
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid fabrication of MOF-based mixed matrix membranes through digital light processing†
Alexey
Pustovarenko
 a,
Beatriz
Seoane
b,
Edy
Abou-Hamad
c,
Helen E.
King
d,
Bert M.
Weckhuysen
b,
Freek
Kapteijn
e and
Jorge
Gascon
*a
a,
Beatriz
Seoane
b,
Edy
Abou-Hamad
c,
Helen E.
King
d,
Bert M.
Weckhuysen
b,
Freek
Kapteijn
e and
Jorge
Gascon
*a
aKing Abdullah University of Science and Technology, KAUST Catalysis Center, Advanced Catalytic Materials, Thuwal 23955, Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa
bUtrecht University, Debye Institute for Nanomaterials Science, Inorganic Chemistry and Catalysis Group, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
cKing Abdullah University of Science and Technology, Core Labs, Thuwal 23955, Saudi Arabia
dDepartment of Earth Sciences, Utrecht University, Princetonlaan 8a, 3584 CB, Utrecht, The Netherlands
eDelft University of Technology, Department of Chemical Engineering, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
First published on 9th March 2021
Abstract
3D printing, also known as additive manufacturing technology, has greatly expanded across multiple sectors of technology replacing classical manufacturing methods by combining processing speed and high precision. The scientific interest in this technology lies in the ability to create solid architectures with customized shapes and predetermined properties through the exploration of formulations enriched with multifunctional microporous additives such as metal–organic frameworks (MOFs). The concept of additive manufacturing involving advanced materials could be fruitfully adapted for MOF-based mixed matrix membrane fabrication to be used in gas separation applications. In this work, a digital light processing (DLP) approach for fast prototyping of MOF-based mixed matrix membranes (MOF-MMMs) with full control over the shape, size and thickness of the resulting composite using a conventionally available 3D printer has been explored. MOF-based printable inks have been formulated from a selection of commercially available acrylate oligomers and MIL-53(Al)–NH2 additive post-synthetically modified with methacrylic functionality. The formulations and resulting composites have been extensively characterized to demonstrate the suitability of the inks for DLP processing into free-standing MOF-based membranes. The MOF filler anchored to the polymeric matrix enhances the overall permeability at constant selectivity when applied for H2/CO2 separation. The obtained results confirm the applicability of the 3D DLP technology for fast prototyping of MOF-based MMMs and provide new opportunities for further development.
1. Introduction
3D printing technology is a rapidly developing field.1–4 Nowadays, fast prototyping is slowly displacing classical manufacturing methods for advanced materials, offering relative operational simplicity and the ability to render a customized product at high speeds and with high precision.5 Bridging the gap between fast prototyping and functionality, additive manufacturing is a very interesting playground for the development of formulations that can enable the fabrication of complex forms of matter with specific properties on demand.6,7To amplify and broaden the applicability prospects of additive compositions, the incorporation of multifunctional components such as microporous solids can be considered to endow the additive formulations with additional properties.8,9 From this perspective, metal–organic frameworks (MOFs) are promising filler candidates.10–15
Conceptually, the integration of a MOF component into additive formulations has been approached and then realized through different 3D printing techniques4 – inkjet,16,17 direct ink writing (DIW),18–27 fused deposition modelling (FDM),28–30 digital light processing (DLP)31 and selective laser sintering (SLS).32,33 For instance, photoluminescent lanthanide MOF-based inks for inkjet printing were reported by da Luz et al.16 In addition to examples of MOF coatings grown on 3D printed objects obtained by stereolithography (SLA)34 or direct ink writing,24 fabrication of monoliths with full integration of the MOF component within a melting matrix of acrylonitrile butadiene styrene (ABS),28,29 poly(lactic acid) (PLA) or thermoplastic polyurethane (TPU)30 has been achieved using fused deposition modelling. However, the FDM processable MOF/thermoplastic composites frequently suffer from a porosity loss that questions their further applicability.28 Unlike in the FDM approach, firm monoliths with accessible internal porosity, suitable for applications involving adsorption, have been obtained via direct ink writing. Moreover, several representative examples of composites with high MOF loading22 suitable for gas separation,18,20,21 catalysis,26 molecule sequestration and release have been reported.19,25 Similarly, tailored shapes with high MOF content32 can be fabricated by means of selective laser sintering (SLS) into self-standing MOF–polymer mixed matrix films.33
From the perspective of using 3D printable MOF-containing additives for gas separation and, particularly, for fast prototyping of MOF-based mixed matrix membranes (MOF-MMMs),35 the digital light processing (DLP) technique deserves particular attention. DLP printing proceeds through the solidification of liquid resins by photoinduced crosslinking on locally illuminated areas, reproducing in this way a 3D shape in a stepwise manner (layer by layer). Thus, the process not only affords fast prototyping but also endows a control over resolution – thickness of the layer, which is an important aspect to consider while dealing with mixed matrix membranes. Building on the work of Halevi et al.31 on DLP processing of a MOF/acrylate admixture into 3D printed shapes and an early study of Zhang et al.36 on copolymerization between post-synthetically functionalized with methyl methacrylate UiO-66-NH2 particles and an acrylate monomer, we are targeting to adapt the previous contributions for fast prototyping of MOF-based mixed matrix membranes (MOF-MMMs).
Herein, we explore a digital light processing technology for fast prototyping of MOF-MMMs. With this approach, MIL-53(Al)–NH2 nanoparticles post-synthetically functionalized with a methacrylic moiety could be covalently integrated within a photopolymerizable matrix of commercially available acrylic oligomers for the further fabrication of MOF-MMMs with a controllable thickness and tailored shape in a matter of minutes without the need for additional solvents or long post-synthetic treatment. Given the fact that the DLP 3D printing technique involving MOF-based inks is currently poorly represented while the approach offers a range of attractive benefits such as rapidness of prototyping with no molds and solvents engaged, herein, we critically evaluate aspects relevant for DLP processing of several acrylate systems with and without MOF-additives into membranes aimed to be applied for the separation of an equimolar H2/CO2 mixture.
2. Experimental section
2.1 Materials and synthetic procedures
2.2 Ink formulations and 3D printing process
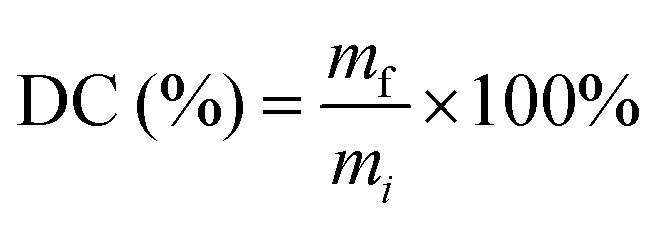 | (1) |
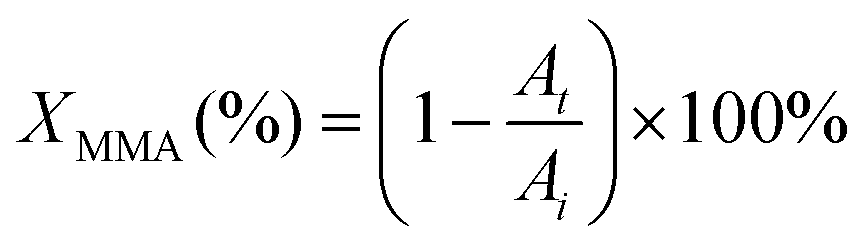 | (2) |
2.3 General characterization
Scanning powder X-ray diffraction (PXRD) patterns were recorded using a Bruker D8 Advance operated with monochromatic Cu-Kα (λ = 1.5418 Å) radiation with a scan speed of 0.5 s per step and a step size of 0.1° over the 5–60° 2θ range.Physisorption isotherms using nitrogen and argon gas probes were recorded at 77 K and at 87 K, respectively, using a Micromeritics ASAP 2040 instrument. Carbon dioxide physisorption isotherms were recorded using a Tristar II 3020 at 273 K. Before the measurement, composite samples were degassed at 150 °C for 8 h. The uptake measurements for MIL-53(Al)–NH2 and MIL-53(Al)–NH2/MMA were conducted on an activated sample treated according to the well-known procedure described elsewhere.37,40,41
Thermogravimetric data (TG) were collected in nitrogen and air atmospheres using a Mettler-Toledo thermal analyzer at a heating rate of 5 °C min−1 in a 25–800 °C temperature range with a gas flow of 25 mL min−1.
Differential scanning calorimetry (DSC) tests were carried out using a Mettler-Toledo TGA/DSC1 Star system by heating 5–10 mg of each sample under a nitrogen flow (50 mL min−1) from 40 up to 230 °C applying a heating rate of 10 °C min−1 and cooling rate of 5 °C min−1. Three consecutive runs were performed. The first cycle was intended to remove any previous thermal history of the samples. Two subsequent cycles were performed using the same protocol.
Transmission electron microscopic (TEM) images were obtained using a JEOL JEM-2100F microscope operated at 120 kV and equipped with a field emission gun (FEG). The samples were dispersed in methanol, sonicated and sprayed on a carbon-coated copper grid and air-dried.
Scanning electron microscopic (SEM) imaging was performed using an FEI TENEO VS microscope equipped with an integrated SDD EDX detector. 3D printed samples were mounted on a 45°/90° holder with the help of double-sided carbon tape and grounded with strokes of conductive silver paint. For cross-sectional evaluation, printed films were cryo-fractured in liquid nitrogen. FIB-SEM cross-sectional studies were carried out using a Helios Nanolab 600 FIB.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 20000 for 13C and was 32 for 1H. An exponential apodization function corresponding to a line broadening of 80 Hz was applied prior to Fourier transformation.
000 and 20000 for 13C and was 32 for 1H. An exponential apodization function corresponding to a line broadening of 80 Hz was applied prior to Fourier transformation.
The 2D 1H–13C heteronuclear correlation (HETCOR) solid-state NMR spectroscopic experiments were conducted using a Bruker AVANCE III spectrometer with a 3.2 mm MAS probe. The experiments were performed according to the following scheme: 90° proton pulse, t1 evolution period, CP to 13C, and detection of the 13C magnetization under TPPM decoupling. For the cross-polarization step, a ramped radio frequency (RF) field centered at 75 kHz was applied to the protons, while the 13C channel RF field was matched to obtain an optimal signal. A total of 64 t1 increments with 2000 scans each were collected. The sample spinning frequency was 15 kHz. Using a contact time of 0.2 ms for the CP step, the polarization transfer in the dipolar correlation experiment was verified to be selective for the first coordination sphere (<5 Å).
Liquid-state nuclear magnetic resonance experiments were performed using a Bruker Advance-400. In the digestion experiment, 10 mg of MIL-53(Al)-NH2/MMA was dissolved in a mixture of DMSO-d6/HF resulting in a clear solution.
Attenuated total reflection–Fourier transform infrared spectroscopic (ATR-FTIR) analyses were performed on a home-built setup that included a Nicolet 8700 FTIR spectrometer equipped with a Harrick Horizon multiple internal reflection accessory, coupled to a cell containing a ZnSe crystal. The photopolymerizable ink formulations were layered on the top of the ZnSe crystal and illuminated using an ACER H5360 LED DLP projector. To observe the spectral changes during the photopolymerization process, real-time ATR spectra were recorded in the range of 2000–1000 cm−1 at 1.93 s intervals. The spectral resolution was 4 cm−1 and 1 scan per time interval was used to achieve the maximum time resolution.
Raman spectroscopic studies were performed using a WITec alpha 300R Raman microscope operated with a 532 nm laser (14 mW). 2D maps of the 3D printed object surface were acquired on a 30 μm × 30 μm area advancing with 1 μm step size along each of the x, y directions.
The stress–strain data were collected using a Zwick Roell tensile testing machine at room temperature. UV polymerized samples were prepared in molds in the form of 55 × 5 × 3 mm blocks and all tested with the applied 150 mN min−1 loading rate.
An optic microscope Leica DM750 equipped with HI Plan EPI 5/10/20/50× objectives and a rotating diascopic polarizer was used to capture the images of 3D printed objects in bright and dark fields.
2.4 Gas permeation measurements
3D printed rounded membranes with an area of 0.2 cm2 were mounted on a flange between Viton® O-rings and placed inside an oven in a home-made permeation setup. The H2/CO2 separation measurements were performed employing an equimolar H2:CO2 gas mixture (20 mL min−1 CO2 and 20 mL min−1 H2) feed. Helium (2.0 mL min−1) was used as a sweep gas at the permeate side. The absolute pressure of the feed stream was adjusted in a range of 1–2 bar using a back-pressure controller at the retentate side, keeping the permeate side at atmospheric pressure. The temperature in the permeation module was adjusted from room temperature to 100 °C through a convection oven. An on-line gas chromatograph (Interscience Compact GC) equipped with a packed Carboxen 1010 PLOT (30 m × 0.32 mm) column and a thermal conductivity detector (TCD) was used to periodically analyze the permeate stream.The permeability of component j (Pj) was calculated as follows (eqn (3)):
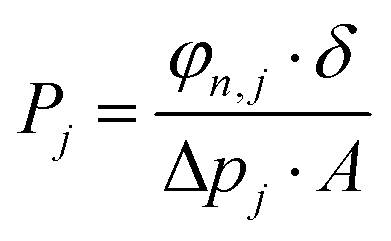 | (3) |
The SI unit for permeability is mol s−1 m−1 Pa−1. However, here, gas permeabilities are reported in the widely used non-SI unit Barrer, where 1 Barrer = 3.35 × 10−16 mol m m−2 Pa−1 s−1.
The separation factor or mixed gas selectivity (α) was calculated as the ratio of the permeability of the faster permeating component (H2) to the permeability of the less permeable component (CO2) as given below (eqn (4)):
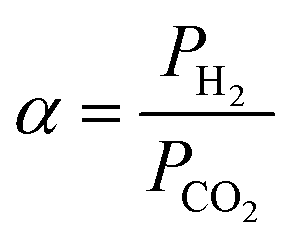 | (4) |
3. Results and discussion
MIL-53(Al)–NH2 framework is a MOF widely used for gas storage and separation purposes and reported to demonstrate remarkable performance when incorporated as a filler into mixed matrix membranes (MMMs).37,41 In particular, the presence of NH2-groups endows this framework with diverse functionalization opportunities (Fig. 1A).42 Thus, the amino groups were brought into play to introduce a photocrosslinkable functionality to MIL-53(Al)–NH2 particles with the aim to improve their processability and integrability into DLP UV-curable inks.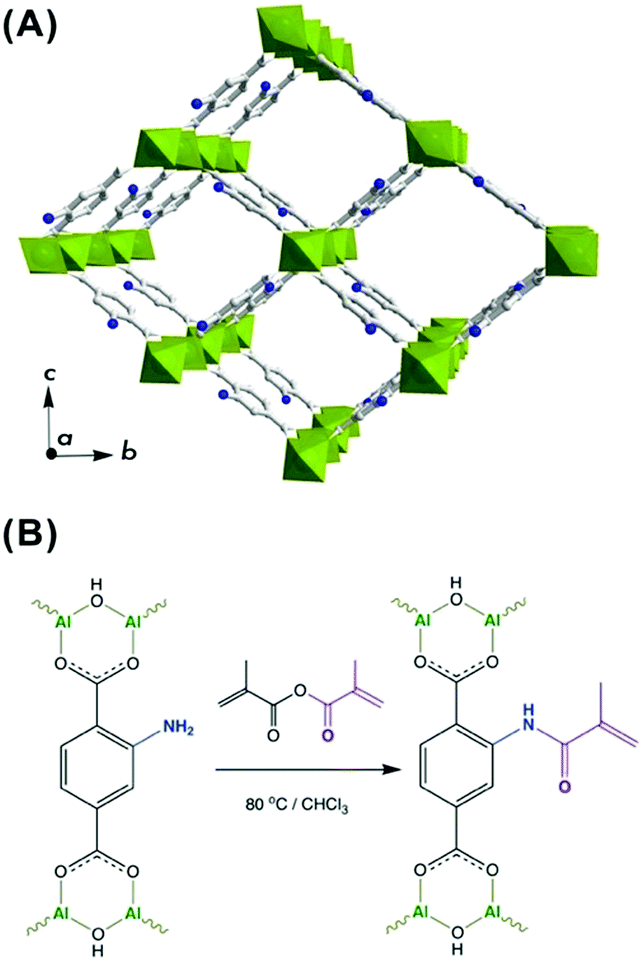 | ||
| Fig. 1 (A) Representation of MIL-53(Al)–NH2 structure in the open pore configuration, where AlO6 polyhedra are depicted in green, and grey and navy are carbon and nitrogen atoms, respectively. (B) Scheme of post-synthetic modification of MIL-53(Al)–NH2 structure with methyl methacrylic anhydride. | ||
To meet the requirements for an effective integrability into a polymer matrix, a MIL-53(Al)–NH2 material was synthesized in the form of nanoparticles with dimensions of about 30–100 nm (Fig. S6 and S7, ESI†). The post-synthetic functionalization was made on a non-activated MOF to assure restricted accessibility of the porous system and to favor preferential attachment to NH2-groups on the surface. Hence, the as-synthesized MIL-53(Al)–NH2 was post-synthetically modified (PSM) through an acylation reaction with a methyl methacrylic anhydride (Fig. 1B). Such modification leads to the framework transformation from np- to lp-conformation,43 as suggested by Fig. 2A. The latter can be attributed to a partial anchoring of NH2-groups inside the pores of the MOF in addition to the modifications occurring on the surface of the particles. Thus, the incorporation of a bulky MMA group into the pore space forces the framework to adopt the lp-configuration, which is in good agreement with the previous reports on PSM of structures exhibiting “breathing behaviour”.40,42
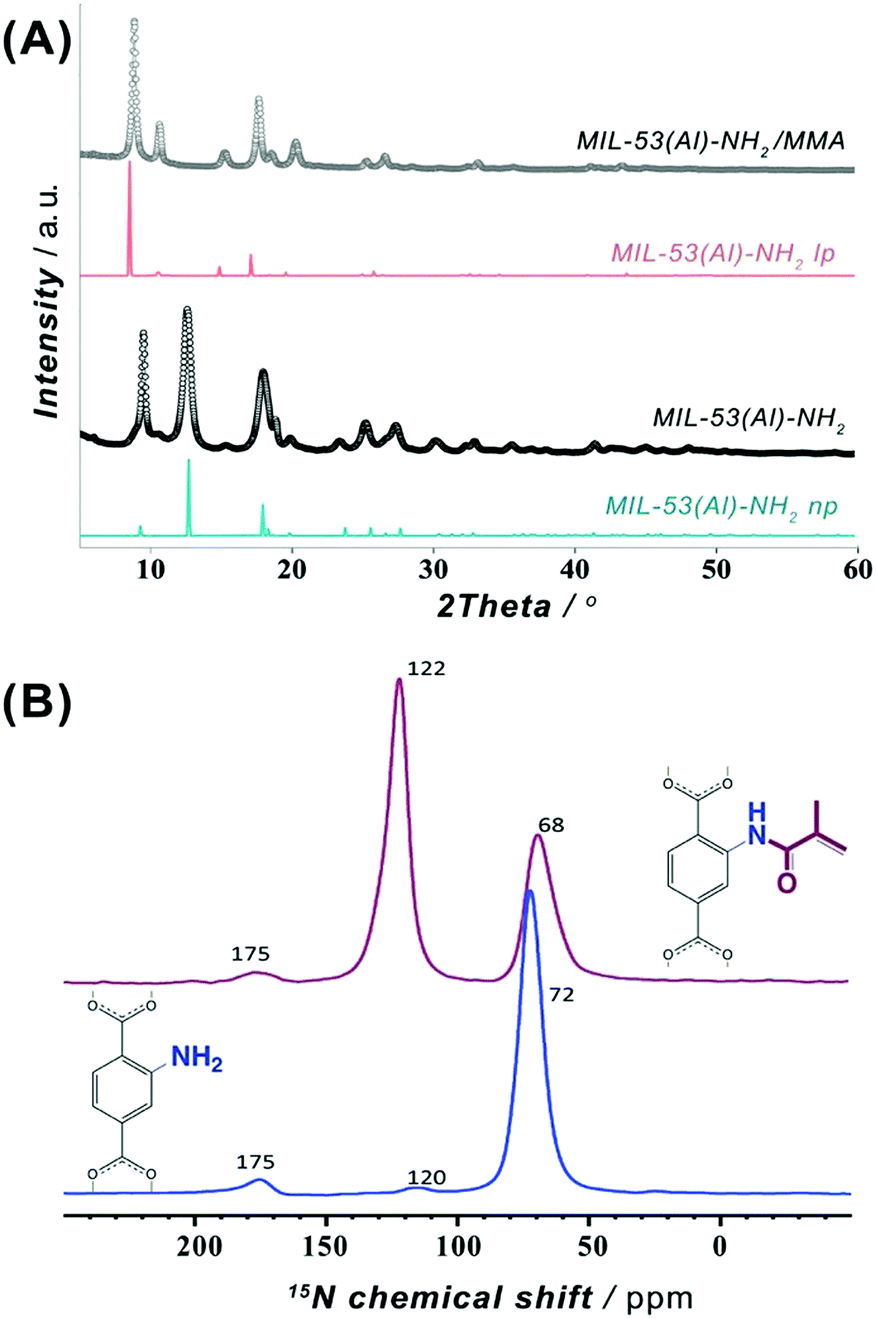 | ||
| Fig. 2 (A) Powder XRD patterns of MIL-53(Al)-NH2 nanoparticles before (black) and after (grey) post-synthetic modification with MMA and compared to open (lp) and narrow pore (np) forms. (B) Solid-state DNP-enhanced 15N NMR spectrum for MIL-53(Al)–NH2 before (blue) and after (wine) post-synthetic modification with MMA. | ||
In order to confirm the covalent character of the post-synthetic modification of MIL-53(Al)–NH2 with MMA functionality, solid-state nuclear magnetic resonance (NMR) spectroscopic studies were carried out on a pristine MIL-53(Al)–NH2 material and its PSM-NH2/MMA version. The 1H MAS NMR spectrum (Fig. S8, ESI†) of the original MIL-53(Al)–NH2 contains four peaks that can be assigned to the aromatic protons of the linker (unresolved peak at 6.9 ppm), protons of the amino group (5.5 ppm) and protons of the bridging μ2-OH group in two possible chemical environments (δiso = 2.8 and 1.9 ppm) as expected.44,45 In the 2D 1H MAS DQ-SQ NMR correlation spectrum (Fig. S9, ESI†), the aromatic proton resonances remain unresolved albeit manifesting two main pairs of correlation peaks: 1H–1H correlations between the (i) aromatic protons and NH2-group (δiso in F2 dimension: 5.5–6.8 ppm) and between the (ii) aromatic protons and bridging OH group (δiso in F2 dimension: 1.9–6.8 ppm). The 13C CPMAS NMR spectrum shows six resonances (Fig. S10, ESI†) attributed to the carbonyl groups in the NH2–BDC linker (δiso = 175.6 ppm) and carbons of the aromatic ring (δiso in 116.4–150.5 ppm range). In addition, the intra-linker coupling interactions between the carbon atoms and aromatic protons were complemented by means of a 2D 1H–13C HETCOR NMR (Fig. S11, ESI†). The 1H MAS NMR spectrum (Fig. S12, ESI†) of the PSM MIL-53(Al)–NH2/MMA material reveals additional weakly resolved resonances of CH3-protons (δiso ∼ 0.7 ppm), CH2 protons (δiso ∼ 5.2 ppm) of the MMA moiety and amide –NH– protons (δiso ∼ 10.7 ppm) that affirm the presence of acylated NH2-groups. Expected changes of the aromatic proton-related chemical shifts upon PSM are also reflected in the presence of a shoulder resonance at δiso ∼ 9.0 ppm. The 2D 1H MAS DQ-SQ NMR spectrum of MIL-53(Al)–NH2/MMA (Fig. S13, ESI†) demonstrates off-diagonal signals that indicate (i) a coupling of geminal –CH2 protons (δiso in F2 dimension: 5.0–6.5 ppm) of the MMA functionality; (ii) a correlation between the bridging μ2-OH of the framework and trans-CH2 of the MMA group (δiso in F2 dimension: 1.6–5.1 ppm) due to their spatial proximity; (iii) a correlation of the methyl-group of MMA (δiso in F2 dimension: 0.7–8.0 ppm) and (iv) the bridging μ2-OH (δiso in F2 dimension: 2.7–8.0 ppm) with the aromatic protons. The appearance of resonances at ∼16.7, 122.7 and 166.0 ppm in 13C CP-MAS NMR spectrum (Fig. S14, ESI†) can be associated with –CH3, –CH2 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbons of the MMA moiety anchored to –NH2 site of the MOF. Additionally, correlations at δiso ∼ (16, 0.6) ppm, (123, 6.0) ppm and (160, 9.9) ppm corresponding to –CH3, –CH2 and C(O)–NH fragments of the MMA functionality can be observed in the 2D 1H–13C CP-MAS HETCOR NMR spectrum (Fig. S15, ESI†).
O carbons of the MMA moiety anchored to –NH2 site of the MOF. Additionally, correlations at δiso ∼ (16, 0.6) ppm, (123, 6.0) ppm and (160, 9.9) ppm corresponding to –CH3, –CH2 and C(O)–NH fragments of the MMA functionality can be observed in the 2D 1H–13C CP-MAS HETCOR NMR spectrum (Fig. S15, ESI†).
To overcome sensitivity constraints of the 15N NMR measurements, DNP-enhanced 15N NMR experiments were carried out on MIL-53(Al)–NH2 and MIL-53(Al)–NH2/MMA samples. As shown in Fig. 2B, the spectrum of the PSM-NH2/MMA material reveals a resonance at ∼122 ppm attributed to the newly formed C(O)–NH functionality in addition to the peak of the –NH2 group at ∼68 ppm, whereas only one strong signal at ∼72 ppm of the –NH2 group is observed in the spectrum of the pristine MOF. Furthermore, a 2D DNP-enhanced 15N–1H CP-MAS HETCOR NMR correlation map exposes two sets of cross-peaks attributed to the couplings between nitrogen atoms in –NH2 and C(O)–NH with the aromatic and amide protons (Fig. S20, ESI†). All observations above indicate the covalent anchoring of MMA in the framework of MIL-53(Al)–NH2.
Additionally, the post-synthetic event in the MIL-53(Al)–NH2/MMA structure was evidenced by thermogravimetric analysis (TGA) in an air atmosphere and argon physisorption measurements (Fig. S6C and D, ESI†). Due to the incorporation of an additional MMA group, the mass loss in the region of 350–600 °C, where a total organic component decomposition takes place, expanded by 12 wt% for the PSM material compared to that of the pristine compound (Table S5, ESI†). Furthermore, the surface measurements reveal that the incorporation of the MMA group leads to a reduction in the intrinsic porosity from 370 m2 g−1 to 303 m2 g−1 (Fig. S6D and Table S5, ESI†). In addition, a comparison of the FTIR spectra of the pristine and modified MIL-53(Al)–NH2/MMA material reveals an increase in the relative intensity of the absorption band situated at ∼1610 cm−1, which is assigned to the CC vinylic group, further suggesting a conceivable PSM occurrence (Fig. S21, ESI†).
In order to determine the percentage of amino groups acylated with MMA, the MIL-53(Al)–NH2/MMA material was digested in a DMSO-d6 and HF mixture and evaluated by liquid-state 1H NMR spectroscopy. The percentage of the modified –NH2 groups was estimated to be 34% by comparing the integrals of the characteristic geminal proton signals of the MMA group (δ = 5.58, 5.95 ppm) and the aromatic protons of the pristine NH2–BDC linker (δ = 6.99–7.75 ppm) (Fig. S22, ESI†). Considering the size of the MIL-53(Al)–NH2 nanoparticles (Fig. S7, ESI†) and the orientation of the unit cell axes relative to the rod-shaped nanocrystal facets (Fig. S23, ESI†),46,47 the estimated amount of -NH2 groups located in the unit cells forming the crystal surface and exposed to PSM was approximated to be around 20%. Thus, this value concedes the percentage of –NH2 acylated with MMA determined from the liquid NMR of the digested MIL-53(Al)–NH2/MMA material, which suggests that the PSM process not only occurs on the surface of the nanoparticles, but also involves part of the porosity.
An essential criterion imposed on any photopolymerizable ink formulation is the velocity of photo-crosslinking events set in motion when exposed to a light source. Thus, the commencing step in the design of a MOF-containing ink formulation was to appraise the suitability of the precursor oligomer mixture for photopolymerization in terms of its behavior under illumination. The corresponding transformation was followed by time-resolved ATR-FTIR on several commercially available acrylate-based systems (Fig. S1 and Table S1, entries 1–6, ESI†), which were intended to form a continuous matrix in printed composites, defining a decrease of ∼1640 cm−1 band area as a merit of methacrylic group conversion (Fig. S24, ESI†).48Fig. 3A summarizes the individual kinetic profiles of T, P, E and U-monomer photopolymerization and demonstrates a rapid MMA transformation in the P-system (with 2.4% s−1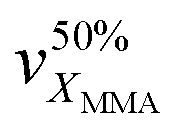 ) with an XMMA conversion reaching 25% compared to more moderate photopolymerization velocities in the T- and E-components (∼1.5% s−1) that demonstrated an XMMA conversion of 27% and 24%, respectively (Fig. 3B). In contrast, the relatively slow behavior of the U-oligomer with a maximum degree of MMA conversion of 15% and a rate of 0.75% s−1 made it less desirable for further involvement in the DLP process. The viscosity of a photopolymerizable formulation is one of the governing factors that determines the kinetics of MMA crosslinking. As observed (Table S6, ESI†), a higher viscosity of the precursor is correlated with slower kinetics of the photopolymerization process. This should not exceed values of about 600 cP for the effective use with a moving build platform.
) with an XMMA conversion reaching 25% compared to more moderate photopolymerization velocities in the T- and E-components (∼1.5% s−1) that demonstrated an XMMA conversion of 27% and 24%, respectively (Fig. 3B). In contrast, the relatively slow behavior of the U-oligomer with a maximum degree of MMA conversion of 15% and a rate of 0.75% s−1 made it less desirable for further involvement in the DLP process. The viscosity of a photopolymerizable formulation is one of the governing factors that determines the kinetics of MMA crosslinking. As observed (Table S6, ESI†), a higher viscosity of the precursor is correlated with slower kinetics of the photopolymerization process. This should not exceed values of about 600 cP for the effective use with a moving build platform.
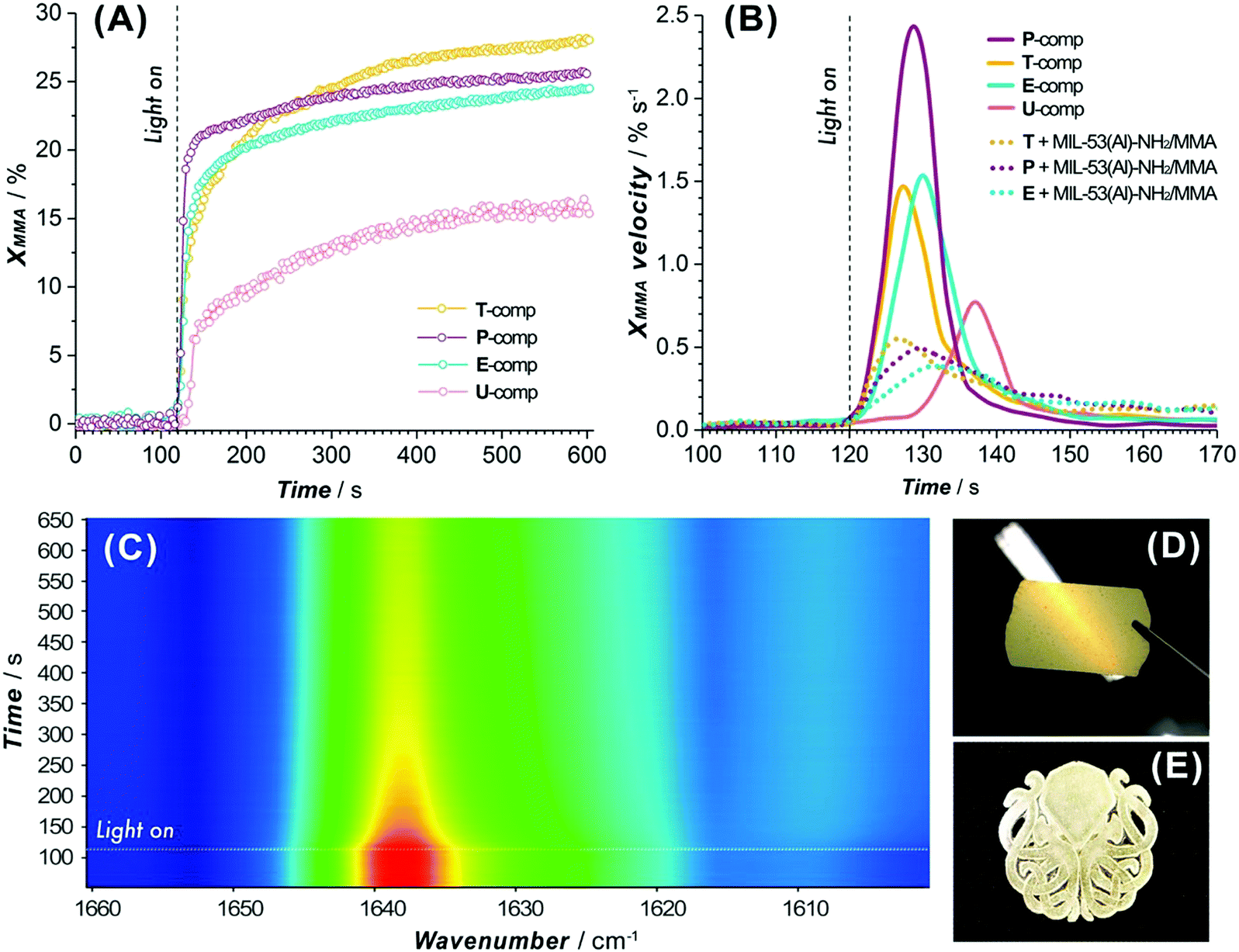 | ||
| Fig. 3 (A) Kinetic profiles of photopolymerization for individual oligomer mixtures (T, P, E and U-components). (B) Comparison of MMA-group conversion velocities for individual oligomers and MOF-contained formulations under the printer light source (turn on at 120 s). (C) 2D-resolved ATR-FTIR spectrum of a T/MIL-53(Al)–NH2/MMA photopolymerization event (lamp on at 120 s). Image of a 50 μm thick printed film viewed against the light (D) and a three-layer printed model (E) both based on T/MIL-53(Al)–NH2/MMA ink. | ||
Subsequently, MOF-based blends with selected commercial oligomers (T, P, E components) were admixed in a certain proportion (Table S1, entries 7, 8 and 10, ESI†) and the kinetics of MMA conversion were elucidated. The polymerization rates of the heterogeneous formulations diminish by a factor of 3–4 compared to those of the systems without the MIL-53-NH2/MMA component (Fig. 3B), still reaching the same conversion levels observed for homogeneous mixtures (Fig. S25 and Table S7, ESI†). Among the tested blends, T/MIL-53-NH2/MMA demonstrated the fastest response to the light (Fig. 3B), as evidenced by a pronounced drop for the ca. 1640 cm−1 band of the MMA group in a 2D ATR-FTIR spectra series (Fig. 3C), and can be used for the DLP printing process to yield solid films and 3D models of desired shapes (Fig. 3D and E).
As a covalent integration of the MIL-53-NH2/MMA nanoparticles into the matrix is an important aspect to consider, the ability of the functionalized MOF component to be grafted to the acrylic moieties of the matrix was elucidated with the help of confocal Raman spectroscopy. Fig. 4A depicts the Raman maps on the 30 × 30 μm2 area of the PSM-MOF-containing 3DP composite membrane in which the contrast scale corresponds to the intensity of the C–(CO)–O symmetrical stretching band (∼600 cm−1) attributed to the MMA moieties involved in copolymerization (Fig. S26D, ESI†). The contrast scale ranges from the strongest (wine) to the weakest (navy) scattering intensities; the sketching zones with a lower crosslinking density of MMA in the dark blue and cyan regions visualize regions where the concentration of crosslinked MMA moieties is higher. Taking into account the previous findings that the MMA conversion in MIL-53-NH2/MMA-acrylate systems usually does not exceed 27–30% in total and considering that the MIL-53-NH2/MMA component and matrix oligomer mixtures both have the crosslinked functionality, relatively homogenous cross-linking in the 3D printed composite despite PSM-MOF additive inclusion can be achieved. Furthermore, the intensity ratios of specific Raman bands related to the acrylic part (both matrix and PSM-NH2/MMA MOF) and the aromatic component of the linker (PSM-NH2/MMA MOF) were compared on 2D contrast maps to provide information on the local chemical composition and distribution of the MOF additive in the accessible depth of the 3DP material. The distribution of MIL-53-NH2/MMA nanoparticles within the acrylic matrix was evaluated from the intensity ratios of the Raman band centered at ∼1450 cm−1 (the aromatic ring C–C chain vibration in the MIL-53(Al) framework) relative to the CO symmetric stretching vibration in the MMA group located at ∼1730 cm−1 (Fig. S26A–C, ESI†). As shown in Fig. 4B, the distinguished regions of the PSM-NH2/MMA MOF component can be mapped where the pixels of colours from cyan to yellow are located, occupying areas of one order of magnitude bigger than the average nanoparticle size of the MIL-53-NH2/MMA additive. The FIB-SEM cross-sectional studies of the 3DP composite membrane further disclose the MIL-53-NH2/MMA clustering and unveil a structural feature of the observed lumps (Fig. 4C). Although the MOF distribution within the polymerized matrix is not perfectly uniform (Fig. 4E and F), individual MIL-53-NH2/MMA nanoparticles are surrounded by the polymer, suggesting a proper integration of the filler without the formation of voids. Notably, the structure of the polyacrylate matrix itself does not possess visible intrinsic porosity compared to that of the conventional polymeric matrices used in the preparation of MMMs (Fig. 4D). The gas adsorption measurements using CO2 as a probe molecule further corroborate this observation (Fig. S27, ESI†). Despite the chemical differences in the acrylate systems employed as a matrix precursor, the shapes of the adsorption isotherms reveal large sorption–desorption hysteresis loops along a wide range of pressures as a common attribute of glassy polymers49 albeit severe divergence in the total adsorption capacity from 0.01 mmol g−1 for the T- to 0.14 mmol g−1 for the U-component and ∼0.17 mmol g−1 for the 2/3T + 1/3U mixture (Fig. S27A, ESI†).
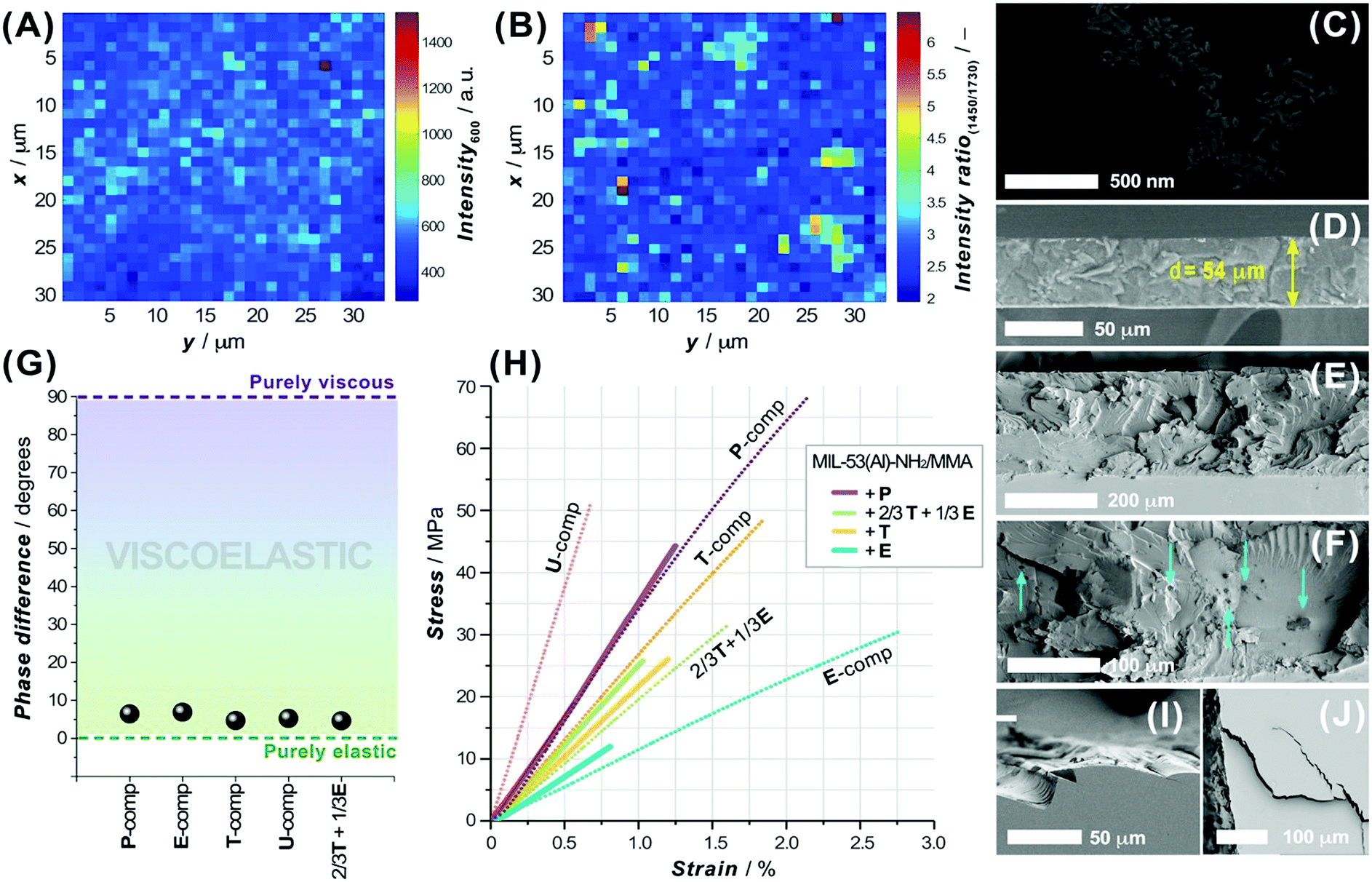 | ||
| Fig. 4 (A and B) Raman maps of a 3D printed membrane (30 × 30 μm2 area, step size of 1 μm) depicting the spatial distribution of (A) the intensity of the characteristic band from MMA functionality and (B) the band intensity ratios of the distinguished frequencies in MIL-53(Al) relative to the MMA component. (C) FIB-SEM cross-section and (D) SEM image of an N2 cracked 54 μm thick T/MIL-53-NH2/MMA 3DP membrane (3DP-M). (E and F) Cross-sectional (liquid N2) views of 3DP-M at different magnifications revealing MIL-53-NH2/MMA lumps (indicated with arrows). (G) Phase difference (δ) between the stress and strain values of pure P, E, T, U-3DP systems and 2/3T + 1/3E mixture on a viscoelastic reference diagram. (H) Stress and strain curves of pure P, E, T, U and 2/3T + 1/3E 3DP materials (dotted lines) and their MOF-based composites (solid lines). Brittle fracture (I) and cracks (J) produced by the fracturing of a T/MIL-53-NH2/MMA 3DP-M. | ||
The hysteresis loop feature is a characteristic aspect of many polymethylmethacrylate systems and is caused by the ability of these systems to undergo CO2-induced swelling associated with a glassy-to-rubbery state transition.50–52 As expected, the adsorption capacity of a printed composite increases with an inclusion of a MIL-53-NH2/MMA component (Fig. S27B, ESI†), which contributes to the boost of the total gas uptake at its microporous surface.
In order to be used in gas separation processes, either purely polymeric or composite membranes require sufficient mechanical stability and flexible resistivity at the conditions close to real operation settings.53 To assess the mechanical properties of the DLP 3D printed materials studied here, specimens of various photopolymerizable systems and blends were tested by applying dynamic mechanical and uniaxial tensile analyses. The dynamic tensile data for a commercially available acrylate ink (P, E, T and U), used as a matrix component in our formulations, and a mixture (2/3T + 1/3E) were compared using a viscoelastic reference diagram (Fig. 4G). Evidently, the elastic nature of these photopolymerizable matrix systems significantly prevails over their viscous counterparts and the phase difference (δ) in the stress–strain responses deviates slightly from one component to another. As anticipated, the MIL-53-NH2/MMA-based composites exhibit a lower damping factor (tanδ) compared to that of a pure polymeric material, agreeing well with the commonly observed trends (Table S8, ESI†).54 Furthermore, the shape of the stress–strain curves for the 3DP polymeric and MOF-based materials suggests the brittle nature of the acrylate crosslinked systems, whose chemical composition defines the mechanical properties expressed in terms of Young's modulus (E), and stress (σR) and strain at break (εR) values (Table S8, ESI†). Hence, the 3DP U-component shows the highest tensile strength (σR = 50.6 MPa, εR = 0.67%) among the other matrix inks, while the E-system, in turn, reveals superior toughness (σR = 30.2 MPa, εR = 2.75%). Certainly, the brittle behaviour of the 3DP composite can also be noted in the corresponding cross-sectional views (Fig. 4E, I and J), where typical brittle fracture patterns and cracks can be observed. It is worth noting that the MOF filler commonly contributes to an earlier rupture of the 3DP composite, presumably due to the formation of discretely localized heterogeneous inclusions and defects that can contribute to the origin and propagation of cracks. Overall, the MIL-53-NH2/MMA-based acrylate 3DP materials display high tensile strengths and are able to withstand the conditions applied in gas separation processes.
Membranes based on a polymer in the glassy state are known to be particularly attractive for the selective separation of H2 from H2/CO2 mixtures.55 Considering the above information, the 3DP acrylate-based membranes were assumed to be applicable in H2/CO2 separation and therefore subjected to mixed gas separation tests to unveil their potential. The selection of the 3DP membrane formulation was rationalized considering the optimal DLP processing parameters, mechanical properties discussed above and glass transition temperatures (Tg) of pure continuous phases and MIL-53-NH2/MMA-based composites (Table S11, ESI†). The U-based formulations were excluded due to their poor DLP processing performance, whereas P-systems show a comparatively low Tg value (Table S11, ESI†) limiting the available temperature range of H2/CO2 gas separation. Thus, the T-component system was selected for further separation of an equimolar mixture of H2 and CO2 at two different temperatures (323 K and 373 K) and 2 bar absolute feed pressure. All the prepared membranes can withstand the permeation test conditions. Fig. 5A summarizes the separation data for the pure T-matrix and T + MIL-53-NH2/MMA composite 3DP membranes.
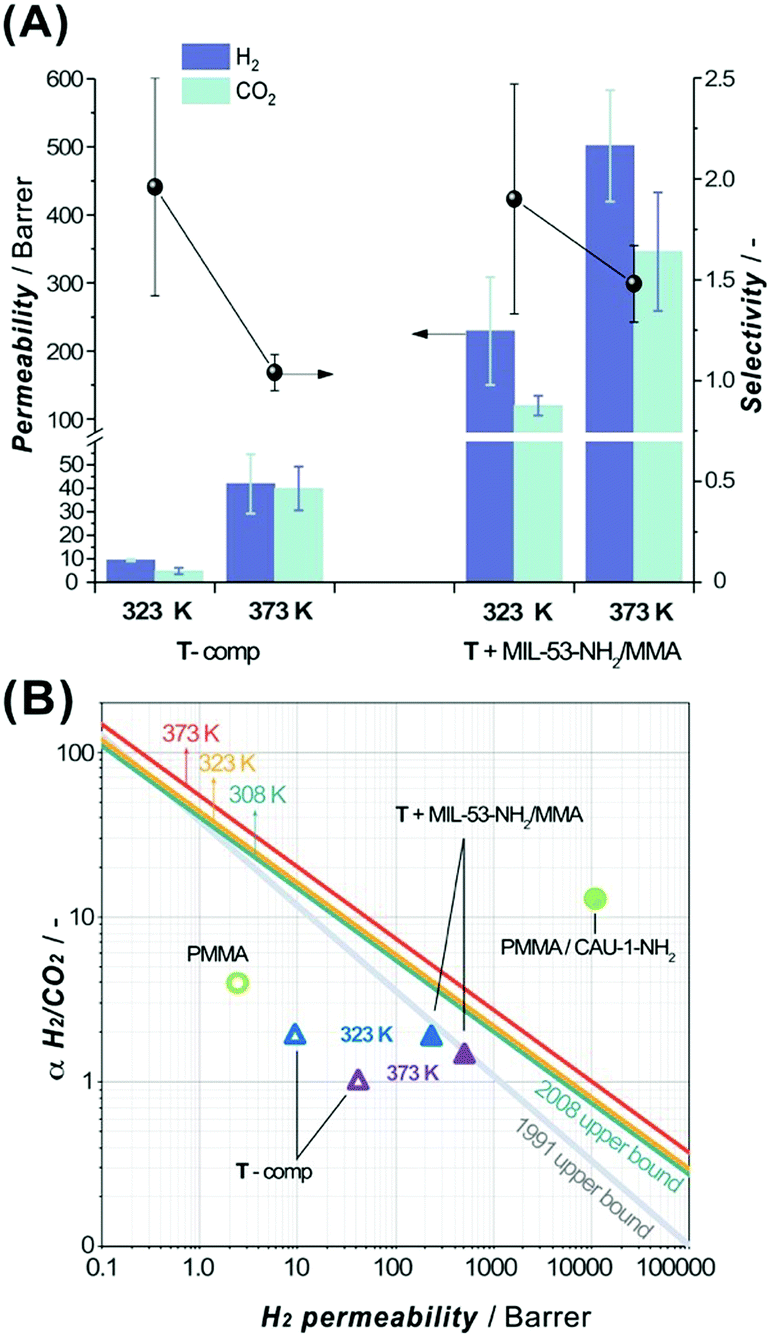 | ||
| Fig. 5 (A) H2 (violet columns, mint error bars) and CO2 (mint columns, violet error bars) permeabilities and H2/CO2 selectivity (black spheres, black error bars) for a 3DP membrane containing pure T-comp or T + MIL-53-NH2/MMA (12 wt% of MOF component) formulation in an equimolar H2/CO2 gas mixture at different temperatures (323 K and 373 K) and measured at 2 bar absolute feed pressure. (B) Comparison of separation performance for T-comp (empty triangle) or T + MIL-53-NH2/MMA (solid triangle) 3DP membrane (at 323 K and 373 K, blue and violet, respectively) with previously reported pure PMMA (2 bar, 303 K, green empty circle)57 and PMMA/CAU-1-NH2 (green solid circle, 3 bar, 298 K, 15 wt% of MOF) composites.56 The lines represent the 1991 Robeson upper bounds (grey line) and updated to 2008, plotted for 308 K (green), 323 K (yellow) and 373 K (red).58,59 | ||
The incorporation of MIL-53-NH2/MMA nanoparticles (12 wt% loading) crosslinked with a continuous matrix leads to a significant increase in H2 and CO2 permeabilities preserving the original T-component membrane selectivity at 323 K. These observations agree with the fact that the porous filler can provide additional pathways for the permeating gases. However, the low H2/CO2 selectivity in the studied membranes compared to that of the other reported relevant MMMs56,57 (Fig. 5B) suggests the presence of non-selective paths for penetrant gas molecules around the filler particles. Upon increasing the temperature up to 373 K, the permeabilities of the bare T-polymer and T + MIL-53-NH2/MMA composites both increase about one order of magnitude (PH2 = 41.9 ± 12.6, PCO2 = 39.9 ± 9.3 versus PH2 = 9.4 ± 0.4, PCO2 = 4.8 ± 1.4 Barrer and PH2 = 501.4 ± 81.9, PCO2 = 345.9 ± 87.1 versus PH2 = 229.3 ± 79.3, PCO2 = 119.6 ± 14.4 Barrer, respectively, Table S12, ESI†), and meanwhile, the H2/CO2 selectivity drops, reflecting a loss of the size-sieving ability, most significantly in the case of pure T-acrylate matrix. The permeability enhancement can be attributed to an increase in the diffusivity of the gases through the polymeric matrix at higher temperatures. It should be noted that the milder drop of the selectivity in the MIL-53-NH2/MMA-grafted T-composite membrane at 373 K indicates a beneficial collaborative interaction between the continuous and dispersed phases, resulting in a selective contribution of the filler to the total H2/CO2 sieving performance. The permeation results of the pure T-matrix and T + MIL-53-NH2/MMA composite membranes at 323 K and 373 K are plotted against the 2008 Robeson upper bounds59 for selected temperatures (Fig. 5B).58 Specifically, the separation performance of the 3DP membranes explored in this study lies below the empirical selectivity-permeability trend line for the H2/CO2 gas pair and occupies a modest position in the graph. Compared to the best performing and chemically related polymethyl methacrylate (PMMA)-based MMM-containing CAU-1-NH2 filler (Fig. 5B), the 3DP T + MIL-53-NH2/MMA membrane demonstrates significantly lower permeability and selectivity. This can be explained in light of chemical, textural and structural differences between these two systems. As 3DP DLP processing requires highly crosslinked methacrylic matrices to enable control over the resulting solid shape, the low permeability is an expected and inevitable outcome, whereas MMMs prepared by a conventional casting method can possess large free volumes that result in high permeabilities.56 Moreover, the preliminary obtained gas separation results for the T + MIL-53-NH2 formulation comprising pristine MOF revealed a non-selective behaviour caused by the poor interaction between the filler and polymer which leads to the formation of defects, and these results were approved using the MOF grafted with methacrylic functionality. Overall, the obtained results suggest that the 3D DLP technology can be used in fast prototyping of MOF-based MMMs, leaving room for further development of ink formulations by varying the content of the polymeric matrix along with the type of the MOF component.
4. Conclusion
In this work, we have demonstrated a fast and convenient approach to manufacture MOF-based polymer composite membranes using a conventional DLP 3D printer. MOF-based photopolymerizable inks formulated from post-synthetically functionalized MIL-53(Al)–NH2/MMA nanoparticles and acrylate oligomers are fully compatible with conventional 3D printing. It was evidenced that the MOF-based photopolymerizable systems undergo a photoinduced crosslinking with the matrix components at a relatively fast rate reaching the degree of crosslinking comparable with the pure oligomer mixtures. Hence, fulfilling the criteria for the DLP process, the MIL-53(Al)–NH2/MMA-containing inks can be transformed into free-standing composite membranes with desirable thickness and shape. The mechanical properties of the resulting 3D printed composites, glassy state of the polymeric media and microporosity provided by the MOF component enable their use as mixed matrix membranes for gas separation applications. The sieving behavior of the 3D printed MOF-MMMs in an equimolar H2/CO2 gas mixture indicates the enhanced permeability of the MIL-53(Al)–NH2/MMA-containing composite in comparison to that observed for the bare polymer.Finally, it was shown that the 3D printing technology involving digital light processing could be successfully applied for fast prototyping of MOF-based mixed membranes and sets a basis for further development in this field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P., E. A.-H. and J. G. acknowledge King Abdullah University of Science and Technology (KAUST) for the financial support. B. S. was supported by her personal VENI grant (The Netherlands National Science Foundation, NWO). The authors gratefully acknowledge Sartomer (Arkema Group) for providing them with the samples of acrylate oligomer mixtures used in this study.References
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212 CrossRef CAS.
- M. R. Hartings and Z. Ahmed, Nat. Rev. Chem., 2019, 3, 305 CrossRef.
- L. D. Tijing, J. R. C. Dizon, I. Ibrahim, A. R. N. Nisay, H. K. Shon and R. C. Advincula, Appl. Mater. Today, 2020, 18, 100486 CrossRef.
- C. Parra-Cabrera, C. Achille, S. Kuhn and R. Ameloot, Chem. Soc. Rev., 2018, 47, 209 RSC.
- R. L. Truby and J. A. Lewis, Nature, 2016, 540, 371 CrossRef CAS PubMed.
- T. D. Ngo, A. Kashani, G. Imbalzano, K. T. Q. Nguyen and D. Hui, Composites, Part B, 2018, 143, 172 CrossRef CAS.
- X. Zhou and C.-j. Liu, Adv. Funct. Mater., 2017, 27, 1701134 CrossRef.
- X. Yun, B. Lu, Z. Xiong, B. Jia, B. Tang, H. Mao, T. Zhang and X. Wang, RSC Adv., 2019, 9, 29384 RSC.
- E. Davoodi, H. Montazerian, R. Haghniaz, A. Rashidi, S. Ahadian, A. Sheikhi, J. Chen, A. Khademhosseini, A. S. Milani, M. Hoorfar and E. Toyserkani, ACS Nano, 2020, 14, 1520 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468 CrossRef CAS PubMed.
- A. Pustovarenko, A. Dikhtiarenko, A. Bavykina, L. E. M. Gevers, A. Ramirez, A. Russkikh, S. Telalovic, A. A. Tapia, J. L. Hazemann, S. Ould-Chikh and J. Gascon, ACS Catal., 2020, 10, 5064 CrossRef CAS.
- B. Seoane, J. Coronas, I. Gascon, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn and J. Gascon, Chem. Soc. Rev., 2015, 44, 2421 RSC.
- J. Yang and Y.-W. Yang, Small, 2020, 16, 1906846 CrossRef CAS PubMed.
- W.-T. Koo, J.-S. Jang and I.-D. Kim, Chem, 2019, 5, 1938 CAS.
- L. L. da Luz, R. Milani, J. F. Felix, I. R. B. Ribeiro, M. Talhavini, B. A. D. Neto, J. Chojnacki, M. O. Rodrigues and S. A. Júnior, ACS Appl. Mater. Interfaces, 2015, 7, 27115 CrossRef CAS PubMed.
- J. L. Zhuang, D. Ar, X. J. Yu, J. X. Liu and A. Terfort, Adv. Mater., 2013, 25, 4631 CrossRef CAS PubMed.
- H. Thakkar, S. Eastman, Q. Al-Naddaf, A. A. Rownaghi and F. Rezaei, ACS Appl. Mater. Interfaces, 2017, 9, 35908 CrossRef CAS PubMed.
- P. Pei, Z. Tian and Y. Zhu, Microporous Mesoporous Mater., 2018, 272, 24 CrossRef CAS.
- H. Thakkar, Q. Al-Naddaf, N. Legion, M. Hovis, A. Krishnamurthy, A. A. Rownaghi and F. Rezaei, ACS Sustainable Chem. Eng., 2018, 6, 15228 CrossRef CAS.
- J. Lefevere, B. Claessens, S. Mullens, G. Baron, J. Cousin-Saint-Remi and J. F. M. Denayer, ACS Appl. Nano Mater., 2019, 2, 4991 CrossRef CAS.
- G. J. H. Lim, Y. Wu, B. B. Shah, J. J. Koh, C. K. Liu, D. Zhao, A. K. Cheetham, J. Wang and J. Ding, ACS Mater. Lett., 2019, 1, 147 CrossRef CAS.
- Z. Lyu, G. J. H. Lim, R. Guo, Z. Kou, T. Wang, C. Guan, J. Ding, W. Chen and J. Wang, Adv. Funct. Mater., 2019, 29, 1806658 CrossRef.
- S. Lawson, Q. Al-Naddaf, A. Krishnamurthy, M. S. Amour, C. Griffin, A. A. Rownaghi, J. C. Knox and F. Rezaei, ACS Appl. Mater. Interfaces, 2018, 10, 19076 CrossRef CAS PubMed.
- S. Sultan, H. N. Abdelhamid, X. D. Zou and A. P. Mathew, Adv. Funct. Mater., 2019, 29, 1805372 CrossRef.
- A. J. Young, R. Guillet-Nicolas, E. S. Marshall, F. Kleitz, A. J. Goodhand, L. B. L. Glanville, M. R. Reithofer and J. M. Chin, Chem. Commun., 2019, 55, 2190 RSC.
- J. Dhainaut, M. Bonneau, R. Ueoka, K. Kanamori and S. Furukawa, ACS Appl. Mater. Interfaces, 2020, 12, 10983 CrossRef PubMed.
- M. Bible, M. Sefa, J. A. Fedchak, J. Scherschligt, B. Natarajan, Z. Ahmed and M. R. Hartings, 3D Print. Addit. Manuf., 2018, 5, 63 CrossRef.
- M. N. Channell, M. Sefa, J. A. Fedchak, J. Scherschligt, M. Bible, B. Natarajan, N. N. Klimov, A. E. Miller, Z. Ahmed and M. R. Hartings, Polym. Adv. Technol., 2018, 29, 867 CrossRef PubMed.
- K. A. Evans, Z. C. Kennedy, B. W. Arey, J. F. Christ, H. T. Schaef, S. K. Nune and R. L. Erikson, ACS Appl. Mater. Interfaces, 2018, 10, 15112 CrossRef CAS PubMed.
- O. Halevi, J. M. R. Tan, P. S. Lee and S. Magdassi, Adv. Sustainable Syst., 2018, 2, 1700150 CrossRef.
- E. Lahtinen, R. L. M. Precker, M. Lahtinen, E. Hey-Hawkins and M. Haukka, ChemPlusChem, 2019, 84, 222 CrossRef CAS PubMed.
- R. Li, S. Yuan, W. Zhang, H. Zheng, W. Zhu, B. Li, M. Zhou, A. Wing-Keung Law and K. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 40564 CrossRef CAS PubMed.
- A. Figuerola, D. A. V. Medina, A. J. Santos-Neto, C. P. Cabello, V. Cerdà, G. T. Palomino and F. Maya, Appl. Mater. Today, 2019, 16, 21 CrossRef.
- J. Dechnik, J. Gascon, C. J. Doonan, C. Janiak and C. J. Sumby, Angew. Chem., Int. Ed., 2017, 56, 9292 CrossRef CAS PubMed.
- Y. Zhang, X. Feng, H. Li, Y. Chen, J. Zhao, S. Wang, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2015, 54, 4259 CrossRef CAS PubMed.
- A. Sabetghadam, B. Seoane, D. Keskin, N. Duim, T. Rodenas, S. Shahid, S. Sorribas, C. Le Guillouzer, G. Clet, C. Tellez, M. Daturi, J. Coronas, F. Kapteijn and J. Gascon, Adv. Funct. Mater., 2016, 26, 3154 CrossRef CAS PubMed.
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848 RSC.
- American Society for Testing and Materials and ASTM, ASTM D2765-11, 2006.
- P. Serra-Crespo, E. Gobechiya, E. V. Ramos-Fernandez, J. Juan-Alcañiz, A. Martinez-Joaristi, E. Stavitski, C. E. A. Kirschhock, J. A. Martens, F. Kapteijn and J. Gascon, Langmuir, 2012, 28, 12916 CrossRef CAS PubMed.
- A. Pustovarenko, M. G. Goesten, S. Sachdeva, M. Shan, Z. Amghouz, Y. Belmabkhout, A. Dikhtiarenko, T. Rodenas, D. Keskin, I. K. Voets, B. M. Weckhuysen, M. Eddaoudi, L. C. P. M. de Smet, E. J. R. Sudhölter, F. Kapteijn, B. Seoane and J. Gascon, Adv. Mater., 2018, 30, 1707234 CrossRef.
- S. Castellanos, K. B. Sai Sankar Gupta, A. Pustovarenko, A. Dikhtiarenko, M. Nasalevich, P. Atienzar, H. García, J. Gascon and F. Kapteijn, Eur. J. Inorg. Chem., 2015, 4648 CrossRef CAS.
- S. Couck, E. Gobechiya, C. E. A. Kirschhock, P. Serra-Crespo, J. Juan-Alcañiz, A. Martinez Joaristi, E. Stavitski, J. Gascon, F. Kapteijn, G. V. Baron and J. F. M. Denayer, ChemSusChem, 2012, 5, 740 CrossRef CAS PubMed.
- S. Devautour-Vinot, G. Maurin, C. Serre, P. Horcajada, D. Paula da Cunha, V. Guillerm, E. de Souza Costa, F. Taulelle and C. Martineau, Chem. Mater., 2012, 24, 2168 CrossRef CAS.
- M. Haouas, C. Volkringer, T. Loiseau, G. Férey and F. Taulelle, Chem. – Eur. J., 2009, 15, 3139 CrossRef CAS PubMed.
- J. M. Chin, E. Y. Chen, A. G. Menon, H. Y. Tan, A. T. S. Hor, M. K. Schreyer and J. Xu, CrystEngComm, 2013, 15, 654 RSC.
- B. Seoane, S. Castellanos, A. Dikhtiarenko, F. Kapteijn and J. Gascon, Coord. Chem. Rev., 2016, 307, 147 CrossRef CAS.
- E. Gulari, K. McKeigue and K. Y. S. Ng, Macromolecules, 1984, 17, 1822 CrossRef CAS.
- R. M. Conforti and T. A. Barbari, Macromolecules, 1993, 26, 5209 CrossRef CAS.
- X.-K. Li, G.-P. Cao, L.-H. Chen, R.-H. Zhang, H.-L. Liu and Y.-H. Shi, Langmuir, 2013, 29, 14089 CrossRef CAS PubMed.
- D. Liu, H. Li, M. S. Noon and D. L. Tomasko, Macromolecules, 2005, 38, 4416 CrossRef CAS.
- A. Rajendran, B. Bonavoglia, N. Forrer, G. Storti, M. Mazzotti and M. Morbidelli, Ind. Eng. Chem. Res., 2005, 44, 2549 CrossRef CAS.
- K. Wang, A. A. Abdala, N. Hilal and M. K. Khraisheh, in Membrane Characterization, ed. N. Hilal, A. F. Ismail, T. Matsuura and D. Oatley-Radcliffe, Elsevier, 2017, p. 259, DOI:10.1016/b978-0-444-63776-5.00013-9.
- V. Panwar and K. Pal, in Clay–Polymer Nanocomposites, ed. K. Jlassi, M. M. Chehimi and S. Thomas, Elsevier, 2017, p. 413, DOI:10.1016/b978-0-323-46153-5.00012-4.
- L. Shao, B. T. Low, T.-S. Chung and A. R. Greenberg, J. Membr. Sci., 2009, 327, 18 CrossRef CAS.
- L. Cao, K. Tao, A. Huang, C. Kong and L. Chen, Chem. Commun., 2013, 49, 8513 RSC.
- C. J. Orme, M. L. Stone, M. T. Benson and E. S. Peterson, Sep. Sci. Technol., 2003, 38, 3225 CrossRef CAS.
- B. W. Rowe, L. M. Robeson, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2010, 360, 58 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical schemes of commercial oligomers; tables with ink formulation composition; DLP 3D printed details and specifications; additional figures; N2 and Ar adsorption data and plots; TEM images and particle size distribution histograms; NMR data and plots; FTIR; viscosity data; ATR-IR, kinetic data of photopolymerization in systems with and without MOF additive; Raman spectra and mapping; mechanical data; optic microscope images; SEM-EDX maps; data on degree of crosslinking; TG and DSC analyses; gas separation performance data. See DOI: 10.1039/d1ma00023c |
| This journal is © The Royal Society of Chemistry 2021 |