
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient construction of a redox responsive thin polymer layer on glassy carbon and gold surfaces for voltage-gated delivery applications†
Anna
Barosi
a,
Avni
Berisha
b,
Claire
Mangeney
 *a,
Jean
Pinson
*c,
Hamid
Dhimane
a and
Peter I.
Dalko
*a
*a,
Jean
Pinson
*c,
Hamid
Dhimane
a and
Peter I.
Dalko
*a
aLaboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, CNRS UMR 8601, Université de Paris, 45 rue des Saints-Pères, F-75270 Paris, France. E-mail: peter.dalko@parisdescartes.fr; claire.mangeney@parisdescartes.fr
bChemistry Department of Natural Sciences Faculty, University of Prishtina rr. “Nëna Tereze” nr. 5, 10000 Prishtina, Kosovo
cITODYS, CNRS, UMR 7086, Université de Paris, 15 rue J-A de Baïf, F-75013 Paris, France. E-mail: jean.pinson@univ-paris-diderot.fr
First published on 11th March 2021
Abstract
A conceptually novel coating strategy is presented based on the selective oligomerisation of picolinium-derived end-tethered polymer chains. The remarkably rapid and nearly quantitative liberation of the covalently tethered ligand makes this system a promising candidate for the construction of densely packed “smart” release interfaces.
Introduction
Research on stimuli-responsive materials1 is among the most active areas in materials science.2 For example, in response to specific internal or external stimuli, the controlled delivery of substances, such as drugs, vitamins, nutrients, contrast agents for imaging, and genes, can be triggered.2 Among the many fields of application, this method may increase drug-targeting efficiency by reducing the side effects/toxicity of the payloads, which are key factors for improving patient compliance in medical health care.3 Electrochemical activation represents probably the most convenient and technologically the simplest approach for an accurate dose-controlled release of substances.4 The large choice of controlling parameters, such as the applied potential, the magnitude of current, the duration of an electrical pulse or the interval between pulses, may allow precise control over the activation process.5 The interest for redox-sensitive smart materials was sustained by the fact that they can be used in electronic circuits or adapted on-chip, or in related techniques. The latest developments have permitted (i) wireless monitoring of the patient's health and (ii) remote-controlled drug delivery for in vivo applications. For instance, integrated microchips were used as implantable and wearable drug-releasing devices.3 The ever-growing need in microelectronics and micro-fabrication emulate the development of such electrically enabled smart materials, with the need for solutions in miniaturization, remote power supply, and wireless control of delivering devices.6 We present here a novel approach for the construction of a redox-sensitive smart system,7 with a sequential growth of the polymer chains attached to the surface, in which electron deficient picoliniums act both as central elements for the formation of end-tethered oligomer chains and as working/fragmenting probes.8 The oligomerisation is based on the Minisci-type reaction of aryl radicals to electron-deficient arenes:9 according to the original observation, protonated pyridine derivatives undergo fast and selective o- and/or p-alkylation under redox alkylation conditions in the presence of in situ generated alkyl radicals (Fig. 1). The scope of this reaction was extended to aryl radicals by Baran using arylboronic acids as radical precursors.10 A chemically analogous transformation can be seen also in recent variants of the Meerwein arylation reaction, a Gomberg–Bachmann-type arylation,11–13 in which the intermediate free-aryl radical is generated either under redox or, more recently, under photoelectron-transfer conditions from aryl diazonium compounds.14,15 This electron-transfer mediated functionalization was advantageously used for the coating of electrode surfaces as well by applying a slight negative electrode potential (Ered = – 0.50 V vs Ag/AgCl) to induce the reduction of the corresponding diazonium salt,16,17,27 thus triggering a reaction with the surface.16,18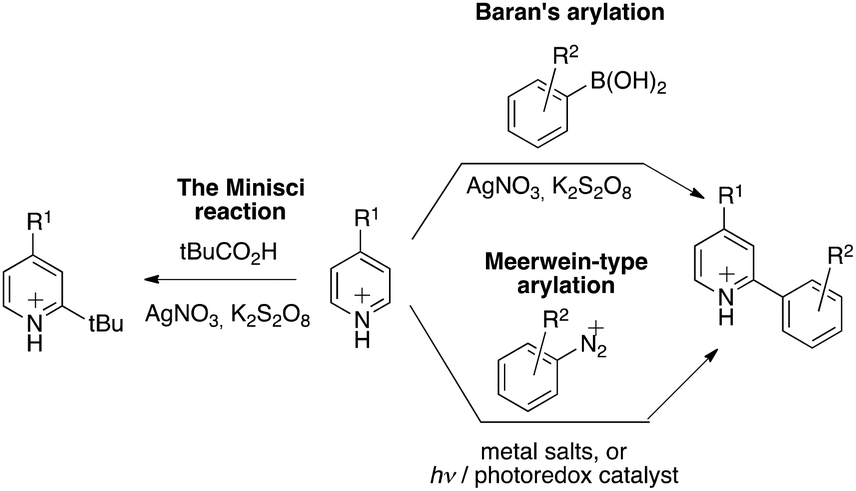 | ||
| Fig. 1 C–H bond arylation of electron-deficient heterocycles. | ||
Results and discussion
It was observed earlier, that N-methyl picolinium esters undergo efficient C–O bond cleavage by electron transfer,23 releasing carboxylic acids, making this system an excellent candidate for the construction of voltage-gated releasing system.18 In order to validate the concept, the synthesis of the smart surface on gold coated wafer (@Au) and glassy carbon electrode (@GC) was initiated by using a drug-mimicking fluorescent reporter (Scheme 1). In this system, the fluorescence of the covalently attached (“caged”) reporter is quenched when attached to the probe, while lights up when liberated, allowing monitoring and quantifying the release. The efficient energy transfer toward picolinium and also to the conductor/semi-conductor surface resulting in fluorescence quenching was reported earlier.23 According to earlier studies23 the fragmentation follows a rather radical ionic than purely radical path thus affording stabilized distonic radical cation and carboxylate anion; the evolution of the reactive intermediate19 follows probably similar path to that described previously.20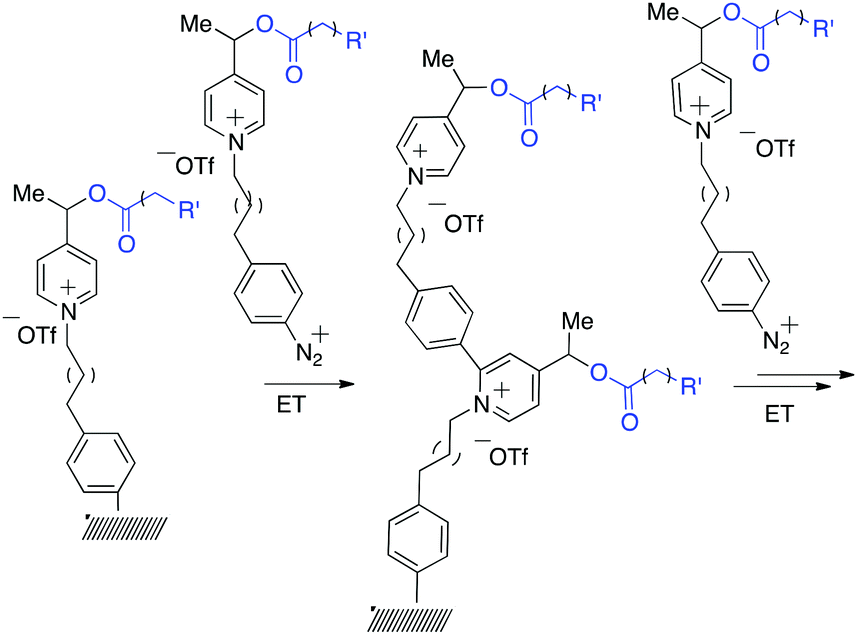 | ||
| Scheme 1 Sequential construction of the brush-like oligomers on gold and glassy carbon surfaces by Minisci-type arylation. | ||
To make this system feasible, several conditions should be satisfied: (1) conditions used for the immobilization of the monomers should be compatible with the presence of the “armed” redox probes; (2) the addition of the aryl radical should be selective to the picolinium ring;21 (3) the chemical modification of the picolinium should not compromise the fragmentation ability; and (4) the electrode potential of the modified probe should be close to that of the unmodified one to avoid the sequential release of the ligand when activated.
Theoretical computations indicated that the electrochemical fragmentation of o-arylated picoliniums can be expected around E = −1.11 V vs. Ag/AgCl providing a convenient gap to find conditions for the electrochemical immobilization and activation (−0.50 V < E < −1.11 V vs. Ag/AgCl) (Scheme 2). Interestingly, according to the computation results, the o-arylation does not modify the electrode-potential of the probe (Scheme 2). Indeed, the prediction of the standard redox potential in solution (DFT) based on the use of the Born–Haber cycle of the most stable conformers22 yielded very similar values for model compound 2 and compound 1 the calculated first reduction potential of which was in good agreement with the experimental value.23
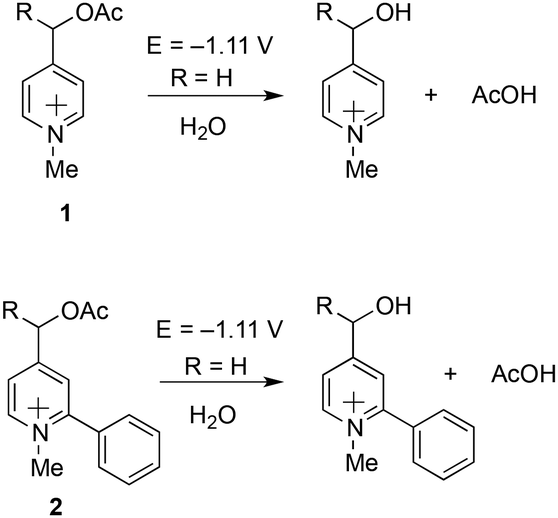 | ||
| Scheme 2 DFT estimation of the first reduction potentials of model compound 2 and comparison with the calculated and experimentally observed value of 1.23 | ||
The retrosynthetic assembly of the redox responsive thin polymer layer-monomer is highlighted in Scheme 3.
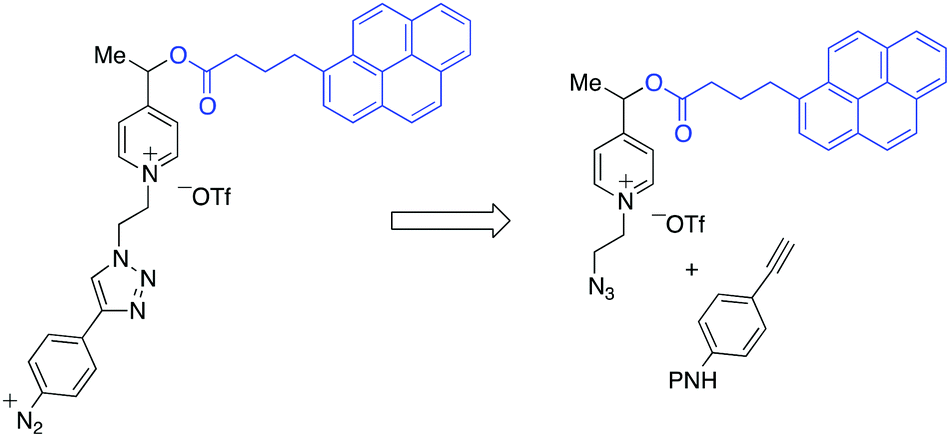 | ||
| Scheme 3 Retrosynthetic analysis of the construction of picolinium-derived monomers. P: protecting group. | ||
The pyrenebutyric acid ester and the azido ethyl chain were installed to picoline 3 by using a standard esterification/quaternisation sequence (Scheme 4). The protected p-ethynyl aniline, 5, was prepared in three steps starting from 4-iodoaniline. The prepared alkyne 5 was “clicked” to the azido-picolinium 4 by using Huisgen conditions affording 6 (75%), and finally the Boc group was removed by TFA. The electrochemical profile of the free aniline 7 was studied in solution by cyclic voltammetry on carbon electrodes, where the redox waves were better defined (ESI†).25 Voltammetric peaks were assigned by comparison with those of purposely-synthesized subunits of the molecule (see in ESI†).
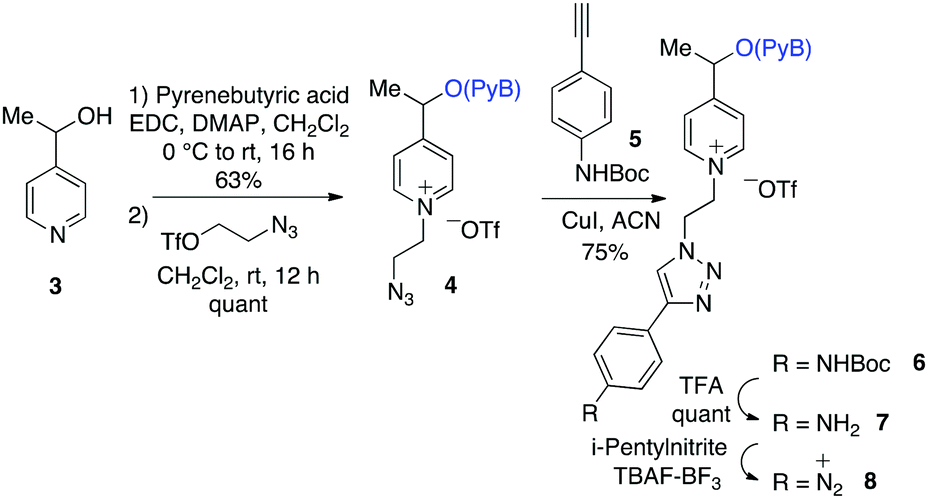 | ||
| Scheme 4 Synthesis of the aryl diazonium monomer (8). | ||
Diazonium salt, 8, was in situ generated from 7 in an electrochemical cell by using i-pentylnitrite as the diazotization reagent and n-Bu4NBF4 (0.1 M) as the supporting electrolyte. The probe was immobilized on gold‡ and GC electrodes, by applying a potential of −0.90 V vs. Ag/AgCl to reduce the diazonium salt, while avoiding the ester cleavage (see ESI†).26 Voltammograms were recorded on GC electrodes, which give better defined voltammograms, while grafting was performed on gold, which permits the recording of IRRAS spectra). The immobilization is considered following earlier described paths: the dissociative ET results in the formation of aryl radical that first binds to the electrode surface, and then participates in chain elongation by consecutive C–H arylation, according to the Minisci path. After electrodeposition, electrodes were rinsed with ethanol under sonication and dried to give the coated materials 9@Au and 9@GC, respectively. Voltammograms before and after deposition of 7/9@GC are presented in Fig. 2.
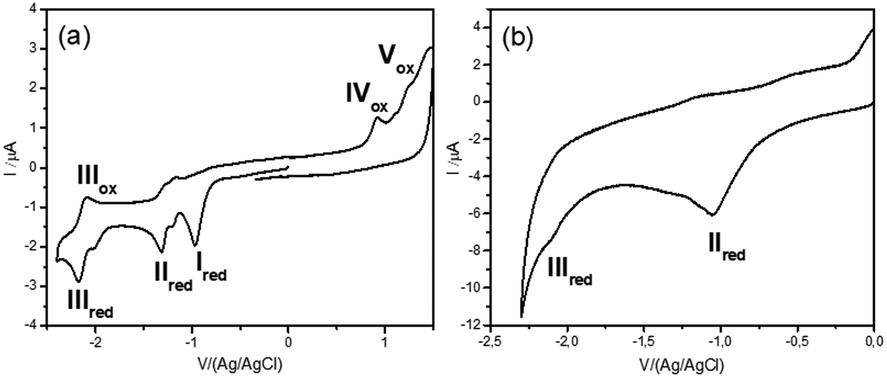 | ||
| Fig. 2 (a) Cyclic voltammogram of 7, before immobilization on a GC electrode (c = 2 mM, v = 0.1 V s−1, ref Ag/AgCl) in ACN + n-Bu4NBF4 (0.1 M). (b) Cyclic voltammogram of 9@GC, after immobilization on a glassy carbon electrode (v = 0.1 V s−1, ref Ag/AgCl) in ACN + n-Bu4NBF4 (0.1 M). | ||
As expected, the peak corresponding to the reduction of the aniline group, Ired, has disappeared. In contrast, peaks IIred and IIIred are detected on the coated electrode, supporting the reduction of the diazonium group of 8 (for the assignment of the fragment peaks see Table S1 in the ESI†). It is noteworthy that the intensity of peak IIIred is lower than that of IIred. This fact can be explained by the fragmentation of the pyridinium derivative during the voltammetric cycle, when the potential reaches ca. −1.2 V. Integration of the voltammogram of 9@GC provided the surface concentration of Γ = 4.0 × 10−10 mol cm−2 of the grafted molecules. This value is in agreement with the Γ = 2.5–5.7 × 10−10 mol cm−2 reported for a monolayer of nitrophenyl groups on a very flat carbon surface.16 The surface modification was characterized by IRRAS by comparing the spectra of 9@Au with that of the synthesized fragments (Fig. S5, ESI†). The most distinctive features are (i) the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band, located at 1743 cm−1, (ii) the triazole signature that appears as two bands at 1690 and 1666 cm−1 for 7, at similar positions as that of grafted 9@Au, (iii) the aromatic ring vibrations at 1646 cm−1 of both picolinium and pyrene, and finally (iv) the C–H out-of-plane vibrations of the aromatic ring, located between 650 and 850 cm−1, particularly the strong band of pyrene at 838 cm−1. The presence of this C–H out of plane vibration band is particularly noteworthy as this band is very sensitive to substitution (this is a method to distinguish isomers); the fact that it is still present is a good indication that there is no attack on the pyrene. The IR spectra thus confirms the grafting of compound 7 on gold surface, without unwanted fragmentation/cleavage of the probe. The immobilization was also analyzed by XPS. The gold signal appeared strongly attenuated after the functionalization, while the carbon and oxygen content increased and a new peak, assigned to picolinium and triazole nitrogens appeared, indicating the coating by a polyaryl layer (see the survey spectrum in Fig. 3).
O band, located at 1743 cm−1, (ii) the triazole signature that appears as two bands at 1690 and 1666 cm−1 for 7, at similar positions as that of grafted 9@Au, (iii) the aromatic ring vibrations at 1646 cm−1 of both picolinium and pyrene, and finally (iv) the C–H out-of-plane vibrations of the aromatic ring, located between 650 and 850 cm−1, particularly the strong band of pyrene at 838 cm−1. The presence of this C–H out of plane vibration band is particularly noteworthy as this band is very sensitive to substitution (this is a method to distinguish isomers); the fact that it is still present is a good indication that there is no attack on the pyrene. The IR spectra thus confirms the grafting of compound 7 on gold surface, without unwanted fragmentation/cleavage of the probe. The immobilization was also analyzed by XPS. The gold signal appeared strongly attenuated after the functionalization, while the carbon and oxygen content increased and a new peak, assigned to picolinium and triazole nitrogens appeared, indicating the coating by a polyaryl layer (see the survey spectrum in Fig. 3).
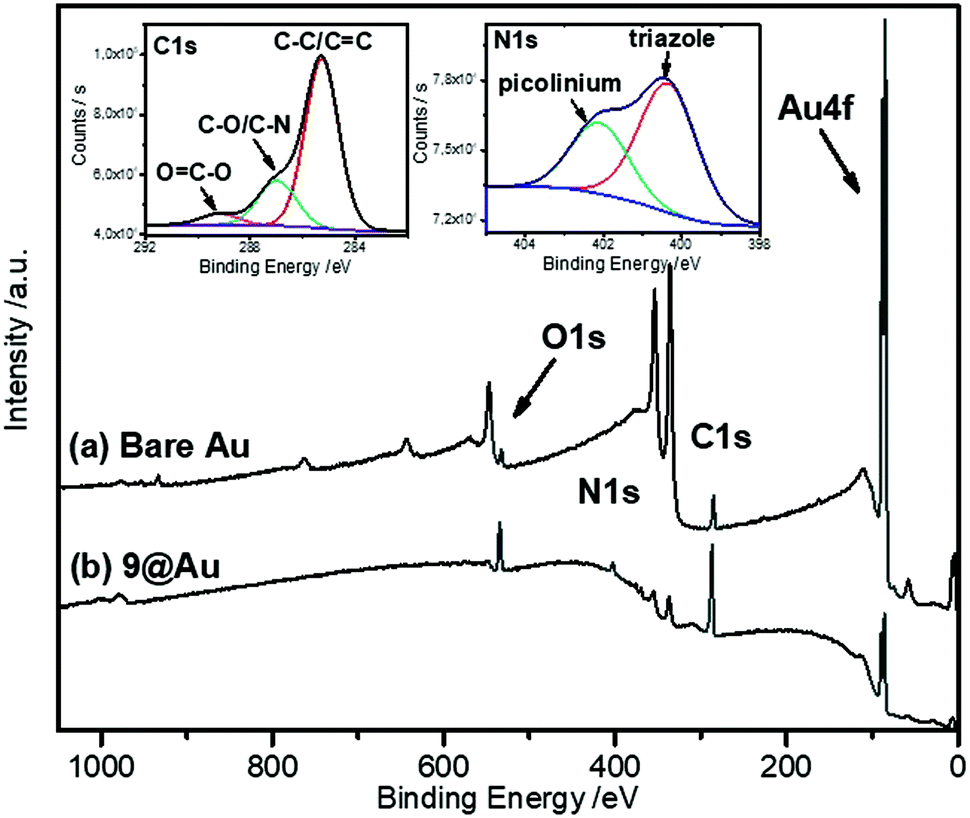 | ||
| Fig. 3 XPS survey spectra of 9@Au and bare Au electrodes. The insets show the C1s and N1s high resolution spectra of 9@Au. | ||
The peak fitting of the C1s signal displayed in the inset of Fig. 3 reveals the presence of all the components contained in the polyaryl layer. The intense peak at 285 eV is assigned to the presence of C–C and CC bonds, the one at 286.8 eV is attributed to the C–O and C–N components, while the peak at 289.3 eV is due to the ester groups, confirming the grafting of the probe, without fragmentation. Moreover, the N1s signal is composed of two components, one at 400.4 eV corresponding to the presence of the triazole, and the one at 402.1 eV corresponding to the picolinium groups. The thickness of the grafted polyaryl layer was estimated by ellipsometry to be 8.8 ± 0.2 nm, thus indicating the formation of oligomers.
The interface between the gold electrode and the grafted probe was studied by using Surface-Enhanced Raman Spectroscopy (SERS). For this study, the probe was grafted on gold nano-island films, prepared by sputtering 5 nm of gold on indium-tin-oxide (ITO) coated-glass substrates. The SERS spectrum (Fig. 4) was recorded after thorough ultrasonic cleaning of the substrate.
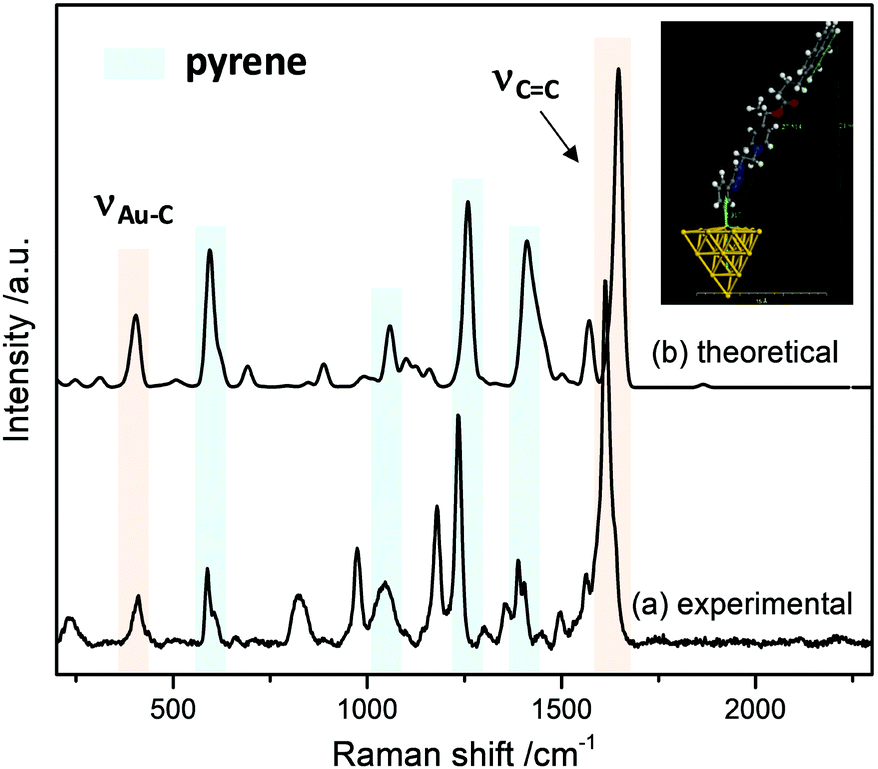 | ||
| Fig. 4 SERS spectrum of 9@Au. Laser excitation at 633 nm. The inset shows the optimized structure of 9@Au20 used to compute the Raman spectrum (on the top) and experimental SERS spectrum of 9@Au (on the bottom). | ||
Several features are noteworthy: the intense N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching of the diazonium group at ca. 2300 cm−1 is not observed any more indicating that the diazonium group of intermediate 8 was electrochemically removed. At the same time, one notices the appearance of a small peak at 410 cm−1, assigned to the Au–C stretching vibration,27 evidencing the covalent grafting of the aryl groups. Also, the strong band related to the stretching of aromatic CC bonds can be observed in the SERS spectra in the 1577–1630 cm−1 range. Other bands associated to the pyrene reporter28 (590 cm−1, skeletal stretch; 1060 cm−1, C–H in-plane bend; 1238 cm−1, C–C stretch/C–H in-plane bend and 1410 cm−1, C–C stretch/ring stretch) can be also recognized.
N stretching of the diazonium group at ca. 2300 cm−1 is not observed any more indicating that the diazonium group of intermediate 8 was electrochemically removed. At the same time, one notices the appearance of a small peak at 410 cm−1, assigned to the Au–C stretching vibration,27 evidencing the covalent grafting of the aryl groups. Also, the strong band related to the stretching of aromatic CC bonds can be observed in the SERS spectra in the 1577–1630 cm−1 range. Other bands associated to the pyrene reporter28 (590 cm−1, skeletal stretch; 1060 cm−1, C–H in-plane bend; 1238 cm−1, C–C stretch/C–H in-plane bend and 1410 cm−1, C–C stretch/ring stretch) can be also recognized.
In order to simulate the SERS spectra of 9@Au, DFT calculations were performed. In this model a gold cluster with 20 atoms was used to simulate 9@Au. Fig. 4 shows the calculated structure and the simulated Raman spectrum; the Au–C bond length is estimated to be 2.317 Å with an Au–Au–C valence angle of 99.72°. A good agreement is observed between the simulated Raman and the experimental SERS spectra, with the presence of both the pyrene bands and the Au–C peak at 409 cm−1, confirming the covalent bonding of the aryl layers on the gold surface. The observed small peak frequency shifts can be attributed to the modification of the electronic structure of the grafted molecules by immobilization on the gold surface.
The modified gold plate and carbon electrodes, 9@GC and 9@Au, were subjected to reduction for 10 min, at −1.2 V/(Ag/AgCl) in ACN, in the presence of n-Bu4BF4 (0.1 M) (Fig. 5). The applied reduction potential corresponds to the reduction peak IIred of the pyridinium group that induced the irreversible fragmentation of the pyrene ester from the probe. By analyzing the electrochemical response of the electrode, one can note the absence of any signal corresponding to the pyrene group (peak IIIred), confirming the cleavage of the ester. It is worth mentioning that the electrochemical response of 7 (in solution) exhibits a reversible IIIred/IIIox peak for pyrene (Fig. 2a), indicating the absence of cleavage of this group in the time lapse of one voltammogram cycle. In contrast, this reversibility is neither observed in 9@Au (Fig. 2b) nor in 9@GC (Fig. 5a), thus indicating that the cleavage is faster than the reverse ET (oxidation). Considering, that there is no diffusion (as probes are immobilized) it is possible to estimate29 the rate of cleavage of the surface bonded pyrene group as ks ∼ 0.3 s−1 from the height of the peaks of the pyrene group in the voltammogram of 9@GC, by using a simple first order kinetic relationship (Fig. 5). Note that the degraded reversibility of the anchored pyrene signal cannot be attributed to the protonation of the radical ionic intermediate by traces of water, as the voltammogram of pyrenebutyric acid is perfectly reversible under the same conditions in solution.
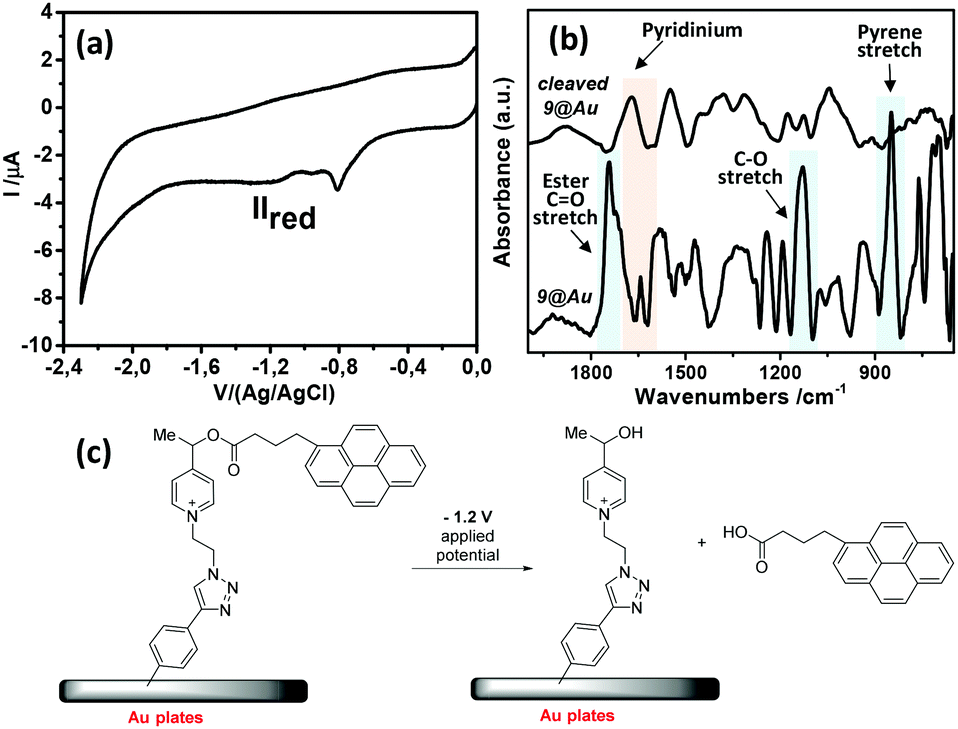 | ||
| Fig. 5 (a) Cyclic voltammogram of the cleaved 9@GC, obtained by applying a reduction potential of −1.2 V to the electrode (10 min); (b) IRRAS spectra of 9@Au before and after cleavage; (c) schematic representation of the fragmentation by electrochemical reduction at −1.2 V (NB: counter ion was omitted for clarity). | ||
The surface was also studied by IRRAS after cleavage. Fig. 5 presents the IRRAS spectra recorded before (bottom) and after (top) electrochemical reduction of 9@Au; the latter clearly shows the disappearance of the 1743 cm−1 (ester CO stretch) and 1130 cm−1 (C–O stretch) bands corresponding to the ester group, as well as the characteristic pyrene band at 847 cm−1. It is noteworthy that the 1650 cm−1 band, assigned to the picolinium group, is still present. This observation is in agreement with the cleavage between the picolinium and the pyrene (corresponding probably to the cleavage of the benzylic C–O bond, as was observed earlier).23 The presence of the free pyrene-derivative in the electrolysate was evidenced by fluorescence spectroscopy (Fig. S6, ESI†) confirming the release of pyrenebutyric acid from the covalently attached probe. The amount of the released pyrene per surface unit (Γ) was calculated by measuring the fluorescence intensity and comparing it to the calibration curve, giving Γ = 1.5 × 10−9 mol cm−2 of released pyrenebutyric acid from the gold electrode. It is noteworthy that the Γ value obtained (on gold) by the fluorescence correlation was larger than the surface concentration, estimated by electrochemistry on GC (Γ = 4 × 10−10 mole cm−2), and was assigned to the formation of oligomers on the gold surface.
Indeed, the Γ value obtained from the fluorescence correlation suggests the presence of n ∼ 4 mers. Likewise, a Γ value of n ∼ 4 mers was deduced in this experiment from ellipsometry, considering the ratio between the film thickness (8.8 nm) and the estimated monolayer thickness from molecular modeling (2.1 nm, see Fig. 4).
These results thus confirm the formation of oligomers on the electrode and also, the efficient fragmentation even at a relatively larger distance from the Au-grafting point.
Conclusions
The construction of “smart“ interfaces on glassy carbon and gold electrodes was presented in which the electron deficient redox probe offers the fragmenting ability and plays key roles in the construction of surface-capped oligomers. Analytical data were consistent with the formation of a short, probably brush-like polyaryl layer of 4 mers, as was deduced from the ellipsometry and fluorescence measurements of the liberated reporter. This novel surface-building strategy can be considered as the first step towards the design of dense “smart” molecular interfaces with improved loading and fragmentation proprieties.Experimental
HPLC analysis was carried out using a Water 515 device with a normal inverse phase column X-Terra MS C18 (length: 75 mm, diameter: 4.6 mm, stationary phase: 2.5 μm) using a Waters 2487 Dual Absorbance Detector (260–360 nm) and an isocratic system of elution (MeOH–MeCN–H2O 7–2–1/H2O AcONH4 (10 mM) pH 4.6). The volume of injection was 10 μL.Electrochemical experiments were performed using an EG&G 263A potentiostat/galvanostat and an Echem 4.30 version software. All potentials are referred to the Ag/AgCl electrode. The electrode for cyclic voltammetry was a glassy carbon (GC) rod (d = 2 mm diameter) sealed in glass. It was polished with different grades of polishing papers and finally with a 0.04 μm alumina slurry on a polishing cloth (DP-Nap, Struers, Denmark), using a Presi Mecatech 234 polishing machine. Grafting was achieved on Au coated (100 nm) Si wafers obtained from Sigma-Aldrich. Before modification, they were rinsed in concentrated sulfuric acid, ultrasonicated in Milli-Q water for 6 min, cleaned with pure ethanol, and dried under a stream of argon. For the formation of the diazonium salt 8, the ammonium salt, 7, was dissolved in ACN + 0.1 M n-Bu4BF4 (c = 2 mM). Isopentylnitrite (c = 2.4 mM) was then added and let to react for 30 min before the experiments started. All the electrochemical experiments were performed in ACN + 0.1 M n-Bu4BF4, deoxygenated with argon.
The IRRAS and ATR Spectra of modified plates were recorded using a purged (low CO2, dry air) Jasco FT/IR-6100 Fourier Transform InfraRed Spectrometer equipped with MCT (mercury–cadmium–telluride) detector. For each spectrum, 1000 scans were accumulated with a spectral resolution of 4 cm−1. The background recorded before each spectrum was that of a clean substrate. ATR spectra were recorded with a germanium ATR accessory (Jasco ATR PR0470-H). X-ray photoelectron spectra were recorded using a Thermo VG Scientific ESCALAB 250 system fitted with a microfocused, monochromatic Al Kα X-ray source (1486.6 eV) and a magnetic lens, which increases the electron acceptance angle and hence the sensitivity. The pass energy was set at 150 and 40 eV for the survey and the narrow regions, respectively. The Avantage Software, version 4.67, was used for digital acquisition and data processing. The spectra were calibrated against C1s set at 285 eV. The thicknesses of the films on Au were measured using a mono wavelength ellipsometer Sentech SE400. The following values were taken for gold ns = 0.17, ks = 3.43; they were measured on the clean surfaces before grafting. The film thicknesses were determined from the same plates after modification, taking ns = 1.46, ks = 0 for the organic layer. Raman spectra were recorded using an XploRA confocal Raman instrument (HORIBA Jobin Yvon) with a 638 nm laser as the source, in backscattering mode. All spectra were taken with a 3 s integration time and recorded within the 120–1750 cm−1 spectral range. The grafted Au20 mono charged gold cluster 9@Au was modeled and used to calculate the Raman spectra. Calculations were performed with Dmol3 through generalized gradient approximations (GGE) using PBE functional30 and DNP basis set (all electron core treatment). Self-consistent iteration method (SCF)31 was used for geometry optimization (1000 iteration steps, using energy convergence of 2.0 e−5 eV per atom).
The theoretical calculation of the standard redox potential in solution is based on the use of the Born–Haber cycle. All theoretical calculations were performed using the Gaussian software.§ The geometry optimizations were performed using B3LYP functional in combination with 6-311+g(d,p) basis set. The dispersion correction was performed based on the Grimme's Dispersion DFT-D3 model. Solvent effects (acetonitrile) were included via the integral equation formalism variant (IEFPCM) approach.32
Synthetic procedures
NMR 1H (CDCl3, 500 MHz): δ 8.57 (2H, dd, J = 4.5, 1.5 Hz), 8.28–8.25 (1H, m), 8.17 (2H, d, J = 8 Hz), 8.10 (2H, t, J = 8 Hz), 8.03 (2H, s), 8.00 (1H, t, J = 7.5 Hz), 7.85 (1H, m), 7.23–7.22 (2H, m), 5.86 (1H, q, J = 6.5 Hz), 3.40 (2H, t, J = 7.5 Hz), 2.54–2.50 (2H, m), 2.22 (2H, qn, J = 6.5 Hz), 1.52 (3H, d, J = 6.5 Hz). NMR 13C (CDCl3, 125 MHz): δ 172.6, 150.6, 150.3, 150.2, 135.6, 131.6, 131.0, 130.2, 129.3, 128.9, 127.6, 127.5, 127.0, 126.0, 125.3, 125.1, 125.0, 123.3, 122.0, 120.8, 70.9, 34.1, 32.8, 26.7, 22.11. MS (ESI): m/z =394.1 [M + H]+. HRMS (ESI): m/z calcd for [C27H23O2N]+ 393.1729, found 393.1735.
NMR 1H (CDCl3, 500 MHz): δ 8.69 (2H, d, J = 6.5 Hz), 8.25 (1H, d, J = 9 Hz), 8.17 (2H, d, J = 8 Hz), 8.11–8.09 (2H, m), 8.03–7.98 (3H, m), 7.84 (1H, d, J = 8 Hz), 7.64 (2H, d, J = 6.5 Hz), 5.7 (1H, q, J = 6.8 Hz), 4.69–4.66 (2H, m), 3.93 (2H, t, J = 5.5 Hz), 3.48–3.40 (2H, m), 2.55 (2H, t, J = 7.5 Hz), 2.24 (2H, qn, J = 7.5 Hz), 1.65-1.55 (2H, m), 1.46 (3H, d, J = 6.5 Hz). NMR 13C (CDCl3, 125 MHz): δ 172.2, 161.9, 145.2, 135.2, 131.5, 131.0, 130.2, 129.4, 128.9, 128.0, 127.7, 127.6, 127.0, 126.2, 125.7, 125.2, 125.1, 125.0, 124.6, 123.3, 69.9, 60.5, 50.7, 33.8, 32.7, 26.4, 21.7. MS (ESI): m/z =463.4 [M]+. HRMS (ESI): m/z calcd for [C29H27O2N4]+ 463.2129, found 463.2137.
NMR 1H (CDCl3, 500 MHz): δ 8.53 (2H, d, J = 6.8 Hz), 8.17 (1H, d, J = 9.5 Hz), 8.10 (2H, d, J = 7.5 Hz), 8.05–8.01 (3H, m), 7.95–7.92 (3H, m), 7.75 (1H, d, J = 8 Hz), 7.57 (2H, d, J = 8.5 Hz), 7.51 (2H, d, J = 6.5 Hz), 7.30 (2H, d, J = 8.5 Hz), 6.73 (1H, s), 5.60 (1H, q, J = 6.5 Hz), 5.15–5.12 (2H, m), 5.00–4.98 (2H, m), 3.37–3.24 (2H, m), 2.44 (2H, t, J = 7 Hz), 2.12 (2H, qn, J = 7 Hz), 1.48 (9H, s), 1.32 (3H, d, J = 6.5 Hz). NMR 13C (CDCl3, 125 MHz): δ 172.1, 161.9, 152.9, 148.0, 146.9, 145.1, 138.8, 135.3, 131.4, 130.9, 130.1, 128.8, 127.6, 127.5, 126.9, 126.4, 126.1, 125.1–124.7 (7C), 123.3, 122.0, 119.4, 69.8, 60.3, 36.4, 33.7, 32.6, 28.5, 26.3, 21.5, 14.9. MS (ESI): m/z =680.1 [M]+. HRMS (ESI): m/z calcd for [C42H42O4N5]+ 680.3231, found 680.3224.
NMR 1H (CDCl3, 250 MHz): δ 8.55 (2H, d, J = 6.8 Hz), 8.32 (1H, s), 8.13 (1H, d, J = 6.8 Hz), 8.05 (2H, d, J = 8.3 Hz), 8.00–7.85 (7H, m), 7.71 (3H, m), 7.41 (2H, d, J = 8.5 Hz), 5.65 (1H, q, J = 6.8 Hz), 5.01 (4H, m), 3.26–3.17 (2H, m), 2.45 (2H, t, J = 7 Hz), 2.04 (2H, qn, J = 7 Hz), 1.35 (3H, d, J = 6.8 Hz). NMR 13C (CDCl3, 125 MHz): δ 173.7, 164.0, 149.4, 146.2, 136.9, 132.8, 132.2, 131.4, 129.9, 128.7, 128.6, 128.4, 127.9, 127.7, 127.3, 127.1, 126.1–125.9 (5C), 124.4, 124.2, 123.1, 122.4, 119.0, 71.4, 61.3, 50.9, 34.5, 33.5, 27.6, 21.7. MS (ESI): m/z =581.1 [M]+. HRMS (ESI): m/z calcd for [C37H35O2N5]+ 581.2780, found 581.2771.
Immobilisation on GC and gold-coated electrodes
The diazonium salt was prepared in situ, inside the electrochemical cell, from the aniline precursor, 7, by adding isopentyl nitrite (1.2 eq.) to the ACN + n-Bu4NBF4 (0.1 M) electrolytic solution. The formed diazonium compound was not isolated and was used without further purification. A GC or gold plate was introduced, respectively, as the cathode in the cell and the solution was thoroughly deoxygenated with argon. 20 voltammograms were recorded between −0.5 and +1.5 V/(Ag/AgCl). Plates were then rinsed with ethanol using sonication for 280 s and dried.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. B. acknowledges the Nano-K grant of the Ile-de-France region. Also, the ANR (Agence Nationale de la Recherche) and CGI (Commissariat à l’Investissement d’Avenir) are acknowledged for their financial support through the Labex SEAM (Science and Engineering for Advanced Materials and devices) ANR 11 LABX 086 and ANR 11 IDEX 05 02 programs.Notes and references
- M. B. Yatvin, J. N. Weinstein, W. H. Dennis and R. Blumenthal, Science, 1978, 202, 1290 CrossRef CAS PubMed.
- Z. X. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590 RSC.
- S. Szunerits, F. Teodorescu and R. Boukherroub, Eur. Polym. J., 2016, 83, 467 CrossRef CAS; E. Cabane, X. Zhang, K. Langowska, C. G. Palivan and W. Meier, Biointerphases, 2012, 7, 9 CrossRef PubMed; H. A. Santos, J. Salonen, L. M. Bimbo, V. P. Lehto, L. Peltonen and J. J. Hirvonen, Drug Delivery Sci. Technol., 2011, 21, 139 CrossRef; Q. He and J. Shi, J. Mater. Chem., 2011, 21, 5845 RSC; C. Viseras, C. Aguzzi, P. Cerezo and M. C. Bedmar, Mater. Sci. Technol., 2008, 24, 1020 CrossRef.
- D. Svirskis, J. Travas-Sejdic, A. Rodgers and S. Garg, J. Controlled Release, 2010, 146, 6 CrossRef CAS PubMed; D. Pan, H. Zhang, T. Fan, J. Chen and X. Duan, Chem. Commun., 2011, 47, 908 RSC; T. Panczyk, T. P. Warzocha and P. J. Camp, J. Phys. Chem. C, 2010, 114, 21299 CrossRef; J. Choubey and A. K. Bajpai, J. Mater. Sci. Mater. Med., 2010, 21, 1573 CrossRef PubMed; N. Ž. Knezević, B. G. Trewyn and V. S.-Y. Lin, Chem. Commun., 2011, 47, 2817 RSC; D. R. Griffin, J. T. Patterson and A. M. Kasko, Biotechnol. Bioeng., 2010, 107, 1012 CrossRef PubMed; W. Zhang, K. Gilstrap, L. Wu, K. C. R. Bahadur, M. A. Moss, Q. Wang, X. Lu and X. He, ACS Nano, 2010, 4, 6747 CrossRef PubMed; W.-K. Fong, T. L. Hanley, B. Thierry, N. Kirby and B. J. Boyd, Langmuir, 2010, 26, 6136 CrossRef PubMed; D. Richard, I. Nguyen, C. Affolter, F. Meyer, P. Schaaf, J.-C. Voegel, D. Bagnard and J. Ogier, Small, 2010, 6, 2405 CrossRef PubMed; L. Yuan, Q. Tang, D. Yang, J. Z. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926 CrossRef; R. Negrini and R. Mezzenga, Langmuir, 2011, 27, 5296 CrossRef PubMed; Z. Zhang, L. Chen, C. Zhao, Y. Bai, M. Deng, H. Shan, X. Zhuang, X. Chen and X. Jing, Polymer, 2011, 52, 676 CrossRef; S. Aryal, C.-M. J. Hu and L. Zhang, ACS Nano, 2010, 4, 251 CrossRef PubMed; L. Wang, M. Liu, C. Gao, L. Ma and D. Cui, React. Funct. Polym., 2010, 70, 159 CrossRef; C. R. Gordijo, K. Koulajian, A. J. Shuhendler, L. D. Bonifacio, H. Y. Huang, S. Chiang, G. A. Ozin, A. Giacca and X. Y. Wu, Adv. Funct. Mater., 2011, 21, 73 CrossRef; A. Guiseppi-Elie, Biomaterials, 2010, 31, 2701 CrossRef PubMed; G. Stevenson, S. E. Moulton, P. C. Innis and G. G. Wallace, Synth. Met., 2010, 160, 1107 CrossRef.
- J. Guo and D. Fan, ChemNanoMat, 2018, 4, 1023 CrossRef CAS; P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512 RSC; D. Svirskis, J. Travas-Sejdic, A. Rodgers and S. Garg, J. Controlled Release, 2010, 146, 6 CrossRef PubMed; Z. Jin, G. Güven, V. Bocharova, J. Halamek, I. Tokarev, S. Minko, A. Melman, D. Mandler and E. Katz, ACS Appl. Mater. Interfaces, 2012, 4, 466 CrossRef PubMed; Z. Jin, A. M. Harvey, S. Mailloux, J. Halámek, V. Bocharova, M. R. Twiss and E. Katz, J. Mater. Chem., 2012, 22, 19523 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef CAS PubMed; B. P. Timko, T. Dvir and D. S. Kohane, Adv. Mater., 2010, 22, 4925 Search PubMed; C. S. Kim, B. Duncan, B. Creran and V. M. Rotello, Nano Today, 2013, 8, 439 CrossRef PubMed; S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef PubMed; Y. Lu, W. Sun and Z. Gu, J. Controlled Release, 2014, 194, 1 CrossRef PubMed; Y. Brudno and D. J. Mooney, J. Controlled Release, 2015, 219, 8 CrossRef PubMed; D. Uppalapati, B. J. Boyd, S. Garg, J. Travas-Sejdic and D. Svirskis, Biomaterials, 2016, 111, 149 CrossRef PubMed; Y. Zhao, A. C. Tavares and M. A. Gauthier, J. Mater. Chem. B, 2016, 4, 3019 RSC; S. Szunerits, F. Teodorescu and R. Boukherroub, Eur. Polym. J., 2016, 83, 467 CrossRef; F. Mastrangelo, G. Fioravanti, R. Quaresima, R. Vinci and E. Gherlone, J. Biomater. Nanobiotechnol., 2011, 2, 533 CrossRef; F. Blobner, P. N. Abufager, R. Han, J. Bauer, D. A. Duncan, R. J. Maurer, K. Reuter, P. Feulner and F. Allegretti, J. Phys. Chem. C, 2015, 119, 15455 CrossRef; B. Mu, P. Liu, P. Du, Y. Dong and C. Lu, J. Polym. Sci., A, 2011, 49, 1969 CrossRef; B. Ergün, L. De Cola, H.-J. Galla and N. Seda Kehr, Adv. Healthcare Mater., 2016, 5, 1588 CrossRef PubMed; J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, Small, 2010, 6, 1952 CrossRef PubMed; F. De Angelis, A. Pujia, C. Falcone, E. Iaccino, C. Palmieri, C. Liberale, F. Mecarini, P. Candeloro, L. Luberto, A. de Laurentiis, G. Das, G. Scala and E. Di Fabrizio, Nanoscale, 2010, 2, 2230 RSC; Y. Zhao, J. L. Vivero-Escoto, I. I. Slowing, B. C. Trewyn and V. S.-Y. Lin, Expert Opin. Drug Delivery, 2010, 7, 1013 CrossRef PubMed.
- J. M. Giussi, M. L. Cortez, W. A. Marmisolle and O. Azzaroni, Chem. Soc. Rev., 2019, 48, 814 RSC.
- K. Matyjaszewski, D. Hongchen, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528 CrossRef CAS PubMed; S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14 RSC.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575 CrossRef CAS; F. Fontana, F. Minisci, M. C. N. Barbosa and E. Vismara, J. Org. Chem., 1991, 56, 2866 CrossRef; J. Tauber, D. Imbri and T. Opatz, Molecules, 2014, 19, 16190 CrossRef PubMed; M. A. J. Duncton, MedChemComm, 2011, 2, 1135 RSC.
- I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194 CrossRef CAS PubMed.
- G. Pratsch, T. Wallaschkowski and M. R. Heinrich, Chem. – Eur. J, 2012, 18, 11555 CrossRef CAS PubMed.
- R. Möhlau and R. Berger, Chem. Ber., 1893, 26, 1196 CrossRef CAS; M. Gomberg and W. E. Bachmann, J. Am. Chem. Soc., 1924, 46, 2339 CrossRef; R. Pschorr, Chem. Ber., 1896, 29, 496 CrossRef.
- F. Vallée, J. J. Mousseau and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 1514 CrossRef PubMed.
- D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem. – Eur. J., 2014, 20, 2960 CrossRef CAS PubMed; J. Zhang, J. Chen, X. Zhang and X. Lei, J. Org. Chem., 2014, 79, 10682 CrossRef PubMed; F. Gomes, V. Narbonne, F. Blanchard, G. Maestri and M. Malacria, Org. Chem. Front., 2015, 2, 464 RSC; L. Zhi, H. Zhang, Z. Yang, W. Liu and B. Wang, Chem. Commun., 2016, 52, 6431 RSC; L. Buglioni, P. Riente, E. Palomares and M. A. Pericas, Eur. J. Org. Chem., 2017, 6986 CrossRef; S. Crespi, S. Jaeger, B. Koenig and M. Fagnoni, Eur. J. Org. Chem., 2017, 2147 CrossRef; D. C. Fabry, Y. A. Ho, R. Zapf, W. Tremel, M. Panthoefer, M. Rueping and T. H. Rehm, Green Chem., 2017, 19, 1911 RSC; S. Zhang, Z. Tang, W. Bao, J. Li, B. Guo, S. Huang, Y. Zhang and Y. Rao, Org. Biomol. Chem., 2019, 17, 4364 RSC; P. K. Vardhanapu, J. Ahmed, A. Jose, B. K. Shaw, T. K. Sen, A. A. Mathews and S. K. Mandal, J. Org. Chem., 2019, 84, 289 CrossRef PubMed; W. Hagui and J.-F. Soulé, J. Org. Chem., 2020, 85, 3655 CrossRef PubMed.
- D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem. – Eur. J, 2014, 20, 2960 CrossRef CAS PubMed.
- M.-C. Bernard, A. Chaussé, E. Cabet-Deliry, M. M. Chehimi, J. Pinson, F. Podvorica and C. Vautrin-Ul, Chem. Mater., 2003, 15, 3450 CrossRef CAS; P. A. Brooksby and A. J. Downard, Langmuir, 2004, 20, 5038 CrossRef PubMed; T. Menanteau, E. Levillain and T. Breton, Chem. Mater., 2013, 25, 2905 CrossRef; T.-C. Kuo, R. L. McCreery and G. M. Swain, Electrochem. Solid State Lett., 1999, 2, 288 CrossRef.
- J. Heinze, Aliphatic and Aromatic Hydrocarbons Reduction, in Organic Electrochemistry, 5th edn, ed. O. Hammerich and B. Speiser, CRC Press, Boca Raton, USA, 2016 Search PubMed.
- M. Delamar, R. Hitmi, J. Pinson and J.-M. Savéant, J. Am. Chem. Soc., 1992, 114, 5883 CrossRef CAS; J. Pinson and F. I. Podvorica, Chem. Soc. Rev., 2005, 34, 429 RSC; A. Berisha, M. M. Chehimi, J. Pinson and F. I. Podvorica, in Electroanalytical Chemistry, ed. A. J. Bard and C. G. Zoski, CRC Press, Boca Raton, FL, 2016 Search PubMed.
- F. M'Halla, J. Pinson and J. M. Savéant, J. Am. Chem. Soc., 1980, 102, 4120 CrossRef.
- C. P. Andrieux, A. Merz, J.-M. Savéant and R. Tomahoghl, J. Am. Chem. Soc., 1984, 106, 1957 CrossRef CAS; F. Hui, J.-M. Noël, P. Poizot, P. Hapiot and J. Simonet, Langmuir, 2011, 27, 5119 CrossRef PubMed; M. Ohkoshi, J. Michinishi, S. Hara and H. Senbok, Tetrahedron, 2010, 66, 7732 CrossRef.
- Hydrogen abstraction of the aryl radical expected to give raise of hydrolytically unstable enol ethers; also, the addition of the radical to the ligand would quench the fluorescence.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- P. Dunkel, A. Barosi, H. Dhimane, F. Maurel and P. I. Dalko, Chem. – Eur. J., 2018, 24, 12920 CrossRef CAS PubMed; A. Barosi, P. Dunkel, E. Guénin, Y. Lalatonne, P. Zeitoun, I. Fitton, C. Journé, A. Bravin, A. Maruani, H. Dhimane, L. Motte and P. I. Dalko, RSC Adv., 2020, 10, 3366 RSC; C. Sundararajan and D. E. Falvey, Org. Lett., 2005, 7, 2631 CrossRef PubMed and references cited.
- Au coated (100 nm) Si wafers (Sigma-Aldrich).
- All experiences were performed in ACN with n-Bu4NBF4 (c = 0.1 M) as supporting electrolyte, a glassy carbon (GC) rod (d = 2 mm) working electrode sealed in glass and an Ag/AgCl reference electrode.
- T. Breton and A. J. Downard, Aust. J. Chem., 2017, 70, 960 CrossRef CAS; S. Gam-Derouich, M. N. Nguyen, A. Madani, N. Maouche, P. Lang, C. Perruchot and M. M. Chehimi, Surf. Interface Anal., 2010, 42, 1050 CrossRef; S. Baranton and D. Bélanger, J. Phys. Chem. B, 2005, 109, 24401 CrossRef PubMed.
- L. Laurentius, S. R. Stoyanov, S. Gusarov, A. Kovalenko, R. B. Du, G. P. Lopinski and M. T. McDermott, ACS Nano, 2011, 5, 4219 CrossRef CAS PubMed; R. Ahmad, L. Boubekeur-Lecaque, M. Nguyen, S. Lau-Truong, A. Lamouri, P. Decorse, A. Galtayries, J. Pinson, N. Felidj and C. Mangeney, J. Phys. Chem. C, 2014, 118, 19098 CrossRef.
- L. Xiao, M. Zhang, Z. Liu, W. Bian, X. Zhang and J. Zhan, Anal. Methods, 2017, 9, 1816 RSC.
- E. Laviron, J. Electroanal. Chem., 1995, 391, 187 CrossRef.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- E. Cances, B. Mennucci and J. J. Tomasi, Chem. Phys., 1997, 107, 3032 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00022e |
| ‡ Gold plated silicon wafers were selected as support due to their reflective properties, facilitating the characterization by infra-red reflection–absorption spectroscopy (IRRAS) and ellipsometry.)24 |
| § All theoretical calculations were performed using the Gaussian software.22 The geometry optimizations were performed using B3LYP functional in combination with the 6-311+g(d,p) basis set. The dispersion correction was performed based on Grimme's Dispersion DFT-D3 model. Solvent effects (acetonitrile) were included via the integral equation formalism variant (IEFPCM) approach. |
| This journal is © The Royal Society of Chemistry 2021 |