
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion effects on the properties of OIPC/PVDF composites†
Frederick
Nti
 a,
George W.
Greene
a,
Haijin
Zhu
a,
Patrick C.
Howlett
a,
Maria
Forsyth
ab and
Xiaoen
Wang
*a
a,
George W.
Greene
a,
Haijin
Zhu
a,
Patrick C.
Howlett
a,
Maria
Forsyth
ab and
Xiaoen
Wang
*a
aInstitute for Frontier Materials, Deakin University, Geelong, VIC 3217, Australia. E-mail: xiaoen.wang@deakin.edu.au
bAustralian Centre of Excellence for Electromaterials Science, Monash University, Clayton, Victoria 3800, Australia
First published on 4th February 2021
Abstract
Organic ionic plastic crystals (OIPCs) are promising solid electrolytes because of their inherent advantages such as non-volatility, non-flammability, good thermal stability, favourable plasticity, and improved electrolyte/electrode interfacial contact. Incorporating nanoparticles into some OIPC matrixes has proven an effective strategy for further increasing conductivity and mechanical integrity. However, the nature of the interaction between the cations and anions of OIPCs with polymers requires further study. In this work, various OIPCs consisting of N-ethyl-N-methyl pyrrolidinium cations ([C2mpyr]+) and three different anions bis(trifluoromethanesulfonyl)imide ([TFSI]−), (bis(fluorosulfonyl)imide ([FSI]−) and tetrafluoroborate, [BF4]−) are selected. The interactions between the OIPCs and poly(vinylidene fluoride) (PVDF) are studied by incorporating different volume fractions of PVDF nanoparticles into the OIPC matrices. Differential scanning calorimetry (DSC), electrochemical impedance spectroscopy (EIS), nuclear magnetic resonance (NMR) and synchrotron X-ray diffraction (XRD) techniques confirm that the OIPCs with different anions interact differently with PVDF. Furthermore, we propose that the degree of the interaction and ionic conductivity enhancement relates to different dipole moments of the OIPC anions. This anion originated interaction induces the formation of disordered OIPC interphases which accounts for the ionic conductivity enhancement.
1. Introduction
Organic ionic plastic crystals (OIPCs), like some crystalline materials, consist of ions located at specific lattice positions, and have ordered structures. They are different from regular crystalline materials because they exhibit temperature dependent solid–solid phase transitions which depict the onset of rotational or translational motion of their molecules/ions.1,2 With each new transition, new conduction modes become accessible, which accounts for the sudden increase in ionic conductivities observed as the material progresses from a more ordered phase to a less ordered phase.3 Also, the phase I of plastic crystals, which is the phase right before plastic crystals melt, is characterized by high degree of freedom for rotational or translational motion of their ions, and this leads to the enhanced conductivity. Plastic crystals can be grouped into; inorganic salts (such as Li2SO4),4 organic molecular species such as succinonitrile5 and organic ionic plastic crystals (OIPCs).6OIPCs are non-volatile and non-flammable safe ionic materials owing to the ionic interactions between their charged species.7–9 They possess a three-dimensional lattice structure similar to inorganic ionic conductors, and yet they are chemically similar to ionic liquids (ILs).1 Their soft characteristic in their plastic phase is appealing because it overcomes the poor point-to-point contact problems existing in most solid-state inorganic fast ion conductors while overcoming the leakage concerns associated with liquid electrolytes.10 Also, their short-range ionic motions allow for fast ion conduction. Similar to ILs, OIPCs allow the design of functionalized electrolytes because of the possibility of selecting choice cations and anions to form the resultant electrolytes.11–15 These properties qualify OIPCs as advanced solid electrolytes for next-generation applications such as batteries, supercapacitors, fuel cells, thermo-cells, dye-sensitised solar cells (DSSCs), etc.6,16,17
In order to enhance the commercial viability of solid electrolytes, OIPCs should not only be safe and environmentally friendly but also, they should have conductivities comparable to liquid ones.18 Unfortunately, in their pristine forms, most OIPCs have relatively low ionic conductivity. N,N-Dimethyl pyrrolidinium bis(fluorosulfonyl)imide ([C2epyr][FSI]), one of the most conductive OIPCs, has an ionic conductivity of 1.9 × 10−5 S cm−1 at 30 °C which is still lower than those of ILs.11 Also, the soft and waxy nature of OIPCs makes the design of free-standing electrolytes a huge challenge. To overcome this challenge, secondary components such as polymer nanoparticles, polymer nanofibres and inorganic nanoparticles have been employed to improve mechanical stability.19–23 In some cases, the introduction of secondary components has been observed to contribute significant enhancement of ionic conductivity. Howlett et al. designed a free-standing membrane based on electrospun poly(vinylidene fluoride) (PVDF) fibres and N-ethyl,N-methyl pyrrolidinium tetrafluoroborate ([C2mpyr][BF4]) for solid-state lithium battery applications. The designed membrane exhibited improved mechanical stability as well as increased conductivity.24 Wang et al. developed PVDF particles coated with Li-doped N-ethyl,N-methyl pyrrolidinium bis(fluorosulfonyl)imide (Li-[C2mpyr][FSI]). Encouragingly, the pressed composite electrolyte pellets (containing only ∼40 wt% of OIPC) showed improved ionic conductivity by one order of magnitude. Also, replacing more than half of the expensive OIPC with cheap PVDF leads to the design of cheaper electrolytes.25
Although enhanced ionic conductivity of OIPCs has been achieved in previous studies, the origin and mechanism of this enhancement is still not clear. Iranipour et al. sought to understand the role of electrospun PVDF nanofibres in the matrix of neat and Li-doped [C2mpyr][BF4].19 They observed that the presence of polymer nanofibres did not change the ion ordering of the OIPC in each crystalline phase but rather, the crystal lattices were expanded. This suggests that the presence of PVDF nanofibres increases the disorder in the OIPC matrix, resulting in increased ionic conductivity even at lower temperatures. In these and more recent works, we have demonstrated the influence of interfacial interaction on the properties (ionic conductivity and phase behaviours) of OIPC/polymer composites.26–28 Recently, we investigated the effect of introducing different volume ratios of two different polymer nanoparticles, PVDF and polystyrene (PS) and studying their effects on the OIPC, [C2mpyr][FSI]. We demonstrated that the intrinsic surface chemistry of the polymer nanoparticles effectively affects the phase behaviours and ion dynamics of OIPC/polymer composite. Particularly, [C2mpyr][FSI]/PVDF composites showed increasing ionic conductivity with increasing volume fractions of PVDF nanoparticles until the percolation threshold was approached, in agreement with previous reports for similar composites.25,27,29 On the other hand, increasing volume fractions of PS particles led to a sharp decline in ionic conductivity of [C2mpyr][FSI]/PS composites because of the increased tortuosity resulting from the clustering of the hydrophobic PS particles.30
It is known that the properties of cations and anions define OIPC properties such as phase behaviour, physical and transport properties of the resulting electrolytes.11 Generally, anions with diffused charges have reduced interaction with their cations compared to anions with localised charges.31 This will subsequently affect the interaction of the anion with secondary components. Yunis et al. reported hexamethylguanidinium cation ([HMG]) based OIPCs with different anions (bis(trifluoromethanesulfonyl)imide [TFSI]−, hexafluorophosphate [PF6]−, [BF4]−, [FSI]− and trifluoromethanesulfonyl(fluorosulfonyl)imide [FTFSI]−). It was observed that smaller symmetrical anions such as [PF6]− and [BF4]− form OIPCs that have high melting points and low ionic conductivities, while the OIPCs with anions having diffused charges show lower melting points and higher ionic conductivities.32
R. Mejri et al. investigated the effects of the ionic species of ionic liquids on the physical and chemical properties of IL/PVDF blends. They observed that the β-phase content of PVDF, known for its piezoelectric properties, was increased upon addition of the ILs. The melt phase of PVDF was observed to be significantly altered depending on the ILs used, suggesting different degrees of interaction. Also, different cation and anion combinations affected the conductivity of the resulting IL/PVDF composites differently.33 In their study of the effects of two phosphonium ILs on poly(vinylidene fluoride-co-chlorotrifluoroethylene) (PVDF-co-CTFE), Yang et al. observed a nanostructuration mechanism which was dependent on the diffusion and degree of interaction of the IL and the polymers. This nanostructuration mechanism could be controlled by altering the chemical structures (e.g. steric effects, polar groups) of the ILs.34 The dipolar Coulombic attraction between hydrogen atoms in PVDF chains and anions in the IL have been observed to promote the formation of β-phase PVDF, thus, making the resulting IL/PVDF composite multifunctional.35,36 The interaction between ILs and PVDF nanofibres alters the morphology as well as the mean diameter of the PVDF nanofibres.37,38
Compared with ILs, OIPCs consist of smaller cations or anions, which provides them different chemical polarity or interactions with their chemical environments. However, the interaction between different anions of OIPCs and PVDF have yet to be fully investigated. In this work, we study the impacts of three anions ([FSI]−, [TFSI]− and [BF4]−) on pyrrolidinium based OIPC/PVDF composites. These anions were selected because of their different chemistries. PVDF was employed in this work due to its different polymorphic phases that have attractive piezoelectric,39 pyroelectric40 and ferroelectric41 properties, and also the potential applications of the resultant composites on batteries.25,42 The interfacial effects observed in the interaction of these OIPCs with PVDF nanoparticles are systematically studied and the origin of these different interactions are discussed.
2. Experimental
2.1 Materials and sample preparation
[C2mpyr][FSI] and [C2mpyr][TFSI] were synthesised by a previously established procedure.43–45 [C2mpyr][BF4] was synthesised by altering a literature method.46 PVDF nanoparticles (KF850, Mw = 3 × 105, Kureha Chemicals, Japan) were dried in a vacuum oven for 2 days at 80 °C before use and methanol (>99%, Sigma-Aldrich) was used as received. Fig. 1 shows the chemical structures of the OIPCs and PVDF used in this work. | ||
| Fig. 1 Chemical structures of; (a) cations used in this work (N-ethyl,N-methyl pyrrolidinium [C2mpyr]+) (b) anions used in this work (bis(fluorosulfonyl)imide) [FSI]−, bis(trifluoromethanesulfonyl)imide [TFSI]− and tetrafluoroborate [BF4]− and (c) repeating units of poly(vinylidene fluoride), (PVDF). | ||
2.2 Composite preparation
The composite materials were prepared by dissolving a calculated amount of OIPC in methanol and dispersing a corresponding amount of PVDF nanoparticles in the resulting solution to form suspensions of OIPC and PVDF nanoparticles. A suspension with uniformly distributed PVDF nanoparticles is formed by sonication followed by stirring for about 30 minutes. After this, the solution is cast on a Petri-dish and quickly dried under flowing Ar. Each dry sample was collected from the Petri-dish and ground in an agate mortar to minimize the particle aggregation. To ensure that the samples are free of any solvents, the samples were further dried on the schlenk line at 50 °C for 24 hours. Finally, the dry samples are stored in Ar-filled glovebox for further characterisation.2.3 Material characterisation
To investigate the effects of different anions on the OIPC/PVDF composites, different volume fractions (10, 30, 50 and 70 vol% which are equivalent to 12, 35, 55 and 75 wt% respectively) of PVDF were added to the OIPCs to form composites. These composite samples were studied by using differential scanning calorimetry (DSC), electrochemical impedance spectroscopy (EIS), nuclear magnetic resonance (NMR) and synchrotron X-ray diffraction (XRD) techniques.The changes in the phase behaviour of the OIPC that occur on the addition of different volume fractions of PVDF were studied using DSC. A Mettler Toledo DSC1 instrument, powered by a STAReV6.10 software was used for all the thermal analysis. About 4–8 mg of sample was sealed in an Al pan inside an Ar-filled glovebox. The thermal history of the samples was treated by cooling to −110 °C and heating the samples to 60 °C, twice, before running the third cycles that are reported in this work. The samples were held at an isothermal temperature −110 °C for 30 minutes before heating. Heating and cooling were controlled at a rate of 2 °C min−1 for all the samples. This was done to ensure that the phase transitions of the samples were completed and to monitor all the possible interactions that occur in the samples. The reported DSC traces have been normalised to the mass of OIPC in the respective composites.
Ionic conductivity was measured by EIS on a Biologic MTZ-35, which runs on an MT-lab software. The composite samples were pressed into pellets in a sealed KBr die under 4 tons of pressure for 10 minutes. For the EIS measurements, the pellet with a diameter of 13 mm was sandwiched between two stainless steel disks and assembled into a coin cell in Ar-glove box to eliminate any moisture contamination. The coin cell was then inserted into a hermetically-sealed barrel cell for conductivity analysis.47 The frequency studied ranged from 10 MHz to 1 Hz with a voltage amplitude of 10 mV within a temperature range of 30 °C to 120 °C at 10 °C steps. The system was allowed to equilibrate for 20 minutes at each target temperature before each measurement was taken. The impedance values were taken at the point at which the semi-circle of the Nyquist plot touched the x-axis at the high-frequency region.
Cation and anion mobilities were studied using variable temperature static 1H and 19F NMR. The samples were packed in 4 mm rotors in an Ar-filled glovebox and inserted into a Bruker AVANCE III 500 MHz wide-bore solid-state spectrometer equipped with a 4 mm H/F-X double resonance probe. The temperature was controlled by a BCU-II temperature control unit. The sample temperature within the probe was calibrated by the isotropic chemical shifts of 207Pb in PbNO3 using the method described in literature.48
Variable temperature XRD was employed to study the diffraction patterns of the neat OIPCs, OIPC/PVDF composites containing 50 vol% of PVDF and PVDF nanoparticles at the Australian Synchrotron. The samples were packed and sealed in 0.3 mm borosilicate glass capillaries (product of Charles Super Company, Massachusetts, USA) in Ar-filled glove box. The wavelength was set at 0.827 Å with a zero error of ±0.006 using a Si(111) double crystal monochromator prior to data collection and the current was ∼200 mA. The samples were cooled using an Oxford Cryosystems Cryostream, which can be heated with a Cyberstar hot-air blower up to 80 K. The ramp rate and equilibration time used for the experiment were 2 °C min−1 and 5 mins respectively. An array of 16 MYTHEN ID microstrip silicon detectors with each module spanning about 5 degrees in 2θ covered data collection over the angular range from 2° to 76°. The data were collected at selected temperatures in each phase of the plastic crystal for 20 s in two detector settings. The data collected from both detectors were merged for each measurement using the Pdviper software and the result plotted and analysed.
3. Results and discussion
3.1 Thermal properties
The thermal behaviours and phase transitions of various OIPC/PVDF composites are investigated and compared by DSC measurements and the results are shown in Fig. 2. Pristine [C2mpyr][TFSI] shows three solid–solid transitions and an intense melting peak (Fig. 2a). It has a crystalline phase I which starts from 46 °C to 89 °C.45 The peaks become distorted, and the intensity of the melting peak is reduced with increasing loading of PVDF. The melting peak appears to develop two components (86 and 89 °C) on addition of PVDF. With 30 vol% of PVDF (70/30), the shoulder peak becomes prominent, making the melt of the OIPC broad. At 50 vol% of PVDF, the shoulder in the melting peak occurring at 86 °C becomes more intense compared to the original melting peak. When 70 vol% of PVDF (30/70) is added, the original melting peak disappears leaving the shoulder peak, which suggests that interaction between the [C2mpyr][TFSI] and PVDF suppresses the crystallinity of the bulk [C2mpyr][TFSI]. Interestingly, there is a new sharp peak formed at 112 °C upon addition of PVDF. Although the origin of this peak is not fully understood, it is proposed to be a semi-crystalline PVDF-rich interphase composed of both PVDF and [C2mpyr][TFSI] cations and anions. To confirm whether this phenomenon is related to the [TFSI]− anion, we tested different PVDF-based composites incorporated with different OIPC or ionic liquids containing [TFSI]− anions. Surprisingly, similar peaks are seen in the DSC heating traces of PVDF based composites (Fig. S1a [P1222][TFSI]/PVDF and Fig. S1b [C3mpyr][TFSI]/PVDF composites, ESI†). This provides evidence that this new peak results from the interaction between [TFSI]− and PVDF. Another interesting observation is that the intensity of this new peak decreases with increasing of PVDF, which is probably due to the insufficient OIPC to interact with PVDF when high loading of PVDF is used. However, the exact phase structure of this new peak is worth investigation in our future work. Also, with increasing vol% of PVDF, two overlapping peaks appear which are shifted to higher temperatures with increasing PVDF loading. These two overlapping peaks (occurring at the 137.5 and 152.2 °C) are similar to those observed in [C2mpyr][FSI]/PVDF composites, as discussed later. The shifting of PVDF melting peaks to higher temperatures suggests that a higher component of less-interacting PVDF remain in the composite on increasing vol% of PVDF. Thus, the overlapping peaks approach the melting peak of the pristine PVDF nanoparticles.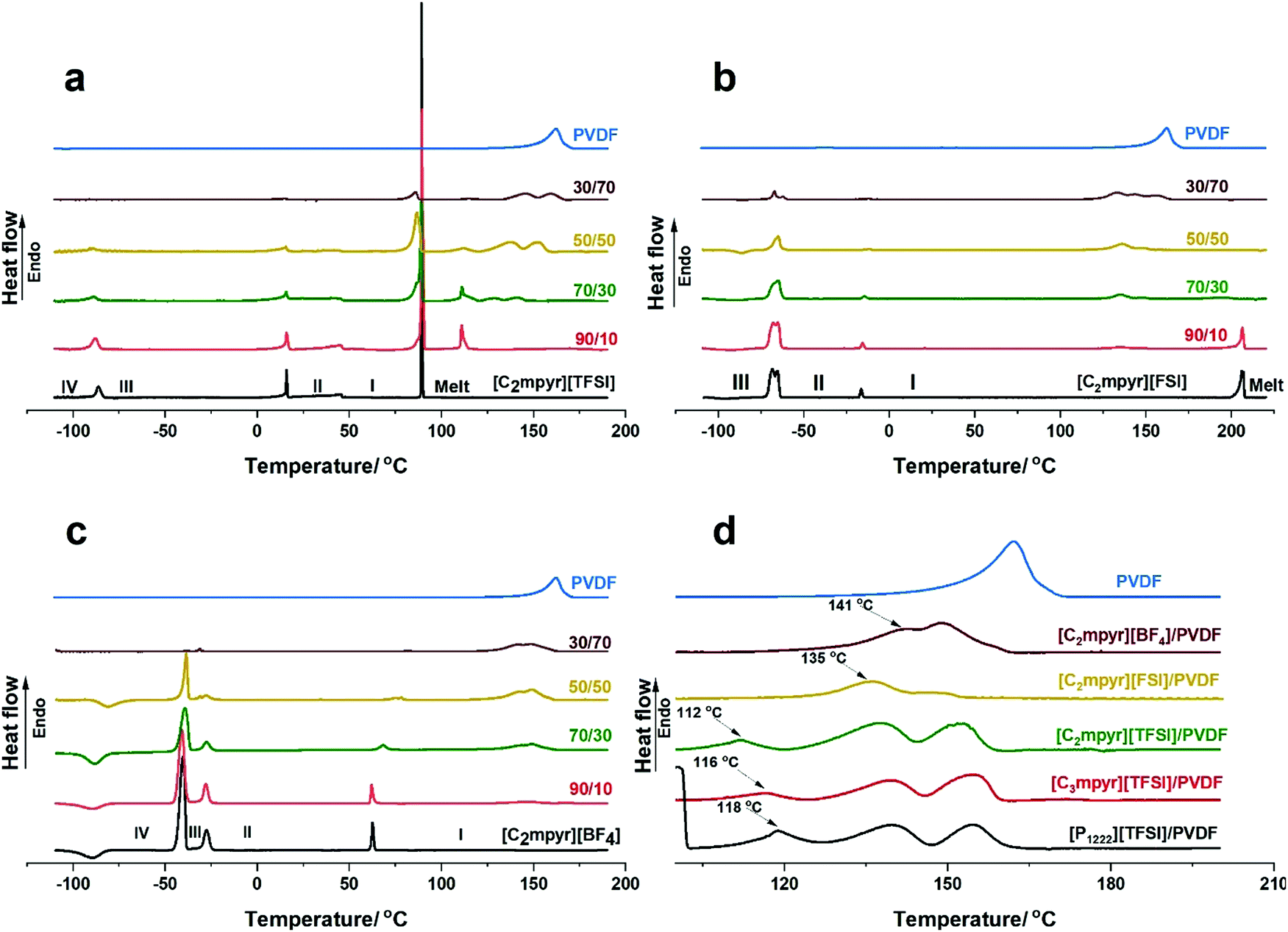 | ||
| Fig. 2 DSC traces of; (a) [C2mpyr][TFSI] (b) [C2mpyr][FSI] (c) [C2mpyr][BF4] containing 10, 30, 50 and 70 vol% of PVDF; (d) comparison of melting phases of PVDF in the 50/50 composition of [C2mpyr][TFSI]/PVDF, [C2mpyr][FSI]/PVDF, [C2mpyr][BF4]/PVDF, [C3mpyr][TFSI]/PVDF and [P1222][TFSI]/PVDF. | ||
Consistent with previous works,42,43 the DSC traces of [C2mpyr][FSI] shows three major transitions, the first two being the first and second solid–solid transitions and the third being the melt of the OIPC (Fig. 2b). [C2mpyr][FSI] has a crystalline phase I starting from −14 °C to 204 °C. On the addition of PVDF particles, the solid–solid transition peaks become distorted while the melting peak begins to disappear (e.g. 50 vol% of PVDF, 50/50). This was attributed to the interaction between [C2mpyr][FSI] and PVDF, which leads to the OIPC becoming less crystalline based on the consideration of enthalpy change.30 Also, this interaction leads to the formation of new overlapping peaks as seen at 135 and 145 °C which are proposed to result from the formation of a PVDF-rich region which contains OIPC ions.33 These overlapping peaks are different from the original melting peak of PVDF which occurs at 164 °C. However, at 70 vol%, there appears a third overlapping peak which is similar to that of the melting peak of the pristine PVDF. This is because at higher loadings of PVDF, there is insufficient OIPC to interact with the PVDF.
Fig. 2c shows the DSC heating traces of [C2mpyr][BF4] and its composites. [C2mpyr][BF4] shows three solid–solid transitions with a broad phase I that starts from 65 °C,46 and a high melting point of 293 °C which falls outside the range of interest for this study (Fig. S1c, ESI†). On the addition of PVDF, the third solid–solid transition peak (phase II to I, around 63 °C) begins to shift to higher temperatures which further increases the phase II range. This is different from the observation in another work where [C2mpyr][BF4] and PVDF nanofibre interactions were studied. The difference could result from the much lower mass of polymer fibres added to the OIPC.28 It should be noted that the melt phase of PVDF observed in the [C2mpyr][BF4]/PVDF composites appear different from those observed in [C2mpyr][TFSI] and [C2mpyr][FSI]. No new phases are observed in the [C2mpyr][BF4]/PVDF composites besides the two peaks that appear at 141 and 148 °C. Similar to the [C2mpyr][TFSI] and [C2mpyr][FSI] composites, the overlapping peaks of the PVDF melt phases occur at temperatures lower than the melt of PVDF (160 °C). This suggests that the melt phases are related to the PVDF-rich interphase formed in [C2mpyr][BF4]/PVDF composites, as explained earlier.
To further understand the anion effects of OIPC on melting of PVDF, we compare the melting phases of PVDF in the 50/50 composition of [C2mpyr][TFSI]/PVDF, [C2mpyr][FSI]/PVDF and [C2mpyr][BF4]/PVDF (Fig. 2d). The PVDF melting peaks of [C3mpyr][TFSI]/PVDF and [P1222][TFSI]/PVDF are also presented. Surprisingly, new peaks similar to the peak occurring at 112 °C for [C2mpyr][TFSI]/PVDF appears at 116 and 118 °C for [C3mpyr][TFSI]/PVDF and [P1222][TFSI]/PVDF respectively. The occurrence of this new peak even when the [TFSI]-based IL ([C3mpyr][TFSI]) is used confirms that its formation is a result of the interaction between [TFSI]− anions and PVDF. The absence of this new peak in [C2mpyr][FSI]/PVDF and [C2mpyr][BF4]/PVDF composites suggests different degrees of interaction between the OIPCs and PVDF. This interaction accounts for the distorted solid–solid transition peaks and the disappearance of the melting peaks observed in Fig. 4(a–c), which suggest that the crystallinity of OIPCs is supressed when they interact with PVDF, as previously reported.10,19,21,22 The presence of the PVDF particles accounts for the loss of crystallinity of the OIPCs, which leads to the formation of disordered OIPC/PVDF interphases.
3.2 Ionic conductivity
Fig. 3a shows the ionic conductivity data for [C2mpyr][TFSI] with different volume ratios of PVDF. [C2mpyr][TFSI] melts at 89 °C (Fig. 2a), thus, the measurement was ended at 80 °C so as to maintain the experimental conditions before melting. The range of the study (30 to 80 °C) covers the phases II and I of [C2mpyr][TFSI]. There is no observed jump in conductivity because the transition from phase II to phase I is a broad transition rather than a sharp one.49 Surprisingly, the conductivity of the composite with 10 vol% PVDF (90/10) improves significantly from 3.87 × 10−9 to 5.86 × 10−8 S cm−1 at 30 °C, which is about an order of magnitude enhancement compared to neat [C2mpyr][TFSI]. And more encouragingly, the conductivity continuously increases to 5.94 × 10−6 S cm−1 at 60 °C with increasing of PVDF up to 70 vol%. Based on the discussion in the DSC section, we assume that interaction between [C2mpyr][TFSI] and PVDF leads to the formation of disordered interphases which accounts for the enhanced ionic conductivity.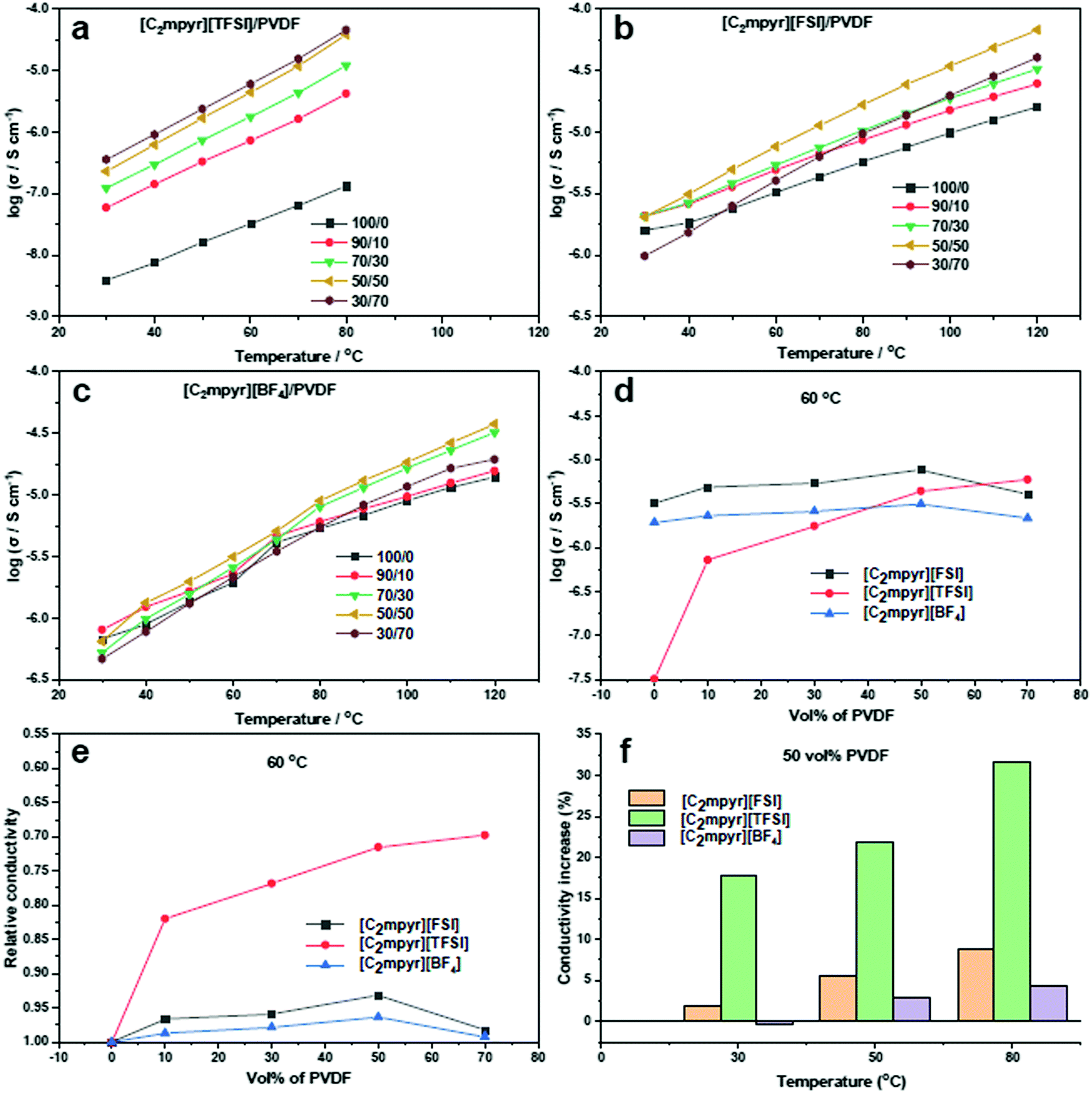 | ||
| Fig. 3 Conductivity data for; (a) [C2mpyr][TFSI] (b) [C2mpyr][FSI] (c) [C2mpyr][BF4] containing 10, 30, 50 and 70 vol% of PVDF nanoparticles; (d) comparison of the conductivity of [C2mpyr][TFSI], [C2mpyr][FSI] and [C2mpyr][BF4] based composites with respect to vol% of PVDF at 60 °C; (e) comparison of the relative conductivity of [C2mpyr][TFSI], [C2mpyr][FSI] and [C2mpyr][BF4] based composites with respect to vol% of PVDF at 60 °C; (f) comparison of percentage increase in ionic conductivity increase with 50 vol% of PVDF at different temperatures (30 °C, 50 °C and 80 °C). | ||
The temperature-related ionic conductivity of the [C2mpyr][FSI] based composites with different volume ratios of PVDF is presented in Fig. 3b. Consistent with DSC results, the conductivity plots do not show irregular jumps in the range between 30 and 120 °C. The ionic conductivity increases with increasing vol% of PVDF until the percolation threshold is approached at 50 vol% (∼55 wt%) of PVDF. This is different from Li0.1[C2mpyr]0.9[FSI]/PVDF composite which percolates on addition of 60 wt% of PVDF particles.25
Different from the above two OIPC/PVDF composite electrolytes, the ionic conductivity plots of [C2mpyr][BF4] (Fig. 3c) show a sharp jump during OIPC transition from phase II to phase I, as seen in the DSC results. This jump in conductivity is characteristic of [C2mpyr][BF4] as shown in previous reports, and furthermore, the OIPC shows increase in conductivity upon addition of PVDF.19,24 However, there appears to be only a slight increase in the conductivity after addition of PVDF nanoparticles. Beyond 50 vol% of PVDF, the conductivity begins to decline again.
In order to further compare the effects of PVDF on the conductivities of different OIPC composites, we compare the conductivity dependence of the composites ([C2mpyr][FSI]/PVDF, [C2mpyr][TFSI]/PVDF and [C2mpyr][BF4]/PVDF) as a function of vol% of PVDF at 60 °C (Fig. 3d). Considering the complexity of phase transitions of the three OIPCs at different temperatures (e.g. [C2mpyr][TFSI] shows a broad phase transition below 40 °C and melts after 80 °C), the 60 °C is selected for comparison. It is seen that ionic conductivity increases with increasing vol% of PVDF nanoparticles for all the temperature points studied. However, whereas there was a sharp drop in conductivity for [C2mpyr][FSI] and [C2mpyr][BF4] based composites after 50 vol% of PVDF is exceeded, [C2mpyr][TFSI]/PVDF composites did not experience a sharp drop but rather the ionic conductivity began to plateau. This suggests that the interaction in the [C2mpyr][TFSI]/PVDF composites probably leads to the formation of a thicker disordered interphase compared to the [C2mpyr][FSI] and [C2mpyr][BF4] composites. Fig. 3e also compares the increase in the conductivity of the OIPC/PVDF composites at 50 vol% of PVDF relative to the conductivity of the pristine OIPCs. Whereas the relative increase in the conductivity of the [C2mpyr][TFSI] is obvious, the relative increase in the conductivity of the [C2mpyr][FSI] and [C2mpyr][BF4] composites are almost similar.
The observed increase in ionic conductivity upon addition of PVDF to the OIPCs is as a result of the formation of a disordered interfacial layer which begins to connect as the vol% of PVDF particles increases. Beyond 50 vol% of PVDF, the PVDF particles begin to percolate and become closely packed. Beyond the percolation threshold, the disordered interphase become isolated islands rather than interconnected networks, thus, the conductivity begins to drop.30 The closely packed PVDF particles lead to increased tortuosity (winding conduction pathway) and hence the decrease in conductivity.
Fig. 3f shows that, for the same vol% of PVDF, [C2mpyr][TFSI] shows a higher increase in conductivity compared to [C2mpyr][FSI] and [C2mpyr][BF4]. Furthermore, the increase in ionic conductivity of the composites follows the order [C2mpyr][TFSI] > [C2mpyr][FSI] > [C2mpyr][BF4]. This difference in ionic conductivity can be attributed to the difference in size, charge distribution and strength of interaction between the anions of the OIPCs and PVDF. [TFSI]− and [FSI]− are large anions that have diffused charges spreading through their S–N–S core, through to their fluorine atoms. This effectively reduces their interaction with the [C2mpyr]+ cation, defining their properties.31 The [TFSI]− anion, with two –CF3 groups, has a more diffused charge which accounts for the lower melting point of [C2mpyr][TFSI] compared to the [C2mpyr][FSI] (two fluorine atoms). Thus, [TFSI]− is less strongly bound to [C2mpyr]+ than [FSI]−. On the other hand, the [BF4]− anion is a small anion with localised charge concentration which results in increased interaction between the [C2mpyr]+ cation and the [BF4]− anion, thus reducing interaction with secondary components.32
3.3 Crystal structure
The synchrotron X-ray diffraction experiments were performed to investigate the crystal structure changes of the pristine OIPCs, their respective PVDF composites, as well as PVDF, measured at the different solid–solid phases (Fig. S2, ESI†). The OIPCs approach crystal orientations with less peaks which suggests increased symmetry as they progress through different solid–solid phases with increasing temperature The diffraction patterns of the OIPC/PVDF composites (Fig. 4a–c) show features similar to those of the pristine OIPC with the diffraction patterns of PVDF embedded in them, which suggests the bulk OIPC is the predominant phase in the composite electrolytes. However, there are notable differences between the peak positions of the pristine OIPCs and the OIPC/PVDF composites. The addition of PVDF nanoparticles to [C2mpyr][TFSI] (Fig. 4a), for example, leads to the shift of the OIPC peaks to higher diffraction angles, which suggests the contraction of the unit cells of the OIPC and the formation of solid solutions.50 The peak positions of [C2mpyr][FSI] composite (Fig. 4b) do not appear to be significantly affected, compared to the pristine OIPCs, in spite of the peak broadening, which suggests the existence of non-uniform strain in the lattice of the [C2mpyr][FSI]/PVDF composite. In the presence of PVDF, [C2mpyr][BF4] (Fig. 4c) peaks appeared to be shifted to lower diffraction angles suggesting that the sizes of the crystals become smaller due to higher lattice strain when PVDF is added to the OIPCs.51 Therefore, we believe that the different peak shifts of the OIPCs on addition of PVDF are related to different degrees of interaction with PVDF. In other words, this different interaction attributed to the different anions, as explained earlier, leads to modifications within the crystal packing.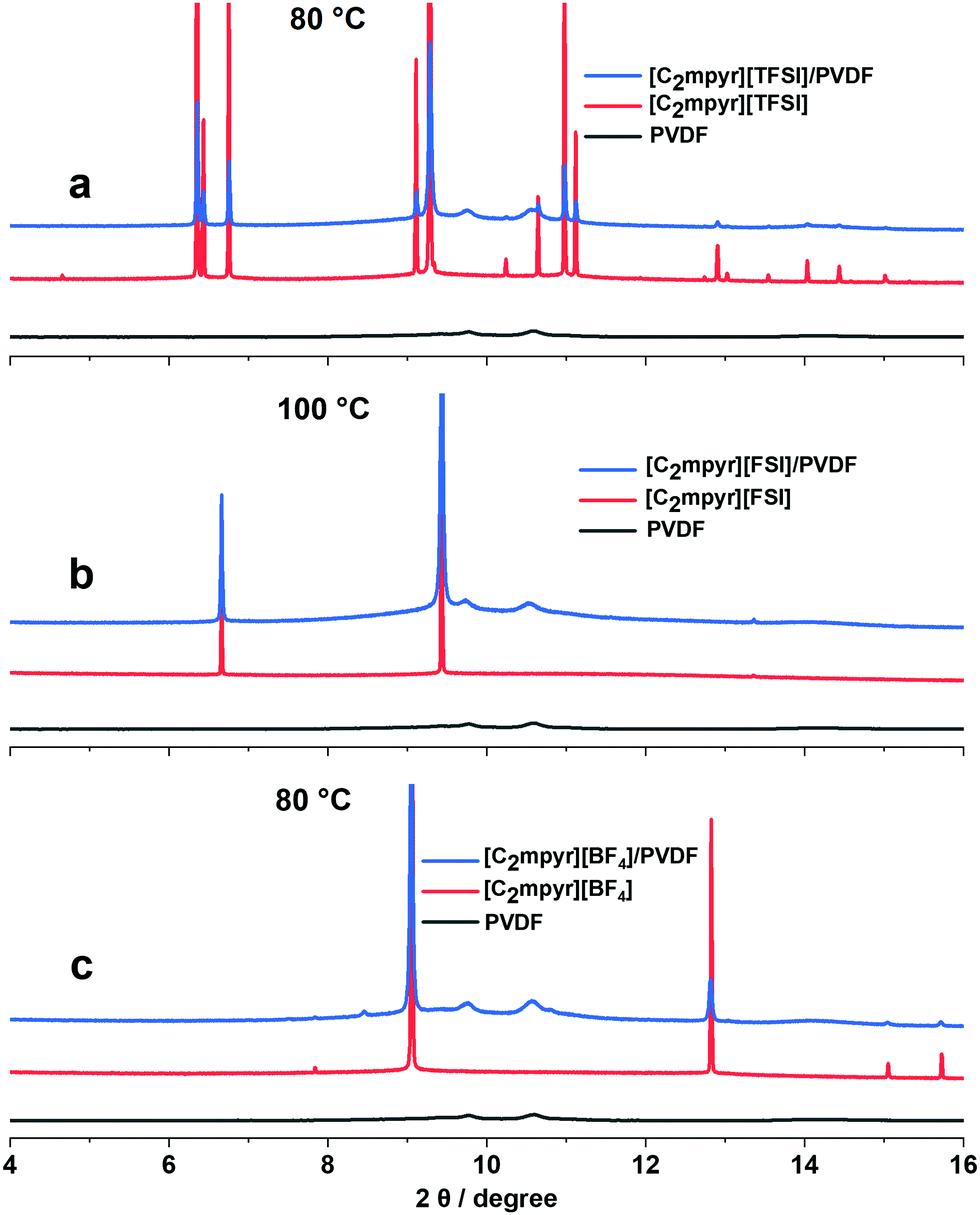 | ||
| Fig. 4 Synchrotron X-ray diffraction patterns for pristine; (a) [C2mpyr][TFSI], (b) [C2mpyr][FSI] and (c) [C2mpyr][BF4], their 50 vol% PVDF composites and PVDF taken in their phase I region. | ||
Furthermore, unlike the diffraction peaks of the pristine OIPCs, the peaks of the composites samples are broad and less defined but the peaks become more defined as temperature increases (Fig. S2b, d and e, ESI†). Considering the DSC results as shown in Fig. 2, it is reasonable to conclude that the broadening of diffraction peaks is due to the formation of disordered or amorphous phases at the interfacial regions between the OIPC and the PVDF nanoparticles.
3.4 Cation and anion dynamics
Investigating the anion and cation dynamics is crucial for further understanding the phase behaviours and transport mechanisms in OIPC/PVDF composites. Solid-state NMR is a particularly useful technique for providing insights of ion dynamics. Fig. 5 presents the static 1H and 19F comparison of the pristine OIPCs ([C2mpyr][TFSI], [C2mpyr][FSI] and [C2mpyr][BF4]), their respective 50 vol% of PVDF composites and pristine PVDF nanoparticles, measured at 60 °C. The composition with 50 vol% of PVDF was used for this study since the PVDF particles approach close-packing at 50 vol% of PVDF. Generally, solid samples have broad lineshapes owing to strong homo-nuclear dipole–dipole interactions for 1H and chemical shift anisotropy (CSA) for 19F. However, enhanced disorder and increased rotational motion are able to suppress the effects of dipole–dipole interactions and CSA leading to the narrowing of the linshapes.52,53 At lower temperatures (e.g. −40 to 20 °C as seen in Fig. S3, ESI†), the lineshapes of the pristine OIPCs and their corresponding composite materials appear similarly broad, which suggests that the [C2mpyr]+ cations, in both the pristine material and the composite, are immobile at lower temperatures. But with increasing temperature, the narrow component becomes prominent in the lineshape of the composites, suggesting enhanced ion dynamics.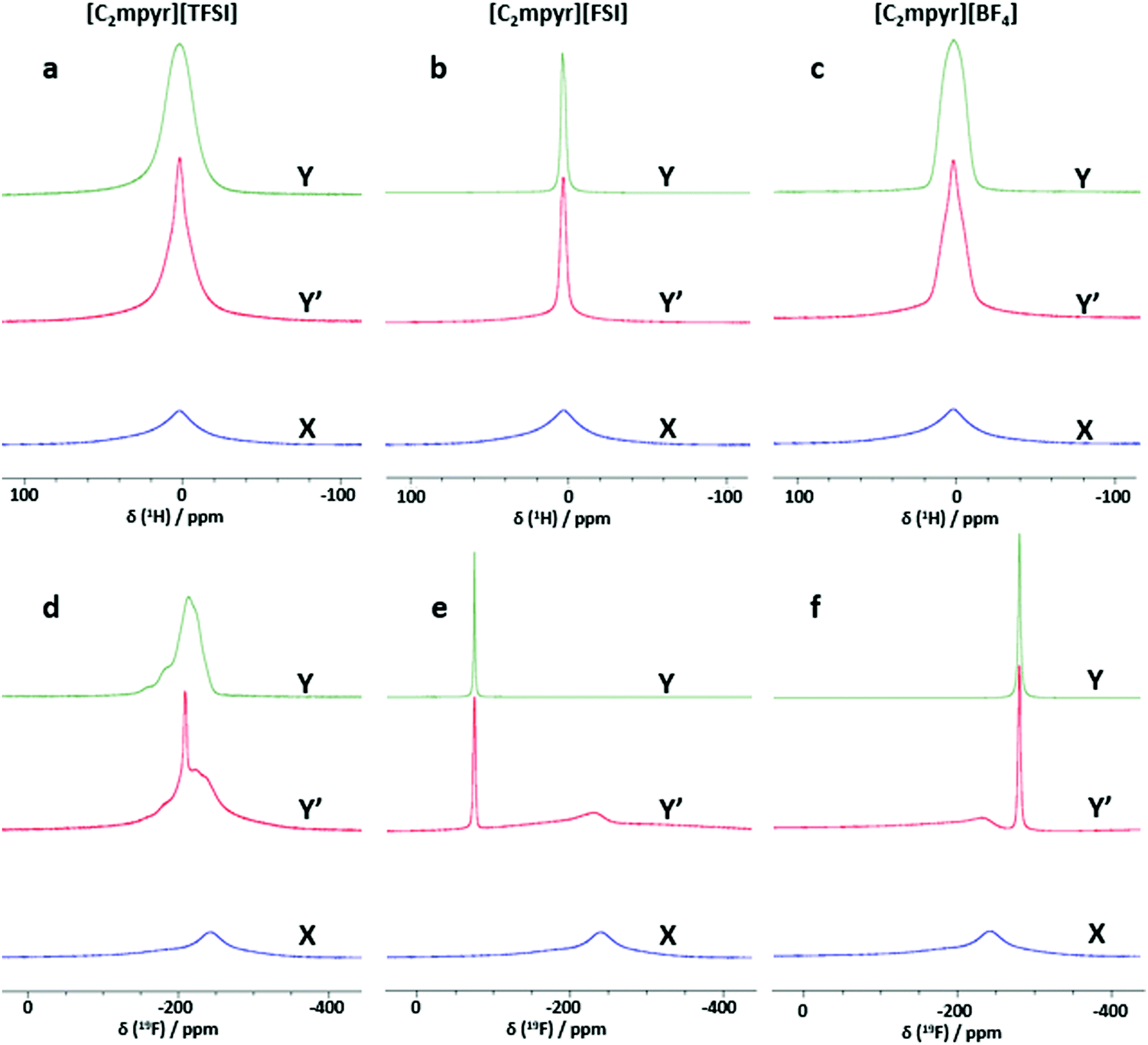 | ||
| Fig. 5 Static 1H NMR spectra of; (a) [C2mpyr][TFSI], [C2mpyr][TFSI]/PVDF and PVDF (Y, Y′ and X); (b) [C2mpyr][FSI], [C2mpyr][FSI]/PVDF and PVDF (Y, Y′ and X); (c) [C2mpyr][BF4], [C2mpyr][BF4]/PVDF and PVDF (Y, Y′ and X); static 19F NMR spectra of; (d) [C2mpyr][TFSI], [C2mpyr][TFSI]/PVDF and PVDF (Y, Y′ and X); (e) [C2mpyr][FSI], [C2mpyr][FSI]/PVDF and PVDF (Y, Y′ and X); (f) [C2mpyr][BF4], [C2mpyr][BF4]/PVDF and PVDF (Y, Y′ and X). All the spectra were obtained at 60 °C. | ||
The 1H NMR lineshapes of pristine ([C2mpyr][TFSI] (Fig. 5a) and [C2mpyr][BF4] (Fig. 5c)) show broad lineshapes suggesting that the cations in the pristine OIPCs are not very mobile. This is different from the 1H NMR lineshape of [C2mpyr][FSI] (Fig. 5b) which is narrow indicating high mobility of cations. However, on addition of PVDF, the 1H NMR lineshapes of the respective composites develop multiple components. The multiple components originate from the interaction of the OIPCs with PVDF (the deconvoluted peaks are shown in Fig. 6d–f). The presence of the narrow component suggests the presence of mobile cations potentially originating from the disordered OIPC formed when the OIPC matrix interacts with the PVDF matrix.
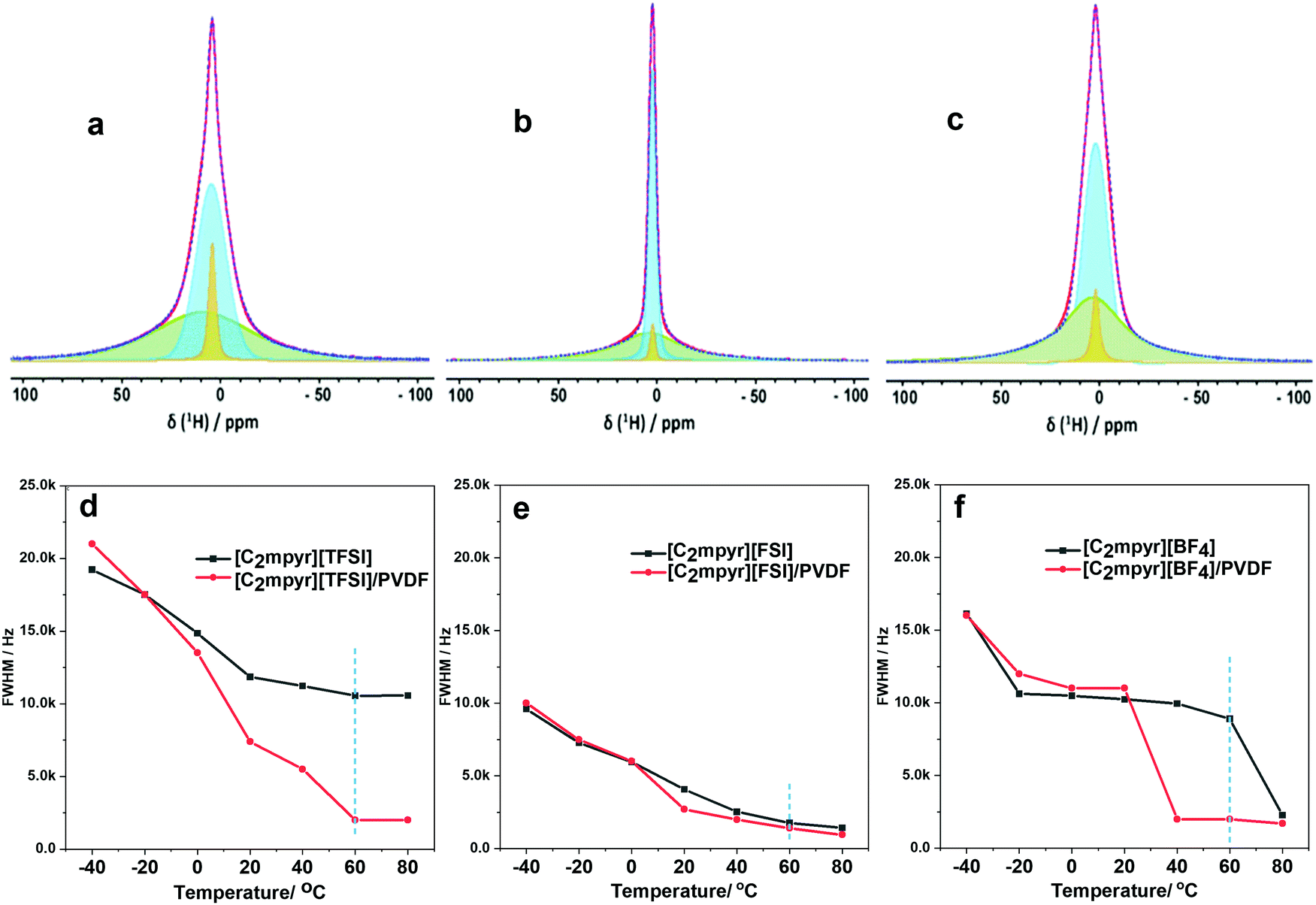 | ||
| Fig. 6 Deconvoluted VT 1H spectrum taken at 60 °C for; (a) [C2mpyr][TFSI]/PVDF; (b) [C2mpyr][FSI]/PVDF; and (c) [C2mpyr][BF4]/PVDF; static 1H full width half maximum of the neat OIPCs and OIPC/PVDF composites (50 vol% of PVDF); (d) [C2mpyr][TFSI] and [C2mpyr][TFSI]/PVDF; (e) [C2mpyr][FSI], [C2mpyr][FSI]/PVDF; (f) [C2mpyr][BF4], [C2mpyr][BF4]/PVDF. | ||
Fig. 5d shows the single pulse static NMR of 19F of [C2mpyr][TFSI] which shows a broad lineshape with strong CSA patterns. In the presence of PVDF, as with the cation lineshapes measured for the composites, the 19F spectra display multiple components (Fig. S3a, ESI†); a broad component at the base with a narrow component superimposed on the broad component. Both PVDF and [C2mpyr][TFSI] have strong CSA patterns which are reflected in the composite. Interestingly, a significant new narrow peak located at −208 ppm is seen in the lineshape of the [C2mpyr][TFSI]/PVDF composite. The presence of the narrow peak indicates the presence of a larger population of highly mobile 19F ions, which could be attributed to the formation of disordered interphases. Furthermore, this narrow component of the 19F becomes prominent with increasing temperature (Fig. S3a, ESI†). In this system the 19F PVDF has a broad lineshape that overlaps with the line shape of 19F [C2mpyr][TFSI] due to their similar chemical shifts.
On the other hand, [C2mpyr][FSI] and [C2mpyr][BF4] have different 19F chemical shifts compared to pristine PVDF as seen from 19F-MAS (Fig. S4b and c, ESI†). The 19F static NMR signal of PVDF appears at −240 ppm as a broad peak whereas the 19F signal of the [C2mpyr][FSI] and [C2mpyr][BF4] appear at −77 ppm and −280 ppm respectively. The 19F peak of PVDF is present in the NMR spectrum of the composite but is shifted to −230 ppm. The shifting of the peak position of PVDF in the composite is indicative of the interactions between the OIPCs and PVDF as suggested by DSC (Fig. 2) and this suggests that the interaction creates different chemical environments for the fluorine atoms of PVDF. Fig. 5f compares the 19F static spectra of [C2mpyr][BF4], [C2mpyr][BF4]/PVDF and PVDF. In their pristine form, the 19F static NMR spectra of both [C2mpyr][FSI] and [C2mpyr][BF4] show line shapes with single components. However, when PVDF is added, the base consists of a broad component which has a narrow component superimposed on it, forming the second component. The presence of the narrow component in the spectra of the composite indicates that some fraction of the anions become mobile upon addition of PVDF to the OIPC. This suggests that the OIPC becomes more disordered in the presence of PVDF.
There are apparent differences of the composites which can be attributed to the different behaviours of the OIPC anions. Whereas the lineshapes of the 1H and 19F spectra of [C2mpyr][TFSI]/PVDF composites appear to be much narrowed, [C2mpyr][FSI]/PVDF and [C2mpyr][BF4]/PVDF composites are less affected relative to their pristine OIPCs. The data also suggest that [C2mpyr][TFSI] strongly interacts with the PVDF particle interface compared to the [C2mpyr][FSI] and [C2mpyr][BF4], and hence, the significant increase in conductivity observed in the [C2mpyr][TFSI]/PVDF composites (Fig. 3d). The lesser effect on ion dynamics by the PVDF in the [C2mpyr][FSI] and [C2mpyr][BF4] materials is consistent with the equivalent relative increase in ionic conductivity in their respective PVDF composites (Fig. 3e).
Fig. 6(a–c) present deconvoluted static 1H NMR lineshapes taken at 60 °C for [C2mpyr][TFSI]/PVDF, [C2mpyr][FSI]/PVDF and [C2mpyr][BF4]/PVDF respectively. The 1H lineshapes of the PVDF-composites of the respective OIPCs appear to have multiple components. The 1H-MAS spectra of PVDF overlaps with those of [C2mpyr][TFSI], [C2mpyr][FSI] and [C2mpyr][BF4], which suggests that PVDF will have chemical shifts similar to the OIPCs (as seen in Fig. S4, ESI†). As such, the broad component seen at the base of the composite materials originates from the rigid PVDF which has homo-nuclear dipolar interaction as its main T2 relaxation mechanism. Another relatively broad component sits on the PVDF base which consists of the OIPC ions that do not interact with the PVDF. Finally, superimposed on this broad component is a narrow component. The presence of this narrow component suggests that a fraction of the cations of the composites become mobile upon addition of PVDF to the OIPCs. This occurs because the dynamics of a fraction of the cations average out the homonuclear dipolar interact ions, leading to the narrowing of the peak superimposed on the broad component.54
To gain more insights on the effects of PVDF on cation dynamics, Fig. 6(d–f) compares the full width half maximum (FWHM) of pristine [C2mpyr][TFSI], [C2mpyr][FSI] and [C2mpyr][BF4] to their composites containing 50 vol% of PVDF (obtained from Fig. S3, ESI†). These comparisons clearly show the influence of the PVDF particles on the ion dynamics, especially at higher temperatures, in the case of [TFSI]−. This is consistent with ionic conductivity analysis. The ion dynamics in pristine [C2mpyr][FSI] are already fast which is seen in the narrow linewidths in the pristine OIPC. Upon addition of PVDF, the decrease in the cation dynamics is only marginal, most likely because the OIPCs is already in its phase I and already very plastic.
[C2mpyr][BF4] shows decreasing linewidths with increasing temperature (Fig. S4, ESI†). At 60 °C, a significant decrease in the linewidth is observed which is consistent with DSC data (Fig. 2c). The composite, however, has a decreased in FWHM linewidth for narrow component overlaid on the broad component indicating that the increase in cation dynamics in the [C2mpyr][BF4]/PVDF composites even in the phase II (e.g. at 40 °C). The sharp drop in FWHM observed in the pristine OIPC is not seen in the composite, and this consistent with the broadening of the phase II–I solid–solid transition evident in the DSC traces (Fig. 2c) that smears out this phase change. These differences in the progressive decrease of the linewidths of the cations can also be attributed to the differences in the interactions of the OIPCs with the PVDF particles. It should be noted that the dynamics of the 19F static NMR of the OIPCs could not be quantified by deconvoluting the peaks due to the strong CSA patterns in the lineshapes of the OIPCS. The 19F NMR of the OIPCs are not ideal Gaussian shapes because of the strong CSA patterns.
Regarding the nature of interaction, there are numerous interactions involved but these are mainly limited to the ‘dispersion’ class of forces (i.e. forces arising from various temporary, induced, and permanent dipoles). While all of these forces exist to one extent or the other, the phase behaviour of the anion and PVDF systems is determined by the strength of the most dominant interaction which varies with each anion. These anion–polymer, dipole–dipole interactions, are consistent with previous reports.33,55 It is known that the electronegativity difference between fluorine and carbon of PVDF (4.0 for fluorine vs. 2.5 for carbon) gives the C–F bond significant polarity/dipole moment.56 Therefore, the –CF2 groups in PVDF easily interact with other available dipoles. The nature of the anions account for the differing strength of their interactions with PVDF. For [TFSI]− and [FSI]− anions, the asymmetrical electron distribution leads to stronger permanent dipole–permanent dipole interactions with PVDF. The strength of the permanent dipole–permanent dipole interaction is dependent on the magnitudes of the dipole moments of the interacting dipoles. Particularly, the strong electron withdrawing –CF3 groups of [TFSI]− increases the polarity of the S=O compared to [FSI]−.57 Thus, the permanent dipole–dipole interaction between [TFSI]− and PVDF is stronger than that between [FSI]− and PVDF. For [C2mpyr][BF4], although the dipole–dipole moment of a single B–F bond is strong, the net dipole moment of [BF4]− is cancelled due to its high symmetry,58 resulting in a weak temporary dipole–permanent dipole interaction with PVDF.
Based on the DSC, EIS, XRD, NMR results and the analysis above, we propose that, the difference in their physical properties, chemical properties and interaction behaviours of the OIPCs and PVDF originate from the different anions of the OIPCs, which accounts for the different degrees of interactions between OIPCs and the PVDF particles. We therefore propose the model in Fig. 7 to define the different phases and interphases formed in the OIPC/PVDF composites. The model suggests that volume of bulk OIPC within which the original ordering is retained, is reduced to form a disordered OIPC/PVDF interphase via the interaction of the OIPCs with the PVDF. The extent of the interaction is dependent on the anion of the OIPC which determines the thickness of the disordered interphase.
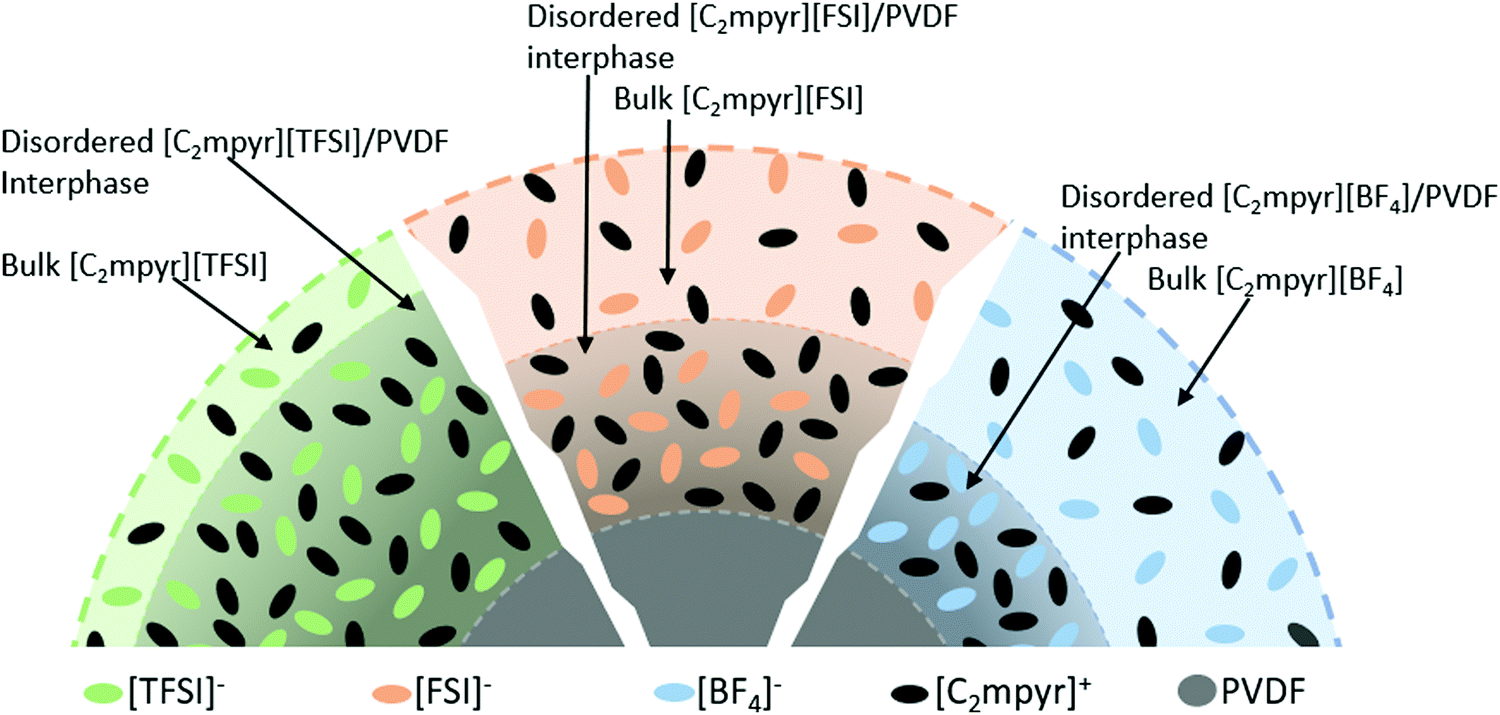 | ||
| Fig. 7 Layered structure showing the different phases and their relative amounts in OIPC/PVDF composites. | ||
4. Conclusion
In this study, three anions ([TFSI]−, [FSI]− and [BF4]−) with different sizes and charge distributions were each coupled with [C2mpyr]+ to form [C2mpyr][TFSI], [C2mpyr][FSI] and [C2mpyr][BF4]. These OIPCs were then combined with different volume fractions of PVDF nanoparticles. The effect of PVDF addition on the thermal properties, ionic conductivity, ion dynamics and crystal structure of the OIPCs/PVDF composites were investigated with respect to the pristine OIPCs so as to understand the effect of the anions in OIPC/PVDF interactions. DSC, EIS, NMR and synchrotron XRD suggest different interactions between the anions and PVDF. In particular, the asymmetrical structure and highly delocalized charge distribution of [TFSI]− induces stronger dipole–dipole interactions than [C2mpyr][FSI] and [C2mpyr][BF4] OIPCs. This subsequently leads to different degrees of enhancement in ionic conductivity and ion dynamics and provides a method to control ionic conductivity in these composite electrolytes. The learnings here can direct the selection of OIPC and polymer pairs that will have strong interfacial interactions leading to a disordered and highly dynamic interphase, that when interconnected results in higher ionic conductivities. Thus, future studies will be focussed on understanding the effects of noncovalent forces, for example by computational simulations, on OIPC/polymer composite behaviours, and optimizing OIPC/polymer interactions by manipulating anion or polymer chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully express their appreciation to the US Army Research Office (ARO) for financial support (W911NF1710560). The Australian Research Council (ARC) is acknowledged for their support through beamtime allocation. A special appreciation to the management and staff of Australian Synchrotron, especially Dr Anita D’Angelo, for beamtime allocation and their support during synchrotron XRD experiments. The authors also acknowledge Prof. Alejandro Müller (POLYMAT), for his help with synchrotron powder diffraction results analysis. Dr Ruhamah Yunis is also acknowledged for her help with material synthesis.References
- J. M. Pringle, P. C. Howlett, D. R. MacFarlane and M. Forsyth, J. Mater. Chem., 2010, 20, 2056–2062 RSC.
- J. Sun, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2002, 148, 145–151 CrossRef CAS.
- P. J. Alarco, Y. Abu-Lebdeh and M. Armand, Solid State Ionics, 2004, 175, 717–720 CrossRef CAS.
- D. Wilmer, K. Funke, M. Witschas, R. D. Banhatti, M. Jansen, G. Korus, J. Fitter and R. E. Lechner, Phys. B, 1999, 266, 60–68 CrossRef CAS.
- W. Haase, Ber. Bunsen-Ges., 1980, 84, 1192 CrossRef.
- M. Forsyth, G. M. A. Girard, A. Basile, M. Hilder, D. R. MacFarlane, F. Chen and P. C. Howlett, Electrochim. Acta, 2016, 220, 609–617 CrossRef CAS.
- X. Wang, R. Kerr, F. Chen, N. Goujon, J. M. Pringle, D. Mecerreyes, M. Forsyth and P. C. Howlett, Adv. Mater., 2020, 32, 1–21 Search PubMed.
- L. Jin, P. C. Howlett, J. M. Pringle, J. Janikowski, M. Armand, D. R. MacFarlane and M. Forsyth, Energy Environ. Sci., 2014, 7, 3352–3361 RSC.
- Z. Bin Zhou and H. Matsumoto, Electrochem. Commun., 2007, 9, 1017–1022 CrossRef.
- J. M. Pringle, Y. Shekibi, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2010, 55, 8847–8854 CrossRef CAS.
- R. Yunis, T. W. Newbegin, A. F. Hollenkamp and J. M. Pringle, Mater. Chem. Front., 2018, 2, 1207–1214 RSC.
- V. Armel, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2011, 4, 2234–2239 RSC.
- Z. Liao, J. Huang, W. Chen, N. Saito, Z. Zhang, L. Yang and S. Hirano, Energy Storage Mater., 2020, 33, 442–451 CrossRef.
- I. S. Klein, Z. Zhao, S. K. Davidowski, J. L. Yarger and C. A. Angell, Adv. Energy Mater., 2018, 8, 1–7 Search PubMed.
- K. Yang, Z. Zhang, Z. Liao, L. Yang and S. ichi Hirano, ChemistrySelect, 2018, 3, 12595–12599 CrossRef CAS.
- P. Wang, Q. Dai, S. M. Zakeeruddin, M. Forsyth, D. R. MacFarlane and M. Grätzel, J. Am. Chem. Soc., 2004, 126, 13590–13591 CrossRef CAS.
- M. Lee, Y. H. Lee, J. H. Park and U. H. Choi, Org. Electron., 2017, 48, 241–247 CrossRef CAS.
- F. Huang, Polym.-Plast. Technol. Eng., 1997, 36, 777–790 CrossRef CAS.
- N. Iranipour, D. J. Gunzelmann, A. Seeber, J. Vongsvivut, C. Doherty, F. Ponzio, L. A. O’dell, A. F. Hollenkamp, M. Forsyth and P. C. Howlett, J. Mater. Chem. A, 2015, 3, 6038–6052 RSC.
- J. Rao, R. Vijayaraghavan, X. Wang, Y. Zhou, P. C. Howlett, D. R. Macfarlane, M. Forsyth and H. Zhu, J. Phys. Chem. C, 2018, 122, 14546–14553 CrossRef CAS.
- J. Adebahr, N. Ciccosillo, Y. Shekibi, D. R. MacFarlane, A. J. Hill and M. Forsyth, Solid State Ionics, 2006, 177, 827–831 Search PubMed.
- Y. Shekibi, S. J. Pas, N. M. Rocher, B. R. Clare, A. J. Hill, D. R. MacFarlane and M. Forsyth, J. Mater. Chem., 2009, 19, 1635–1642 RSC.
- Y. Zhou, X. Wang, H. Zhu, M. Yoshizawa-Fujita, Y. Miyachi, M. Armand, M. Forsyth, G. W. Greene, J. M. Pringle and P. C. Howlett, ChemSusChem, 2017, 10, 3135–3145 CrossRef CAS.
- P. C. Howlett, F. Ponzio, J. Fang, T. Lin, L. Jin, N. Iranipour and J. Efthimiadis, Phys. Chem. Chem. Phys., 2013, 15, 13784–13789 RSC.
- X. Wang, H. Zhu, G. W. Greene, Y. Zhou, M. Yoshizawa-Fujita, Y. Miyachi, M. Armand, M. Forsyth, J. M. Pringle and P. C. Howlett, Adv. Mater. Technol., 2017, 2, 1–6 CAS.
- G. W. Greene, F. Ponzio, N. Iranipour, H. Zhu, A. Seeber, M. Forsyth and P. C. Howlett, Electrochim. Acta, 2015, 175, 214–223 CrossRef CAS.
- N. Iranipour, PhD Thesis, Deakin University, 2016, vol. 1, pp. 1–283.
- X. Wang, H. Zhu, G. W. Greene, J. Li, N. Iranipour, C. Garnier, J. Fang, M. Armand, M. Forsyth, J. M. Pringle and P. C. Howlett, J. Mater. Chem. A, 2016, 4, 9873–9880 RSC.
- Y. Zhou, X. Wang, H. Zhu, M. Armand, M. Forsyth, G. W. Greene, J. M. Pringle and P. C. Howlett, Energy Storage Mater., 2018, 0–1 Search PubMed.
- F. Nti, L. Porcarelli, G. W. Greene, H. Zhu, F. Makhlooghiazad, D. Mecerreyes, P. C. Howlett, M. Forsyth and X. Wang, J. Mater. Chem. A, 2020, 8, 5350–5362 RSC.
- J. M. Pringle, J. Golding, K. Baranyai, C. M. Forsyth, G. B. Deacon, J. L. Scott and D. R. MacFarlane, New J. Chem., 2003, 27, 1504–1510 RSC.
- R. Yunis, A. F. Hollenkamp, C. Forsyth, C. M. Doherty, D. Al-Masri and J. M. Pringle, Phys. Chem. Chem. Phys., 2019, 21, 12288–12300 RSC.
- R. Mejri, J. C. Dias, A. C. Lopes, S. Bebes Hentati, M. M. Silva, G. Botelho, A. Mão De Ferro, J. M. S. S. Esperança, A. Maceiras, J. M. Laza, J. L. Vilas, L. M. León and S. Lanceros-Mendez, Eur. Polym. J., 2015, 71, 304–313 CrossRef CAS.
- J. Yang, S. Pruvost, S. Livi and J. Duchet-Rumeau, Macromolecules, 2015, 48, 4581–4590 CrossRef CAS.
- F. Wang, A. Lack, Z. Xie, P. Frübing, A. Taubert and R. Gerhard, Appl. Phys. Lett., 2012, 100, 062903 CrossRef.
- C. Xing, M. Zhao, L. Zhao, J. You, X. Cao and Y. Li, Polym. Chem., 2013, 4, 5726–5734 RSC.
- I. Aldalur, X. Wang, A. Santiago, N. Goujon, M. Echeverría, M. Martínez-Ibáñez, M. Piszcz, P. C. Howlett, M. Forsyth, M. Armand and H. Zhang, J. Power Sources, 2020, 448, 227424 CrossRef CAS.
- C. Xing, J. Guan, Y. Li and J. Li, ACS Appl. Mater. Interfaces, 2014, 6, 4447–4457 CrossRef CAS.
- C. Chang, V. H. Tran, J. Wang, Y. K. Fuh and L. Lin, Nano Lett., 2010, 10, 726–731 CrossRef CAS.
- P. C. A. Hammes and P. P. L. Regtien, Sens. Actuators, A, 1992, 32, 396–402 CrossRef CAS.
- S. J. Kang, Y. J. Park, I. Bae, K. J. Kim, H. C. Kim, S. Bauer, E. L. Thomas and C. Park, Adv. Funct. Mater., 2009, 19, 2812–2818 CrossRef CAS.
- Y. Zhou, X. Wang, H. Zhu, M. Armand, M. Forsyth, G. W. Greene, J. M. Pringle and P. C. Howlett, Phys. Chem. Chem. Phys., 2017, 19, 2225–2234 RSC.
- M. Yoshizawa-Fujita, E. Kishi, M. Suematsu, T. Takekawa and M. Rikukawa, Chem. Lett., 2014, 43, 1909–1911 CrossRef.
- P. M. Dean, B. R. Clare, V. Armel, J. M. Pringle, C. M. Forsyth, M. Forsyth and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 334–340 CrossRef CAS.
- D. R. MacFarlane, P. Meakin, J. Sun, N. Amini and M. Forsyth, J. Phys. Chem. B, 1999, 103, 4164–4170 CrossRef CAS.
- S. Forsyth, J. Golding, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2001, 46, 1753–1757 CrossRef CAS.
- A. Fdz De Anastro, N. Casado, X. Wang, J. Rehmen, D. Evans, D. Mecerreyes, M. Forsyth and C. Pozo-Gonzalo, Electrochim. Acta, 2018, 278, 271–278 CrossRef CAS.
- A. Bielecki and D. P. Burum, J. Magn. Reson., Ser. A, 1995, 116, 215–220 CrossRef CAS.
- M. Forsyth, T. Chimdi, A. Seeber, D. Gunzelmann and P. C. Howlett, J. Mater. Chem. A, 2014, 2, 3993–4003 RSC.
- H. Chen, A. Sayari, A. Adnot and F. Larachi, Appl. Catal., B, 2001, 32, 195–204 CrossRef CAS.
- B. D. Cullity and S. R. Stock, Elements of X-ray Diffraction, Pearson, 2001 Search PubMed.
- M. Yoshizawa-Fujita, H. Yamada, S. Yamaguchi, H. Zhu, M. Forsyth, Y. Takeoka and M. Rikukawa, Batteries Supercaps, 2020, 3, 884–891 CrossRef CAS.
- J. M. Pringle, J. Adebahr, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2010, 12, 7234–7240 RSC.
- J. Rao, R. Vijayaraghavan, Y. Zhou, P. C. Howlett, D. R. Macfarlane, M. Forsyth and H. Zhu, Phys. Chem. Chem. Phys., 2018, 20, 4579–4586 Search PubMed.
- L. He, J. Sun, X. Wang, C. Wang, R. Song and Y. Hao, Polym. Int., 2013, 62, 638–646 CrossRef CAS.
- N. Matsunaga, D. W. Rogers and A. A. Zavitsas, J. Org. Chem., 2003, 68, 3158–3172 CrossRef CAS.
- S. Asha, K. P. Vijayalakshmi and B. K. George, Int. J. Quantum Chem., 2019, 119, 1–12 Search PubMed.
- Q. Zhang, D. Liu, Q. Li, X. Zhang and Y. Wei, J. Chem. Eng. Data, 2018, 63, 1256–1265 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00992j |
| This journal is © The Royal Society of Chemistry 2021 |