
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual detection of nafcillin using a molecularly imprinted polymer-based platform coupled to thermal and fluorescence read-out†
Alexander D.
Hudson
a,
Oliver
Jamieson
a,
Robert D.
Crapnell
 b,
Knut
Rurack
c,
Thais C. C.
Soares
d,
Francesco
Mecozzi
b,
Alex
Laude
e,
Jonas
Gruber
f,
Katarina
Novakovic
a and
Marloes
Peeters
*a
b,
Knut
Rurack
c,
Thais C. C.
Soares
d,
Francesco
Mecozzi
b,
Alex
Laude
e,
Jonas
Gruber
f,
Katarina
Novakovic
a and
Marloes
Peeters
*a
aNewcastle University, School of Engineering, Merz Court, Claremont Road, NE1 7RU, Newcastle Upon Tyne, UK. E-mail: marloes.peeters@newcastle.ac.uk
bManchester Metropolitan University, Faculty of Science and Engineering, John Dalton Building, Chester Street, M1 5GD, Manchester, UK
cChemical and Optical Sensing Division, Bundesanstalt für Materialforschung und -prüfung (BAM), Richard-Willstätter-Straße 11, 12489 Berlin, Germany
dDepartamento de Engenharia Química, Escola Politécnica, Universidade de São Paulo, Avenida Prof. Luciano Gualberto, trav. 3, 380, CEP 05508-900, São Paulo, SP, Brazil
eNewcastle University, Bioimaging Unit, Leech Building, Framlington Place, NE2 4HH, Newcastle Upon Tyne, UK
fDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, CEP 05508-000, São Paulo, SP, Brazil
First published on 23rd June 2021
Abstract
Reported here is the production of molecularly imprinted polymer (MIP) films, integrating a fluorescent moiety that serves as both an element for template interaction and signalling, for the thermal and optical detection of the beta-lactam antibiotic nafcillin. Fluorescein methacrylate (FluMa) was synthesized and introduced during the molecular imprinting process as the sole monomer and in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with methacrylic acid (MAA), allowing to draw first conclusions on the MIP formation potential of such a rather large and rigid monomer. At first, MIP microparticles containing FluMa were prepared by free radical polymerisation. Optical batch rebinding experiments revealed that FluMa can act as a functional monomer for selective detection of nafcillin; however, the addition of MAA as co-monomer significantly improved performance. Subsequently, thin MIP films containing FluMa were deposited onto functionalised glass slides and the influence of porogen, drying time, and monomer composition was studied. These MIP-functionalised glass electrodes were mounted into a customised 3D-printed flow cell, where changes in the liquid were either evaluated with a thermal device or using fluorescence bright field microscopy. Thermal analysis demonstrated that multiple MIP layers enhanced sensor specificity, with detection in the environmentally relevant range. The fluorescence bright field microscope investigations validated these results, showing an increase in the fluorescence intensity upon exposure of the MIP-functionalised glass slides to nafcillin solutions. These are promising results for developing a portable sensor device that can be deployed for antibiotics outside of a dedicated laboratory environment, especially if sensor design and fluorophore architecture are optimised.
:1 mixture with methacrylic acid (MAA), allowing to draw first conclusions on the MIP formation potential of such a rather large and rigid monomer. At first, MIP microparticles containing FluMa were prepared by free radical polymerisation. Optical batch rebinding experiments revealed that FluMa can act as a functional monomer for selective detection of nafcillin; however, the addition of MAA as co-monomer significantly improved performance. Subsequently, thin MIP films containing FluMa were deposited onto functionalised glass slides and the influence of porogen, drying time, and monomer composition was studied. These MIP-functionalised glass electrodes were mounted into a customised 3D-printed flow cell, where changes in the liquid were either evaluated with a thermal device or using fluorescence bright field microscopy. Thermal analysis demonstrated that multiple MIP layers enhanced sensor specificity, with detection in the environmentally relevant range. The fluorescence bright field microscope investigations validated these results, showing an increase in the fluorescence intensity upon exposure of the MIP-functionalised glass slides to nafcillin solutions. These are promising results for developing a portable sensor device that can be deployed for antibiotics outside of a dedicated laboratory environment, especially if sensor design and fluorophore architecture are optimised.
Introduction
Antibiotics have revolutionised modern medicine and agriculture but it is accepted that their use in veterinary or clinical practice exerts a selective pressure, which accelerates the emergence of antimicrobial resistance (AMR).1 According to a recent report2 by the World Health Organisation (WHO), AMR is considered a global threat comparable to terrorism and climate change. Deaths linked to AMR are currently estimated at 700000 annually worldwide but the number is projected to catapult to 10 million by 2050 if no intervention occurs. Tackling AMR requires a multi-faceted approach, which range from antibiotic stewardship to development of novel antibiotics, responsible manufacturing practices, and monitoring of antibiotic residual levels in the environment.
Penicillins and tetracyclines cover more than 50% of the total human antibiotics' consumption worldwide,3 with penicillin being the most used antibiotic within veterinary care.4 The maximum residual levels (MRLs) of antibiotics in food and feed products are governed by European Commission regulation 37/2010, where MRLs in milk for penicillins vary between 4–30 μg kg−1.5 In contrast, there is no limit set on the antibiotic traces in drinking and surface water. In India, one of the world's leading pharmaceutical producers, point-source-pollution leads to 58000 newborn deaths per year.6 Even in developed countries, there are serious implications associated with excessively high concentrations of pharmaceuticals in drinking water. The risk of these deadly drug resistance infections is significantly higher in developing countries due to the lack of optimal wastewater treatment and high costs associated with vigilant monitoring of pharmaceuticals in wastewater.
Current tests that are used for on-site detection of antibiotics are either colorimetric bacterial inhibition tests or lateral flow immunoassays.7–10 The drawback of the colorimetric bacterial inhibition test is its long measurement time11 whereas dipstick assays are not able to selectively determine antibiotics and solely provide semi-quantitative information whether concentrations are above or below accepted MRL levels. A recent review on current developments in antibiotic screening has considered the use of Molecularly Imprinted Polymers (MIPs) to replace natural receptors, including enzymes and antibodies, as recognition elements.12 The technique of molecular imprinting relies on the creation of specific cavities in a 3D-polymeric network, which are complementary to the size, shape, and chemical functionality of the target species.13,14 Compared to their natural counterparts, these synthetic ligands offer the advantages of low-cost, robustness, and ability to tailor towards the target of interest. There are few reports of MIPs produced for nafcillin, the beta-lactam antibiotic of interest for this study, available in literature.15–17 Nafcillin is selected because of its importance in the treatment of Staphylococcus infections due to its resistance to penicillinase compared to more common antibiotics.18Staphylococcus aureus is one of the most common pathogens in healthcare, which can cause invasive infections, sepsis, and has been associated with ∼20000 annual deaths in the US. The structural similarity to other beta-lactam antibiotics, such as amoxicillin, also means inferences can be made on how this technology will translate to the detection of these other drugs.
The main focus of studies on antibiotic-imprinted polymers has been improving the process of producing the polymers and their selectivity. MIPs for beta-lactam antibiotics have been produced in a range of different forms, including microparticles,19,20 hybrid inorganic–organic particles,21 sol–gel coated quantum dots,22 electropolymerized layers on electrodes,23 and nanoparticles.24,25 In each case, a high degree of binding and low limits of detection were obtained, even with a wide variety of detection methods being used, showing how effective MIPs can be for these antibiotics.26 There are two main drawbacks to the methods outlined in these studies: the focus is in improving the performance of the polymer in terms of sensitivity and selectively and leaves little room for adapting it in a more portable way, and the detection methods used are either laboratory-based or require laboratory-based equipment for validation. Addressing these issues is especially important in wastewater treatment as point-source detection is vital to reducing antibiotic concentrations before entering the water treatment facilities. It is critical to have quick, accurate and reliable sensors that can be brought into the field to be used by personnel without specialized training and provide analytical results in a time as short as possible. Attempts to address the first issue have been done by utilizing electrosensing27 but require further optimization of the imprinted polymers that are used in conjunction.28
While there are examples of commercial MIPs available for purification and separation, the difficulty to integrate MIPs into sensor platforms29 and lack of straightforward and low-cost analysis techniques prevent them from entering the market as analytical tools. Previous work by our group30 paved the path towards using MIPs in combination with thermal analysis for biomolecule detection. While this heat transfer-based technique was capable of detecting biomolecules in a fast and low-cost manner at physiologically relevant concentrations in complex samples, including saliva,31 tap water32 and serum,33 drawbacks of the technology include (i) difficult integration into a portable sensor set-up and (ii) the need for a temperature gradient in relation to the ambient temperature. Within this manuscript, we introduce a second detection method to the platform that addresses these issues and provides built-in validation to the results. This was achieved by introducing a fluorescent moiety into the MIPs, fluorescein methacrylate (FluMa), which shows a change in its fluorescence intensity when the target rebinds to the polymer. Previous studies have achieved this in MIPs using various fluorescent elements, including quantum dots34 and specifically-tailored fluorophores,35 and achieved high selectivity and sensitivity with a number of different targets.36 The approach presented here is focused on using a more readily available fluorophore as HTM relies on the presence of a large number of binding sites to achieve a sufficiently strong change in the thermal resistance. Therefore, it would be highly expensive and inefficient to have a more complex fluorophore present in each site. We compare the performance of fluorescent MIPs in the form of microparticles and thin films in terms of rebinding capacity,37 and fluorescent response in the presence of the target. Within this analysis, we explore and optimise the procedure for producing MIPs as thin films in a reproducible and consistent manner. To the best of our knowledge, this is the first study that utilizes a dual detection method for antibiotics while utilizing low-cost recognition elements. The ability to perform validation of results within the same sensor platform would greatly increase the accuracy and efficiency of testing on-site samples.
Experimental
Equipment and reagents
Experiments were carried out in an environment of 20 ± 1 °C unless stated otherwise. Phosphate buffered saline (PBS) tablets were purchased from Sigma Aldrich (Gillingham, UK) and used to maintain a constant pH level (pH = 7.4) and ionic strength of the solutions that were used during the experimentation. Dimethyl sulfoxide (DMSO) was sourced from TCI (Oxford, UK). Fluorescein free acid, trimethylamine, methacryloyl chloride, methacrylic acid (MAA), trimethylolpropane trimethacrylate (TRIM), nafcillin sodium salt, azobisisobutyronitrile (AIBN), magnesium sulphate, tetrahydrofuran (THF), chloroform, dichloromethane, methanol, and N,N-dimethylformamide (DMF) were sourced from Sigma Aldrich (Gillingham, UK). An Agilent 8453 UV-vis spectrophotometer (Santa Clara, US) was used for all UV-vis analyses. A Stuart mini orbital shaker SSM1 (Staffordshire, UK) was utilised throughout the research. A Polytec UV LC-5 light source (λmax = 365 nm, Karlsbad, Germany) was used to initiate the polymerisation reaction. A VWR INCU-Line Digital Mini Incubator (Lutterworth, UK) was used to maintain the ambient temperature of the thermal measurements.Synthesis of FluMa (1)
Fluorescein free acid (2) (1.00 g, 3.01 mmol) was dissolved in dry THF (250 mL) and then charged with trimethylamine (0.7064 g, 5.069 mmol). The reaction was cooled to 0 °C. A solution of methacryloyl chloride (3) (0.343 g, 3.535 mmol) in dry THF (10 mL) was then added slowly to the mother reaction solution. The reaction was kept at 0 °C for 90 min and then left for 12 hrs at RT. The reaction was monitored by TLC (schematic reaction shown in Fig. 1) and left overnight to reach completion, then quenched by the addition of water (40 mL). Evaporation under reduced pressure was performed followed by extraction with CHCl3 (150 mL) three times. The organic layer was dried with MgSO4, filtered, and the solvent removed as above. The resulting residue was purified by silica gel column chromatography using CH2Cl2 (100):CH3OH (5) v/v as eluant, affording FluMa (1) characterised by 1H NMR (Fig. A1, ESI†). The resulting product was an orange solid and a yield of 0.3 g (24.9%) was obtained. 1H NMR (CDCl3, 400 MHz) d (ppm): 7.98–8.00 (1H, d), 7.52–7.60 (2H, m), 7.25 (2H, s), 7.15–7.16 (1H, d), 6.77 (2H, s), 6.75 (1H, s), 6.59–6.62 (2H, m), 6.28 (1H, s), 5.75 (1H, s), 2.01 (3H, s).Preparation of bulk MIP microparticles and layers
MIP microparticles were synthesized for nafcillin with two different monomer compositions, either with pure FluMa (100 mg, 0.25 mmol) or a 1:1 mixture of FluMa (50 mg, 0.13 mmol) with MAA (50 mg, 0.58 mmol). The general functionalisation procedure includes dissolving the template nafcillin sodium salt (25 mg, 0.057 mmol) together with functional monomers in 4.0 mL of DMF. Subsequently, the crosslinker TRIM (500 mg, 1.48 mmol), and initiator AIBN (25 mg, 0.15 mmol) were added and the solution was degassed with nitrogen for several minutes. Polymerisation was initiated by increasing the temperature to 65 °C where the mixture was kept for overnight to ensure the reaction had completed. The same procedure was followed for the NIPs except nafcillin sodium salt was not added. Polymers containing only FluMa as the only monomer are denoted as A, whereas polymers with mixed monomers are denoted as B. The resulting polymer blocks were ground to obtain fine particles, from which the template was removed by Soxhlet extraction using a 1:1 solution of methanol and chloroform at 70 °C. The extraction procedure was repeated daily (average of three days) until no traces of the template or remaining monomers were detected in the filtrate by UV-vis analysis.
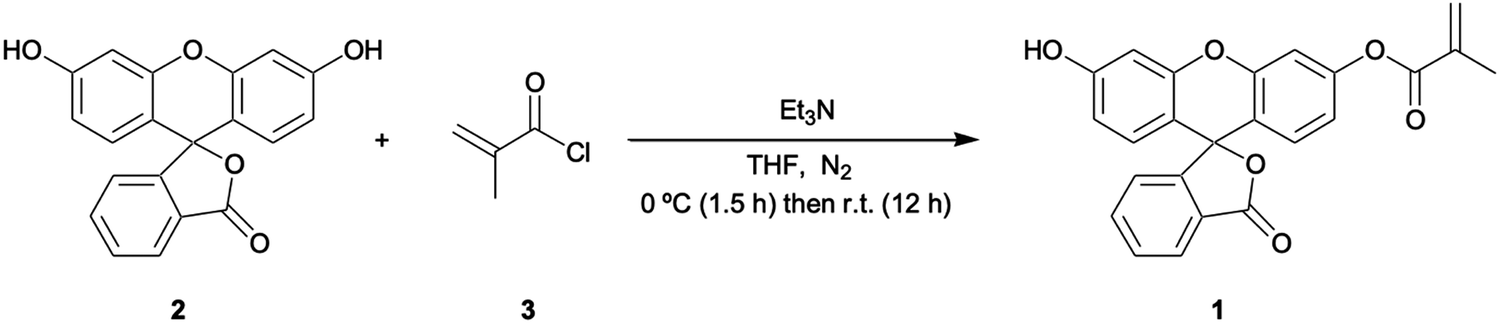 | ||
| Fig. 1 Synthesis of fluorescein methacrylate (FluMa) (1). | ||
Thin film polymer layers were generated with a similar composition as described above. Three different monomer mixtures were used to produce polymers: FluMa only (12.2 mg, 0.030 mmol), MAA only (10.3 μL, 0.122 mmol), or a mixture of the two at various molar ratios (ranging from 1:1 to 1:20). 2.0 mg (0.012 mmol) of AIBN and 13.8 mg (0.032 mmol) of nafcillin sodium salt were mixed with the monomer(s). 194 μL (0.609 mmol) of TRIM was then added to the mixture, and all the components were dissolved in 100 μL of solvent. 2:1 v:v DMF:DMSO was used as the solvent for single layer film formation, whereas THF was used for multilayer. Non-imprinted layers were formed with solutions that did not contain the template (nafcillin). The solution was vortexed until all the solid particulates had dissolved. The mixture was covered in aluminium foil to protect it from light.
The layers were formed on modified glass slides, which were cleaned and functionalized in a multistep process (Fig. B1, ESI†). The glass was first cut into 1 cm × 1 cm square chips. The chips were sonicated in water, methanol, and acetone sequentially for 5 min each, then allowed to completely dry. Next, they were sonicated in a solution of ammonium hydroxide, hydrogen peroxide, and water in a 1:1:5 volume ratio for 15 min at ∼60 °C. The glass was rinsed thoroughly with deionized water. They were then sonicated in a solution of hydrochloric acid, hydrogen peroxide, and water in a 1:1:6 volume ratio for 15 min at ∼60 °C. They were again rinsed then dried with N2. The chips were submerged into a 4% v/v solution of 3-(trimethoxysilyl)propyl methacrylate in toluene in a sealed container and left to functionalise overnight. The glass was removed from the solution, rinsed with methanol and dried.
To form the polymer layers, 5 μL of the pre-polymer mixture were deposited onto each of the functionalised chips. An unmodified glass slide was then placed on top of the chip to evenly distribute the mixture and minimize oxygen exposure. The UV light source was positioned 5 cm away from the chip, giving an intensity of 0.1 mW cm−2, to initiate the polymerisation. The samples were irradiated for 3 min, then the glass slide was removed, and the chips were washed in chloroform for 10 s and left to dry for 2 h. This cycle was repeated up to two more times to form thicker layers on the chips. Extraction of the template for imprinted films was conducted by placing the glass slides in 3 mL of chloroform on an orbital shaker (155 rpm) for 4 h, followed by 3 mL of methanol overnight.
Batch rebinding experiments analysed with UV-vis absorption spectroscopy
The rebinding of the target molecule nafcillin to the polymer particles was evaluated using UV-vis absorption spectroscopy and compared against that of other antibiotics including cephalexin and tetracycline (Fig. 2). Initially, calibration curves were constructed in PBS, where the absorbance was measured at λ = 330 nm for nafcillin, λ = 264 nm for cephalexin, and λ = 362 nm for tetracycline. | ||
| Fig. 2 Chemical structures of the three antibiotic drugs studied for rebinding. | ||
For these experiments, polymer particles (10 mg) were incubated with 5 mL PBS solutions (0–0.30 mM) of the molecule of interest and left on the orbital shaker (160 rpm) for 30 min at room temperature. After filtrating the suspensions with a 0.45 μm syringe filter, the absorbance of the remaining solution was measured and converted to the free concentration of the target (Cf) in solution using the calibration curve. Binding isotherms are then plotted where the free concentration is plotted vs the amount of antibiotic (substrate bound (Sb) in μmol g−1) that has bound per gram of polymer.
Additive manufacturing/3D-printing of flow cells
The flow cell STL files were designed using Fusion 360 (Autodesk, Portland, US) and converted into build files using PreForm (FormLabs, Somerville, US). The flow cells were printed on a FORM 2 stereolithography (SLA) 3D-Printer (FormLabs, Somerville, US) using FORM 2 Clear Resin (GPCLO4). The flow cells were post-processed by sonicating with isopropanol (Fisher Scientific, Loughborough, UK) for 15 min to remove any excess resin. After washing, the printing supports were detached, and the rough edges were removed with Wetordry paper (P2000). Post-curing was performed at 60 °C for 10 min using a FormLabs FORM Cure (405 nm, FormLabs, Somerville, US). The screw holes to fix the copper block to the flow cell were tapped in house using a M3 tapping kit.Thermal measurements of nafcillin
The functionalised electrodes were mounted onto the copper block of the flow cell and coupled to a heat-transfer device as described in previous work.30 The flow cell used was redesigned to have the top of the chamber replaced with optically transparent glass that was sealed with silicone glue (see Fig. 3). The temperature of the copper block, defined as T1, was steered by adapting the voltage of the power resistor (22 Ω) with in-house designed Lab View software and a proportional-integral-derivative (PID) controller. The PID parameters used for all measurements were as follows: P = 1, I = 18, D = 0.3. In all sensing experiments presented in this manuscript the temperature of the heat sink was set to 37.00 ± 0.02 °C to mimic in vivo conditions. The thermocouple was inserted into the flow cells 1.7 mm above the electrode surface and the temperature measured in the liquid by these thermocouples is defined as T2. The entire set-up was contained in an incubator where the ambient temperature was monitored and maintained at 25 °C.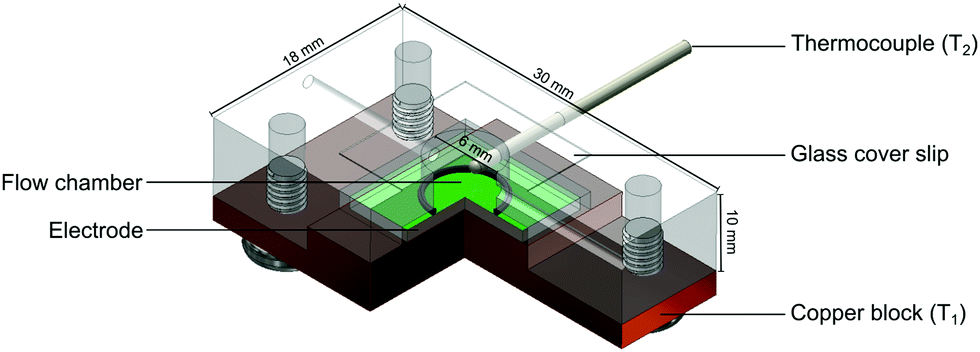 | ||
| Fig. 3 Schematic of the 3D printed flow cell used for fluorescence and thermal measurements. | ||
All measurements were conducted using PBS (pH = 7.4) that had been degassed by vigorous stirring at 65 °C for a minimum of 1 h to remove the influence of air bubbles on the signal. The thermal resistance (Rth) was calculated for each measurement by dividing the temperature gradient (T1 − T2) over the power required to keep the heat sink at 37 °C. Prior to calculating the average Rth and standard deviation, the signal was stabilised for 10 min after each injection. From this, dose–response curves were constructed and used to calculate the limit of detection (LOD).
Fluorescence measurements of nafcillin
Fluorescence measurements were obtained using two different methods: a fluorescence spectrometer, or a wide field fluorescence inverted microscope. For the fluorometer, the electrode was mounted onto the bottom of a polystyrene cuvette at ∼30° to the detector. The solution was then added to the cuvette and measurements were taken to both determine excitation/emission wavelengths from the respective spectra. Intensity changes were then measured over time with the excitation wavelength set to 460 nm and the emission recorded at 515 nm.For measurements using the microscope (Nikon Eclipse Ti-E, Nikon, Surbiton, UK), the electrode was mounted into the HTM flow cell which was then positioned onto the microscope holder using a custom 3D printed mount (Fig. C1, ESI†). Each electrode had a small area (∼1 × 1 mm) of polymer removed for focusing and background subtraction. Using a 4× objective lens (Plan Apochromat, Lambda, 0.2NA, Nikon, Surbiton, UK), the microscope was focused on the polymer layer within the flow cell after PBS was injected. The sample was illuminated by a white light lamp (Sola SE, Lumencor, Beaverton, US) and filtered using a filter cube (Chroma 49002 ET, λex = 450–490 nm, λem = 500–550 nm, λmirror = 495 nm). Images were taken using a sCMOS camera (Andor Zyla 4.2, Andor, Belfast, UK) every 10 s, with an exposure time of 0.5 s, unless stated otherwise. The resulting images were then processed using our bespoke Python script for both visualization and analysis. For the analysis, the summed intensity in three regions of the polymer and one region with no polymer, with each region being 100000 μm2 in area, was calculated. The values for the polymer regions were then averaged, and the background region was subtracted from this average (Fig. D1, ESI†).
Results and discussion
Synthesis of FluMa (1)
The synthesis of FluMa reported in literature38 was modified as follows: fluorescein free acid and the other reactants were dissolved in THF instead of chloroform because of solubility issues; quenching with water was introduced to prevent excess methacryloyl chloride from reacting further; purification via column chromatography was carried out with CH2Cl2 (100):CH3OH (5) v:v as it provided better separation based on polarity.
Batch rebinding results analysed by UV-vis absorption spectroscopy
Batch rebinding measurements were done with polymer microparticles to determine the selectivity and binding capacity of an imprinted polymer with FluMa as the functional monomer. Initially, the two polymer compositions listed in the experimental section were used for the rebinding of nafcillin (Fig. 4). Both imprinted polymers showed a significantly higher degree of binding compared to their non-imprinted analogues, suggesting that FluMa-containing MIPs are able to selectively bind the target. However, NIP B showed significantly less binding compared to NIP A. It was also seen that MIP A was able to bind more of the target at lower concentrations but became saturated more quickly compared to MIP B. Furthermore, MIP B was able to bind a greater total amount of target per gram. These results suggest that FluMa is able to act as a functional monomer for imprinting, but the overall polymer performance is enhanced by the addition of a co-monomer. This is further illustrated when comparing the imprint factor (IF) for each composition. The IF was calculated by taking the ratio of Sb (the number of moles of template bound per gram of the polymer) for MIP:NIP of each composition at a specific final concentration. The Sb values were determined using an allometric fit for all data set. The IF of MIP B was three times that of MIP A at 0.05 mM (IFA = 2.0, IFB = 6.0) and this difference increased at higher concentrations. Imprinted polymers for beta-lactam antibiotics have typically used smaller functional monomers (such as MAA) and shown high binding capacities (>10 μg mg−1) and excellent selectivity.39,40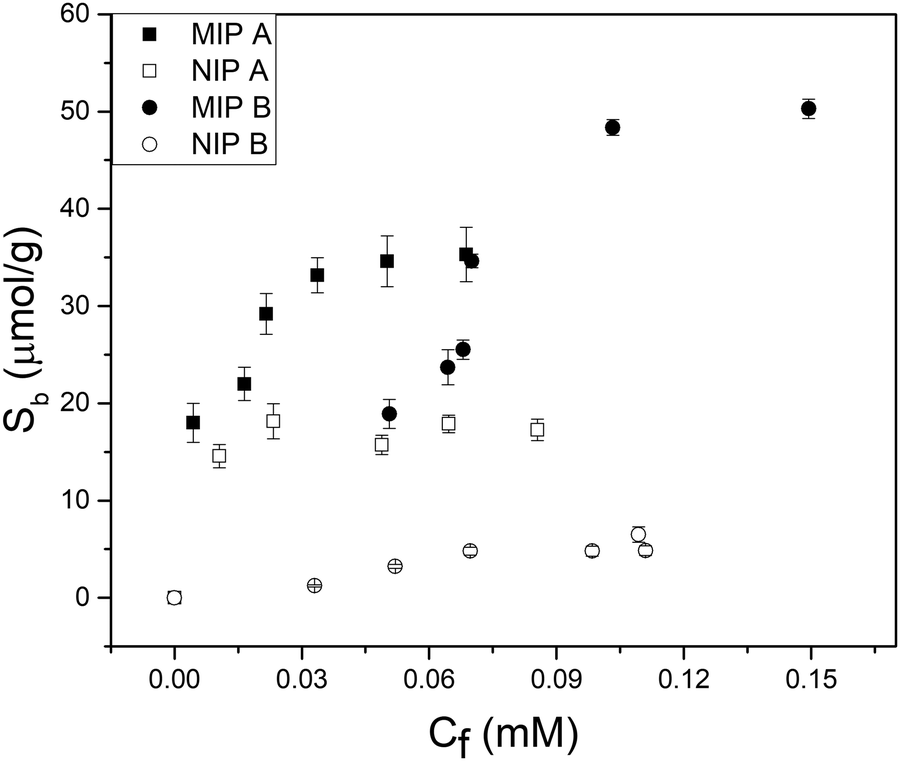 | ||
| Fig. 4 Batch rebinding measurements using UV-vis absorption spectroscopy for two polymer compositions using solutions of nafcillin sodium salt in PBS (pH = 7.4). | ||
In terms of selectivity, MIP A was used to determine the specific effect of FluMa as the functional monomer. Optical batch rebinding experiments were performed using nafcillin along with two other antibiotics: cephalexin, a similarly structured beta-lactam, and tetracycline. A wide concentration range was studied since selectivity is key for future use of the sensors in real samples, where the target will be present alongside a plethora of (abundant) interferents. The results of these measurements are shown in Fig. E1 (ESI†). For both drugs, the degree of binding was less than nafcillin at all concentrations and showed no preferential binding when comparing the MIP and NIP. This can be seen using a selectivity factor, which is calculated by comparing the Sb, as determined by allometric fit, of the MIP at a given concentration for nafcillin compared to a competitor molecule (Sb nafcillin/Sb competitor at Cf = X). A value greater than 1 shows a preference for the imprinted drug over competing species. At 0.05 mM, the selectivity factor for cephalexin and tetracycline were determined with an allometric fit and were 12.8 and 8.4, respectively. Overall, this suggests that FluMa-based MIPs are capable of forming selective binding sites for this class of antibiotics. Interestingly, however, tetracycline showed both a higher amount of binding and a slight preference for the imprinted polymer at higher concentrations compared to cephalexin. This can be seen in the apparent specificity factor (aSF), calculated as the ratio of Sb for each drug using a nafcillin-imprinted and non-imprinted polymer at a specific concentration. At 0.1 mM, the aSF for tetracycline is 1.8, whereas aSF for cephalexin is 0.8. Although, at first, this seems unexpected based on the chemical classes of the three drugs, with tetracycline being significantly different from the other two, a closer look to the chemical structure provides a possible explanation. Like nafcillin, tetracycline bears an aromatic unit that is more planar, and the entire molecule is more elongated to better interact with the dominant moiety of FluMa, the xanthene fragment, via π–π interactions than cephalexin can. Because we also refrained from using other functional monomers to target the carboxylate group of the beta-lactams, other highly conjugated systems (such as tetracycline) might indeed lead to increased non-specific binding. However, this provides valuable insights into further tuning features of the system besides factors such as particle size or MIP formulation.
Optimization of transparent polymer films on glass substrate
In order to obtain reliable measurements for both thermal and fluorescence analysis, the imprinted polymer films needed to be reproducible and homogeneous in both transparency and smoothness. All films produced were between 2 and 3 μm in thickness, as determined by white light profilometry (Fig. F1, ESI†). The height was determined by making a scratch in the layer and comparing the bare electrode to one covered with polymer layer. A number of factors were discovered to have a significant impact on the final state of the films and required optimization. These included the volume of solution deposited on the glass, the irradiation time, the porogen's volatility, and the washing and extracting parameters. In each case, the aim was to generate a thin, transparent film with minimum to no cracking, while retaining a consistent fluorescence response across all samples (both MIPs and NIPs) when measured on the fluorescence microscope.A range of volumes were initially tested when generating these polymer films. From 1 to 50 μL, polymerization occurred on the chip, however 5 and 10 μL provided the best results as they provided sufficient coverage without excess solution being pushed over the edges of the chip. This was further narrowed down to 5 μL to not only minimize the amount of solution per chip but also to allow for the easiest release from the cover glass. In terms of film quality, however, there was minimal difference within this range. These volumes were fairly consistent across the range of porogens that were tested, with greater volumes (typically double) used when the components’ solubility was decreased. For the irradiation time, it was found that 3 min was optimal as shorter times would produce a softer film that would deform upon cover glass removal or washing, and longer times would cause cracking or opaque regions to form as the degree of crosslinking increased. There were small variations in this time (±30 s) when changes to the composition (specifically with AIBN and FluMa) were made as these will directly influence the amount of light absorbed and therefore, the rate of polymerization.
The porogens used for the generation of films include methanol, ethanol, chloroform, DMF, DMSO, 2:1 v:v DMF:DMSO, and tetrahydrofuran (THF). Volatility was the most critical property which dictated the viability of the porogen; higher volatility would cause the solvent to rapidly evaporate both during and after film formation. This led to increased strain on the film as it was hardening via crosslinking and would often result in crack formation or increased opacity (Fig. 5). Conversely, low volatility porogens required longer drying times and even control of additional conditions (increased temperature, vacuum drying) to sufficiently remove it from the films. This factor becomes important in the washing and extraction steps as significant solvent exchange caused defects to form in the film. These steps require the use of more volatile solvents and the rapid drying of the polymer network can introduce strain if the film is not fully cured. There were additional considerations for porogen choice, including the effect on binding as examined in previous studies.41–43 Based on scanning electron microscopy images that provide insight into the surface structure, 2:1 v:v DMF:DMSO was chosen for single layers as it gave the most consistent films and THF was used for triple layers as it gave comparable consistency with significantly shorter drying times.
 | ||
| Fig. 5 Scanning electron micrographs of the polymer films using (a) chloroform and (b) methanol as the porogen. | ||
The specific parameters of the washing and extraction steps were examined and showed a significant effect on both the film quality and degree of template extraction. As was the case for the porogen, sufficient drying was required to avoid cracks forming in the polymer film. Chloroform was used to wash the films after polymerization and methanol was used to extract the template. Two washing steps took place: a short initial wash to remove excess monomer solution to maintain the smooth surface of the film, and a longer wash to extract any remaining unreacted components out of the film. Chloroform was chosen as the washing solvent as it was conveniently able to solubilize every component other than nafcillin, allowing the imprints to fully form. For extraction, methanol was able to readily extract nafcillin from the polymer films without causing significant defects. Methanol solutions of either water (1:1) or acetic acid (1:1) were tested but showed no significant improvement in extraction, imprint formation or film quality.
Thermal measurements
The effect of introducing the fluorescent monomer on the thermal measurements was first investigated to confirm the results obtained in the batch rebinding. Thin film chips were prepared using imprinted and non-imprinted versions of three polymers: one with FluMa as the functional monomer, one with MAA as the functional monomer, and one with a combination of the two. The thermal resistance (Rth) was measured after injections of buffered nafcillin solutions with increasing concentration, and the percentage change between the injections was calculated. The results of these measurements are shown in Fig. 6. Spikes in the Rth for the raw data correspond to injections of the solutions as they are at room temperature.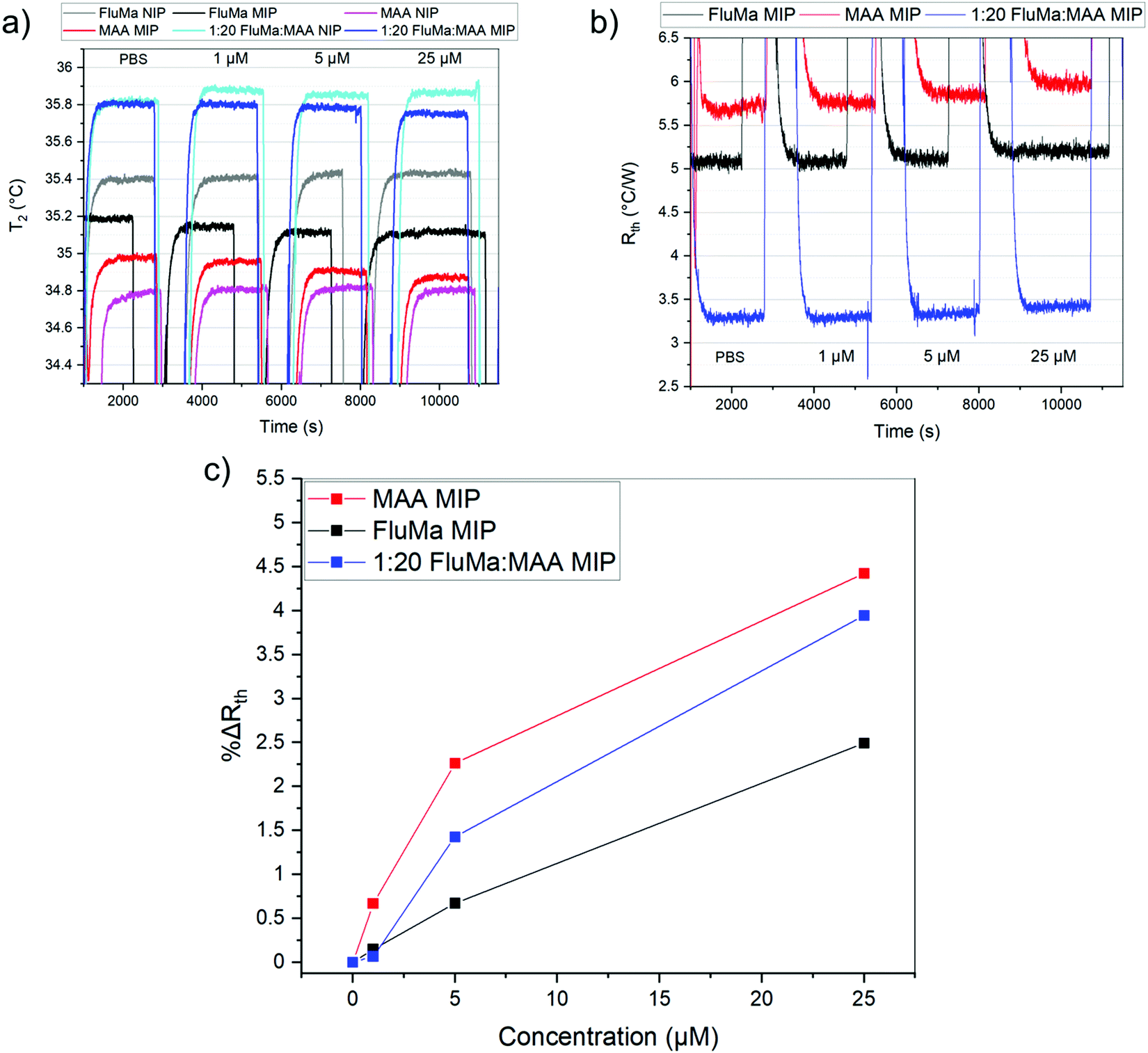 | ||
| Fig. 6 Binding capability of polymer thin films with different monomers analysed via thermal measurements. (a) Temperature change within the flow cell over time for 6 polymer films. (b) Thermal resistance measurement for MIP films using different functional monomers. (c) Percent change in thermal resistance after each injection, calculated using the 10 minutes of stable signal before the following injection. Error bars were calculated over 600 points but are not visible because they are smaller than symbol size. | ||
Interestingly, the polymer layer containing FluMa, either as the only monomer or mixed, did not show the same increased affinity as the microparticles at low concentrations when compared to the polymer with only MAA. However, this may be a result of the difference in concentration range (μM vs. mM), mass (>1 mg vs. 10 mg), or simply the nature of films vs. microparticles and would be interesting to investigate in the future. The main conclusion is that the MAA-based polymer continued to show a higher binding capacity and specificity compared to the FluMa-based polymer. This is further illustrated with the 1:20 FluMa:MAA film, which shows a slight decrease in binding compared to the pure MAA film. This confirms that using a combination of the two monomers is preferred to optimize the performance of the imprinted polymer while retaining the fluorescence functionality of FluMa. Various monomer ratios (ranging from 1:1 to 1:20 of FluMa:MAA) were tested in terms of film formation and fluorescence intensity and it was determined that 1:4 gave the most consistent results. The optimized polymer was then used to measure across a larger range of concentrations, with the thermal results shown in Fig. 7. From 1 μM to 50 μM, an increase in the thermal resistance was seen for the imprinted polymer layer, whereas the non-imprinted film showed no definitive increase across the same range. At 50 μM, the thermal resistance (Rth) had increased by an average of 0.12 ± 0.03 °C W−1 for imprinted films, significantly higher than the noise of the signal and the increase seen in the non-imprinted films.
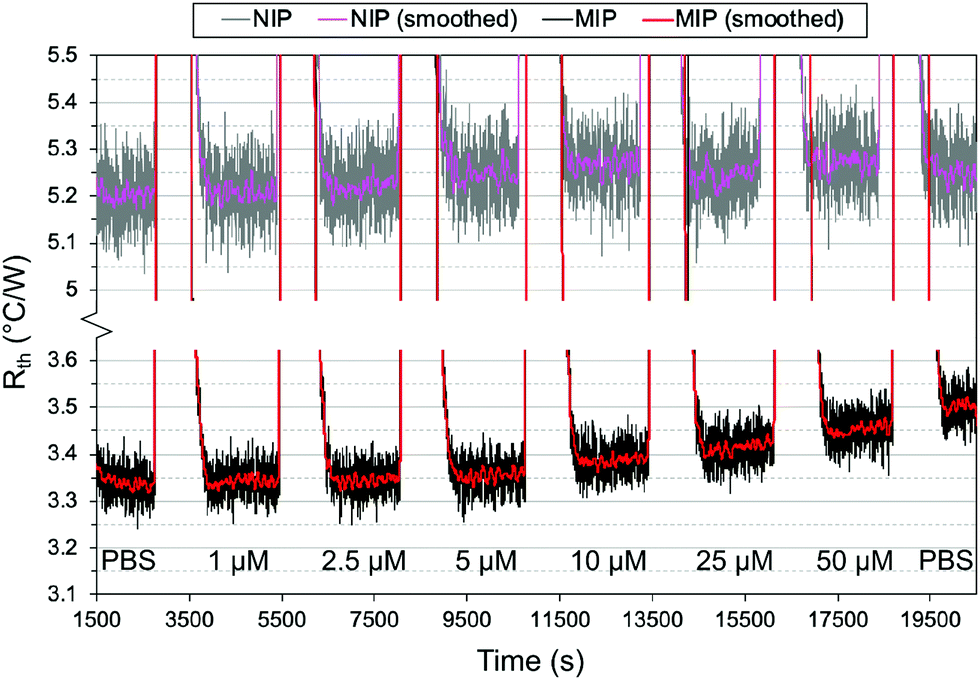 | ||
| Fig. 7 Thermal measurement of nafcillin-imprinted (black) and non-imprinted (grey) polymer layers with injections of nafcillin solutions with increasing concentration. The red and magenta lines correspond to a gentle median filter (60 s) applied to the raw data for the MIP and NIP, respectively. | ||
It was noted, however, that the observed increase was smaller than expected based on previous measurements with MIPs and HTM, with an Rth increase of 0.5 °C W−1 obtained in the nM range.37 One possible explanation was the total mass of polymer on the glass substrates. For a single layer of polymer, the mass of polymer was ∼0.3 mg (based on gravimetric measurements) which is significantly lower than the microparticles. A lower amount of polymer means there is a lower number of binding sites present and thus, less of the drug will rebind. Therefore, two changes were introduced to account for this: additional layers were deposited on top of the chips to increase the mass of polymer, and a lower concentration range of nafcillin was used.
As previously discussed, THF was used as the porogen and each layer was sufficiently dried before the addition of a new one to avoid cracking. The triple layered films showed a higher amount of nafcillin extracted compared to the single layers, suggesting the polymer network was connected and that the target in the first layer could still be extracted; the combined polymer layers had a total mass of 1.0 mg (opposed to 0.3 mg for the single layer). The thermal measurements for these films used 50 nM to 5000 nM (5 μM) concentrations of nafcillin and are shown in Fig. 8 (the raw measurements can be found in Fig. G1, ESI†). The same trends as the single layers were observed: preferential binding to the imprinted polymer, higher binding without FluMa but increased non-specific binding when FluMa is present. Interestingly, a comparable increase in Rth for solutions containing 50 μM nafcillin with single layers was seen with only 5 μM (0.22 ± 0.07 °C W−1 with MAA only, 0.21 ± 0.08 °C W−1 with 1:4 FluMa:MAA), with significant increases even seen at 50 and 500 nM. This means that the limit of detection was improved by one order of magnitude by increasing the mass of polymer on the chips, meaning layer optimisation is key to improve the specificity. A higher mass also means more of the fluorescein is present, which is important for the subsequent fluorescent analysis.
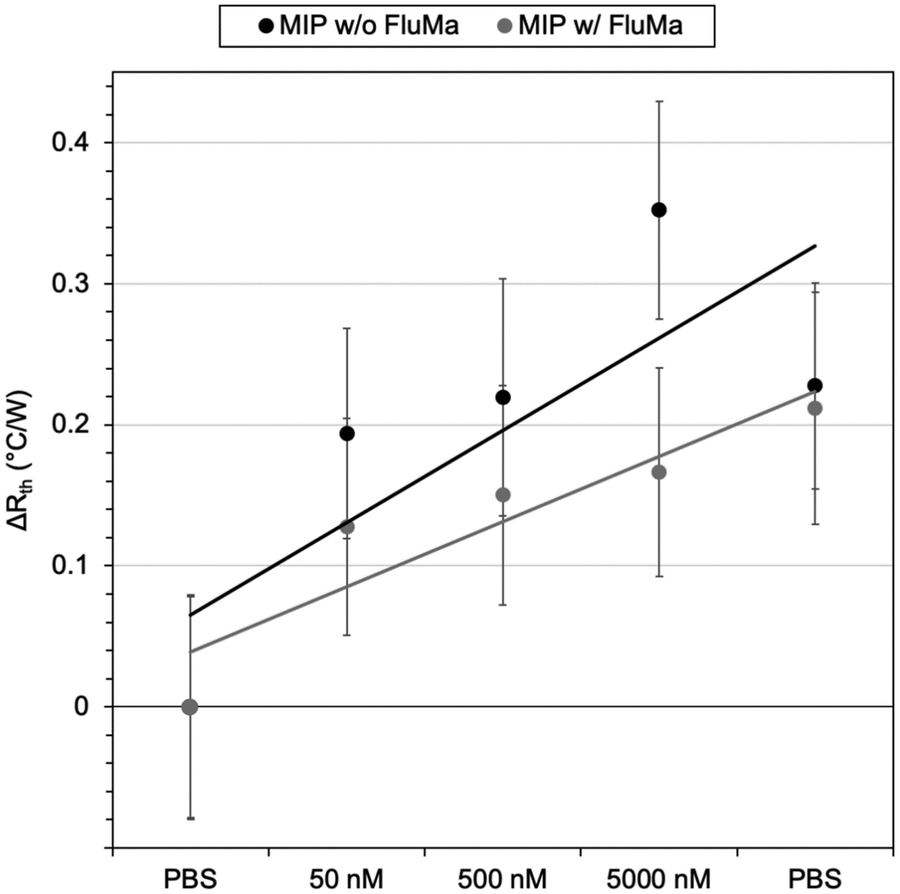 | ||
| Fig. 8 Thermal measurement of nafcillin solutions with imprinted polymer films with three layers. Black data points represent films containing no FluMa in the composition, whereas grey represent films with 1:4 FluMa:MAA. Each point is the change in Rth after the initial PBS injection, with values calculated from the average over 10 minutes of stabilization after injection of the corresponding solution and error bars representing the noise. Linear trend lines were added for each dataset. | ||
Fluorescence analysis
The excitation and emission wavelengths (460 nm and 515 nm, respectively) used for fluorescence measurements were chosen on the basis of the absorption and fluorescence spectra, i.e., 460 nm is ideal for exciting the monoanionic form of fluorescein44 and sufficiently blue-shifted from the maximum of the fluorescence band at ca. 515 nm.45 It should be noted that because of the linker introduced through one of the terminal xanthene oxygens, the highest fluorescent dianionic form of fluorescein cannot be generated here, simplifying the spectroscopic response. Furthermore, the fluorescence wavelengths for the FluMa-containing polymer films were measured and found to be 463 nm for excitation and 518 nm for emission (Fig. H1, ESI†). The location of the acrylate group does not significantly interfere with the resonance structure of the molecule and thus, does not cause a large shift in the fluorescence wavelengths.Using these wavelengths, the fluorescence response of the polymer films was monitored over time in the presence of nafcillin. This was initially done with a fluorometer to determine how the fluorescence would be affected by nafcillin. Changes in fluorescence can be attributed to interactions between the target and the FluMa in the film as both the drug in solution and films that did not contain FluMa did not show a response at the measured wavelengths. When immersed in a PBS solution without nafcillin, the polymer films showed no change in fluorescence over 30 min. When the imprinted film was then introduced into a 50 μM solution of nafcillin, a 7% increase in fluorescence was seen after 10 minutes (Fig. 9 – experiment performed in triplicate). This result may be unexpected as previous studies on fluorescent imprinted polymers has required specific tailoring of the fluorescent moiety to achieve signal enhancement.46 To understand the underlying reasons for this response, we need to look at the local chemistry occurring in the imprints. Despite the conjugated nature of both the antibiotic drug and the fluorescent moiety, it is unlikely that the change in fluorescence intensity is due to π–π stacking-mediated charge-transfer interactions. This is based on studies that were done using UV-vis spectroscopy to examine whether any interactions occurred when the target, nafcillin, was introduced into a solution containing FluMa. There was no discernible shift in the bands for FluMa, which would be expected in case of such π–π interactions. Therefore, the change in fluorescence is more likely to be attributed to specific microenvironmental changes within the cavities where FluMa resides. Normally, fluorescein has a rich pH chemistry that can complicate assays when not adequately buffered.44,47 However, here, the equilibrium between the monoanion (precisely FluH−) and the dianion (precisely Flu2−) of fluorescein which is characterized by pKa3 = 6.4 does not play a role because of the point of introduction of the methacrylate moiety. Since the next pKa for FluH2/FluH− is found at pKa2 = 4.3, it can be safely assumed that in the present case with solutions buffered at pH = 7.4 the monoanionic form is prevalent. When considering the three antibiotics, it is apparent that nafcillin exist exclusively in its (mono)anionic form at neutral pH (pKa = 2.7, conversion of NAFH into NAF−) whereas cephalexin and tetracycline exist as mixtures of neutral and (mono)anionic forms (decisive pKa2 = 7.3 for CEPH/CEP− in cephalexin and 7.7 for TETH2/TETH− in tetracycline).48 The latter two could thus in principle engage in (re-)protonation interactions with FluMa. Although buffering should prevent such interactions, it should be noted that buffering can be different in cavities in a less hydrophilic polymer network like a MIP than for bulk solution, which in turn can have an impact on the microscopic protonation equilibria.49 However, based on the sequence of pKa data of the antibiotics, such protonation-induced fluorescence modulations cancel out. Further studies of this specific interaction are required to fully understand the effect of local pH changes, including solvent effects and target size.
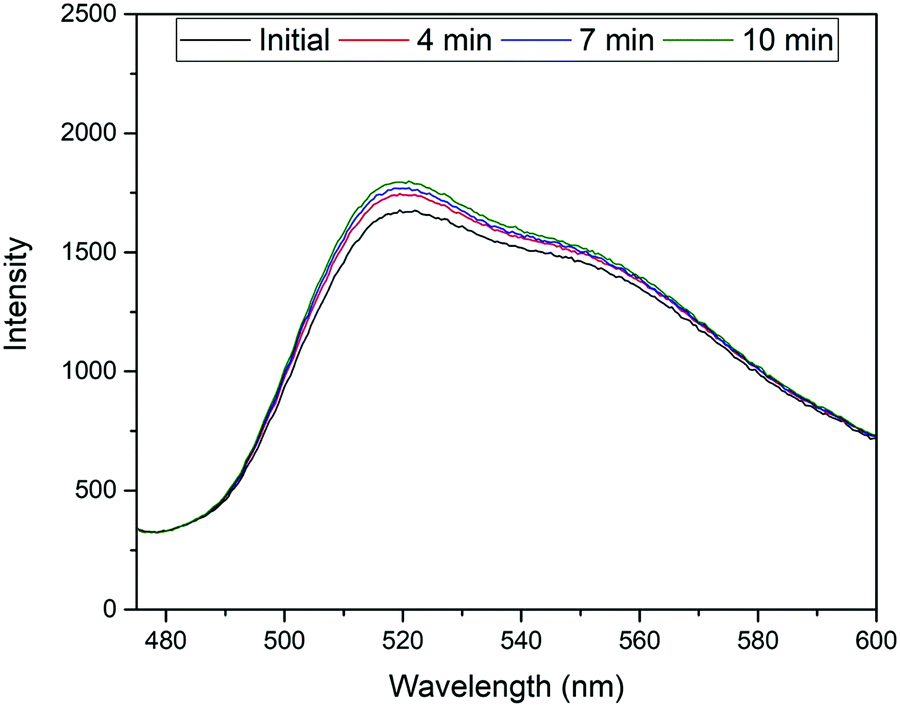 | ||
| Fig. 9 Temporal measurement of fluorescence response from MIP film in the presence of a 50 μM solution of nafcillin sodium salt. | ||
With this knowledge, the analysis process was altered to allow for fluorescence and thermal measurements to be obtained simultaneously. Two changes were implemented to achieve this: the polymer films were measured while in the glass-top flow cell used for the thermal measurements rather than a cuvette, and a fluorescence upright microscope was used in place of the fluorometer. This set-up is notable as it allows thermal analysis to be conducted at the same time, opening up the possibility for dual detection. The response of the film in the presence of solutions with different concentrations of nafcillin in this set-up are shown in Fig. 10. This process was repeated three times, with each time a similar pattern was observed. The fluorescent reading is an arbitrary unit (AU).
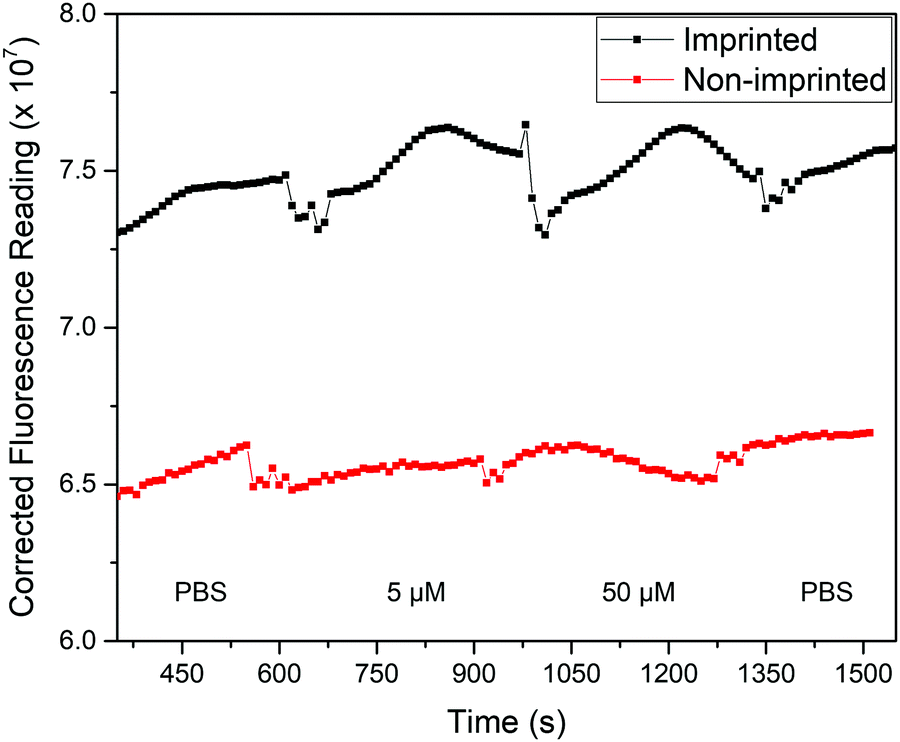 | ||
| Fig. 10 Background-corrected fluorescence response (AU) of polymer films with injections of various solutions of nafcillin. Spikes in the response correspond to the solution injections. | ||
The average fluorescence signal from the film was background corrected to minimize fluctuations caused by changes in the liquid within the flow cell. Each solution was left in the flow cell for at least 5 min as the previous measurements showed the enhancement began to stabilize after this time. Measurements were taken with stabilization times up to 15 min and showed comparable changes in fluorescence, although had less variability between experiments. Therefore, further optimization of the chip design would be required to reduce measurement time. When comparing the imprinted and non-imprinted films, a small increase in the fluorescence is seen for the imprinted after the introduction of nafcillin that is not present for the non-imprinted. Fig. 10 shows the distinctly higher fluorescence response at 50 μM for the imprinted films. There was no distinct difference in the polymer's response at concentrations lower than 50 μM, although increasing the amount of polymer may improve this as measurements with multilayer films showed a similar trend with solutions containing 0.5 μM nafcillin (1.2% increase for MIP, 2.3% decrease for NIP). Nonetheless, these results showed a specific fluorescence response in the presence of the drug target that can be used in combination with the measured thermal response. The next step for this work would be to carry out simultaneous thermal and fluorescent analysis to develop a truly dual sensing platform.
Conclusions
In summary, fluorescence and thermal analysis has been successfully integrated into a molecularly imprinted polymer-based system for the detection of antibiotics. A fluorescent monomer was synthesized and incorporated into the polymer network. The imprinted polymer was generated as both microparticles and thin films, with the monomer ratio affecting the binding of nafcillin for each form differently. A detailed breakdown of the factors affecting film quality was examined, with porogen and drying time found to be the most crucial elements to the process. Increases in the thermal resistance were seen for imprinted polymer films with drug concentrations in the micromolar range (3.6%@50 μM for single layer, 3.5%@5 μM for triple layer). These same films showed a specific fluorescence increase in the presence of the drug, showing the capability of dual detection methods with this sensor system. The proposed explanation for this increase is based on local pH changes in the imprinted sites. Future studies can look to expand the analysis library for this technology by exploring different target drugs and fluorophores. Furthermore, fundamental studies examining the underlying phenomena that generate the fluorescence change would help guide future changes and optimizations. Overall, these results show the potential to design a portable sensor device with simple and fast readout based on functionalized MIPs and thermal analysis. The device can be used for rapid screening of samples, allowing to pinpoint areas with high antibiotic pressure in a wide range of milieus such as the waterways, in the food industry and agriculture settings, and finally within hospital environments. This is an elegant approach that tackles accelerated development of AMR from its source, which will provide fundamental insight into the influence of selective pressure on microorganisms. It also provides the option to introduce self-validation into a sensor platform and could be expanded into other detection methods in the future.Conflicts of interest
We have no conflicts of interest to report for this manuscript.Acknowledgements
MP and AH would like to acknowledge the EPSRC for funding under grant number EP/R029296/2. JG acknowledges funding from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) through grants 424027/2018-6 and 307501/2019-1. Funding from Innovate UK (KTP Reference: 11473) is acknowledged for salary of FM. Data, including raw data, is available from: https://data.ncl.ac.uk.References
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- Interagency Coordination Group on Antimicrobial Resistance, 2019. No time to wait: securing the future from drug-resistant infections report to the secretary-general of the United Nations, World Health Organisation. https://www.who.int/antimicrobial-resistance/interagency-coordination-group/IACG_final_report_EN.pdf?ua=1.
- World Health Organization, 2018. WHO Report on Surveillance of Antibiotic Consumption. https://www.who.int/docs/default-source/documents/no-time-to-wait-securing-the-future-from-drug-resistant-infections-en.pdf?sfvrsn=5b424d7_6.
- N. De Briyne, J. Atkinson, L. Pokludová and S. P. Borriello, Veterinary Record, Br. Vet. Assoc., 2014, 173, 325–330 Search PubMed.
- European Commission, Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off. J. Eur. Union L15, 2010, pp. 1–72.
- J. Fick, H. Söderström, R. H. Lindberg, C. Phan, M. Tysklind and D. G. J. Larsson, Environ. Toxicol. Chem., 2009, 28, 2522–2527 CrossRef CAS PubMed.
- M. A. Abedalwafa, Y. Li, C. Ni and L. Wang, Anal. Methods, 2019, 11, 2836–2854 RSC.
- C. Dincer, R. Bruch, E. Costa-Rama, M. T. Fernández-Abedul, A. Merkoçi, A. Manz, G. A. Urban and F. Güder, Adv. Mater., 2009, 31, 1806739 Search PubMed.
- J. R. Dolan, D. Forster, M. Dunthorn, D. Bass, K. Bittner, C. Boutte, R. Christen, J. Claverie, J. Decelle, B. Edvardsen, E. Egge, W. Eikrem, W. H. C. F. Kooistra, R. Logares, R. Massana, M. Montresor, F. Not, H. Ogata, J. Pawlowski, M. C. Pernice, S. Romac, K. Shalchian-tabrizi, D. Sarno, N. Simon, T. A. Richards, R. Siano, D. Vaulot, P. Wincker, A. Zingone, C. Vargas, C. T. Stoeck, M. Csic and P. Mar, FEMS Microbiol. Ecol., 2016, 94, 1–11 Search PubMed.
- R. Parthasarathy, C. E. Monette, S. Bracero and M. Saha, FEMS Microbiol. Ecol., 2018, 94, fiy105 CrossRef CAS PubMed.
- M. G. Pikkemaat, Anal. Bioanal. Chem., 2009, 395, 893–905 CrossRef CAS PubMed.
- V. Gaudin, Biosens. Bioelectron., 2017, 90, 363–377 CrossRef CAS PubMed.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed.
- E. Turiel and A. M. Esteban, Molecularly imprinted polymers, Solid-Phase Extraction, John Wiley & Sons, Ltd, Chichester, UK, 2019, pp. 215–233 Search PubMed.
- A. Fernández-González, R. Badía Laíño, M. E. Diaz-García, L. Guardia and A. Viale, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 804, 247–254 CrossRef PubMed.
- C. Giovannoli, L. Anfossi, F. Biagioli, C. Passini and C. Baggiani, Microchim. Acta, 2013, 180, 1371–1377 CrossRef CAS.
- L. Guardia, R. Badía and M. E. Díaz-García, Biosens. Bioelectron., 2006, 21, 1822–1829 CrossRef CAS PubMed.
- A. J. Wright, The penicillins, Mayo Clinic Proceedings, Elsevier Ltd, 1999, pp. 290–307 Search PubMed.
- J. L. Urraca, M. C. Moreno-Bondi, G. Orellana, B. Sellergren and A. J. Hall, Anal. Chem., 2007, 79, 4915–4923 CrossRef CAS PubMed.
- J. Yin, Z. Meng, M. Du, C. Liu, M. Song and H. Wang, J. Chromatogr. A, 2010, 1217, 5420–5426 CrossRef CAS PubMed.
- A. G. Ayankojo, J. Reut, A. Öpik, A. Furchner and V. Syritski, Biosens. Bioelectron., 2018, 118, 102–107 CrossRef CAS PubMed.
- K. Chullasat, P. Nurerk, P. Kanatharana, F. Davis and O. Bunkoed, Sens. Actuators, B, 2018, 254, 255–263 CrossRef CAS.
- G. Moro, F. Bottari, N. Sleegers, A. Florea, T. Cowen, L. M. Moretto, S. Piletsky and K. De Wael, Sens. Actuators, B, 2019, 297, 126786 CrossRef CAS.
- N. Caro, T. Bruna, A. Guerreiro, P. Alvarez-Tejos, V. Garretón, S. Piletsky, J. González-Casanova, D. Rojas-Gómez and N. Ehrenfeld, Nanomaterials, 2020, 10, 306 CrossRef CAS PubMed.
- S. Korposh, I. Chianella, A. Guerreiro, S. Caygill, S. Piletsky, S. W. James and R. P. Tatam, Analyst, 2014, 139, 2229–2236 RSC.
- J. Cederfur, Y. Pei, M. Zihui and M. Kempe, J. Comb. Chem., 2003, 5, 67–72 CrossRef CAS PubMed.
- S. Pilehvar, K. Gielkens, S. A. Trashin, F. Dardenne, R. Blust and K. De Wael, Crit. Rev. Food Sci. Nutr., 2016, 56, 2416–2429 CrossRef CAS PubMed.
- F. Bottari, R. Blust and K. De Wael, Curr. Opin. Electrochem., 2018, 10, 136–142 CrossRef CAS.
- O. S. Ahmad, T. S. Bedwell, C. Esen, A. Garcia-Cruz and S. A. Piletsky, Trends Biotechnol., 2019, 37, 294–309 CrossRef CAS PubMed.
- M. Peeters, P. Csipai, G. Geerets, A. Weustenraed, B. Van Grinsven, R. Thoelen, J. Gruber, W. De Ceuninck, T. J. Cleij, F. J. Troost and P. Wagner, Anal. Bioanal. Chem., 2013, 405, 6453–6460 CrossRef CAS PubMed.
- F. Canfarotta, J. Czulak, K. Betlem, A. Sachdeva, K. Eersels, B. Van Grinsven, T. J. Cleij and M. Peeters, Nanoscale, 2018, 10, 2081–2089 RSC.
- K. Betlem, I. Mahmood, R. D. Seixas, I. Sadiki, R. L. D. Raimbault, C. W. Foster, R. Crapnell, S. Tedesco, C. E. Banks, J. Gruber and M. Peeters, Chem. Eng. J., 2019, 359, 505–517 CrossRef CAS.
- R. D. Crapnell, F. Canfarotta, J. Czulak, R. Johnson, K. Betlem, F. Mecozzi, M. P. Down, K. Eersels, B. van Grinsven, T. J. Cleij, R. Law, C. E. Banks and M. Peeters, ACS Sens., 2019, 4, 2838–2845 CrossRef CAS PubMed.
- L. Zhang and L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 16248–16256 CrossRef CAS PubMed.
- W. Wan, M. Biyikal, R. Wagner, B. Sellergren and K. Rurack, Angew. Chem., Int. Ed., 2013, 52, 7023–7027 CrossRef CAS PubMed.
- S. Wagner, J. Bell, M. Biyikal, K. Gawlitza and K. Rurack, Biosens. Bioelectron., 2018, 99, 244–250 CrossRef CAS PubMed.
- O. Jamieson, T. C. C. Soares, B. A. de Faria, A. Hudson, F. Mecozzi, S. J. Rowley-Neale, C. E. Banks, J. Gruber, K. Novakovic, M. Peeters and R. D. Crapnell, Chemosensors, 2020, 8, 5–13 CrossRef CAS.
- Z. Huang, X. Zhang, X. Zhang, S. Wang, B. Yang, K. Wang, J. Yuan, L. Tao and Y. Wei, RSC Adv., 2015, 5, 65884–65889 RSC.
- M. Díaz-Bao, R. Barreiro, J. M. Miranda, A. Cepeda and P. Regal, J. Anal. Methods Chem., 2015, 959675 Search PubMed.
- N. Wu, Z. Luo, Y. Ge, P. Guo, K. Du, W. Tang, W. Du, A. Zeng, C. Chang and Q. Fu, J. Pharm. Anal., 2016, 6, 157–164 CrossRef PubMed.
- F. Horemans, A. Weustenraed, D. Spivak and T. J. Cleij, J. Mol. Recognit., 2012, 25, 344–351 CrossRef CAS PubMed.
- X. Song, J. Wang and J. Zhu, Mater. Res., 2009, 12, 299–304 CrossRef CAS.
- Y. Zhang, X. Qu, F. F. Wang, G. Wu, J. Li, H. Hong and C. Liu, RSC Adv., 2015, 5, 83619–83627 RSC.
- R. Sjöback, J. Nygren and M. Kubista, Spectrochim. Acta, Part A, 1995, 51, L7–L21 CrossRef.
- X. F. Zhang, J. Zhang and L. Liu, J. Fluoresc., 2014, 24, 819–826 CrossRef CAS PubMed.
- Q. Yang, J. Li, X. Wang, H. Peng, H. Xiong and L. Chen, Biosens. Bioelectron., 2018, 112, 54–71 CrossRef CAS.
- D. Margulies, G. Melman and A. Shanzer, Nat. Mater., 2005, 4, 768–771 CrossRef CAS PubMed.
- D. W. Newton and R. B. Kluza, Drug Intell. Clin. Pharm., 1978, 12, 546–554 CrossRef CAS.
- N. Klonis, A. H. A. Clayton, E. W. Voss and W. H. Sawyer, Photochem. Photobiol., 1998, 67, 500–510 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00192b |
| This journal is © The Royal Society of Chemistry 2021 |