
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaleimide-functionalized metal–organic framework for polysulfide tethering in lithium–sulfur batteries†
David A.
Burns
 a,
Angelica
Benavidez
b,
Jessica L.
Buckner
c and
V. Sara
Thoi
*ad
a,
Angelica
Benavidez
b,
Jessica L.
Buckner
c and
V. Sara
Thoi
*ad
aDepartment of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, USA. E-mail: sarathoi@jhu.edu
bDepartment of Chemical and Biological Engineering, University of New Mexico, Albuquerque, New Mexico 87131, USA
cAir Force Research Lab, Kirtland Air Force Base, Albuquerque, New Mexico 87117, USA
dDepartment of Materials Science and Engineering, Johns Hopkins University, Baltimore, Maryland 21218, USA
First published on 22nd March 2021
Abstract
Lithium–sulfur (Li–S) batteries have great potential as next generation energy storage devices. However, the redox chemistry mechanism involves the generation of solubilized lithium polysulfides, which can lead to leaching of the active material and, consequently, passivated electrodes and diminished capacities. Chemical tethering of lithium polysulfides to materials in the sulfur cathode is a promising approach for resolving this issue in Li–S batteries. Borrowing from the field of synthetic chemistry, we utilize maleimide functional groups in a Zr-based metal–organic framework to chemically interact with polysulfides through the Michael Addition reaction. A combination of molecular and solid-state spectroscopies confirms covalent attachment of Li2Sx to the maleimide functionality. When integrated into Li–S cathodes, the maleimide-functionalized framework exhibits notable performance enhancements over that of the unfunctionalized material, revealing the promise of polysulfide anchors for Li–S battery cycling.
Introduction
The perpetual growth in demand for energy storage devices to power portable devices and electric vehicles necessitates a search for affordable high-capacity batteries beyond lithium ion (Li-ion) technologies. Lithium sulfur (Li–S) batteries are receiving tremendous attention due to their high theoretical specific energy (2680 W h kg−1) and energy density (2199 W h L−1).1 The ability to deliver such high amounts of energy is derived from the series of steps in the discharge mechanism in which elemental sulfur (S8) is electrochemically converted to lithium polysulfides (Li2Sx, x ≤ 8) at the cathode.1–3 Many of these Li2Sx species are electrochemically reduced to shorter chain lengths before being deposited as insoluble Li2S2 and Li2S species at the end of discharge. However, the diffusion of dissolved Li2Sx away from the surface of the cathode is the source of many problems, including electrode passivation and extensive capacity fade over time.In pursuit of designing Li–S cells worthy of competing with Li-ion technologies, a number of materials have been explored to minimize leaching of Li2Sx from the sulfur cathode, such as the use of metal–organic frameworks (MOFs).4–13 MOFs are porous, crystalline materials with finely controllable chemical and physical properties. The high surface area of porous MOFs has been taken advantage of to physically adsorb soluble Li2Sx and mitigate their diffusion away from the cathode.6–8 Additionally, MOFs with chemisorption sites provide stronger association between Li2Sx and the internal surface of the material and have been shown to be effective for enhancing the cycle life of Li–S batteries.9–14 Although these adsorption methods offer improvements in performance, they still do not sufficiently solve polysulfide dissolution during long term cycling.
Our research group has previously demonstrated functionalized Zr-MOFs are versatile platforms for studying polysulfide adsorption.15–17 One of the more effective methods of retaining Li2Sx is to chemically bond them to the cathode. We recently illustrated the feasibility of this technique by integrating lithium thiophosphate (Li3PS4) moieties into a Zr-MOF to create a material capable of chemically tethering Li2Sx.17 The covalent anchoring sites were effective for enhancing both sulfur utilization and retention of Li2Sx. Similar chemical tethering approaches have been used in other classes of materials as well, which further shows the promise of the technique.18–23
Taking advantage of the synthetic versatility of MOFs, we explored novel methods to enhance the number of covalent anchor sites. Our strategy to accomplish this goal is incorporate the anchor site on the organic linker to ensure its prevalence throughout the MOF. Inspired by its previous use in a MOF for fluorescent chemical sensing of thiols,24 the maleimide (Mi) moiety was selected for facile integration into the organic struts. Mi groups can selectively react with thiols through the Michael Addition mechanism. This reactivity has strong literature precedence in synthetic chemistry and biochemistries14,25 as well as polymer and materials chemistries.26–29 Notably, base-catalyzed Michael addition involves the deprotonation of thiols to create the more reactive thiolate anion for faster reactions.30 We anticipated that the maleimide functionality would react with the anionic polysulfides generated during Li–S cycling in a similar manner to this base-catalyzed mechanism. In this work, we use maleimide-functionalized UiO-66 (Mi-UiO-66) as a test platform for understanding Mi reactivity towards polysulfides (Scheme 1). We present spectroscopic evidence that demonstrate the ability of Mi to chemically tether Li2Sx through covalent linkage and their promise for enhancing cycling performance. Importantly, this simple organic functionalization can be translated to other cathode materials for Li–S batteries.
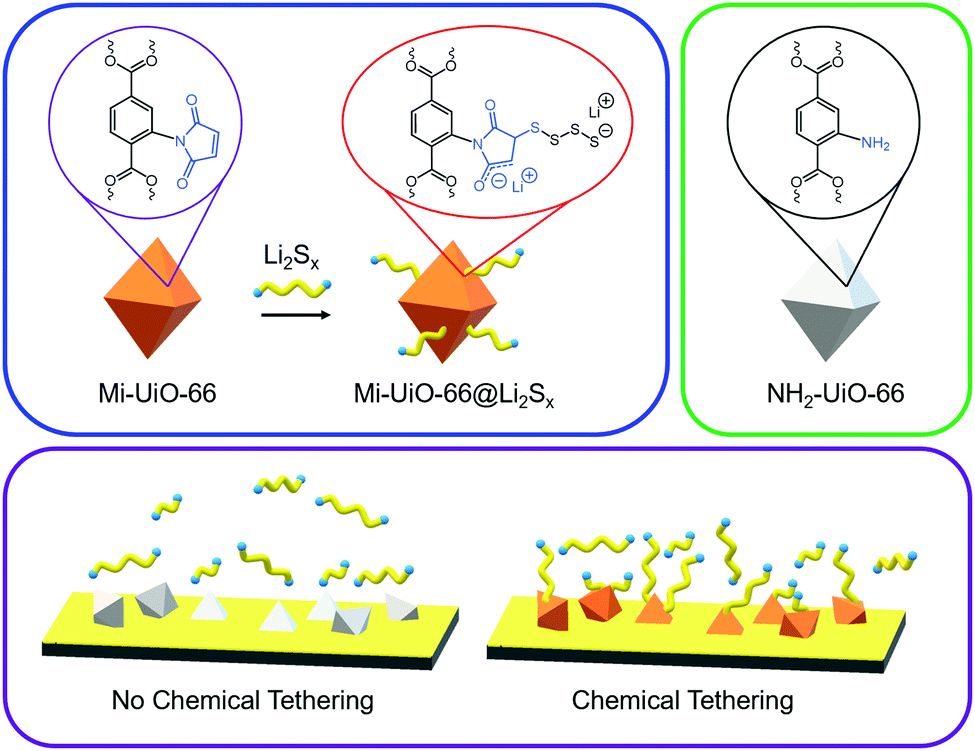 | ||
| Scheme 1 The postulated structure of Mi-UiO-66 (orange polyhedra) upon the introduction of lithium polysulfides (Li2Sx, yellow/blue), the control NH2-UiO-66 (grey polyhedra), and a representation of the chemical tethering strategy in a MOF composite electrode (bottom). | ||
Results and discussion
Molecular reactivity of maleimides with Li2Sx
We first validated the reactivity of Mi in a series of model reactions between Mi-functionalized organic molecules and polysulfides (Li2Sx) in an environment similar to that of an Li–S battery. Li2Sx solutions were made in the common Li–S electrolyte solvent system, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME). The methyl ester analog (Mi-BDOMe) of the Mi-functionalized benzene dicarboxylate linker (Mi-BDC) was a convenient model compound. Upon adding the white Mi-BDOMe powder to the orange Li2Sx (x ≤ 8) solution, an immediate color change to deep red was observed, which is consistent with the rapid formation of the maleimide enolate.31 UV-Vis absorption spectra (Fig. 1a) illustrate the color change with a redshift of 30 nm in the absorption of the solution. Fluorescence spectroscopy further confirms the chemical connectivity between Mi-BDOMe and Li2Sx (Fig. 1b). Evidence of the adduct is shown by the shift of the peak emission wavelength, λmax, to ∼490 nm, which agrees with the λmax of the Mi-UiO-66 following Michael addition of thiols.24 Thus, we anticipate the Mi-BDC linker is capable of chemically tethering Li2Sx in the DOL/DME solvent environment.
:1 ratio of 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME). The methyl ester analog (Mi-BDOMe) of the Mi-functionalized benzene dicarboxylate linker (Mi-BDC) was a convenient model compound. Upon adding the white Mi-BDOMe powder to the orange Li2Sx (x ≤ 8) solution, an immediate color change to deep red was observed, which is consistent with the rapid formation of the maleimide enolate.31 UV-Vis absorption spectra (Fig. 1a) illustrate the color change with a redshift of 30 nm in the absorption of the solution. Fluorescence spectroscopy further confirms the chemical connectivity between Mi-BDOMe and Li2Sx (Fig. 1b). Evidence of the adduct is shown by the shift of the peak emission wavelength, λmax, to ∼490 nm, which agrees with the λmax of the Mi-UiO-66 following Michael addition of thiols.24 Thus, we anticipate the Mi-BDC linker is capable of chemically tethering Li2Sx in the DOL/DME solvent environment.
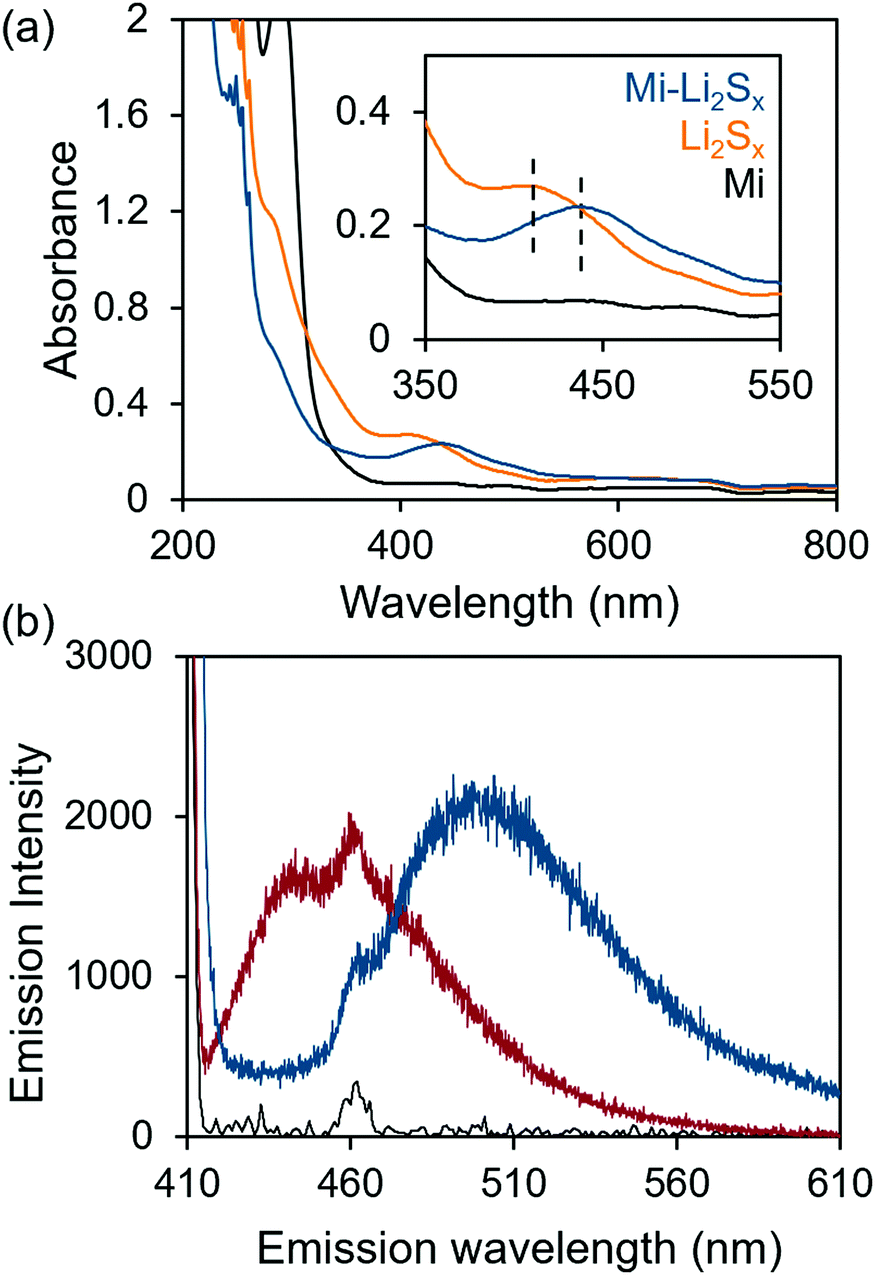 | ||
| Fig. 1 (a) UV-Vis absorption spectra solutions of Li2Sx (orange), Mi-BDOMe (Mi, black), and Li2Sx + Mi-BDOMe (Mi-Li2Sx, blue) in 1:1 DOL/DME. The inset shows the absorbance change upon addition of Mi-BDOMe to Li2Sx. (b) Fluorescence emission spectra of Mi-BDOMe (red) and the Mi-BDOMe-Li2Sx (blue) solutions excited with 405 nm light. The background fluorescence of the solvent mixture is shown in black. | ||
In search of structural confirmation of the reaction products, N-phenyl maleimide (NPM) was used as a proxy for the Mi linker in 1H and 13C NMR experiments. Upon exposing a solution of NPM to Li2S, significant changes are observed in the 1H NMR spectrum (Fig. 2), in addition to a change to a deep red color (Fig. S3, ESI†). Importantly, the peak associated with the alkene protons in the NPM spectrum disappears in the NPM + Li2S spectrum, which is consistent with the anticipated loss of the alkene after the Michael addition. Meanwhile, two other features of equal integration appear at 5.89 ppm and 2.77 ppm. Formerly in the alkene, these protons have been assigned to the enolate (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–C) and CH–S, respectively. It is worth noting that the broad features from 6–8 ppm and 3–5 ppm, as well as the additional features under 3 ppm, have previously been assigned to the homopolymerization of maleimide groups (Fig. S4, ESI†).31,32 The polymerization is propagated by the enolate product, which supports the formation of the enolate following the addition of Li2Sx to the maleimide moiety.
CH–C) and CH–S, respectively. It is worth noting that the broad features from 6–8 ppm and 3–5 ppm, as well as the additional features under 3 ppm, have previously been assigned to the homopolymerization of maleimide groups (Fig. S4, ESI†).31,32 The polymerization is propagated by the enolate product, which supports the formation of the enolate following the addition of Li2Sx to the maleimide moiety.
 | ||
| Fig. 2 1H NMR spectra of NPM (bottom) and NPM + Li2Sx (top) collected in CD3CN. NPM alkene protons are highlighted in orange, and the enolate product peaks are highlighted in blue and green. The asterisk (*) indicates peaks from residual DOL/DME. | ||
Likewise, 13C NMR spectra show the peaks associated with the carbonyls at 169.6 ppm and the maleimide alkene at 134.0 ppm shift to new positions upon addition of Li2Sx to the solution of NPM. The carbonyls, now inequivalent, are observed at 158.3 ppm and 157.3 ppm. The 13C shift from the alkene has notably decrease in intensity and a new peak at 52.3 ppm has been assigned to the carbon in the C–S bond. Other features, including near 175 ppm and below 50 ppm arise from the maleimide homopolymer byproduct as observed in the 1H NMR spectrum.32 The polymerization side reactions are specific to these soluble molecular models and are not expected to take place in the Mi-UiO-66 due to the fixed positions of the MOF linkers. Corroborating the NMR results, the Michael addition was also confirmed in Fourier transform infrared absorption spectroscopy (Fig. S7, ESI†) by the reduction of the strong C–H alkene peak at 829 cm−1, as well as the shifting of the imide peak from 1144 cm−1 to 1179 cm−1 following the reaction.32
Reactivity of Mi-functionalized MOF with Li2Sx
Building upon the findings of our molecular studies, we translated the reactivity of maleimide towards Li2Sx to the solid state in Mi-functionalized UiO-66 MOF (Mi-UiO-66). UiO-66 is a convenient platform for studying Mi reactivity in the solid state because of the diversity of chemical functionalities that can be integrated into the organic linker without changing the overall structure of the framework. Mi-UiO-66 was synthesized according to literature procedure as detailed in the ESI,† and powder X-ray diffraction and IR confirm the structural and chemical composition, respectively (Fig. S8 and S9, ESI†).24 Integration of maleimide into the MOF was further demonstrated by digesting Mi-UiO-66 in D2SO4/D2O. 1H NMR spectroscopy of the resulting solution verified successful incorporation of the maleimide functional group in the framework (Fig. S10, ESI†).To investigate the reactivity between Mi-UiO-66 and Li2Sx, we turned to X-ray photoelectron spectroscopy (XPS). Mi-UiO-66 and a control sample, UiO-66, were immersed in solutions of Li2S8 overnight prior to the measurement to obtain Mi-UiO-66@Li2Sx and UiO-66@Li2Sx. The presence of Li2Sx was observed in scans of both MOFs, suggesting adsorption of the Li2Sx to the MOF (Fig. 3). Prominent S2p3/2 and S2p1/2 peaks for mid-chain S atoms of Li2Sx were seen in high resolution scans of both samples at 163.5 eV and 164.8 eV at the expected separation of 1.2 eV and 2:1 intensity ratio.33 Less conspicuous peaks assigned to the S2p3/2 and S2p1/2 of the terminal S atoms in Li2Sx were observed at 162.0 eV and 163.2 eV in both samples as well. Most importantly, S2p3/2 and S2p1/2 peaks assigned to an S atom in the C–S moiety at 163.0 eV and 164.2 eV were observed in the Mi-UiO-66@Li2Sx sample but not in UiO-66@Li2Sx. These values agree with other literature containing maleimide Michael addition adducts.26,28 Overall, these results align well with our solution phase experiments, and together strongly suggest that the Mi-UiO-66 is capable of chemically bonding to Li2Sx species generated during cycling.
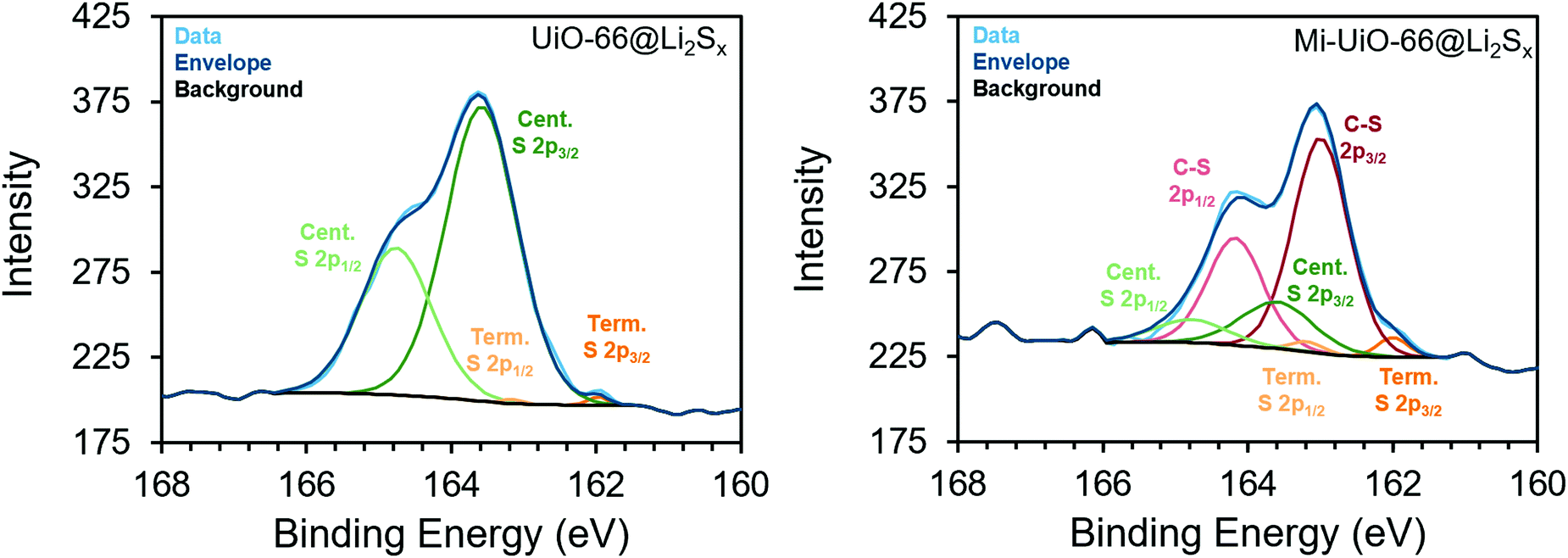 | ||
| Fig. 3 XPS spectra of the S2p region of UiO-66@Li2Sx (left) and Mi-UiO-66@Li2Sx (right). | ||
After establishing the reactivity of Mi-UiO-66 towards Li2Sx, we utilized the framework material as a cathode additive for Li–S batteries. Battery preparation procedures can be found in detail in the ESI.† Briefly, the composite cathodes containing a physical mixture of the 30 wt% MOF, 45 wt% sulfur, 15 wt% Super P carbon, and 10 wt% of poly(vinylidene fluoride) (PVDF) were assembled into 2032 coin cells using a Li metal anode and a Celgard separator in the presence of a standard ether electrolyte. The coin cells were cycled galvanostatically for one cycle at a rate of C/10 (168 mA g−1 S) followed by 100 cycles at C/2 (840 mA g−1 S). As shown in Fig. 4a, the initial discharge capacity at C/10 of Mi-UiO-66 is 1247 ± 107 mA h g−1 compared to 968 ± 99 mA h g−1 for NH2-UiO-66. Although both Mi-UiO-66 and NH2-UiO-66 have similarly stable capacity fade, capacity retention of Mi-UiO-66 cells after 100 cycles at C/2 is notably greater than that of the NH2-UiO cells (Fig. 4b and Fig. S11, ESI†). This observation suggests greater sulfur utilization was achieved likely due to the enhanced electrochemical accessibility of Mi-tethered polysulfides restrained to the electrode surface. The galvanostatic cycling curves for these experiments (Fig. S12, ESI†) reveal that both the charge and discharge voltages are more favorable in the Mi-UiO-66 cells, indicating reduced cell polarization. The reduction of polarization compared to NH2-UiO-66 suggests that the maleimide group in Mi-UiO-66 improves the reaction kinetics for sulfur conversion.34 Electrochemical impedance spectroscopy (EIS) of discharged cells provides similar insights into performance differences (Fig. S13 and Table S1, ESI†). The higher charge transfer resistance, R3, in the Mi-UiO-66 cell compared to that of NH2-UiO-66 suggests a more complete conversion to the insulating Li2S at the end of the discharge cycle.
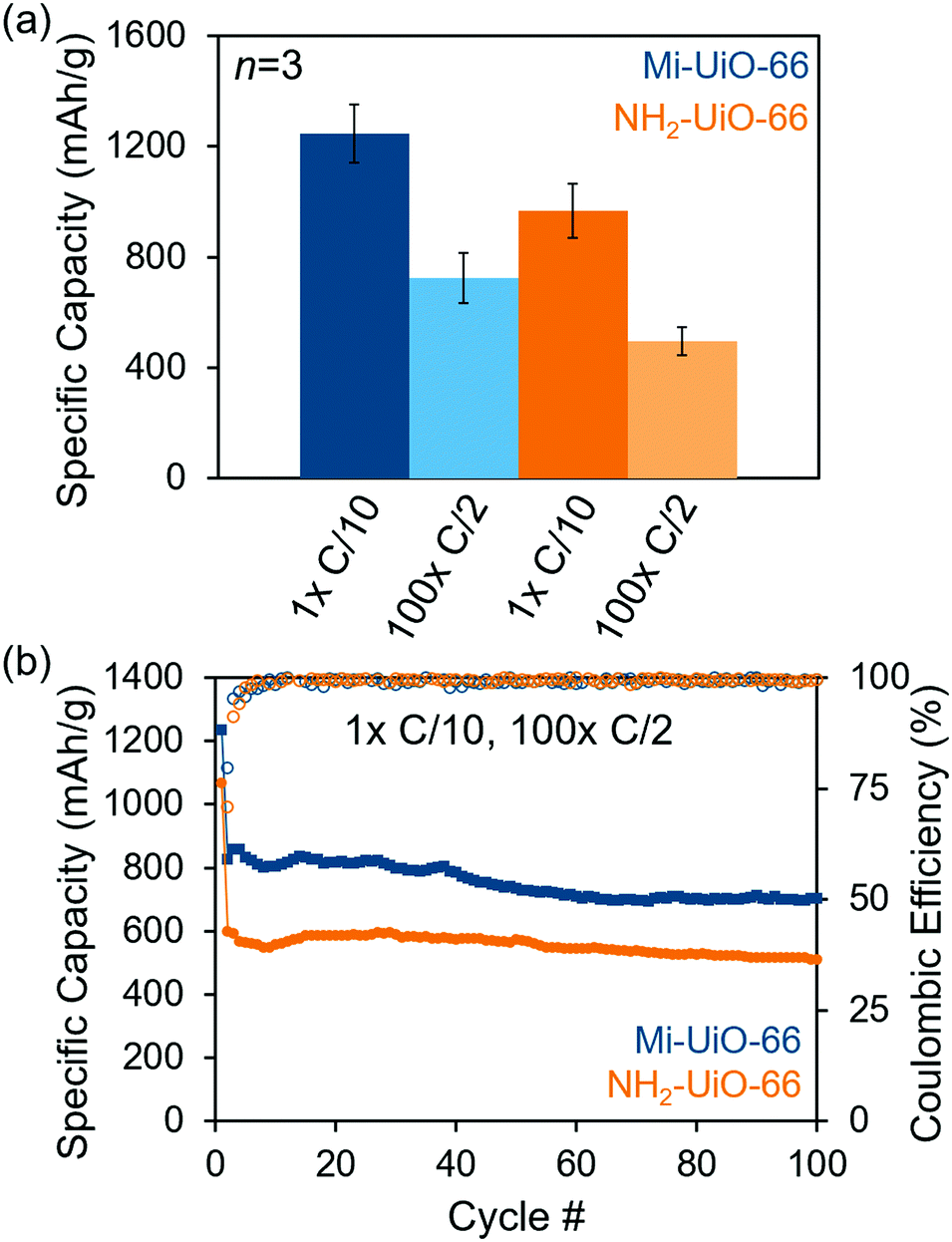 | ||
| Fig. 4 Capacity retention of Mi-UiO-66 (blue) and NH2-UiO-66 (orange) cells cycled at C/10 for 1 cycle and C/2 for 100 cycles. (a) Average initial (dark) and final (light) capacities of each type of cell (n = 3). (b) Specific capacity (filled squares) and coulombic efficiency (open circles) for representative cells over 100 cycles. | ||
The benefits of Mi-UiO-66 are also maintained across varied charge and discharge rates (Fig. 5). At all C-rates, the Mi-UiO-66 cells exhibit higher specific capacities compared to NH2-UiO-66, although both types of cell show deteriorating performance at the highest C-rates. Galvanostatic charge and discharge curves (Fig. S14, ESI†) additionally illustrate that the NH2-UiO-66 cells experience much stronger polarization and struggle to produce stable plateaus. Ultimately, the enhanced performance of Li–S cells containing Mi-UiO-66 as a cathode additive demonstrates the benefits of installing chemical anchoring sites within a sulfur cathode. We postulate that the pendant polysulfides tethered to the MOF stabilize solubilized Li2Sx species while maintaining their electrochemical accessibility at the cathode surface. We note a related study has shown electrochemical accessibility in similar MOFs is limited to near the external surface of the MOF particles. Thus, we expect the largest contributions of the Mi moiety occur at such locations rather than within the internal MOF structure.35 We anticipate enhancements could be found with expanded pore architectures that would increase accessibility to maleimide moieties within the framework.
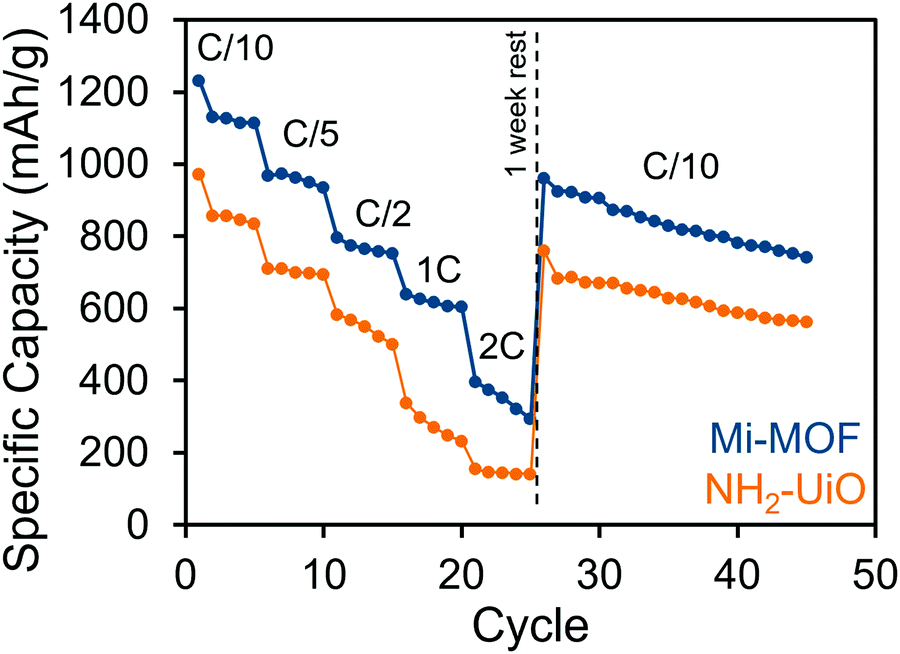 | ||
| Fig. 5 Rate performance of Mi-UiO-66 (blue) and NH2-UiO-66 (orange) cells cycled at C/10, C/5, C/2, 1C and 2C. | ||
Conclusions
Inspired by its well-known reactivity with thiolates, the maleimide functional group was selected as a candidate for chemical anchoring of Li2Sx to improve Li–S battery performance. We confirm this reactivity in solution phase experiments as well as in maleimide-functionalized UiO-66 as a model platform. When added to Li–S cathodes, the Mi-UiO-66 provided enhanced capacity, cyclic stability, and reduced cell polarization relative to the control MOF additive. Using electrochemistry and spectroscopy, this proof-of-concept study reveals the polysulfide tethering capability of the maleimide group, which leads to greater electrochemical Li2Sx accessibility and facilitates faster kinetics for sulfur redox. We expect our chemical tethering approaches can be translated to other classes of cathode materials, such as polymers and conductive carbons, and result in further improvements in Li–S batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the Division of Materials Research at the National Science Foundation (Award #1945114) and Johns Hopkins University through a generous start-up package. D. A. B. also thanks the Department of Chemistry for the Harry and Cleio Greer Fellowship and the Air Force Research Lab Scholars Program through the Universities Space Research Association. Finally, D. A. B. would like to thank Sahil Aggarwhal at Johns Hopkins University for helpful discussions on thiolate chemistry and Dr Thomas Peng at the Air Force Research Lab for generously providing lab space. AFRL: approved for public release; distribution is unlimited. Public Affairs release approval #AFMC-2020-0251.Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- K. Sun, N. Li, D. Su and H. Gan, J. Electrochem. Soc., 2019, 166, A50–A58 CrossRef CAS.
- H. Bin and Wu and X. W. Lou, Sci. Adv., 2017, 3, 1–17 Search PubMed.
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Chem. Commun., 2019, 2, 86 CrossRef.
- J. Zhou, R. Li, X. Fan, Y. Chen, R. Han, W. Li, J. Zheng, B. Wang and X. Li, Energy Environ. Sci., 2014, 7, 2715 RSC.
- H. Jiang, X.-C. Liu, Y. Wu, Y. Shu, X. Gong, F.-S. Ke and H. Deng, Angew. Chem., Int. Ed., 2018, 57, 3916–3921 CrossRef CAS PubMed.
- R. Demir-Cakan, M. Morcrette, F. Nouar, C. Davoisne, T. Devic, D. Gonbeau, R. Dominko, C. Serre, G. Férey and J. M. Tarascon, J. Am. Chem. Soc., 2011, 133, 16154–16160 CrossRef CAS PubMed.
- H. Park and D. J. Siegel, Chem. Mater., 2017, 29, 4932–4939 CrossRef CAS.
- Z. Wang, B. Wang, Y. Yang, Y. Cui, Z. Wang, B. Chen and G. Qian, ACS Appl. Mater. Interfaces, 2015, 7, 20999–21004 CrossRef CAS PubMed.
- A. E. Baumann, G. E. Aversa, A. Roy, M. L. Falk, N. M. Bedford and V. S. Thoi, J. Mater. Chem. A, 2018, 6, 4811–4821 RSC.
- X.-F. Liu, X.-Q. Guo, R. Wang, Q.-C. Liu, Z.-J. Li, S.-Q. Zang and T. C. W. Mak, J. Mater. Chem. A, 2019, 7, 2838–2844 RSC.
- J. H. Park, K. M. Choi, D. K. Lee, B. C. Moon, S. R. Shin, M.-K. Song and J. K. Kang, Nat. Publ. Gr., 2016, 6, 25555 CAS.
- S. M. Heilmann and J. K. Rasmussen, Comprehensive Heterocyclic Chemistry, Elsevier, 1st edn, 1984, pp. 269–315 Search PubMed.
- A. E. Baumann, D. A. Burns, C. D. Jose and V. S. Thoi, ACS Appl. Mater. Interfaces, 2019, 11(2), 2159–2167 CrossRef CAS PubMed.
- B. Liu, A. E. Baumann and V. S. Thoi, Polyhedron, 2019, 170, 788–795 CrossRef CAS.
- A. E. Baumann, X. Han, M. M. Butala and V. S. Thoi, J. Am. Chem. Soc., 2019, 141, 17891–17899 CrossRef CAS PubMed.
- D. R. Deng, F. Xue, C.-D. Bai, J. Lei, R. Yuan, M. Sen Zheng and Q. F. Dong, ACS Nano, 2018, 12, 11120–11129 CrossRef CAS PubMed.
- F. Xu, S. Yang, X. Chen, Q. Liu, H. Li, H. Wang, B. Wei and D. Jiang, Chem. Sci., 2019, 10, 6001–6006 RSC.
- R. Fang, J. Xu and D.-W. Wang, Energy Environ. Sci., 2020, 13, 432–471 RSC.
- C. Yang, Q. Du, Z. Li, M. Ling, X. Song, V. Battaglia, X. Chen and G. Liu, J. Power Sources, 2018, 402, 1–6 CrossRef CAS.
- S. Zeng, L. Li, J. Yu, N. Wang and S. Chen, Electrochim. Acta, 2018, 263, 53–59 CrossRef CAS.
- X. Liu, S. Wang, A. Wang, Z. Wang, J. Chen, Q. Zeng, P. Chen, W. Liu, Z. Li and L. Zhang, J. Mater. Chem. A, 2019, 7, 24515–24523 RSC.
- Y.-A. Li, C.-W. Zhao, N.-X. Zhu, Q.-K. Liu, G.-J. Chen, J.-B. Liu, X.-D. Zhao, J.-P. Ma, S. Zhang and Y.-B. Dong, Chem. Commun., 2015, 51, 17672–17675 RSC.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- T. N. Gevrek, T. Bilgic, H.-A. Klok and A. Sanyal, Macromolecules, 2014, 47, 7842–7851 CrossRef CAS.
- S. S. Nagane, S. S. Kuhire, U. A. Jadhav, S. A. Dhanmane and P. P. Wadgaonkar, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 630–640 CrossRef CAS.
- P. Gobbo, M. C. Biesinger and M. S. Workentin, Chem. Commun., 2013, 49, 2831 RSC.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- A. Kamimura, N. Murakami, F. Kawahara, K. Yokota, Y. Omata, K. Matsuura, Y. Oishi, R. Morita, H. Mitsudera, H. Suzukawa, A. Kakehi, M. Shirai and H. Okamoto, Tetrahedron, 2003, 59, 9537–9546 CrossRef CAS.
- Y. Yoshida and T. Endo, Polym. Chem., 2016, 7, 6770–6778 RSC.
- J. Wu, S. Sun and B. M. Novak, J. Appl. Polym. Sci., 2019, 48909 Search PubMed.
- M. Fantauzzi, B. Elsener, D. Atzei, A. Rigoldi and A. Rossi, RSC Adv., 2015, 5, 75953–75963 RSC.
- J. Yan, X. Liu and B. Li, Adv. Sci., 2016, 3, 1600101 CrossRef.
- R. H. Palmer, J. Liu, C.-W. Kung, I. Hod, O. K. Farha and J. T. Hupp, Langmuir, 2018, 34(16), 4707–4714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00084e |
| This journal is © The Royal Society of Chemistry 2021 |