
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PET waste as organic linker source for the sustainable preparation of MOF-derived methane dry reforming catalysts†
Leila
Karam
ab,
Arianna
Miglio‡
c,
Stefania
Specchia
 c,
Nissrine
El Hassan
a,
Pascale
Massiani
b and
Julien
Reboul
*b
c,
Nissrine
El Hassan
a,
Pascale
Massiani
b and
Julien
Reboul
*b
aDepartment of Chemical Engineering, Faculty of Engineering, University of Balamand, P.O. Box 33 Amioun, El Koura, Lebanon
bSorbonne Université, Campus UPMC, CNRS UMR-7197, Laboratoire de Réactivité de Surface, 4 Place Jussieu, 75005 Paris, France. E-mail: julien.reboul@sorbonne-universite.fr
cPolitecnico di Torino, Department of Applied Science and Technology, Corso Duca degli Abruzzi 24, 10129 Torino, Italy
First published on 25th March 2021
Abstract
A catalyst made of Ni0 nanoparticles highly dispersed on a lamellar alumina support was prepared by an environmentally-friendly route. The latter involved the synthesis of an aluminum-containing metal–organic framework (MOF) MIL-53(Al) in which the linkers were derived from the depolymerization of polyethylene terephthalate (PET) originating from plastic wastes. After demonstrating the purity and structure integrity of the PET-derived MIL-53(Al), this MOF was impregnated with nickel nitrate salt and then calcined to form a lamellar Ni–Al2O3 mixed metal oxide with a high surface area (SBET = 1276 m2 g−1, N2 sorption). This mixed oxide consisted of nickel aluminate nanodomains dispersed within amorphous alumina, as revealed by PXRD and TPR analyses. Subsequent reduction under H2 resulted in the formation of well-dispersed 5 nm Ni0 nanoparticles homogeneously occluded within the interlamellar porosity of the γ-alumina matrix, as attested by electron microscopy. This waste-derived catalyst displayed catalytic performances in the reaction of dry reforming of methane (DRM) as good as its counterpart made from a MOF obtained from commercial benzene-1,4-dicarboxylic acid (BDC). Thus, under similar steady state conditions, at 650 °C and 1 bar, the PET-derived catalyst led to CH4 and CO2 conversions as high as those on the BDC-derived catalyst, and its catalytic stability and selectivity towards DRM were excellent as well (no loss of activity after 13 h and H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO products ratio remaining at 1). Moreover, both catalysts were much better than those of a reference nickel alumina catalyst prepared by conventional impregnation route. This work therefore demonstrates the possibility of using plastic wastes instead of commercial chemicals to prepare efficient porous nickel–alumina DRM catalysts from MOFs, fostering the concept of circular economy.
:CO products ratio remaining at 1). Moreover, both catalysts were much better than those of a reference nickel alumina catalyst prepared by conventional impregnation route. This work therefore demonstrates the possibility of using plastic wastes instead of commercial chemicals to prepare efficient porous nickel–alumina DRM catalysts from MOFs, fostering the concept of circular economy.
1. Introduction
Dry reforming of methane (DRM) is a promising approach for carbon dioxide valorization and syngas generation. This process uses CH4 and CO2 (greenhouse gases) originating from natural or renewable sources (biomass, municipal solid wastes) and converts them into the valuable carbon monoxide and hydrogen gases according to the equation: CH4(g) + CO2(g) = 2CO(g) + 2H2(g).1–3 The H2/CO ratio close to 1 makes DMR more interesting for the production of high-added products such as oxo-alcohols and light olefins via the Fischer–Tropsch synthesis, compared with methane steam reforming or partial oxidation.4–7 Despite its environmental and economic advantages, DRM is still not industrially applied. In the literature, there are only few cases of industrial demonstration, for example a plant able to transform up to 60 tons per day of CO2via the autothermal dry reforming of CO2-rich natural gas for the production of linear C2–C15 olefins, with a selectivity higher than 90%.8 The main challenge that hinders the widespread applications of this virtuous process resides in the performances of the required catalyst. Catalysts with supported noble metals such as platinum, palladium, rhodium or ruthenium were found to be very active and stable9–11 but they are expensive, and recent efforts are rather dedicated to the development of nickel-based catalysts due to the lower price and good activity of this transition metal. Although promising, the application of Ni0 as active phase for DRM is however hampered by stability issues related to the sensitivity of nickel towards sintering and to the frequent formation of carbonaceous deposits under the high reaction temperatures required for the DRM to proceed. Efforts have therefore been put to enhance the activity and stability of the catalyst by several ways like the formation of Ni0 nanoparticles with reduced sizes, the addition of a second metal or promoter,12–14 altering the type of support used15–17 or enhancing the interaction between Ni and its carrier.18–21In previous reports, our group described methods to obtain highly stable DRM catalysts based on Ni nanoparticles highly dispersed on silica and alumina supports.20,22–25 In our latter contribution, the aluminum-based metal–organic framework (MOF) MIL-53(Al), a highly porous MOF composed of one-dimensional infinite aluminum hydroxyl chains connected to one another by benzendicarboxylate (BDC) linkers (its chemical formula is [Al(OH)BDC]x), was employed as a hybrid alumina precursor. Its high porosity was exploited to disperse nickel cation precursors in close proximity to the inorganic cornerstones of the framework through an impregnation step.26 Calcination of the Ni-impregnated MIL-53(Al), carried out to remove the organic phase, resulted in a porous alumina phase within which Ni cations were homogeneously dispersed in the form of thermally stable nickel aluminate nanodomains. The formation of these nickel aluminate nano-crystallites is believed to be at the origin of the particularly high stability and small size of the nickel metal nanoparticles obtained after reduction. It may explain the remarkable performance of the prepared MOF-derived catalyst in DRM (CO and H2 yield above 90% and CO:H2 close to 1 for 100 h at 650 °C).26,27
However, the benefit of this method, and more generally of any approach relying on the use of MOFs as sacrificial precursors for the production of porous carbons or metal oxides, is hampered by the irremediable loss of the organic part after the calcination step. De facto, although potentially promising in a range of applications and particularly in catalysis,28 sacrificial-MOFs-derived approaches thus suffer from the relatively high economic and environmental cost associated to the organic linkers production and to their degradation. Specifically, terephthalic acid (C8H4O4, also termed benzene dicarboxylic acid, BDC), one of the most widely employed organic linkers in MOF synthesis, is industrially produced by the costly oxidation of petroleum-derived xylenes used as starting materials. This energy-intensive and multi-step process relies on several expensive metal catalysts and product purification operations.29 Strategies towards more sustainable synthesis of MOF-derived materials are still rare in the literature. One of them could be to use MOFs made from cheap linker sources derived from recycled organic feedstocks (such as domestic plastics), with the advantage of not only reducing the cost of MOF production but also to address issues related to waste remediation.30
Polyethylene terephthalate (PET) is a thermoplastics of the polyester family with chemical formula (C10H8O4)n. It is synthesized from ethylene glycol (C2H6O2) and terephthalic acid (named BDC, in the following). It is commonly used in the production of plastic bottles and containers as well as fibers for clothing and carpets. PET is one of the main plastics source in urban waste, with global production of 30.3 × 106 tons in 2017.31 Because of its large production volume, its short useful life as a domestic package and its non-biodegradable property, recycling of PET becomes a growing environmental concern.32 Moreover, recycling plastic wastes is going in the direction of increasing the recycling rates, expected to reach 55% by 2030 for plastic packages, as imposed by the EU Commission, thus fostering the growing concept of circular economy.33,34 In this context, synthesizing MOFs from BDC issued from the depolymerization of PET was recently demonstrated to be a very attractive strategy to simultaneously lower the cost and the ecological footprint of MOF production and recycle the terephthalate component of PET.35 In this process, the depolymerization of PET occurs in the aqueous MOF synthesis medium by the hydrolysis of the ester bond that hold PET together, generating in turn the elementary BDC bricks. To our knowledge, this strategy was however not yet applied in the context of the preparation of carbonaceous or inorganic materials derived from the degradation of MOFs.
Herein, we demonstrate the feasibility of performing Ni0–Al2O3 DRM catalysts derived from an aluminum-terephthalate MIL-53(Al) synthesized by replacing commercial BDC with waste-sourced PET flakes within the hydrothermal synthesis mixture. The synthesis of MOF using PET is first validated by comparing the physicochemical characteristics of the MIL-53(Al) obtained from PET to those of a reference material prepared from the commercial BDC source. After Ni-impregnation, calcination and reduction, the catalytic performance in DRM of the thus produced PET-derived Ni0–Al2O3 catalyst is monitored and compared to that of the BDC-derived Ni0–Al2O3 catalyst as well as to that of a particularly active ad stable MIL-53(Al)-derived Ni0–Al2O3 catalyst produced (from commercial BDC) by microwave from our previous work.26
2. Experimental section
Materials preparation
Samples labelled as Ni0–Al2O3–BDCMW and Ni0–Al2O3-COM were synthesized for comparison purposes following a preparation procedure elaborated in details in our previous paper.26 Briefly, Ni0–Al2O3–BDCMW was synthesized following the same procedure as for Ni0–Al2O3–PET except that MIL-53 was synthesized under microwave irradiations. Ni0–Al2O3-COM was synthesized by incipient wetness impregnation of a commercial aluminum hydroxide (AlOOH).
Catalytic tests
The catalytic performance of the prepared catalysts was studied in a fixed bed reactor (Hastelloy X, I.D. = 9 mm, L = 30.5 cm) situated inside a microactivity reference catalytic reactor unit (MAR, PID Eng and Tech, Spain). Typically, 50 mg of catalyst was loaded into the reactor and reduced in situ under 5 vol% H2/Ar (30 ml min−1) at 800 °C for 2 h (heating rate 5 °C min−1). This protocol was chosen based on our previous study.26 The reactor was then cooled down to 200 °C and the reducing gas was replaced by a gas composed of an equimolar amount of CH4 and CO2 diluted in Ar (molar ratio 5/5/90) introduced at a gas hourly space velocity (GHSV) of 72 L g−1 h−1 and atmospheric pressure. The temperature was increased stepwise from 200 °C till 800 °C to investigate the activity profiles of the catalysts. Stability measurements were finally performed at 650 °C after cooling the reactor to this temperature. Chosen experiments were repeated to ensure reproducibility of catalytic results under the used conditions. The catalytic performance was established from the gas composition at the exit of the reactor monitored with an INFICON micro GC equipped with a TCD and two parallel channels (Molecular sieve and Plot U). The data are reported from 0 to 13 h of reaction (stability test) in terms of:– reactants conversions, estimated from the equation “X conversion (%) = (Cin − Cout)/Cin”, where Cin and Cout represent the inlet and outlet concentrations of CH4 or CO2 gases, respectively,
– “H2:CO (mole ratio) = MH2:MCO”, where MH2 and MCO are the molar concentrations of the formed H2 and CO products.
Carbon balance was systematically verified during the tests and was always above 98%.
Physicochemical characterization techniques
Powder X-ray diffraction patterns (PXRD) were recorded on a BRUKER type D8 diffractometer equipped with a Cu Kα irradiation source (λ = 1.5405 nm) and operating at 30 kV and 10 mA. The acquisitions were done in a 2θ range of 5 to 90°. Crystalline phase identification was based on comparison with standard PXRD files published by the international center for diffraction data (ICDD).Thermogravimetric analysis was performed on a TA SDT Q600 instrument working in horizontal mode. One of the two alumina pan was filled with about 20 mg of sample while the other (reference) pan was kept empty. The analysis was carried out in air flow (50 ml min−1) from room temperature to 900 °C with a ramping rate of 5 °C min−1.
Textural properties were evaluated from nitrogen adsorption–desorption isotherms recorded at −198 °C with a BELSORP-max apparatus. The specific surface areas of the samples were determined with the standard BET procedure considering the relative equilibrium pressure interval of 0.05–0.3. The samples were degassed before measurements at 120 °C under vacuum for 6 h.
Transmission electron microscopy (TEM) observations were done on ultrathin sections of solids to correctly visualize the dispersed nickel nanoparticles (and eventual carbon deposits) and their location inside or outside the porous alumina grains. The sections were prepared as follows: a few milligrams of powder were mixed with an EPON 812 embedding resin in a beam capsule. Polymerization of the mixture took place at 60 °C for 48 h, then the polymerized blocks were cut with a diamond knife in slices (50–70 nm in thickness) that were deposited on copper grids covered with a carbon membrane layer. TEM images were taken on a JEOL-JEM 200 electron microscope operating at 200 keV (LaB6gun). Average metallic Ni0 particle sizes were estimated considering at least 450 particles present in the grains imaged by TEM. Scanning Electron Microscopy (SEM) images were taken on a Hitachi SU-70 SEM-FEG microscope with an electron acceleration tension of 7 kV. After careful grinding, the samples were deposited on the analysis support and observed in mixed mode (70% of secondary electrons and 30% of retro-diffused signals).
3. Results and discussion
Validation of the PET-derived MIL-53(Al) synthesis
The quality of the MIL-53-PET synthesized by employing the depolymerization of PET flakes with a mean size of 5 × 5 mm manually cut from domestic water bottles was attested by comparing its physicochemical characteristics with those of the reference MIL-53-BDC obtained from the commercial BDC powder conventionally used to synthesize MIL-53(Al). At the end of the hydrothermal synthesis, the as-synthesized MIL-53-PET appears as a white precipitate dispersed in a brownish solution, whose color likely originates from the decomposition of the ethylene glycol formed during the PET hydrolysis. After thorough washing and recovery, no residual PET flakes were visually detected in the MIL-53-PETw powder. The X-ray diffractogram of MIL-53-PETw after drying at 80 °C is similar to that of MIL-53-BDCw dried in the same conditions and corresponds to the pure as-synthesized form of MIL-53(Al) (Fig. 1A(a′ and b′)).36 After activation at 330 °C for 3 days and partial rehydration (storage few days in room atmosphere), the diffractograms of the two materials (MIL-53-PETAC and MIL-53-BDCAC) are again similar (Fig. 1A(a and b)). They both correspond mainly to the hydrated narrow-pore form of MIL-53(Al), characterized by the (200) and (110) reflections at 2θ = 9.3° and 12.5°, respectively, and they contain a small portion of still dehydrated large-pore phase, characterized by the (101) reflection at 2θ = 8.7°.36 The N2 sorption isotherms of both activated MIL-53-PETAC and MIL-53-BDCAC (dehydrated in situ before the measurement) are almost similar as well, corresponding to the Type I shape expected for highly microporous MOFs (Fig. 1B(a and b)). Their BET specific surfaces (S = 1412 and 1397 m2 g−1, respectively) as well as their pore volumes (V = 0.59 and 0.58 cm3 g−1, respectively) are almost the same and in good correspondence with those commonly determined for pure MIL-53.36 The thermograms of the two activated samples are also very close, showing a first small weight loss below 100 °C due to dehydration, then one main weight loss equal to 72% at ca. 600 °C that corresponds to the BDC degradation (Fig. 1C(a and b)). The plateau at 22% observed for T > 600 °C gives the estimation of the amount of Al2O3 formed at the end of the analysis. This profile is in good agreement with the AlOH(BDC) formula of MIL-53(Al).36 The fact that no additional weight loss corresponding to remaining PET is detected on the thermograms of MIL-53-PET confirms that the PET hydrolysis occurred until its total consumption during the PET-derived synthesis. Accordingly, the MIL-53(Al) synthesis reaction yields calculated for MIL-53-PET and MIL-53-BDC were almost identical, and equal to 28% and 29% for the PET-derived and BDC-based syntheses, respectively. Therefore, using PET as a BDC source does not limit the extent of the MIL-53(Al) synthesis reaction. In addition, SEM analyses of the two MOFs were obtained as large micrometer scaled crystals (Fig. 2a and b). MIL-53-PETAC crystals display a rod-like morphology while MIL-53-BDCAC crystals are less elongated and exhibit an ill-defined morphology. All these features attest the purity of the MIL-53(Al) obtained from recycled PET and therefore the possibility to use it as a promising alumina precursor for the synthesis of Ni0–Al2O3 catalysts. | ||
| Fig. 1 (A) X-ray diffraction patterns, (B) N2 sorption isotherms, and (C) thermogravimetric profiles of (a′) washed as-synthesized MIL-53-PETw and (b′) MIL-53-BDCw, (a) activated MIL-53-PETAC and (b) MIL-53-BDCAC, (c) impregnated Ni-MIL-53-PETImp and (d) calcined Ni–Al2O3–PET. The star and the black triangles indicate the main peaks positions corresponding to the open and closed forms of the MIL-53(Al) structure, respectively (as reported in ref. 36). | ||
 | ||
| Fig. 2 (a–f) SEM and (d′–f′) TEM images of activated (a) MIL-53-PETAC, (b) MIL-53-BDCAC, (c) MIL-53-BDCMWAC (from ref. 26) and of impregnated and calcined (d and d′) Ni–Al2O3–PET, (e and e′) Ni–Al2O3–BDC and (f and f′) Ni–Al2O3–BDCMW (from ref. 26). The TEM images were recorded on microtome cuts of the samples. | ||
The TG profiles prompted us to set the calcination temperature of impregnated Ni-MIL-53-PETImp (and of reference Ni-MIL-53-BDCImp) at 500 °C in order to form subsequently the porous Ni–Al mixed oxide intermediate phases (labeled Ni–Al2O3–PET and Ni–Al2O3–BDC, respectively). This temperature is indeed appropriate to guarantee the total degradation of the linkers while preserving a significant porosity and a large surface area in the calcined material.26 A first effect of the calcination at 500 °C is the transformation of MOF crystals into materials with a rod-like shape clearly deriving from the original MOF shapes (Fig. 2d). Independently of the PET-derived or BDC-derived synthesis route, the sudden departure of the organics from MOF crystals during calcination induces a drastic volume contraction of the whole material on which cracks and cavities are already clearly seen by SEM (Fig. 2d and e). They are better visualized on the TEM images of microtomic cuts of the samples made perpendicularly to the lamellae (Fig. 2d′ and e′), where both impregnated and then calcined MOFs look like each other. Micrographs taken at higher magnification of Ni–Al2O3–PET (Fig. 3a) and Ni–Al2O3–BDC (Fig. S2A, ESI†) show that the crystals are actually formed of corrugated lamellae packed along a main axis. The formation of interlamellar pores is also confirmed by N2 sorption analysis that displays a gradual increase of adsorbed N2 on the P/P0 range from 0.6 to 1 and a slit-like hysteresis characteristic of interlamellar spaces (Fig. 1B(d) for Ni–Al2O3–PET and Fig. S3A(b) (ESI†) for Ni–Al2O3–BDC). Pore size distributions of all samples are shown in Fig. S4 (ESI†). The related specific surface area and pore volume are equal to 356 (resp. 352) m2 g−1 and 1.12 (resp. 0.99) cm3 g−1 for the PET-derived (resp. BDC-derived) Ni–alumina material.
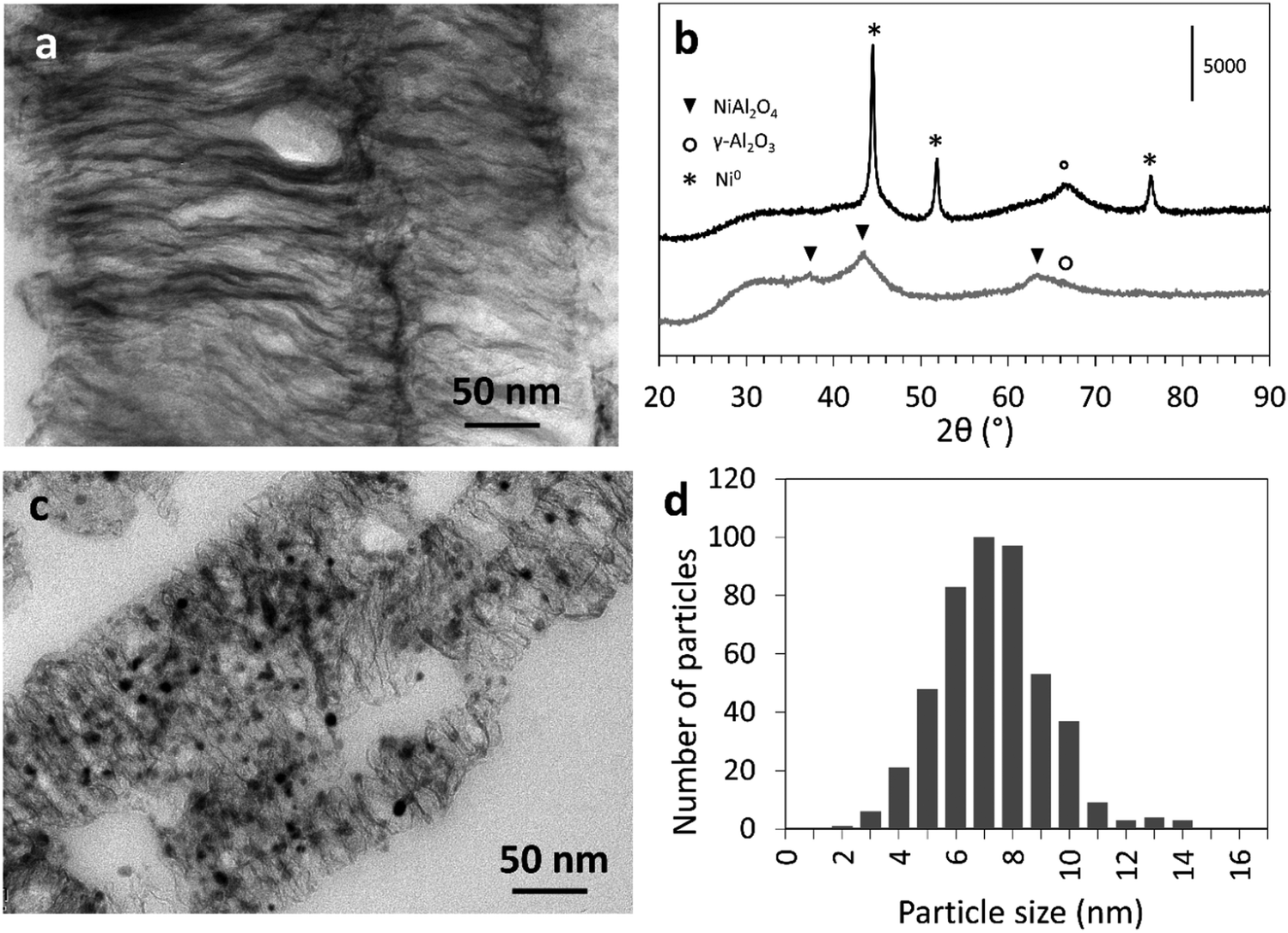 | ||
| Fig. 3 (a) TEM image of a microtome cut of Ni–Al2O3–PET, (b) PXRD patterns of Ni–Al2O3–PET (grey line, bottom) and of Ni0–Al2O3–PET (black line top), (c) TEM image of a microtome cut of Ni0–Al2O3–PET and (d) histogram established from at least 450 Ni0 nanoparti cles in typical TEM images of Ni0–Al2O3–PET. | ||
The PXRD pattern of the lamellar Ni–Al2O3–PET (Fig. 3b, bottom curve, see Fig. S3B (ESI†) for the PXRD pattern of Ni–Al2O3–BDC) exhibits three main very broad peaks corresponding to a cubic spinel NiAl2O4 phase with (311), (400) and (440) planes giving main peaks at 2θ = 37.0°, 45.0° and 65.0°.26 The broad peaks at 37° and 45° are asymmetric and may also contain overlaid peaks of γ-Al2O3, which suggests that the lamellae are made of NiAl2O4 nano-crystallites dispersed within poorly crystalline alumina. The very high nickel dispersion is also illustrated by the absence of any detectable nickel oxide nanoparticles in the TEM images. In addition, the strong interaction of the nickel cations with the aluminum oxide phase is supported by the high reduction temperature (T > 700 °C) determined by TPR analysis (Fig. S5, ESI†).
Reduction of the Ni–Al2O3–PET at T = 800 °C under H2, leading to the Ni0–Al2O3–PET catalyst, induced the formation of Ni0 nanoparticles occluded within the interlamellar spaces (Fig. 3c) and characterized by a narrow size distribution centered on 7.5 nm ((Fig. 3d), top curve). The simultaneous appearance of new PXRD peaks assignable to the (111), (200) and (220) planes of crystalline Ni0 (at 2θ = 44.5°, 51.9° and 76°, respectively) and the shift of the peak at 2θ = 65.0° (compared to that before reduction) towards a position characteristic of pure γ-Al2O3 attest the Ni extraction from the oxide matrix towards the lamellae surface (Fig. 3b, top curve). Application of the Scherrer's equation to the Ni0 reflection at 2θ = 51.9° leads to a mean Ni0 nanoparticle size of 5 nm, in the same range even if slightly smaller than the value estimated by TEM. The sizes of the Ni0 nanoparticles are slightly less homogeneous in the BDC-derived Ni0–Al2O3–BDC catalyst (Fig. S2B and C, ESI†). This may be due to a lower control (e.g., imperfect stirring) during preparation of this sample that was one of the first to be prepared using this new MOF-based strategy.
 | ||
| Fig. 4 (A) CH4 and (B) CO2 conversions during DRM upon continuous increase of temperature from 400 °C till 650 °C over in situ reduced (800 °C/2 h) catalysts: Ni0–Al2O3–PET (green tilted stripes), Ni0–Al2O3–BDC (purple diamond grid), Ni0–Al2O3–BDCMW26 (blue square grid), and Ni0–Al2O3-COM26 (orange horizontal stripes). | ||
Remarkably, the conversion of both reactants starts to be observed on Ni0–Al2O3–PET, Ni0–Al2O3–BDC and Ni0–Al2O3–BDCMW from 400 °C, while it appears only at 500 °C on commercial Ni0–Al2O3-COM. This starting of conversion at lower temperature reveals a higher activity, also attested by the systematically much higher CH4 and CO2 conversions on MOF-derived catalysts, compared to Ni0–Al2O3-COM, whatever the reaction temperature. More importantly, the conversion on Ni0–Al2O3-PET is as good – or even better – as that on Ni0–Al2O3–BDC prepared by following the exact same route, except for the BDC origin (either waste- or chemical-derived). It is however slightly lower than that on Ni0–Al2O3–BDCMW, especially at T > 450 °C, due to its distinct morphology as will be further discussed below. In addition, Ni0–Al2O3–PET displays a very high stability, with no change in conversions for 13 h at 650 °C under reactant stream, as for the other MOF-derived catalyst and much better than on Ni0–Al2O3-COM (Fig. 5A and B). Ni0–Al2O3–PET selectivity towards DRM reaction is excellent as well as indicated by the obtained H2:CO molar ratio that keeps always very close to 1 (0.99), as for Ni0–Al2O3–BDC (Fig. 5C).
 | ||
| Fig. 5 Catalytic performance and stability of the catalysts during 13 h of DRM reaction: Ni0–Al2O3–PET (green spheres), Ni0–Al2O3–BDC (purple diamonds), Ni0–Al2O3–BDCMW (blue spheres) and Ni0–Al2O3-COM (orange triangles): (A) CH4 conversion, (B) CO2 conversion and (C) H2:CO ratios. Black continued lines are the calculated values at the thermodynamic equilibrium for CH4 and CO2 conversions. | ||
In view of the structural purity of the MIL-53(Al) sacrificial phase obtained above from the PET wastes source (sample MIL-53-PETAC), the remarkable performance of the Ni0–Al2O3–PET catalyst, higher than that of the standard catalyst (Ni0–Al2O3-COM) and similar to those of the catalysts made from commercial BDC (Ni0–Al2O3–BDC and Ni0–Al2O3–BDCMW), were expected. Notably, the production of Ni0–Al2O3 from PET-derived MIL-53 occurs via the generation of the same nickel aluminate intermediate species than in the synthesis of Ni0–Al2O3–BDCMW (or Ni0–Al2O3–BDC), which we previously identified as key intermediates for the production of a highly active and stable Ni0–Al2O3 catalyst.27 Indeed, we recently proposed that the improved stability of the Ni2+ ions isolated within nickel aluminate nanodomains in the calcined materials slows down their extraction from the support during the reducing step, thus contributing to the formation of highly stable small nanoparticles. The strong interaction between Ni0 nanoparticles and the Al2O3 support, and thereby the improved stability of the catalyst, is assumed to be due to remaining NiAl2O4 phases acting as a “glue” at the nanoparticle/Al2O3 interface. This strong interaction is believed to inhibit not only nickel sintering but also the potential formation of carbon nanotubes,26,39 which growth is caused by the presence of weakly attached nickel particles able to move far away from the support itself by being embedded at the tip of the nanotubes while these nanotubes form.
The better performance observed with Ni0–Al2O3–BDCMW compared to those obtained with MIL-53-PET and MIL-53-BDC can be likely related to the nanoscopic size of the MIL-53(Al) crystals from which this catalyst derives. In fact, there is an inversely proportional relationship between the size of nanoparticles and the CH4 conversion. MIL-53-BDCMW crystal size ranges from 50 to 100 nm while the sizes of crystals of MIL-53-PET and MIL-53-BDC are both in the macroscopic regime, being comprised between 1 μm and more than 10 μm (Fig. 2a–c). Two hypotheses may explain why the nanosize of MIL-53-BDCMW crystals favors the catalytic performances of Ni0–Al2O3–BDCMW. First, downsizing MIL-53(Al) crystals may enhance the dispersion of the Ni precursor during the impregnation step, resulting after calcination, in a better dispersion of Ni2+ within the Al2O3 matrix and, in turn, in slightly smaller Ni0 nanoparticles after reduction. A slightly lower size of Ni0 nanoparticles were indeed detected in Ni0–Al2O3–BDCMW compared to Ni0–Al2O3–PET and Ni0–Al2O3–BDC (size = 6.8 nm26 against 7.5 nm in Ni0–Al2O3–PET, Fig. 3). A decrease of the size of metal Ni nanoparticles is expected to increase the number of working active Ni0 sites and hence to enhance the catalytic activity in DRM reaction with higher turnover frequency values.40 Moreover, the presence of Ni0 nanoparticles can favor also the stability of the catalyst, since it is well-know that large Ni particle cause severe coke deposition.41,42 In this regard, it is noted that carbon deposits (graphitic and disordered carbon deposits) were detected by RAMAN spectroscopy on the spent Ni0–Al2O3–PET catalyst (Fig. S6, ESI†), which was not the case for the spent Ni0–Al2O3–BDCMW.21 Second, combustion of the nanosized Ni-impregnated MIL-53-BDCMW crystals results after combustion in Ni-containing alumina nanosheets with size of few tenths of nanometers (Fig. 2f′ and ref. 26) that are much smaller as well as more curved and interwoven than in Ni0–Al2O3–PET and Ni0–Al2O3–BDC (Fig. 2d′ and e′). These mesostructure differences most likely imply differences in the surface chemistry properties (regarding the coordinated environment of Al3+ and the quantity of surface hydroxyl groups) of the γ-Al2O3 supports obtained after the Ni2+ reduction step. The effect of the alumina support properties were recently shown to drastically alter the catalytic activity of alumina supported catalysts, including Ni0–Al2O3.43,44 One can for instance assume that the nanosheets of Ni0–Al2O3–BDCMW possess more basic sites, which readily and stably adsorb CO2 to form bicarbonate and thereby facilitate the DRM reaction. In view of its interest, work is going on this aspect that was however not the central scope of the present work.
Conclusion
In this work we demonstrated the possibility of lowering the economic and environmental impact of the synthesis of Ni0–Al2O3 catalysts made from MOFs by employing recycled PET, originating from plastic wastes, as organic linker source. The recycling of PET can be envisaged in the growing interest towards the circular economy. Physico-chemical properties as well as the synthesis yield of the MIL-53(Al) made from PET were first shown to be identical to those of the same MOF made from commercial benzene dicarboxylic acid, confirming that the depolymerization process providing BDC during the course of MOF synthesis does not affect the crystallization of MIL-53(Al). Calcination followed by reduction of the nickel-impregnated MIL-53(Al) derived from PET led to the formation of a mesostructured Ni0–Al2O3 catalyst composed of uniform Ni0 nanoparticles homogeneously dispersed on interwoven alumina lamellae. The performance of this catalyst in DRM reaction, both in terms of CH4 and CO2 conversions, selectivity and stability, was as good as those of Ni0–Al2O3 catalysts obtained from commercial BDC, and much better than a conventionally prepared equivalent catalyst. This study therefore shows that MOFs synthesized from cheap recycled organic precursors can be transformed into materials with properties as good as those of materials obtained from commercial sources. The environmental and economic cost related to the loss of BDC during the transformation of MIL-53(Al) into the catalyst is compensated by the benefits provided by the recycling of PET waste, namely the remediation of plastic wastes accumulation in the environment and the avoidance of industrially produced organic linkers from petroleum-derived molecules. Furthermore, also replacing the metal salts commonly used for MOF synthesis (chloride or nitrate metal salts) by more sustainable metal sources, such as natural or recycled metal oxides or metal sources derived from domestic wastes, would enable the implementation of even more sustainable routes for the synthesis of MOF-derived catalysts, enhancing the circular economy. This aspect is currently under study in our lab.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors sincerely acknowledge the ERANET EU-FP7 initiative, the national ANR (France), CNRS-L (Lebanon) and MIUR (Italy) agencies for their support and funding through the SOL-CARE (Energy-065) project (JC-ENERGY-2014 first call). The University of Balamand Research Council is also thanked for the BIRG 02/2016 contribution. Sandra Casale is warmly thanked for her invaluable contribution to the electron microscopy observations. Jean-Marc Krafft is warmly thanked for his contribution to the RAMAN spectroscopy analyses of the spent catalysts. AM gratefully acknowledges the Erasmus + Programme Countries scholarship for her stay at the Sorbonne University.References
- W. L. Luyben, J. Process Control, 2016, 39, 77–87 CrossRef CAS.
- Z. Navas-Anguita, P. L. Cruz, M. Martín-Gamboa, D. Iribarren and J. Dufour, Fuel, 2019, 235, 1492–1500 CrossRef CAS.
- Y. Gao, J. Jiang, Y. Meng, F. Yan and A. Aihemaiti, Energy Convers. Manage., 2018, 171, 133–155 CrossRef CAS.
- J. E. A. Graciano, A. D. Carreira, R. Giudici and R. M. B. Alves, Production of Fuels from CO2-rich Natural Gas using Fischer-Tropsch Synthesis Coupled to Trireforming Process, Elsevier Masson SAS, 2017, vol. 40 Search PubMed.
- E. Akiki, D. Akiki, C. Italiano, A. Vita, R. Abbas-Ghaleb, D. Chlala, G. Drago Ferrante, M. Laganà, L. Pino and S. Specchia, Int. J. Hydrogen Energy, 2020, 45, 21392–21408 CrossRef CAS.
- T. V. Choudhary and V. R. Choudhary, Angew. Chem., Int. Ed., 2008, 47, 1828–1847 CrossRef CAS.
- L. Pino, C. Italiano, A. Vita, M. Laganà and V. Recupero, Appl. Catal., B, 2017, 218, 779–792 CrossRef CAS.
- Q. Chen, D. Wang, Y. Gu, S. Yang, Z. Tang, Y. Sun and Q. Wu, Energy Convers. Manage., 2020, 205, 112348 CrossRef CAS.
- Z. Hou, P. Chen, H. Fang, X. Zheng and T. Yashima, Int. J. Hydrogen Energy, 2006, 31, 555–561 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev.: Sci. Eng., 1999, 41, 1–42 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- L. Karam and N. El Hassan, J. Environ. Chem. Eng., 2018, 6, 4289–4297 CrossRef CAS.
- S. Damyanova, B. Pawelec, K. Arishtirova, J. L. G. Fierro, C. Sener and T. Dogu, Appl. Catal., B, 2009, 92, 250–261 CrossRef CAS.
- J. Horlyck, C. Lawrey, E. C. Lovell, R. Amal and J. Scott, Chem. Eng. J., 2018, 352, 572–580 CrossRef CAS.
- R. Zhang, G. Xia, M. Li, Y. Wu, H. Nie and D. Li, J. Fuel Chem. Technol., 2015, 43, 1359–1365 CrossRef CAS.
- S. E. Evans, J. Z. Staniforth, R. J. Darton and R. M. Ormerod, Green Chem., 2014, 16, 4587–4594 RSC.
- W. J. Jang, H. M. Kim, J. O. Shim, S. Y. Yoo, K. W. Jeon, H. S. Na, Y. L. Lee, D. W. Jeong, J. W. Bae, I. W. Nah and H. S. Roh, Green Chem., 2018, 20, 1621–1633 RSC.
- K. Tao, L. Shi, Q. Ma, D. wang, C. Zeng, C. Kong, M. Wu, L. Chen, S. Zhou, Y. Hu and N. Tsubaki, Chem. Eng. J., 2013, 221, 25–31 CrossRef CAS.
- X. Lv, J. Chen, Y. Tan and Y. Zhang, Catal. Commun., 2012, 20, 6–11 CrossRef CAS.
- L. Karam, S. Casale, H. El Zakhem and N. El Hassan, J. CO2 Util., 2017, 17, 119–124 CrossRef CAS.
- Y. Zhao, Y. Kang, H. Li and H. Li, Green Chem., 2018, 20, 2781–2787 RSC.
- M. N. Kaydouh, N. El Hassan, A. Davidson, S. Casale, H. El Zakhem and P. Massiani, C. R. Chim., 2015, 18, 293–301 CrossRef CAS.
- M. N. Kaydouh, N. El Hassan, A. Davidson, S. Casale, H. El Zakhem and P. Massiani, Microporous Mesoporous Mater., 2015, 220, 99–109 CrossRef.
- S. Andraos, R. Abbas-Ghaleb, D. Chlala, A. Vita, C. Italiano, M. Laganà, L. Pino, M. Nakhl and S. Specchia, Int. J. Hydrogen Energy, 2019, 44, 25706–25716 CrossRef CAS.
- L. Karam, M. Armandi, S. Casale, V. El Khoury, B. Bonelli, P. Massiani and N. El Hassan, Energy Convers. Manage., 2020, 225, 113470 CrossRef CAS.
- L. Karam, J. Reboul, S. Casale, N. El Hassan and P. Massiani, ChemCatChem, 2019, 12, 373–385 CrossRef.
- L. Karam, J. Reboul, N. El Hassan, J. Nelayah and P. Massiani, Molecules, 2019, 24, 4107–4120 CrossRef CAS PubMed.
- H. Konnerth, B. M. Matsagar, S. S. Chen, M. H. G. Prechtl, F. K. Shieh and K. C. W. Wu, Coord. Chem. Rev., 2020, 416, 213319 CrossRef CAS.
- A. Saffer and R. S. Barker, US Pat., 2833816, 1958 Search PubMed.
- E. S. M. El-Sayed and D. Yuan, Green Chem., 2020, 22, 4082–4104 RSC.
- T. S. Gomes, L. L. Y. Visconte and E. B. A. V. Pacheco, J. Polym. Environ., 2019, 27, 533–548 CrossRef CAS.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, J. Cleaner Prod., 2019, 225, 1052–1064 CrossRef CAS.
- R. Meys, F. Frick, S. Westhues, A. Sternberg, J. Klankermayer and A. Bardow, Resour., Conserv. Recycl., 2020, 162, 105010 CrossRef.
- G. Lonca, P. Lesage, G. Majeau-Bettez, S. Bernard and M. Margni, Resour., Conserv. Recycl., 2020, 162, 105013 CrossRef.
- S. Manju, P. K. Roy, A. Ramanan and C. Rajagopal, Mater. Lett., 2013, 106, 390–392 CrossRef.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- A. Wolf, A. Jess and C. Kem, Chem. Eng. Technol., 2016, 298, 1040–1048 CrossRef.
- G. Kolb, T. Baier, J. Schürer, D. Tiemann, A. Ziogas, S. Specchia, C. Galletti, G. Germani and Y. Schuurman, Chem. Eng. J., 2008, 138, 474–489 CrossRef CAS.
- L. D. Vella and S. Specchia, Catal. Today, 2011, 176, 340–346 CrossRef CAS.
- J. W. Han, J. S. Park, M. S. Choi and H. Lee, Appl. Catal., B, 2017, 203, 625–632 CrossRef CAS.
- S. Tang, L. Ji, J. Lin, H. C. Zeng, K. L. Tan and K. Li, J. Catal., 2000, 194, 424–430 CrossRef CAS.
- D. Chen, K. O. Christensen, E. Ochoa-Fernández, Z. Yu, B. Tøtdal, N. Latorre, A. Monzón and A. Holmen, J. Catal., 2005, 229, 82–96 CrossRef CAS.
- M. Trueba and S. P. Trasatti, Eur. J. Inorg. Chem., 2005, 3393–3403 CrossRef CAS.
- D. Shen, M. Huo, L. Li, S. Lyu, J. Wang, X. Wang, Y. Zhang and J. Li, Catal. Sci. Technol., 2020, 10, 510–516 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00984a |
| ‡ Present address: TWM – Technologies for Waste Management, Via Assietta 27, 10128 Torino, Italy. |
| This journal is © The Royal Society of Chemistry 2021 |