 7) for 30 min, and +10 V was supplied using a DC power supply (Model: 2231A-30-3, Keithley, a Tektronix company, USA). During the electrochemical exfoliation, the partially oxidized graphene nanosheets (GR) were exfoliated and settled in PBS (Fig. S1, ESI†). After the reaction completed, the as-synthesized GR flakes were washed thoroughly using Milli-Q water (through vacuum filtration) to remove the unwanted ions. After that, GR flakes were ultra-sonicated in DI water for 60 min and centrifuged at 3000 rpm to obtain the supernatant GR dispersion. Finally, the GR dispersion (top supernatant solution) was collected and stored for further use.
2.4 Preparation of the PEDOT/GR-based sensor
The surface of GCE was cleaned by polishing with a series of alumina particle slurry (Al2O3, particle sizes of ∼1 μm, 0.3 μm and 0.05 μm in sequence) on a polishing pad. Thereafter, GCE was washed with ultrapure Milli-Q water and dried at room temperature. The homogeneous GR suspension (6 μL from 5 mg mL−1) was placed on the GCE surface and dried. Next, GR/GCE was rinsed using distilled water to remove the unbound particles. To prepare the PEDOT/GR nanocomposite-modified electrode, EDOT was electro-polymerized on GR/GCE or GR/ITO/PET (indium tin oxide-coated polyethylene terephthalate) by potential cycling between −0.3 and 1.4 V for 10 cycles at a scan rate of 50 mV s−1 in 10 mL acetonitrile containing 0.01 M EDOT and 0.1 M LiClO4 (Scheme S1, ESI†). After the electrochemical polymerization, a dark blue film was observed on the electrode surface, which is the natural color of the PEDOT film (Fig. S2, ESI†).52,53 These results confirmed that PEDOT was successfully deposited on the surface of the GR-modified electrode. The as-prepared PEDOT/GR/GCE was thoroughly rinsed with acetonitrile to remove any loosely bound monomers present on the electrode surface. For the control experiments, PEDOT/GCE (without GR) was also prepared by using a similar procedure.
2.5 Preparation of tobacco samples
To prepare the real samples, commercially available cigarettes (10 brands) were purchased from a local departmental shop. First, the filters and rolling papers of these cigarettes were completely removed and the tobacco leaves were collected by mixing and grinding (Fig. S3, ESI†). After that, 500 mg of tobacco powder was taken in a 25 mL beaker and mixed with 10 mL of Milli-Q water. This mixture was ultra-sonicated for 60 min.54 Finally, the yellowish solution was filtered through a cellulose nitrate filter paper (pore size = 0.45 μm), and the filtrate was collected and used for further study.
3. Results and discussion
3.1 Electrochemical polymerization of EDOT on GR/GCE
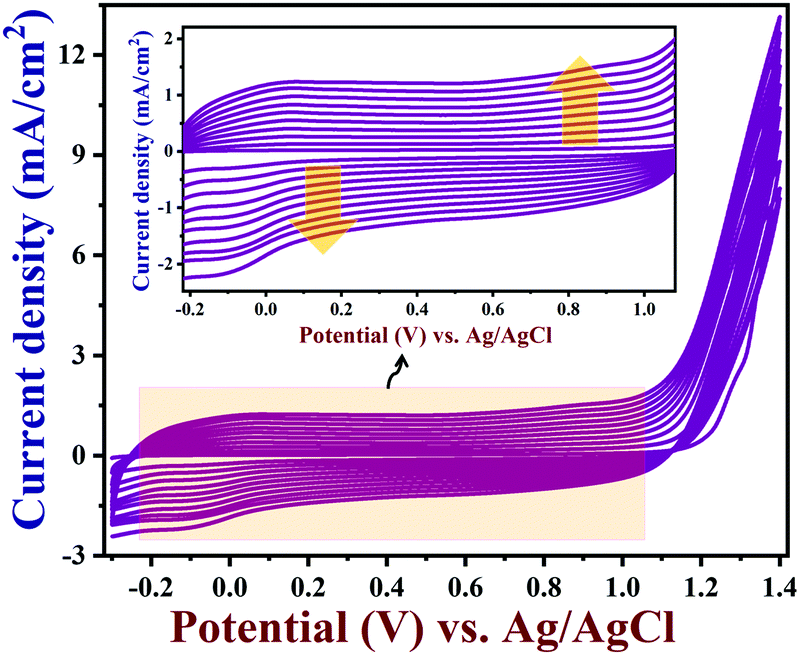
Cyclic voltammograms (CVs) were recorded using GR/GCE in the potential window between −0.3 V and +1.4 V for 10 cycles in acetonitrile containing 0.01 M EDOT and 0.1 M LiClO4 (Fig. 1). In this way, PEDOT was deposited on the surface of GR/GCE. The positive potential range was used up to +1.4 V for the polymerization in order to avoid any overoxidation on the PEDOT film, which was observed earlier at a higher positive potential.55,56Fig. 1 (inset) shows the uniform deposition of PEDOT on the GR/GCE surface for up to 10 cycles. During the electro-polymerization, the capacitance currents were increased with respect to each cycle, which confirmed the gradual deposition of the PEDOT conducting polymer film on the GR/GCE surface. This was in good agreement with the reported polymerization methods of EDOT in LiClO4/acetonitrile solution.57
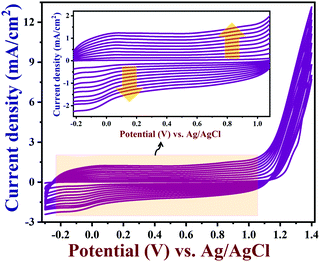 |
| | Fig. 1 The electrochemical polymerization of EDOT on GR/GCE from acetonitrile containing 0.01 M EDOT and 0.1 M lithium perchlorate at a scan rate of 0.05 V s−1 (10 cycles) (inset: enlarged view). | |
3.2 HR-TEM, FE-SEM and Raman characterizations
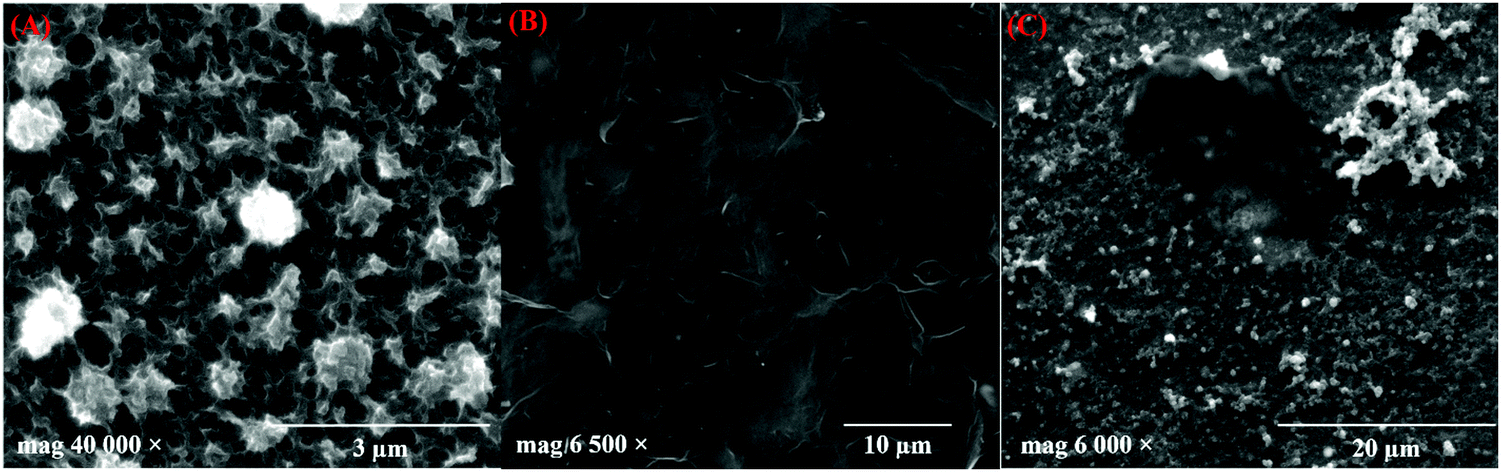
TEM images of the prepared GR are shown in Fig. 2A, which reveals the presence of graphene sheets with a length of ∼1.8 μm. The number of layers in GR was observed by the HR-TEM image analysis (Fig. 2B). As can be seen, a thin GR sheet was observed with few layers, and the interlayer spacing was 0.34 nm, which is in agreement with the lattice distance of graphitic carbon (002).58 In addition, the selected area electron diffraction (SAED) patterns of the prepared GR exhibited a six-fold diffraction pattern, which is relevant to the lattice orientation of GR (Fig. 2B, inset).59 Furthermore, Fig. 2C shows the TEM images of PEDOT/GR, which confirm the successful deposition of PEDOT on the GR sheets. The surface morphology changes on the electrode surface after deposition of PEDOT and PEDOT/GR were analyzed using FE-SEM. Fig. 3A depicts the surface morphology of the PEDOT, which indicated the formation of a unique 3D porous network structure. After the coating of GR on the electrode surface, it was found that the GR film uniformly covered the electrode surface (Fig. 3B). Interestingly, after the electro-deposition of PEDOT on the GR-modified electrode, a 3D porous network was clearly observed on the GR surface (Fig. 3C). In addition, PEDOT/GR exhibited the elemental composition of C (38.97%), O (46.8%) and S (14.23%) by energy-dispersive X-ray spectroscopy (EDS). It was confirmed that a hybrid film composed of PEDOT and GR was obtained. The EDS spectra of the GR, PEDOT and PEDOT/GR are provided in Fig. S4 (ESI†).
 |
| | Fig. 2 (A) TEM image of GR and (B) HR-TEM image of GR with corresponding lattice fringes (inset shows the SAED pattern). (C) TEM image of the PEDOT/GR surface. | |
 |
| | Fig. 3 FE-SEM images of (A) PEDOT, (B) GR film and (C) PEDOT/GR film. | |
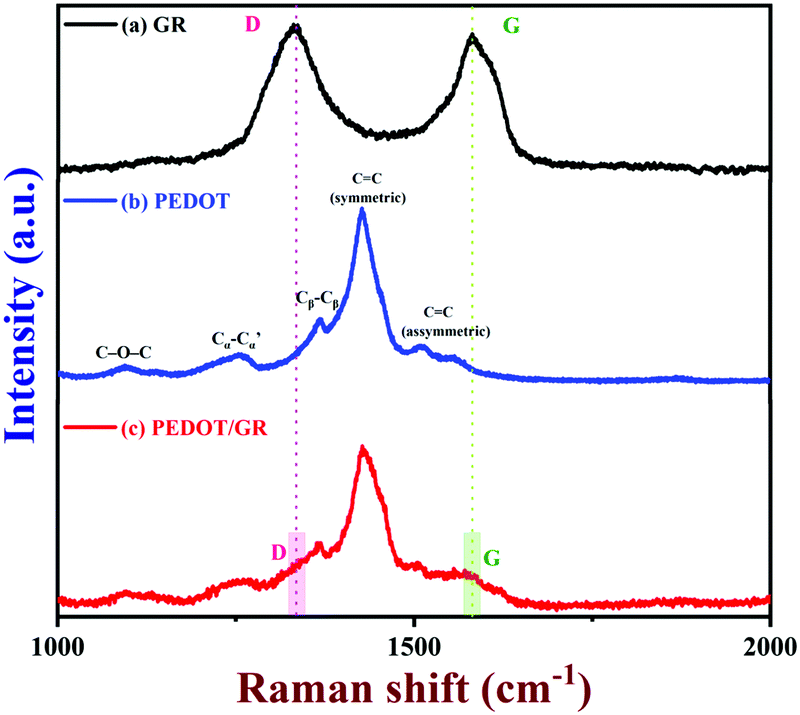
Next, the Raman spectrum of GR was recorded (Fig. 4, curve a). The GR exhibited both D and G bands at 1334 and 1581 cm−1, respectively.60 Also, the ID/IG ratio of the GR was found to be 1.07, which indicated that GR was partially oxidized.37 Furthermore, the Raman spectra of the PEDOT and PEDOT/GR composite were recorded; they showed similar bands corresponding to PEDOT and GR, which confirmed the formation of the PEDOT/GR nanocomposite (Fig. 4, curves b and c). The Raman bands at 1427 cm−1 and 1507 cm−1 indicated the presence of symmetric and asymmetric stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (curves b and c).61–63 In addition, the other Raman bands observed at 1367, 1259, 1095 and 985 cm−1 denoted the corresponding Cβ–Cβ stretching, Cα–Cα′ inter-ring stretching, C–O–C deformation and oxyethylene ring deformation of PEDOT (curves b and c).64 However, it is worth mentioning that the D and G bands of GR were not seen clearly in the Raman spectrum of PEDOT/GR, which was due to the overwhelming Raman bands of the PEDOT film present on GR. However, Raman bands at 1339 and 1585 cm−1 were present in the spectrum of the hybrid film, which indicated that the high-porosity GR sheets were embedded in the PEDOT film (Fig. 4, curve c).44
C (curves b and c).61–63 In addition, the other Raman bands observed at 1367, 1259, 1095 and 985 cm−1 denoted the corresponding Cβ–Cβ stretching, Cα–Cα′ inter-ring stretching, C–O–C deformation and oxyethylene ring deformation of PEDOT (curves b and c).64 However, it is worth mentioning that the D and G bands of GR were not seen clearly in the Raman spectrum of PEDOT/GR, which was due to the overwhelming Raman bands of the PEDOT film present on GR. However, Raman bands at 1339 and 1585 cm−1 were present in the spectrum of the hybrid film, which indicated that the high-porosity GR sheets were embedded in the PEDOT film (Fig. 4, curve c).44
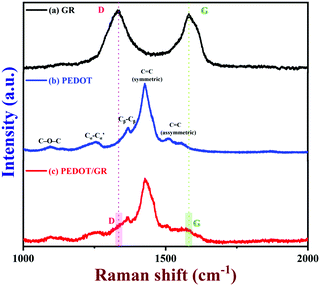 |
| | Fig. 4 Raman spectra of (a) GR, (b) PEDOT and (c) PEDOT/GR film (a 633 nm excitation laser was used). | |
3.3 Electrochemical characterization of PEDOT/GR/GCE
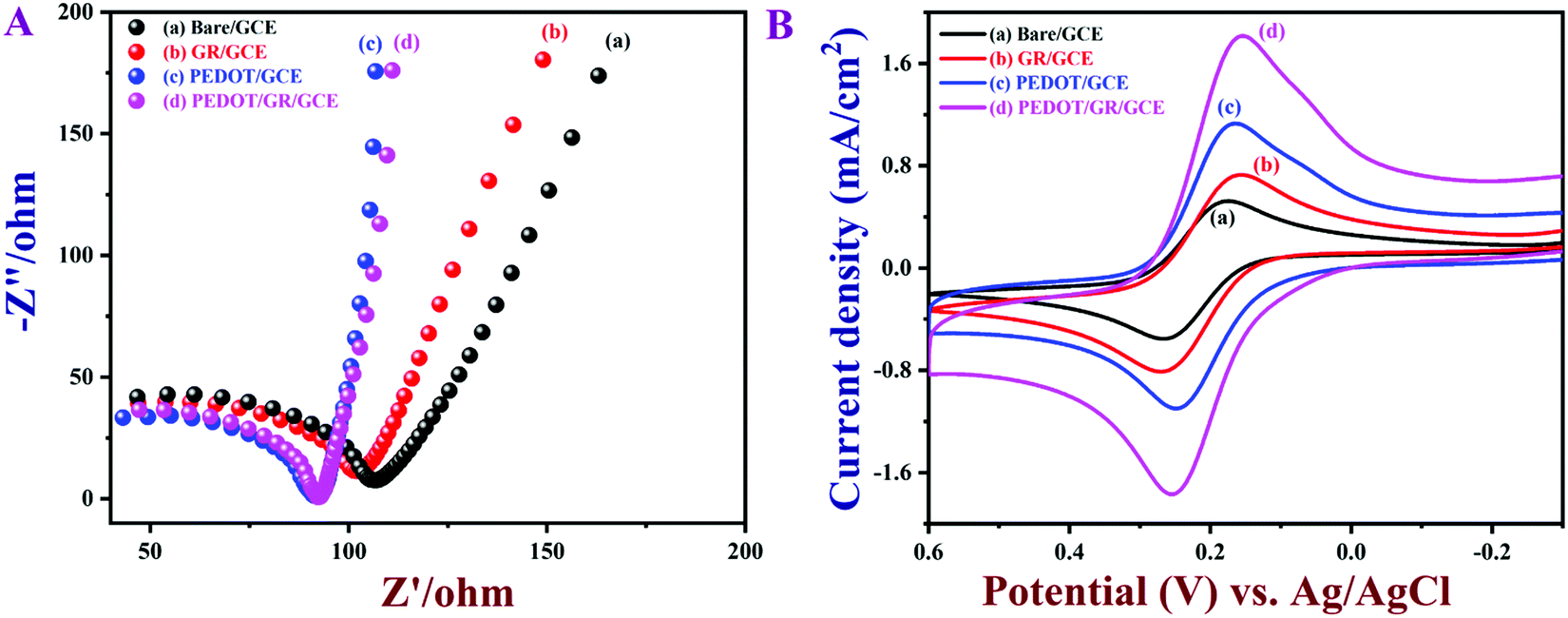
The electrochemical properties of PEDOT/GR/GCE were investigated by EIS and CV in 0.1 M KCl containing 5 mM [Fe(CN)6]3−/4−. EIS is one of the most efficient techniques to investigate the interfacial electron transfer resistance (Rct) of electrochemical sensors.65 The Nyquist plots of bare/GCE, GR/GCE, PEDOT/GCE and PEDOT/GR/GCE were recorded (Fig. 5A). For bare/GCE, GR/GCE and PEDOT/GCE, Rct values of 70, 63 and 50 Ω were obtained, respectively. However, after the deposition of PEDOT on the GR/GCE surface, the Rct decreased to 41 Ω, which clearly indicated that the PEDOT improved the conductivity of the modified electrode (PEDOT/GR/GCE). In addition, the lowest Rct value indicated the higher electron transfer efficiency of the electrode.65
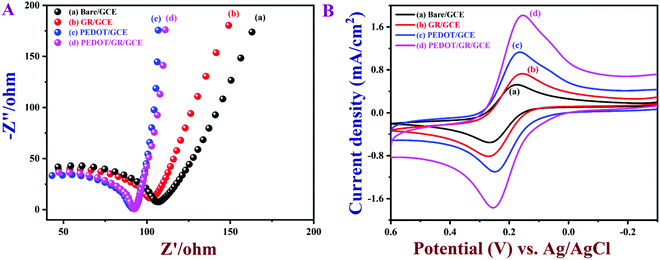 |
| | Fig. 5 (A) Nyquist plots and (B) CVs of bare/GCE, GR/GCE, PEDOT/GCE and PEDOT/GR/GCE in 0.1 M KCl containing 5 mM [Fe(CN)6]3−/4−. Scan rate = 0.02 V s−1. | |
Fig. 5B shows CVs of the [Fe(CN)6]3−/4− on various modified electrodes recorded in 0.1 M KCl containing 5 mM [Fe(CN)6]3−/4− at a scan rate of 0.02 V s−1. It was obvious that PEDOT/GR/GCE showed a much higher redox peak current (1.81 mA cm−2) with a lower peak separation (ΔEp) of 99 mV compared to all the other electrodes, which suggested a quasi-reversible process.66 For comparison, ΔEp values were calculated from the CVs of [Fe(CN)6]3−/4− recorded using bare/GCE (97 mV), GR/GCE (112 mV) and PEDOT/GCE (82 mV). These electrodes showed lower redox peak currents compared to PEDOT/GR/GCE, which indicated that the rate of the electron transfer process has been increased due to the synergistic effect between PEDOT and GR. Graphical representations of the different modified electrodes and their ΔEp and Rct values are shown in Fig. S5 (ESI†).
The electrochemically active surface areas of the modified electrodes were evaluated by CVs recorded in 0.1 M KCl containing [Fe(CN)6]3−/4− at different scan rates from 10 to 100 mV s−1. The CVs of PEDOT/GR/GCE showed a linear response between the redox peak currents and the square roots of the scan rate (ν1/2) (Fig. S6A and B, ESI†). Using the Randles–Sevcik eqn (1), the electro-active surface areas (A) of the modified electrodes were calculated.67
| | 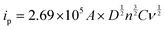 | (1) |
where
ip is the peak current (
A) and
n is the number of electrons involved in the redox reaction.
D,
C and
A have their usual meanings. From the slope value (Fig. S6B, ESI
†), the electro-active surface area of PEDOT/GR/GCE was determined as 0.34 cm
2, which was higher than the bare/GCE (0.07 cm
2), GR/GCE (0.09 cm
2) and PEDOT/GCE (0.22 cm
2). Subsequently, the rates of charge transfer (
Ks) of bare/GCE, GR/GCE, PEDOT/GCE and PEDOT/GR/GCE were calculated using the following
eqn (2).
68,69| | 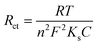 | (2) |
where
Ks is the rate of charge transfer,
n is the number of electrons,
C is the concentration of [Fe(CN)
6]
3−/4− and
T,
F, and
R have their usual meanings. The
Ks value of PEDOT/GR/GCE was calculated as 1.29 × 10
−6 cm s
−1, which indicated the higher catalytic activity of PEDOT/GR/GCE compared to that of bare/GCE (7.6 × 10
−7 cm s
−1), GR/GCE (8.45 × 10
−7 cm s
−1) and PEDOT/GCE (1.06 × 10
−6 cm s
−1). Finally, it was found that PEDOT/GR/GCE exhibited a higher electro-active surface area, higher current density, lower Δ
Ep and higher charge transfer rate compared to PEDOT/GCE and GR/GCE.
3.4 Electrochemical oxidation of nicotine
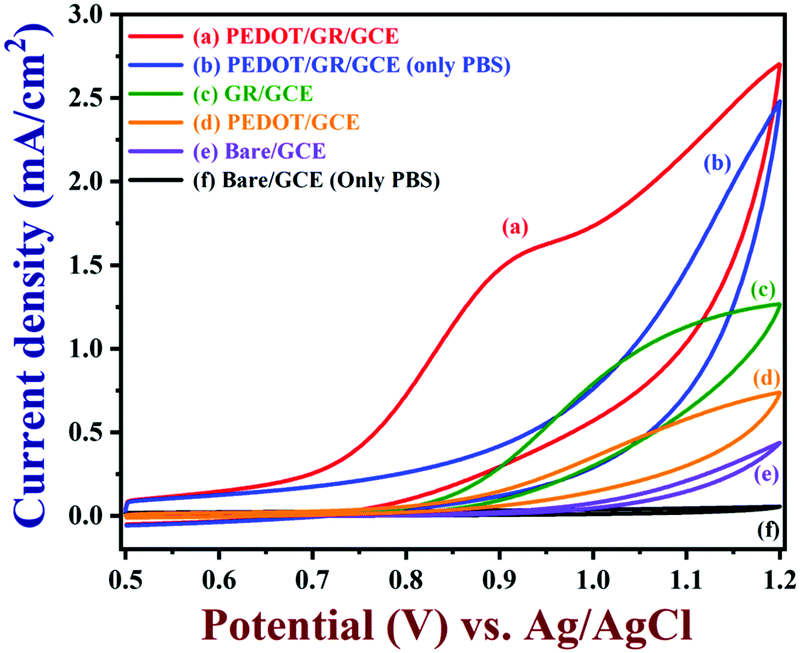
CVs were recorded to evaluate the electro-catalytic activity of PEDOT/GR-modified GCE in the presence of 100 μM nicotine in 0.1 M PBS (pH 7) (Fig. 6, curve a). A highly enhanced oxidation peak was observed at +0.91 V for nicotine due to the efficient electro-catalytic oxidation on PEDOT/GR/GCE. At the same time, bare/GCE (curve e), PEDOT/GCE (curve d) and GR/GCE (curve c) were utilized to investigate the oxidation of 100 μM nicotine at a scan rate of 0.02 V s−1 (Fig. 6). In the absence of nicotine, no irreversible oxidation peaks were observed in 0.1 M PBS (Fig. 6, curves f and b) on bare/GCE and PEDOT/GR/GCE. However, after the addition of 100 μM nicotine to 0.1 M PBS, bare/GCE showed an oxidation peak at +1.12 V (Fig. 6, curve e) with a low peak current of 0.248 mA cm−2. This indicated that the bare/GCE required a high voltage for nicotine oxidation. Interestingly, both GR/GCE and PEDOT/GCE showed enhanced nicotine oxidation peak currents at lower potentials of +1.11 V and +1.04 V, respectively (Fig. 6, curves c and d). For comparison, a graph was plotted for the modified electrodes and their respective nicotine oxidation potentials and catalytic currents (Fig. S7, ESI†). Evidently, the PEDOT/GR-based sensor showed a high oxidation current (1.51 mA cm−2) for nicotine at lower potential (+0.91 V) (Fig. 6, curve a and Fig. S7, ESI†). This indicated the potential use of PEDOT/GR for the electro-catalytic oxidation of nicotine. The enhanced electro-catalytic activity may arise due to the synergistic effect between the negatively charged GR and positively charged PEDOT.
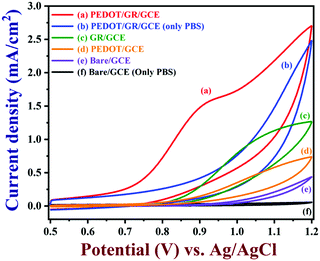 |
| | Fig. 6 CVs of PEDOT/GR/GCE (a), GR/GCE (c), PEDOT/GCE (d) and bare/GCE (e) recorded in 0.1 M PBS (7 pH) with 100 μM nicotine. CVs of PEDOT/GR/GCE (b) and bare/GCE (f) are shown in 0.1 M PBS (7 pH) without nicotine (scan rate = 0.02 V s−1). | |
3.5 The effect of scan rate on nicotine oxidation
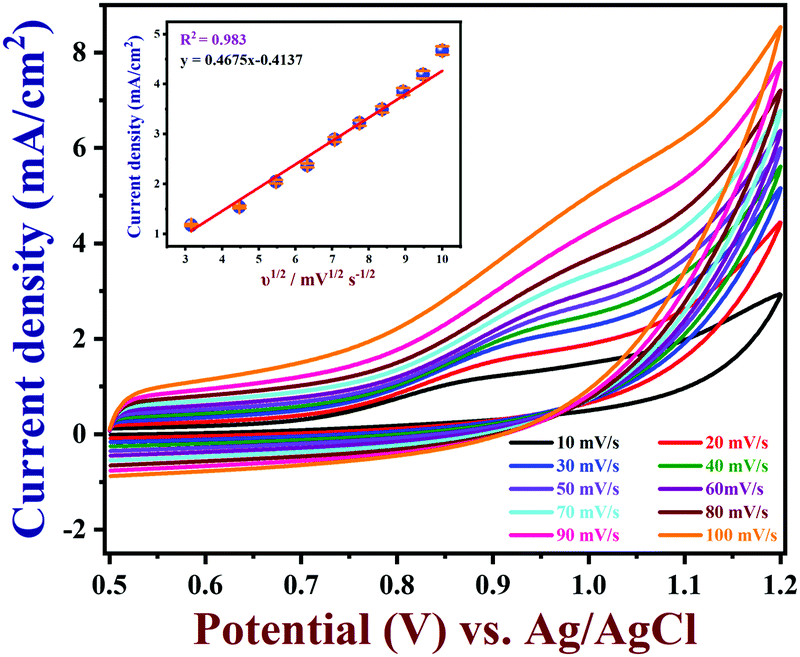
The effects of different scan rates on the oxidation of 100 μM nicotine were studied using PEDOT/GR/GCE in 0.1 M PBS (Fig. 7). The obtained CVs indicated that the nicotine oxidation peak currents linearly increased with the square root of the scan rate from 10 to 100 mV s−1. In addition, the oxidation peak potentials gradually shifted to a high positive potential, which indicated the kinetic limitation of the reaction.70 The relationship between the nicotine oxidation current and square root of the scan rate (inset of Fig. 7) displayed a linear equation of y = 0.4675x − 0.4137 with a correlation coefficient (r2) of 0.983. These results suggested that nicotine oxidation on PEDOT/GR/GCE was a diffusion-controlled process.71,72 Next, a graph was plotted between the natural logarithm of the scan rate (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ν) and the oxidation potential (Epa) of nicotine, which showed a linearity (y = 0.0298x + 1.0340) (Fig. S8, ESI†). Using the relationship between Epa and lnν, the number of electrons involved in the irreversible oxidation of nicotine can be determined using the following Laviron eqn (3).73
ν) and the oxidation potential (Epa) of nicotine, which showed a linearity (y = 0.0298x + 1.0340) (Fig. S8, ESI†). Using the relationship between Epa and lnν, the number of electrons involved in the irreversible oxidation of nicotine can be determined using the following Laviron eqn (3).73| | 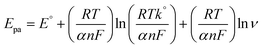 | (3) |
where α is the co-efficient of charge transfer, n is the number of electrons involved in the reaction, R is the gas constant, T is the temperature and F is the Faraday constant. Here, the ‘αn’ was calculated as 0.86. Because the nicotine oxidation is an irreversible process, the charge transfer coefficient was assumed as 0.5. Thus, ‘n’ was calculated as 1.72, which revealed that during the nicotine oxidation, two electrons were involved in the electro-oxidation process on PEDOT/GR/GCE. The electro-oxidation of nicotine occurred due to the hydroxylation on the n-methyl group (Scheme 2).74,75
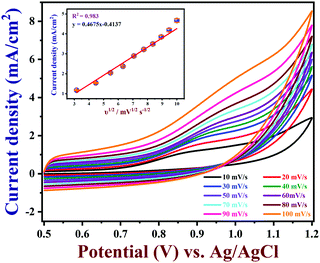 |
| | Fig. 7 Effect of scan rate on 100 μM nicotine oxidation on PEDOT/GR/GCE (inset: the linear relationship between the oxidation peak current and square root of the scan rate (n = 3)). | |
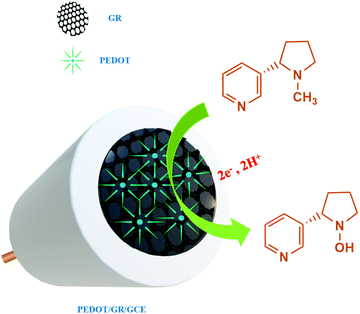 |
| | Scheme 2 Electrochemical oxidation mechanism of nicotine on PEDOT/GR/GCE. | |
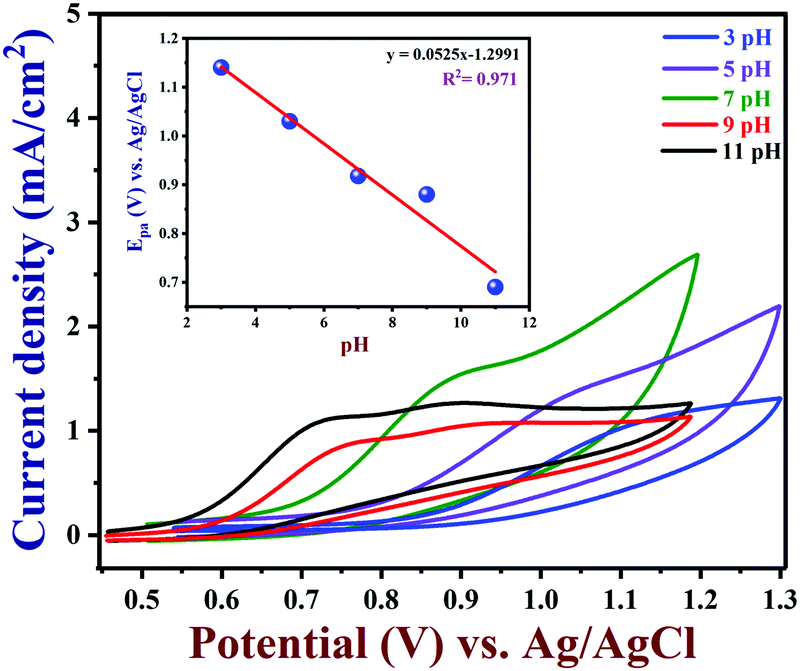
3.6 Effect of pH on nicotine oxidation
The electrochemical activity of PEDOT/GR/GCE was tested in different pH solutions. CVs were recorded in various pH buffer solutions (from 3 to 11) with 100 μM nicotine on PEDOT/GR/GCE. The corresponding nicotine oxidation peak potentials and peak currents were recorded at different pH values (pH 3 to 11), as shown in Fig. 8. When the pH was increased from 3 to 11, the nicotine oxidation potential shifted to the less positive side due to the involvement of the same number of protons and electrons. Furthermore, a non-linear graph was plotted between the pH and nicotine oxidation peak currents (Fig. S9, ESI†). The maximum nicotine oxidation peak current was observed at pH 7. It should be mentioned that at pH 9, the overpotential of nicotine decreased, and at the same time, the oxidation peak current decreased. Therefore, pH 7 was selected as the optimum pH for further experiments. A linear graph was drawn from the plot of different pH values vs. the oxidation potential of nicotine as y = 0.0525x − 1.2991 (x = pH; y = V) with r2 of 0.971 (inset of Fig. 8). The scientific relationship between pH and Epa can be expressed by the following Nernst eqn (4).76| | 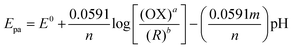 | (4) |
where ‘m’ and ‘n’ represent the number of protons and electrons involved in the electrochemical reaction and ‘a’ and ‘b’ are the oxidant and reductant coefficients in the reaction. In this case, the calculated slope value was 0.0525 V, which indicated that the same number of protons and electrons were involved in the oxidation of nicotine (Scheme 2).
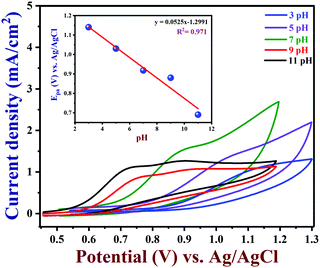 |
| | Fig. 8 Effect of pH on the oxidation of 100 μM nicotine in 0.1 M PBS with various pH (from 3 to 11) on a PEDOT/GR/GCE at a scan rate of 0.02 V s−1 (inset: a linear plot of the Epa of nicotine oxidation vs. pH). | |
3.7 Optimization of GR loading and EDOT polymerization
The electrochemical activity of the modified electrode can be affected by the amount of GR present in the composite. To study the effect of GR concentration, GR dispersions were prepared with different concentrations (from 1 to 9 mg mL−1). From the each concentrations, 6 μL of GR dispersion was taken and coated on the GCE surface for the electro-oxidation of nicotine (100 μM) in 0.1 M PBS (pH 7). A graph was plotted between the nicotine oxidation peak currents and concentration of GR present on the GCE, as shown in Fig. S10 (ESI†). As can be seen, the oxidation peak currents of nicotine gradually increased with increasing GR concentration on GCE from 1 to 5 mg mL−1, and they started to decrease (from 6 to 9 mg mL−1) after that. This may be due to saturated GR film formation on the electrode surface, which may inhibit the interaction between nicotine and the electrode.77,78 From this study, 6 μL of 5 mg mL−1 GR concentration was found to be the optimum value, and it was used to prepare the sensor for the detection of nicotine.
The PEDOT thickness on the GR/GCE surface may also influence the nicotine oxidation peak current. To determine the optimum PEDOT thickness, PEDOT polymer film was deposited on GR/GCE by varying the electro-polymerization cycles (from 3 to 20 cycles). As-prepared PEDOT/GR/GCE electrodes with varied PEDOT thicknesses were used to oxidize 100 μM nicotine in 0.1 M PBS (pH 7). A graph was plotted between the nicotine oxidation peak currents and the number of EDOT polymerization cycles (Fig. S11, ESI†). Initially, the nicotine oxidation peak currents increased with the thickness of the PEDOT films. However, after the 10th cycle, the nicotine oxidation peak current started to decrease. Hence, the PEDOT film deposited with 10 cycles was used to prepare the sensor for the detection of nicotine.
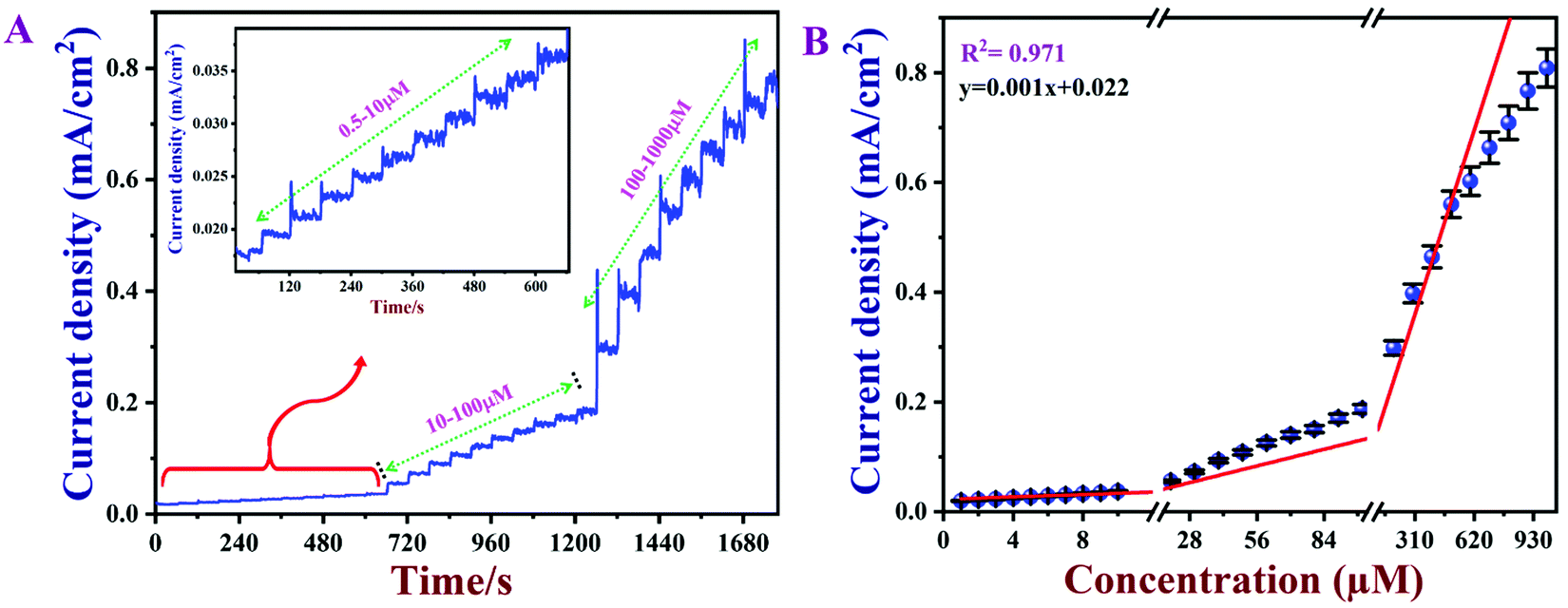
3.8 Calibration graph and limit of detection (LOD)
Amperometry can be used to prepare calibration graphs because it offers high sensitivity, good reproducibility and a higher limit of detection (LOD).4Fig. 9A shows the amperograms recorded with different nicotine concentrations (each addition made after every 60 s) in 0.1 M PBS (pH 7) at an applied potential of +0.91 V. Moreover, the standard deviations (SD) and error bars were estimated by three replicate measurements. Fig. 9B displays the calibration graph of nicotine, which was made by an amperometry curve recorded using different concentrations of nicotine and the corresponding oxidation currents. The oxidation currents increased linearly with additions of various concentrations of nicotine from 0.5 to 1000 μM. The linear equation was obtained as y = 0.001x + 0.022 with r2 of 0.971. The PEDOT/GR/GCE sensor response time was ∼2 s for nicotine. The LOD was calculated using the following eqn (5).| | 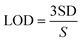 | (5) |
where SD is the standard deviation of the blank (1.52 × 10−5) and S is the slope of the calibration graph (1.09 × 10−3). The calculated LOD was listed with the other reported electrochemical sensors for nicotine in Table S1 (ESI†).1,4,17,18,74,79–86 This new sensor showed a lower LOD, high selectivity and wide linear range of detection for nicotine due to the π–π interaction of the PEDOT/GR with the nicotine.87 In addition, the higher electro-active surface area of the PEDOT/GR may have provided a 3D porous network with more active sites towards favorable nicotine oxidation.
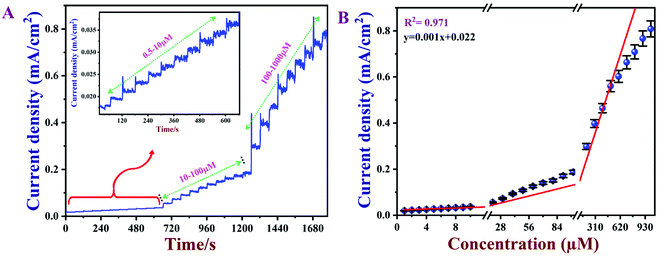 |
| | Fig. 9 (A) Amperometric response of PEDOT/GR/GCE for consecutive additions of nicotine (each 60 s) from 0.5–1000 μM in 0.1 M PBS (pH 7) at an applied potential of +0.91 V. The electrolyte was stirred at 1000 rpm. (B) Calibration plot between the oxidation peak currents vs. the concentration of nicotine (n = 3). | |
3.9 Real sample analysis
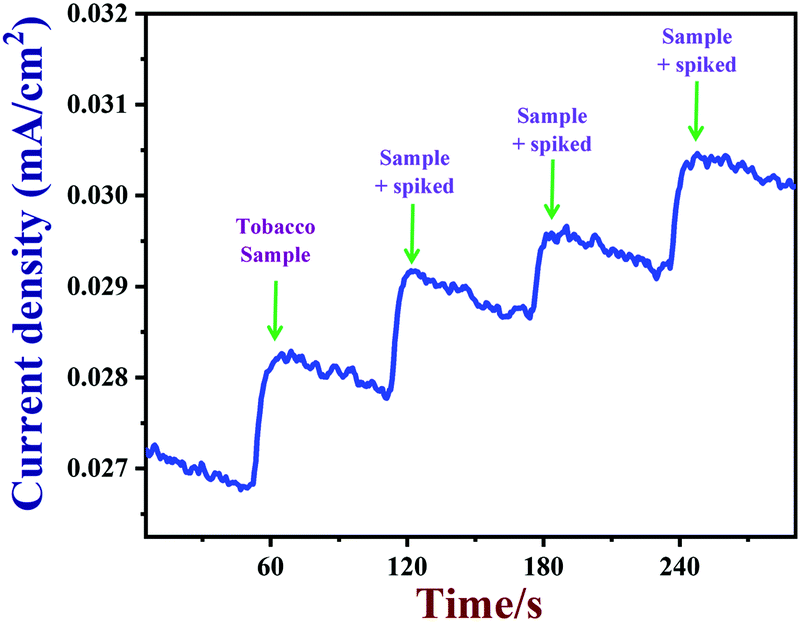
Next, PEDOT/GR/GCE was employed to detect nicotine levels in tobacco samples. The amperograms were recorded using a PEDOT/GR/GCE as a working electrode in 0.1 M PBS (pH 7) with subsequent additions of tobacco samples (see the Experimental section for the preparation of the tobacco samples) (Fig. 10). In addition, standard nicotine solutions (1 to 3 μM) were spiked into 0.1 M PBS with tobacco samples, and the concentrations were determined by the same PEDOT/GR/GCE sensor. Finally, the nicotine concentrations in tobacco samples and the spiked nicotine concentrations were calculated by the standard addition method.88,89 The nicotine recovery values were in the range between 99.3% and 102.6% (Table 1). This indicated that our proposed PEDOT/GR/GCE-based sensor may be a valuable transducer for the accurate detection of nicotine content in tobacco samples.
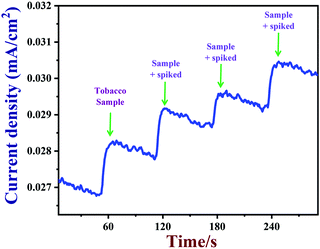 |
| | Fig. 10 Amperometric response of PEDOT/GR/GCE in 0.1 M PBS with successive addition of tobacco samples (real samples) containing different concentrations of nicotine at an applied potential of +0.91 V. The solution was stirred at a rate of 1000 rpm. | |
Table 1 Detection of nicotine in tobacco samples before and after spiking of nicotine using PEDOT/GR/GCE as a transducer
| S. no. |
Sample |
Added (μM) |
Found (μM) |
RSD%a |
Recovery % |
|
Mean value of triplicate results.
|
| 1 |
Tobacco sample |
— |
4.90 ± 0.005 |
2.45 |
— |
| 2 |
Tobacco sample + spiked |
1 |
5.99 ± 0.004 |
2.23 |
101.6 |
| 3 |
Tobacco sample + spiked |
2 |
6.85 ± 0.001 |
1.00 |
99.3 |
| 4 |
Tobacco sample + spiked |
3 |
8.13 ± 0.003 |
2.77 |
102.6 |
3.9.1 Reproducibility, stability and selectivity.
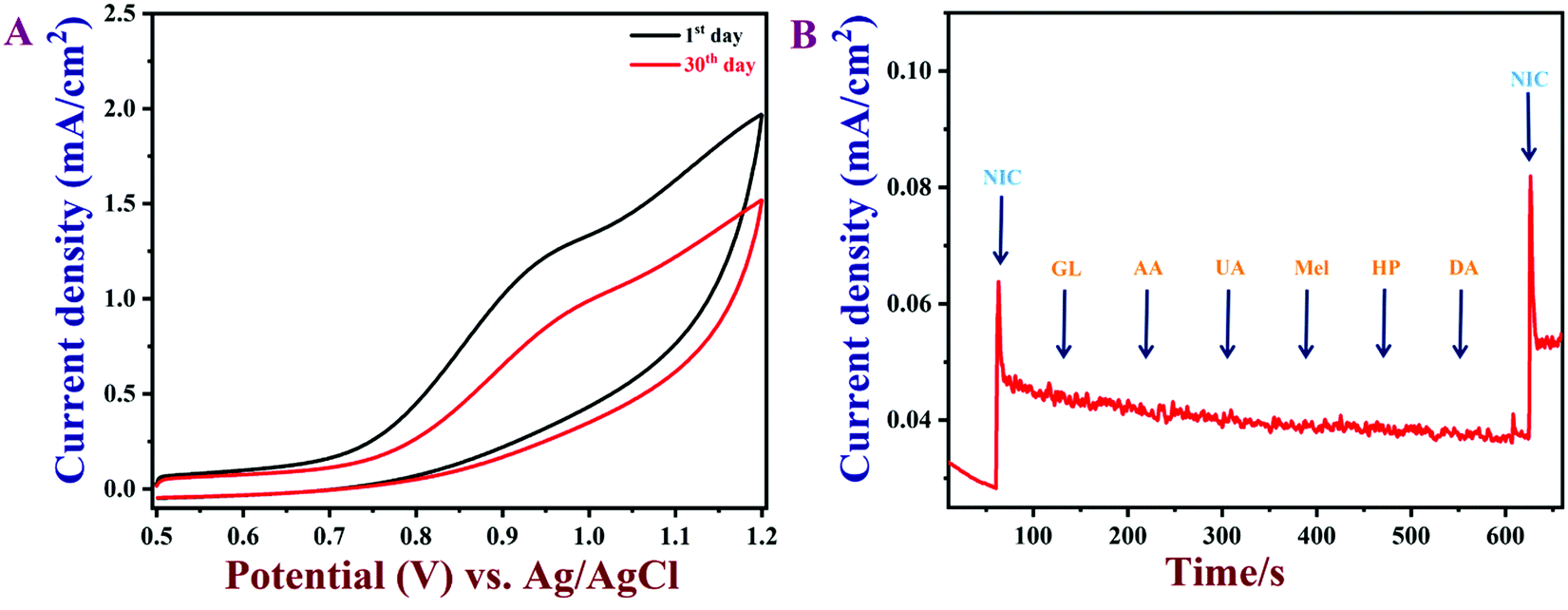
The reproducibility of the PEDOT/GR/GCE was also examined. It was found that the as-prepared PEDOT/GR-modified GCE could be reused after rinsing with PBS. Five individually prepared PEDOT/GR/GCE samples were tested against nicotine oxidation (50 μM) and showed average current responses of 96.6% with a relative standard deviation, RSD, of 2.7% (Fig. S12, ESI†). Next, the reusability of the PEDOT/GR-modified electrode was carried out by detecting nicotine (100 μM) at given regular time intervals in 0.1 M PBS (pH 7) (Fig. 11A). After each measurement, the PEDOT/GR/GCE was stored at 4 °C. After storage for 30 days, the PEDOT/GR/GCE sensor retained about 77% of its electro-activity, which suggested that the proposed sensor was highly stable and could be used for repeated detection of nicotine.
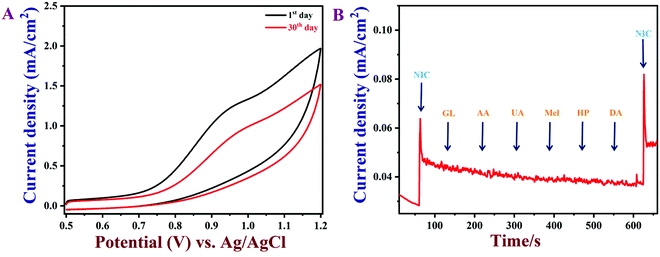 |
| | Fig. 11 (A) CVs of PEDOT/GR/GCE in 0.1 M PBS (pH 7) containing 100 μM nicotine with different storage times. (B) The amperometric response of PEDOT/GR/GCE with the additions of various interfering molecules (GL, AA, UA, Mel, HP, DA; concentration of each compound was 30 μM) with 5 μM of nicotine at an applied potential of +0.91 V. The PBS was stirred at 1000 rpm. | |
To ascertain the selectivity of the sensor, amperograms were recorded in 0.1 M PBS (pH 7) with 5 μM nicotine and subsequent additions of common interfering molecules, such as anhydrous dextrose (GL), ascorbic acid (AA), uric acid (UA), melamine (MA), hydrogen peroxide (HP) and dopamine (DA) (the concentration of each compound was 30 μM). These molecules were selected for the selectivity study because of their widespread availability in human body fluids and environmental samples.18,90 Furthermore, the interferences due to other alkaloids that exist in tobacco were not tested because of their lower content (0.2–0.5%), which may not affect the oxidation of nicotine.18,85,91,92 As shown in Fig. 11B, the nicotine oxidation peak current was stable even after the addition of the interfering biomolecules. This result indicated that these molecules did not produce any current signals and that the PEDOT/GR composite showed high selectivity for nicotine over these molecules. The above studies indicated that PEDOT/GR/GCE can be used to detect nicotine with high selectivity.
4. Conclusion
In summary, GR was prepared by a simple electrochemical exfoliation method using graphite sheets. The as-prepared GR was characterized by HR-TEM to confirm its layered structure. Furthermore, PEDOT was electro-deposited on a GR/GCE surface, and the PEDOT/GR hybrid film formation was confirmed by Raman spectroscopy. In addition, the surface morphology of the hybrid film was investigated by FE-SEM. The as-prepared PEDOT/GR/GCE showed enhanced electrochemical activity and excellent electro-catalytic activity for nicotine oxidation. PEDOT/GR/GCE linearly responded to nicotine from 0.5–1000 μM, and the LOD was found to be 47 nM. In addition, the PEDOT/GR-based sensor showed several advantages, such as high reproducibility, high selectivity, rapid response, and high stability, when used for the detection of nicotine. Finally, PEDOT/GR/GCE was successfully used to detect nicotine concentrations in tobacco samples with high recoveries. We believe that our proposed PEDOT/GR/GCE sensor can be used to detect nicotine with accuracy in real-world samples.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Department of Science & Technology (International Bilateral Cooperation Division) for financial support through the “INDO-RUSSIA Project (File No.: INT/RUS/RFBR/385)”. J. R thanks SRM-IST for the PhD student fellowship. We acknowledge the HRTEM FACILITY at SRMIST which was supported by the project from MNRE (Project No. 31/03/2014-15/PVSE-R&D), Government of India. We also acknowledge the SRM Institute of Science and Technology for providing “micro-Raman facility”.
References
- A. Levent, Y. Yardim and Z. Senturk, Electrochim. Acta, 2009, 55, 190–195 CrossRef CAS.
- B. Lin, J. Chen, Y. Zeng, L. Li, B. Qiu, Z. Lin and L. Guo, ACS Sens., 2019, 4, 1844–1850 CrossRef CAS PubMed.
- A. L. A. M. Stolker, W. Niesing, E. A. Hogendoorn, A. Bisoen Rambali and W. Vleeming, J. Chromatogr. A, 2003, 1020, 35–43 CrossRef CAS PubMed.
- R. Jerome and A. K. Sundramoorthy, Anal. Chim. Acta, 2020, 1132, 110–120 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- P. A. Newhouse, A. Potter, M. Kelton and J. Corwin, Biol. Psychiatry, 2001, 49, 268–278 CrossRef CAS PubMed.
- G. K. Lloyd and M. Williams, J. Pharmacol. Exp. Ther., 2000, 292, 461–467 CAS.
- J. I. Seeman, J. A. Fournier, J. B. Paine and B. E. Waymack, J. Agric. Food Chem., 1999, 47, 5133–5145 CrossRef CAS PubMed.
- F. Moriya, Y. Hashimoto and J. Furumiya, Forensic Sci. Int., 2007, 168, 102–105 CrossRef CAS PubMed.
- D.-S. Tang, L. Zhang, H.-L. Chen, Y.-R. Liang, J.-L. Lu, H.-L. Liang and X.-Q. Zheng, Sep. Purif. Technol., 2007, 56, 291–295 CrossRef CAS.
- M. C. Acosta, A. R. Buchhalter, A. B. Breland, D. C. P. Hamilton and T. Eissenberg, Nicotine Tob. Res., 2004, 6, 615–620 CrossRef CAS PubMed.
- M. Page-Sharp, T. W. Hale, L. P. Hackett, J. H. Kristensen and K. F. Ilett, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 796, 173–180 CrossRef CAS PubMed.
- A. W. Abu-Qare and M. B. Abou-Donia, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 757, 295–300 CrossRef CAS.
- J. J. Langone, H. B. Gjika and H. Van Vunakis, Biochemistry, 1973, 12, 5025–5030 CrossRef CAS PubMed.
- M. S. Lin, J. S. Wang and C. H. Lai, Electrochim. Acta, 2008, 53, 7775–7780 CrossRef CAS.
- A. M. Hossain and S. M. Salehuddin, Arabian J. Chem., 2013, 6, 275–278 CrossRef CAS.
- M. Stočes and I. Švancara, Electroanalysis, 2014, 26, 2655–2663 CrossRef.
- H. Kassa, A. Geto and S. Admassie, Bull. Chem. Soc. Ethiop., 2013, 27, 321–328 Search PubMed.
- M. R. Miah, J. Masud and T. Ohsaka, Electrochim. Acta, 2008, 54, 316–321 CrossRef CAS.
- T. F. Manny, R. Miah, F. Islam, D. Sen and R. Mahmud, Russ. J. Electrochem., 2020, 56, 570–577 CrossRef CAS.
- M. R. Miah, M. T. Alam and T. Ohsaka, Anal. Chim. Acta, 2010, 669, 75–80 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
-
A. K. Sundramoorthy, T. H. Vignesh Kumar and S. Gunasekaran, in Graphene Bioelectronics, Advanced Nanomaterials, ed. A. B. T.-G. B.Tiwari, Elsevier, 2018, pp. 267–306 Search PubMed.
- A. K. Sundramoorthy and S. Gunasekaran, TrAC,
Trends Anal. Chem., 2014, 60, 36–53 CrossRef CAS.
- R. D. Nagarajan and A. K. Sundramoorthy, Sens. Actuators, B, 2019, 301, 127132 CrossRef CAS.
- N. Murugan, A. Sundaramurthy, S.-M. Chen and A. K. Sundramoorthy, Mater. Res. Express, 2017, 4, 124005 CrossRef.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- K. Nakada, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- T. H. V. Kumar, S. K. Yadav and A. K. Sundramoorthy, J. Electrochem. Soc., 2018, 165, B848–B861 CrossRef.
-
Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun’Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 Search PubMed.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O’Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O’Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624 CrossRef CAS PubMed.
- R. Sakamoto, K. Hoshiko, Q. Liu, T. Yagi, T. Nagayama, S. Kusaka, M. Tsuchiya, Y. Kitagawa, W.-Y. Wong and H. Nishihara, Nat. Commun., 2015, 6, 6713 CrossRef CAS PubMed.
- Y. Wang, L. Li, L. Yan, X. Gu, P. Dai, D. Liu, J. G. Bell, G. Zhao, X. Zhao and K. M. Thomas, Chem. Mater., 2018, 30, 3048–3059 CrossRef CAS.
- J. Rajendran, A. N. Reshetilov and A. K. Sundramoorthy, RSC Adv., 2021, 11, 3445–3451 RSC.
- A. K. Sundramoorthy, Y. Wang, J. Wang, J. Che, Y. X. Thong, A. C. W. Lu and M. B. Chan-Park, Sci. Rep., 2015, 5, 1–13 CrossRef PubMed.
- N. G. Yasri, A. K. Sundramoorthy, W.-J. Chang and S. Gunasekaran, Front. Mater., 2014, 1, 33 CrossRef.
- P. Garrigue, M.-H. Delville, C. Labrugère, E. Cloutet, P. J. Kulesza, J. P. Morand and A. Kuhn, Chem. Mater., 2004, 16, 2984–2986 CrossRef CAS.
- H. Ba, L. Truong-Phuoc, C. Pham-Huu, W. Luo, W. Baaziz, T. Romero and I. Janowska, ACS Omega, 2017, 2, 8610–8617 CrossRef CAS PubMed.
- F. Parnianchi, M. Nazari, J. Maleki and M. Mohebi, Int. Nano Lett., 2018, 8, 229–239 CrossRef CAS.
- X. Luo, C. L. Weaver, S. Tan and X. T. Cui, J. Mater. Chem. B, 2013, 1, 1340–1348 RSC.
- S. Nambiar and J. T. W. Yeow, Biosens. Bioelectron., 2011, 26, 1825–1832 CrossRef CAS PubMed.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- W. Si, W. Lei, Z. Han, Y. Zhang, Q. Hao and M. Xia, Sens. Actuators, B, 2014, 193, 823–829 CrossRef CAS.
- F. Jiang, R. Yue, Y. Du, J. Xu and P. Yang, Biosens. Bioelectron., 2013, 44, 127–131 CrossRef CAS PubMed.
- E. Spain, T. E. Keyes and R. J. Forster, Biosens. Bioelectron., 2013, 41, 65–70 CrossRef CAS PubMed.
- C. Zhu, J. Zhai, D. Wen and S. Dong, J. Mater. Chem., 2012, 22, 6300–6306 RSC.
- S. Ameen, M. S. Akhtar and H. S. Shin, Sens. Actuators, B, 2012, 173, 177–183 CrossRef CAS.
- A. K. Sundramoorthy, B. S. Premkumar and S. Gunasekaran, ACS Sens., 2016, 1, 151–157 CrossRef CAS.
- O. Sadak, W. Wang, J. Guan, A. K. Sundramoorthy and S. Gunasekaran, ACS Appl. Nano Mater., 2019, 2, 4386–4394 CrossRef CAS.
- O. Sadak, A. K. Sundramoorthy and S. Gunasekaran, Carbon, 2018, 138, 108–117 CrossRef CAS.
- Y. Zhu, M. T. Otley, A. Kumar, M. Li, X. Zhang, C. Asemota and G. A. Sotzing, Chem. Commun., 2014, 50, 8167–8170 RSC.
- J.-H. Kang, S.-M. Paek, Y. Bin Choy, S.-J. Hwang and J.-H. Choy, J. Nanosci. Nanotechnol., 2007, 7, 4131–4134 CrossRef CAS PubMed.
- Y. Jing, X. Yuan, Q. Yuan, K. He, Y. Liu, P. Lu, H. Li, B. Li, H. Zhan and G. Li, Sci. Rep., 2016, 6, 29230 CrossRef CAS PubMed.
- A. I. Melato, M. H. Mendonça and L. M. Abrantes, J. Solid State Electrochem., 2009, 13, 417–426 CrossRef CAS.
- J. Heinze, A. Rasche, M. Pagels and B. Geschke, J. Phys. Chem. B, 2007, 111, 989–997 CrossRef CAS PubMed.
- S. Berchmans, Sci. Rep., 2019, 9, 19184 CrossRef PubMed.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- M. Wang, T. Zhang, D. Mao, Y. Yao, X. Zeng, L. Ren, Q. Cai, S. Mateti, L. H. Li, X. Zeng, G. Du, R. Sun, Y. Chen, J.-B. Xu and C.-P. Wong, ACS Nano, 2019, 13, 7402–7409 CrossRef CAS PubMed.
- G. Jiang, Z. Lin, C. Chen, L. Zhu, Q. Chang, N. Wang, W. Wei and H. Tang, Carbon, 2011, 49, 2693–2701 CrossRef CAS.
- M. S. Cho, Y. Y. Yun, J. D. Nam, Y. Son and Y. Lee, Synth. Met., 2008, 158, 1043–1046 CrossRef CAS.
- F. Tran-Van, S. Garreau, G. Louarn, G. Froyer and C. Chevrot, J. Mater. Chem., 2001, 11, 1378–1382 RSC.
- S. Garreau, G. Louarn, J. P. Buisson, G. Froyer and S. Lefrant, Macromolecules, 1999, 32, 6807–6812 CrossRef CAS.
- S. Aeiyach, J. J. Aaron, M. Jouini, K. I. Chane-Ching, J. C. Lacroix, J. Tanguy and P. C. Lacaze, ThinPolyheteroaromatic Films Synthesized in Organized Media: Structures and Properties, ACS Symposium Series, ACS Publications, 2003, 38–51 CAS.
- S. Dhillon and R. Kant, J. Chem. Sci., 2017, 129, 1277–1292 CrossRef CAS.
- M. C. Granger and G. M. Swain, J. Electrochem. Soc., 1999, 146, 4551 CrossRef CAS.
-
A. J. Bard and L. R. Faulkner, Fundamentals and applications, WileyNew York, 2001, vol. 2 Search PubMed.
-
E. Barsoukov and J. R. Macdonald, Impedance spectroscopy: theory, experiment, and applications, John Wiley & Sons, New York, 2005 Search PubMed.
- E. N. Primo, P. Cañete-Rosales, S. Bollo, M. D. Rubianes and G. A. Rivas, Colloids Surf., B, 2013, 108, 329–336 CrossRef CAS PubMed.
- N. Murugan, R. Jerome, M. Preethika, A. Sundaramurthy and A. K. Sundramoorthy, J. Mater. Sci. Technol., 2020, 72, 122–131 CrossRef.
- E. J. Ukpong, I. A. Udoetok and N. W. Akpanudo, J. Appl. Chem., 2013, 5, 50–55 CAS.
- M. Opitz, J. Yue, J. Wallauer, B. Smarsly and B. Roling, Electrochim. Acta, 2015, 168, 125–132 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1974, 52, 355–393 CrossRef CAS.
- X. Li, H. Zhao, L. Shi, X. Zhu, M. Lan, Q. Zhang and Z. H. Fan, J. Electroanal. Chem., 2017, 784, 77–84 CrossRef CAS.
- T. W. B. Lo, L. Aldous and R. G. Compton, Sens. Actuators, B, 2012, 162, 361–368 CrossRef CAS.
- V. Erady, R. J. Mascarenhas, A. K. Satpati, S. Detriche, Z. Mekhalif, J. Delhalle and A. Dhason, Mater. Sci. Eng., C, 2017, 76, 114–122 CrossRef CAS PubMed.
- A. Uzunoglu, A. S. Ahsen, F. Dundar, A. Ata and O. Ozturk, J. Appl. Electrochem., 2017, 47, 139–155 CrossRef CAS.
- Y. Tabe, M. Nishino, H. Takamatsu and T. Chikahisa, J. Electrochem. Soc., 2011, 158, B1246 CrossRef CAS.
- A. Karthika, P. Karuppasamy, S. Selvarajan, A. Suganthi and M. Rajarajan, Ultrason. Sonochem., 2019, 55, 196–206 CrossRef CAS PubMed.
- Y. Jing, B. Yu, P. Li, B. Xiong, Y. Cheng, Y. Li, C. Li, X. Xiao, M. Chen and L. Chen, Sci. Rep., 2017, 7, 14332 CrossRef PubMed.
- L. Highton, R. O. Kadara, N. Jenkinson, B. Logan Riehl and C. E. Banks, Electroanalysis, 2009, 21, 2387–2389 CrossRef CAS.
- M. J. Sims, N. V. Rees, E. J. F. Dickinson and R. G. Compton, Sens. Actuators, B, 2010, 144, 153–158 CrossRef CAS.
- C.-T. Wu, P.-Y. Chen, J.-G. Chen, V. Suryanarayanan and K.-C. Ho, Anal. Chim. Acta, 2009, 633, 119–126 CrossRef CAS PubMed.
- S.-J. Wang, H.-W. Liaw and Y.-C. Tsai, Electrochem. Commun., 2009, 11, 733–735 CrossRef CAS.
- Ľ. Švorc, D. M. Stanković and K. Kalcher, Diamond Relat. Mater., 2014, 42, 1–7 CrossRef.
- A. Geto, M. Amare, M. Tessema and S. Admassie, Electroanalysis, 2012, 24, 659–665 CrossRef CAS.
- Y. Xu, W. Lei, J. Su, J. Hu, X. Yu, T. Zhou, Y. Yang, D. Mandler and Q. Hao, Electrochim. Acta, 2018, 259, 994–1003 CrossRef CAS.
- K. Cinková, L. Dianová, M. Vojs, M. Marton and Ľ. Švorc, Acta Chim. Slovaca, 2015, 8, 166–171 CrossRef.
- K. Liu, W.-Z. Wei, J.-X. Zeng, X.-Y. Liu and Y.-P. Gao, Anal. Bioanal. Chem., 2006, 385, 724–729 CrossRef CAS PubMed.
- E. Mehmeti, T. Kilic, C. Laur and S. Carrara, Microchem. J., 2020, 158, 105155 CrossRef CAS.
- J. M. Garrigues, A. Pérez-Ponce, S. Garrigues and M. de la Guardia, Anal. Chim. Acta, 1998, 373, 63–71 CrossRef CAS.
- H. Xiong, Y. Zhao, P. Liu, X. Zhang and S. Wang, Microchim. Acta, 2010, 168, 31–36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00974a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a