
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalide-driven formation of lead halide perovskites: insight from ab initio molecular dynamics simulations†
Waldemar
Kaiser
 ab,
Edoardo
Mosconi
*ac,
Asma A.
Alothman
c,
Daniele
Meggiolaro
a,
Alessio
Gagliardi
b and
Filippo
De Angelis
*ade
ab,
Edoardo
Mosconi
*ac,
Asma A.
Alothman
c,
Daniele
Meggiolaro
a,
Alessio
Gagliardi
b and
Filippo
De Angelis
*ade
aComputational Laboratory for Hybrid/Organic Photovoltaics (CLHYO), Istituto CNR di Scienze e Tecnologie Chimiche “Giulio Natta” (CNR-SCITEC), Via Elce di Sotto 8, 06123 Perugia, Italy. E-mail: edoardo@thch.unipg.it; filippo@thch.unipg.it
bDepartment of Electrical and Computer Engineering, Technical University of Munich, Arcisstrasse 21, 80333 Munich, Germany
cChemistry Department, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
dDepartment of Chemistry, Biology and Biotechnology, University of Perugia, Via Elce di Sotto 8, 06123 Perugia, Italy
eCompuNet, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy
First published on 4th May 2021
Abstract
Controlling the crystallization mechanism of metal halide perovskites is of utmost importance to grow defect-less perovskite layers for efficient solar cells and optoelectronic devices. Despite its relevance, there is a lack of microscopic understanding of the nucleation and crystallization processes during the formation of the perovskite phase from its precursors. To unveil the electronic and atomistic features of this process we carry out ab initio molecular dynamics simulations on a model system which consists of a stoichiometric layered lead iodide (PbI2)·methylammonium iodide (MAI) structure, characteristic of intermediate phases observed in sequential deposition methods. Our results show clear evidence of halide-driven chemistry: MAI iodine ions attack lead ions in the PbI2 layers and cause a nucleophilic substitution of Pb–I bonds with a subsequent breaking of the PbI2 layer. Undercoordinated [PbIn]2−n complexes are initially formed which create the 3D perovskite framework mediated by additional nucleophilic attacks. The relatively fast rearrangement of [PbIn]2−n complexes followed by motion of MA cations limits the perovskite growth. Our results provide insight into the key steps of the perovskite formation on a microscopic scale, providing hitherto inaccessible details on the factors limiting the perovskite growth and on the effect of different halides on the kinetics of crystal formation.
1. Introduction
Metal halide perovskites (MHPs) have emerged in the last decade as the most promising material class for efficient solar energy conversion in photovoltaic devices. MHPs show favorable optoelectronic properties such as a direct band gap,1–3 large dielectric constants,4 low carrier effective masses,5,6 sharp optical absorption with low Urbach energies,7 and low non-radiative recombination.8,9 In combination with low-cost and large-scale processing techniques such as spin-coating from solution or vapor phase deposition, MHPs fulfill all requirements to compete with established photovoltaic technologies. A drawback of MHPs is their propensity to defect formation due to the weak metal-halide bonds.10,11 Related to this, variations in the local structure in form of grain boundaries or incomplete crystallization12,13 may give rise to accelerated degradation and loss of efficiency.The basic chemical reaction for MHP synthesis is surprisingly simple: mixing a solution of a metal−halide precursor (e.g. PbI2) with a halide salt (e.g. methylammonium iodide, hereafter MAI) leads to thin film formation by solvent evaporation. The formation process of the perovskite phase is, however, far more complex and it evolves through various intermediate phases.14,15 Controlling the reaction kinetics is crucial to form a homogeneous, defect-free photoactive layer for efficient solar cells. Within solution-processing techniques, dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) are commonly employed solvents to control the formation of highly crystalline perovskites.16,17 These coordinating solvents serve the function of dissolving the precursors and of forming iodoplumbate complexes (such as PbI2, PbI3− and PbI42− coordinated by various solvent molecules) which facilitate perovskite film formation.16,18–26 Interestingly, a strong correlation between the concentration of PbI42− species in the precursor solution and the density of charge recombination centers in the perovskite thin film has been reported.18 Further insight into the nucleation mechanism of MAPbI3 in solution was recently reported by Röthlisberger and coworkers.27 Using classical molecular dynamics they showed the presence of a PbI2-like phase made of clusters of Pb2+ and I− before any nucleation is observed. These clusters are finally converted into the perovskite phase by the diffusion of MA cations into the aggregates. MHPs can also be grown by sequential disposition methods, which first introduce PbI2 from solution and subsequently expose the pre-formed PbI2 film to a MAI solution.28 Alberti and coworkers reported that the crystallization of the perovskite phase from PbI2 occurs through layered intermediates that are formed upon MAI intercalation in PbI2.29 Song et al. observed crystalline precursor phases within spin-coated MAPbI3 films prepared using different metal-halide routes (PbI2, PbCl2), highlighting the importance of annealing duration to obtain defect-less perovskites.30 Grätzel and coworkers demonstrated an accelerated crystallization process by illumination during sequential deposition.31 This observation is linked to the compensation of surface traps within the PbI2 film by photo-generated holes, which improves the migration of the I− from MAI into the PbI2 film. Illuminated precursor solutions show an increased amount of high-order iodoplumbates which allows uniform perovskite film formation with low defect density and high reproducibility.32 Seok and coworkers observed a highly crystalline, self-organized MAI–PbI2–DMSO intermediate phase within fully solution-processed MAPb(I1−xBrx)3 perovskites by rapid intercalation of MAI and DMSO in between the weakly bonded PbI2 layers.33 Interestingly, such intermediate phases are also observed during thermally induced degradation of MAPbI3 perovskites.34 These studies further highlight the relevance of a stable intermediate phase for the formation of a uniform and dense perovskite layer upon thermal annealing.
A different route to perovskite formation is vapor-phase synthesis.35–40 Vapor-phase deposition techniques are of special interest as they allow large scale and volume production and are widely used in the semiconductor industry. In this case, the precursors are evaporated and the perovskite formation occurs by direct reaction. As early as in 1998 Era et al. fabricated layered perovskite structures by dual-source vapor deposition.41 They showed that vaporized organic ammonium iodide intercalates into the van der Waals gaps of vacuum-deposited PbI2 layers to form self-organizing perovskite layers. Sequential vapor-phase deposition facilitates the formation of highly uniform MHP films for efficient solar cells in situ by vapor deposition of MAI in vapor-deposited metal–halide films.38,39,42–44 Vapor-phase deposition has been recently used to fabricate wide-bandgap mixed-cation and mixed-halide perovskite films with suppressed halide segregation by simultaneous sublimation of four precursors.45 Perovskite crystallization by vapor-phase deposition occurs by a deconstruction-reconstruction mechanism due to high thermal energies, while solution-processed perovskites crystallize by a dissolution-reconstruction mechanism.46 Brenner et al. proposed that the crystallization process within vapor-phase deposition is initiated by the attack to PbI2 sheets by highly energetic MAI vapor that forms randomly oriented crystal seeds; additional energy (e.g. temperature or light) induces an entropy-driven reorganization of the octahedra during crystallization.46 The final morphology shows a strong orientational and structural relationship with the initial PbI2 crystallites. Intermediate techniques have also been experimented, such as the vapor-assisted solution synthesis.47,48
Despite the relevance of the perovskite crystal growth on the final material optoelectronic quality there is still a lack of microscopic understanding of its fundamental mechanisms. Electronic and atomistic simulations may provide a powerful tool to gain insight into the fundamental aspects of chemical processes on an atomic scale. Understanding the key steps during the crystallization of the perovskite phase from the initial precursors can open new perspectives for improved control of the perovskite morphology for controlled materials properties. In this work, we report results from ab initio molecular dynamics simulations on the formation kinetics of MAPbI3 perovskite from MAI and PbI2 precursors. In particular, we focus the attention on the early stages of perovskite crystallization mechanism to provide a unique and experimentally inaccessible insight. We start from a layered structure of PbI2 intercalated with MAI in the absence of solvent. Through the dynamics, we provide insight into the early stages of the reaction kinetics during perovskite formation. We find in particular that the reaction is initiated and driven by a series of halide nucleophilic attacks to PbI2 layers, leading to formation of iodoplumbate complexes and breaking of the PbI2 network. Migration of the so formed iodoplumbate complexes bypasses the high activation energy related to Pb ion migration making lead available for the subsequent creation of vertical (i.e. across PbI2 layers) connectivity. The slower MA dynamics followed by reorientation of connected [PbIn]2−n polyhedral (not observed on our time scales) finishes the process and leads to perovskite formation. Our results allow us to gain insight into the factors limiting the perovskite growth, offering at the same time a view on the role of different halides (e.g. Cl and Br) as precursors and additives in the kinetics of crystal formation.
2. Model and computational details
Car–Parrinello molecular dynamics (CPMD)49 simulations have been carried out with Quantum Espresso50 using the GGA–PBE51 functional. Electron–ion interactions were described by scalar relativistic ultrasoft pseudopotentials with electrons from I 5s, 5p; N and C 2s, 2p; H 1s; and Pb 6s, 6p, 5d shells explicitly included. Plane-wave basis set cutoffs for the smooth part of the wave functions and the augmented density were 25 and 200 Ry, respectively. CPMD simulations have been performed using an integration time step of 10 au for a total simulation time of ca. 250 ps. The fictitious masses of the electronic degrees of freedom are set to 1000 au. We set the atomic masses of all atoms to an identical value of 5 amu to enhance the dynamical sampling during the CPMD simulations. The atomic positions are randomized initially to reach a temperature of 450 ± 30 K without further applying any thermostat.To properly define the PbI2·MAI we initially modelled the isolated PbI2 and MAI species adopting the experimental P3m1 structure52 for PbI2 and the P4/nmm:2 structure for MAI.53 The initial configuration is made of a layered PbI2·MAI structure of two crystalline PbI2-layers intercalated with MAI layers with a stoichiometric 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (see Fig. S1, ESI†). Then we increase the distance between the PbI2 layers and insert MAI with PbI2:MAI = 1:1 stoichiometry. The supercell consists of 8 Pb ions, 8 MA molecules, and 24 I ions in total with a = b = 9.1 Å, c = 28.47 Å and γ = 120°. Throughout the manuscript, we distinguish between iodide ions originating from MAI, labeled as I*, and iodide ions originating from PbI2, labeled as I.
:1 ratio (see Fig. S1, ESI†). Then we increase the distance between the PbI2 layers and insert MAI with PbI2:MAI = 1:1 stoichiometry. The supercell consists of 8 Pb ions, 8 MA molecules, and 24 I ions in total with a = b = 9.1 Å, c = 28.47 Å and γ = 120°. Throughout the manuscript, we distinguish between iodide ions originating from MAI, labeled as I*, and iodide ions originating from PbI2, labeled as I.
3. Results and discussion
To initiate the dynamics we chose a starting configuration made of PbI2 layers oriented along the 〈001〉 direction, which is the preferential growth direction of PbI2,46 intercalated by a stoichiometric MAI amount in its P4/nmm:2 crystal structure.53 This starting structure is mimicking the intermediate phase experimentally characterized by Alberti et al.29 and it can be associated with those obtained in sequential deposition methods (two-step method), whereby exposure of MAI solutions to deposited PbI2 layers leads to an intercalation of MAI in between PbI2 planes to form MA–Pb–I intermediates.29,54 This underlines that the initial configuration of our model is potentially close to intermediate situations encountered during perovskite synthesis.Fig. 1 visualizes snapshots of the key steps during the system dynamical evolution. We summarize here the main steps. Starting from the ordered PbI2·MAI arrangement, Fig. 1a, iodine ions readily migrate from MAI towards the PbI2 layers, with a consequent nucleophilic attack to Pb ions. The crystalline PbI2 layers break and PbIx complexes are formed close to the initial MAI positions, Fig. 1b. These initial fast halide-driven structural changes are followed by a slow rearrangement of the inorganic complexes and of the organic cations, Fig. 1c. A series of nucleophilic attacks combined with the slow rearrangement of the inorganic complexes transform the structure from the initially layered system towards a 3D perovskite framework, Fig. 1d. Additionally, the mutual orientation of the organic cations approaches the expected alignment in bulk MAPbI3, Fig. 1d.
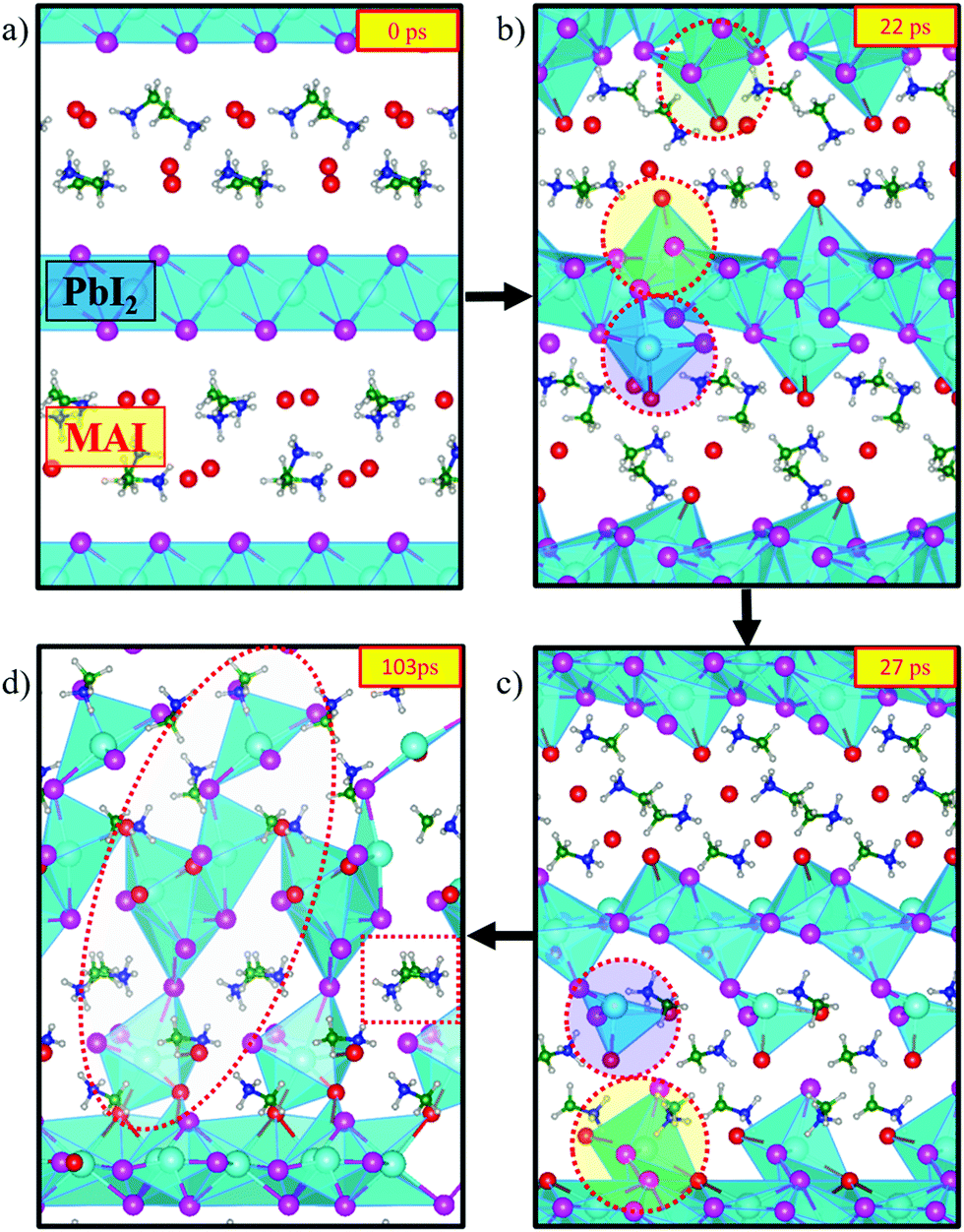 | ||
| Fig. 1 Time evolution of the MAPbI3 creation starting from a stochiometric, layered PbI2·MAI supercell from CPMD. The colors of the ions are as follows: Pb (cyan), iodide from PbI2 (purple), iodide from MAI (red), N (green), C (blue), H (gray). The supercell has been duplicated for improved visibility. Dotted regions correspond to selected areas of interest, see text. The VESTA software package has been used for visualization.55 | ||
We start our detailed analysis by investigating the mechanism that induces the breaking of the PbI2 layer. We distinguish iodine originating from PbI2 (hereafter I) from iodine originating from MAI (hereafter I*). Fig. 2a shows the temporal evolution of the average Pb/I coordination number, see Fig. S2 (ESI†) for individual Pb/I contributions. Within 25 ps of simulation time significant changes in both coordination numbers are observed. The Pb/I* coordination number (representing MAI iodine binding to lead) increases by ∼1 while the Pb/I coordination number simultaneously drops by ∼1. This is a clear indication of MAI iodine attacking the PbI2 layers, with simultaneous breaking of one Pb–I bond from PbI2. Basically, the electron-rich I* attacks the electrophilic lead center and replaces one Pb–I bond. The average Pb–I distance decreases substantially from the initial value of 5.6 Å to 3.2 Å (see Fig. S3, ESI†) and it approaches the expected Pb–I bond length of ∼3.19 Å of MAPbI3.56 This iodine attack breaks the crystalline PbI2 structure and lead-iodide complexes start to detach from the PbI2 layer, Fig. 2b. In the remainder of the trajectory, multiple changes in the number of coordinated I and I* for the individual Pb ions are observed (Fig. S2, ESI†). It is worth to analyze the PbI2 breaking by the nucleophilic attacks of I* to Pb ions for one specific Pb center, see representative structures in Fig. 2b. Each individual iodine ion is labeled consistently to capture the dynamics during the nucleophilic attacks. Initially, Pb is sixfold coordinated with I; the distance to the nearest I* ion, I*(A), is larger than 5 Å. Within 2 ps, we observe a first nucleophilic attack of a I*(A) to Pb. Simultaneously, the Pb–I(5) bond is broken. I*(A) attacks the positively charged Pb center, which reduces the Pb–I*(A) distance from 5.2 Å to 3.2 Å (see Fig. S4b, ESI†). This induces the breaking of the Pb–I(5) bond, as captured by the distance increase between Pb and I(5), see Fig. S4c (ESI†). At this point, the Pb ion is still within the starting PbI2 position. The first nucleophilic attack mainly causes structural disorder in the PbI2 layer while to complete breaking of the PbI2 layer a second nucleophilic attack is required. At ∼21 ps, I*(B) attacks the Pb ion, Fig. 2b; simultaneously, two Pb–I bonds, Pb–I(1) and Pb–I(4), are broken. The second nucleophilic attack eventually opens the structure and pulls the Pb center towards the region initially occupied by MAI. The structure now consists of undercoordinated Pb ions in the MAI region and excess iodine in the PbI2 region. The final snapshot in Fig. 2b visualizes the fivefold coordinated Pb ion entering the interlayer region.
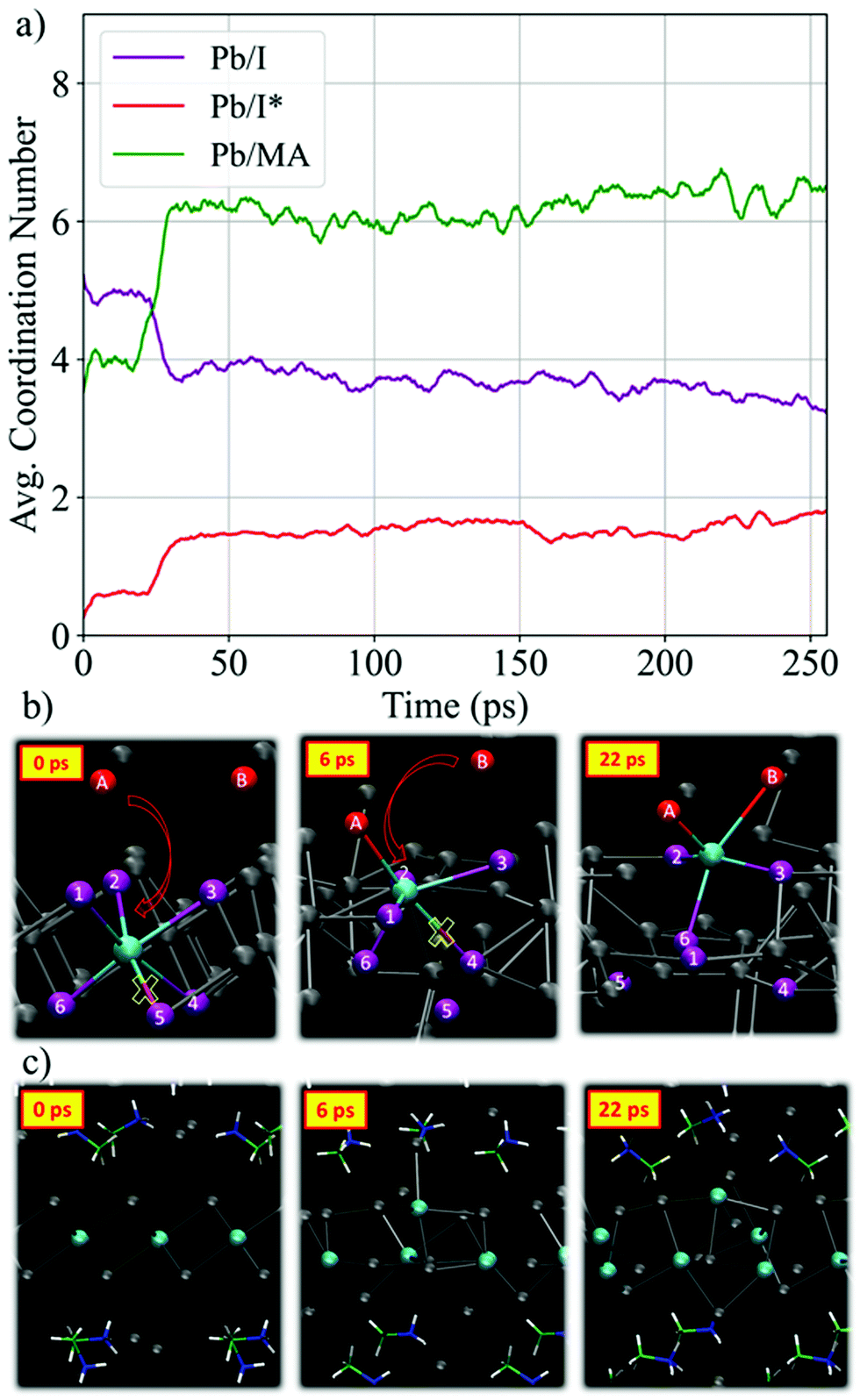 | ||
| Fig. 2 (a) Time evolution of the average Pb/I and Pb/MA coordination number using a cutoff distance of 3.7 Å and 7 Å, respectively. We distinguish coordination of Pb with PbI2 iodine (purple) and MAI iodine (red). All curves show moving averages; see Fig. S6 (ESI†), for raw data. (b) Snapshots showing the breaking of the PbI2 layer by the nucleophilic attack of MAI iodine for a particular Pb center. (c) Snapshots showing the motion of MA cations and Pb ions during the PbI2 breaking. | ||
It is also interesting to investigate the time evolution of MA cations motion in terms of Pb/MA coordination number, in Fig. 2a (see Fig. S5 (ESI†), for each individual Pb ion). The average coordination number increases within the first 30 ps from ∼3.5 to ∼6.5 but it remains considerably below 8, the expected value in MAPbI3, during the remaining trajectory. After breaking of the PbI2 layers, Pb ions move towards the starting MAI positions, while MA cations remain located roughly in their starting position, Fig. 2c. This reduces the distance between Pb and MA and explains the increase in Pb/MA coordination. These observations can be reasoned by two aspects: (i) complexation of metals with an s2 electron configuration, such as Pb2+, with halide ions was shown to occur easily with low activation energy;16,57 (ii) iodine from MAI has strong donor capabilities and readily forms Lewis adducts with PbI2.17,58 Additionally, it is well known that iodine ions have the lowest activation energy barriers for migration and consequently show higher diffusivity compared to MA cations.59 I* ions are fully negatively charged, while the effective charge of I in the PbI2-layers is partially screened due to the presence of the positively charged Pb centers. Thus, Pb tends to form new bonds with the I*. Such intercalation reactions of layered PbI2 with iodine ions have been proposed in the early years due to low activation energies.60
We now move to study the initial formation of the 3D perovskite framework. Fig. 3a visualizes the key steps of the reaction mechanism that forms the inorganic framework connecting the initial layers. In the first snapshot, additional nucleophilic attacks are highlighted. Starting at 22 ps, I* attacks lead ion Pb(A) and breaks the upper PbI2 layer. Additional formation and breaking of bonds of Pb(B) with iodine is observed. Subsequently, the inorganic complex formed by Pb(B) migrates into the interlayer region. Pb(B) remains bonded by one iodine ion to Pb(C) in the lower PbI2 region. Simultaneously, the distance between Pb(B) and Pb(A) decreases from initially 13 Å to 5 Å, Fig. 3b. The number of coordinated iodine to Pb(B), Fig. 3c, indicates that Pb(B)–I bonds are released and another Pb–I* bond is formed. At 32 ps, only two Pb–I bonds are present, while up to three Pb–I* bonds are formed. After 45 ps, the distances between the individual lead ions approach a value of ∼6.0 to 6.5 Å, which is close to Pb–Pb distance of ∼6.26 Å in the MAPbI3 perovskite.61 Two Pb(B)–I* bonds and three Pb(B)–I bonds are kept such that Pb(B) remains undercoordinated. The highlighted Pb ions bonded by iodine ions form roughly a straight line connecting the initial PbI2 regions, as known from the perovskite phase. Throughout the remaining trajectory the number of fivefold coordinated Pb ions remains roughly constant (see Fig. S7, ESI†). The analysis also visualizes the presence of fourfold coordinated Pb ions, which suggests the presence of iodine deficient regions. Also interesting, the Pb/MA coordination number increase from ∼6 to ∼8 concerted to the Pb-halide migration, Fig. 3c, indicating a local MA environment close to that of the perovskite product, see also Fig. S8 (ESI†).
 | ||
| Fig. 3 (a) Snapshots showing the formation of the 3D perovskite framework. Lead ions are consistently labeled to track their dynamics. (b) Temporal evolution of the distance between Pb ions. (c) Temporal evolution of the coordination number of Pb(B) ion with PbI2 iodine (purple) and with MAI iodine (red). The green curve shows the Pb(B)/MA coordination number. | ||
A salient feature revealed by our ab initio molecular dynamics simulations is the observation of Pb-migration taking place as Pb-halide complexes, not as individual Pb ions. Ionic migration of Pb2+ in the perovskite lattice (through a Pb-vacancy hopping pathway) has high activation energies,59 which may imply that Pb is the limiting element to perovskite growth. Our simulations show, however, that halide coordination to Pb allows facile migration pathways in the early stages of perovskite nucleation through the effective formation of [PbIn]2−n complexes, in line with the results reported by Kamat and coworkers.22,62 The Pb/I coordination strongly decreases before the Pb–halide complex enters the initial MAI region due to several nucleophilic attacks. In perovskite bulk, Pb-vacancies move along the side of the square formed by four Pb and four I ions (see ref. 59, Fig. 1c) being also undercoordinated during migration while the detailed Pb/I coordination was not reported.
The remaining questions to be answered are how close our structure gets to the perovskite phase and, consequently, what factors may limit the perovskite growth. As already noted above, the average Pb/I coordination number remains below the expected value of 6, which means that undercoordinated Pb ions remain present throughout the simulation. In the final 50 ps of the trajectory, the Pb/I* coordination number approaches 2, with a simultaneous decrease in the Pb/I coordination number to 3. These changes are also visible in the time evolution of the I/Pb coordination number (see Fig. S6c, ESI†). The average number of Pb ions bonded to I* shows a notable increase to a value of 2 during the final 50 ps, while the number of Pb ions bonded to I slowly drops towards 1. This change in coordination is mediated by the migration of iodine ions as visible by the jumps in the time evolution of the Pb/I and Pb/I* coordination number for the individual Pb ions, Fig. S2 (ESI†). Undercoordinated iodine is mainly located in the starting MAI region, while undercoordinated Pb is observed within both the MAI and the PbI2 region. Undercoordinated iodine indicates the presence of lead vacancies during the structural evolution. Lead and iodine vacancies belong to the most stable defects in all growth-conditions.11,63 In the 2-charge state, lead vacancies provide shallow trap states for holes, with high migration barriers.63 As iodine ions from MAI initialize the crystallization process and fully coordinate with Pb ions, an excess of MAI may improve both the crystallization and reduce the amount of undercoordinated Pb. This agrees with experimental studies, which report increased coordination of Pb ions with iodine at increased MAI concentration.21 This might be further improved with the usage of hydriodic acid (HI) as additive.21
To understand the limiting factor in the perovskite growth, we analyze the radial distribution functions (RDF) between the different fragments and compare them to the RDF of the orthorhombic phase of MAPbI3.64Fig. 4 shows the cumulative RDF for (a) Pb–I, (b) Pb–MA, (c) MA–MA, and (d) Pb–Pb; the corresponding RDFs are shown in Fig. S10 (ESI†). The RDF of Pb–I (Fig. S9, ESI†) shows an increased signal for Pb–I* and a simultaneous decrease for Pb–I at 3.2 Å, capturing the nucleophilic substitution. The Pb–I RDF, Fig. 4a, shows slightly lower coordination than that of bulk perovskite at the typical Pb–I bond distance of 3.2 Å, with the remaining signal at ∼3 Å due to the remaining PbI2-rich regions in the initial PbI2 position. The Pb–MA RDF changes within the first 25 ps approaching the expected RDF of MAPbI3 but it remains quite far apart from the perovskite one, Fig. 4b. Interestingly, the MA–MA and Pb–Pb RDFs, Fig. 4c and d, show slow changes in the remaining trajectory after a fast change in the early simulation stages. These RDFs show an intermediate phase between the initial PbI2·MAI and the perovskite phase that is made of a PbI2-rich region in the initial PbI2 position and the 3D perovskite framework forming across the PbI2-layers (see also Fig. 1d).
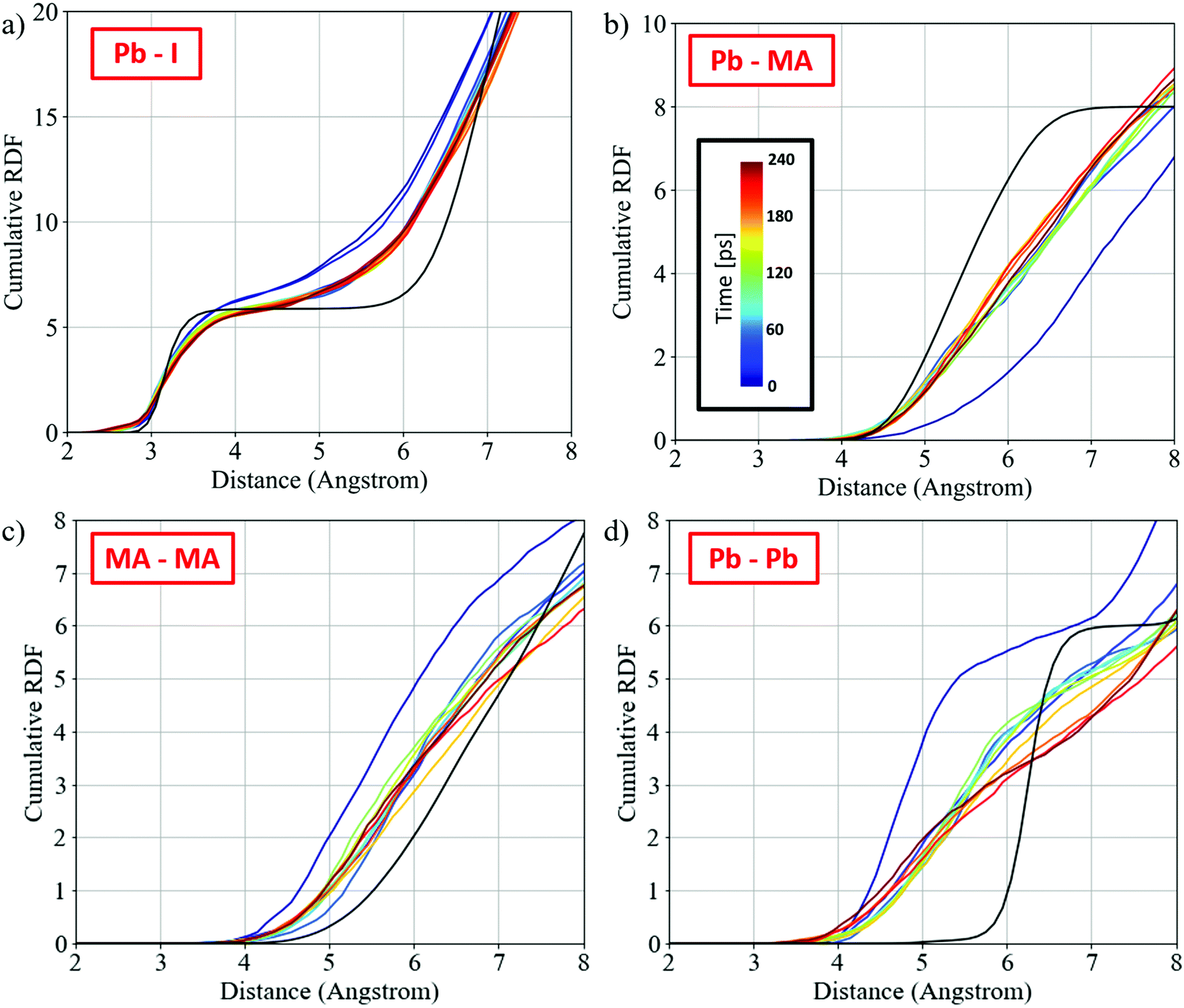 | ||
| Fig. 4 Cumulative radial distribution function (RDF) for (a) Pb–I, (b) Pb–MA, (c) MA–MA, and (d) Pb–Pb. Nitrogen has been used as a reference for the MA cation. The black curves show the expected cumulative RDF for the orthorhombic phase of MAPbI3. The colors capture the time information of the RDF: time increases from blue to red (see color scale in (b)). All curves show averages across ∼25 ps time slices. | ||
To reach the perovskite phase, MA and Pb ions need to rearrange within the structure. Both components have higher activation energies than I ions,59 which may slow down the rearrangement limiting in turn the perovskite growth. As initially shown in Fig. 1, undercoordinated Pb ions rearrange following several nucleophilic attacks to form the 3D perovskite framework across the starting MAI region. This process only requires hundreds of ps and it occurs inhomogeneously. Thus, regions with faster perovskite growth are observed. Previously, Röthlisberger and coworkers demonstrated a slow transition of edge-sharing octahedra to the perovskite corner-sharing octahedra after 180 ns by metadynamics.27 This slow dynamical evolution testify the incomplete perovskite formation observed at our time scales, pointing at a potentially significant effect of post-synthetic annealing treatment on the perovskite crystal and optoelectronic quality.65
A mechanistic picture of the perovskite crystallization from one-step solution processing was discussed by Röthlisberger and coworkers.27 Their main findings show that nucleation starts by aggregates of Pb2+ and I− ions which evolve into clusters of edge-sharing [PbI6]4− octahedra, which are comparable to the PbI2 layers in the two-step deposition method (see Fig. 1a). The transformation from these edge-sharing clusters to corner-sharing perovskites was shown to occur by MA+ cation diffusion into the Pb–I clusters, while details of reaction chemistry of the [PbI6]4− complexes during perovskite crystallization were not addressed. Our findings add a novel perspective on the role of the halide on the perovskite nucleation and present unexpected fast halide-mediated migration of [PbIn]2−n complexes. Moreover, the observation of undercoordinated Pb centers during perovskite crystallization is of utmost importance to accurately engineer the precursor composition to obtain defect-free perovskites.
Scheme 1 summarizes the main mechanistic findings of our study. The initial stage of perovskite formation is the nucleophilic attack of reactive iodine ions from MAI to PbI2, Scheme 1a, leading to breaking of the edge-sharing connectivity and detachment of PbIn polyhedra from PbI2, Scheme 1b. Migration of PbIn polyhedra then occurs, bridging different PbI2 layers and creating out of plane connectivity, Scheme 1c. Slow polyhedral rearrangement, not captured by our simulations, eventually leads to 3D perovskite formation, Scheme 1d.
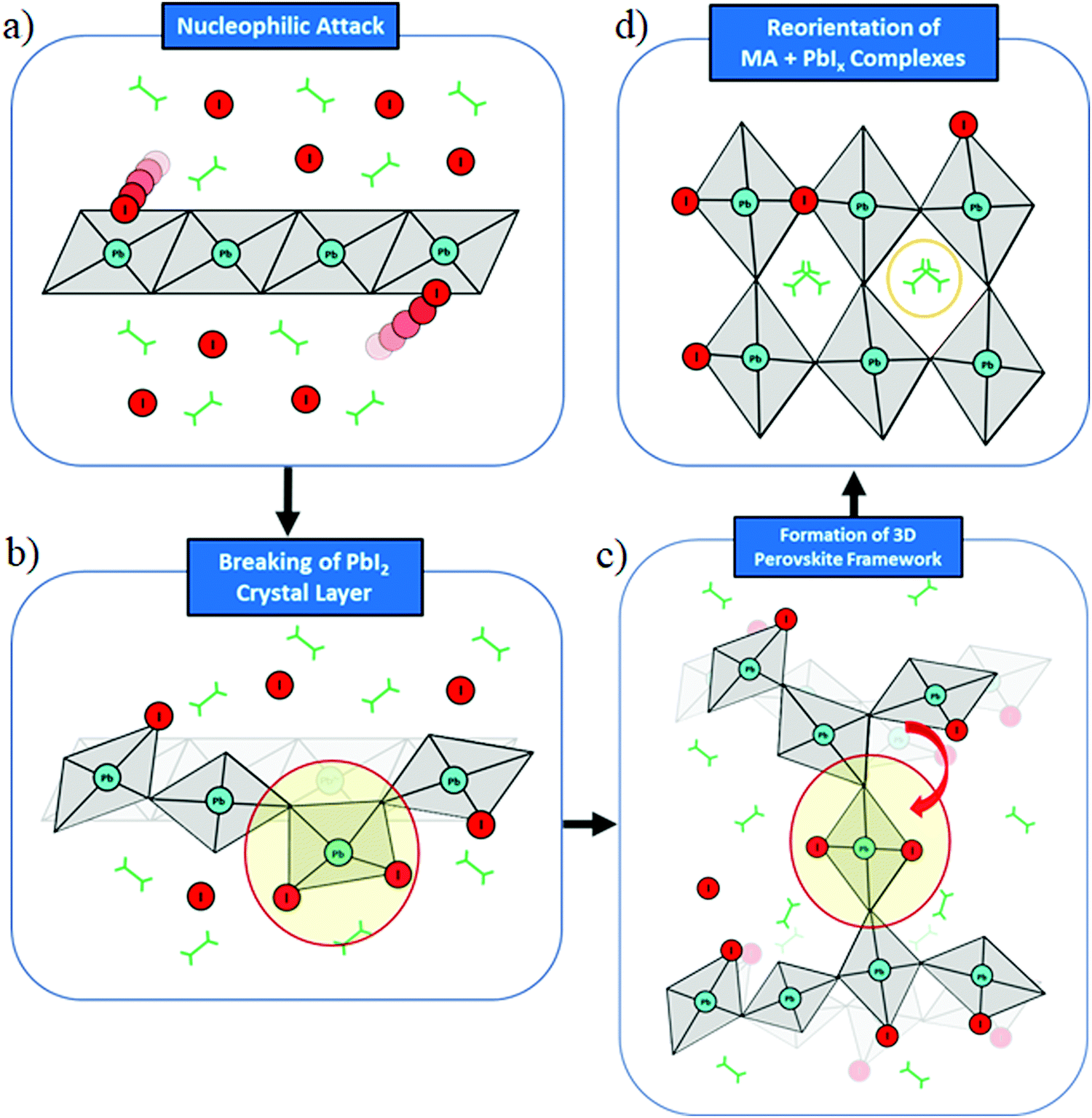 | ||
| Scheme 1 Schematic representation of the main steps in the formation of methylammonium lead iodide perovskite from MAI and PbI2 precursors. | ||
The mechanism highlighted by our ab initio molecular dynamics simulations highlights some critical factors potentially influencing the defect chemistry associated to the growing crystal. The crystal growth is activated and driven by halides. The nucleophilic attack is rather fast, as well as the formation of Pb–I bonds to form bridges between PbI2 layers. Halides other than iodine can have significant effect on the kinetics of this initial perovskite formation step. Typically using chlorine in precursors or as an additive together with iodine was shown to have a beneficial effect on the lead-iodide perovskite growth.66–69 Chlorine is typically not retained into the crystal lattice of lead-iodide perovskites beyond a few % due to the limited miscibility of Cl and I.70 Thus its effect on the final lead-iodide perovskite quality is mainly related to crystal growth kinetics.66
Based on our mechanistic analysis we can speculate on what the effect of different precursor conditions is on the perovskite growth. Referring to the standard stoichiometric perovskite synthesis (eqn (1) below)
| PbI2 + MAI → MAPbI3 | (1) |
MAI rich conditions can activate PbI2 exfoliation and the formation of 3D network, improving kinetics of crystallization. As explained above, a slow step in our mechanism is the reorganization of iodoplumbate clusters to form 3D framework. To this aim, the incorporation of MA in the forming complex cage is essential. If the formation of the 3D inorganic network is quite fast (all is driven by iodine), it is likely that the slow motion of MA cations could determine the defect chemistry in the early stage of perovskite formation. In the limited timescale of dynamics only Pb and I species are moving to build the 3D network, while evidently MA cations rearrange in longer timescales. The limited availability of MA cations can be at the origin of the low coordination of Pb (n = 5) in the MAI interlayer, due to lack of positive counter-charge. This slow MA motion can potentially lead to the formation of MAI-poor and MAI-rich portions, and overall a high density of I and MA Frenkel defects in the crystal.11 Slowing crystallization through stronger lead coordinating solvents may help to keep stoichiometry during cluster formation and reduce the disorder.
With reference to the use of different halides (e.g. Br and Cl) as precursors or additives to lead-iodide perovskites synthesis, two different situations related to the “chlorine route” can be highlighted, corresponding to using a PbCl2 salt as lead source (eqn (2) below) or a MACl additive as chlorine source (eqn (3) below):
| PbCl2 + 3MAI → MAPbI3 + 2MACl | (2) |
| PbI2 + MACl + MAI → MAPbI3 + MACl | (3) |
In (1) iodine is both the nucleophile and the leaving group. In other words, iodine is both the attacking and leaving species, with iodine coming from MAI more nucleophilic (thus more active) than iodine from PbI2, as discussed above.
In (2) Cl (and to a lesser extent Br) form stronger bonds with Pb in PbCl2 than iodine. At the same time Cl (and Br) is generally a worse leaving group than iodine due to its smaller size and poor solvation in organic solvents. We thus expect reaction (2) to be significantly slower than (1) in all the four schematic reaction steps highlighted in Scheme 1. This is consistent with early observations by Jen and coworkers.66 Stone et al. showed that this chlorine route forms a stable precursor with slow crystallization which can be accurately controlled by gradual MACl evaporation.71
In (3) MACl may look as a spectator. Its role is, however, that of promoting Cl nucleophilic attack to PbI2 due to its stronger donor capability compared to iodine and the formation of stronger Pb–Cl bonds. Thus, the initial stage of PbI2 network breaking, (a) and (b) in Scheme 1, is facilitated likely leading to formation of mixed [PbClmIn]2−n−m complexes. The subsequent step of polyhedra migration with formation of the vertical perovskite connectivity, (c) in Scheme 1, should also be facilitated. Being a worse leaving group than iodine, the final chlorine expulsion to form MAPbI3, (d) in Scheme 1, is definitely slowed down, overall leading to a slower and likely more effective crystal growth. Consistently, Xu et al. observed an increased reaction of PbI2 in presence of MACl into an intermediate MAPbI2Cl phase, while the conversion into MAPbI3 and MACl by adding MAI is strongly decelerated.72 MACl is stabilizing the intermediate phase which finally is converted to perovskite by thermal annealing, with simultaneous sublimation of residual MACl.58,73,74 Finally, Xie et al. observed a recrystallization process leading to vertical perovskite growth,75 and Kim et al. observed an improvement in morphology and phase crystallinity with increased grain size using MACl as precursor.76
4. Summary and conclusions
We have reported results from ab initio molecular dynamics simulations of the key steps of the formation of metal-halide perovskites. Our starting configuration is made of a layered structure of PbI2 and MAI, representing the intermediate phase likely encountered in sequential deposition techniques. Our analysis allows us to cast some insight into the initial steps of MAPbI3 formation. We find that the determining step to achieve a material with high crystallinity is related to breaking the PbI2 layers. The breaking of PbI2 is initiated by the nucleophilic attack of iodine ions coming from MAI to lead ions. After breaking the PbI2 layers, undercoordinated Pb ions in the form of iodoplumbate complexes migrate into the starting MAI region and create the “vertical” interconnection between the initial PbI2 layers. Further nucleophilic attacks mediate the formation of this 3D perovskite framework. The limiting process in the perovskite growth is the slow rearrangement of the polyhedra and of the organic MA cations, which is reflected in slow changes in the radial distribution functions approaching (but not reaching, within the explored time scale) the final perovskite structure.Based on the proposed reaction mechanism we can expand our view to speculate on the mechanistic implications related to the use of different halides (e.g. Br and Cl) as precursors or additives to lead-iodide perovskites synthesis. Considering the chlorine effect in the lead source (e.g. PbCl2) or as an additive (e.g. MACl) the proposed reaction mechanisms implies in both cases a slower kinetics due to breaking stronger Pb–Cl bonds in different reaction stages. Such knowledge should be helpful to deepen our control on perovskite synthesis, leading to further optimized materials in terms of stability and optoelectronic quality.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 764047 of the ESPRESSO project. This work was also supported by the Distinguished Scientist Fellowship Program (DSFP) of King Saud University, Riyadh, Saudi Arabia. This research was funded by PON Project “Tecnologia per celle solari bifacciali ad alta Efficienza a 4 terminali per utility scale (BEST-4U), of the Italian Ministry MIUR (CUP B88D19000160005)”. W. K. and A. G. acknowledge the TUM International Graduate School of Science and Engineering (IGSSE, GSC 81) by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) for funding. The Ministero dell'Istruzione dell'Università e della Ricerca (MIUR) and Università degli Studi di Perugia are acknowledged for financial support through the program “Dipartimenti di Eccellenza 2018–2022” (Grant AMIS) to F. D. A.References
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Lead iodide perovskite sensitized all-solid-state submicron thin film mesoscopic solar cell with efficiency exceeding 9%, Sci. Rep., 2012, 2, 591, DOI:10.1038/srep00591
.
- Y. H. Chang, C. H. Park and K. Matsuishi, First-Principles Study of the Structural and the Electronic Properties of the Lead-Halide-Based Inorganic–Organic Perovskites (CH3NH3)PbX3 and CsPbX3 (X = Cl, Br, I), J. Korean Phys. Soc., 2004, 44(4), 889–893 CAS
- P. Umari, E. Mosconi and F. De Angelis, Relativistic GW calculations on CH3NH3PbI3 and CH3NH3SnI3 perovskites for solar cell applications, Sci. Rep., 2014, 4, 4467, DOI:10.1038/srep04467
- E. J. Juarez-Perez, R. S. Sanchez, L. Badia, G. Garcia-Belmonte, Y. S. Kang, I. Mora-Sero and J. Bisquert, Photoinduced Giant Dielectric Constant in Lead Halide Perovskite Solar Cells, J. Phys. Chem. Lett., 2014, 5(13), 2390–2394, DOI:10.1021/jz5011169
- F. Brivio, K. T. Butler, A. Walsh and M. van Schilfgaarde, Relativistic quasiparticle self-consistent electronic structure of hybrid halide perovskite photovoltaic absorbers, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89(15), 155204, DOI:10.1103/PhysRevB.89.155204
- K. Galkowski, A. Mitioglu, A. Miyata, P. Plochocka, O. Portugall, G. E. Eperon, J. T.-W. Wang, T. Stergiopoulos, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Determination of the exciton binding energy and effective masses for methylammonium and formamidinium lead tri-halide perovskite semiconductors, Energy Environ. Sci., 2016, 9(3), 962–970, 10.1039/C5EE03435C
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, Organometallic Halide Perovskites: Sharp Optical Absorption Edge and Its Relation to Photovoltaic Performance, J. Phys. Chem. Lett., 2014, 5(6), 1035–1039, DOI:10.1021/jz500279b
- M. B. Johnston and L. M. Herz, Hybrid Perovskites for Photovoltaics: Charge-Carrier Recombination, Diffusion, and Radiative Efficiencies, Acc. Chem. Res., 2016, 49(1), 146–154, DOI:10.1021/acs.accounts.5b00411
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber, Science, 2013, 342(6156), 341–344, DOI:10.1126/science.1243982
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D’Haen, L. D’Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. Angelis and H.-G. de; Boyen, Intrinsic Thermal Instability of Methylammonium Lead Trihalide Perovskite, Adv. Energy Mater., 2015, 5(15), 1500477, DOI:10.1002/aenm.201500477
- D. Meggiolaro and F. de Angelis, First-Principles Modeling of Defects in Lead Halide Perovskites: Best Practices and Open Issues, ACS Energy Lett., 2018, 3(9), 2206–2222, DOI:10.1021/acsenergylett.8b01212
- J.-P. Correa-Baena, M. Anaya, G. Lozano, W. Tress, K. Domanski, M. Saliba, T. Matsui, T. J. Jacobsson, M. E. Calvo, A. Abate, M. Grätzel, H. Míguez and A. Hagfeldt, Unbroken Perovskite: Supp Inf, Adv. Mater., 2016, 28(25), 5031–5037, DOI:10.1002/adma.201600624
- N. Phung and A. Abate, The Impact of Nano- and Microstructure on the Stability of Perovskite Solar Cells, Small, 2018, 14(46), e1802573, DOI:10.1002/smll.201802573
- S. Sanchez, U. Steiner and X. Hua, Phase Evolution During Perovskite Formation—Insight from Pair Distribution Function Analysis, Chem. Mater., 2019, 31(9), 3498–3506, DOI:10.1021/acs.chemmater.9b00748
- J. Zhang, Z. Wang, A. Mishra, M. Yu, M. Shasti, W. Tress, D. J. Kubicki, C. E. Avalos, H. Lu, Y. Liu, B. I. Carlsen, A. Agarwalla, Z. Wang, W. Xiang, L. Emsley, Z. Zhang, M. Grätzel, W. Guo and A. Hagfeldt, Intermediate Phase Enhances Inorganic Perovskite and Metal Oxide Interface for Efficient Photovoltaics, Joule, 2020, 4(1), 222–234, DOI:10.1016/j.joule.2019.11.007
- J. S. Manser, M. I. Saidaminov, J. A. Christians, O. M. Bakr and P. V. Kamat, Making and Breaking of Lead Halide Perovskites, Acc. Chem. Res., 2016, 49(2), 330–338, DOI:10.1021/acs.accounts.5b00455
- N. Ahn, D.-Y. Son, I.-H. Jang, S. M. Kang, M. Choi and N.-G. Park, Highly Reproducible Perovskite Solar Cells with Average Efficiency of 18.3% and Best Efficiency of 19.7% Fabricated via Lewis Base Adduct of Lead(II) Iodide, J. Am. Chem. Soc., 2015, 137(27), 8696–8699, DOI:10.1021/jacs.5b04930
- R. J. Stewart, C. Grieco, A. V. Larsen, G. S. Doucette and J. B. Asbury, Molecular Origins of Defects in Organohalide Perovskites and Their Influence on Charge Carrier Dynamics, J. Phys. Chem. C, 2016, 120(23), 12392–12402, DOI:10.1021/acs.jpcc.6b03472
- K. Yan, M. Long, T. Zhang, Z. Wei, H. Chen, S. Yang and J. Xu, Hybrid halide perovskite solar cell precursors: colloidal chemistry and coordination engineering behind device processing for high efficiency, J. Am. Chem. Soc., 2015, 137(13), 4460–4468, DOI:10.1021/jacs.5b00321
- Q. Hu, L. Zhao, J. Wu, K. Gao, D. Luo, Y. Jiang, Z. Zhang, C. Zhu, E. Schaible, A. Hexemer, C. Wang, Y. Liu, W. Zhang, M. Grätzel, F. Liu, T. P. Russell, R. Zhu and Q. Gong, In situ dynamic observations of perovskite crystallisation and microstructure evolution intermediated from PbI64- cage nanoparticles, Nat. Commun., 2017, 8, 15688, DOI:10.1038/ncomms15688
- A. Sharenko, C. Mackeen, L. Jewell, F. Bridges and M. F. Toney, Evolution of Iodoplumbate Complexes in Methylammonium Lead Iodide Perovskite Precursor Solutions, Chem. Mater., 2017, 29(3), 1315–1320, DOI:10.1021/acs.chemmater.6b04917
- K. G. Stamplecoskie, J. S. Manser and P. V. Kamat, Dual nature of the excited state in organic–inorganic lead halide perovskites, Energy Environ. Sci., 2015, 8(1), 208–215, 10.1039/C4EE02988G
- E. Radicchi, E. Mosconi, F. Elisei, F. Nunzi and F. De Angelis, Understanding the Solution Chemistry of Lead Halide Perovskites Precursors, ACS Appl. Energy Mater., 2019, 2(5), 3400–3409, DOI:10.1021/acsaem.9b00206
- J. C. Hamill, J. Schwartz and Y.-L. Loo, Influence of Solvent Coordination on Hybrid Organic–Inorganic Perovskite Formation, ACS Energy Lett., 2018, 3(1), 92–97, DOI:10.1021/acsenergylett.7b01057
- M. Jung, S.-G. Ji, G. Kim and S. I. Seok, Perovskite precursor solution chemistry: from fundamentals to photovoltaic applications, Chem. Soc. Rev., 2019, 48(7), 2011–2038, 10.1039/c8cs00656c
- B. Li, D. Binks, G. Cao and J. Tian, Engineering Halide Perovskite Crystals through Precursor Chemistry, Small, 2019, 15(47), 1903613, DOI:10.1002/smll.201903613
- P. Ahlawat, M. I. Dar, P. Piaggi, M. Grätzel, M. Parrinello and U. Rothlisberger, Atomistic Mechanism of the Nucleation of Methylammonium Lead Iodide Perovskite from Solution, Chem. Mater., 2020, 32(1), 529–536, DOI:10.1021/acs.chemmater.9b04259
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Sequential deposition as a route to high-performance perovskite-sensitized solar cells, Nature, 2013, 499(7458), 316–319, DOI:10.1038/nature12340
- G. Pellegrino, S. D’Angelo, I. Deretzis, G. G. Condorelli, E. Smecca, G. Malandrino, A. La Magna and A. Alberti, From PbI2 to MAPbI3 through Layered Intermediates, J. Phys. Chem. C, 2016, 120(35), 19768–19777, DOI:10.1021/acs.jpcc.6b03798
- T.-B. Song, Z. Yuan, M. Mori, F. Motiwala, G. Segev, E. Masquelier, C. V. Stan, J. L. Slack, N. Tamura and C. M. Sutter-Fella, Revealing the Dynamics of Hybrid Metal Halide Perovskite Formation via Multimodal In Situ Probes, Adv. Funct. Mater., 2020, 30(6), 1908337, DOI:10.1002/adfm.201908337
- A. Ummadisingu, L. Steier, J.-Y. Seo, T. Matsui, A. Abate, W. Tress and M. Grätzel, The effect of illumination on the formation of metal halide perovskite films, Nature, 2017, 545(7653), 208–212, DOI:10.1038/nature22072
- S. Lee, S. Y. Lim, T. Kim, J. H. Lee, Y. Kim, H. Ahn, K. K. Hong, H. Cheong, H. Shin, D. H. Wang and J. H. Park, High-Valent Iodoplumbate-Rich Perovskite Precursor Solution via Solar Illumination for Reproducible Power Conversion Efficiency, J. Phys. Chem. Lett., 2021, 1676–1682, DOI:10.1021/acs.jpclett.0c03849
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Solvent engineering for high-performance inorganic–organic hybrid perovskite solar cells, Nat. Mater., 2014, 13(9), 897–903, DOI:10.1038/nmat4014
- N.-K. Kim, Y. H. Min, S. Noh, E. Cho, G. Jeong, M. Joo, S.-W. Ahn, J. S. Lee, S. Kim, K. Ihm, H. Ahn, Y. Kang, H.-S. Lee and D. Kim, Investigation of Thermally Induced Degradation in CH3NH3PbI3 Perovskite Solar Cells using In situ Synchrotron Radiation Analysis, Sci. Rep., 2017, 7(1), 4645, DOI:10.1038/s41598-017-04690-w
- M. Liu, M. B. Johnston and H. J. Snaith, Efficient planar heterojunction perovskite solar cells by vapour deposition, Nature, 2013, 501(7467), 395–398, DOI:10.1038/nature12509
- D. Pérez-Del-Rey, P. P. Boix, M. Sessolo, A. Hadipour and H. J. Bolink, Interfacial Modification for High-Efficiency Vapor-Phase-Deposited Perovskite Solar Cells Based on a Metal Oxide Buffer Layer, J. Phys. Chem. Lett., 2018, 9(5), 1041–1046, DOI:10.1021/acs.jpclett.7b03361
- J. Ávila, C. Momblona, P. Boix, M. Sessolo, M. Anaya, G. Lozano, K. Vandewal, H. Míguez and H. J. Bolink, High voltage vacuum-deposited CH3NH3PbI3–CH3NH3PbI3 tandem solar cells, Energy Environ. Sci., 2018, 11(11), 3292–3297, 10.1039/C8EE01936C
- J. Ávila, C. Momblona, P. P. Boix, M. Sessolo and H. J. Bolink, Vapor-Deposited Perovskites: The Route to High-Performance Solar Cell Production?, Joule, 2017, 1(3), 431–442, DOI:10.1016/j.joule.2017.07.014
- M. Sessolo, C. Momblona, L. Gil-Escrig and H. J. Bolink, Photovoltaic devices employing vacuum-deposited perovskite layers, MRS Bull., 2015, 40(8), 660–666, DOI:10.1557/mrs.2015.170
- L. K. Ono, M. R. Leyden, S. Wang and Y. Qi, Organometal halide perovskite thin films and solar cells by vapor deposition, J. Mater. Chem. A, 2016, 4(18), 6693–6713, 10.1039/C5TA08963H
- M. Era, K. Maeda and T. Tsutsui, Self-organization approach to organic/inorganic quantum-well based on metal halide-based layer perovskite, Thin Solid Films, 1998, 331, 285–290, DOI:10.1016/S0040-6090(98)00932-8
- C.-W. Chen, H.-W. Kang, S.-Y. Hsiao, P.-F. Yang, K.-M. Chiang and H.-W. Lin, Efficient and uniform planar-type perovskite solar cells by simple sequential vacuum deposition, Adv. Mater., 2014, 26(38), 6647–6652, DOI:10.1002/adma.201402461
- H. Hu, D. Wang, Y. Zhou, J. Zhang, S. Lv, S. Pang, X. Chen, Z. Liu, N. P. Padture and G. Cui, Vapour-based processing of hole-conductor-free CH3NH3PbI3 perovskite/C60 fullerene planar solar cells, RSC Adv., 2014, 4(55), 28964–28967, 10.1039/C4RA03820G
- S. Ngqoloda, C. J. Arendse, T. F. Muller, P. F. Miceli, S. Guha, L. Mostert and C. J. Oliphant, Air-Stable Hybrid Perovskite Solar Cell by Sequential Vapor Deposition in a Single Reactor, ACS Appl. Energy Mater., 2020, 3(3), 2350–2359, DOI:10.1021/acsaem.9b01925
- L. Gil-Escrig, C. Dreessen, F. Palazon, Z. Hawash, E. Moons, S. Albrecht, M. Sessolo and H. J. Bolink, Efficient Wide-Bandgap Mixed-Cation and Mixed-Halide Perovskite Solar Cells by Vacuum Deposition, ACS Energy Lett., 2021, 827–836, DOI:10.1021/acsenergylett.0c02445
- T. M. Brenner, Y. Rakita, Y. Orr, E. Klein, I. Feldman, M. Elbaum, D. Cahen and G. Hodes, Conversion of Single Crystalline PbI2 to CH3NH3PbI3: Structural Relations and Transformation Dynamics, Chem. Mater., 2016, 28(18), 6501–6510, DOI:10.1021/acs.chemmater.6b01747
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H.-S. Duan, H.-H. Wang, Y. Liu, G. Li and Y. Yang, Planar heterojunction perovskite solar cells via vapor-assisted solution process, J. Am. Chem. Soc., 2014, 136(2), 622–625, DOI:10.1021/ja411509g
- W. Soltanpoor, C. Dreessen, M. C. Sahiner, I. Susic, A. Z. Afshord, V. S. Chirvony, P. P. Boix, G. Gunbas, S. Yerci and H. J. Bolink, Hybrid Vapor-Solution Sequentially Deposited Mixed-Halide Perovskite Solar Cells, ACS Appl. Energy Mater., 2020, 3(9), 8257–8265, DOI:10.1021/acsaem.0c00686
- R. Car and M. Parrinello, Unified Approach for Molecular Dynamics and Density-Functional Theory, Phys. Rev. Lett., 1985, 55, 2471–2474, DOI:10.1103/PhysRevLett.55.2471
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. Gironcoli, S. de; Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21(39), 395502, DOI:10.1088/0953-8984/21/39/395502
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Semiconducting tin and lead iodide perovskites with organic cations: phase transitions, high mobilities, and near-infrared photoluminescent properties, Inorg. Chem., 2013, 52(15), 9019–9038, DOI:10.1021/ic401215x
- R. T. Wang, A. F. Xu, J. Y. Chen, L. W. Yang, G. Xu, V. Jarvis and J. F. Britten, Reversing Organic–Inorganic Hybrid Perovskite Degradation in Water via pH and Hydrogen Bonds, J. Phys. Chem. Lett., 2019, 10(22), 7245–7250, DOI:10.1021/acs.jpclett.9b02972
- R. Kang, J.-S. Yeo, H. J. Lee, S. Lee, M. Kang, N. Myoung, S.-Y. Yim, S.-H. Oh and D.-Y. Kim, Exploration of fabrication methods for planar CH3NH3PbI3 perovskite solar cells, Nano Energy, 2016, 27, 175–184, DOI:10.1016/j.nanoen.2016.06.052
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276, DOI:10.1107/S0021889811038970
- M. T. Weller, O. J. Weber, P. F. Henry, A. M. Di Pumpo and T. C. Hansen, Complete structure and cation orientation in the perovskite photovoltaic methylammonium lead iodide between 100 and 352 K, Chem. Commun., 2015, 51(20), 4180–4183, 10.1039/c4cc09944c
- O. Horváth and I. Mikó, Spectra, equilibrium and photoredox chemistry of tri- and tetraiodoplumbate(II) complexes in acetonitrile, J. Photochem. Photobiol., A, 1998, 114, 95–101, DOI:10.1016/S1010-6030(98)00214-7
- Z. Wang, Y. Zhou, S. Pang, Z. Xiao, J. Zhang, W. Chai, H. Xu, Z. Liu, N. P. Padture and G. Cui, Additive-Modulated Evolution of HC(NH2)2PbI3 Black Polymorph for Mesoscopic Perovskite Solar Cells, Chem. Mater., 2015, 27(20), 7149–7155, DOI:10.1021/acs.chemmater.5b03169
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. de Angelis, Defect migration in methylammonium lead iodide and its role in perovskite solar cell operation, Energy Environ. Sci., 2015, 8(7), 2118–2127, 10.1039/C5EE01265A
- O. E. Lanford and S. J. Kiehl, The Solubility of Lead Iodide in Solutions of Potassium Iodide-Complex Lead Iodide Ions, J. Am. Chem. Soc., 1941, 63(3), 667–669, DOI:10.1021/ja01848a010
- W.-W. Wang, J.-S. Dang, R. Jono, H. Segawa and M. Sugimoto, A first-principles prediction on the “healing effect” of graphene preventing carrier trapping near the surface of metal halide perovskites, Chem. Sci., 2018, 9(13), 3341–3353, 10.1039/c7sc04837h
- J. S. Manser, B. Reid and P. V. Kamat, Evolution of Organic–Inorganic Lead Halide Perovskite from Solid-State Iodoplumbate Complexes, J. Phys. Chem. C, 2015, 119(30), 17065–17073, DOI:10.1021/acs.jpcc.5b05898
- D. Meggiolaro, S. G. Motti, E. Mosconi, A. J. Barker, J. Ball, C. Andrea Riccardo Perini, F. Deschler, A. Petrozza and F. De Angelis, Iodine chemistry determines the defect tolerance of lead-halide perovskites, Energy Environ. Sci., 2018, 11(3), 702–713, 10.1039/C8EE00124C
- C. Quarti, E. Mosconi and F. De Angelis, Interplay of Orientational Order and Electronic Structure in Methylammonium Lead Iodide: Implications for Solar Cell Operation, Chem. Mater., 2014, 26(22), 6557–6569, DOI:10.1021/cm5032046
- Y. Yamada, M. Endo, A. Wakamiya and Y. Kanemitsu, Spontaneous Defect Annihilation in CH3NH3PbI3 Thin Films at Room Temperature Revealed by Time-Resolved Photoluminescence Spectroscopy, J. Phys. Chem. Lett., 2015, 6(3), 482–486, DOI:10.1021/jz5026596
- S. T. Williams, F. Zuo, C.-C. Chueh, C.-Y. Liao, P.-W. Liang and A. K.-Y. Jen, Role of chloride in the morphological evolution of organo-lead halide perovskite thin films, ACS Nano, 2014, 8(10), 10640–10654, DOI:10.1021/nn5041922
- K. H. Stone, A. Gold-Parker, V. L. Pool, E. L. Unger, A. R. Bowring, M. D. McGehee, M. F. Toney and C. J. Tassone, Transformation from crystalline precursor to perovskite in PbCl2-derived MAPbI3, Nat. Commun., 2018, 9(1), 3458, DOI:10.1038/s41467-018-05937-4
- N. D. Pham, V. T. Tiong, P. Chen, L. Wang, G. J. Wilson, J. Bell and H. Wang, Enhanced perovskite electronic properties via a modified lead(II) chloride Lewis acid–base adduct and their effect in high-efficiency perovskite solar cells, J. Mater. Chem. A, 2017, 5(10), 5195–5203, 10.1039/C6TA11139D
- L. Fan, Y. Ding, J. Luo, B. Shi, X. Yao, C. Wei, D. Zhang, G. Wang, Y. Sheng, Y. Chen, A. Hagfeldt, Y. Zhao and X. Zhang, Elucidating the role of chlorine in perovskite solar cells, J. Mater. Chem. A, 2017, 5(16), 7423–7432, 10.1039/C7TA00973A
- S. Colella, E. Mosconi, P. Fedeli, A. Listorti, F. Gazza, F. Orlandi, P. Ferro, T. Besagni, A. Rizzo, G. Calestani, G. Gigli, F. Angelis and R. de; Mosca, MAPbI3−xClx Mixed Halide Perovskite for Hybrid Solar Cells: The Role of Chloride as Dopant on the Transport and Structural Properties, Chem. Mater., 2013, 25(22), 4613–4618, DOI:10.1021/cm402919x
- K. H. Stone, A. Gold-Parker, V. L. Pool, E. L. Unger, A. R. Bowring, M. D. McGehee, M. F. Toney and C. J. Tassone, Transformation from crystalline precursor to perovskite in PbCl2-derived MAPbI3, Nat. Commun., 2018, 9(1), 3458, DOI:10.1038/s41467-018-05937-4
- Y. Xu, L. Zhu, J. Shi, S. Lv, X. Xu, J. Xiao, J. Dong, H. Wu, Y. Luo, D. Li and Q. Meng, Efficient hybrid mesoscopic solar cells with morphology-controlled CH3NH3PbI3−xClx derived from two-step spin coating method, ACS Appl. Mater. Interfaces, 2015, 7(4), 2242–2248, DOI:10.1021/am5057807
- C. Mu, J. Pan, S. Feng, Q. Li and D. Xu, Quantitative Doping of Chlorine in Formamidinium Lead Trihalide (FAPbI3−xClx) for Planar Heterojunction Perovskite Solar Cells., Adv. Energy Mater., 2017, 7(6), 1601297, DOI:10.1002/aenm.201601297
- V. Thampy and K. H. Stone, Solution-Phase Halide Exchange and Targeted Annealing Kinetics in Lead Chloride Derived Hybrid Perovskites, Inorg. Chem., 2020, 59(18), 13364–13370, DOI:10.1021/acs.inorgchem.0c01732
- F. Xie, C.-C. Chen, Y. Wu, X. Li, M. Cai, X. Liu, X. Yang and L. Han, Vertical recrystallization for highly efficient and stable formamidinium-based inverted-structure perovskite solar cells, Energy Environ. Sci., 2017, 10(9), 1942–1949, 10.1039/C7EE01675A
- M. Kim, G.-H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Methylammonium Chloride Induces Intermediate Phase Stabilization for Efficient Perovskite Solar Cells, Joule, 2019, 3(9), 2179–2192, DOI:10.1016/j.joule.2019.06.014
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00371b |
| This journal is © The Royal Society of Chemistry 2021 |