
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFull-colour solvatochromic fluorescence emitted from a semi-aromatic imide compound based on ESIPT and anion formation†
Atsuko
Tabuchi
 a,
Teruaki
Hayakawa
b,
Shigeki
Kuwata
a,
Ryohei
Ishige
a and
Shinji
Ando
*a
a,
Teruaki
Hayakawa
b,
Shigeki
Kuwata
a,
Ryohei
Ishige
a and
Shinji
Ando
*a
aDepartment of Chemical Science & Engineering, Tokyo Institute of Technology, Ookayama 2-12-1-E4-5, Meguro-ku, Tokyo 152-8552, Japan. E-mail: ando.s.aa@m.titech.ac.jp; Tel: +81-3-5734-2137
bDepartment of Materials Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, Tokyo 152-8552, Japan
First published on 22nd June 2021
Abstract
A multi-colour solvatochromic fluorophore was developed by substituting a fully organic 3-amino-N-cyclohexyl phthalimide group with a p-toluenesulfonyl (tosyl) group. This compound is essentially colourless in the visible region and exhibits reddish fluorescence through excited-state intramolecular proton transfer (ESIPT) in the crystalline state with a considerably large Stokes shift of 9786 cm−1, as well as bright fluorescence in the full-colour range, from purple to red, when dissolved in 10 different organic solvents. Excitation wavelength measurements, lifetime measurements, and time-dependent density functional theory (TD-DFT) calculations clarified that the full-colour solvatochromism of the fluorescence results from the combination of the fluorescence emissions from the three different excited states: locally excited (N*), intramolecular proton-transferred (T*), and anion states (A*). This unique solvatochromic fluorescence property is expected to be applied in organic electronic devices and biological imaging tools.
1. Introduction
Organic molecules which absorb short-wavelength ultraviolet (UV) light and emit long-wavelength light from violet to the near-infrared (NIR) region with a visible full-colour range are attracting considerable attention for their potential applications in solar spectral converters, fluorescent probes and labels in biology, and emitting materials for organic electronic devices, such as organic light-emitting diodes (OLEDs).1–3 Of particular interest are fluorescent dyes with luminescent properties that are sensitive to physical changes in the local environment.4 For instance, solvatochromic phosphors exhibit environmentally sensitive emission properties.5 This dynamic behaviour is essential for the application of the dyes as imaging tools to provide information on the state of proteins.6 To tune the fluorescence colour, molecules exhibiting dual or multiple emissions are useful. For instance, dual emission can be obtained one emitter with two emitting states, two independent emitters, or two correlated emitters, and their mechanisms and the electronic or geometrical conditions have been investigated for molecular design and applications. Such materials have been of great interest over the past few years.7,8In recent years, aromatic imide/amide molecules have attracted significant attention because of their excellent photothermal and chemical stabilities with rigid structures, high-fluorescence quantum yields, and characteristic electron-transporting properties.9 Since the lowest energy electronic transition (LET) of phthalimide is a locally excited (LE) n–π* transition derived from a carbonyl group,10 phthalimide and N-alkylphthalimide exhibit only weak fluorescence with minimal oscillator strengths. N-Alkylphthalimides without substituents on the benzene ring do not exhibit fluorescence; however, those substituted with electron-donating or electron-withdrawing substituents exhibit bright fluorescence because the LET can be changed to an LE π–π* transition with a large oscillator strength.11 To achieve bright fluorescence, it is also important to suppress the aggregation caused quenching (ACQ) because the formation of aggregates induced by π–π stacking or charge transfer interactions in aromatic molecules frequently reduces the fluorescence quantum yields. The introduction of bulky functional groups12,13 and helical structures14 is known to be effective to suppress or reduce the ACQ.
To achieve high UV absorption followed by full-colour emission with high efficiency, long-wavelength fluorescence with a very large Stokes shift (ν) is a prerequisite. An enhancement of Stokes shift helps to prevent self-absorption of emitted light, which leads to an increase in fluorescence quantum yield. We focused on the use of excited-state intramolecular proton transfer (ESIPT), a process in which the excited state is relaxed by the structural relaxation (tautomerisation) from the normal (N*) form to the tautomer (T*) form through proton transfer in the excited state, and it can be used for white light emission and sensing probes.15–17 There are two types of ESIPT: hydroxyl (–OH) and amino (>NH) types, in which the proton donors are O and N, respectively. The >NH type has been found to undergo ESIPT more efficiently than the –OH type;18 it has a great advantage in that it can be substituted with various groups, such as –NH–R. In addition, ESIPT is sensitive to the ambient environment and can be easily tuned by changing the electron-donating or electron-withdrawing substituents,11,19 pH,20 and solvent polarity.5,21–23 Thus far, by changing the chemical structures with adjusted electron-withdrawing substituents and extending the π-conjugated framework, various types of fluorophores that emit light covering the full-colour range from blue to the NIR region have been achieved.24–26 To achieve full-colour solvatochromic photoluminescence, blue, green, and red colour emissions (light's three primary colours) are essential. Especially, red fluorescence is difficult to achieve by excitation in the UV region due to the very larger Stokes shift. Okamoto et al. reported the fluorescent properties of 3-aminophthalimides substituted with electron-withdrawing group (EWGs) of –SO2CH3, –SO2CF3, –COCH3 and –COCF3, which showed fluorescence at longer wavelengths in solutions.11 Only the one substituted with –SO2CH3 showed red emission via ESIPT, though its quantum yield was medium (Φ = 0.06). Besides, Chou et al. introduced another EWG of p-toluenesulfonyl (tosyl) substituent into 2-(2′-aminophenyl)benzothiazole and observed red emission with a relatively small Φ value of 0.0086.19
However, as far as we know, no compound has been reported to contain a single molecule that exhibits colourlessness and transparency under solar light and full-colour photoluminescence (PL) over the entire visible spectrum, from purple to red, under UV irradiation, depending on the solvent. In this study, we designed and synthesised a new fluorophore of the N-cyclohexylphthalimide group substituted with a tosyl group, which exhibits long-wavelength fluorescence via ESIPT with a considerably large Stokes shift. In addition, we clarified the mechanism of the full-colour fluorescence of this molecule dissolved in organic solvents using UV-vis absorption, excitation, and emission spectroscopy; lifetime measurements; and quantum chemical calculations.
2. Experimental
2.1. Materials
The compound, 3-nitrophthalic anhydride (1), purchased from Kanto Chemical Co., Inc., was used as received. Cyclohexylamine, Pd/C (55% water), and p-toluenesulfonyl chloride, purchased from Tokyo Kasei Co., Ltd., were used as received.2.2. Synthesis
The synthesis of 3-tosylamino-N-cyclohexylphthalimide (3TsAPI) is shown in Scheme 1. | ||
| Scheme 1 Synthesis route of 3-tosylamino-N-cyclohexylphthalimide (3TsAPI). | ||
Anal. calculated for C21H22N2NaO4S: C, 63.30; H, 5.56; N, 7.03; O, 16.06; S, 8.03. Found: C, 63.16; H, 5.61; N, 6.86; O, 16.00; S, 8.11.
Synthetic procedures for other N-cyclohexylphthalimide compounds with different EWGs and those 1H NMR spectra are presented in ESI† (Fig. S4–S6).
2.3. Measurements
The PL quantum yields were measured using a calibrated integrating sphere (C9920, Hamamatsu) connected to a multichannel analyser (C7473, Hamamatsu) using an optical fibre link.
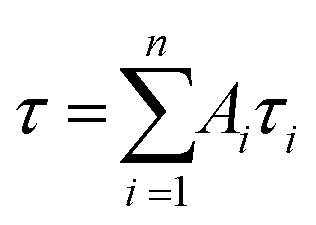 , where Ai is the pre-exponential for lifetime τi.
, where Ai is the pre-exponential for lifetime τi.
2.4. Quantum chemical calculations
Density-functional theory (DFT) calculations were conducted using the Gaussian-16 software (Rev.C.01),27 as described in our previous studies.28,29 Geometry optimisation was independently performed with the B3LYP and CAM-B3LYP functionals with the 6-311G(d) basis set for the ground (S0) and excited (S1) states, respectively. The 6-311++G(d,p) basis set was used to calculate the vertical excitation wavelengths and oscillator strengths (f) for each of the S0 and S1 geometries. The S0 → S1 transition at the optimised S0 geometry corresponds to the optical absorption of the ground state. Contrarily, that at the optimised S1 geometry corresponds to the fluorescent emission according to Kasha's rule. Each calculated transition was replaced by a Gaussian broadening function with a width of 0.10 eV, producing the shapes of the calculated spectra. The solvent effects in the ground and excited states were incorporated based on the polarisable continuum model (PCM) implemented in the Gaussian software.2.5. Crystallography
A single crystal suitable for X-ray analysis was mounted on a fibre loop. Diffraction experiments were performed on a Rigaku Saturn CCD area detector with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Intensity data (6° < 2θ <55°) were corrected for the Lorentz-polarisation effects and absorption. Structure solution and refinements were carried out using the CrystalStructure (2000–2018) program package (Rigaku Corp., Tokyo Japan). The heavy-atom positions were determined by a direct methods program (SIR92), and the remaining non-hydrogen atoms were found by subsequent Fourier syntheses and refined by full-matrix least-squares techniques against F2 using the SHELXL-2014/7 programme.30 The position of the NH hydrogen atom was refined, whereas the remaining hydrogen atoms were included in the refinements using a riding model. Crystal data: C21H22N2O4S, M = 398.48, triclinic, a = 5.3459(13), b = 13.292(3), c = 14.387(3) Å, α = 70.586(8), β = 81.639(9), γ = 84.599(10)°, U = 952.8(4) Å3, T = 93 K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), Z = 2, 11
(no. 2), Z = 2, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 858 reflections measured, 4336 unique (Rint = 0.0623), which were used in all calculations. The final wR(F2) was 0.1176 (all data). CCDC 2067299 contains the supplementary crystallographic data for this study.†
858 reflections measured, 4336 unique (Rint = 0.0623), which were used in all calculations. The final wR(F2) was 0.1176 (all data). CCDC 2067299 contains the supplementary crystallographic data for this study.†
3. Results and discussion
3.1. Molecular design for 3TsAPI
The design of a new fluorophore, which is transparent and colourless in the visible region and exhibits reddish fluorescence under UV irradiation, is based on the TD-DFT calculations. To achieve red fluorescence with a high Φ, introduction of bulky substituents could be helpful to reduce intermolecular interactions and suppress ACQ in solution and the solid state. By combining the molecular design concepts of the strong EWG and the bulky substituents, we consider that the introduction of tosylamino and N-cyclohexyl groups into a phthalimide (3TsAPI) is promising owing to the ESIPT fluorescence from the proton-transferred T* form and the bulky phenylsulfonyl and cyclohexyl groups. The calculated absorption and emission spectra of 3TsAPI in toluene are shown in Fig. 1(a). The longest wavelength absorption of the N form and the N* and T* emission peaks appear at 338 nm, 412 nm, and 605 nm, respectively. Since the calculated energy difference between T* and N* (δET*–N*) is −11.5 kJ mol−1, the tautomerisation from N* to T* should proceed smoothly in the excited state. Thus, 3TsAPI was predicted to emit red fluorescence via ESIPT with a very large Stokes shift (ν = 13066 cm−1). The predicted photophysical processes are shown schematically in Fig. 1(c). In addition, the optical absorption and the emission spectra of the N* and T* forms of N-cyclohexylphthalimide substituted with amino (–NH2), acetylamino (–NHCOCH3), trifluoroacetylamino (–NHCOCF3), methylsulfonylamino (–NHSO2CH3), and tosylamino (–NHSO2C6H4–CH3) groups in toluene solutions were calculated based on the TD-DFT theory as displayed in Fig. S7 (ESI†). The predicted fluorescence wavelengths from the N*/T* states in toluene are 433/655, 402/585, 350/545, 401/598 and 412/605 nm, and the values of δET*–N* are +40.7, +24.3, +9.2, −15.5, and −11.5 kJ mol−1, respectively, which means that ESIPT is not likely to occur in the amino and acetylamino-substituted compounds, and only the strong EWGs of methylsulfonylamino and tosylamino groups can afford reddish fluorescence via ESIPT.
 | ||
| Fig. 1 Calculated UV-vis absorption (dotted line) and emission (solid line) spectra of 3TsAPI in (a) normal form (N and N*, blue) and tautomer form (T*, red) in toluene, and (b) anionic form (A and A*, green) in DMSO. (c, d) Photophysical processes relating to these forms. | ||
Since the electron-withdrawing tosyl group may induce the dissociation of the hydrogen atom in the amide group,31 the absorption and emission spectra and the photophysical processes calculated for the anionic forms in the ground (A) and excited (A*) states in DMSO are shown in Fig. 1(b) and (d), in which the absorption and fluorescence peaks appear at 429 nm and 513 nm, respectively. Notably, compared with those of the N and N* forms, the absorption and emission of the anion show significant bathochromic shifts of 91 and 102 nm, respectively, which may cause a yellowish colouration under white light and green fluorescence under UV irradiation.
The optimised conformations of 3TsAPI in the N and N* forms calculated under vacuum conditions were compared with those in the crystal lattice in Fig. S8(a)–(c) (ESI†). Moreover, the calculated absorption spectra of the N form and the fluorescence spectra of the N* form of 3TsAPI in various solvents are shown in Fig. S9 and S10 (ESI†), respectively. Intriguingly, the experimentally determined crystal structure of 3TsAPI is quite similar to the optimised N and N* structures, and the conformational change caused by one-electron excitation from N to N* is quite small, which leads to a small ν value of N* emission (5322 cm−1 in toluene). In addition, the solvent-dependence of the N absorption and N* fluorescence peaks is also insignificant owing to their less polarisable electronic structures. Fig. S11 (ESI†) shows the spatial distributions of the HOMOs and LUMOs in the optimized geometries for the ground state (N), the excited state (N*), the excited proton transferred state (T*), and the excited anionic state (A*) of 3TsAPI. The very similar electronic structures of the N and N* states support the characters of the calculated absorption and emission spectra.
In contrast, the optimised conformations of the T*, A, and A* forms are quite different from those of N and N*, as displayed in Fig. S8(d)–(f) (ESI†). The dihedral angles of C5–N2–S1–C15 are almost 180°, and the benzene ring of the tosyl group in the T* form is located orthogonally to the phthalimide plane, whereas it is located in the same plane in the A form. The large conformational change in T* significantly enhances the Stokes shift, whereas the limited change in A* reduces it. For instance, the calculated ν value for the emission from T* in toluene (13066 cm−1) is 3.4 times larger than that from A* (3816 cm−1). However, as seen in Fig. S11 (ESI†), it is intriguing to note that the electronic structures of T* and A* look very similar to each other, but totally different from those of N and N*. The HOMOs and LUMOs are mainly localised at the phthalimide moiety, and both the excited states can emit bright fluorescence at longer wavelengths via LE(π*–π) transitions once T* or A* are generated.
The calculated T* spectra of 3TsAPI in various organic solvents are shown in Fig. S12 (ESI†). The shortest and longest fluorescences were predicted to appear at 572 nm in MeOH/MeCN and 608 nm in cyclohexane, respectively, owing to the dielectric effects (effect of the polarity of solvents) on the ESIPT process. The predicted order of the T* emission agrees well with the relative permittivity values of the solvents adopted in the Gaussian software. It is well known that the T* fluorescence shows a bathochromic shift in less polar solvents, such as toluene, and a hypsochromic shift in more polar solvents, such as EtOH.5
Moreover, the calculated absorption and emission spectra of A and A* of 3TsAPI in various solvents are shown in Fig. S13 and S14 (ESI†), respectively. The solvent effects and the predicted order of the solutions are similar to those of the calculated T* emission, which indicates that the anion absorptions and emissions also reflect the dielectric effects of the solvents. Fig. S15 (ESI†) shows the relationship between the relative permittivity (εr) and the calculated deprotonation energy of 3TsAPI under vacuum and in various solvents. The inverse proportion indicates that the anion of 3TsAPI can be formed readily in polar solvents that are between acetone (εr = 20.49) and DMSO (46.83) in the figure. Notably, the optimised conformation of the A form gradually changes depending on the polarity of the solvent as shown in Fig. S16 (ESI†). The coplanar conformation between the benzene ring of the tosyl group and the phthalimide plane is stable in less polar solvents, such as toluene and tetrahydrofuran (THF), and under vacuum, whereas more twisted conformations become preferable in polar solvents, such as acetone, MeCN, and DMSO.
3.2 Crystal Structure of 3TsAPI
The crystalline structure of 3TsAPI, analysed by single-crystal X-ray diffraction, is shown in Fig. 2. The needle-shaped crystals obtained from the ethanol solution were used for the measurement. The sulfonamide N2 atom lies in the phthalimide plane in close contact with the imide carbonyl oxygen O1 atom (2.849(2) Å), indicating a strong intramolecular hydrogen bond in the quasi-6-membered ring. Meanwhile, the S1 atom deviated slightly from the phthalimide plane (0.525(2) Å), and the two benzene rings are orthogonal to each other with an interplane angle of 85.17(7)°. Thus, the skeletal structure of 3TsAPI has a bent ‘L-shape’ in the crystal.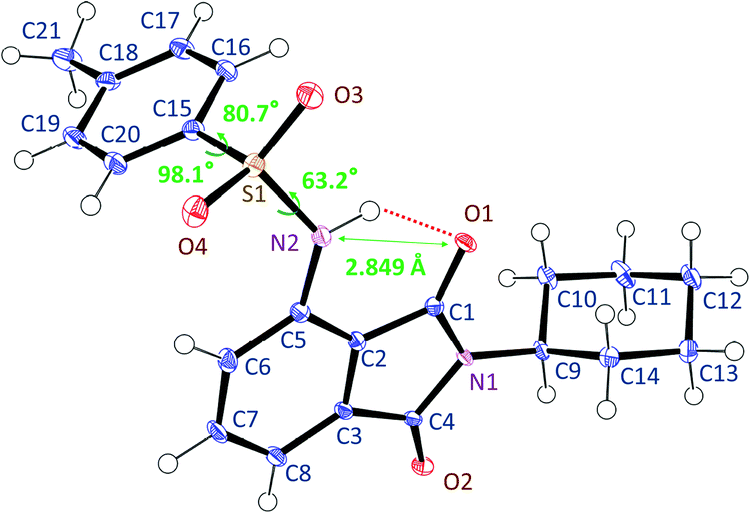 | ||
| Fig. 2 Conformation of 3TsAPI in the crystal lattice determined by single-crystal X-ray diffraction analysis. | ||
3.3. Optical properties of 3TsAPI crystal
Fig. 3 shows the excitation and emission spectra of 3TsAPI in the crystalline state at 20 °C. The excitation and emission peaks are observed at 370 nm and 580 nm, respectively. Therefore, the crystal appears colourless under white light and exhibits bright orange fluorescence under UV irradiation at 365 nm. Based on the large Stokes shift (ν = 9786 cm−1) and the considerably short fluorescence lifetime (τ = 1.1 ns, Fig. S17, ESI†), the observed emission is attributable to the T* fluorescence (Fig. 1(a) and (c)) via ESIPT. Moreover, a high fluorescence quantum yield (Φ = 0.22) was observed despite the large Stokes shift. We have also synthesized N-cyclohexylphthalimide compounds substituted at the 3-position with –NH2, –NHCOCH3, –NHCOCF3, –NHSO2CH3 groups (the synthesis procedures are shown in the ESI,† as stated in Section 2.2), and their experimental excitation and emission spectra in the crystalline state are shown in Fig. S18 (ESI†). In addition, the excitation and emission wavelengths (λex, λem) and quantum yield (Φ) are summarised in Table S1 (ESI†). As predicted by the TD-DFT calculations, the compounds substituted with –NH2 and –NHCOCH3 show green fluorescence at 503 nm and blue fluorescence at 456 nm, respectively, without ESIPT emission, while the compound with –NHCOCF3 shows yellow fluorescence at 542 nm and the one with –NHSO2CH3 demonstrates red fluorescence at 585 nm. These results indicate that the emission colour can be well tuned by the selection of EWGs. It should be noted that the Φ value of 3TsAPI is 2.75 times as high as another red-emitting compound with –NHSO2CH3. This should be originated from the bulky tosyl and cyclohexyl groups that reduce intermolecular aggregation and suppress ACQ. Accordingly, 3TsAPI could be the best compound exhibiting red fluorescence via ESIPT with a large Stokes shift and a high quantum yield.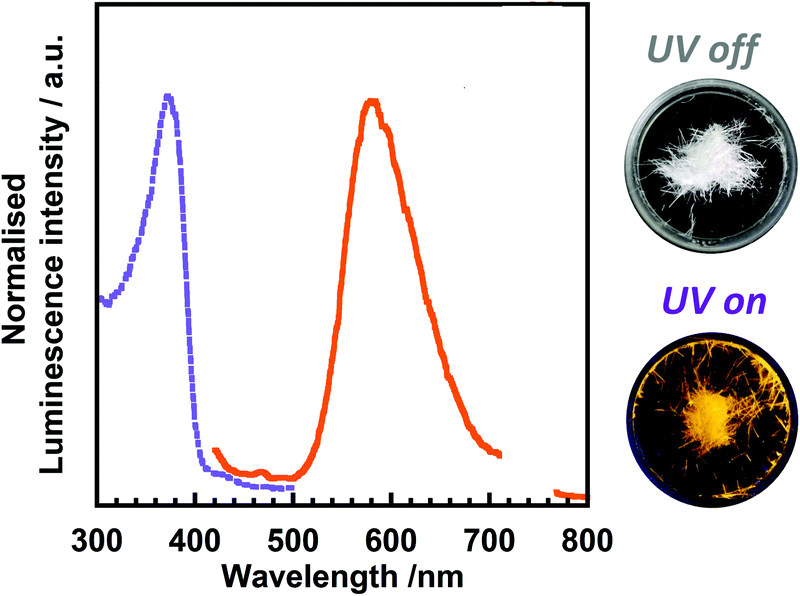 | ||
| Fig. 3 Excitation (dotted line)/emission (solid line) spectra of 3TsAPI in the crystalline state (λex = 370 nm, λem = 580 nm), and photographs under white light (UV off) and UV (λ = 365 nm) irradiation (UV on). | ||
3.4. Optical properties of 3TsAPI in solutions
Fig. 4 shows the UV-vis absorption spectra of 3TsAPI dissolved in 10 different organic solvents with a wide polarity range. Strong absorption peaks are observed at around 340 nm for all solvents (Table 1). This wavelength correlates with the calculated highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) transition at 338 nm (Fig. 1(a) and (c)). As depicted in Fig. S11 (ESI†), the N* form mainly contributes to the locally excited state (N*) generated by one-electron excitation from the HOMO to the LUMO. In addition, relatively weak absorption peaks were observed between 390 and 480 nm in solvents with proton-accepting abilities, such as DMSO, MeOH, and EtOH. Fig. 5 shows photographs of these solutions under white light. The nine solutions, except for DMSO, are almost colourless and transparent, although the DMSO solution shows a pale yellow colour, which is caused by the broad absorption at 419 nm. Since the calculated absorption of the anion in DMSO at 429 nm (Fig. 1(b) and (d)) is very close to this wavelength, these weak absorption bands may originate from the partly proton-dissociated anionic form. As predicted by the calculation, deprotonation is likely to occur in solvents that are more polar than THF. The significant dependence of the optimised conformations of the anionic state on the polarity of the solvents may account for the characteristic solvatochromism in the absorption spectra. | ||
| Fig. 4 (a) UV-vis absorption spectra for 3TsAPI dissolved in toluene, acetone, ethanol (EtOH), methanol (MeOH), dimethyl sulfoxide (DMSO), ethyl acetate (EtOAc), acetonitrile (MeCN), ethylene glycol (EG), chloroform (CHCl3), and cyclohexane (Cyclohex). The concentration of 3TsAPI was set to 2.5 × 10−4 mol L−1. (b) Expansion of the spectra (350 nm < λ < 550 nm). | ||
| Solvent | λ abs/nm | λ ex/nm | λ em/nm | ν/cm−1 | τ/ns | Φ |
|---|---|---|---|---|---|---|
| Toluene | 342 | 346 | 602 |
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_i_char_2009.gif) 288 288
|
0.4 | 0.06 |
| 361 | 409 | 3263 | 4.8 | 0.07 | ||
| Acetone | 337 | 351 | 599 |
11796
|
0.2 | 0.01 |
| 351 | 420 | 4681 | 1.6 | |||
| 400 | 496 | 4822 | 11.2 | 0.19 | ||
| EtOH | 339 | 395 | 507 | 5585 | 7.1 | 0.15 |
| MeOH | 337, 400 | 392 | 508 | 5825 | 9.7 | 0.09 |
| DMSO | 339, 419 | 419 | 528 | 4927 | 5.6 | 0.21 |
| EtOAc | 338 | 350 | 600 |
11905
|
0.1 | 0.02 |
| 400 | 490 | 4592 | 12.6 | 0.31 | ||
| MeCN | 337 | 345 | 597 |
12235
|
0.1 | 0.02 |
| 395 | 493 | 5049 | 7.7 | 0.14 | ||
| EG | 337 | 355 | 592 |
11277
|
0.9 | 0.02 |
| 355 | 427 | 4750 | 0.4 | |||
| CHCl3 | 341 | 350 | 601 |
11927
|
0.3 | 0.04 |
| Cyclohex | 341 | 346 | 598 |
12168
|
0.3 | 0.03 |
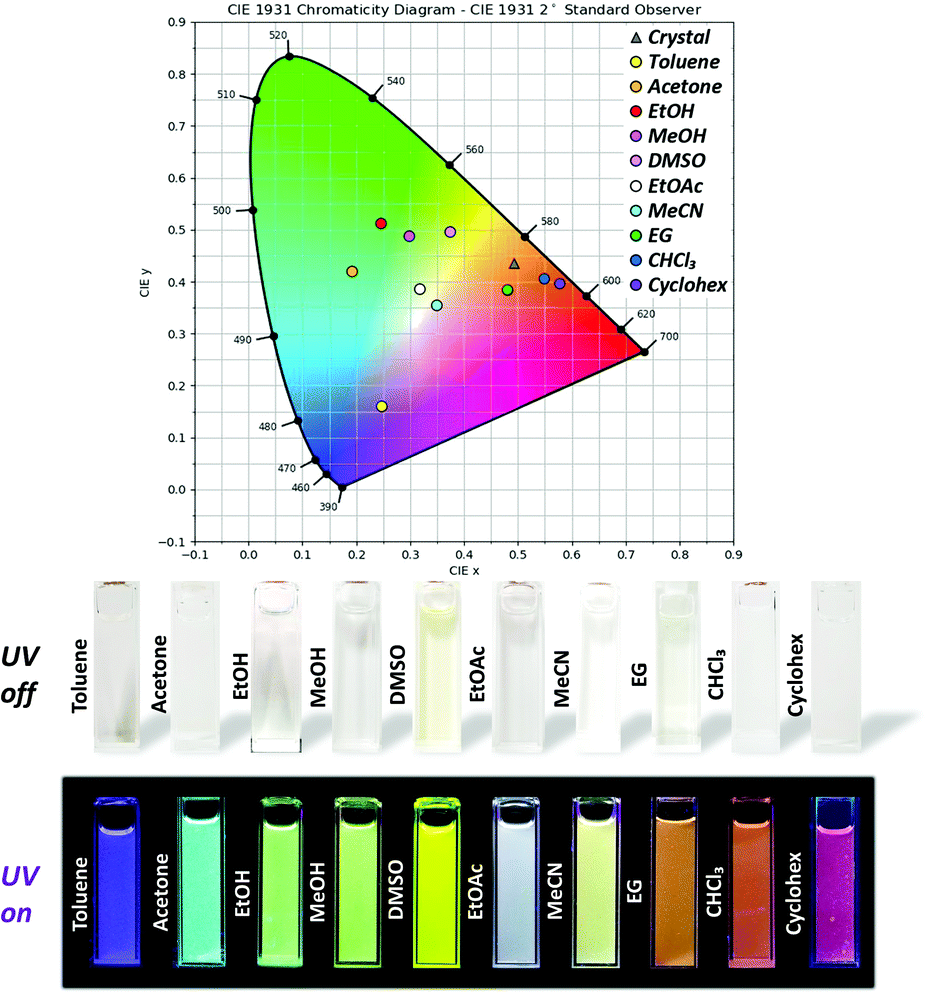 | ||
| Fig. 5 International Commission on Illumination (CIE) coordinates for the fluorescence colours of 3TsAPI in the crystalline state and in various organic solvents, and photographs of these solutions under white light (UV off) and UV (λ = 365 nm) irradiation (UV on). | ||
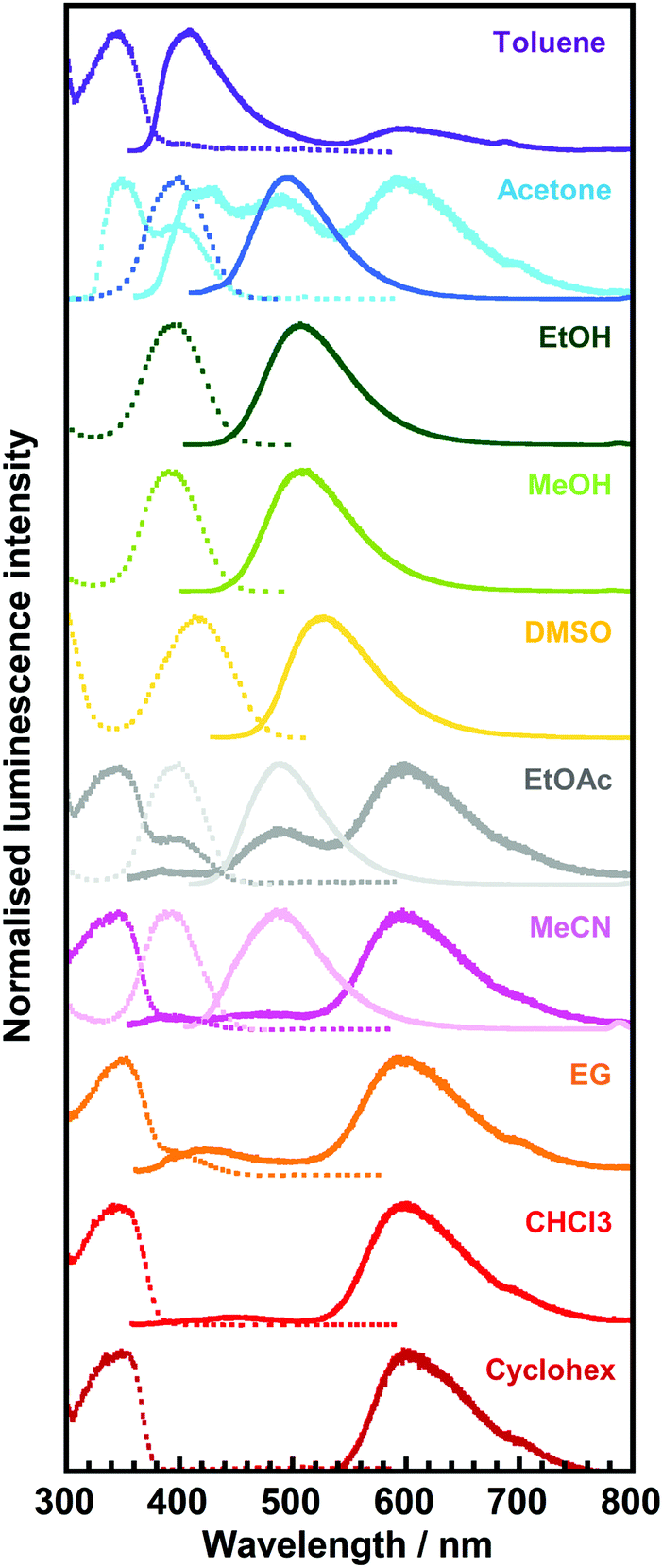 | ||
| Fig. 6 Excitation (dotted line)/emission (solid line) spectra of 3TsAPI in toluene, acetone, ethanol (EtOH), methanol (MeOH), dimethyl sulfoxide (DMSO), ethyl acetate (EtOAc), acetonitrile (MeCN), ethylene glycol (EG), chloroform (CHCl3), and cyclohexane (Cyclohex) solutions. The excitation and emission wavelengths (λex, λem) are summarised in Table 1. | ||
For the solvents, except for EtOH, MeOH, and DMSO, long-wavelength fluorescence is observed at approximately 600 nm when excited at approximately 350 nm. This is attributable to the T* fluorescence via ESIPT because of the considerably large Stokes shifts (ν = 11277–12288 cm−1) and the considerably short lifetimes (τ < 1.0 ns, Table 1). The observed emission wavelengths adequately reflect the calculated value for T* fluorescence (Fig. 1(a), (c) and Fig. S12, ESI†). Conversely, the short-wavelength fluorescence observed at approximately 400 nm in toluene, acetone and EG can be attributed to the N* fluorescence due to the small Stokes shifts (ν = 3263–4750 cm−1) and the moderately short fluorescence lifetimes (1.0 < τ < 5.0 ns, Table 1). These wavelengths are well predicted by the calculated N* fluorescence (Fig. 1(a), (c) and Fig. S10, ESI†). The differences in the intensity ratio between T* and N* fluorescence can be explained by the solvent dependence of the energy barriers for the tautomerisation between N* and T*.32
In addition to the fact that the T* fluorescence via ESIPT was not observed in EtOH, MeOH, and DMSO, other fluorescence bands with small Stokes shifts (4592 cm−1 < ν < 5825 cm−1) were observed between 490 nm and 528 nm in acetone, EtOAc, and MeCN when excited at approximately 400 nm (392 nm < λex < 419 nm). Moreover, their excitation wavelengths, λex, were longer than those of N* (345 nm < λex < 361 nm), and the fluorescence lifetimes (τ) were significantly elongated up to > 5.0 ns compared with those of N* and T* (τ < 5.0 ns), which are quite different from the N* and T* emissions.
To investigate the origin of the fluorescence characterised by long excitation wavelengths and long lifetimes, a small portion of trifluoroacetic acid (TFA) was added to the solutions. Fig. 7 shows the emission spectra of 3TsAPI in acetone and EtOH before and after the addition of TFA, with their photographs under UV irradiation. In addition, the excitation and emission wavelengths (λex, λem), Stokes shifts (ν), fluorescence lifetimes (τ), and quantum yields (Φ) are summarised in Table S2 (ESI†). As shown in Fig. 7(a), the addition of TFA to the acetone solution effectively suppresses the fluorescence at 496 nm with a long τ (11.9 ns), and the fluorescence from T* via ESIPT at 605 nm increases when excited at 351 nm (Fig. S20, ESI†). The fluorescence colour under UV irradiation immediately changed from light blue to orange upon the addition of TFA. As shown in Fig. 7(b), the addition of TFA effectively suppressed the fluorescence at 507 nm in EtOH with a long τ (7.1 ns), and subsequently, the fluorescence from T* at 601 nm increased upon excitation at 350 nm. The fluorescence colour changed from green to orange. These changes in the emission spectra and lifetimes clearly indicate that the original state of 3TsAPI in these solutions included the anionic form, implying that deprotonation occurred in the ground state. The experimental λex and λem values correspond well with the calculated values for the anionic forms, as shown in Fig. 1(b) and (d) (λex = 429 nm, λem = 513 nm in DMSO). The lifetimes of the fluorescence at short-wavelengths of 414 nm in acetone/TFA and 400 nm in EtOH/TFA are very short (1.7 ns and 0.4 ns, respectively), and the emission wavelengths correlate with the calculated wavelengths for the N*. Only A* exhibited fluorescence in EtOH, MeOH, and DMSO without T* fluorescence because the tosyl-substituted amide group of 3TsAPI forms strong hydrogen bonds with these proton-accepting solvents, as shown in Fig. S21 (ESI†), thus suppressing the ESIPT process.31,33–35 As shown in Fig. 4, these solutions demonstrated partial absorption of the A form in the visible region, also indicating that 3TsAPI forms anions in solvents with a strong proton-accepting ability.
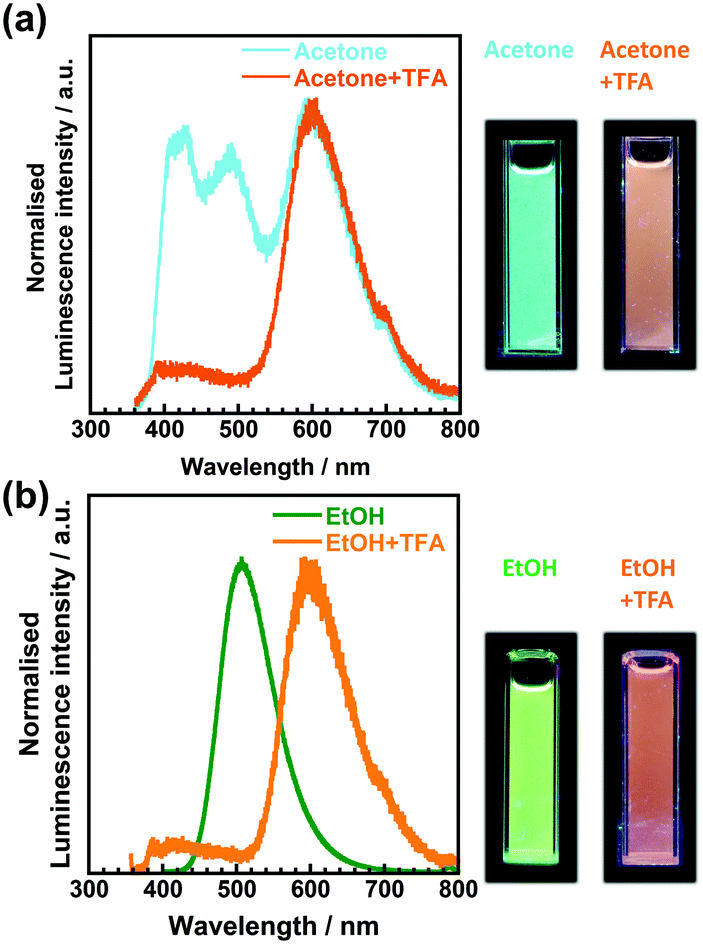 | ||
| Fig. 7 Emission spectra of 3TsAPI dissolved (a) in acetone before (λex = 351 nm, light blue) and after adding TFA (λex = 350 nm, orange). The concentration of TFA was set to 1.0 × 10−2 mol L−1. (b) In EtOH (λex = 395 nm, green) and a mixed solvent of EtOH/TFA (λex = 347 nm, orange). The concentration of TFA was set to 2.0 × 10−4 mol L−1. | ||
Finally, the origin of the full-colour fluorescence represented in the CIE coordinates and the photographs of the 3TsAPI solutions (Fig. 5) is discussed. In the acetone solution, the fluorescence bands of the anionic form and T* were observed in addition to the N* fluorescence, and the combination of these emissions led to a light blue colour. Although only the anion fluorescence was observed in EtOH, MeOH, and DMSO, different fluorescence colours were achieved, ranging from green to yellow. Considering the order of the relative permittivity (εr dielectric constants) of these solvents: DMSO (εr = 46.8) > MeOH (32.6) > EtOH (24.9) (Fig. S15, ESI†), the excited state of the anionic forms is more stable in high εr solvents, which results in long-wavelength emissions resulting in different colours. As shown in Table 1, anion fluorescence was observed in polar solvents of EtOAc, acetone, EtOH, MeOH, MeCN and DMSO, which coincides well with the order of εr in Fig. S15 (ESI†) except EG. In the EtOAc and MeCN solutions, T* fluorescence was observed in addition to the anion fluorescence, after which white and pale yellow fluorescence colours were observed. In particular, due to the good balance between the relative fluorescence intensities of the anionic and T* forms, the emission spectrum covered the entire visible region, which endowed the EtOAc solution with white fluorescence (Fig. S22, ESI,†λex = 360 nm). In the EG, CHCl3, and Cyclohex solutions, the intense T* fluorescence led to the characteristic orange-red fluorescence because the anionic form is destabilised in non-polar solvents with small εr values (4.7 for CHCl3 and 2.0 for Cyclohex) (Fig. S15, ESI†). However, the reason for the orange fluorescence in EG with a high εr of 40.2 is unclear at present.
In summary, the novel imide compound, 3TsAPI, exhibits both small and large Stokes shifted fluorescence derived from N* and T*, as well as another fluorescence derived from the anionic form. These three types of fluorescence compete through interactions in various solvents, resulting in multi-colour fluorescence in the entire visible region. The compound, 3TsAPI, is an extraordinary fluorophore that exhibits bright fluorescence in the full-colour range, from purple to red, owing to the exquisite balance of the three fluorescence species generated in various organic solvents.
4. Conclusions
We developed a new fluorophore that absorbs only UV light and emits fluorescence in the long-wavelength region of visible light. The TD-DFT calculations predicted that an imide compound with an –R–N–H…O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– type intramolecular hydrogen bond substituted with a strong electron-withdrawing tosyl group (3TsAPI) would absorb only UV light and exhibit reddish fluorescence through ESIPT. This compound exhibited a high quantum yield and a considerably large Stokes shift (9786 cm−1) in the crystalline state. The high quantum yield originated from the suppression of ACQ, resulting from the bulky tosyl and cyclohexyl groups. This compound shows an extremely unique solvatochromic fluorescence that exhibits a full-colour range from purple to red in 10 different organic solvents. Excitation wavelength measurements, lifetime measurements, and TD-DFT calculations clarified that the full-colour fluorescence results from the combination of the fluorescence emitted from the three states, namely N*, T*, and A*. The significant colour change caused by solvatochromic fluorescence and the characteristic dependence on the excitation wavelength is caused by the different stabilities and subtle balance among the states of N*, T*, and A*, arising from the interactions between the molecules and various solvents with different polarities. These properties have great potential for application in organic electronic devices, such as OLEDs and imaging tools in biology.
C– type intramolecular hydrogen bond substituted with a strong electron-withdrawing tosyl group (3TsAPI) would absorb only UV light and exhibit reddish fluorescence through ESIPT. This compound exhibited a high quantum yield and a considerably large Stokes shift (9786 cm−1) in the crystalline state. The high quantum yield originated from the suppression of ACQ, resulting from the bulky tosyl and cyclohexyl groups. This compound shows an extremely unique solvatochromic fluorescence that exhibits a full-colour range from purple to red in 10 different organic solvents. Excitation wavelength measurements, lifetime measurements, and TD-DFT calculations clarified that the full-colour fluorescence results from the combination of the fluorescence emitted from the three states, namely N*, T*, and A*. The significant colour change caused by solvatochromic fluorescence and the characteristic dependence on the excitation wavelength is caused by the different stabilities and subtle balance among the states of N*, T*, and A*, arising from the interactions between the molecules and various solvents with different polarities. These properties have great potential for application in organic electronic devices, such as OLEDs and imaging tools in biology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Prof. Vidmantas Gulbinas and Dr Aurimas Vysniauskas at the Center for Physical Sciences and Technology in Lithuania for their valuable discussions concerning this study. We also wish to thank Dr Naiqiang Liang, Ms Mayuko Nara, Mr Ryoji Orita and Mr Takehiko Kambara at the Tokyo Institute of Technology for their help for the synthesis and measurements of fluorescent imide compounds. This work was financially supported by JSPS KAKENHI Grant Number 17H03112 and 21H01995.References
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, Multicolor conjugated polymer dots for biological fluorescence imaging, ACS Nano, 2008, 2, 2415–2423, DOI:10.1021/nn800590n.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Fluorescent analogs of biomolecular building blocks: Design, properties, and applications, Chem. Rev., 2010, 110, 2579–2619, DOI:10.1021/cr900301e.
- R. Sun, S. Feng, D. Wang and H. Liu, Fluorescence-Tuned Silicone Elastomers for Multicolored Ultraviolet Light-Emitting Diodes: Realizing the Processability of Polyhedral Oligomeric Silsesquioxane-Based Hybrid Porous Polymers, Chem. Mater., 2018, 30, 6370–6376, DOI:10.1021/acs.chemmater.8b02514.
- G. S. Loving, M. Sainlos and B. Imperiali, Monitoring protein interactions and dynamics with solvatochromic fluorophores, Trends Biotechnol., 2010, 28, 73–83, DOI:10.1016/j.tibtech.2009.11.002.
- O. A. Kucherak, P. Didier, Y. Mèly and A. S. Klymchenko, Fluorene analogues of prodan with superior fluorescence brightness and solvatochromism, J. Phys. Chem. Lett., 2010, 1, 616–620, DOI:10.1021/jz9003685.
- D. M. Shcherbakova, M. A. Hink, L. Joosen, T. W. J. Gadella and V. V. Verkhusha, An orange fluorescent protein with a large Stokes shift for single-excitation multicolor FCCS and FRET imaging, J. Am. Chem. Soc., 2012, 134, 7913–7923, DOI:10.1021/ja3018972.
- J. Gierschner, S. K. Behera and S. Y. Park, Dual Emission: Classes, Mechanisms and Conditions, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202009789.
- N. A. Kukhta and M. R. Bryce, Dual emission in purely organic materials for optoelectronic applications, Mater. Horizons, 2021, 8, 33–55, 10.1039/d0mh01316a.
- Y. Qin, G. Li, T. Qi and H. Huang, Aromatic imide/amide-based organic small-molecule emitters for organic light-emitting diodes, Mater. Chem. Front., 2020, 4, 1554–1568, 10.1039/d0qm00084a.
- V. Wingtens, P. Valat, J. Kossanyi and L. Biczok, Spectroscopic properties of aromatic dicarboximides, J. Chem. Soc., Faraday Trans., 1994, 90, 411–421, 10.1039/FT9949000411.
- H. Okamoto, K. Itani, M. Yamaji, H. Konishi and H. Ota, Excited-state intramolecular proton transfer (ESIPT) fluorescence from 3-amidophthalimides displaying RGBY emission in the solid state, Tetrahedron Lett., 2018, 59, 388–391, DOI:10.1016/j.tetlet.2017.12.049.
- M. Stolte, T. Schembri, J. Süβ, D. Schmidt, A.-M. Krause, M. O. Vysotsky and F. Würthner, 1-Mono- and 1,7-Disubstituted Perylene Bisimide Dyes with Voluminous Groups at Bay Positions: In Search for Highly Effective Solid-State Fluorescence Materials, Chem. Mater., 2020, 32, 6222–6236, DOI:10.1021/acs.chemmater.0c02115.
- D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Highly Emissive Organic Single-Molecule White Emitters by Engineering o-Carborane-Based Luminophores, Angew. Chem., Int. Ed., 2017, 56, 11370–11374, DOI:10.1002/anie.201703862.
- K. T. Wong, Y. Y. Chien, R. T. Chen, C. F. Wang, Y. T. Lin, H. H. Chiang, P. Y. Hsieh, C. C. Wu, C. H. Chou, Y. O. Su, G. H. Lee and S. M. Peng, Ter(9,9-diarylfluorene)s: Highly efficient blue emitter with promising electrochemical and thermal stability, J. Am. Chem. Soc., 2002, 124, 11576–11577, DOI:10.1021/ja0269587.
- I. E. Serdiuk, White Light from a Single Fluorophore: A Strategy Utilizing Excited- State Intramolecular Proton-Transfer Phenomenon and Its Veri fi cation, 2017 DOI:10.1021/acs.jpcc.7b00137.
- L. G. T. A. Duarte, J. C. Germino, J. F. Berbigier, C. A. Barboza, M. M. Faleiros, D. De Alencar Simoni, M. T. Galante, M. S. De Holanda, F. S. Rodembusch and T. D. Z. Atvars, White-light generation from all-solution-processed OLEDs using a benzothiazole-salophen derivative reactive to the ESIPT process, Phys. Chem. Chem. Phys., 2019, 21, 1172–1182, 10.1039/c8cp06485g.
- A. C. Sedgwick, L. Wu, H. Han, S. D. Bull, X. He, T. D. James and J. L. Sessler, Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents, Chem. Soc. Rev., 2018, 47, 8842–8880, 10.1039/c8cs00185e.
- A. Bhattacharyya, S. K. Mandal and N. Guchhait, Imine-Amine Tautomerism vs Keto-Enol Tautomerism: Acceptor Basicity Dominates over Acceptor Electronegativity in the ESIPT Process through a Six-Membered Intramolecular H-Bonded Network, J. Phys. Chem. A, 2019, 123, 10246–10253, DOI:10.1021/acs.jpca.9b08646.
- H. W. Tseng, J. Q. Liu, Y. A. Chen, C. M. Chao, K. M. Liu, C. L. Chen, T. C. Lin, C. H. Hung, Y. L. Chou, T. C. Lin, T. L. Wang and P. T. Chou, Harnessing excited-state intramolecular proton-transfer reaction via a series of amino-type hydrogen-bonding molecules, J. Phys. Chem. Lett., 2015, 6, 1477–1486, DOI:10.1021/acs.jpclett.5b00423.
- Z. C. Wen, J. A. B. Ferreira and S. M. B. Costa, Novel pH tunable fluorescent sensor with dual recognition mode, J. Photochem. Photobiol., A, 2008, 199, 98–104, DOI:10.1016/j.jphotochem.2008.05.012.
- Y. Niko, S. Kawauchi and G. I. Konishi, Solvatochromic pyrene analogues of prodan exhibiting extremely high fluorescence quantum yields in apolar and polar solvents, Chem. – Eur. J., 2013, 19, 9760–9765, DOI:10.1002/chem.201301020.
- S. Chatterjee, K. Ahire and P. Karuso, Room-Temperature Dual Fluorescence of a Locked Green Fluorescent Protein Chromophore Analogue, J. Am. Chem. Soc., 2020, 142, 738–749, DOI:10.1021/jacs.9b05096.
- X. Liu, A. Li, W. Xu, Z. Ma and X. Jia, An ESIPT-based fluorescent switch with AIEE, solvatochromism, mechanochromism and photochromism, Mater. Chem. Front., 2019, 3, 620–625, 10.1039/C8QM00633D.
- Y. Chen, Y. Fang, H. Gu, J. Qiang, H. Li, J. Fan, J. Cao, F. Wang, S. Lu and X. Chen, Color-Tunable and ESIPT-Inspired Solid Fluorophores Based on Benzothiazole Derivatives: Aggregation-Induced Emission, Strong Solvatochromic Effect, and White Light Emission, ACS Appl. Mater. Interfaces, 2020, 12, 55094–55106, DOI:10.1021/acsami.0c16585.
- L. Wang, M. Fujii, M. Namba, M. Yamaji and H. Okamoto, Fluorescence properties of amido-substituted 2,3-naphthalimides: Excited-state intramolecular proton transfer (ESIPT) fluorescence and responses to Ca2+ ions, Tetrahedron Lett., 2019, 60, 151189, DOI:10.1016/j.tetlet.2019.151189.
- S. H. Hwang and T. L. Choi, Iridium-Catalyzed Direct C-H Amidation Producing Multicolor Fluorescent Molecules Emitting Blue-to-Red Light and White Light, Org. Lett., 2020, 22, 2935–2940, DOI:10.1021/acs.orglett.0c00618.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Ando, T. Fujigaya and M. Ueda, Density Functional Theory Calculations of Photoabsorption Spectra of Organic Molecules in the Vacuum Ultraviolet Region, Jpn. J. Appl. Phys., 2002, 41, L105–L108, DOI:10.1143/JJAP.41.L105.
- S. Ando and M. Ueda, DFT calculations of photoabsorption spectra for alicyclic and heterocyclic compounds in the VUV region, J. Photopolym. Sci. Technol., 2003, 16, 537–544, DOI:10.2494/photopolymer.16.537.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122, DOI:10.1107/S0108767307043930.
- K. Togasaki, T. Arai and Y. Nishimura, Effect of Moderate Hydrogen Bonding on Tautomer Formation via Excited-State Intermolecular Proton-Transfer Reactions in an Aromatic Urea Compound with a Steric Base, J. Phys. Chem. A, 2020, 124, 6617–6628, DOI:10.1021/acs.jpca.0c05045.
- P. Zhou, M. R. Hoffmann, K. Han and G. He, New insights into the dual fluorescence of methyl salicylate: Effects of intermolecular hydrogen bonding and solvation, J. Phys. Chem. B, 2015, 119, 2125–2131, DOI:10.1021/jp501881j.
- V. V. Shynkar, A. S. Klymchenko, E. Piémont, A. P. Demchenko and Y. Mély, Dynamics of Intermolecular Hydrogen Bonds in the Excited States of 4‘-Dialkylamino-3-hydroxyflavones. On the Pathway to an Ideal Fluorescent Hydrogen Bonding Sensor, J. Phys. Chem. A, 2004, 108, 8151–8159, DOI:10.1021/jp047990l.
- A. J. G. Strandjord, P. F. Barbara and P. Sloan, Proton-Transfer Kinetics of 3-Hydroxyflavone: Solvent Effects the actual charge, 1985, pp. 2355–2361 DOI:10.1021/j100257a041.
- R. Das, A. S. Klymchenko and M. Yves, Unusually slow proton transfer dynamics of a 3-hydroxychromone dye in protic solvents†, Photochem. Photobiol. Sci., 2009, 1583–1589, 10.1039/b906710h.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2067299. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00308a |
| This journal is © The Royal Society of Chemistry 2021 |